

Abstract
DNA mismatch repair is initiated by the recognition of mismatches by MutS proteins. The mechanism by which MutS searches for and recognizes mismatches and subsequently signals repair remains poorly understood. We used single-molecule analyses of atomic force microscopy images of MutS-DNA complexes, coupled with biochemical assays, to determine the distributions of conformational states, the DNA binding affinities, and the ATPase activities of wild type and two mutants of MutS, with alanine substitutions in the conserved Phe-Xaa-Glu mismatch recognition motif. We find that on homoduplex DNA, the conserved Glu, but not the Phe, facilitates MutS-induced DNA bending, whereas at mismatches, both Phe and Glu promote the formation of an unbent conformation. The data reveal an unusual role for the Phe residue in that it promotes the unbending, not bending, of DNA at mismatch sites. In addition, formation of the specific unbent MutS-DNA conformation at mismatches appears to be required for the inhibition of ATP hydrolysis by MutS that signals initiation of repair. These results provide a structural explanation for the mechanism by which MutS searches for and recognizes mismatches and for the observed phenotypes of mutants with substitutions in the Phe-Xaa-Glu motif.
DNA mismatch repair (MMR)4 corrects DNA synthesis errors post-replicatively and is essential for the stability of the genome. MutS homologs, which are highly conserved throughout prokaryotes and eukaryotes, are responsible for the initiation of MMR and are also involved in DNA damage-induced activation of cell cycle checkpoints and apoptosis (1-3). To initiate MMR, MutS must first locate and recognize base pair mismatches or base insertion/deletions (IDLs) in duplex DNA. MutS proteins are dimers with DNA binding and ATPase activities, both of which are essential for MMR (1, 3). In the presence of ATP, binding of MutS to a mismatch or IDL, but not to homoduplex DNA, results in a conformational change in MutS, such that it forms a mobile clamp state that can move along the DNA (1-7). This state in turn promotes the protein-protein interactions that signal excision and resynthesis of the nascent DNA strand (1-4).
In MMR, MutS has one of the most difficult tasks of all DNA repair enzymes. It must find mismatches or IDLs among the vast excess of correctly paired DNA in a timely manner. Unlike most DNA repair enzymes, such as DNA glycosylases and AP endonucleases that recognize chemical modifications of the DNA base or backbone, MutS must recognize normal bases that differ only in their noncovalent interactions with the complementary strand. This task is further complicated by the requirement that MutS must recognize multiple different mismatches, and the most common mismatches, such as GT, AC, or single IDLs, do not destabilize or distort double-stranded DNA significantly (8-10).
The crystal structures of Thermus aquaticus and Escherichia coli MutS and the human homolog MutSα (Msh2-Msh6) bound to heteroduplex DNAs containing several different mismatches (4, 5, 11-17) have shed light onto the interactions that govern mismatch recognition. The majority of contacts between MutS and DNA are nonspecific, with only two amino acids from one subunit of the dimer making specific interactions with the mismatch. These residues belong to a Phe-Xaa-Glu motif that is conserved in prokaryotes and in the Msh6 subunit of eukaryotic MutSα, and mutation of either Phe or Glu to alanine results in defective MMR in vivo (18-23). All of the MutS mismatched DNA structures show DNA kinked with a 45-60° bend angle. The conserved Phe stacks with a mismatched or inserted base, which is rotated out into the minor groove by ∼3 Å, and the Glu forms a hydrogen bond with the N-3 of a mismatched thymine or the N-7 of mismatched purines (11, 12, 16). These structural data led to the idea that local flexibility of a mismatch or IDL site underlies mismatch recognition by MutS (2). A recent study using atomic force microscopy (AFM) to visualize directly MutS proteins bound to mismatches and to homoduplex DNA (9) demonstrated that MutS-DNA complexes exhibit a single population of conformations in which the DNA is bent at homoduplex sites (nonspecific complexes), but two populations of conformations, bent and unbent, at mismatch sites (specific complexes). These findings led to a model of MutS action in which the kinked conformation observed in the crystal structures represents an initial recognition complex (IRC) that is an intermediate in the pathway toward an unbent conformation, which is the ultimate recognition complex (URC) that signals repair (3, 9). The proposed unbent URC provides a structural explanation for the observed inverse correlation between the ease with which a mismatch is bent (or kinked) and the efficiency with which it is repaired (3, 9). However, several important questions still remain about the process of mismatch recognition and signaling of repair. How does MutS find mismatches or IDLs in DNA? What roles do the conserved Phe and Glu residues play in the formation of bent and unbent complexes at mismatches and at homoduplex sites on DNA? How do the distributions of bent and unbent MutS-DNA complexes correlate with mismatch or IDL recognition and initiation of repair by MutS?
