

Abstract
We describe a new method for encoded synthesis, efficient on-resin screening, and rapid unambiguous sequencing of combinatorial peptide libraries. An improved binary tag system for encoding peptide libraries during synthesis was designed to facilitate unequivocal assignment of isobaric residues by MALDI-TOF MS analysis. The improved method for encoded library synthesis was combined with a new versatile on-resin screening strategy that permitted multiple stages and types of screening to be employed successively on one library under mild conditions. The new method facilitated a combinatorial study of transglutaminase (TGase) enzyme substrate peptides, revealing new details of the effect of amino acid composition on TGase substrates. The approach was first demonstrated for an encoded library (130 321 compounds) of lysine pentapeptide substrates of TGase, synthesized using the “split-mix” method. The library was reacted on-resin with TGase enzyme and a soluble desthiobiotin labeled glutamine substrate. Initial screening was performed by adsorbing streptavidin-coated magnetic microparticles onto library beads, followed by magnetic separation. The differential binding affinities of desthiobiotin and biotin for streptavidin were exploited to release the magnetic microparticles and regenerate the desthiobiotin-labeled resin beads for further screening by flow-cytometry-based automated bead sorting, resulting in 345 beads that were sequenced by MALDI-TOF MS analysis. A second library consisted of encoded glutamine hexapeptide substrates, which was reacted on-resin with TGase enzyme and a soluble desthiobiotin-labeled cadaverine. Two-stage screening identified 267 glutamine peptides as TGase-reactive, of which 21 were further analyzed by solution-phase enzyme kinetics. Kinetic results indicated that the peptide PQQQYV from the library has a 68-fold greater substrate specificity than the best known glutamine substrate QQIV. The new encoding and screening strategies described here are expected to be broadly applicable to synthesis and screening of combinatorial peptide libraries in the future.
Combinatorial chemistry has become an important tool for new ligand identification and lead compound optimization since its introduction in 1991.1–3 However, on-resin screening and structure determination of library compounds on selected beads are still challenging tasks for the efficient application of this tool. In a typical on-resin screening assay, a library of perhaps millions of compound beads is screened against specific molecular targets, and individual positive beads are picked manually under a microscope using a pipet, a time-consuming and labor-intensive process. Structure determination of the compound on selected beads further adds to the experimental burden and in some cases may be inconclusive.4 To determine the structure of compounds on beads more efficiently and conclusively, several encoding methods have been developed.5,6 Generally, a chemical or physical tag is added during or after coupling of the library building block to the bead. These tags record the reaction history on individual beads and, hence, the structures of the compounds they encode. Chemical tags are released from the bead after biological screening and analyzed by a highly sensitive analytical technique, such as GC/MS, RP-HPLC, and MS.7–9 Such methods often require orthogonal chemistries for tagging and, therefore, additional synthetic steps as well as additional time to release and analyze the tags.
Another encoding method, the so-called ladder synthesis approach, has been reported for peptide and peptidomimetic libraries.10–12 In this approach, a small amount (~10 mol %) of a terminating or capping reagent is added into the coupling solution along with a building block during stepwise library synthesis, generating the full-length products plus a small percentage of sequence-truncated products. After screening, full-length products with the truncated fragments released from individual beads were analyzed by MALDI-TOF MS sequencing, providing the complete peptide sequence of the full-length product. However, with existing tag systems, it remains difficult to uniquely identify isobaric residues or residues with mass differences of a few mass units by MALDI-MS analysis.12
In this paper, we describe a new method for efficient synthesis, on-resin screening, and unambiguous structural identification of combinatorial peptide libraries. A new binary tag coding system for ladder synthesis was developed and demonstrated by construction of a pentapeptide library of lysine substrates for transglutaminase (TGase) enzyme. As illustrated in Figure 1, the lysine substrate library was reacted on-resin with TGase and a soluble desthiobiotin-labeled glutamine substrate, after which good substrate peptides were isolated by a new two-stage screening method involving magnetic separation followed by fluorescent separation of streptavidin-labeled beads. Rapid and unambiguous sequencing of selected beads by MALDI-TOF MS was facilitated by the binary tag coding system. We used a similar strategy to synthesize and screen an encoded glutamine hexapeptide library by reaction with a soluble desthiobiotin-labeled cadaverine. Two-stage screening of this library identified 21 glutamine peptides that were studied in greater detail by enzyme kinetics, which revealed that the peptide PQQQYV has a 68-fold-greater substrate specificity than the best-known glutamine substrate QQIV.13 Although this method is described for screening of peptide substrates of TGase, an important enzyme that is widely distributed in mammalian connective tissues, it is expected to be broadly applicable to rapid screening and identification of ligands from combinatorial libraries of peptides and other compounds.
Figure 1.

Schematic illustration of the strategy for on-resin enzyme reaction, library screening, and sequencing of selected beads.
EXPERIMENTAL SECTION
1. Lysine Peptide Library
Materials and General Methods
PEGA amine resin (0.2 mmol/g, 770 000–1 270 000 beads/g, 300–500 μm) was purchased from Polymer Laboratories Inc. (Amherst, MA). Fmoc-photocleavable linker, Fmoc-Val Wang resin (0.51 mmol/g), O-(N-Boc-2-aminoethyl)-O′-(N-diglycolyl-2-aminoethyl)-hexaethyleneglycol (Boc-OEG7 acid), O-(N-Fmoc-2-aminoethyl)-O′-(2-carboxyethyl)-undecaethylene glycol (Fmoc-OEG11 acid), and protected amino acids were purchased from Novabiochem (La Jolla, CA), except for Ac-Asp(OtBu)-OH, Ac-Dab(Boc)-OH, Ac-Gln(Trt)-OH and Boc-N-Me-Ile-OH, which were from Bachem (Torrance, CA). Benzotriazole-1-yloxytris(dimethylamino)-phosphonium hexafluorophosphate (BOP), N-hydroxybenzotriazole (HOBt), diisopropylethylamine (DIEA), and N-methylpyrrolidinone (NMP) were purchased from Advanced ChemTech (Louisville, KY). Transglutaminase from guinea pig liver, bovine serum albumin (BSA), MOPS, Tween-20, (+)-biotin and D-desthiobiotin were purchased from Sigma Chemical Company (St. Louis, MO). Piperidine and triisopropylsilane (TIS) were purchased from Aldrich (Milwaukee, WI). Acetonitrile was purchased from Burdick and Jackson. TFA was purchased from J. T. Baker. α-Cyano-4-hydroxycinnamic acid (CHA) was obtained from Fluka (Milwaukee, WI). MagnaBind Streptavidin beads (1–4 μm) were purchased from Pierce Biotechnology (Rockford, IL). Streptavidin MicroBeads (50 nm) were purchased from Miltenyi Biotec (Auburn, CA). SPHEROStreptavidin Magnetic Particles (8.0–9.9 μm) were purchased from Spherotech Inc. (Libertyville, IL). Rhodamine B-streptavidin was purchased from Molecular Probes (Eugene, Oregon). ESI-MS analysis of peptides was performed on a LCQ LC-MS system (Finnigan, Thermoquest, CA).
