

Abstract
Interleukin-6 (IL-6) is a pleiotropic cytokine that regulates diverse cell functions including proliferation and differentiation. Within the liver IL-6 signaling plays a central role during normal hepatic growth and regeneration yet can inhibit the proliferation of hepatocellular carcinoma (HCC) cells. The aim of the current study was to identify underlying mechanisms whereby IL-6 induces cell cycle arrest in HCC cells. These studies demonstrate that IL-6 inhibits cell cycle progression at the G0/G1 interface through inhibition of cyclin dependent kinase (cdk) 2 and cdk4 activity in the absence of changes in total cyclin (A, D1, D3 and E) or cdk (cdk2, 4 and cdc2 p34) expression. Inhibition of signal transduction pathways associated with IL-6 receptor activation demonstrate that IL-6-dependent inhibition of G0-G1 progression occurs via Janus tyrosine kinase-signal transducers and activators of transcription-3 (Jak-STAT3)-dependent induction of p21waf1/cip1 and is independent of ERK-MAPK signaling. These data demonstrate that, while IL-6 plays a central role in hepatocyte priming and proliferation in vivo, the pronounced inhibition of proliferation observed in HCC cells occurs due to IL-6-STAT3-dependent regulation of cdk2/cdk4 activity and p21waf1/cip1 expression.
Keywords: Hepatocellular carcinoma, Interleukin-6, Mitogenesis, growth arrest, cyclin dependent kinases
INTRODUCTION
Hepatocellular carcinoma (HCC) accounts for approximately 85% of primary malignant tumors of the liver (1–3). Globally, HCC is the fifth most common malignancy and the third most common cause of cancer related death accounting for >600,000 deaths per year (2–4). At present surgical resection or complete hepatic transplant remains the most effective treatment for HCC (2, 3, 5). However, the insidious nature of HCC, late detection and/or metastasis coupled with a lack of availability of transplantable organs limits the therapeutic options available (2, 3, 5). Unlike other organs the healthy liver demonstrates the ability to undergo regeneration following a reduction in functional volume. In order for regeneration to successfully proceed following decreased hepatic mass it is essential that diverse cell signaling pathways are coordinated (6). In contrast to regeneration, HCC is characterized by abnormal responsiveness to cytokines and growth factors and this has led to interest in determining changes that occur in the signal transduction mechanisms of transformed hepatic cells for pathways normally associated with the regeneration process (1–3).
Interleukin-6 (IL-6) regulates diverse biological functions and has particular significance in the liver in regulating acute phase protein production, protection against liver injury and stimulation of liver regeneration (6–8). During hepatic regeneration IL-6 plays a pivotal role in priming hepatocytes for proliferation through sensitization to growth factors (6–8). Reduced liver mass results in elevated TNF-α that, in turn, stimulates IL-6 expression (8, 9). IL-6 then binds to an IL-6 receptor-α (IL-6Rα) subunit leading to the formation of a high-affinity hexamer complex comprising two molecules each of IL-6, IL-6Rα and the signal transducer gp130 (10). Signal transduction proceeds via gp130-associated Janus tyrosine kinase (Jak) and the recruitment/activation of signal transducers and activators of transcription (STAT’s) (10, 11). In addition to the Jak-STAT pathway, IL-6 has been demonstrated to activate intracellular mitogen activated protein kinase (MAPK) signaling cascades. In this instance Src homology protein 2 tyrosine phosphatase-2 (SHP-2) binds to activated gp130 and stimulates the small G-protein signaling molecule p21ras. Following p21ras activation, via GTP displacement of GDP activation of intracellular MAPKs (MAPK) signaling cascades, including p42/p44 extracellular signal regulated kinase (p42/44 ERK) (10–13). Active STAT 3, in collaboration with other transcription factors, enhances the expression of nuclear factors enabling resting hepatocytes to respond to other growth factors and proliferate (10, 11). More recently, the ability of IL-6 to act as a complete mitogen during liver growth has become apparent. Double transgenic mice expressing IL-6 and soluble IL-6R (sIL-6R) demonstrate nodular regenerative hyperplasia and adenoma development (14, 15) while supra-physiological IL-6 levels in nude mice cause dramatic hepatomegaly in the absence of liver injury (16).
Currently the role of IL-6 in tumor formation and/or progression remains ambiguous, IL-6 being demonstrated to act as a growth factor for several cancers including renal cell carcinoma and multiple myeloma while inhibiting proliferation of early stage melanoma cells, breast carcinoma cells and a number of leukaemia/lymphoma cell lines (17–20). Similarly, IL-6 appears to have multiple effects in the development and/or progression of HCC. Interleukin-6 acts as an autocrine growth factor in the IL-6 producing HCC-M cell line (21), a survival factor during TGF-β-induced apoptosis in human Hep3B cells (17) and inhibits proliferation in specific human and rat HCC cell lines (22–24). Previous studies by our group have identified altered expression of IL-6 signaling components in a rat model of HCC in vitro and in vivo. Furthermore, treatment of these HCC cells with IL-6 inhibits cellular proliferation (13). The aim of the current study is to determine the cell cycle regulatory effects of IL-6 in mediating inhibition of proliferation in this rat model of HCC.
MATERIALS AND METHODS
Materials
Recombinant human IL-6 (rhIL-6) was purchased from Biosource Int. (Camarillo, CA). Antisera specific against STAT 3, ERK1/2, p21waf1/cip1, cdk2 and 4, cdc2 p34, cyclin A, GAPDH and KNRK nuclear extract were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Antisera specific against pSTAT 3 (Tyr705), pERK1/2, p27Kip1, cyclin D3, D1 and E, the Rb-C fusion protein, β-actin, GADPH and the MEK-ERK inhibitor PD98059 were purchased from Cell Signaling Technology (Beverly, MA). The STAT 3 inhibitor AG490 and Histone H1 (calf thymus) were purchased from Calbiochem (San Diego, CA). The ECL chemiluminesence detection system was purchased from Pierce Biotech. Inc. (Rockford, IL).
Cell culture and animal model of HCC
The rat H4IIE hepatoma cell line was obtained from ATCC (Bethesda, MD) and maintained as previously (24). To maintain cell tumorgenicity cells (≈1×106) were routinely (2–4 passages) inoculated directly into the left hepatic lobe of male ACI rats with a 30-gauge needle. This technique results in a reproducible tumor mass forming 14–16 days post-inoculation from which fresh H4IIE cells are isolated (24).
Preparation of cell lysates and immunoblotting
Quiescent H4IIE cells (serum depleted media, 36 hours) were treated with rhIL-6 (50ng/ml, 0–24 hours), rinsed with PBS (4°C), lysed using a kinase lysis buffer (25mM HEPES, 300mM NaCl, 1.5mM MgCl2, 200µM EDTA, 1% Triton X-100 (v/v), 20mM β-glycerophospate, 0.1% SDS (w/v), 0.5% sodium deoxycholate (v/v), 0.5mM DTT, 100mM Na3VO4, 100µg/ml PMSF, 2µg/ml leupeptin, 2µg/ml aprotinin, pH7.5, 4°C) and stored at −80°C prior to analysis. Protein concentrations were equalized and samples boiled with Laemmli buffer. Samples were then analyzed by Western blot as previously (13). Primary antibodies were used at a dilution of 1:1000 with the exception of anti-pSTAT 3 (1:2000). Secondary antibodies were used at a dilution of 1:5000. Detection was performed using ECL and X-ray film as previously (13).
Inhibition of p42/p44-MAPK and Jak-STAT pathways
H4IIE cells were made quiescent for 36 hours in serum-depleted media. PD98059 (20µM) and AG490 (100µM) were dissolved in DMSO and serially diluted in serum free media prior to addition 1 hour before rhIL-6 treatment (50ng/mL). Following rhIL-6 treatment cells were collected in lysis buffer (4°C), sonicated and stored at −80°C prior to analysis. Control cells were treated with DMSO (0.1% v/v) 1 hour prior to treatment with rhIL-6.