To address these questions, we used AFM to characterize the binding affinities and conformational properties of wild type MutS (wtMutS) and two MutS mutants, each with a single mutation in the Phe-Xaa-Glu motif, interacting with specific (mismatches and IDLs) and nonspecific (homoduplex) sites on DNA (see Fig. 1). We have recently developed methods to determine protein-DNA binding constants and specificities (ratio of the affinity for a specific site to nonspecific sites) from AFM images, by analyzing the occupancy of a protein on DNA as a function of its position on the DNA (24). We previously used this method to measure the binding affinities and specificities of wild type T. aquaticus MutS for a T-bulge and a GT mismatch and demonstrated that the binding parameters determined using this method are consistent with those from fluorescence anisotropy measurements (24). This AFM method is particularly useful for proteins, such as MutS, that have high affinities for DNA ends, which can prevent accurate determination of the specificity for a mismatch using bulk methods (24). Comparison of the binding affinities and conformations of specific and nonspecific MutS-DNA complexes of wild type and mutant proteins provides information on the mechanism by which MutS finds and recognizes a mismatch. In addition to these AFM studies, we examined the ATPase activities of the mutant MutS proteins in the presence of homoduplex DNA or DNA containing a mismatch or an IDL, which allows a detailed comparison of structure, stability, and function, thereby providing a better understanding of how these conserved residues contribute to the ability of MutS to initiate DNA mismatch repair.
FIGURE 1.

DNA substrates and AFM images of MutS-DNA complexes. a, schematic diagram of DNA substrates showing the position of the mismatch: T-bulge (left rectangle) and GT mismatch (right rectangle). b-d, AFM images of 10 nm wtMutS (b), 20 nm MutS-F39A (c), and 10 nm MutS-E41A (d) incubated and deposited with the 783T-bulge DNA substrate (2-5 nm). The images are 1 μm × 1 μm with a height scale of 2 nm. The arrows in the images indicate protein-DNA complexes at different DNA sites: DNA ends (yellow), specific complex at a T-bulge (red), and nonspecific complexes (pink).
MATERIALS AND METHODS
MutS Proteins and DNA Substrates—T. aquaticus MutS F39A and E41A were purified as described previously for wtMutS except that a gentle procedure of heat denaturation was used during purification (25, 26). DNA substrates for AFM were created by ligating three fragments as described previously (9). The sequences of the individual strands of the fragment middle pieces containing the mismatches are: 1) 5′-GTG TCG GGT TCG CGT CAT ATG GCG GGT GTC GGG GCT GGC TTA AGG TGT GAA ATA CCT CAT CTC GAG (G)CG TGC CGA TAT TTC-3′ and 2) 5′-ATA TCG GCA CGT CTC GAG ATG AGG TAT TTC ACA CCT TAA GCC AGC CCC GAC ACC CGC CAT ATG ACG CGA ACC CGA CAC TAC-3′. For generating 783-bp T-bulge DNA (783T-bulge), the middle G underlined in the sequence of oligonucleotide 1 was omitted (indicated by the base being in brackets in the sequence). Oligonucleotides 1 and 2 were annealed to produce the middle fragment for 783T-bulge and for the 783-bp GT mismatch DNA (783GT). In the 783-bp DNA fragments, both the T-bulge and the GT mismatch are located 27% of the total DNA length (213 bp) from one of the fragment ends. Fig. 1a shows a schematic of the DNA fragments used in the AFM experiments. The ATPase assays were performed with 23-bp duplexes formed by annealing: 1) 5′-GCG CGA CGG TAT ATA GCT GCC GG-3′ and 2) 5′-CC GGC AGC TAT XTA CCG TCG CGC-3′, where X was absent, G, or A to form T-bulge, GT mismatch, and AT homoduplex substrates, respectively.