Library Synthesis
PEGA amine resin (300–500 μm, 1 g, 0.2 mmol) was washed with MeOH × 3 and NMP × 3. Fmoc-photocleavable linker (Fmoc-PCL) (312 mg, 0.6 mmol, 3 equiv), BOP (265 mg, 0.6 mmol), and HOBt (92 mg, 0.6 mmol) were dissolved in NMP (7 mL), followed by addition of DIEA (105 μL, 0.6 mmol). After 10 min at room temperature with occasional stirring, the solution was added into the resin and rocked overnight. A ninhydrin test showed the coupling reaction was complete. After removal of Fmoc in 20% piperidine in NMP for 20 min, NH2-OEG7-Pro-Tyr(tBu)-Phe-Arg(Pbf)-Val-PCL-PEGA resin was synthesized by Fmoc solid-phase peptide synthesis strategy. Each coupling reaction was performed for 30 min or 2 h for Boc-OEG7 acid with a 10-min preactivation of 5 equiv of Fmoc-amino acid or Boc-OEG7 acid/BOP/HOBt/DIEA (1:1:1:1) at room temperature. Fmoc was removed by 20% piperidine in NMP for 20 min, and Boc was removed by 10% TFA in DCM for 30 min × 2. After Boc removal, Fmoc-Gly and Boc-Gly (19:1) were coupled to the resin using the above coupling protocol.
The resin was suspended in methanol and dispensed evenly into 19 syringe reaction vessels of a Multiblock manual library synthesizer (CSPS Pharmaceuticals, CA). The resin was washed with methanol and NMP, and Fmoc was removed as described above. For the synthesis of position X4, each Fmoc amino acid (0.19 mmol, 20 equiv) (19 total) and its counterpart Boc-amino acid (0.01 mmol) or another Boc-amino acid (0.01 mmol) for the binary tag according to Table S2 (Supporting information) were dissolved in NMP (0.5 mL) and mixed with BOP (0.2 mmol) and HOBt (0.2 mmol) in NMP (0.5 mL), followed by addition of DIEA (0.2 mmol) in NMP (0.5 mL). After 10 min, each solution was added to one reaction vessel (19 total), and shaken for 2 h. The randomization step was carried out in methanol by mixing the resins from all 19 reaction vessels and then uniformly redistributing them back into the reaction vessels. The Fmoc protecting group was removed after the split step to prevent the resin beads from sticking to the cover of the synthesis apparatus during randomization. This procedure was repeated for syntheses of positions X3, X2, and X1. For the synthesis of lysine residue between positions X3 and X2, the same procedures as above were used, except that Fmoc-Lys(Boc)-OH was used in all of the reaction vessels and the randomization step was omitted. After removal of N-terminal Fmoc from position X1, the peptide resins were pooled and treated with TFA-TIS-H2O (95:2.5:2.5) overnight. The beads were washed with NMP, DCM, MeOH and H2O.
Synthesis of Desthiobiotin-OEG11-QQIV (DOQ)
The solid-phase synthesis of this conjugate was carried out manually on Fmoc-Val-Wang resin (0.5 g, 0.51 mmol/g) by an Fmoc strategy with a side-chain-unprotected Fmoc-Gln. Fmoc was removed in 20% piperidine in NMP. Fmoc-amino acid (1 mmol), BOP (442 mg, 1 mmol), and HOBt (135 mg, 1 mmol) were dissolved in NMP (5 mL) and followed by addition of DIEA (174 μL, 1 mmol). After 10 min, the solution was added to the resin and rocked for 60 min. Desthiobiotin and Fmoc-OEG11 acid were treated as Fmoc-amino acid, but the coupling reaction of Fmoc-OEG11 acid was rocked overnight. After completion of the synthesis, the resin was treated with TFA-TIS-H2O (96:2:2) for 30 min × 3. The crude product was obtained by concentration of the TFA solution and addition of diethyl ether. The crude product was purified using a semi-preparative C-18 RP-HPLC column (250 × 22 mm, 10 μm, Vydac) with an acetonitrile gradient of 14–30% in 120 min. ESI-MS of the purified product gave peaks at m/z 1282.6 and 1299.2.
On-Resin Enzymatic Reaction
The library beads were washed with glacial acetic acid × 3, H2O × 3, and 0.1 M MOPS buffer (pH 7.2) × 3 and drained. A 20-mL portion of 0.1 M MOPS buffer solution (pH 7.2) containing 20 mM CaCl2 and 2 mM DOQ was added to the beads and rocked for 10 min, followed by addition of 20 mL of 0.1 M MOPS buffer solution (pH 7.2) containing 2 mM EDTA, 20 mM DTT, and TGase (10 units). The reaction mixture was rocked at room temperature for 30 min and drained. The beads were washed with glacial acetic acid × 3, H2O × 3, MeOH × 3, H2O × 3, and PBS (pH 7.2) × 3.
Two-Stage Library Screening
The beads were washed with PBS (pH 7.2) × 3 and drained. PBS (pH 7.2) and 0.1% BSA were added to the resin beads to a total 30 mL of volume. Streptavidin-magnetic particles (2 mL) were added to the beads, and the mixture was incubated at room temperature for 2 h. PBS (pH 7.2) containing 0.3% BSA was added to dilute the resin beads and separated using a magnet (Magnetic Particle Concentrator, Dynal Biotech) × 3. Several sources of magnetic streptavidin microbeads (50 nm, Miltenyi Biotec; 4 μm, Pierce Biotechnology; 8.0–9.9 μm, Spherotech) were investigated. Preliminary experiments indicated that 8.8–9.0-μm streptavidin-magnetic particles specifically labeled biotin-PEGA resin beads but did not label biotin-free PEGA resin.
The magnetically selected beads (about 1/10 of the original volume) were treated with 50 mM biotin in PBS (pH 7.2) for 2 h and washed with H2O × 3, NMP × 3, H2O × 3, PBS (pH 7.2) × 3, 0.3% BSA in PBS (pH 7.2) × 3, 0.1% Tween-20 in H2O × 3, H2O× 3, PBS (pH 7.2) × 3, and drained. BSA (0.3%), 0.1% Tween-20 in PBS (pH 7.2) (10 mL), and 0.1 mg of rhodamine B-streptavidin were added to the beads and incubated at room temperature for 30 min. The beads were then washed with PBS (pH 7.2) × 3, 0.3% BSA in PBS × 3, and 0.1% Tween-20 in PBS × 3. The beads were diluted with 0.1% Tween-20 and 0.3% BSA in PBS and sorted using a COPAS bead sorter (Union Biometrica, MA) with a sheath solution of 0.1% Tween-20 in PBS at a rate of 30–40 beads per second. The selected beads were collected into BSA-treated 96-well plates. As negative controls, a small portion (~10 mg) of the beads not selected during magnetic screening were also treated as described above and imaged by fluorescence microscopy.
MALDI-TOF MS Analysis of a Single Bead
The individual beads collected by the COPAS bead sorter were selected according to their fluorescence intensity and washed with H2O. The MALDI-TOF mass spectra were obtained as follows: (1) The matrix solution was prepared by dissolving 10 mg of α-cyano-4-hydroxycinnamic acid (CHA) in 100 μL of 3% TFA in H2O, 300 μL of H2O, and 600 μL of acetonitrile. (2) A 2-μL portion of acetonitrile-H2O (1:1) was added to the sample spot of the MALDI-TOF sample plate containing a single bead and irradiated for 15 min using a BLAK-RAY LAMP (model UVL-56, long wave UV 365 nm, Upland, CA). (3) A 1-μL portion of matrix solution was applied onto the sample spot containing the bead and allowed to air-dry. MALDI-TOF MS analysis was performed on a Voyager-DE Pro mass spectrometer (PerSeptive Biosystems).
2. Glutamine Peptide Library
Library Synthesis and Screening
Details of the materials and general methods, library synthesis, synthesis of desthiobiotin-PEG600-cadaverine (DPC), on-resin enzymatic reaction, two-stage library screening, and an example MALDI-TOF MS analysis of a single glutamine library bead are described in the Supporting Information.