Cyclin dependent kinase (cdk) assays
H4IIE cells were grown to ≈80% confluence and made quiescent for 36 hours in serum depleted media. Cells were next treated with rhIL-6 (50ng/mL, 0–24 hours), lysates prepared and incubated with antibodies against either cdk2 or cdk4 (1:50 dilution) for 4 hours prior to incubation in 20µL protein A agarose beads overnight (4°C). Beads were collected by centrifugation and washed once in kinase extraction buffer (4°C) followed by PBS (x2, 4°C). A final wash was carried out in 50mM HEPES/1mM DTT solution (4°C) and the beads were resuspended in 10µL of 50mM HEPES, 1mM DTT. 20µL of 3x Kinase Reaction buffer containing either 2mg/mL of Histone H1 (cdk2) or Rb-C fusion protein (cdk4) was then added and the reaction performed for 30 minutes (30°C) prior to termination by the addition of 15µL of Laemmli buffer (100°C). Samples were then resolved by SDS-PAGE, transferred to nitrocellulose membranes and exposed to X-ray film (12–18 hours, −80°C).
Cell cycle analysis
H4IIE cells were isolated, cultured and grown to 80% confluence in high serum media (10% FBS, v/v). Cells were then synchronized in SFM (replaced every 48 hours) for 7 days. At the end of this period cells were treated with rhIL-6 (50ng/ml) for 2 hours. SFM was then removed and replaced with either 2.5% (v/v) FBS media or 2.5% (v/v) FBS media containing 50ng/ml rhIL-6. For cell cycle analysis cells were detached, pelleted and resuspended in PBS containing 0.1% BSA. 1.5×106 cells/mL were then fixed in ethanol (4°C), resuspended in RNase A (0.7mg/mL) and incubated at 37°C. Propidium iodide was then added (50µg/mL) and the cells incubated in darkness (RT). Cells were then analyzed using a BD FACScalibur flow cytometer (BD Biosciences, San Jose, CA) and data modeled using Modfit LT analysis software.
Data Analysis and Statistics
Densitometeric analysis of signal intensity was analyzed using the EDAS-290 system (Kodak, Rochester, NY) as previously (13). In the instance of antibodies against phosphorylated proteins (pERK1/2, pSTAT 3) signal intensity was corrected to total protein detection (ERK1/2, STAT 3). Cumulative densitometric data was then calculated as the fold change in phosphorylated protein signal intensity versus untreated (t=0 minutes) cells. For experiments in which no active protein was detected the signal intensity was given the numerical value 0. For all other experiments membranes were stripped and probed with a loading control antibody (β-actin), signal intensity corrected accordingly and fold changes in expression following treatment calculated. Tests of statistical significance were performed using a Students t-test and a p value of <0.05 was taken as significant.
RESULTS
Interleukin-6-dependent changes in p21waf1/cip1 expression are STAT 3 and not ERK1/2 dependent
We have previously reported rhIL-6 activates p42/p44-ERK and STAT3 signaling in HCC cells in vitro (13). To control for the use of DMSO as a diluent for pharmacological inhibitors of p42/p44-ERK and STAT3 signaling H4IIE HCC cells were treated with DMSO (0.1% (v/v)) 1 hour prior to rhIL-6 treatment (50ng/ml, 0–120 minutes [ERK1/2 and STAT signaling] and 0–24 hours [p21waf1/cip1 or p27Kip1 expression]. Western blot analysis demonstrated that in the absence of DMSO rhIL-6 stimulated ERK1/2 in a biphasic manner (13). However, in the presence of DMSO only the second peak of ERK1/2 activity was observed 20–60 minutes after rhIL-6 addition, a maximal effect being observed 40 minutes after addition (Figure 1a, 5.09 ± 0.26 fold increase at 40 minutes versus DMSO alone, n=4 separate experiments, p<0.05). No significant changes in total p42/p44-ERK expression were detected at any of the time points assayed (Figure 1a). In contrast, the profile of STAT 3 activity following rhIL-6 treatment was identical to that previously reported in the absence of DMSO (13) in which a significant increase was detected 10 minutes after addition increasing to a maximum 40–60 minutes after treatment (Figure 1b, 7.62 ± 0.87 fold increase versus untreated at 40 minutes, n=4 separate experiments, p<0.05). No significant changes in total p42/p44-ERK or total STAT 3 protein expression were detected at any of the time points assayed (Figure 1a and b).
Figure 1.
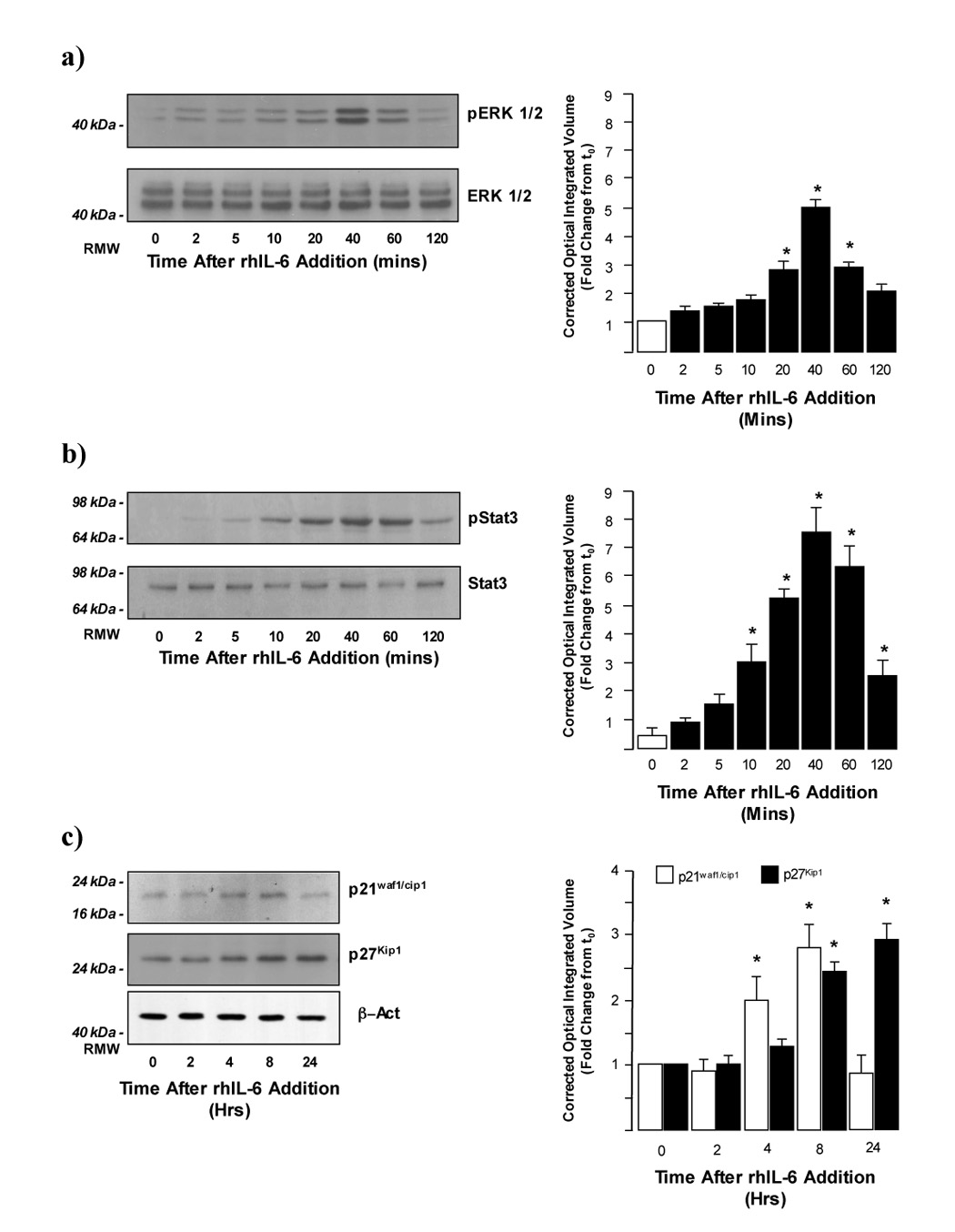
a) rhIL-6 stimulates ERK1/2 activity in H4IIE cells. Representative Western blot analysis of active (phosphorylated; pERK1/2) and total ERK1/2 expression in H4IIE cells following treatment with rhIL-6 (50ng/ml, 0–120 minutes). Cells were pretreated with DMSO (0.1% (v/v) 60 minutes prior to rhIL-6 treatment. Cumulative densitometric analysis of pERK1/2 expression was then performed and pERK1/2 values corrected to total ERK1/2 values. *p<0.05 rhIL-6 treated versus untreated (t=0 minutes), n=4 separate experiments.
