


Abstract
HIV-1 integrase (IN) oligomerization and DNA recognition are crucial steps for the subsequent events of the integration reaction. Recent advances described the involvement of stable intermediary complexes including dimers and tetramers in the in vitro integration processes, but the initial attachment events and IN positioning on viral ends are not clearly understood. In order to determine the role of the different IN oligomeric complexes in these early steps, we performed in vitro functional analysis comparing IN preparations having different oligomerization properties. We demonstrate that in vitro IN concerted integration activity on a long DNA substrate containing both specific viral and nonspecific DNA sequences is highly dependent on binding of preformed dimers to viral ends. In addition, we show that IN monomers bound to nonspecific DNA can also fold into functionally different oligomeric complexes displaying nonspecific double-strand DNA break activity in contrast to the well known single strand cut catalyzed by associated IN. Our results imply that the efficient formation of the active integration complex highly requires the early correct positioning of monomeric integrase or the direct binding of preformed dimers on the viral ends. Taken together the data indicates that IN oligomerization controls both the enzyme specificity and activity.
INTRODUCTION
The HIV-1 integration of viral DNA into the cellular genome catalyzed by integrase (IN) is a key step in the biological cycle of the virus. In the first step, IN removes two nucleotides from both 3′-ends of the blunt-ended DNA generated by reverse transcriptase (RT). In the second step, the resulting 3′ OH ends, through a trans-sterification reaction, integrate the processed viral molecule into the target DNA with a cleavage site separated by 5 bp in the case of HIV. In the cells, the viral DNA is part of a large nucleoprotein complex called the pre-integration complex (PIC) (1). Integrase and other viral and cellular factors are associated with the viral DNA in the PIC and transported to the nucleus where integration takes place. PIC purified from infected cells can integrate the viral DNA in vitro with a high fidelity and display the hallmarks of HIV-1 integration (2–4).
Both 3′ processing and strand transfer can be partially reproduced in vitro by using DNA substrates mimicking the viral ends and pure recombinant IN (5–7). However, in these assays, only one viral end is cleaved and/or integrated (half site integration, HSI) in contrast to the two viral LTR integration process observed in vivo (full-site integration, FSI). More recent studies have shown that in vitro recombinant IN is able to catalyze concerted integration in the absence of other cofactors (8–10). However, in addition to the requirement of nonphysiological compounds such as PEG and DMSO, HSI always appeared more tolerant to the reaction conditions than FSI. This suggests that in vitro IN does not fold in a proper active structure. Some parameters are known to improve FSI, such as the use of unprocessed blunt-ended substrates (11). But, despite the numerous data obtained concerning the concerted integration reaction, the different requirements for FSI and HSI remains still obscure.
Functional analysis of the INċDNA complexes using chemical crosslink, time-resolved fluorescence anisotropy and two-dimensional gel electrophoresis revealed specific reaction intermediates (12–14). Data indicate that, retroviral DNA integration occurs in the context of a series of highly stable nucleoprotein complexes called stable synaptic complex (SSC) and strand transfer complex (STC), leading to a tetrameric IN complex bound to both viral and target DNA (13).
This sequence of events leading to the formation of the integration complex remains unclear, particularly the initial association between IN and DNA leading to the tetramer associated with both viral ends. At least three questions remain to be answered: (i) How the positioning of IN takes place on the viral ends? (ii) Does the IN monomer play a role in this early step of the integration process? (iii) Does the IN dimerize after binding to the LTR leading to an association into the tetrameric form?
Previously, multimerization of IN following interaction with DNA was revealed by small angle X-ray scattering (SAXS) and time-resolved fluorescence anisotropy (15–17). Those results strongly suggest the involvement of different IN oligomers depending on the nature of the DNA substrate.
Here we provide a functional study of the different IN oligomers populations found in solution. Comparison of IN activity using enzymes at different oligomerization states showed that the integration activity was highly dependent both on viral end sequences for the formation of specific IN oligomers, or the presence of preformed catalytically active oligomers in solution. Our data also demonstrate that multimerization of IN on nonspecific DNA leads to the formation of a new functional variant of IN able to generate nonspecific random double-strand breaks. Taken together our findings strongly suggest that (i) enzyme specificity is determined by its oligomerization and (ii) the initial binding of the enzyme to the viral extremities, and thus its correct positioning on the LTRs, constitutes a prerequisite step in the formation of active complexes.
MATERIALS AND METHODS
HIV-1 IN
Standard purification was performed essentially as previously described (18). The soluble fraction containing the HIV-1 IN obtained from JSC 310 (IN), expressing IN protein was loaded on a Hitrap butyl-sepharose 4B column (1 ml, Pharmacia-LKB), washed with LSC buffer (50 mM HEPES pH 7.6, 0.2 M NaCl, 0.1 mM EDTA, 1 mM DTT, 7 mM CHAPS, 10% glycerol) and equilibrated with 5 volumes HSC buffer (50 mM HEPES pH 7.6, 0.2 M NaCl, 1 M ammonium sulfate, 0.1 mM EDTA, 1 mM DTT, 7 mM CHAPS). Proteins were eluted by a decreasing ammonium sulfate gradient (1–0 M). Fractions containing IN activity were pooled and 7 mM CHAPS was added. Pooled fractions were 1/3 diluted with 50 mM HEPES pH 7.6, 0.1 mM EDTA, 1 mM DTT, 10% glycerol, 7 mM CHAPS and loaded on a Hitrap Heparine Sepharose CL-4B column (1 ml, Pharmacia-LKB), washed with 5 volumes HS buffer (50 mM HEPES pH 7.6, 1 M NaCl, 0.1 mM EDTA, 1 mM DTT, 10% glycerol, 7 mM CHAPS) and equilibrated with a linear NaCl gradient (0–1 M NaCl). Fractions containing IN activity (eluted at 300 mM NaCl) were pooled and either concentrated or not by ultrafiltration (Centricon Millipore), followed by addition of 7 mM CHAPS. ZnSO4, 50 µM, was added if necessary in the stock fraction. Purified IN was kept at −80°C in 300 mM NaCl. Proteins were analyzed by electrophoresis in a 12% SDS–PAGE and western blotting using a polyclonal anti-IN antibody (Invitrogen, Carlsbad, CA, USA). Dilutions of the IN protein were carried during various times reported in the results section in dilution buffer without cation (HEPES 20 mM, pH 7.6) or with either Mg++ (7.5 mM MgCl2, HEPES 20 mM, pH 7.6) or Mn++ (7.5 mM MnCl2, HEPES 20 mM, pH 7.6, standard conditions).
Detergent free enzyme was purified following the new purification procedure leading to INHybrid enzyme. For that purpose CHAPS 7 mM was used during extraction but omitted during the final purification steps and ZnSO4 50 µM was added in all the chromatography buffers described above.
Determination of oligomerization state
Gel filtration chromatography
Purified IN was diluted in 1 ml loading solution (50 mM HEPES pH 7.5, 7 mM CHAPS, 1 mM DTT, 150 mM NaCl, 0.1 mM EDTA) to a final enzyme concentration of 150 nM and chromatographed through a Smart Superose 12 (Pharmacia-LKB) on the Ettan LC system. The void volume was determined with blue dextran (>2000 kDa) and the column was calibrated with aldolase (158 kDa), bovine serum albumin (67 kDa), ovalbumin (43 kDa) and chymotrypsinogen A (25 kDa) (Pfizer, Kirkland, Québec). Proteins were eluted with a flow rate of 0.04 ml/min and recorded by continuously monitoring the absorbance at 280 nm. Prior to chromatography, samples were centrifuged for 10 min at 10 000 r.p.m. to remove large protein aggregates.
Disuccinimidyl suberate crosslink
One picomole of purified IN was incubated for 30 min at 22°C (standard conditions) with 0.8 µg of Disuccinimidyl suberate (DSS) in a HEPES 50 mM pH 7.5 buffer and at a final NaCl concentration of 30 mM in a total volume of 20 µl. The crosslinked products were separated on 12% SDS–PAGE gel and detected by western blotting using a polyclonal anti-IN antibody (Invitrogen).
