

Abstract
The classical model of adaptive evolution in an asexual population postulates that each adaptive clone is derived from the one preceding it1. However, experimental evidence suggests more complex dynamics2-5 with theory predicting the fixation probability of a beneficial mutation as dependent on the mutation rate, population size, and the mutation's selection coefficient6. Clonal interference has been demonstrated in viruses7 and bacteria8, but has not been demonstrated in a eukaryote and a detailed molecular characterization is lacking. Here we use different fluorescent markers to visualize the dynamics of asexually evolving yeast populations. For each adaptive clone within one of our evolving populations, we have identified the underlying mutations, monitored their population frequencies and used microarrays to characterize changes in the transcriptome. These data provide the most detailed molecular characterization of an experimental evolution to date, and provide direct experimental evidence supporting both the clonal interference and the multiple mutation models.
In his seminal paper in 1932, Muller proposed that in asexual populations, adaptive events occur in succession1. This now classical model of evolution by clonal replacement has been the basis for experimental and theoretical work in microbial population genetics for three quarters of a century9,10. Support for this theory has come from studies of E. coli populations in which the data are consistent with the complete replacement of the population with an adaptive clone prior to the appearance of subsequent ones11. Furthermore, neutral marker frequency in evolving populations has been used to infer the occurrence and fixation of adaptive events, known as adaptive sweeps12,13. However, theory suggests that the chance of fixation of an adaptive mutation is dependent on multiple factors6,14 and mounting experimental evidence suggests that the classical model of clonal replacement may require substantial modification2,3,5,8,15. However, owing to the difficulty of establishing the exact provenance of newly arising adaptive clones, prior studies have focused on the end clones from experimental evolutionse.g. 3,16, and no empirical study has clearly demonstrated the dynamics of and characterized the molecular changes underlying adaptive events during evolution. Here, we report the use of different fluorescent markers to visualize the occurrence of specific adaptive events in evolving populations of Saccharomyces cerevisiae, which we use to facilitate the detailed molecular characterization of each.
To determine the population structure during adaptive evolution in asexual populations growing in continuous culture, we seeded glucose-limited chemostat cultures with equal numbers of three fluorescently marked otherwise isogenic haploid yeast strains, and monitored their relative population sizes by FACS. The data (Figure 1A) show that subpopulations expand at various times during the in vitro evolution, suggesting the presence of an adaptive clone within a particular expanding subpopulation. However, these expansion events did not lead to clonal replacement; instead, we observed that a differently colored subpopulation would subsequently expand, and the previously expanding population would contract. In additional experiments (Figure 1B) we observed the same phenomenon, whereby the expansion of subpopulations rarely resulted in clonal replacement and adaptive events typically did not fix in the population.