b) rhIL-6 stimulates STAT 3 activity in HCC cells. Representative Western blot analysis of active (phosphorylated; pSTAT 3) and total STAT 3 expression in H4IIE cells following treatment with rhIL-6 (50ng/ml, 0–120 minutes). Cells were pretreated with DMSO (0.1% (v/v) 60 minutes prior to rhIL-6 treatment. Cumulative densitometric analysis of pSTAT 3 was then performed and pERK1/2 values corrected to total ERK1/2 values. *p<0.05 rhIL-6 treated versus untreated (t=0 minutes), n=4 separate experiments.
c) rhIL-6 stimulates p21waf1/cip1 and p27Kip1 expression in H4IIE cells Representative Western blot analysis of p21waf1/cip1 (upper panel) and p27Kip1 (middle panel) expression in H4IIE cells following treatment with rhIL-6 (50ng/ml, 0–24 hours). Cells were pretreated with DMSO (0.1% (v/v) 60 minutes prior to rhIL-6 treatment. Cumulative densitometric analysis of p21waf1/cip1 (open bars) and p27Kip1 (closed bars) expression was then performed and values corrected for b-actin expression (lower panel). *p<0.05 rhIL-6 treated versus untreated (t=0 minutes), n=4 separate experiments.
Analysis of p21waf1/cip1 and p27Kip1 expression following rhIL-6 treatment in the presence of DMSO also demonstrated similar patterns of expression to those observed in the absence of DMSO (13). rhIL-6 significantly stimulated p21waf1/cip1 4 hours after treatment rising to a maximum at 8 hours before returning to baseline at 24 hours and significantly stimulating p27Kip1 8 hours after treatment, an effect maintained up to 24 hours post-treatment (Figure 1c, n=4 separate experiments, p<0.05).
To assess the relative contribution of p42/p44-ERK and STAT 3 signaling in the regulation of p21waf1/cip1 and p27Kip1 expression we next employed the MEK specific inhibitor PD98059 (PD; 20µM) or the Jak-STAT3 specific inhibitor AG490 (AG) diluted in DMSO (0.1% (v/v) final concentration). Pretreatment of H4IIE cells for 1 hour with PD abolished rhIL-6 stimulated p42/p44-ERK activity at all time points assayed in the absence of significant changes in total p42/p44-ERK expression (Figure 2a, upper panels, and Figure 2c, n=4 separate experiments). In contrast, pretreatment with PD (20µM) did not significantly affect the temporal profile of STAT 3 activation in H4IIE cells although the magnitude of the maximal response to rhIL-6 was lower than that observed in the presence of DMSO alone (Figure 2a (lower panels) and 2c, n=4 separate experiments, p<0.05 versus untreated).
Figure 2.
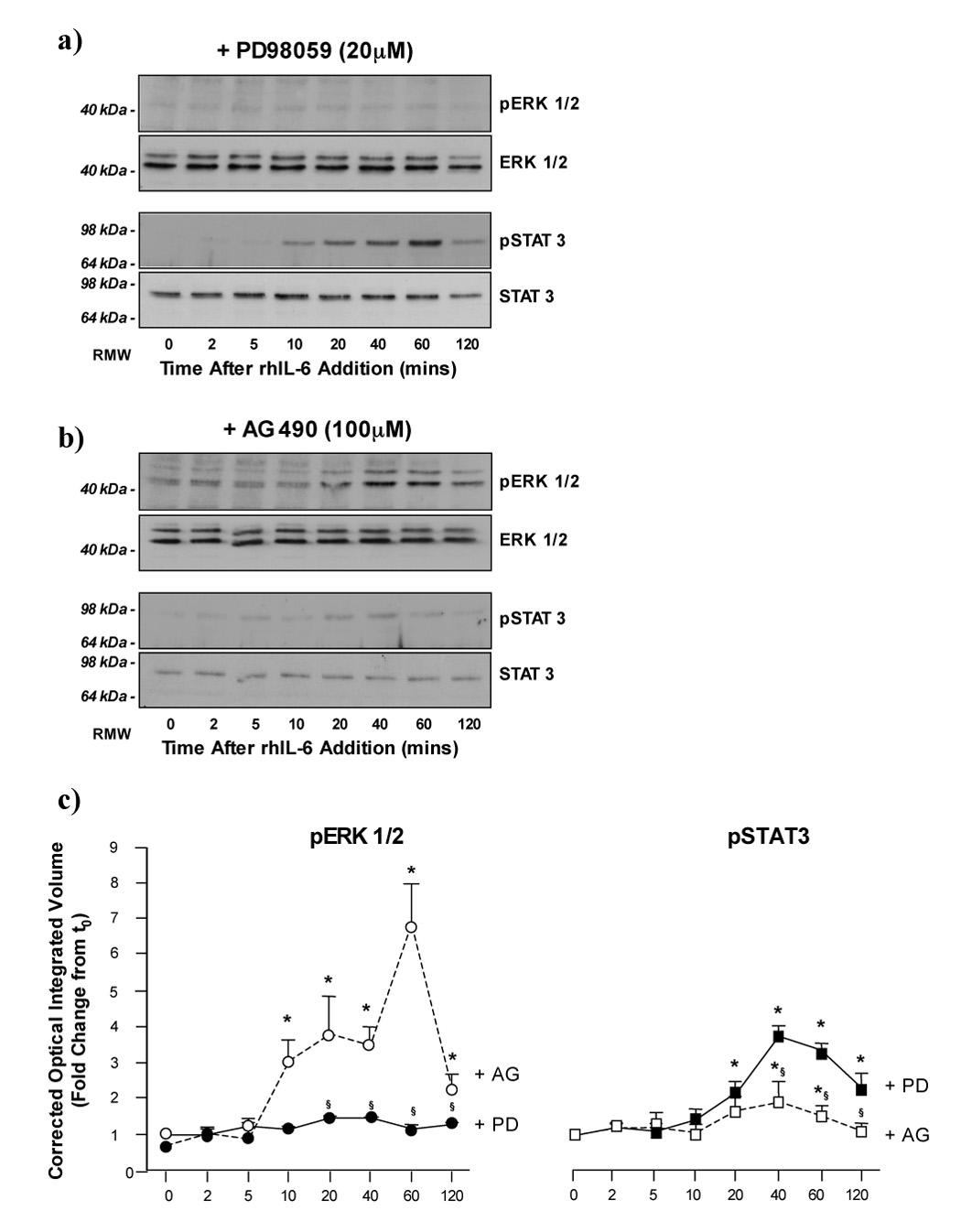
a) PD98059 inhibits rhIL-6 dependent ERK1/2 signaling but not STAT 3 signaling in H4IIE cells. Representative Western blot analysis of active (phosphorylated; pERK1/2) and total ERK1/2 expression (upper panels) in H4IIE cells following treatment with rhIL-6 (50ng/ml, 0–120 minutes) in the presence of PD98059 (20µM). Representative Western blot analysis of active (phosphorylated; pSTAT 3) and total STAT 3 expression (lower panels) in H4IIE cells following treatment with rhIL-6 (50ng/ml, 0–120 minutes) in the presence of PD98059 (20µM).