Parallel Synthesis of Selected Glutamine Peptide Substrates
Two-stage screening of the glutamine peptide library identified 267 glutamine peptides, of which 21 were selected for manual parallel synthesis by the standard Fmoc solid-phase peptide strategy on a Wang resin. In addition, three control peptides, acetyl-Gly-Gln-Gln-Gln-Leu-Gly (Q3);14 Gln-Gln-Ile-Val peptide (Q2)13 from fibronectin; and a gliadin protein peptide, Pro-Gln-Gln-Gln-Phe-Pro (PQ3),15 were synthesized. Purification of peptides was performed using a Waters HPLC system (Waters, Milford, MA) on a Vydac 218TP C18 reversed-phase column (250 × 22 mm, 10 μm) with a gradient of acetonitrile in water containing 0.1% TFA and UV detection at 215 nm. Their molecular weights were confirmed by MALDI-TOF MS analysis. The peptides were then frozen and lyophilized. Substance P peptide was purchased from Sigma (St. Louis, MO).
Transglutaminase-Catalyzed Enzymatic Reactions
Enzymatic reactions were carried out in 50 mM Tris-HCl buffer containing 7.5 mM CaCl2, 2.5 mM DTT, 1 mM EDTA, 1 mM dansyl cadaverine (DC), varying amounts of a glutamine peptide substrate, and purified guinea pig liver TGase (Sigma) (0.08 U/mL) in a total volume of 300 μL of reaction mixture at pH 8.0, 25 °C. To terminate the reaction at predetermined time intervals (3, 6, 9, and 15 min), aliquots of the reaction mixture were removed and added to an equal volume of 2% TFA in water containing 0.2 mM εN-dansyl-L-lysine (Sigma) as the internal standard.
RP-HPLC Analysis of Enzymatic Reaction Mixture
All reaction products were analyzed at room temperature using a Waters HPLC system (Waters, Milford, MA) on a Vydac 218TP C18 reversed-phase column (250 × 4.6 mm, 10 μm) with a gradient of 5–40% acetonitrile in 0.1% TFA-water (v/v) over 30 min at a flow rate of 1 mL/min with photodiode array detection from 200 to 400 nm, after injection of 10 μL of sample via an autosampler. For quantification purposes, εN-dansyl-L-lysine was used as an internal standard to calibrate the UV absorbance response at 288 nm.
Characterization of Enzymatic Cross-Linked Products
Cross-linked products of all glutamine peptides were confirmed by MALDI-TOF MS analysis. To determine the Gln position involved in the TGase reaction with DC, the product peak was subjected to MALDI-TOF post-source decay (PSD) analysis (Figure S8).
Enzyme Kinetic Study of Glutamine Peptide Substrate and Analysis of Kinetic Data
Rate studies were carried out as described above with an optimized enzyme concentration and varying amounts of peptide substrate. For each peptide, the concentration range of glutamine peptide substrate was adjusted such that the product formation was linear over the first 6 min. Kinetic constants were calculated using a curve-fitting program (SigmaPlot 9.0 with Enzyme Kinetics Module, SPSS Inc., IL) by fit of the initial velocities versus substrate concentration to the Michaelis-Menten equation. A molecular weight of 77 kDa was used to calculate the enzyme concentration.
RESULTS AND DISCUSSION
Binary Tag Coding System and MALDI-TOF MS Sequencing
In the original ladder synthesis method, a small amount of Ac-Ala was added into an Fmoc-amino acid solution (1:9) as a capping reagent to partially terminate the peptide chain at each coupling step.10 The resulting truncated peptides record the history of peptide synthesis and can be decoded by MS analysis to provide the full-length peptide sequence by analyzing their MS peaks. However, this approach was complicated by the fact that Ac-Ala and Fmoc-amino acids exhibited widely different coupling rates. The method was subsequently improved through the use of a Fmoc-amino acid/Boc-amino acid (9:1) mixture, in which the counterpart Boc-protected amino acid with the same side-chain protection served as a terminating reagent.11,12 In addition to providing similar reaction rates, this method simplified the interpretation of mass spectra in that the mass difference of neighboring peaks represents the residual molecular weight of the encoded amino acid. To distinguish isobaric residues by MS, a mixture of Ac-Ala and propionyl-Ala10 or Ac-Ala and Ac-Gly16 were used as capping reagents at the coupling step to encode Ile and Gln residues, and later, Fmoc-Gly was proposed to encode Gln and Ile in a bilayer-bead synthesis approach.17
However, despite the improvements in the encoded ladder synthesis method, our preliminary results (not shown) indicated that unambiguous identification of isobaric residues or residues with mass differences of one or two Da using MALDI-TOF MS sequencing remains challenging. Among the 19 DNA-encoded amino acids used in this study, there are three groups of such amino acids (see Table 1). To facilitate unambiguous identification of these residues, we designed a new binary tag coding system. Our ladder synthesis employed an Fmoc-amino acid/Boc-amino acid (19:1) method, with the Boc-amino acid acting as capping reagent to partially terminate the peptide chain and as a primary ladder tag. For most amino acids, the mass difference of two neighboring tags is simply the residual molecular weight of the encoded amino acid. For residues of similar or the same mass, addition of a secondary tag was necessary (Table 1 and Supporting Information). For these residues, both primary and secondary tags were added to the coupling mixture at 5% of the Fmoc amino acid (19:1:1), resulting in unique mass differences that allowed identification of the residues by MALDI-TOF MS analysis.
Table 1.
Amino Acids with Isobaric or Similar Molecular Weights Requiring a Binary Tag Systema
| primary tag | MWb | secondary tag | MWb | PDc |
|---|---|---|---|---|
| Gln | 128.06 | Ac-Gln | 170.08 | +42 |
| Glu | 129.04 | |||
| Lys | 128.09 | Ac-Dab | 142.08 | +14 |
| Asn | 114.04 | |||
| Asp | 115.03 | Ac-Asp | 157.05 | +42 |
| Ile | 113.08 | Me-Ile | 127.08 | +14 |
| Leu | 113.08 | γ-Abu | 85.05 | −28 |
| Pro | 97.05 | |||
| Val | 99.01 | Ile | 113.08 | +14 |
| Thr | 101.05 |
Details of primary and secondary tag systems for these amino acids are given in Table S2.
Residual molecular weight.
Molecular weight difference from primary tag.
The binary tag encoding method and MALDI-TOF MS sequencing approach was first verified with the peptide Gln-Gln-Ile-Val (QQIV) synthesized on solid phase by ladder synthesis approach (Scheme 1) using the protected amino acids shown in Table S1. This peptide was selected because it is a known TGase substrate13,18 and contains an Ile residue suitable for testing the efficacy of the binary tag system. First, a photocleavable linker (PCL) was included between the solid support and the peptides to facilitate sequencing by MALDI-TOF MS. Although methionine-containing linkers have been used for this purpose in the past, complications arise with methionine-containing compounds in peptide libraries. Instead, we chose an o-nitrobenzyl PCL since it is cleaved by exposure to UV light (365 nm) and may be more compatible with MALDI-TOF MS sequencing.19 The PCL was added to the PEGA resin (0.2 mmol/g, 300–500 μm, 770 000–1 270 000 beads/g) by Fmoc strategy with a BOP-HOBt preactivation method.
Scheme 1. Synthesis of QQIV Using Binary Encoding Methoda.

a (a) Fmoc-amino acid/BOP/HOBt/DIEA, 30 min; piperidine/NMP, 20 min; (b) Fmoc-Ahx/Boc-Abu-OH (19:1)/BOP/HOBt/DIEA, 30 min; piperidine/NMP, 20 min; (c) Fmoc-AA/Boc-AA(19:1)/BOP/HOBt/DIEA, 2 h; 20% piperidine in NMP for Val and Gln; (d) Fmoc-Ile/Boc-Ile/Boc-Abu-OH(19:1:1)/BOP/HOBt/DIEA, 2 h, 20% piperidine in NMP for Ile; and (e) TFA-TIS-H2O (95:2.5:2.5) overnight.