Fig 1.
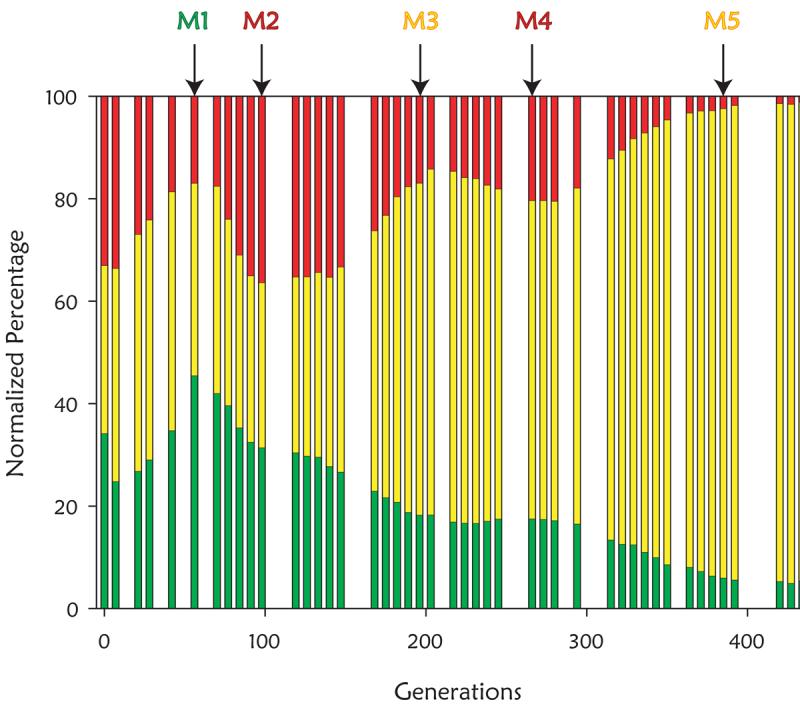
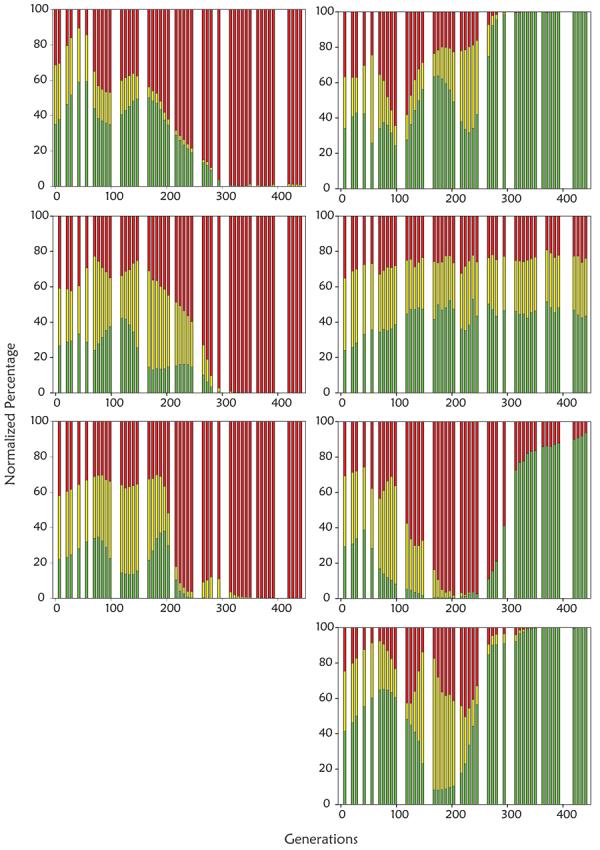
Population dynamics during evolution. The green, red, and yellow bars represent the proportions of each subpopulation of the population as a whole at each generation. A. Experimental population (size of approximately 109 cells) from which adaptive clones marked M1-M5 were isolated and subsequently characterized. The times when putative adaptive subpopulations reach their maxima are marked with arrows. Consistent with previous reports13, these adaptive events occur in the glucose-limited population approximately every 50-100 generations. B. Population dynamics of seven additional populations evolved under glucose limitation. The populations on the right contain approximately108 cells, while the populations on the left contain approximately 109 cells.
To further establish the existence of adaptive mutations in the expanding subpopulations, we sorted samples from the population depicted in Figure 1A by FACS from generations where an expanding subpopulation reached its maximum proportion. Using pairwise fitness assays, the most fit adaptive clones from the expanding red, green, or yellow subpopulations from generations 56, 91, 196, 266, 385, as indicated in Figure 1A, were identified (see Methods for details). These we refer to as M1 through M5 (Supplementary Table 1). Fitness coefficients were used to estimate whether each adaptive subpopulation was comprised solely of their respective adaptive clones (Figure 2). The fitness coefficients showed each adaptive subpopulation, other than that containing M2, isolated at generation 91, to be an impure mixture of adaptive clones and wild-type or less fit clones, because there were significant differences between the fitness of the adaptive clone and the adaptive subpopulation in one or both of the competition experiments shown in Figure 2. While M2 was not more fit than the previous adaptive clone, M1, it was fitter than the original parents. Thus, while M2 was able to outcompete the residual parental population, it was not able to out compete M1 itself in a direct competition, which is consistent with the data shown in Figure 1A.
Fig 2.

Fitness coefficients of adaptive clones (in brown) and adaptive subpopulations (the color of the subpopulation is designated by the color of the generation number under each measurement) from which the adaptive clones were isolated (in orange) versus A) the original parents and B) the immediately preceding adaptive clone, measured in triplicate. Error bars are +/− one standard deviation from the mean. Fitness coefficients of adaptive clones that are statistically different from the adaptive subpopulation they were isolated from, as determined using a one tailed t-test with p-value cut off of 0.05, are denoted with an asterisk.