b) AG490 inhibits rhIL-6 dependent STAT 3 signaling but not ERK1/2 signaling in H4IIE cells. Representative Western blot analysis of active (phosphorylated; pERK1/2) and total ERK1/2 expression (upper panels) in H4IIE cells following treatment with rhIL-6 (50ng/ml, 0–120 minutes) in the presence of AG490 (100µM). Representative Western blot analysis of active (phosphorylated; pSTAT 3) and total STAT 3 expression in H4IIE cells following treatment with rhIL-6 (50ng/ml, 0–120 minutes) in the presence of AG490 (100µM).
c) Densitometric analysis of ERK1/2 and STAT 3 activity in the presence of PD98059 and AG490. Cumulative densitometric analysis was performed in which active (phosphorylated; pERK1/2) was corrected for total ERK1/2 expression in the presence of PD98059 (+PD (20µM),●—●) or AG490 (+AG (100µM),○—○), left panel. *p<0.05 rhIL-6 versus treated versus untreated (time = 0), §p<0.05 rhIL-6 treated +AG versus rhIL-6 treated +PD, n=4 separate experiments. Cumulative densitometric analysis was performed in which active (phosphorylated; pSTAT 3) was corrected for total STAT 3 expression in the presence of PD98059 (+PD (20µM),■—■) or AG490 (+AG (100µM),□—□) right panel. *p<0.05 rhIL-6 versus treated versus untreated (time = 0), §p<0.05 rhIL-6 treated +AG versus rhIL-6 treated +PD, n=4 separate experiments.
In preliminary studies using AG, inhibition of IL-6-dependent STAT 3 activation was observed at a range of 50–150µM (data not shown). However, at doses >100µM altered cellular morphology was observed leading to our experiments being performed at this dose. Treatment of H4IIE HCC cells with rhIL-6 in the presence of AG (100µM) led to significantly increased p42/p44-ERK activity versus untreated cells in the presence of AG alone (Figure 2b (upper panels) and 2c, n=4 separate experiments, p<0.05). In addition, both the magnitude and duration of p42/p44-ERK activity as compared to rhIL-6 treatment in the presence of DMSO alone (Figure 1a and Figure 2b). In contrast, pre-treatment with AG significantly blunted STAT 3 activation following IL-6 treatment versus treatment with IL-6 alone (Figure 1b and Figure 2b (lower panels)). Unlike pretreatment with PD, treatment with AG was unable to completely block the effects of rhIL-6 on STAT 3 activation, significantly increased pSTAT 3 detection occurring 40–60 minutes after rhIL-6 treatment, although the level of STAT 3 activity was significantly lower than that detected for rhIL-6 stimulation in the presence of DMSO or PD (Figure 1a and Figure 2a).
Finally, analysis of p21waf1/cip1/p27Kip1 expression was performed in H4IIE HCC cells pre-treated with either PD or AG followed by rhIL-6 treatment. These data demonstrated that inhibition of ERK1/2 signaling did not significantly affect either p21waf1/cip1/p27Kip1 expression following rhIL-6 treatment (Figure 3a). In contrast, while inhibition of Jak/STAT 3 signaling did not significantly affect the rh-IL-6 stimulated p27Kip1 expression in H4IIE HCC cells, pretreatment with AG abrogated the rhIL-6-dependent increases in p21waf1/cip1 expression (Figure 3b).
Figure 3.
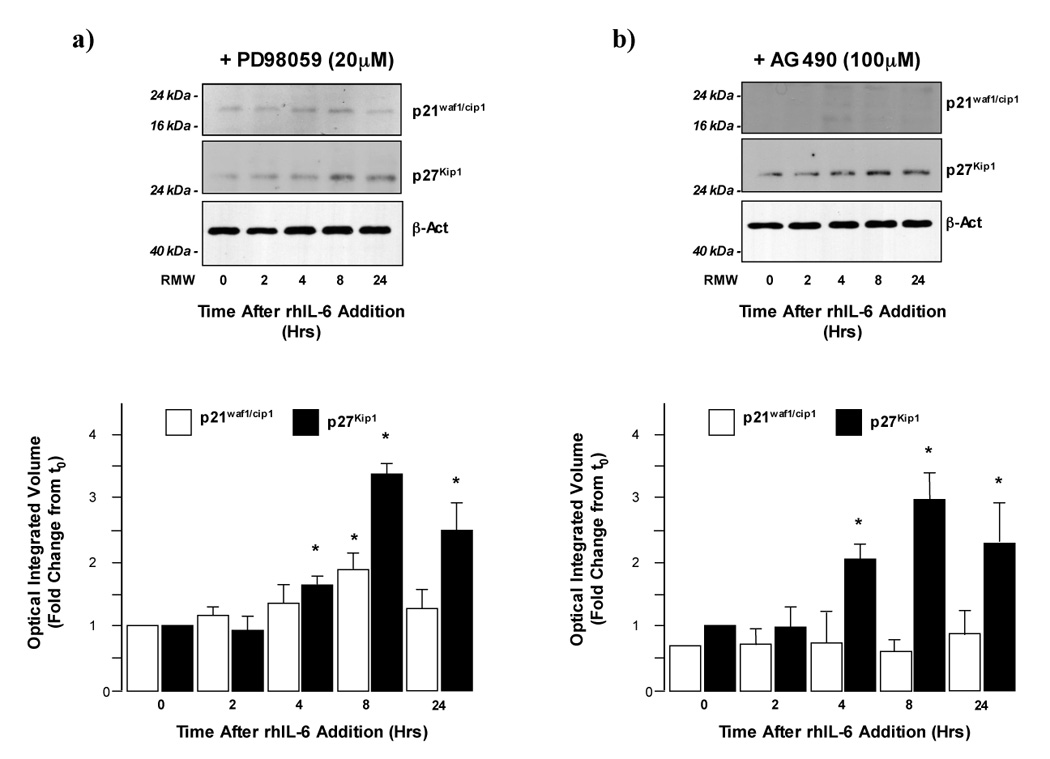
a) Inhibition of ERK1/2 signaling does not affect IL-6 dependent p21waf1/cip1 or p27Kip1 expression in H4IIE cells. Representative Western blot analysis of p21waf1/cip1 and (upper panel) and p27Kip1 expression (lower panel) in H4IIE cells pretreated with PD98059 (20µM) following treatment with rhIL-6 (50ng/ml; 0–24 Hrs). Cumulative densitometric analysis of p21waf1/cip1 (open bars) and p27Kip1 (closed bars) expression was then performed and values corrected for b-actin expression (lower panel). *p<0.05 rhIL-6 treated versus untreated (t=0), n=4 separate experiments.
b) Inhibition of STAT 3 signaling inhibits IL-6 dependent p21waf1/cip1 but not p27Kip1 expression in H4IIE cells. Representative Western blot analysis of p21waf1/cip1 and (upper panel) and p27Kip1 expression (lower panel) in H4IIE cells pretreated with AG490 (100µM, 60 minutes) following treatment with rhIL-6 (50ng/ml; 0–24 Hrs). Cumulative densitometric analysis of p21waf1/cip1 (open bars) and p27Kip1 (closed bars) expression was then performed and values corrected for b-actin expression (lower panel). *p<0.05 rhIL-6 treated versus untreated (t=0), n=4 separate experiments.
rhIL-6 does not alter G1 dependent cyclin or cdk expression in H4IIE cells
To determine the effects of rhIL-6 on the cell cycle we next assayed the expression of cyclins involved in progression through the G1 restriction point in H4IIE cells. H4IIE cells were treated with rhIL-6 (50ng/ml, 0–24 hours) and Western blot analysis performed using antibodies specific against cyclin A, D1, D3 and E. No significant differences in cyclin A, D1, D3 or E expression were detected in H4IIE cells in response to rhIL-6 versus untreated cells (Figure 4, n=4 separate experiments). Having established the expression profile of cyclins in H4IIE cells in response to rhIL-6, we next determined the expression of cyclin dependent kinases (cdk’s) demonstrated to be critical during transition through the G1 cell cycle restriction point (cdk2 and cdk4) and the G2/M stage of the cell cycle (cdc2 p34). These data demonstrated rhIL-6 did not cause any significant change in cdk2, cdk4 or cdc2 p34 expression in H4IIE cells or hepatocytes (Figure 4b, n=4 separate experiments).