Because certain amino acid residues in a peptide are more MALDI-TOF-sensitive than others,10,18 following the PCL, a de novo designed MALDI-TOF MS sensitive sequence (MSS) peptide, Pro-Tyr-Phe-Arg-Val, was added to ensure similar signal sensitivity of full-length peptide and truncated ladder peptides under one MALDI-TOF condition. Following addition of the PCL and MSS peptide, a 6-aminohexanoic acid (Ahx) linker with Boc-4-aminobutyric acid (Boc-Abu) (19:1) as the first ladder tag (their residue molecular weights have a 28-Da difference) was added, and finally, QQIV was added using a 19:1 Fmoc-AA/Boc-AA mixture for Gln and Val, and a 19:1:1 Fmoc-Ile/Boc-Ile/Boc-Abu mixture for the Ile position (Table S1).
After removal of the protecting groups, several resin beads were loaded onto a MALDI sample plate and irradiated at 365 nm in acetonitrile-water (1:1) (2 μL) for 15 min, and then an aliquot of matrix solution was pipetted onto the beads. After the samples were air-dried, MALDI-TOF MS analysis of an individual bead generated a mass spectrum consisting of a series of peptide ladders (truncated sequences) as well as the full-length peptide sequence, as shown in Figure 2. Interpretation of the mass spectrum proved straightforward, with the sequence of the full-length peptide simply determined by calculating mass differences between peaks, as shown in the figure. The presence of the secondary tag by addition of Boc-Abu-OH during addition of the second residue resulted in an additional mass peak located 28 m/z units less than the mass of Ile, demonstrating the practical value of the binary tag coding method.
Figure 2.

MALDI-TOF MS spectrum of QQIV peptide synthesized using binary tag coding system and coupled to PEGA resin via photocleavable linker and MALDI sensitive sequence (MSS).
Library Design and Synthesis
To demonstrate the method in library form, we first synthesized a combinatorial library of TGase lysine peptide substrates consisting of 130 321 members (19 × 19 × 19 × 19). The design and construction of the encoded library using ladder synthesis approach is outlined in Scheme 2. PEGA resin, known for its ability to fully swell in aqueous media and allow permeation of enzymes,20–22 was chosen as the solid support to allow resin-bound lysine peptides to be assayed for enzymatic reactivity. The PCL and MSS peptide were synthesized as described above. The MSS peptide was separated from the library peptides by a monodisperse oligoethylene glycol (OEG7) spacer to prevent possible interference of the MSS peptide with bioactivity of the library peptides.12 The OEG7 spacer was coupled to the resin using Boc-OEG7 acid using the same coupling procedure as above, and the Boc protecting group was removed with treatment of DCM-TFA (9:1) at room temperature (30 min× 2). Under these conditions, the Pbf protecting group of the Arg residue in MSS was found to be stable, whereas the Boc protecting group of Boc-OEG7 acid was removed completely. Fmoc-Gly/Boc-Gly (19:1) was then coupled to the OEG7 spacer to generate the first ladder tag. At this stage, some beads were treated to remove the protecting groups and prepared for MALDI-TOF MS analysis, which indicated that the synthesis of the linker was satisfactory.
Scheme 2. Construction of Encoded Lysine Peptide Library Using Ladder Methoda.

a (a) Boc-OEG7 acid/BOP/HOBt/DIEA, 2h; TFA/DCM (9:1), 30 min × 2; (b) Fmoc-Gly/Boc-Gly(19:1)/BOP/HOBt/DIEA, 2 h, 20% piperidine in NMP, 20 min; (c) Fmoc-AA/Boc-AA (19:1)/BOP/HOBt/DIEA, 2 h, 20% piperidine in NMP for X′1, X′2, X′3, and X′4; (d) Fmoc-Lys(Boc)-OH/BOP/HOBt/DIEA, 2 h, 20% piperidine in NMP, 20 min; and (e) TFA-TIS-H2O (95:2.5:2.5) overnight.
The lysine pentapeptide library was synthesized using the combined split-mix and ladder synthesis method. The lysine residue at position 3 was fixed while the residues at positions 1, 2, 4, and 5 were randomized using all DNA-encoded amino acids except cysteine, which was excluded from the library for the sake of simplicity to eliminate disulfide cross-linking and possible interference with the cysteine residue at the active site of TGase. At each randomized position, a mixture of Fmoc-amino acid and Boc-amino acid (5%) was used for coupling. Additionally, for the residues shown in Table 1, a second protected amino acid (5%) was added in the coupling mixture as the secondary tag. A complete description of the primary and secondary tags used for constructing the library is included in Table S2. After addition of the final residue and removal of the Fmoc protecting group, the peptide resins were treated with TFA-TIS-H2O (95:2.5:2.5) to remove all protecting groups without cleaving the peptides from the resin. For the glutamine peptide library, we employed a similar strategy to synthesize an encoded glutamine hexapeptide library containing two position-fixed contiguous Gln in a hexapeptide (Scheme S2, Supporting Information).
A New Two-Stage Library Screening
On-resin screening of combinatorial libraries have previously utilized an ELISA-like assay system using biotinylated biological targets, followed by visualization through avidin/streptavidin conjugates.4,23,24 To perform the on-resin TGase enzyme assay and screening on the lysine peptide library, we designed and synthesized the desthiobiotinylated glutamine peptide conjugate desthiobiotin-OEG11-Gln-Gln-Ile-Val (DOQ) (Scheme 3). Desthiobiotin is an analogue of biotin that binds less tightly to streptavidin, such that desthiobiotin-streptavidin complexes can be disrupted by competitive biotin/desthiobiotin exchange in the presence of excess biotin.25 DOQ was prepared by solid-phase synthesis using Fmoc strategy and Fmoc-OEG11 acid as the spacer building block. DOQ was purified by preparative RP-HPLC, and its structure was confirmed by ESI-MS.
Scheme 3.

Desthiobiotin-OEG11-QQIV (DOQ)
Analysis of the peptide library began with TGase-catalyzed incorporation of DOQ onto resin-bound Lys peptides by incubating the synthesized lysine library (~770 000) in a buffer solution of 100 mM MOPS, pH 7.2; 1 mM DOQ; 10 mM CaCl2; 10 mM DTT; and 1 mM EDTA with TGase (0.25 U/mL) at room temperature for 30 min. This step resulted in the labeling of individual resin beads with desthiobiotin according to the ability of the bound Lys peptide to act as a TGase substrate. Thus, the amount of desthiobiotin coupled to each resin bead reflected the substrate properties of the bound peptide. Screening of the peptide library was designed to take advantage of the differential affinities of desthiobiotin and biotin toward streptavidin, wherein streptavidin-based labeling compounds can be easily released from the resin beads under mild conditions by addition of biotin. This is an important feature of our approach, which in principle permits rescreening or the use of multiple screening strategies successively on the same library. Here, we demonstrate this with a two-stage screening process (Scheme 4) involving magnetic separation followed by further labeling with rhodamine B-streptavidin and separation according to fluorescence intensity.
Scheme 4.