SAXS
SAXS experiments were performed as described by Baranova et al. (17). SAXS patterns were obtained with a Siemens diffractometer (Germany) by step-by-step scanning using a goniometer and an X-ray scintillation detector. Small-angle roentgenograms were measured in the angular range h = 0.013–0.22 Å−1, where h = 4π sinθ/λ, 2θ, is the scattering angle, and λ is the X-ray wavelength. The first step in mathematical processing of the SAXS data and computational checks of functions for size distribution of spherical particles were performed using the computer program and algorithms described earlier (19) as well as optimization programs (20).
In vitro activities
Processing and strand transfer
Standard assays were performed as described previously (21) in 20 mM HEPES pH 7.5, 10 mM DTT, 7.5 mM MnCl2, 0.05% NP40 in a total volume of 20 µl. The final NaCl concentration was adjusted at 30 mM in all reactions. The reaction mixture was incubated at 37°C for 1 h in the presence of IN (50 nM) and radiolabeled oligonucleotides (50 nM) and the incubation was stopped by adding 10 µl of loading buffer (95% formamide, 20 mM EDTA, 0.05% bromophenol blue) and heating at 90°C for 5 min (standard conditions). The reaction products were analyzed by electrophoresis on 15% polyacrylamide gels with 7 M urea in Tris–borate–EDTA pH 7.6 and autoradiographed. The sequence of the ODNs used to perform the processing and strand transfer assays were the following:
ODN 70: 5′GTGTGGAAAATCTCTAGCAGT3′,
ODN 71: 5′GTGTGGAAAATCTCTAGCA3′,
ODN 72: 5′ACTGCTAGAGATTTTCCACAC3′.
To perform the 3′ processing assay, the 5′ radiolabeled ODN 70 hybridized to ODN 72 was used as a substrate while the 5′ radiolabeled ODN 71 hybridized to ODN 72 was used as a substrate in the strand transfer reaction.
The unlabeled 21 bp hybrid 71–72 was used as specific ODN for the reassociation experiments. As a control of specificity a 21 bp random ODN was also used and obtained by hybridization of the following ODNs:
Random21_5′: 5′CGTAAGGTCATTTCAACTGAT3′.
Random21_3′: 3′ATCAGTTGAAATGACCTTACG3′.
Concerted integration DNA substrates
Standard concerted integration reactions were performed as described previously (22), except that no cellular proteins were added. Briefly, purified HIV-1 IN (50 nM) was pre-incubated with both the 5′-end-labeled donor DNA (10 ng) containing the 3′-processed U3 and U5 LTR sequences and the target DNA plasmid pBSK+ (100 ng) at 0° C for 20 min in a total volume of 5 µl. Then the reaction mixture (20 mM HEPES pH 7.5, 10 mM DTT, 7.5 mM MgCl2, 10% DMSO, 8% PEG, 30 mM NaCl) was added and the reaction proceeded for 90 min at 37°C. Incubation was stopped by adding a phenol/isoamyl alcohol/chloroform mix (24/1/25 v/v/v). The aqueous phase was loaded on a vertical 1% agarose gel in the presence of 1% bromophenol blue and 1 mM EDTA. After separation of the products, the gel was treated with 5% TCA for 20 min, dried and autoradiographed. All IN activities were quantified by scanning of the bands after gel electrophoresis and autoradiography using the Image J software. Both target and donor plasmids were kind gifts from Dr Karen Moreau (Université Claude Bernard-Lyon I, France). The target corresponds to the plasmid pBSK+ (Stratagene, La Jolla, California) carrying the zeocin resistance encoding gene. The 294 bp preprocessed donor substrate was obtained as described previously (12) and contains after cleavage by NdeI the supF tRNA gene flanked by two pre-cleaved extremities mimicking the 3′-processed U3 and U5 LTR sequences. The unprocessed donor was generated by cloning a donor containing ScaI ends into a PGem-T vector (Promega, Charbonnieres, France) as previously described (11). The PGem-T-SupFScaI resulting vector was cleaved by ScaI and the substrate fragment was purified.
Cloning of integration products for FSI quantification
The same protocol described previously (12) was used. Briefly, after concerted integration the products were purified on a DNA purification system column (Promega) as described by the supplier and then introduced into a MC1060/P3 Escherichia coli strain which contained ampicillin, tetracycline and kanamycin resistance genes. Both ampicillin and tetracycline resistance genes carry an amb mutation. These proteins are thus expressed only in the presence of the supF gene products. Integration clones carrying both zeocin-resistance and supF genes were therefore selected in the presence of 40 µg/ml ampicillin, 10 µg/ml tetracycline, 15 µg/ml kanamycin and 25 µg/ml zeocin. Plasmids were isolated from quadruple resistant colonies and checked by PCR sequencing (ABI Prism big dye terminator cycle sequencing ready reaction kit, Applied Biosystems) using the U3 primer (5′-TATGGAAGGGCTAATTCACT-3′) and the U5 primer (5′-TATGCTAGAGATTTTCCACA-3′).
Nonsequence-specific DNA endonuclease assay
Standard reactions were performed as described previously (23). Substrate of the non-sequence-specific endonuclease activity of HIV-1 integrase was the bacterial pUC19 DNA plasmid from Gibco. Purified integrase (1 pmol) was incubated with 200 ng of pUC19 DNA in a reaction mixture of 10 µl containing 20 mM HEPES pH 7.5, 10 mM DTT, 0.05% NP40, 30 mM NaCl and MnCl2 or MgCl2 (7.5 mM). The reaction mixture was incubated up to 1 h at 37°C and stopped by addition of 2 µl of 95% formamide, 20 mM EDTA, 0.05% bromophenol blue stop solution (standard conditions). Samples were analyzed on a 1% agarose minigel containing ethidium bromide (0.5 µg/ml). Electrophoresis was carried out for 30 min at 100 V at room temperature. DNA was detected by fluorescence upon exposure to UV light. Activity was evaluated by quantification of the bands corresponding to the different topological forms of the plasmid using the Image J software after scanning.
RESULTS
Purification of differently associated IN
In order to analyze the dynamic association of IN with DNA, our first aim was to obtain different forms of the associated enzyme in the absence of a crosslink agent. Several parameters can affect IN oligomerization such as enzyme concentration, presence of detergent and Zn ions (15,16,24,25). Our standard preparations of enzyme were all obtained in the presence of CHAPS but differ in their protein concentrations and the addition of Zn or not (Materials and Methods section). Different enzymes were obtained: (i) INLC stored at initial protein concentration <1 µM; (ii) INHC stored at initial protein concentration 5 µM; and (iii) INZn stored at initial protein concentration 12.5 μm and 50 µM Zn.
The oligomerization state of each sample was checked by size exclusion (gel filtration) chromatography. As shown in Figure 1A, we obtained in the presence of Zn an IN solution containing monomers, dimers and tetramers. In contrast, all the IN preparations obtained in the absence of Zn and in the presence of CHAPS were dissociated (Supplementary Material 1). The oligomerization equilibrium of the associated preparation was further checked by crosslink with DSS followed by SDS–PAGE and western blot. As shown in Figure 1B, the data obtained using chromatography were confirmed since monomers, dimers and tetramers were detected using this approach with similar proportions than those obtained after gel filtration. Longer crosslink incubation times did not change the oligomerization profile, indicating that DSS was able to crosslink only preformed oligomers without inducing further multimerization. Same results were obtained when using the SAXS methods (data not shown). Consequently, we assumed that the crosslink analysis reflects the proportion of oligomers in the IN solutions, and thus, this method was used to evaluate all the IN preparations throughout this work.
Figure 1.