Figure 4.

a) rhIL-6 treatment does not alter Cyclin A, D1, D3 or E expression in H4IIE cells. Representative Western blot analysis of Cyclins A, D1, D3 and E and the house keeper protein GAPDH (lower panel) in untreated (−) or rhIL-6 (50ng/ml, 0–24 hours) treated (+) H4IIE cells.
b) rhIL-6 does not alter cdk2, cdk4 or cdc2p34 expression in H4IIE cells. Representative Western blot analysis of cdk2, cdk4 and cdc2p34 expression in untreated H4IIE cells or H4IIE cells treated with rhIL-6 (50ng/ml, 0–24 hours). GAPDH expression is shown to demonstrate protein loading.
rhIL-6 inhibits cdk2 and cdk4 activity in H4IIE cells
Having determined the relative contribution of ERK and STAT 3 signaling in H4IIE cells on p21waf1/cip1/p27Kip1 expression in response to rhIL-6 and a lack of effect of rhIL-6 on cyclin and cdk expression we next assessed the effects of rhIL-6 on cdk activity using substrate-kinase reactions. These data demonstrated rhIL-6 significantly inhibited cdk2 activity as demonstrated by decreased substrate (histone H1) phosphorylation at 8 and 24 hours versus untreated controls (Figure 5a, n=3 separate experiments, p<0.05). Similarly, rhIL-6 significantly inhibited cdk4 activity, as determined by decreased Rb-C fusion protein phosphorylation 8 and 24 hours after rhIL-6 treatment versus untreated control (Figure 5b, n=3 separate experiments, p<0.05).
Figure 5.
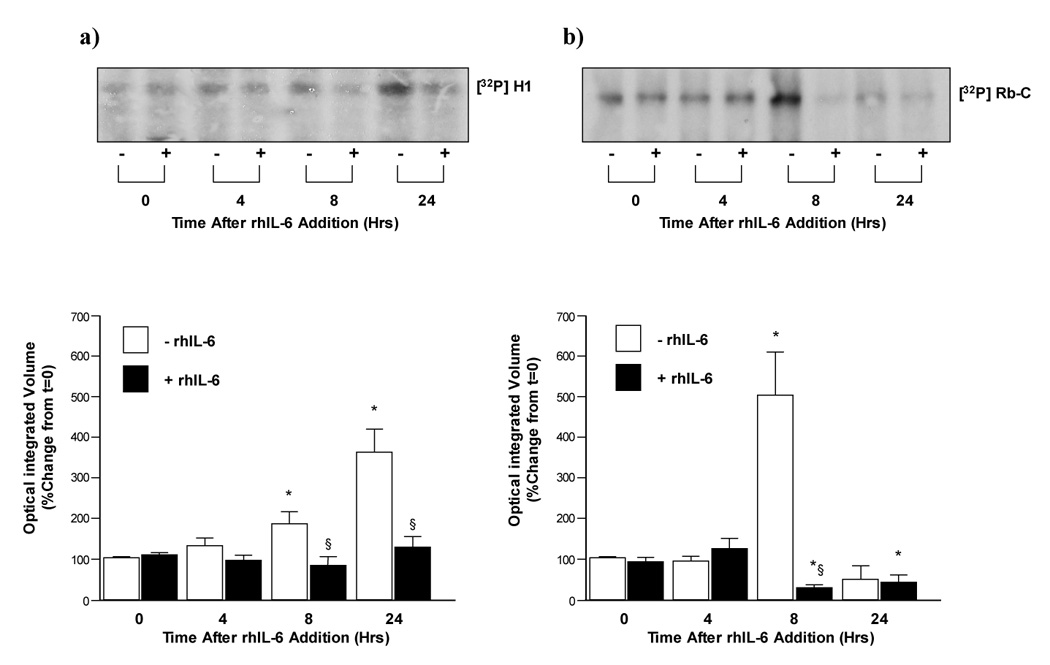
a) rhIL-6 inhibits cdk2 activity in H4IIE cells. cdk2 activity was assessed by a kinase reaction using histone H1 as a substrate for immunoprecipitated cdk2 in untreated (−) and rhIL-6 treated (+) H4IIE cells. Cumulative densitometric analysis of cdk2 activity/histone H1 phosphorylation ([32P]H1) following treatment with rh-IL-6 (lower panel). n=3 separate experiments, *p<0.05 versus time = 0 hours, §p<0.05 rhIL-6 treated (closed bars) versus untreated (open bars).
b) rhIL-6 inhibits cdk4 activity in H4IIE cells. cdk4 activity was assessed by a kinase reaction using retinoblastoma-C (Rb-C) protein as a substrate for immunoprecipitated cdk4 in untreated (−) and rhIL-6 treated (+) H4IIE cells (0–24 hours). Cumulative densitometric analysis of cdk4 activity/Rb-C phosphorylation ([32P]Rb) following treatment with rh-IL-6 (lower panel). n=3 separate experiments, *p<0.05 versus time = 0 hours, §p<0.05 rhIL-6 treated (closed bars) versus untreated (open bars).
IL-6 induces G0/G1 growth arrest in H4IIE cells
To determine the effect of IL-6 on cell cycle progression, synchronized H4IIE cells were pre-treated with rhIL-6 (50ng/ml) followed by FBS (2.5% v/v) and stained with propidium iodide (PI) prior to flow cytometry analysis. These data demonstrate FBS treatment induces H4IIE cell exit from the G0/G1 phase of the cell cycle, a significant decrease in G0/G1 cell population being observed at 20h (15.09 ± 0.35% decrease in G0/G1 population, n=4, p<0.05 versus time 0; Figure 6b) increasing to a maximum 28 hours after FBS addition (19.72 ± 0.02% decrease in G0/G1 population, n=4, p<0.05 versus time 0; Figure 6a and b). This change in G0/G1 population occurs concomitant with an increase in the S phase population (Figure 6a, n=4). Pretreatment of H4IIE cells with rhIL-6 (50ng/mL) followed by FBS (2.5% v/v) resulted in significantly decreased exit of cells from the G0/G1 phase of the cell cycle at 20 hours versus FBS alone (Figure 6b, 9.86 ± 0.16% (rhIL-6 + FBS) versus 15.09 ± 0.35% (FBS alone), n=4, p<0.05). No significant difference in the percentage of cells exiting the G0/G1 phase of the cell cycle was detected in HCC cells following FBS treatment in the presence of rhIL-6 between 20 hours and the later time points assayed. In contrast, at all the time points assayed (20–32 hours) the presence of rhIL-6 significantly inhibited cells exiting the G0/G1 phase as compared to FBS alone (Figure 6).
Figure 6. rhIL-6 induces G0/G1 growth arrest in H4IIE cells.