Atomic Force Microscopy—Protein-DNA complexes were formed by incubating 10-20 nm T. aquaticus MutS F39A or E41A dimers with 1-5 nm of the DNA substrates for 1-10 min at room temperature in binding buffer (20 mm Hepes-NaOH, pH 7.8, 50 mm NaCl, 5 mm MgCl2) in a total volume of 20 μl. The reaction was deposited onto freshly cleaved mica (Spruce Pine Mica Company) at room temperature. After <1 min of incubation, the mica surface was rinsed with high pressure liquid chromatography grade water, blotted dry, and then dried under a stream of nitrogen. Although the samples are “dried,” the surface, the protein, and the DNA remain hydrated because mica is extremely hygroscopic. The images were captured in air with a Nanoscope IIIa (Digital Instruments) microscope in tapping mode. Pointprobe® tapping mode silicon probes (Molecular Imaging Corporation) with spring constants of ∼50 n·m-1 and resonance frequencies ∼170 kHz were used for all imaging. The images were collected at a speed of 3 Hz, a size of 1 μm × 1 μm, and a resolution of 512 × 512 pixels.
Image Analysis—Either NANOSCOPE software (Digital Instruments, Veeco, Santa Barbara, CA) or Scion Image was used to measure the DNA contour lengths, the position of the proteins on the DNA, and the MutS-induced DNA bend angles, as described previously (9). The program Origin (Microcal Inc.) was used for statistical analysis of the data. For each data set, images from at least three independent experiments were analyzed, compared, and pooled.
The positions of MutS binding on the DNA fragments were determined by measuring the distance from the bound MutS protein to each end. The binding position is defined as the ratio of the length of the shorter DNA tract divided by the total contour length. Complexes with centers within one standard deviation of the expected mismatch position were categorized as specific complexes. We did not end label the DNA to unequivocally identify the DNA ends, which means that some nonspecific complexes will be counted as specific complexes but not vice versa. For wtMutS and MutS-E41A, less than 1% of the nonspecific complexes will be counted as specific because the specific complexes are significantly more stable than the nonspecific complexes (see Table 1 and Ref. 9). DNA bending was determined by measuring the angle, θ, at the intersection of the two DNA arms at the position of the DNA-bound protein. The DNA bend angle is defined as 180° - θ. DNA molecules having MutS bound to more than one internal site or to more than one DNA fragment were not included in the statistical analysis of bend angles.
TABLE 1.
DNA dissociation constants and specificities
|
wtMutSa
|
MutS-F39A
|
MutS-E41A
|
||||
|---|---|---|---|---|---|---|
| Kd | Specificity | Kd | Specificity | Kd | Specificity | |
| nM | nM | nM | ||||
| GT | 77 ± 8 | 300 ± 36 | >2800b | <10b | 79 ± 25c | 294 ± 72c |
| T-bulge | 21 ± 2 | 1660 ± 220 | >2800b | <10b | 13 ± 3 | 1560 ± 420 |
| DNA endsd | 53 ± 9 | 538 ± 95 | 73 ± 6 | 386 ± 74 | 117 ± 58 | 200 ± 80 |
| Nonspecificd | 28,900 ± 5,600 | 28,000 ± 2,200 | 23,300 ± 7,300 | |||
The data for wtMutS are taken from Ref. 24.