Oligomerization state (A and B) and functional analyses (C and D) of the associated INZn preparation. One hundred and fifty picomole of IN purified in presence of 50 µm ZnSO4 (INZn, 10 µM) were submitted to gel filtration chromatography. (A) The nature of the IN peaks was determined by comparing profiles obtained with proteins of known molecular weights aldolase (158 kDa), bovine serum albumin (67 kDa), ovalbumin (43 kDa) and chymotrypsinogen A (25 kDa). One picomole of INZn was submitted to DSS crosslink (0.8 µg DSS, 50 mM HEPES pH 7.5, 30 mM NaCl) for 15 min and 60 min at 22°C and then 12% SDS–PAGE gel followed by western blot using polyclonal anti-IN antibodies. (B) Monomer (Mo), dimer (Di) and tetramer (Te) positions were determined by comparison with a molecular weight marker (MQ). Concerted integration assay (C) was performed without IN (lane -IN) or with 1 pmol of INzn (same final protein and NaCl concentrations of respectively 50 nM and 30 mM) using 100 ng of acceptor DNA (3000 bp) and 10 ng of 32P 5′-labelled donor pre-processed DNA (296 bp). The reaction products were either loaded on 1% agarose gel or cloned in MC1060/P3 E. coli strain. The position and the structure of the different products obtained after half-site (HSI), full-site (FSI) and donor/donor integration (d/d) are reported. The number of resistant selected colonies obtained in absence of IN (-IN) or after integration reaction carried by INZn (mean ± SD of three independent experiments) and the structure of the integration loci from 20 clones in each condition are reported in D.
Integrase dissociation inhibits in vitro integration activity
The in vitro activity of the INZn preparation was checked using the concerted integration assay. Standard reaction was initially performed using a 294 bp DNA substrate containing the 21 nt pre-processed HIV-1 viral ends at each extremity, as described previously (12). Figure 1C shows that the INZn enzyme was highly active since all the expected integration products were detected. In order to precisely monitor the full site integration events the circular forms of integration were cloned and quantified. As reported in Figure 1D analysis of the integration loci structure indicated that the 5 bp duplications was found confirming that the enzyme catalyzed concerted integration with a good efficiency and fidelity. In contrast, the dissociated enzymes mentioned above were found less active and the IN monomers were found totally inactive (Supplementary Material 1).
In order to determine the relationship between the activity and the oligomerization state of IN, we analyzed the activity of the INZn preparation before and after dissociation (strategy described in Supplementary Material 2). The highly concentrated enzyme was diluted at a protein concentration ranging from 12.5 μm to 0.125 µM and the oligomerization state was checked by DSS crosslink and quantified by densitography of the western blot analysis. DSS crosslink analysis shown in Figure 2 indicated that the dilution led to the dissociation of IN. Samples at concentrations below 1.5 µM appeared mainly as monomers under these conditions while dimers appeared when the concentrations were increased. Tetramers were observed only at the highest concentration of 12.5 µM of the initial stock solution, in agreement with the results obtained by exclusion size chromatography and crosslinking (Figure 1).
Figure 2.

Effect of enzyme initial concentrations on IN oligomerization in solution. One picomole of each diluted fraction was submitted to DSS crosslinking. Then monomer, dimer and tetramer bands were quantified using the Image J software. The percentage of each oligomer was plotted versus the initial IN concentration.
Diluted samples were tested in vitro for concerted integration under standard conditions (7.5 mM MnCl2, HEPES 20 mM, pH 7.6), using pre-processed substrate and FSI was quantified by selection of integrants clones. The final concentration of IN and NaCl were adjusted respectively to 50 nM and 30 mM for each experiment. As shown in Figure 3A and quantification in Figure 3B, no integration activity was detected with the dissociated enzyme appearing as monomers in the previous DSS analysis (lanes 6–7). The integration activity was found highly dependent on the amount of oligomers in the initial IN solutions, especially dimers. Increasing the proportion of oligomers also improved the level of FSI activity as shown by the results of integrants cloning (figure 3C), without affecting the quality of the integration since no significant changes were observed regarding the proportion of 5 bp duplication as seen in Figure 3D. The same effect was observed when IN was diluted in presence of Mg++ or in the absence of cations (data not shown).
Figure 3.

Effect of initial IN concentrations on in vitro integration of the pre-processed donor DNA. Concerted integration assay was performed with 1 pmol of IN from the different diluted preparations of INZn as shown (D): 12.5 µM (lane 1), 6.25 µM (lane 2), 3.125 µM (lane 3), 1.56 µM (lane 4), 0.78 µM (lane 5), 0.3 µM (lane 6) and 0.125 µM (lane 7). The final NaCl concentration was adjusted to 30 mM in all assays. The reaction products were either loaded on 1% agarose gel (A) or cloned in MC1060/P3 E. coli strain (C). The position and the structure of the different products obtained after half-site (HSI), full-site (FSI) and donor/donor integration (d/d) are shown (A). (B) corresponds to the densitometry estimation of the FSI and FSI+HIS bands of experiments shown in A. The different integration products were quantified using the Image J software. Values are the mean ± SD (error bars) of three independent experiments. The number of resistant selected colonies (C) obtained after integration reaction carried by the different diluted IN and the structure of the integration loci (D) from 20 clones in each condition are reported.
The viral end structure was previously found to affect the in vitro concerted integration activity (11). To determine whether the inhibition of the activity concomitant with IN dissociation was dependent on the viral end structure from the donor DNA, we used a DNA carrying the intact unprocessed 21 nt LTR ends. The substrate was generated using a DNA fragment containing the ScaI restriction site as reported previously (11). Using this substrate a higher proportion of FSI was detected (Figure 4). Analysis of the integration loci indicated that the use of intact viral ends improved both, the efficiency and the quality of the integration (Figure 4C and D, to be compared with Figure 3C and D), thus confirming a previous report (11). However, as observed with the pre-processed substrate, IN dissociation led to the inhibition of integration without affecting the quality of the integration (no change was observed in the integration loci structure as seen in Figure 4D). The profile obtained with the unprocessed substrate remained similar to that observed with the pre-processed DNA except the inhibition rate that was found different since FSI and HSI appeared less tolerant to the dissociation step (compare Figure 4B to 3B).
Figure 4.

Effect of initial IN concentrations on in vitro integration of the unprocessed donor DNA. Concerted integration assay was performed with 1 pmol of IN from the different diluted preparations of INZn as shown in Figure 3: 12.5 μm (lane 1), 6.25 μm (lane 2), 3.125 μm (lane 3), 1.56 µM (lane 4), 0.78 μm (lane 5), 0.3 μm (lane 6) and 0.125 µM (lane 7). The final NaCl concentration was adjusted to 30 mM in all reaction solutions. The reaction products were either loaded on 1% agarose gel (A) or cloned in MC1060/P3 E. coli strain (C). The position and the structure of the different products obtained after half-site (HSI), full-site (FSI) and donor/donor integration (d/d) are shown in (A). (B) corresponds to densitometry of the FSI and FSI+HIS bands of experiments shown in A. The different integration products were quantified using the Image J software. Values are the mean ± SD (error bars) of three independent experiments. The number of resistant selected colonies (C) obtained after integration reaction carried by the different diluted IN and the structure of the integration loci (D) from 20 clones in each condition are reported.
This indicates that the initial oligomerization state of IN in solution affects the efficiency of the integration reaction independently of the viral end structure. Since the quality of the integration was found to be highly dependent on the formation of the synaptic IN tetramer complex on viral ends (12), our data suggest that only the initial attachment step was affected by the dissociation and not the final active complex structure. Furthermore, since integration activity appeared only in the presence of oligomers in solution we concluded that either preformed active oligomers can exist in solution, or that preformed oligomers in solution are required to be activated on DNA. The striking result obtained with the IN samples dissociated into monomers suggested that, under our conditions, monomers of IN are inactive probably because they were unable to associate on DNA to form the active oligomers. Therefore, it was tempting to speculate that preformed oligomers (dimers or tetramers) could bind directly to DNA to form the active INċDNA complexes.
The inactivity of the IN monomers on a long DNA substrate used for the concerted integration assay could be due to their inability to bind DNA, to catalyze the integration reaction and/or to actively oligomerize on viral ends. To answer this question we further analyzed the functional association of dissociated preparations of IN focusing on short ODN mimicking the viral end.