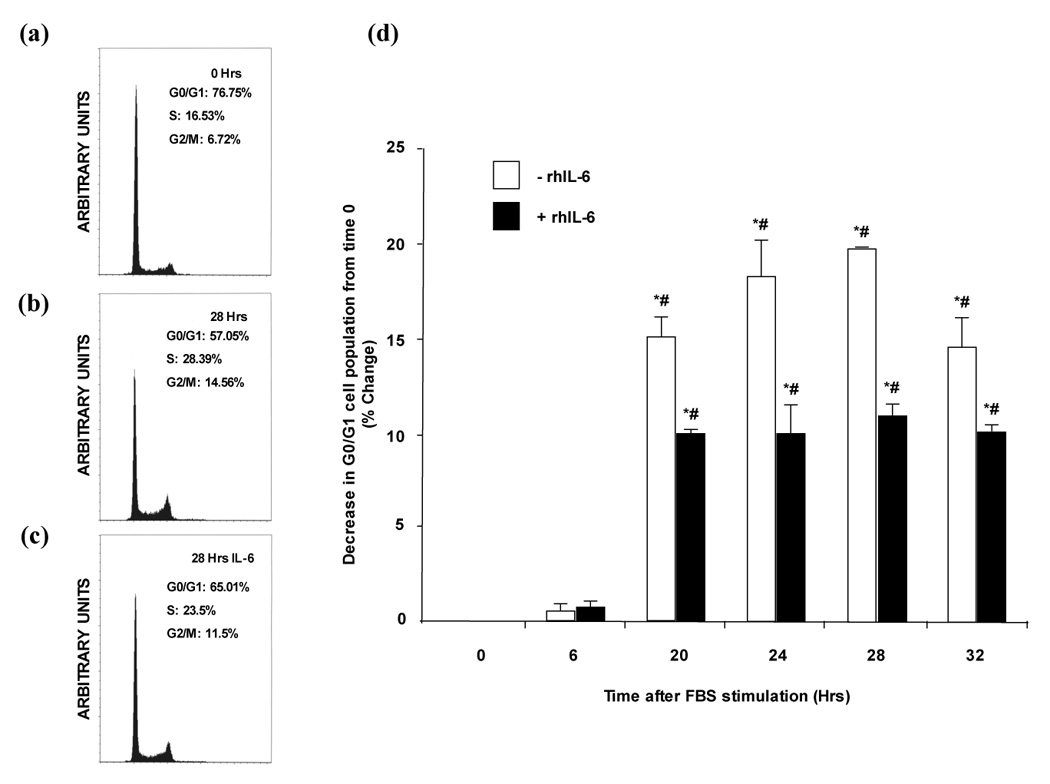
a) Representative FACS analysis demonstrating cell cycle phase of synchronized H4IIE cells prior to rhIL-6 treatment (t=0 hours).
b) Representative FACS analysis demonstrating cell cycle phase of H4IIE cells at 28 hours following FBS (2.5% v/v) treatment in the absence of rhIL-6.
c) Representative FACS analysis demonstrating cell cycle phase of H4IIE cells at 28 hours following FBS (2.5% v/v) treatment in the presence of rhIL-6.
d) Cumulative FACS analysis of H4IIE cells treated with FBS (2.5% v/v) in the presence or absence of rhIL-6 (50ng/ml). n=4 separate experiments, *p<0.05 versus 6 hours, #p<0.05 rhIL-6 treated versus untreated.
DISCUSSION
During the process of liver regeneration normal hepatocytes are primed to enter the G1-phase of the cell cycle and, in doing so, acquire increased responsiveness to growth factors allowing passage through the G1 restriction point (6, 8, 25). Decreased hepatic mass leads to increased serum IL-6 and hepatic IL-6R activation. Downstream activation of STAT 3, in collaboration with additional transcription factors, acts to enhance the expression of genes critical to repopulation (6, 8). The significance of IL-6 during these events is highlighted in studies utilizing IL-6−/− mice in which STAT 3 activation and early gene expression is absent and defective regeneration occurs (26). IL-6 can also act as a complete hepatic mitogen when present in excess (6, 14). In contrast this study identifies an inhibitory effect of rhIL-6 on G1 phase progression in transformed hepatic cells. IL-6 treatment leading to decreased cdk2 and cdk4 activity associated with a STAT 3 dependent increase in the cdk inhibitor p21waf1/cip1 and increased expression of p27Kip1.
Previous studies by our group report rhIL-6 stimulates ERK activity in a biphasic manner, an initial spike of pERK detection occurring 2–5 minutes after addition followed by a second sustained period of ERK activity 20–60 minutes later in H4IIE cells (24). In the current studies in which inhibitors of ERK (PD98059) or STAT 3 (AG490) were employed it was of interest that pre-treatment of cells with drug vehicle (DMSO; 0.1% v/v) abolished early (2–5 minute) ERK activity, whilst not affecting the second peak of ERK activity or STAT 3 activity. These data are all the more unusual as DMSO has been used as a vehicle for PD98059 during the study of ERK activity stimulated by a variety of cytokines in numerous cell lines including those of hepatic origin without apparently affecting ERK activity (27–29). Following IL-6 receptor binding a hexameric signaling complex forms (10, 11, 30). In order for ERK to become activated a series of scaffold proteins are assembled on the intracellular portion of the gp130 component of the receptor (10, 11). As such several possible explanations exist as to why DMSO inhibits early ERK activity whilst not affecting the later sustained ERK activation. Given the necessity for IL-6 to bind to gp80 for receptor complex formation to occur it is possible that the presence of DMSO in the culture medium delays the ability of IL-6 to bind to gp80 and initiate the receptor complex formation and abrogates early ERK activation. However, if this were the case it might be anticipated that a decrease in total ERK activity might occur, an effect that was not observed. Alternatively, the solvent properties of DMSO may act to alter plasma membrane fluidity and in doing so inhibit or slow the ability of gp130 subunits to associate and propagate the downstream intracellular signaling cascades. In this instance later signaling events such as the second period of ERK activation, STAT3 activity and increased p21waf1/cip1/p27Kip1 expression may not be affected while earlier events do not occur. Whether these events are unique to IL-6 signaling or also occur for other cytokines that utilize the gp130 receptor (e.g. Oncostatin-M, Leukemia inhibitory factor, IL-11) (30) and/or the use of DMSO, as opposed to other solvent vehicles, remains to be tested experimentally.
Progression through the cell cycle is regulated by cdks (31). Cyclins associate with cdks are the primary regulators of cdk activity (31, 32). The activation of cdk4 plays an important role in the passage through the G1 restriction point when the cell becomes committed to proceed through the cell cycle, while cdk2 activation plays an essential role in the transition into S phase and DNA synthesis. Cyclin D1 expression, which is induced during G1 phase in response to mitogens, complexes with and activates cdk4. Cyclin dependent kinase-2 (cdk2) is regulated primarily by cyclin E and cyclin A during G1/S transition and S phase respectively (31, 32). The retinoblastoma protein (Rb) is the major target for cyclin dependent kinase activity and is phosphorylated by both cyclin D dependent kinases and by cdk2. Hyperphosphorylation of the Rb protein disrupts its association with E2F transcription factors which, once released, stimulate the expression of a series of genes required for DNA synthesis (31, 32). In the present study rhIL-6 treatment resulted in decreased cdk2 and cdk4 activity while analysis of G1/S-phase cyclin and cdk expression demonstrated that total levels of these proteins were unchanged. Increased expression of the cdk inhibitors p21waf1/cip1 and p27Kip1 act as the most probable mediators of diminished activity. p21waf1/cip1 and p27Kip1 are members of the Cip/Kip family of cdk inhibitors that affect cyclin D-, E- and A-dependent kinase activity through binding of both the cyclin and cdk subunits (32). The IL-6 induced expression of p21waf1/cip1 and p27Kip1 in H4IIE cells correlates with decreases in cdk activity. Overall, increased cdk inhibitor expression, leading to decreased cdk activity and decreased Rb phosphorylation in response to rhIL-6, gives a direct mechanism for G1 growth arrest in H4IIE cells. Indeed, several reports have identified a similar inhibitory effect of IL-6 in transformed cells that are associated with increased p27Kip1/p21waf1/cip1 expression (19, 23, 33).