Although we do not detect any preference of MutS-F39A for a GT or a T-bulge (supplemental Fig. S1), the resolution of the position distributions limits our detection of binding preferences to specificities ≤10. Consequently the binding affinity of MutS-F39A to a GT or T-bulge could be as much as 10-fold tighter than to nonspecific sites.
The values obtained with the width of the distribution being held to the width determined from the fits of the T-bulge data.
All of the standard deviations given in the table reflect variations between repeated experiments on either GT or T-bulge DNA. The values for nonspecific sites and DNA ends represent the averages (and standard deviations) of the binding affinities to these sites on the 783GT and 783Tbulge DNA fragments.
Binding Constants and Specificities—Binding specificities and constants were calculated as described previously (24). Briefly, the analysis method is based on the determination of the average occupancy of MutS at nonspecific and specific (mismatch) sites on the DNA as well as at the DNA ends. Knowledge of the position of the mismatch in the DNA fragment (at 27% of DNA length for 783GT and 783T-bulge) allows us to determine the frequencies of MutS bound to specific and nonspecific sites. It is important to note that each DNA fragment contains one specific site, two DNA ends, and 769 nonspecific sites (24). Consequently, at 1 nm DNA fragment, the concentrations of specific sites, DNA ends, and nonspecific sites are 1, 2, and 769 nm, respectively. The high concentration of nonspecific sites makes it possible to determine the binding affinity of MutS for homoduplex DNA, even though it is weak (24). From the AFM images, only DNA fragments with lengths within one standard deviation from the average measured length were considered in the analysis. A Gaussian fit to the distribution of positions of MutS on the DNA provides the specificity of the protein for the mismatch (24). Typically, the resolution of the positions of protein on the DNA allows detection of mismatch specificities ≥10. The binding constants for DNA mismatches, DNA homoduplex sites, and DNA ends are calculated from the specificities and fractional DNA occupancies, as described previously (24).
ATPase Assays—Stopped flow phosphate (Pi) release assays using E. coli phosphate-binding protein labeled with MDCC-PBP were performed with wild type and mutant MutS proteins, as described previously (27). Briefly, 40 μl of 2 μm MutS dimer, in the absence or presence of 8 μm DNA, was mixed with 40 μl of 16 μm MDCC-PBP and 1 mm ATP in 20 mm Hepes-NaOH, pH 7.7, 40 mm NaCl, 5 mm MgCl2, at 40 °C, and the change in fluorescence upon MDCC-PBP binding to Pi was measured by excitation at 425 nm and emission at >450 nm. The burst kinetics traces were fit to a an exponential + linear equation,
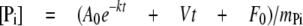 |
(Eq.1) |
where [Pi] corresponds to phosphate concentration, A0 is the amplitude, k is the observed burst rate constant, V is the velocity of the linear phase, F0 is the initial fluorescence intensity, t is time, and mPi is the slope of the Pi standard curve measured under the same conditions.
RESULTS
For the AFM studies, we employed two 783-bp heteroduplex DNA substrates containing either a single GT mismatch (783GT) or a single T-bulge (783Tbulge) 213 bp (27%) from one end (Fig. 1a). Because we know the position of the mismatch along the DNA, we can discriminate between MutS bound to a mismatch or to homoduplex DNA by measuring the distance of the complex from the ends of the DNA fragment (see “Materials and Methods”). The T. aquaticus MutS mutants each possess a single mutation in the Phe-Xaa-Glu motif: Phe39 to Ala (F39A) and Glu41 to Ala (E41A). Representative AFM images of heteroduplex DNA deposited in the presence of wtMutS, MutS-F39A, and MutS-E41A are shown in Fig. 1. From a large number of AFM images, we determined the occupancies of the proteins as a function of position on the DNA to calculate the binding affinities and specificities (24) (see “Materials and Methods”), and we measured the MutS-induced DNA bend angles to characterize the conformations of the complexes.