Integrase monomers can actively associate on short ODN mimicking the viral ends to catalyze 3′ processing and strand transfer activities
Dissociated samples after dilution were tested for their folding into active complexes on short DNA carrying only the final 21 bp of viral LTR or nonspecific sequences. Surprisingly, dissociated INzn (0.125 µM) was able to catalyze the processing (Figure 5A) and the strand transfer reactions (Figure 5B). However, this activity was revealed only when incubations were performed at times longer than the standard conditions (2 h instead of 1). This suggests that IN monomers can fold into active complexes when bound to short specific DNA in contrast to the situation with the longer substrate used for concerted integration assay.
Figure 5.

IN activity and oligomerization on short ODN mimicking the viral ends. 1 pmole of associated (12.5 µM initial concentration) and dissociated (0.125 µM) INZn were analyzed for 3′ processing (A) and strand transfer activities (B). Reactions were performed under standard conditions (7.5 mM MnCl2, 20 mM HEPES pH 7.6) for 0 to 120 min. The final NaCl concentration was adjusted to 30 mM in all reaction solutions. IP, integration product; P, processing product. 3′ processing activity detected after 120 min of reaction with both enzyme preparations was analyzed using either MgCl2 or MnCl2 (7.5 mM) and the percentage of activity recovered was reported in C. Results are the mean ± SD (error bars) of three independent experiments.
However, standard processing reactions were performed using Mn++ while concerted integration assays were done in the presence of Mg++ or not. Since different metal ions are known to affect IN conformational changes (26–28), we performed new processing experiments using Mg++ cations. As reported in Figure 5C, the use of Mg++ decreased the activity for both associated and dissociated IN with a more dramatic effect on enzyme monomers. Indeed, no activity was observed after 2 h using monomers while a significant activity of the associated IN activity was observed. Since the associated IN activity was also affected by the nature of the cations, it remains difficult to conclude about the effect of Mg++ on the active complexes. However, it has previously been shown that the half-life of INċDNA complexes is dependent upon the cations (29,30) and lower in the presence of Mg++. One explanation why the 3′ processing catalyzed by monomers was more severely impaired in presence of Mg++ could be the lower stability of the complexes formed from the sequential recruitment of a second monomer by a first one already bound to DNA. This also suggests the emergence of a more stable complex issued from the binding of IN oligomers explaining why the associated IN remains active even in presence of Mg++.
The hypothesis of differences in the kinetic and stability of the complexes depending of the cations was (i) confirmed by the detection of some processing activity for IN monomers in experiments performed after longer reactions time (more than 4 h) in presence of Mg++ (Supplementary Material 3) and (ii) strongly supported by the following reassociation experiments.
Reactivation of integrase monomers by short specific ODNs
IN monomers appeared inactive on long DNA while they present 3′ processing and strand transfer activity on short specific viral sequences. This indicates that they can bind DNA and actively oligomerize on the viral end. Thus, the inactivity of monomers on long DNA substrate could be due to their inefficiency to be positioned on the viral ends in contrast to the associated forms which could bind more specifically the end sequence to form the active complexes. If this hypothesis is correct, the reassociation of monomers into dimers should lead to the reactivation of the dissociated preparation of IN. It has been described previously that the preincubation of IN with short ODNs leads to its oligomerization and to the activation of 3′ processing reaction (17,18), suggesting that active oligomers could be released from the INċDNA complex. To verify this point, we performed a reassociation experiment of IN monomer preparations.
One picomole of dissociated INZn was incubated overnight with 1 pmole of the 21 nt ODN presenting the LTR ends or random nonviral ODN under our standard conditions (7.5 mM MnCl2, HEPES 20 mM, pH 7.6). The self association of the protein was analyzed using DSS crosslink before performing concerted integration assays. Results in Figure 6 indicate that preincubation of the dissociated enzyme with either short specific viral ODN or random ODN led to its reassociation into oligomers (mainly dimers). These data have been validated by SAXS experiments (data not shown) confirming that monomers can bind either to specific or nonspecific DNA and associate on it. In order to determine whether the oligomers formed on both ODN were functionally similar, we tested the reassociated fractions in concerted integration assays. Results are reported in Figure 7A and show that the incubation of IN monomers with the 21 nt viral ends led to recovery of integration (lane 3 compared to lane 2). In contrast, no integration was detected after incubation with nonviral ODN (lane 4), indicating that, in this case, the multimer enzyme was inactive. Selection of integrants carrying the circular form of FSI products indicated that this reaction was also recovered, at least partially, when the dissociated enzyme was pre-incubated with specific ODN but not with random sequences (Figure 7B). Sequencing of the integration loci showed a similar profile with associated and reassociated IN. The effect of the viral end structure was also analyzed using the blunt substrate for the concerted integration assay. As reported in Figure 7C and D, the same reactivation was observed under these conditions, confirming that the enzyme dissociation affected the early steps of IN attachment to DNA and not the formation of the final synaptic complex.
Figure 6.

Effect of specific and nonspecific ODN on integrase self association. One picomole of INzn 12.5μm or 0.125 µM (lanes 1 and 2) pre-incubated with specific or nonspecific 21 bp ODN (lanes 3 and 4) were submitted to DSS crosslink for 30 min at 22°C and loaded on 12% SDS–PAGE before western-blotting with polyclonal anti-IN antibody. In all reactions the final NaCl concentration was adjusted to 30 mM. Monomer (Mo), dimer (Di) and tetramer (Te) positions were determined by comparison with a molecular weight marker (MQ).
Figure 7.

Effect of reassociation of INZn on in vitro integration activity. Assays were performed with 100 ng receptor DNA and 10 ng pre-processed (A) or unprocessed donor (C) with 1 pmol of INzn 12.5μm or 0.125 µM diluted in standard dilution buffer (7.5 mM MnCl2, HEPES 20 mM, pH 7.6) without preincubation (respectively lanes 1 and 2) or after preincubation with 1 pmol of specific 21 bp viral ODN (lanes 3) or random 21 bp ODN (lane 4) under standard conditions (7.5 mM MnCl2, HEPES 20 mM, pH 7.6). The reaction products were then loaded on 1% agarose gel. The position and the structure of the different products obtained after half-site (HSI), full-site (FSI) and donor/donor integration (d/d) are reported in the figure. Effect of reassociation on full site integration. Reaction products obtained with the pre-processed (B) or the unprocessed (D) donor were cloned in MC1060/P3 E. coli strain as described in Materials and methods section. The number of resistant selected colonies obtained after integration reaction under the different conditions is reported in the figure. Results are the mean ± SD (error bars) of three independent experiments.
Interestingly, the reassociated monomers obtained after preincubation with the specific ODN were also found more active for 3′ processing especially when reaction was performed in presence of Mg++ (Supplementary material 3). This confirmed that the differences of activity observed between Mg++ and Mn++ conditions were due to differences in kinetics. In addition, the same re-association and reactivation effects were observed when the experiments were performed with IN diluted in presence of Mg++ (7.5 mM MgCl2, HEPES 20 mM, pH 7.6) (data not shown) demonstrating that the reassociation of monomers into oligomers and their reactivation were not only Mn++ dependent. Furthermore, the reassociation experiments performed with other IN preparation obtained with different purification procedures described in Materials and methods section led to the same results demonstrating that the reassociation of monomers into active oligomers was not dependent of the purification method (Supplementary material 4).
In order to obtain more information on the mechanism of reactivation, we examined the kinetic of reactivation of the dissociated INZn enzyme in presence of Mg++ or Mn++. As shown in Figure 8, the kinetic of reactivation was found different when comparing both conditions. Indeed, the activation detected in presence of Mg++ was observed after 2 h and was maximal at 6 h. In contrast, when Mn++ was used the activation was not detected before 10 h. These results are consistent with the half-life of the INċDNA complexes determined before in the presence of both cations (31). Taken together these results indicate that monomer reactivation was mainly due to IN reassociation on short ODNs followed by the release of active oligomeric complexes. But the previous demonstration of the displacement of viral DNA termini from stable IN nucleoprotein complexes induced by the secondary DNA binding interaction (31) meant that we could not rule out a possible tertiary complex between IN, the short ODN and the concerted integration substrate as a possible mechanism of activation.
Figure 8.