Both the Jak-STAT and ERK-MAPK pathways are implicated in increased p21waf1/cip1 and p27Kip1 expression in response to IL-6- mediated STAT 3/ERK 1/2 activation (19, 23, 33). In H4IIE cells inhibition of IL-6 stimulated ERK 1/2 activity did not affect p21waf1/cip1 or p27Kip1 expression. In contrast, STAT 3 inhibition ablated IL-6 stimulated p21waf1/cip1 expression. However despite the effects of AG490 on decreasing p21waf1/cip1 expression, AG490 failed to alter rhIL-6-stimulated p27Kip1 expression. One possible explanation for this observation may be that it was not possible to completely inhibit STAT 3 in these cells and this low basal STAT 3 activity is sufficient to induce p27Kip1. Alternatively, AG490 prolonged rhIL-6 stimulated ERK1/2 activity. Similar changes in ERK 1/2 activation have been reported following inhibition of STAT 3 using either AG490 or genetic means in other models, this effect being linked to decreased SOCS and/or phosphatase expression (34, 35). While STAT 3 and MAPK signaling are the primary downstream cascades activated following IL-6R stimulation, other pathways may also be activated. STAT 1 (19), STAT 5 (23) and PI3K (17) have all been reported to be activated following IL-6R activation in other cell types. Furthermore, both PI3K and STAT 5 can modulate p27Kip1 expression (23, 36) raising the possibility of multiple effector pathways mediating IL-6 dependent signaling.
Analysis of a variety of human and rat HCC cell lines demonstrates IL-6 inhibits proliferation in human and rat HCC cell lines (13, 22, 23). Conversely a role for IL-6 as an autocrine growth factor in human HCC-M cells (21) and a survival factor in Hep3B HCC cells (17) has been reported. These reports highlight distinct biological outcomes of IL-6 signaling in different HCC cell types. In addition to HCC, IL-6 is reported to act as a paracrine growth inhibitor in certain prostate cancer cell lines while acting as an autocrine growth stimulator in others (37). Interestingly, IL-6 has been reported to “switch” from acting as a paracrine growth inhibitor to an autocrine growth stimulator during the progression of human melanoma (38, 39). Taking into consideration that IL-6 is produced and secreted by H4IIE cells (13), these data suggest a potential novel role for IL-6 in HCC where it acts as an autocrine growth inhibitor in HCC, effects similar to that observed in human lung cancer cells (40). In this case, the cells have a relatively low IL-6 sensitivity when compared with noncarcinogenic human bronchial epithelial cells suggesting that escape from growth regulation of this inhibitory factor may be involved in lung oncogenesis. Similarly, H4IIE cells are characterized by decreased expression of IL-6 receptor components (13) supporting potentially evasive cell mechanisms from IL-6 growth inhibition. Autocrine negative growth regulation by other cytokines, such as TGF-β, in other cancer models has been established while the development of multicytokine resistance during oncogenesis is linked to clonal dominance of primary tumours by metastatically competent cells (41, 42).
In conclusion, the current studies demonstrate that inhibition of proliferation in H4IIE HCC cells occurs due to IL-6-STAT 3-dependent regulation of cdk2/cdk4 activity and p21waf1/cip1 expression. This mechanism in turn inhibits the progression through the G0/G1 interface of the cell cycle effectively halting the mitogenic process. In addition, our data demonstrate that the use of DMSO inhibits the early responsiveness of H4IIE cells to IL-6-dependent ERK activation but does not affect later events including ERK and STAT activation and increased p27Kip1/p21waf1/cip1 expression. Further studies are required to determine the mechanism(s) by which early IL-6-ERK activation are inhibited by DMSO. Finally, it will be of considerable interest to determine whether IL-6-dependent inhibition of HCC growth observed in these studies in vitro translates to the in vivo setting.
Acknowledgments
Financial Support: This work was supported in part by a grant from the National nstitutes of Health (CA90895 [IHM])
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Macdonald GA. Pathogenesis of hepatocellular carcinoma. Clin Liver Dis. 2001;5:69–85. doi: 10.1016/s1089-3261(05)70154-9. [DOI] [PubMed] [Google Scholar]
- 2.McKillop IH, Moran DM, Jin X, Koniaris LG. Molecular pathogenesis of hepatocellular carcinoma. J Surg Res. 2006;136:125–135. doi: 10.1016/j.jss.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 3.Okuda K. Hepatocellular carcinoma--history, current status and perspectives. Dig Liver Dis. 2002;34:613–616. doi: 10.1016/s1590-8658(02)80200-6. [DOI] [PubMed] [Google Scholar]
- 4.Bosch FX, Ribes J, Cleries R, Diaz M. Epidemiology of hepatocellular carcinoma. Clin Liver Dis. 2005;9:191–211. doi: 10.1016/j.cld.2004.12.009. v. [DOI] [PubMed] [Google Scholar]
- 5.Befeler AS, Di Bisceglie AM. Hepatocellular carcinoma: diagnosis and treatment. Gastroenterology. 2002;122:1609–1619. doi: 10.1053/gast.2002.33411. [DOI] [PubMed] [Google Scholar]
- 6.Koniaris LG, McKillop IH, Schwartz SI, Zimmers TA. Liver regeneration. J Am Coll Surg. 2003;197:634–659. doi: 10.1016/S1072-7515(03)00374-0. [DOI] [PubMed] [Google Scholar]
- 7.Ramadori G, Christ B. Cytokines and the hepatic acute-phase response. Seminars in Liver Disease. 1999;19:141–155. doi: 10.1055/s-2007-1007106. [DOI] [PubMed] [Google Scholar]
- 8.Streetz KL, Luedde T, Manns MP, Trautwein C. Interleukin 6 and liver regeneration. Gut. 2000;47:309–312. doi: 10.1136/gut.47.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamada Y, Kirillova I, Peschon JJ, Fausto N. Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc Natl Acad Sci U S A. 1997;94:1441–1446. doi: 10.1073/pnas.94.4.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fukada T, Hibi M, Yamanaka Y, Takahashi-Tezuka M, Fujitani Y, Yamaguchi T, Nakajima K, Hirano T. Two signals are necessary for cell proliferation induced by a cytokine receptor gp130: involvement of STAT3 in anti-apoptosis. Immunity. 1996;5:449–460. doi: 10.1016/s1074-7613(00)80501-4. [DOI] [PubMed] [Google Scholar]
- 12.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 13.Moran DM, Mayes N, Koniaris LG, Cahill PA, McKillop IH. Interleukin-6 inhibits cell proliferation in a rat model of hepatocellular carcinoma. Liver Int. 2005;25:445–457. doi: 10.1111/j.1478-3231.2005.01083.x. [DOI] [PubMed] [Google Scholar]
- 14.Maione D, Di Carlo E, Li W, Musiani P, Modesti A, Peters M, Rose-John S, Della Rocca C, Tripodi M, Lazzaro D, Taub R, Savino R, Ciliberto G. Coexpression of IL-6 and soluble IL-6R causes nodular regenerative hyperplasia and adenomas of the liver. EMBO Journal. 1998;17:5588–5597. doi: 10.1093/emboj/17.19.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schirmacher P, Peters M, Ciliberto G, Blessing M, Lotz J, Meyer zum Buschenfelde KH, Rose-John S. Hepatocellular hyperplasia, plasmacytoma formation, and extramedullary hematopoiesis in interleukin (IL)-6/soluble IL-6 receptor double-transgenic mice. American Journal of Pathology. 1998;153:639–648. doi: 10.1016/S0002-9440(10)65605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zimmers TA, McKillop IH, Pierce RH, Yoo JY, Koniaris LG. Massive liver growth in mice induced by systemic interleukin 6 administration. Hepatology. 2003;38:326–334. doi: 10.1053/jhep.2003.50318. [DOI] [PubMed] [Google Scholar]