Single nucleotide mutations, chromosomal rearrangements, gene duplications, and transposon insertions are important evolutionary mechanisms by which adaptation occurs, and examples of all of these have been found in experimental evolution3,15,17-20. To investigate the underlying genome changes in our adaptive clones, we utilized Affymetrix yeast tiling arrays, as described by Gresham et al21, with modifications (see Methods section). A total of nine point mutations, one frameshift mutation, two amplifications, and one insertion was observed in clones M1 through M5 (Table 1). Clones M2 and M4 share an identical nucleotide change on chromosome 7, strongly suggesting that a succession of adaptive events occurred within the red subpopulation. In the yellow subpopulation, however, the later adaptive clone did not arise from the earlier one, as the mutation observed in M3 was not shared by M5. Thus, just as adaptive events do not always sweep the entire population (Figure 1), the same is true within a subpopulation. Amplifications of the HXT6/7 locus were observed in the independent lineages M4 and M5. Amplification of this locus has been observed previously17,18, suggesting that this mutation is adaptive and relatively frequent.
Table 1.
Genotyping results for adaptive clones analyzed.
| Clone | Genome modifications | Gene | Mutation | Amino acid residue change |
Comment | |
|---|---|---|---|---|---|---|
| Chromosome | Position | |||||
| M1 Green |
4 | 1014687 | MTH1 | C to T | Glu338 to Stop |
Negative regulator of the glucose- sensing signal transduction pathway |
| M2 Red |
7 | 617107 | COX18 | T to A | Leu59 to His |
Mitochondrial inner membrane protein |
| M3 Yellow |
2 | 521875 | IRA1 | G to A | Arg1583 to Lys |
GTPase-activating protein that negatively regulates Ras |
| M4 Red |
2 | 616441 | TAF5 | G to T | Gly693 to Val |
Subunit (90 kDa) of TFIID and SAGA complexes |
| 4 | HXT6/HXT7 | amplification | High-affinity glucose transporter | |||
| 6 | 73427 | RIM15 | Single base pair deletion |
frame shift on codon 333 |
Glucose-repressible protein kinase | |
| 7 | 617107 | COX18 | T to A | Leu59 to His |
Mitochondrial inner membrane protein | |
| 11 | 64698 | MNN4 | A to G | Lys924 to Glu |
Putative positive regulator of mannosylphosphate transferase |
|
| 16 | 912523 | T to G | intergenic | |||
| M5 Yellow |
1 | 39602 | GPB2 | insertion of ∼340bp from Ty1 LTR |
Multistep regulator of cAMP-PKA signaling |
|
| 4 | HXT6/HXT7 | amplification | High-affinity glucose transporter | |||
| 15 | 893332 | SLY41 | G to T | Trp253 to Leu |
Protein involved in ER-to-Golgi transport |
|
| 16 | 422266 | MUK1 | C to A | Ser441 to stop |
Protein of unknown function; computational analysis of large-scale protein-protein interaction data suggests a possible role in transcriptional regulation |
|
Several mutations observed in the adaptive clones were in genes involved in glucose signaling, either via glucose transport or the protein kinase A (PKA) signaling pathway; this is not surprising, considering that these cultures were grown under glucose-limited conditions. Two independent mutations involved in the PKA signaling pathway were found in adaptive clones M3 and M5. The IRA1 missense mutation found in M3 changes an Arg residue to a Lys; it is unknown how this might affect the activity of the gene product. The GPB2 mutation in M5 was caused by the insertion and of a Ty1 element in the coding region of the gene, likely inactivating its gene product. In addition, a frame shift mutation was observed in RIM15 in M4, also likely rendering its gene product nonfunctional; RIM15 is a protein kinase conserved in the Saccharomyces sensu stricto that acts downstream of and is inhibited by PKA, and is involved in nutrient signaling in yeast. From the population frequency data, it is unclear whether the RIM15 mutation is adaptive (see below). If the IRA1 mutation is hypomorphic, then both the GPB2 and IRA1 mutations would be expected to result in a phenotype that reproduces some aspects of increased signaling through the PKA pathway. Indeed, transcriptome data suggest an increase in PKA signaling in M5 (see Supplementary Materials). In M1, a nonsense mutation was found in the conserved gene MTH1, which is involved in the PKA-independent glucose-signaling pathway, and negatively regulates transcription of the hexose transporters. The transcriptional profile of M1 showed an increase in expression of the hexose transporters and also suggests an increase in PKA signaling in this clone; a possible cause and effect scenario is shown in Supplementary Figure 1.