Effet of cations on the kinetic of reactivation of dissociated INZn. One picomole of dissociated INzn 0.125 µM was preincubated with specific 21 bp ODN for 0–24 h in presence of Mg++ or Mn++ (7.5 mM) and then tested for concerted integration. Enzyme activity was compared to that of the dissociated INZn incubated under the same condition but without ODN. The percentage of activation was reported.
Our major observation is that both ODNs led to the formation of oligomers, mostly dimers, from monomers, but only specific viral DNA led to active integration complexes. We conclude that active dimers can be formed from monomers only after oligomerization of the enzyme on the specific 21 nt viral ends. Furthermore the close association profile obtained in both preincubation conditions (specific or nonspecific ODN) and the difference of in vitro activity summarized for INZn in Table 1 strongly indicate that the dimeric complexes fold on each type of DNA are functionally different.
Table 1.
Summary of the association and activity analyses of the different enzyme preparations
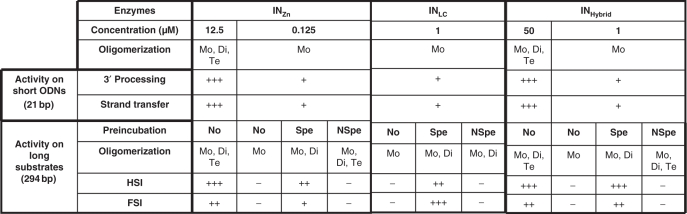 |
The INZn, INLC (purified in presence of 7 mM CHAPS in addition or not of 50 μM ZnSO4) were compared for self association, 3′ processing, strand transfer, HSI and FSI products after pre-incubation or not (No) with specific (Spe) or nonspecific (NSpe) 21 bp ODN. The presence of oligomers was evaluated by DSS crosslink and western blotting, monomers (Mo), dimers (Di) and tetramers (Te) are reported. In vitro activities were quantified using Image J software or counting of FSI integrant clones. More than 20% of 3′ processing and strand transfer (+++), 5–20% (+), 15–25% of HSI (+++), 5–15% (++), no activity (−), Above 100 FSI integrant clones (+++), 50–100 (++), 10–50 (+). All the activities reported here were obtained under standard conditions (7.5 MnCl2 for 3′ processing and strand transfer; 7.5 MgCl2 for concerted integration). Pre-incubations with ODN were performed in presence of MgCl2 during 16 h.
Taken together these data strongly suggest that the inefficiency of monomers to catalyze integration on long DNA substrate was mainly due to their inability to become adequately positioned on the viral ends, where their association should lead to active dimers. Since, under in vitro integration assay conditions in presence of long substrates, IN mainly interacts with nonspecific DNA, we further studied the function of the INċDNA complexes formed on these nonviral sequences.
IN monomers association on nonspecific DNA leads to double-strand endonuclease activity
Previous reports indicated that IN displays an endonucleolytic non-sequence-specific activity on heterologous DNA (23, 32). However, neither the role of this activity nor the nature of IN forms catalyzing the cleavage has been determined. We investigated the activity of the oligomers induced by the binding to nonspecific ODNs. The activity of the associated and dissociated preparations of IN (INZn, 12.5μm and 0.125 µM) were compared using the nonspecific DNA cleavage assay previously described (23). The enzyme displaying a mixture of monomers, dimers and tetramers presented a classical profile of single-strand non-sequence-specific DNA (Figure 9A, quantified in Figure 9B). Interestingly, the monomers showed a different profile characterized by the formation of linear molecules indicating a double-strand DNA endonuclease activity (Figure 9A quantified in Figure 9C). Both single-strand and double-strand DNA cleavages were inhibited by the IN-specific ODN inhibitor 93 del (33,34), indicating that the breaks were due to intrinsic IN activity.
Figure 9.

Non-sequence-specific endonuclease activity of IN preparations (A quantified in B and C). One pmole of associated (12.5 µM initial concentration) and dissociated (0.125 µM) INZn were incubated 0–60 min with 200 ng of pUC19 plasmid in standard conditions (7.5 MnCl2, 20 mM HEPES pH 7.6). The final NaCl concentration was adjusted to 30 mM in all reaction solutions. Same reactions were performed for 1 h in presence of 10 nM of the 93del inhibitor aptamer. The Close Circular (CC), Open Circular (OC) and Linear (L) forms of the plasmid are shown and quantified. The percentage of each form was plotted versus time for each reaction conditions performed with the 12.5 µM (B) and 0.125 µM (C) INZn. Values are the mean ± SD (error bars) of three independent experiments. Effect of cations on the single strand DNA cuts catalyzed by associated INZn (D) and double strand DNA cuts catalyzed by dissociated INZn (E). One picomol of dissociated INZn (0.125 µM) was incubated 0–120 min with 200 ng of pUC19 plasmid in standard conditions and in the presence of either 7.5 mM MnCl2 (standard conditions) or 7.5 mM MgCl2. The final NaCl concentration was adjusted to 30 mM in all reaction solutions. The Close Circular (CC), Open Circular (OC) and Linear (L) forms of the plasmid were quantified and the percentages of single and double strand cuts were reported. Values are the mean ± SD (error bars) of two independent experiments.
Since Mg++ and Mn++ have been shown to influence the specificity of the enzyme (27,28,30), we further studied the effect of both cations on the nonspecific cleavage catalyzed by IN. Figure 9D and E show that double strand DNA cuts catalyzed by the dissociated enzyme were less tolerant to the cations used in the assay. Indeed Mg++ decreased the double strand cleavage activity without inducing significant changes in the single strand cuts catalyzed by the associated IN. These results support the data obtained in the specific processing cleavage (Figure 5C) indicating that the complexes issued from the association of monomers on DNA may require Mn++ to be efficient in contrast to the INċDNA complexes obtained from the association of preformed IN oligomers on the substrate. This could be linked to the better stability of these complexes in presence of Mn++ as previously observed (29).
Since monomers were able to oligomerize on nonviral DNA without displaying specific IN integration activity, we may propose that the dimers formed from monomers on unspecific DNA (i) acquired a new non-sequence-specific endonucleolytic activity and (ii) were functionally and probably structurally different from the dimers folded on viral ends.
DISCUSSION
The initial interaction of IN with its substrate is a crucial step for the specific recognition of the viral ends and determines the following phases of the integration reaction. It has been proposed that during integration, changes in the oligomerization state of IN may lead to the formation of several forms displaying different functions (12,17). Even if all the data point to the requirement of a stable synaptic complex involving an IN tetramer and both viral LTR (12,13), the precise folding pathway of this complex on the viral ends remains to be established. The use of enzyme preparations with different oligomerization profiles, described in this work, sheds new light on the early events of integration in vitro and the role of IN oligomers in the initial steps. Functional and structural analyses of the different IN preparations are summarized in Table 1.
Formation of active SSC INċDNA complexes depends on dimers binding to viral sequences or early dimerization on viral ends
All the preparations of IN monomers used in our work and obtained following different procedures were found inactive for integration using a 294 bp DNA containing the 21 nt viral ends processed or unprocessed at both ends. In contrast, IN oligomers were able to display integration activity. SAXS and crosslink experiments showed that IN monomers were able to oligomerize both on short specific and nonspecific DNA (Figure 6). However, oligomers generated either on specific or nonspecific substrates differed in their activity (Figure 7).
Analysis of monomer activities on a short ODN mimicking the viral ends showed that they retained low but significant processing and strand transfer activities (Figure 5 and Supplementary Material 3). In addition, integration activity was recovered after reassociation on specific viral ODN in contrast to nonspecific ODN (Figure 7). Thus, the lack of monomers activity on longer DNA mimicking the viral substrate was not due to an oligomerization or a catalytic deficiency but to inadequate positioning of IN on the specific viral ends, leading to incorrect binding in internal positions on the DNA molecule competing with the shorter specific viral ends. Consequently (i) the formation of active IN complexes on DNA directly depends on the correct positioning of IN on viral ends and (ii) dimers folded on specific or nonspecific DNA are not functionally similar. This is strongly supported by our previous structural analysis using SAXS method of the INċDNA complexes formed on both type of ODN leading to the identification of dimers differing in their radii of gyration [type 1 on specific DNA and type 2 on nonspecific DNA, Ref. (17)].