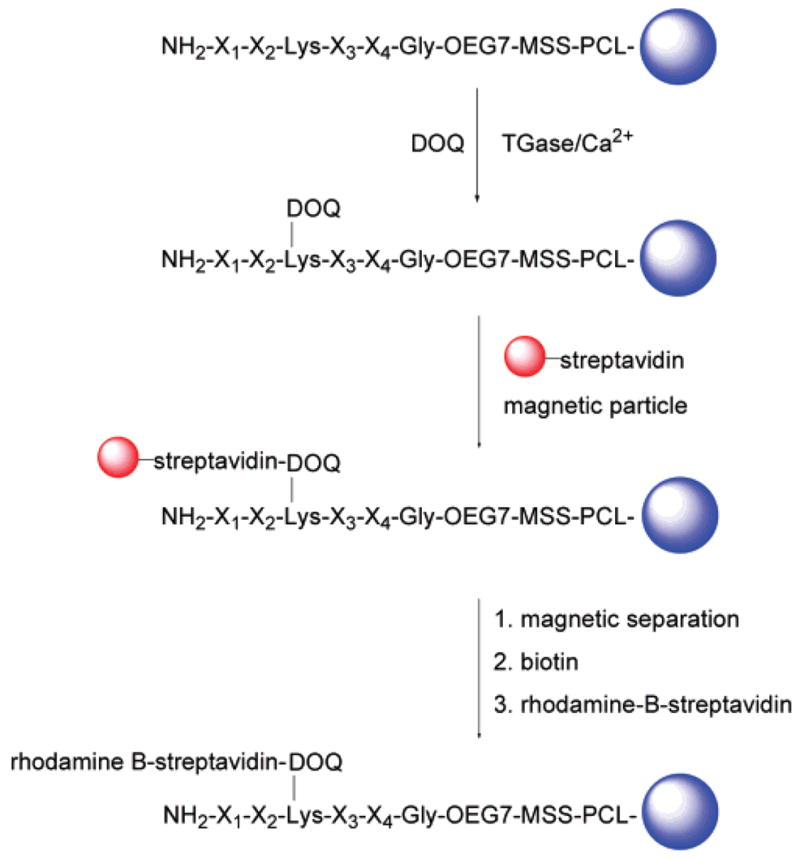
On-Resin Enzyme Assay and Two-Stage Screening of Encoded Lys Peptide Library
Screening of the glutamine peptide library was performed in a similar fashion, except with the use of desthiobiotin-PEG600-cadaverine (DPC) as a desthiobiotinylated amine donor for the on-resin TGase enzyme assay (Scheme S3).
First-Stage Magnetic Screening
Magnetic separation of live cells and DNA fragment mixtures using magnetic particle conjugates has proven to be a simple and efficient separation method.26–28 We took advantage of magnetic separation for screening of resin-bound peptides by incubating PEGA resin beads with magnetic streptavidin microbeads (8.0–9.9 μm, Spherotech), followed by separation of magnetically labeled beads using a commercial magnet (Magnetic Particle Concentrator, Dynal Biotech).
After exhaustive washes following the TGase reaction, the library was incubated with 2 mL of streptavidin-magnetic particles (8.8–9.0 μm) in 30 mL PBS, pH 7.2, 0.1% BSA for 2 h. Magnetic separation was afforded by placing a polypropylene conical tube (15 or 50 mL) in the Magnetic Particle Concentrator. The tube was filled to 2/3 volume with PBS, pH 7.2 and 0.3% BSA, and the resin suspension was added dropwise into the tube from above. Magnetic particle labeled beads were attracted to the wall of the tube by the magnetic field, while unlabeled beads fell to the bottom of the tube. Subsequent analysis showed that 1/10 of the beads (~77 000 beads) were magnetically isolated, with selected beads exhibiting a darker color than unselected beads due to the presence of magnetic iron oxide. A control experiment performed under identical conditions allowed us to conclude that the magnetic labeling of the resin-bound peptide library was desthiobiotin-specific. The glutamine peptide library was subject to the same magnetic screen process as above; however, fewer magnetically labeled beads (<10 000 beads) were isolated due to a more stringent substrate requirement of TGase toward glutamine peptides.
Second-Stage Fluorescence Screening
The population of beads selected by magnetic separation was further screened by removal of magnetic particles and relabeling with rhodamine B-streptavidin conjugate. Removal of magnetic particles from the surface of desthiobiotin-labeled resin was facilitated by taking advantage of the differential affinities of biotin and desthiobiotin for streptavidin.25 Thus, when magnetically selected resin beads were suspended in a solution containing excess biotin, the higher-affinity biotin complexed with streptavidin, displacing desthiobiotin and releasing the peptide-bound resin beads from the magnetic particles. After extensive washing to remove excess biotin and magnetic particles, the resulting desthiobiotin-peptide beads were fluorescently labeled by incubating with 0.1 mg of rhodamine B-streptavidin in 10 mL of PBS, pH 7.2; 0.1% Tween-20; and 0.3% BSA at room temperature for 30 min. Examination of the beads using a fluorescence microscope revealed a range of fluorescence intensities (Figure 3a) that presumably correlate to the amount of desthiobiotin and, therefore, peptide substrate properties. Fluorescent beads exhibited a characteristic halo appearance, with fluorescence intensity greatest at the bead surface and lowest in the core. We hypothesize this halo effect to be due to the inability of the enzyme to fully penetrate the PEGA resin during the enzyme reaction, resulting in TGase reaction only near the surface. In a parallel experiment, some beads not selected by magnetic screening were treated as described above and observed under a fluorescence microscope. These beads appeared dark under identical illumination (data not shown), indicating that the fluorescent labeling was desthiobiotin-specific. MALDI-TOF MS spectra showed the PCL linker to be stable for at least 2 h of illumination under these conditions, indicating that the possibility of cleavage of resin-bound peptides by illumination in the microscope was minimal.
Figure 3.

Fluorescence microscope images of selected Lys peptide library beads (bead size 300–500 μm) with a Leica DM IRB microscope (a) after first stage magnetic screening and relabeling with fluorescent streptavidin, 5× objective; and (b) after second stage fluorescence screening using automated bead sorter, 10× objective.
After magnetic screening of the glutamine peptide library, the number of selected beads was dramatically reduced, allowing 267 individual positive beads to be selected manually during second-stage screening using a fluorescence microscope and a micropipette. Although the number of beads in the lysine library was reduced by ~90% during magnetic screening, due to the broad specificity of TGase toward amine substrates and the use of a relatively long enzyme reaction time, the number of remaining beads proved too cumbersome to further screen manually using the fluorescence microscope. To accelerate the secondary screening process, a flow cytometry-based bead-sorting instrument (COPAS bead sorter, Union Biometrica) was used. The fluorescently labeled beads were passed through the sorting instrument three successive times at a rate of 30–40 beads/s, and beads meeting the following increasingly selective criteria were retained: first pass, top 1% beads sorted by global total red fluorescence (TR) (Figure S2); second pass, top 0.2% of beads sorted by red fluorescence peak height (PH) (Figure S3); and third pass, top 0.1% of beads sorted by normalized global total red fluorescence (NTR) (Figure S4). This process resulted in selection of 973 beads (~0.12% of the library beads), which were collected in BSA-treated 96-well plates (one bead/well). Examination of selected beads by fluorescence microscopy revealed intense fluorescent labeling near the surface (Figure 3b).
MALDI-TOF MS Sequencing of Selected Library Beads
To evaluate the library synthesis and the modified encoding system, 26 beads were randomly selected from the lysine library prior to screening. Each individual bead was analyzed by MALDI-TOF MS after exposure to UV light (365 nm), as described above. Peptide sequences were easily obtained by MALDI-TOF MS analysis for all 26 beads, and all amino acid building blocks used in the library synthesis were identified. No sequence deletion or residue modifications were detected. The MS spectra indicated that the residues in Table 1 could be identified clearly and unambiguously.
From the pool of 973 individual beads selected during two-stage screening of the lysine library, we conducted MALDI-TOF MS analysis on only those that exhibited the highest individual fluorescence values. Individual beads were first washed with water to remove buffer salts, detergent, and BSA. Each bead was placed onto a sample position on the sample plate and irradiated at 365 nm in acetonitrile-water (1:1) for 15 min, the matrix solution was added, and the bead was air-dried. In this way, ~1/3 of the selected beads (345 total, Table S3) were analyzed, and all of their peptide sequences were unequivocally assigned by spectral interpretation, as illustrated in Figure 4. Among individual positive beads selected during the second-stage screening of the glutamine peptide library, 267 glutamine peptide sequences were identified (Table S4).