To determine population frequencies for the mutations, the order of occurrence of the mutations, and potentially which mutations are adaptive, we performed allele specific RT-PCR22 on population samples drawn from across the evolutionary time course (Figure 3). These data suggest that in clone M4 the observed mutations in TAF5, RIM15, and the intergenic mutation on chromosome 16 are either not adaptive, or were lost to drift, as their frequencies never reached a detectable threshold. Sequencing of six additional clones at those loci from the generation at which M4 was isolated also showed no evidence of these mutations being present. The population dynamics of the other mutations in M4 suggest that they occurred in the following order: COX18, HXT6/7 amplification, and then MNN4. In the case of clone M5, it appears that the HXT6/7 amplification was the first adaptive event, followed by the mutation in MUK1, which is currently a gene of unknown function, yet appears strongly adaptive under these conditions. The GPB2 and SLY41 mutations started to expand in the population towards the end of the evolution, however, it is currently unclear whether either or both of these mutations are adaptive. Interestingly, RT-PCR revealed that the HXT6/7 amplification also arose in the green subpopulation, and thus arose independently in all three fluorescently labeled subpopulations. The data strongly suggest that towards the ends of the evolution, the HXT6/7 amplification had fixed in all three subpopulations, indicating 3 independent occurrences of this amplification event
Fig 3.
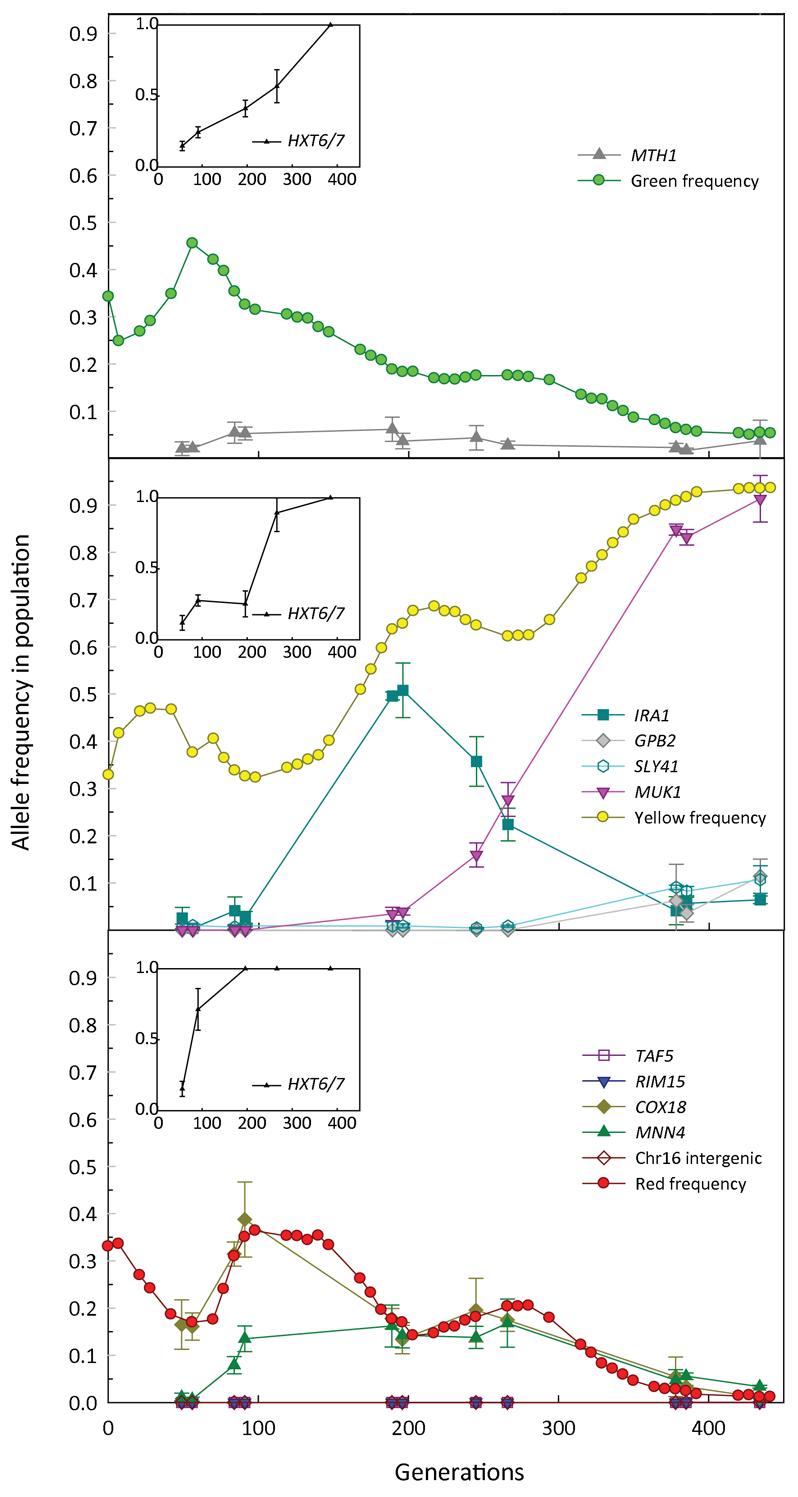
The frequencies of the observed alleles in the entire population, and the frequencies of HXT amplifications within each of the subpopulations; the green subpopulation shown in top panel, yellow subpopulation in the middle panel, and the red subpopulation in the bottom panel. Since the HXT6/7 amplification estimation was not as accurate as for the other mutations, and is a measure of the mean copy number within the subpopulation (see Supplementary Materials), it is plotted separately from the other allele frequencies, in the inset boxes. The red, yellow and green circles denote the frequency of those fluorescently labeled subpopulations.