Effect of Phe39 on DNA Affinity and Bending—Inspection of the position distributions of MutS-F39A bound to DNA substrates containing either a GT mismatch or a T-bulge reveals that unlike wtMutS (24), MutS-F39A does not exhibit any significant preference for mismatch or IDL sites relative to homoduplex DNA (see supplemental Fig. S1 and Table 1). The resolution of the position distributions limits our detection of binding preferences to specificities ≥10. Consequently the binding affinity of MutS-F39A to a GT mismatch or T-bulge could be up to 10-fold tighter than to nonspecific sites (Table 1). Although there is a loss of specificity for the mismatch, the binding affinity of MutS-F39A to homoduplex DNA is the same as wtMutS, and the binding affinity for DNA ends is only slightly reduced relative to wtMutS (Table 1). These results are consistent with previous biochemical studies that reported a dramatic loss of affinity of MutS-F39A for mismatches and some apparent loss of binding to homoduplex DNA (19, 28, 29).
To examine the effect of the F39A mutation on the conformation of MutS-DNA complexes, we measured the bend angles induced by MutS-F39A at homoduplex sites and specific sites (GT mismatch and T-bulge). Although MutS-F39A does not exhibit significant specificity for the mismatches, we can still determine the bend angles at these sites because we know their positions in the DNA. The distributions of DNA bend angles (Fig. 2, d-f) show that MutS-F39A exhibits a single population of complexes with an average bend angle of ∼60° for the GT mismatch and T-bulge, as well as for homoduplex DNA. Like wtMutS, MutS-F39A induces DNA bending on homoduplex DNA and at mismatches; however, MutS-F39A no longer exhibits a peak at 0° at a T-bulge or a GT mismatch. Because of the loss of binding specificity of MutS-F39A, a relatively small number of specific complexes were analyzed (Fig. 2, e and f), resulting in greater error in the fits to these bend angle distributions; however, it is clear that there is no significant population of unbent MutS-F39A complexes with a T-bulge or a GT mismatch.
FIGURE 2.

Histograms of DNA bend angles induced by wtMutS (top row), MutS-F39A (middle row), and MutS-E41A (bottom row). Distributions of bend angles are shown for wtMutS bound to homoduplex (nonspecific) DNA (a), a GT mismatch (b), and a T-bulge (c); for MutS-F39A bound to nonspecific DNA (d), a GT mismatch (e), and a T-bulge (f); and for MutS-E41A bound to nonspecific DNA (g), a GT mismatch (h), and a T-bulge (i). The number of complexes (n) analyzed for each distribution is shown on the y axis. The distributions for the nonspecific DNA are determined from MutS proteins bound to homoduplex sites on the 783GT and 783T-bulge DNA fragments. The data for the distribution for wtMutS bound to a T-bulge are taken from reference (9). Complexes in which more than one MutS protein was bound internally to the DNA were not included in these distributions. The curves drawn in a and d--g are single Gaussian fits to the data, and those drawn in b, c, h, and i are double Gaussian fits to the data. For the single Gaussian fits, fitting the bend angle distributions to a sum of two Gaussians does not significantly improve the fits, and a binomial distribution analysis shows no peak at 0° for these data sets (a and d--g). In contrast, a binomial distribution analysis of each of the plots in b, c, and i as well as of the plot in h indicates that the peak at 0° (in b, c, and i) and the peak at 15° (in h) are significant with p = 5 × 10-10, 6 × 10-8, 5 × 10-6, and 7 × 10-12, respectively. In addition, for complexes bound at the specific GT mismatch site, the bend angle shift from 0° for wtMutS to 15° for MutS-E41A is very significant (p = 2 × 10-32, based on normal distribution significance test). For MutS-F39A, a relatively small number of complexes are observed at specific sites (e and f) because of the loss of binding specificity, resulting in greater error in these fits; however, there is clearly no significant population of unbent complexes.