Figure 4.

Representative MALDI-TOF MS spectrum of a selected lysine peptide library bead. The binary tag system allowed unambiguous identification of the Gln, Val, and Leu residues and the full-length peptide Gln-Tyr-Lys-Val-Leu. The peak at m/z 1818.91 was assigned to Gln, which is known to undergo cyclization during MALDI when present at the N-terminal position, resulting in a mass loss of m/z 17.
Results of Library Screening: TGase Substrate Characteristics
TGases (protein-glutamine/amine γ-glutamyltransferase, EC 2.3.2.13) are calcium-dependent enzymes that catalyze a posttranslational acyl-transfer reaction between the γ-carboxamide groups of peptide-bound glutaminyl residues and the ε-amino groups of lysine residues in proteins, or certain primary amino groups.29,30 This reaction results in the cross-linking of proteins through the formation of ε-(γ-glutamyl)lysine isopeptide side-chain bridges. TGases are widely distributed in many cell types and tissues31 and participate in a variety of biological processes, including stabilization of fibrin clots during hemostasis,32 membrane stiffening of erythrocytes,33 formation of the cornified cell envelope in terminally differentiated epidermal keratinocytes,34 and production of the vaginal plug by post-ejaculatory clotting of rodent seminal plasma.35
The reaction catalyzed by TGases involves two substrates: an acyl donor (glutamine) peptide and an acyl acceptor (lysine) peptide (Scheme S1). Previous investigations of substrate specificity on acyl donor peptides show that the substrate requirement for glutamine substrates is highly selective and stringent.36–42 Two previous combinatorial studies of TGase substrates have been reported; however, both have employed phage display approaches and have only investigated acyl donor peptides.43,44 In contrast, transglutaminases are generally considered to have a broad specificity toward acyl acceptor (lysine) substrates.45,46 Earlier studies suggested that lysine-containing peptides and a variety of synthetic primary amines mimicking the side chain of lysine residue could serve as acyl acceptor substrates for TGases.47–49 It has been shown that the composition and sequence of the amino acids adjacent to lysine residues in peptide and protein substrates can have an effect on the amine specificity.14,50–52 TGase can also be used as a biological cross-linking agent to develop new biomimetic hydrogels as an alternative to the chemical and physical cross-linking approaches normally employed to form hydrogels.14,53–56
Results of Library Screening: Lysine Peptide Substrates of TGase
Following on-resin TGase assay and two-stage screening, 345 lysine peptide sequences were identified by MALDI-TOF MS analysis as described above. Complete sequence data are included as Supporting Information (Table S3), from which the position frequency for all amino acids was calculated. The results are shown in Figure 5 and indicate broad tolerance of amino acid composition in Lys substrates of TGase. This finding is consistent with the widely accepted notion of broad specificity of TGase toward amine substrates.29,31,52 Nevertheless, some new information emerged from analysis of the selected beads. For example, it was noted that the acidic residues Glu and Asp as well as the aromatic residue Trp were found to occur in low frequency at nearly every position. Another interesting observation relates to the high frequency of Arg found at each position. The effect of Arg on Lys substrate specificity has not been previously recognized and, in the case of position X2, is somewhat surprising, given that a previous study indicated that hydrophobic residues in this position (flanking residue to Lys on N-terminal side) enhance the substrate properties of Lys.52 More follow-up studies using combinatorial or conventional enzyme assays, or both, may further illuminate the effects of Glu, Asp, Trp, and Arg residues on Lys substrate activity.
Figure 5.

Analysis of frequency data for amino acids found in positions (A) X1, (B) X2, (C) X3, and (D) X4 of sequenced peptides identified by two-stage screening of a lysine peptide library. Primary sequence data for each individual peptide is included in Table S3.
Results of Library Screening: Glutamine Peptide Substrates of TGase
Following on-resin TGase assay and two-stage screening, 267 glutamine peptide sequences were identified by MALDI-TOF MS analysis, as described above. Complete sequence data are included as Supporting Information (Table S4), from which the position frequency for all amino acids was calculated. The results are shown in Figure S6 and indicate a more stringent requirement of amino acid composition in Gln substrates of TGase. This result is consistent with the existing knowledge about the specificity of TGase. Overall, Arg, Ile, Phe, and Pro occurred at high frequency. The flanking residues have important impact on the substrate specificity. At positions X2 and X5 (flanking residue to Gln), Leu, Glu, Asp, Asn, and Trp were found to occur in low frequency. Another interesting observation relates to the high frequency of Arg found at each position. This was also the case for the lysine peptide library, leading us to speculate that Arg may be important for solubility of the peptides during the on-resin TGase reaction.
To more fully characterize the specificity of glutamine peptides toward TGase, 21 glutamine peptides were selected on the basis of the results of library screening and synthesized, and the kinetic constants of the cross-linked product formation were directly measured (Table 2). As an example, typical time courses of the enzyme-catalyzed reactions of incorporation of dns-ε-aca-cadaverine into Val-His-Gln-Gln-Gln-Arg (0.1 mM) are shown in Figure S7. From the HPLC data, kinetic constants for the 21 peptides and 4 peptide controls were determined and are listed in Table 2. Overall, most of the selected peptides have a significantly higher specificity than the best known glutamine peptide, QQIV. One peptide in particular, PQQQYV, has a 68-fold-higher substrate specificity than peptide QQIV, demonstrating that our method is effective to screen and identify good enzyme substrates. These findings may lead to better understanding of these enzymes and to further development of enzyme-cross-linked hydrogels. In addition, we believe that this facile methodology will be useful for rapid screening and identification of highly specific ligands for other biological targets from a large combinatorial library or a mixture of compounds.
Table 2.