The MTH1 mutation found in clone M1 was present in the population at low frequency, and sequencing of six additional clones from the green population did not identify another MTH1 mutant. This suggests that the observed decline in the green population after generation 56, when the COX18 mutation was in a higher frequency in the population than the MTH1 mutation was most likely due to the loss of residual parental green subpopulation. However, it also suggests that the initial expansion of green was not entirely due to the MTH1 mutation, as that expansion cannot be solely due to the subpopulation carrying the MTH1 mutant allele. By the end of the evolution, of the five observed putative adaptive mutations, and the additional three independently derived hexose transporter amplifications, only half would likely have survived in the population, those in MUK1, SLY41 and GPB2, and the HXT6/7 amplification in the yellow population (though the adaptive significance of the SLY41 and GPB2 mutations is currently unknown), of which only two approach fixation. The other observed adaptive mutations would likely have been lost to clonal interference.
We identified genes whose expression significantly changed between the adaptive clones and the original parents using microarrays (see Supplementary Materials). We compared our data to those of Wang et al23, who characterized transcriptional changes in response to increased PKA signaling. In two of our adaptive clones, M1 and M5, the data are consistent with an increase in PKA signaling, as the genes whose expression is either significantly increased or decreased in those clones also show increased or decreased expression respectively in their data. An increase in PKA signaling in M5 would be expected, given that Gpb2 is an inhibitor of this pathway, and M5's mutation in GPB2 is an insertion that is likely inactivating. However, no mutations were found in the components of the PKA signaling pathway in M1. A possible mechanism may be that the induced hexose transporter gene expression, as a result of the missense mutation in MTH1, leads to increased transport of glucose into the cell, which in turn increases the activity of adenylate cyclase, resulting in a more active PKA (Supplementary Figure 1). The occurrence of independent mutations that lead to increased PKA signaling suggests that mutations in this pathway may be readily adaptive under glucose-limited conditions.
In large asexual populations, the occurrence of competing beneficial mutations in the population will interfere with the expansion of and may result in the elimination of an existing adaptive lineage. Indeed, this may be the basis for a “speed limit” that constrains the pace of adaptive evolution in such populations, even when the mutation rate is increased by the presence of so-called “mutators”24,25. All things being equal, clonal interference should increase the complexity of population structure, and place limits on the selective advantage enjoyed by adaptive clones within an evolutionary succession. Clonal interference has been recently incorporated into theoretical models of microbial evolution6,26,27, and its effects either inferred or observed in experimental studies of E. coli and viruses7,24. The non-serial occurrences of adaptive events and the incomplete adaptive sweeps observed in our data provide direct support for clonal interference as an important phenomenon in yeast evolving asexually.
A recent multiple mutation model developed by Desai et al28,29, proposed that subsequent adaptive mutations arise within the currently most adaptive clone, even in the presence of clonal interference. Under this multiple mutations model, we might expect later arising adaptive clones to contain at least one beneficial mutation that occurred on top of a previously observed adaptive clone. Our data indicate that the second observed red adaptive clone, M4, which is the penultimate clone isolated in our experiment, clearly arose from a prior red adaptive clone, M2. However, while the final adaptive clone that we isolated, M5, contains multiple mutations, the only other observable expansion in the yellow subpopulation, M3, is not the founding lineage (Table 1). However, the population frequency data clearly establish the presence of multiple adaptive mutations within clone M5, whose expansion was not obvious from the FACS data. The presence of multiple adaptive mutations in clones M4 and M5 supports the theory that multiple mutations are important for adaptive evolution in large populations. In summary, our data provide the most detailed molecular characterization of adaptive evolution in a eukaryote to date and unequivocally demonstrate that in sufficiently large evolving asexual populations of S. cerevisiae, the trajectory across the adaptive landscape is determined by clonal interference, rather than clonal replacement.