Effect of Glu41 on DNA Affinity and Bending—In contrast to MutS-F39A, the position distributions of MutS-E41A on mismatch and IDL-containing DNA fragments show clearly that this mutant retains specificity for both the GT mismatch and the T-bulge (supplemental Fig. S1). Indeed, the affinities and specificities of MutS-E41A for these sites differ only slightly from those of wtMutS (Table 1). These results are consistent with biochemical studies, which show no significant differences between T. aquaticus wtMutS and MutS-E41A affinities or specificities for GT mismatch or T-bulge containing DNA (21); however, the corresponding mutant from E. coli exhibited a modest increase in nonspecific binding and a decrease in specificity (21, 22).
Comparison of the DNA bend angle distributions of MutS-E41A and wtMutS at specific and nonspecific DNA sites reveals intriguing differences between the two proteins (Fig. 2). For nonspecific complexes, the bend angle distribution for MutS-E41A is broader than that of wtMutS, with a significant percentage of complexes exhibiting lesser extents of DNA bending (Fig. 2, g versus a). This result suggests that Glu41 is involved in inducing DNA bending at nonspecific sites. For the specific complexes, the distributions of DNA bend angles exhibit two populations (Fig. 2) and are fit significantly better by double Gaussians than by single Gaussians (data not shown). Notably, however, the positions of the two peaks are different for MutS-E41A bound to a T-bulge versus a GT mismatch. The bend angle distribution for MutS-E41A complexed with the T-bulge resembles that of wtMutS (Fig. 2, i versus c), as evidenced by binomial distribution analysis, which yields a significance of the population at 0° of p = 5 × 10-6 and p = 6 × 10-8 for MutS-E41A and wtMutS, respectively. In contrast, both peaks in the bend angle distribution for MutS-E41A bound to the GT mismatch are shifted to higher angles relative to wtMutS. The peak for the population of complexes that are “unbent” is shifted significantly from an angle of 0° for wtMutS to an angle of 15° for MutS-E41A (Fig. 2, b versus h), and the peak of the “bent” population has shifted from ∼ 40° for wtMutS to ∼ 58° for MutS-E41A. A binomial distribution analysis gives a significance of p = 7 × 10-12 for the population at 15° for MutS-E41A and a significance of p = 5 × 10-10 for the population at 0° for wtMutS. The significance of the difference in the positions of the two peaks for wtMutS (0°) and MutS-E41A (15°) is p = 2 × 10-32 according to a normal distribution based significance test of two averages. The relevance of this finding is explored further below.
Effect of MutS-DNA Conformation on the Next Step in Mismatch Repair—The similarity in wtMutS and MutS-E41A interactions with a T-bulge and the differences in their interactions with a GT mismatch presented us with an opportunity to assess the functional relevance of the various MutS-DNA complex conformations detected by AFM analysis. Previous studies have shown that binding of MutS to mismatched or IDL-containing DNA inhibits its rapid ATP hydrolysis activity, leading to a long-lived ATP-bound state (27, 29, 30) that likely signals MMR (3, 5, 31). Accordingly, to determine how MutS-DNA conformations relate to function, we measured the ATP hydrolysis activities of wild type and mutant MutS proteins in the presence of homoduplex DNA or DNA containing a T-bulge or a GT mismatch.