| no. | sequence | concn range (mM) | Vmax (μM/min) | kcat (min−1) | Km(app) (mM) | kcat/Km(app) (min−1 mM−1) |
|---|---|---|---|---|---|---|
| 1 | Pro-Gln-Gln-Gln-Gln-Met | 0.073–1.1 | 4.5 | 17.0 | 0.27 | 64 |
| 2 | Val-Gln-Gln-Gln-Gln-Asn | 0.11–5.4 | 27.0 | 100.0 | 2.4 | 42 |
| 3 | Arg-Ile-Gln-Gln-Gln-Arg | 0.097–4.4 | 16.6 | 62.0 | 1.3 | 49 |
| 4 | Gly-Pro-Gln-Gln-Gln-Phe | 0.41–2.3 | 8.4 | 32.0 | 0.65 | 48 |
| 5 | Pro-Gln-Gln-Gln-Phe-Phe | 0.097–2.3 | 17.0 | 64.0 | 0.71 | 91 |
| 6 | Asn-Pro-Gln-Gln-Phe-Phe | 0.13–8.6 | 23.0 | 86.0 | 2.9 | 30 |
| 7 | Val-His-Gln-Gln-Gln-Arg | 0.83–13 | 20.0 | 76.0 | 3.0 | 25 |
| 8 | Pro-His-Gln-Gln-Gln-Phe | 0.17–5.3 | 35.0 | 130.0 | 10.0 | 13 |
| 9 | Pro-Gln-Gln-Gln-Tyr-Val | 0.12–5.0 | 25.0 | 95.0 | 0.21 | 450 |
| 10 | Pro-Gln-Gln-Gln-Tyr-Arg | 0.12–5.0 | 4.8 | 18.0 | 0.47 | 38 |
| 11 | Met-Arg-Gln-Gln-Tyr-Arg | 0.42–13 | 11.0 | 41.0 | 6.0 | 6.8 |
| 12 | Thr-Phe-Gln-Gln-Arg-Arg | 0.096–3.6 | 3.7 | 14.0 | 1.3 | 11 |
| 13 | Asn-Phe-Gln-Gln-Tyr-Arg | 0.093–3.4 | 26.0 | 96.0 | 1.4 | 69 |
| 14 | Asn-Arg-Gln-Gln-Arg-His | 0.71–9.5 | 24.0 | 90.0 | 4.5 | 20 |
| 15 | Asn-Ile-Gln-Gln-Arg-Tyr | 0.20–6.3 | 13.0 | 50.0 | 1.5 | 33 |
| 16 | Thr-Phe-Gln-Gln-Arg-Phe | 0.070–4.4 | 12.0 | 44.0 | 0.12 | 370 |
| 17 | Leu-Phe-Gln-Gln-Arg-Ser | 0.10–5.7 | 9.4 | 35.0 | 1.5 | 24 |
| 18 | Pro-Phe-Gln-Gln-Arg-Arg | 0.19–2.4 | 4.9 | 18.0 | 1.2 | 16 |
| 19 | Ile-Phe-Gln-Gln-Arg-Phe | 0.12–3.8 | 12.0 | 44.0 | 0.26 | 180 |
| 20 | Arg-Ser-Gln-Gln-Ser-Arg | 0.44–13 | 15.0 | 96.0 | 9.5 | 10 |
| 21 | Phe-Ile-Gln-Gln-Gln-Phe | insoluble | ||||
| Q2 | Gln-Gln-Ile-Val | 0.18–3.6 | 0.69 | 2.6 | 0.39 | 6.6 |
| Q3 | Ac-Gly-Gln-Gln-Gln-Leu-Gly | 0.073–1.5 | 0.69 | 2.6 | 0.059 | 44 |
| PQ3 | Pro-Gln-Gln-Gln-Phe-Pro | 0.22–3.3 | 23.0 | 86.0 | 1.2 | 72 |
| SP | substance P, RPKPQQFFGLM-NH2 | 0.079–2.4 | 3.0 | 11.0 | 0.15 | 73 |
Bold Gln indicates site(s) of tTG isopeptide bond.
Underline indicates that one of those Gln is crosslinked to dansyl-cadaverine but could not be elucidated by MALDI-TOF PSD MS.
CONCLUSIONS
We designed an improved binary tag system for encoding peptide libraries. This new approach facilitated rapid and efficient on-resin screening and sequencing by MALDI-TOF MS, as demonstrated by a new two-stage screening procedure for assaying enzyme activity of resin-bound peptide targets. The first stage screening utilized magnetic separation, resulting in removal of most of the library compounds. Further screening was facilitated by exploiting differences in streptavidin affinity for desthiobiotin and biotin, allowing release of magnetic particles from the peptide resin beads and relabeling with fluorescent streptavidin for either manual or automated bead selection. Sequencing of selected beads was performed by MALDI-TOF MS analysis with unequivocal assignment of identical or similar mass made possible through the use of an improved binary tag system during library synthesis. The approach was demonstrated for libraries of lysine and glutamine substrate peptides of TGase. For the lysine substrate peptide library, the resulting fluorescently labeled beads were screened in a bead sorter to yield a total of 973 beads, of which 345 were sequenced. Screening of the glutamine peptide library revealed 267 glutamine substrate peptides, and enzyme kinetic analysis of a subset of these revealed several peptides with high specificity toward TGase, with the peptide PQQQYV having a 68-fold-higher substrate specificity than the best known glutamine substrate QQIV and 5-fold higher than the reported value of β-casein.37
Supplementary Material
Details of protected amino acids and tag compounds used in ladder synthesis, complete sequence data of all TGase amine substrate peptides selected during library screening, and details of glutamine peptide library synthesis and analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors gratefully acknowledge Dr. Bo Liu and Kathleen Barnhart at Union Biometrica for bead sorting and support from the NIH (DE 13030 and EB 003806).
References
- 1.Furka A, Sebestyen F, Asgedom M, Dibo G. Int J Pept Protein Res. 1991;37:487–493. doi: 10.1111/j.1399-3011.1991.tb00765.x. [DOI] [PubMed] [Google Scholar]
- 2.Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. Nature. 1991;354:82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- 3.Houghten RA, Pinilla C, Blondelle SE, Appel JR, Dooley CT, Cuervo JH. Nature. 1991;354:84–86. doi: 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]
- 4.Lam KS, Lebl M, Krchnak V. Chem Rev. 1997;97:411–448. doi: 10.1021/cr9600114. [DOI] [PubMed] [Google Scholar]
- 5.Czarnik AW. Curr Opin Chem Biol. 1997;1:60–66. doi: 10.1016/s1367-5931(97)80109-3. [DOI] [PubMed] [Google Scholar]
- 6.Braeckmans K, De Smedt SC, Leblans M, Pauwels R, Demeester J. Nat Rev Drug Discovery. 2002;1:447–456. doi: 10.1038/nrd817. [DOI] [PubMed] [Google Scholar]
- 7.Ohlmeyer MHJ, Swanson RN, Dillard L, Reader JC, Asouline G, Kobayashi R, Wigler M, Still WC. Proc Natl Acad Sci USA. 1993;90:10922–10926. doi: 10.1073/pnas.90.23.10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ni ZJ, Maclean D, Holmes CP, Murphy MM, Ruhland B, Jacobs JW, Gordon EM, Gallop MA. J Med Chem. 1996;39:1601–1608. doi: 10.1021/jm960043j. [DOI] [PubMed] [Google Scholar]
- 9.Fitzgerald MC, Harris K, Shevlin CG, Siuzdak G. Bioorg Med Chem Lett. 1996;6:979–982. [Google Scholar]
- 10.Youngquist RS, Fuentes GR, Lacey MP, Keough T. J Am Chem Soc. 1995;117:3900–3906. [Google Scholar]
- 11.StHilaire PM, Lowary TL, Meldal M, Bock K. J Am Chem Soc. 1998;120:13312–13320. [Google Scholar]
- 12.Tornoe CW, Sanderson SJ, Mottram JC, Coombs GH, Meldal M. J Comb Chem. 2004;6:312–324. doi: 10.1021/cc020085v. [DOI] [PubMed] [Google Scholar]
- 13.Parameswaran KN, Velasco PT, Wilson J, Lorand L. Proc Natl Acad Sci USA. 1990;87:8472–8475. doi: 10.1073/pnas.87.21.8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu BH, Messersmith PB. J Am Chem Soc. 2003;125:14298–14299. doi: 10.1021/ja038593b. [DOI] [PubMed] [Google Scholar]
- 15.Qiao SW, Bergseng E, Molberg O, Jung G, Fleckenstein B, Sollid LM. J Immunol. 2005;175:254–261. doi: 10.4049/jimmunol.175.1.254. [DOI] [PubMed] [Google Scholar]