Methods
Strains
Yeast strains (Supplementary Table 2) were derived from FY2, a derivative of S288c. Fluorescently tagged strains were constructed by integrating plasmids pGS62, pGS63, and pGS64 into FY2 (see Supplementary Methods).
Evolution experiments
30 ml chemostat vessels were inoculated with equal numbers of each fluorescently marked yeast strain in 20ml. The populations were evolved at steady-state under glucose-limited conditions (0.08% glucose in Delft minimal medium) at 30°C with a dilution rate of 0.2 hr−1. The proportion of each fluorescently marked sub population was monitored by FACS. We determined that the FACS data were reproducible (Supplementary Figure 2) and that none of the different colored populations had a significant advantage or disadvantage in either serial batch or short-term continuous cultures (Supplementary Figure 3).
Identification and Isolation of each adaptive mutant
Based on the population dynamics (Figure 1A), we determined which of our samples should have a substantial fraction of an adaptive clone in one of the fluorescently marked sub-populations, which was then separated out using FACS. Seven independent colonies were picked from each sorted sample for fitness measurements.
Each clone was competed in continuous culture against the previous adaptive mutant and the clone with highest fitness coefficient was chosen as the adaptive clone. In the case of the red subpopulation from generation 91, none of the clones showed a fitness advantage compared to the preceding green-marked adaptive clone. We compared these red clones against the original parent and determined that they were all similarly fitter, and picked one arbitrarily as the adaptive clone.
Pairwise competition experiments
Chemostat experiments in 0.08% glucose Delft minimal medium were started with equal numbers of each of the two clones to be competed, which had different fluorescent markers at a dilution rate of 0.2 hr−1 at 30°C. In the case of competition involving the adaptive subpopulation, a mixture of cells patched on 2% glucose minimum medium plates were used. Samples were taken every ∼8-15 hours and the culture compositions were measured by FACS. The rate of expansion of the fitter strain was plotted on a semi-log scale, and the fitness coefficient calculated at the linear range of expansion as:
where x is the normalized proportion of the fitter clone, y is the normalized proportion of the less fit clone, Δt is the difference in generations between t2 and t1, and s is the differential growth rate per generation between the two clones. Relative fitness coefficient is (1 + s)/1. Experiments were repeated at least three times.
Mutation analysis
Affymetrix Yeast tiling v1.0R arrays were used for finding mutations in the evolved strains, by comparing the evolved strains' and parent strains' data. Only the perfect match (PM) data from the resulting CEL files were used for analysis. Probes that span repeated regions or transposable elements were filtered out. Normalization factors were computed for each chromosome by averaging the signals for all the probes (after filtering) in each strain. The difference between the log2 transformed average signal for each chromosome in the two strains was used as a normalization factor. For each unfiltered probe, the log2 ratios of the evolved strain over parent strain intensities were calculated, and normalized using the difference calculated above for the appropriate chromosome. For each chromosome, a value, j, for chromosome position i was calculated by averaging the normalized log2 ratios of probes between positions i+6 to i+18. Potential single base mutations or deletions were identified by looking for at least 4 consecutive j's whose values were less than the mean minus 3 standard deviations (the standard deviation was calculated for the whole chromosome). Potential amplifications were identified by a long stretch of consecutive j's, whose values were greater than the mean plus 3 standard deviations.
Sequencing
Potential mutations were validated by Sanger sequencing using the parental strains as negative controls (primers are listed in Supplementary Table 3).