Correct positioning on viral ends is dependent on IN oligomerization or viral sequences accessibility to monomer binding
The results obtained with associated preparations of IN and the reactivation of monomers by preincubation with specific ODN (see summary in Table 1) strongly suggest that preformed active oligomers, especially dimers, can bind the viral ends as functional complexes while monomers can not. This is firmly supported by the demonstration that purified cross-linked oligomers retain in vitro activity in contrast to monomers (12).
Previous binding studies did not show any significant difference of IN affinity for either the specific 21 nt viral derivative or for nonspecific random ODNs (18). In addition, we and others observed that retroviral IN protected a larger region above 100 bp beyond the 21 nt specific LTR sequence on the integration substrate in vitro (35–37). Thus, during in vitro assays there is a higher probability for IN to bind the longer nonspecific internal region of the substrate and thus to form inactive oligomers, as reported above.
Consequently we propose two ways to form active complexes: (i) by direct binding of preformed active (or ‘activable’) dimers on viral ends; (ii) by binding of IN monomers and further dimerization on viral ends. In the first case, the formation of the active complexes is highly dependent of the oligomerization state of the enzyme interacting with DNA, as supported by the difference of activity observed for dissociated and associated IN. Our data obtained with IN monomers clearly indicate that in this case the binding to viral ends is poorly efficient, probably due to the low affinity of IN for viral sequences in comparison to unspecific DNA. This is strongly supported by recent analysis from Deprez and co-workers revealing a nonspecific DNA binding mode of the enzyme and an optimal activity leading to DNA binding and cooperativity (38), confirming the requirement for dimers assembling on viral ends.
Additional nonspecific DNA binding factors may optimize the IN positioning on its substrate. Vpr, a component of the PIC, has been previously shown to be a nonspecific DNA binding protein (39,40). In addition, the Ct terminal (52–96) domain of the protein was shown to stimulate in vitro homologous strand transfer of mini-viral DNA (41). The HIV-1 nucleocapsid protein, NCp7, has been shown to activate in vitro integration by an undetermined mechanism (42). Our data raise the question of whether such DNA binding proteins if present in the PIC should be required to help the correct positioning of IN by binding to nonspecific DNA sequences. Since no data indicate an important role for such proteins in integration during infection, the eventual physiological relevance and the mechanism of IN positioning by these factors remain to be established.
IN monomers oligomerization on nonspecific DNA leads to complexes having double-strand nonsequence-specific DNA cleavage activity
Single-strand endonuclease activity has been previously described for HIV-1 IN (23,32). This activity was assumed to reflect an inefficient 3′ processing reaction on an aberrant DNA substrate. As shown in Figure 9, dissociated IN displayed a new double-strand DNA cleavage activity on nonspecific DNA in contrast to the associated forms of the enzyme. Even if the physiological relevance of this activity remains to be elucidated some aspects of the structure of the INċDNA complexes can be highlighted. Our main conclusion is that dimers formed on viral ends and those formed on unspecific DNA are functionally and probably structurally different as supported by the demonstration by SAXS of types 1 and 2 dimers folding on respectively specific and nonspecific DNA and differing in their radii of gyration (17).
The 3′ processing and the single-strand DNA nuclease activity observed with associated IN are catalytically similar but differ in specificity suggesting that the same dimers (type 1) are involved in both reactions and could fold on both viral ends to form the active complexes performing single-strand 3′ processing of the LTR. In addition, a recent study reports that a two-LTR junction could be cleaved on both strands by a tetramer of IN probably formed by the juxtaposition of two type 2 dimers cutting each strand at the specific CA sequence (43). Some structural data support this hypothesis: the C-terminal domain of one monomer acts with the central catalytic domain from another monomer at each viral DNA end. In this dimer one monomer could carry the catalytic property as well as the substrate capture (44) while the other monomer could be involved in the correct folding of the complex. This would result in an asymmetric dimer, as suggested by crosslink experiments indicating that no trimers of IN could be detected, which in turn suggested that intra-dimer and inter-dimer contacts in the tetramer are not equivalent (12,13). Thus, in asymmetric dimers (type 1), one catalytic site could be positioned for the DNA cleavage on one strand and especially for the 3′-end processing of the LTR, as described for other proteins from the same family where this asymmetry defines the single strand cut [Mu, Cre transposases (45,46)]. More recently modelling analyses on the complex between the IN active site and viral DNA also suggest that only the active sites from two central monomers in contrast to distal ones could be involved in the 3′ processing leading to asymmetrical dimers in the active tetramer (47).
On the basis of this model, we propose that the IN type 1 dimers could differ from type 2 IN dimers formed on the nonspecific sequence by the symmetrical positioning of both catalytic sites on the complex. Consequently, the nature of the tetramer resulting from the contact between two dimers depends on the nature of each dimer. A recent single-image reconstitution of a tetramer of IN bound to DNA showed that the complex probably involves asymmetrical interactions (48). This confirms that the final active SSC complex is probably folded from different types of dimers.
A model for the in vitro positioning of HIV-1 IN on viral ends
Our in vitro data led us to propose a model described in Figure 10 for the initial attachment of IN on viral DNA. IN can bind DNA under all the different oligomeric forms including monomers. Interaction between IN monomers and DNA might induce oligomerization of the enzyme, leading to dimers and tetramers whose structures and functions depend of the bound DNA sequence. Interaction between monomers and nonspecific DNA induce the formation of dimers inactive for processing, strand transfer and integration activities (type 2) but displaying a non-sequence-specific double strand endonuclease activity (way A). The interaction of monomers with the specific 21 nt viral DNA end sequence would allow the formation of type 1 dimers able to catalyze processing and strand transfer reactions in the so-called Strand Transfer Complex (way B), (13).
Figure 10.

Model of the in vitro IN positioning on viral ends. Interaction of monomers with short nonspecific nonviral DNA induces the formation of dimers inactive for processing, strand transfer and integration activities (type 2 dimers, way A). The interaction of monomers with the specific 21 nt viral DNA ends allow the formation dimers able to catalyze the processing and strand transfer reactions (type 1 dimers, way B). In the presence of longer DNA mimicking the viral genome and containing both short specific DNA regions and larger unspecific domains and in the absence of targeting, IN (standard in vitro conditions) will bind more frequently the nonspecific sequences (way A1). This will lead to the major formation of inactive type 2 dimers (A2). In solution and especially in associated IN preparations (concentrated one or enzyme purified in absence of detergent, in presence of Zn++), preformed dimers (including types 1 and 2) are present. Type 1 dimers can bind the viral ends (B1) allowing the formation of the active tetrameric synaptic complex (SSC, B2).
In the presence of longer DNA containing both short specific DNA regions and larger unspecific domains and in the absence of targeting to the specific ends (standard in vitro conditions), IN would bind with higher frequency to nonspecific sequences. This would lead mainly to the formation of inactive type 2 dimers (way A). In solution and especially in associated IN preparations, preformed dimers (including types 1 and 2) are present. Type 1 dimers bind the viral ends better than nonspecific DNA [the type 1 dimer association with the specific viral ends was found to be more stable, Ref. (17)] and perform the integration reaction either without or after structural changes, thereby allowing the formation of the active tetramer synaptic complex (SSC), (way B). In contrast to monomers a better interaction of associated IN with viral ends can be explained by the closed structure adopted by IN and revealed by single-image reconstitution of a tetramer of IN bound to DNA (48). This study suggests the presence of a central channel resembling a variety of other DNA-binding proteins wrapped around their substrate. In the preformed dimer the only way to interact with DNA could be the entry of the DNA substrate end inside the channel. That process would favour the specific binding of the enzyme to the viral ends. In contrast the monomer could bind the DNA at an internal position of the substrate before recruiting another monomer to wrap the DNA around the channel favouring interactions with multiple nonspecific binding sites. This mechanism could require a better stability of the INċDNA complex as suggested by the better activity observed for dissociated IN preparations in all assays performed with Mn++, cations known to increase the half-life of these complexes (29, Figures 5 and 8).