- 16.Beebe KD, Wang P, Arabaci G, Pei DH. Biochemistry. 2000;39:13251–13260. doi: 10.1021/bi0014397. [DOI] [PubMed] [Google Scholar]
- 17.Wang XB, Peng L, Liu RW, Gill SS, Lam KS. J Comb Chem. 2005;7:197–209. doi: 10.1021/cc049887b. [DOI] [PubMed] [Google Scholar]
- 18.Valero ML, Giralt E, Andreu D. Lett Pept Sci. 1999;6:109–115. [Google Scholar]
- 19.Holmes CP, Jones DG. J Org Chem. 1995;60:2318–2319. [Google Scholar]
- 20.Kress J, Zanaletti R, Amour A, Ladlow M, Frey JG, Bradley M. Chem Eur J. 2002;8:3769–3772. doi: 10.1002/1521-3765(20020816)8:16<3769::AID-CHEM3769>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 21.Kohli RM, Walsh CT, Burkart MD. Nature (London, United Kingdom) 2002;418:658–661. doi: 10.1038/nature00907. [DOI] [PubMed] [Google Scholar]
- 22.Tolborg JF, Petersen L, Jensen KJ, Mayer C, Jakeman DL, Warren RAJ, Withers SG. J Org Chem. 2002;67:4143–4149. doi: 10.1021/jo0163445. [DOI] [PubMed] [Google Scholar]
- 23.Vagner J, Barany G, Lam KS, Krchnak V, Sepetov NF, Ostrem JA, Strop P, Lebl M. Proc Natl Acad Sci USA. 1996;93:8194–8199. doi: 10.1073/pnas.93.16.8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ronn LCB, Olsen M, Ostergaard S, Kiselyov V, Berezin V, Mortensen MT, Lerche MH, Jensen PH, Soroka V, Saffells JL, Doherty P, Poulsen FM, Bock E, Holm A. Nat Biotechnol. 1999;17:1000–1005. doi: 10.1038/13697. [DOI] [PubMed] [Google Scholar]
- 25.Hirsch JD, Eslamizar L, Filanoski BJ, Malekzadeh N, Haugland RP, Beechem JM. Anal Biochem. 2002;308:343–357. doi: 10.1016/s0003-2697(02)00201-4. [DOI] [PubMed] [Google Scholar]
- 26.Pankhurst QA, Connolly J, Jones SK, Dobson J. J Phys D: Appl Phys. 2003;36:R167–R181. [Google Scholar]
- 27.Kodituwakku AP, Jessup C, Zola H, Roberton DM. Immunol Cell Biol. 2003;81:163–170. doi: 10.1046/j.1440-1711.2003.01152.x. [DOI] [PubMed] [Google Scholar]
- 28.Melnik K, Nakamura M, Comella K, Lasky LC, Zborowski M, Chalmers JJ. Biotechnol Prog. 2001;17:907–916. doi: 10.1021/bp010079r. [DOI] [PubMed] [Google Scholar]
- 29.Lorand L, Conrad SM. Mol Cell Biochem. 1984;58:9–35. doi: 10.1007/BF00240602. [DOI] [PubMed] [Google Scholar]
- 30.Lorand L, Graham RM. Nat Rev Mol Cell Biol. 2003;4:140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 31.Folk JE, Finlayson JS. Adv Protein Chem. 1977;31:1–133. doi: 10.1016/s0065-3233(08)60217-x. [DOI] [PubMed] [Google Scholar]
- 32.Pisano JJ, Finlayso Js, Peyton MP. Science. 1968;160:892–893. doi: 10.1126/science.160.3830.892. [DOI] [PubMed] [Google Scholar]
- 33.Siefring GE, Apostol AB, Velasco PT, Lorand L. Biochemistry. 1978;17:2598–2604. doi: 10.1021/bi00606a022. [DOI] [PubMed] [Google Scholar]
- 34.Rice RH, Green H. J Cell Biol. 1978;76:705–711. doi: 10.1083/jcb.76.3.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams-Ashman HG, Pabalan SS, Notides AC, Lorand L. Proc Natl Acad Sci USA. 1972;69:2322–2325. doi: 10.1073/pnas.69.8.2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Folk JE, Cole PW. J Biol Chem. 1965;240:2951–2960. [PubMed] [Google Scholar]
- 37.Gorman JJ, Folk JE. J Biol Chem. 1981;256:2712–2715. [PubMed] [Google Scholar]
- 38.Gorman JJ, Folk JE. J Biol Chem. 1984;259:9007–9010. [PubMed] [Google Scholar]
- 39.Pastor MT, Diez A, Perez-Paya E, Abad C. FEBS Lett. 1999;451:231–234. doi: 10.1016/s0014-5793(99)00572-4. [DOI] [PubMed] [Google Scholar]
- 40.Hohenadl C, Mann K, Mayer U, Timpl R, Paulsson M, Aeschlimann D. J Biol Chem. 1995;270:23415–23420. doi: 10.1074/jbc.270.40.23415. [DOI] [PubMed] [Google Scholar]
- 41.Ohtsuka T, Ota M, Nio N, Motoki M. Biosci, Biotechnol, Biochem. 2000;64:2608–2613. doi: 10.1271/bbb.64.2608. [DOI] [PubMed] [Google Scholar]
- 42.Kahlem P, Terre C, Green H, Djian P. Proc Natl Acad Sci USA. 1996;93:14580–14585. doi: 10.1073/pnas.93.25.14580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keresztessy Z, Csosz E, Harsfalvi J, Csomos K, Gray J, Lightowlers R, Lakey J, Balajthy Z, Fesus L. Protein Sci. 2006;15:2466–2480. doi: 10.1110/ps.051818406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sugimura Y, Hosono M, Wada F, Yoshimura T, Maki M, Hitomi K. J Biol Chem. 2006;281:17699–17706. doi: 10.1074/jbc.M513538200. [DOI] [PubMed] [Google Scholar]
- 45.Ohtsuka T, Sawa A, Kawabata R, Nio N, Motoki M. J Agric Food Chem. 2000;48:6230–6233. doi: 10.1021/jf000302k. [DOI] [PubMed] [Google Scholar]
- 46.Sobel JH, Gawinowicz MA. J Biol Chem. 1996;271:19288–19297. doi: 10.1074/jbc.271.32.19288. [DOI] [PubMed] [Google Scholar]
- 47.Lorand L, Rule NG, Ong HH, Furlanet R, Jacobsen A, Downey J, Oner N, Bruner-Lorand J. Biochemistry. 1968;7:1214–1223. doi: 10.1021/bi00843a043. [DOI] [PubMed] [Google Scholar]
- 48.Lorand L, Parameswaran KN, Stenberg P, Tong YS, Velasco PT, Jonsson NA, Mikiver L, Moses P. Biochemistry. 1979;18:1756–1765. doi: 10.1021/bi00576a019. [DOI] [PubMed] [Google Scholar]
- 49.Schrode J, Folk JE. J Biol Chem. 1979;254:653–661. [PubMed] [Google Scholar]
- 50.Grootjans JJ, Groenen P, Dejong WW. J Biol Chem. 1995;270:22855–22858. doi: 10.1074/jbc.270.39.22855. [DOI] [PubMed] [Google Scholar]
- 51.Groenen P, Smulders R, Peters RFR, Grootjans JJ, Vandenijssel P, Bloemendal H, Dejong WW. Eur J Biochem. 1994;220:795–799. doi: 10.1111/j.1432-1033.1994.tb18681.x. [DOI] [PubMed] [Google Scholar]
- 52.Gross M, Whetzel NK, Folk JE. J Biol Chem. 1977;252:3752–3759. [PubMed] [Google Scholar]
- 53.Sanborn TJ, Messersmith PB, Barron AE. Biomaterials. 2002;23:2703–2710. doi: 10.1016/s0142-9612(02)00002-9. [DOI] [PubMed] [Google Scholar]
- 54.Sperinde JJ, Griffith LG. Macromolecules. 1997;30:5255–5264. [Google Scholar]
- 55.Hennink WE, van Nostrum CF. Adv Drug Delivery Rev. 2002;54:13–36. doi: 10.1016/s0169-409x(01)00240-x. [DOI] [PubMed] [Google Scholar]
- 56.McHale MK, Setton LA, Chilkoti A. Tissue Eng. 2005;11:1768–1779. doi: 10.1089/ten.2005.11.1768. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details of protected amino acids and tag compounds used in ladder synthesis, complete sequence data of all TGase amine substrate peptides selected during library screening, and details of glutamine peptide library synthesis and analysis. This material is available free of charge via the Internet at http://pubs.acs.org.