Population allelic frequency determination
The population frequencies of identified mutations were determined by kinetic PCR22 using either the iCycler (Bio-Rad Laboratories) or the ABI 7900HT (Applied Biosciences Inc.). Each reaction mixture consisted of 20 ng (iCycler) or 10 ng (ABI 7900HT) of total genomic DNA, 0.5 μM primers, and either iQ SYBR Green Supermix (iCycler; Bio-Rad) or iTaq SYBR Green Supermix with ROX (ABI 7900HT; Bio-Rad) in final reaction volume of 20 μl (iCycler) or 10 μl (ABI 7900HT). The reaction conditions are: iCycler (95°C 3 minutes followed by 40 cycles of 95°C 20 seconds, 63°C 20 seconds, 72°C 20 seconds) and ABI 7900HT (95°C 3 minutes followed by 40 cycles of 95°C 15 seconds, 60°C 1 minute). The approximate frequency of the HXT6/7 amplification was only determined from the sorted subpopulations. The calculations for the frequencies of the HXT6/7 amplification in the subpopulations were based on a copy number of 8 for this region. The primers used are listed in Supplementary Table 3. Control experiments demonstrating the abilities of the primers to distinguish between the two different alleles are shown in the Supplementary Figure 3.
RNA purification and gene expression analysis
Single colonies were used to inoculate overnight cultures in 2% glucose Delft minimal medium grown at 30°C, which were then used to inoculate glucose-limited chemostat cultures. Cells were harvested approximately 48 hours after inoculation, filtered using 0.45μm analytical test filter funnels (Nalgene), snap-frozen in liquid nitrogen, and stored at −80°C for RNA purification at a later time. Each experiment was repeated 3 times.
RNA was extracted using a hot acid phenol method and total RNA was eventually resuspended in nuclease-free water. Using the Low RNA Input Linear Amplification Kit, Two-Color (Agilent), 375ng of purified total RNA and RNA Spike-In control (Agilent) were labeled with either 6nmol of Cy3-CTP or Cy5-CTP (GE Healthcare) and hybridized to Agilent 8×15k yeast catalog arrays, which were then washed and scanned. Feature Extraction v 9.1.5 (Agilent) was used to normalize the data using the Spike-In controls.
To identify differentially expressed genes, each set of data was analyzed using the software Significance Analysis of Microarray30 in a one-class analysis. A delta value was chosen to obtain a set of statistically differentially regulated genes with a false discovery rate of approximately 1%.
Supplementary Material
Acknowledgements
This work was supported by NIH grants R01 HG003328 (GS) and F32 GM079113 (KK). We thank Julia Ying for help with sample handling, Elizabeth Tanner for carrying out some of the gene expression microarray experiments, Hua Tang for discussions on experimental design, Joe Horecka for help with RT-PCR, and Frank Rosenzweig, Barbara Dunn and Arend Sidow for comments on the manuscript.
Footnotes
Accession Numbers
The microarray expression data are available at GEO with accession GSE11071.
The authors declare no competing financial interests.
References
- 1.Muller HJ. Some genetic aspects of sex. American Naturalist. 1932;66:118–138. [Google Scholar]
- 2.Helling RB, Vargas CN, Adams J. Evolution of Escherichia coli during Growth in a Constant Environment. Genetics. 1987;116:349–358. doi: 10.1093/genetics/116.3.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Notley-McRobb L, Ferenci T. The generation of multiple co-existing mal-regulatory mutations through polygenic evolution in glucose limited populations of Escherichia coli. Environmental Microbiology. 1999;1:45–52. doi: 10.1046/j.1462-2920.1999.00003.x. [DOI] [PubMed] [Google Scholar]
- 4.Notley-McRobb L, Ferenci T. Experimental analysis of molecular events during mutational periodic selections in bacterial evolution. Genetics. 2000;156:1493–1501. doi: 10.1093/genetics/156.4.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenzweig RF, Sharp RR, Treves DS, Adams J. Microbial Evolution in a Simple Unstructured Environment - Genetic Differentiation in Escherichia coli. Genetics. 1994;137:903–917. doi: 10.1093/genetics/137.4.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerrish PJ, Lenski RE. The fate of competing beneficial mutations in an asexual population. Genetica. 1998;103:127–144. [PubMed] [Google Scholar]
- 7.Miralles R, Gerrish PJ, Moya A, Elena SF. Clonal interference and the evolution of RNA viruses. Science. 1999;285:1745–1747. doi: 10.1126/science.285.5434.1745. [DOI] [PubMed] [Google Scholar]
- 8.Hegreness M, Shoresh N, Hartl D, Kishony R. An equivalence principle for the incorporation of favorable mutations in asexual populations. Science. 2006;311:1615–1617. doi: 10.1126/science.1122469. [DOI] [PubMed] [Google Scholar]