Consequently, we propose that IN oligomerization controls both the enzyme specificity and its activity on DNA. Despite some differences observed in the kinetics performed in presence of Mg++ or Mn++ similar conclusions were raised indicating that our in vitro observations could also be relevant in the case of infected cells. In the cell the formation of active type 1 dimers and integration must be optimal. This is especially important since we show here that the dimerization IN on non-LTR sequences leads to the formation of dimer complexes having a double-strand DNA cleavage activity able to digest the viral genome if unprotected. Viral factors of the PIC, displaying nonspecific DNA-binding properties, may be involved in IN to the specific viral ends by masking IN nonspecific DNA binding sites and protecting the viral genome. Strategies aimed to stimulate genome destruction on the basis of this proposal appear as a new therapeutic axe to be developed.
The precise structure of each oligomer variant remains to be resolved. A priority should be to conduct structural studies of the IN complexed to the two viral ends. Studies of the enzyme structure in complex with nonspecific DNA and with one specific DNA end should provide interesting data on types 1 and 2 dimers, respectively, and should confirm their structural differences. The detailed biochemical study of the conditions leading to the formation of specific active oligomers of HIV-1 IN, as described here, should be useful to accomplish these structural studies.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The French Agence Nationale de Recherche contre le SIDA (ANRS); the Centre National de la Recherche Scientifique (CNRS); University Victor Segalen Bordeaux 2 (to V.P. and M.A); The Molecular and Cell Biology Program of the Presidium of the Russian Academy of Sciences (project no. 10.5, partial); the Russian Foundation for Basic Research (project no. 03-04-49781, partial); Siberian Division of the Russian Academy of Sciences (to G.N., partial). Funding for open access charge: The French Agence Nationale de Recherche contre le SIDA (ANRS).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are deeply grateful to Professors J. Pageze and R. Cooke (University Bordeaux 2) for proof-reading the manuscript. We thank also Dr M. Benleulmi for fruitful discussion.
REFERENCES
- 1.Bowerman B, Brown PO, Bishop JM, Varmus HE. A nucleoprotein complex mediates the integration of retroviral DNA. Genes Dev. 1989;3:469–478. doi: 10.1101/gad.3.4.469. [DOI] [PubMed] [Google Scholar]
- 2.Brown PO, Bowerman B, Varmus HE, Bishop JM. Correct integration of retroviral DNA in vitro. Cell. 1987;49:347–356. doi: 10.1016/0092-8674(87)90287-x. [DOI] [PubMed] [Google Scholar]
- 3.Ellison V, Abrams H, Roe T, Lifson J, Brown P. Human immunodeficiency virus integration in a cell-free system. J. Virol. 1990;64:2711–2715. doi: 10.1128/jvi.64.6.2711-2715.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farnet CM, Haseltine WA. Integration of human immunodeficiency virus type 1 DNA in vitro. Proc. Natl Acad. Sci. USA. 1990;87:4164–4168. doi: 10.1073/pnas.87.11.4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bushman FD, Craigie R. Activities of human immunodeficiency virus (HIV) integration protein in vitro: specific cleavage and integration of HIV DNA. Proc. Natl Acad. Sci. USA. 1991;88:1339–1343. doi: 10.1073/pnas.88.4.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sherman PA, Fyfe JA. Human immunodeficiency virus integration protein expressed in Escherichia coli possesses selective DNA cleaving activity. Proc. Natl Acad. Sci. USA. 1990;87:5119–5123. doi: 10.1073/pnas.87.13.5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engelman A, Mizuuchi K, Craigie R. HIV-1 DNA Integration: Mechanism of viral DNA Cleavage and strand transfer. Cell. 1991;67:1211–1221. doi: 10.1016/0092-8674(91)90297-c. [DOI] [PubMed] [Google Scholar]
- 8.Hindmarsh P, Leis J. Retroviral DNA integration. Microbiol. Mol. Biol. Rev. 1999;63:836–843. doi: 10.1128/mmbr.63.4.836-843.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sinha S, Pursley MH, Grandgenett DP. Efficient concerted integration by recombinant human immunodeficiency virus type 1 integrase without cellular or viral cofactors. J. Virol. 2002;76:3105–3113. doi: 10.1128/JVI.76.7.3105-3113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sinha S, Grandgenett DP. Recombinant human immunodeficiency virus type 1 integrase exhibits a capacity for full-site integration in vitro that is comparable to that of purified preintegration complexes from virus-infected cells. J. Virol. 2005;79:8208–8216. doi: 10.1128/JVI.79.13.8208-8216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li M, Craigie R. Processing of viral DNA ends channels the HIV-1 integration reaction to concerted integration. J. Biol. Chem. 2005;280:29334–29339. doi: 10.1074/jbc.M505367200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faure A, Calmels C, Desjobert C, Castroviejo M, Caumont-Sarcos A, Tarrago-Litvak L, Litvak S, Parissi V. HIV-1 integrase crosslinked oligomers are active in vitro. Nucleic Acids Res. 2005;33:977–986. doi: 10.1093/nar/gki241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li M, Mizuuchi M, Burke TR, Craigie R. Retroviral DNA integration: reaction pathway and critical intermediates. EMBO J. 2006;25:1295–1304. doi: 10.1038/sj.emboj.7601005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guiot E, Carayon K, Delelis O, Simon F, Tauc P, Zubin E, Gottikh M, Mouscadet JF, Brochon JC, Deprez E. Relationship between the oligomeric status of HIV-1 integrase on DNA and enzymatic activity. J. Biol. Chem. 2006;281:22707–22719. doi: 10.1074/jbc.M602198200. [DOI] [PubMed] [Google Scholar]
- 15.Deprez E, Tauc P, Leh H, Mouscadet JF, Auclair C, Brochon JC. Oligomeric states of the HIV-1 integrase as measured by time-resolved fluorescence anisotropy. Biochemistry. 2000;39:9275–9284. doi: 10.1021/bi000397j. [DOI] [PubMed] [Google Scholar]
- 16.Deprez E, Tauc P, Leh H, Mouscadet JF, Auclair C, Hawkins ME, Brochon JC. DNA binding induces dissociation of the multimeric form of HIV-1 integrase: a time-resolved fluorescence anisotropy study. Proc. Natl Acad. Sci. USA. 2001;98:10090–10095. doi: 10.1073/pnas.181024498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baranova S, Tuzikov FV, Zakharova OD, Tuzikova NA, Calmels C, Litvak S, Tarrago-Litvak L, Parissi V, Nevinsky GA. Small-angle X-ray characterization of the nucleoprotein complexes resulting from DNA-induced oligomerization of HIV-1 integrase. Nucleic Acids Res. 2007;35:975–987. doi: 10.1093/nar/gkl1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caumont A, Jamieson G, de Soultrait VR, Parissi V, Fournier M, Zakharova OD, Bayandin R, Litvak S, Tarrago-Litvak L, Nevinsky GA. High affinity interaction of HIV-1 integrase with specific and non-specific single-stranded short oligonucleotides. FEBS Lett. 1999;455:154–158. doi: 10.1016/s0014-5793(99)00859-5. [DOI] [PubMed] [Google Scholar]
- 19.Feigin LA, Svergun DI. Structure analysis by small-angle X-ray and neutron scattering. NY: Plenum Press; 1987. [Google Scholar]
- 20.Gill PE, Murray W, Wrighe MH. Practical optimization. London, NY, Toronto, Sydney, San Francisco: Academic Press; 1981. [Google Scholar]