- 17.Chen RH, Chang MC, Su YH, Tsai YT, Kuo ML. Interleukin-6 inhibits transforming growth factor-beta-induced apoptosis through the phosphatidylinositol 3-kinase/Akt and signal transducers and activators of transcription 3 pathways. J Biol Chem. 1999;274:23013–23019. doi: 10.1074/jbc.274.33.23013. [DOI] [PubMed] [Google Scholar]
- 18.Kawano M, Hirano T, Matsuda T, Taga T, Horii Y, Iwato K, Asaoku H, Tang B, Tanabe O, Tanaka H, et al. Autocrine generation and requirement of BSF-2/IL-6 for human multiple myelomas. Nature. 1988;332:83–85. doi: 10.1038/332083a0. [DOI] [PubMed] [Google Scholar]
- 19.Kortylewski M, Heinrich PC, Mackiewicz A, Schniertshauer U, Klingmuller U, Nakajima K, Hirano T, Horn F, Behrmann I. Interleukin-6 and oncostatin M-induced growth inhibition of human A375 melanoma cells is STAT-dependent and involves upregulation of the cyclin-dependent kinase inhibitor p27/Kip1. Oncogene. 1999;18:3742–3753. doi: 10.1038/sj.onc.1202708. [DOI] [PubMed] [Google Scholar]
- 20.Miki S, Iwano M, Miki Y, Yamamoto M, Tang B, Yokokawa K, Sonoda T, Hirano T, Kishimoto T. Interleukin-6 (IL-6) functions as an in vitro autocrine growth factor in renal cell carcinoma. FEBS Letters. 1989;250:607–610. doi: 10.1016/0014-5793(89)80805-1. [DOI] [PubMed] [Google Scholar]
- 21.Kumagai N, Tsuchimoto K, Tsunematsu S, Toda K, Takeuchi O, Saito H, Morizane T, Tsuchiya M, Ishii H. Inhibition of growth of human hepatoma cells by dual-function antisense IL-6 oligonucleotides. Hepatol Res. 2002;22:119–126. doi: 10.1016/s1386-6346(01)00128-0. [DOI] [PubMed] [Google Scholar]
- 22.Kim H, Baumann H. Dual signaling role of the protein tyrosine phosphatase SHP-2 in regulating expression of acute-phase plasma proteins by interleukin-6 cytokine receptors in hepatic cells. Molecular & Cellular Biology. 1999;19:5326–5338. doi: 10.1128/mcb.19.8.5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klausen P, Pedersen L, Jurlander J, Baumann H. Oncostatin M and interleukin 6 inhibit cell cycle progression by prevention of p27kip1 degradation in HepG2 cells. Oncogene. 2000;19:3675–3683. doi: 10.1038/sj.onc.1203707. [DOI] [PubMed] [Google Scholar]
- 24.Moran DM, Koniaris LG, Jablonski EM, Cahill PA, Halberstadt CR, McKillop IH. Microencapsulation of engineered cells to deliver sustained high circulating levels of interleukin-6 to study hepatocellular carcinoma progression. Cell Transplant. 2006;15:785–798. doi: 10.3727/000000006783981477. [DOI] [PubMed] [Google Scholar]
- 25.Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol. 2004;5:836–847. doi: 10.1038/nrm1489. [DOI] [PubMed] [Google Scholar]
- 26.Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V, Taub R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 1996;274:1379–1383. doi: 10.1126/science.274.5291.1379. [DOI] [PubMed] [Google Scholar]
- 27.Huynh H, Nguyen TT, Chow KH, Tan PH, Soo KC, Tran E. Over-expression of the mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK in hepatocellular carcinoma: its role in tumor progression and apoptosis. BMC Gastroenterol. 2003;3:19. doi: 10.1186/1471-230X-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park HJ, Kim BC, Kim SJ, Choi KS. Role of MAP kinases and their cross-talk in TGF-beta1-induced apoptosis in FaO rat hepatoma cell line. Hepatology. 2002;35:1360–1371. doi: 10.1053/jhep.2002.33205. [DOI] [PubMed] [Google Scholar]
- 29.Sengupta TK, Talbot ES, Scherle PA, Ivashkiv LB. Rapid inhibition of interleukin-6 signaling and Stat3 activation mediated by mitogen-activated protein kinases. Proc Natl Acad Sci U S A. 1998;95:11107–11112. doi: 10.1073/pnas.95.19.11107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bravo J, Heath JK. Receptor recognition by gp130 cytokines. EMBO Journal. 2000;19:2399–2411. doi: 10.1093/emboj/19.11.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–134. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 32.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 33.Bellido T, O'Brien CA, Roberson PK, Manolagas SC. Transcriptional activation of the p21(WAF1,CIP1,SDI1) gene by interleukin-6 type cytokines. A prerequisite for their pro-differentiating and anti-apoptotic effects on human osteoblastic cells. J Biol Chem. 1998;273:21137–21144. doi: 10.1074/jbc.273.33.21137. [DOI] [PubMed] [Google Scholar]
- 34.Ernst M, Inglese M, Waring P, Campbell IK, Bao S, Clay FJ, Alexander WS, Wicks IP, Tarlinton DM, Novak U, Heath JK, Dunn AR. Defective gp130-mediated signal transducer and activator of transcription (STAT) signaling results in degenerative joint disease, gastrointestinal ulceration, and failure of uterine implantation. J Exp Med. 2001;194:189–203. doi: 10.1084/jem.194.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandberg EM, Ma X, VonDerLinden D, Godeny MD, Sayeski PP. Jak2 tyrosine kinase mediates angiotensin II-dependent inactivation of ERK2 via induction of mitogen-activated protein kinase phosphatase 1. J Biol Chem. 2004;279:1956–1967. doi: 10.1074/jbc.M303540200. [DOI] [PubMed] [Google Scholar]
- 36.Banerji L, Glassford J, Lea NC, Thomas NS, Klaus GG, Lam EW. BCR signals target p27(Kip1) and cyclin D2 via the PI3-K signalling pathway to mediate cell cycle arrest and apoptosis of WEHI 231 B cells. Oncogene. 2001;20:7352–7367. doi: 10.1038/sj.onc.1204951. [DOI] [PubMed] [Google Scholar]
- 37.Spiotto MT, Chung TD. STAT3 mediates IL-6-induced growth inhibition in the human prostate cancer cell line LNCaP. Prostate. 2000;42:88–98. doi: 10.1002/(sici)1097-0045(20000201)42:2<88::aid-pros2>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 38.Florenes VA, Lu C, Bhattacharya N, Rak J, Sheehan C, Slingerland JM, Kerbel RS. Interleukin-6 dependent induction of the cyclin dependent kinase inhibitor p21WAF1/CIP1 is lost during progression of human malignant melanoma. Oncogene. 1999;18:1023–1032. doi: 10.1038/sj.onc.1202382. [DOI] [PubMed] [Google Scholar]
- 39.Lu C, Kerbel RS. Interleukin-6 undergoes transition from paracrine growth inhibitor to autocrine stimulator during human melanoma progression.[erratum appears in J Cell Biol 1993 Apr;121(2):following 477] J Cell Biol. 1993;120:1281–1288. doi: 10.1083/jcb.120.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takizawa H, Ohtoshi T, Ohta K, Yamashita N, Hirohata S, Hirai K, Hiramatsu K, Ito K. Growth inhibition of human lung cancer cell lines by interleukin 6 in vitro: a possible role in tumor growth via an autocrine mechanism. Cancer Res. 1993;53:4175–4181. [PubMed] [Google Scholar]
- 41.Kerbel RS. Expression of multi-cytokine resistance and multi-growth factor independence in advanced stage metastatic cancer. Malignant melanoma as a paradigm. American Journal of Pathology. 1992;141:519–524. [PMC free article] [PubMed] [Google Scholar]
- 42.Lu C, Vickers MF, Kerbel RS. Interleukin 6: a fibroblast-derived growth inhibitor of human melanoma cells from early but not advanced stages of tumor progression. Proc Natl Acad Sci U S A. 1992;89:9215–9219. doi: 10.1073/pnas.89.19.9215. [DOI] [PMC free article] [PubMed] [Google Scholar]