- 9.Crow JF, Kimura M. Evolution in Sexual and Asexual Populations. American Naturalist. 1965;99:439–450. [Google Scholar]
- 10.Paquin C, Adams J. Relative fitness can decrease in evolving asexual populations of S. cerevisiae. Nature. 1983;306:386–371. doi: 10.1038/306368a0. [DOI] [PubMed] [Google Scholar]
- 11.Atwood KC, Schneider LK, Ryan FJ. Periodic selection in Escherichia coli. Genetics. 1951;37:146–55. doi: 10.1073/pnas.37.3.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novick A, Szilard L. Experiments with the Chemostat on Spontaneous Mutations of Bacteria. Proceedings of the National Academy of Sciences of the United States of America. 1950;36:708–719. doi: 10.1073/pnas.36.12.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paquin C, Adams J. Frequency of fixation of adaptive mutations is higher in evolving diploid than haploid yeast populations. Nature. 1983;302:495–500. doi: 10.1038/302495a0. [DOI] [PubMed] [Google Scholar]
- 14.Kim Y, Orr HA. Adaptation in sexuals vs. asexuals: Clonal interference and the Fisher-Muller model. Genetics. 2005;171:1377–1386. doi: 10.1534/genetics.105.045252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adams J, Oeller PW. Structure of Evolving Populations of Saccharomyces cerevisiae - Adaptive-Changes Are Frequently Associated with Sequence Alterations Involving Mobile Elements Belonging to the Ty Family. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:7124–7127. doi: 10.1073/pnas.83.18.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herring CD, et al. Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale. Nat Genet. 2006;38:1406–12. doi: 10.1038/ng1906. [DOI] [PubMed] [Google Scholar]
- 17.Brown CJ, Todd KM, Rosenzweig RF. Multiple duplications of yeast hexose transport genes in response to selection in a glucose-limited environment. Mol Biol Evol. 1998;15:931–42. doi: 10.1093/oxfordjournals.molbev.a026009. [DOI] [PubMed] [Google Scholar]
- 18.Dunham MJ, et al. Characteristic genome rearrangements in experimental evolution of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2002;99:16144–9. doi: 10.1073/pnas.242624799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Segre AV, Murray AW, Leu JY. High-resolution mutation mapping reveals parallel experimental evolution in yeast. Plos Biology. 2006;4:1372–1385. doi: 10.1371/journal.pbio.0040256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blanc VM, Adams J. Evolution in Saccharomyces cerevisiae: identification of mutations increasing fitness in laboratory populations. Genetics. 2003;165:975–83. doi: 10.1093/genetics/165.3.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gresham D, et al. Genome-wide detection of polymorphisms at nucleotide resolution with a single DNA microarray. Science. 2006;311:1932–1936. doi: 10.1126/science.1123726. [DOI] [PubMed] [Google Scholar]
- 22.Germer S, Holland MJ, Higuchi R. High-throughput SNP allele-frequency determination in pooled DNA samples by kinetic PCR. Genome Res. 2000;10:258–66. doi: 10.1101/gr.10.2.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, et al. Ras and Gpa2 mediate one branch of a redundant glucose signaling pathway in yeast. Plos Biology. 2004;2:610–622. doi: 10.1371/journal.pbio.0020128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Visser JA, Rozen DE. Clonal interference and the periodic selection of new beneficial mutations in Escherichia coli. Genetics. 2006;172:2093–100. doi: 10.1534/genetics.105.052373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Visser JAGM, Zeyl CW, Gerrish PJ, Blanchard JL, Lenski RE. Diminishing returns from mutation supply rate in asexual populations. Science. 1999;283:404–406. doi: 10.1126/science.283.5400.404. [DOI] [PubMed] [Google Scholar]
- 26.Gerrish P. The rhythm of microbial adaptation. Nature. 2001;413:299–302. doi: 10.1038/35095046. [DOI] [PubMed] [Google Scholar]
- 27.Rozen DE, de Visser JAGM, Gerrish PJ. Fitness effects of fixed beneficial mutations in microbial populations. Current Biology. 2002;12:1040–1045. doi: 10.1016/s0960-9822(02)00896-5. [DOI] [PubMed] [Google Scholar]
- 28.Desai MM, Fisher DS. Beneficial mutation-selection balance and the effect of linkage on positive selection. Genetics. 2007;176:1759–1798. doi: 10.1534/genetics.106.067678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Desai MM, Fisher DS, Murray AW. The speed of evolution and maintenance of variation in asexual populations. Curr Biol. 2007;17:385–94. doi: 10.1016/j.cub.2007.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response (vol 98, pg 5116, 2001) Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10515–10515. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.