- 21.Parissi V, Calmels C, De Soultrait VR, Caumont A, Fournier M, Chaignepain S, Litvak S. Functional interactions of human immunodeficiency virus type 1 integrase with human and yeast HSP60. J. Virol. 2001;75:11344–11353. doi: 10.1128/JVI.75.23.11344-11353.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moreau K, Faure C, Violot S, Verdier G, Ronfort C. Mutations in the C-terminal domain of ALSV (Avian Leukemia and Sarcoma Viruses) integrase alter the concerted DNA integration process in vitro. Eur. J. Biochem. 2003;270:4426–4438. doi: 10.1046/j.1432-1033.2003.03833.x. [DOI] [PubMed] [Google Scholar]
- 23.Parissi V, Caumont A, de Soultrait VR, Desjobert C, Calmels C, Fournier M, Gourgue G, Bonneu M, Tarrago-Litvak L, Litvak S. The lethal phenotype observed after HIV-1 integrase expression in yeast cells is related to DNA repair and recombination events. Gene. 2003;322:157–168. doi: 10.1016/j.gene.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 24.Lee SP, Xiao J, Knutson JR, Lewis MS, Han MK. Zn2+ promotes the self-association of human immunodeficiency virus type- 1 integrase in vitro. Biochemistry. 1997;36:173–180. doi: 10.1021/bi961849o. [DOI] [PubMed] [Google Scholar]
- 25.Zheng R, Jenkins TM, Craigie R. Zinc folds the N-terminal domain of HIV-1 integrase, promotes multimerization, and enhances catalytic activity. Proc. Natl Acad. Sci. USA. 1996;93:13659–13664. doi: 10.1073/pnas.93.24.13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leh H, Brodin P, Bischerour J, Deprez E, Tauc P, Brochon JC, LeCam E, Coulaud D, Auclair C, Mouscadet JF. Determinants of Mg2+-dependent activities of recombinant human immunodeficiency virus type 1 integrase. Biochemistry. 2000;39:9285–9294. doi: 10.1021/bi000398b. [DOI] [PubMed] [Google Scholar]
- 27.Asante-Appiah E, Skalka AM. A metal-induced conformational change and activation of HIV-1 integrase. J. Biol. Chem. 1997;272:16196–16205. doi: 10.1074/jbc.272.26.16196. [DOI] [PubMed] [Google Scholar]
- 28.Yi J, Asante-Appiah E, Skalka AM. Divalent cations stimulate preferential recognition of a viral DNA end by HIV-1 integrase. Biochemistry. 1999;38:8458–8468. doi: 10.1021/bi982870n. [DOI] [PubMed] [Google Scholar]
- 29.Pemberton IK, Buckle M, Buc H. The metal ion-induced cooperative binding of HIV-1 integrase to DNA exhibits a marked preference for Mn(II) rather than Mg(II) J. Biol. Chem. 1996;271:1498–1506. doi: 10.1074/jbc.271.3.1498. [DOI] [PubMed] [Google Scholar]
- 30.Engelman A, Craigie R. Efficient magnesium-dependent human immunodeficiency virus type 1 integrase activity. J. Virol. 1995;69:5908–5911. doi: 10.1128/jvi.69.9.5908-5911.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pemberton IK, Buc H, Buckle M. Displacement of viral DNA termini from stable HIV-1 integrase nucleoprotein complexes induced by secondary DNA-binding interactions. Biochemistry. 1998;37:2682–2690. doi: 10.1021/bi971893j. [DOI] [PubMed] [Google Scholar]
- 32.Katzman M, Sudol M. Nonspecific alcoholysis, a novel endonuclease activity of human immunodeficiency virus type 1 and other retroviral integrases. J. Virol. 1996;70:2598–2604. doi: 10.1128/jvi.70.4.2598-2604.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Soultrait VR, Lozach PY, Altmeyer R, Tarrago-Litvak L, Litvak S, Andreola ML. DNA aptamers derived from HIV-1 RNase H inhibitors are strong anti- integrase agents. J. Mol. Biol. 2002;324:195–203. doi: 10.1016/s0022-2836(02)01064-1. [DOI] [PubMed] [Google Scholar]
- 34.Phan AT, Kuryavyi V, Ma JB, Faure A, Andreola ML, Patel DJ. An interlocked dimeric parallel-stranded DNA quadruplex: a potent inhibitor of HIV-1 integrase. Proc. Natl Acad. Sci. USA. 2005;102:634–639. doi: 10.1073/pnas.0406278102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen H, Engelman A. The barrier-to-autointegration protein is a host factor for HIV type 1 integration. Proc. Natl Acad. Sci. USA. 1998;95:15270–15274. doi: 10.1073/pnas.95.26.15270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei SQ, Mizuuchi K, Craigie R. A large nucleoprotein assembly at the ends of the viral DNA mediates retroviral DNA integration. EMBO J. 1997;16:7511–7520. doi: 10.1093/emboj/16.24.7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei SQ, Mizuuchi K, Craigie R. Footprints on the viral DNA ends in moloney murine leukemia virus preintegration complexes reflect a specific association with integrase. Proc. Natl Acad. Sci. USA. 1998;95:10535–10540. doi: 10.1073/pnas.95.18.10535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guiot E, Carayon K, Delelis O, Simon F, Tauc P, Zubin E, Gottikh M, Mouscadet JF, Brochon JC, Deprez E. Relationship between the oligomeric status of HIV-1 integrase on DNA and enzymatic activity. J. Biol. Chem. 2006;281:22707–22719. doi: 10.1074/jbc.M602198200. [DOI] [PubMed] [Google Scholar]
- 39.Zhang S, Pointer D, Singer G, Feng Y, Park K, Zhao LJ. Direct binding to nucleic acids by Vpr of human immunodeficiency virus type 1. Gene. 1998;212:157–166. doi: 10.1016/s0378-1119(98)00178-4. [DOI] [PubMed] [Google Scholar]
- 40.de Rocquigny H, Caneparo A, Delaunay T, Bischerour J, Mouscadet JF, Roques BP. Interactions of the C-terminus of viral protein R with nucleic acids are modulated by its N-terminus. Eur. J. Biochem. 2000;267:3654–3660. doi: 10.1046/j.1432-1327.2000.01397.x. [DOI] [PubMed] [Google Scholar]
- 41.Bischerour J, Tauc P, Leh H, de Rocquigny H, Roques B, Mouscadet JF. The (52–96) C-terminal domain of Vpr stimulates HIV-1 IN-mediated homologous strand transfer of mini-viral DNA. Nucleic Acids Res. 2003;31:2694–2702. doi: 10.1093/nar/gkg364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poljak L, Batson SM, Ficheux D, Roques BP, Darlix JL, Kas E. Analysis of NCp7-dependent activation of HIV-1 cDNA integration and its conservation among retroviral nucleocapsid proteins. J. Mol. Biol. 2003;329:411–421. doi: 10.1016/s0022-2836(03)00472-8. [DOI] [PubMed] [Google Scholar]
- 43.Delelis O, Parissi V, Leh H, Mbemba G, Petit C, Sonigo P, Deprez E, Mouscadet JF. Efficient and specific internal cleavage of a retroviral palindromic DNA sequence by tetrameric HIV-1 integrase. PLoS ONE. 2007;2:e608. doi: 10.1371/journal.pone.0000608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diamond TL, Bushman FD. Division of labor within human immunodeficiency virus integrase complexes: determinants of catalysis and target DNA capture. J. Virol. 2005;79:15376–15387. doi: 10.1128/JVI.79.24.15376-15387.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo F, Gopaul DN, van Duyne GD. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature. 1997;389:40–46. doi: 10.1038/37925. [DOI] [PubMed] [Google Scholar]
- 46.Kobryn K, Watson MA, Allison RG, Chaconas G. The Mu three-site synapse: a strained assembly platform in which delivery of the L1 transposase binding site triggers catalytic commitment. Mol. Cell. 2002;10:659–669. doi: 10.1016/s1097-2765(02)00596-8. [DOI] [PubMed] [Google Scholar]
- 47.Chen X, Tsiang M, Yu F, Hung M, Jones GS, Zeynalzadegan A, Qi X, Jin H, Kim CU, Swaminathan S, Chen JM. Modeling, analysis, and validation of a novel HIV integrase structure provide insights into the binding modes of potent integrase inhibitors. J. Mol. Biol. 2008;380:504–519. doi: 10.1016/j.jmb.2008.04.054. [DOI] [PubMed] [Google Scholar]
- 48.Ren G, Gao K, Bushman FD, Yeager M. Single-particle Image Reconstruction of a Tetramer of HIV Integrase Bound to DNA. J. Mol. Biol. 2006;366:286–294. doi: 10.1016/j.jmb.2006.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.