Pre-steady state analyses of phosphate release, which reports the ATP hydrolysis step in the reaction, show that both wtMutS (Fig. 3A) and MutS-E41A (Fig. 3B) catalyze a rapid burst of ATP hydrolysis with a rate of 8 ± 0.5 s-1 followed by a slow turnover rate of 0.4-0.5 s-1 in the presence of homoduplex DNA. These two proteins (and MutS-F39A) exhibit the same ATPase activity profile in the absence of DNA as well (29) (data not shown). As reported previously, the presence of T-bulge DNA inhibits the burst of ATP hydrolysis catalyzed by wtMutS, stabilizing it in an ATP-bound state (Fig. 3A) (29). T-bulge DNA also suppresses the burst of ATP hydrolysis by MutS-E41A (Fig. 3B), suggesting that both proteins achieve the same conformation in complex with this IDL, which is consistent with the AFM data (Fig. 2, c and i). In striking contrast, DNA containing a GT mismatch suppresses ATP hydrolysis by wtMutS (Fig. 3A), but not by MutS-E41A (Fig. 3B), which displays the same burst of ATP hydrolysis as in the absence of DNA or presence of homoduplex DNA. Notably, the ATPase data completely parallel the AFM data. Specifically, on a T-bulge, MutS-E41A and wtMutS induce similar DNA conformations (Fig. 2, c versus i) and exhibit similar ATPase kinetics (Fig. 3, A versus B), whereas on a GT mismatch, MutS-E41A induces different DNA conformations (Fig. 2, b versus h) and shows altered ATPase kinetics relative to wtMutS (Fig. 3, A versus B). These observations suggest that the MutS-DNA complex may need to form a specific unbent conformation before it can be stabilized in a functionally relevant ATP-bound state and that this conformation depends on both Glu41 and the type of discrepancy in the double helix.
FIGURE 3.

ATPase activity of wtMutS (A) and MutS-E41A (B) in the presence of homoduplex (black), T-bulge (light gray), and GT mismatch DNA (dark gray). For reactions that show biphasic kinetics, the rate constants of the burst phase and the linear steady-state phase are shown on the plots.
DISCUSSION
Phe39 Does Not Induce DNA Bending but Rather It Induces the Formation of the Unbent Complexes at Mismatches—The inability of the MutS-F39A mutant to recognize either a GT mismatch or a T-bulge is consistent with previous biochemical studies (19, 28) and indicates that the conserved Phe is a major contributor to specific mismatch recognition. Interestingly, however, MutS-F39A still induces DNA bending similar to wtMutS on the GT mismatch and the T-bulge as well as on homoduplex DNA (Fig. 2, d-f). We have previously demonstrated that in the absence of MutS, the bend angle distributions for the GT mismatch and the T-bulge, as well as for homoduplex DNA, are all half-Gaussian distributions centered at 0° with similar breadths, indicating that none of the substrates exhibit any significant intrinsic DNA bending (9). These results indicate that Phe39 is not responsible for inducing DNA bending at specific or nonspecific sites, although it is clearly involved in stabilizing the kinked IRC at specific sites. In addition, the loss of specificity, taken together with the wild type-like DNA bending, suggests that the relative ease of bending of mismatches compared with homoduplex DNA does not contribute a significant amount of stability to the MutS mismatch complex. The observation that Phe39 is not necessary for MutS-induced DNA bending is quite surprising, given that phenylalanines are generally involved in inducing DNA bending (32-35). Notably, it is the unbent population that is absent from the specific complexes formed with MutS-F39A (Fig. 2, e and f), indicating that Phe39 is essential for the formation of the unbent URC at specific sites. Both the loss of binding specificity and the loss of the formation of the URC can explain the observation that mutation of this conserved Phe in MutS homologs results in a loss of DNA MMR in vivo (18, 19, 28).
Although the binding affinities of MutS-F39A to the mismatches are reduced by >40-fold (Table 1), the affinity and bend angle distribution for homoduplex DNA are very similar to those of wtMutS (Table 1 and Fig. 2, a and d). Assuming that mutation of Phe39 to Ala does not cause significant conformational changes in the protein, these results strongly suggest that the conserved Phe in the Phe-Xaa-Glu motif of MutS homologs does not interact with nonspecific DNA and only interacts with the mismatched (or IDL) base after some mismatch-induced conformational change in the MutS-DNA complex (see below).
Glu41-dependent Formation of the Unbent URC Is Necessary to Signal Repair—In contrast to mutation of the conserved Phe residue, mutation of the conserved Glu to Ala in T. aquaticus MutS (E41A) or E. coli MutS (E38A) has little effect on the binding affinities and specificities for a GT mismatch or a T-bulge (Table 1) (21, 22). In addition, the crystal structures of wild type and E38A E. coli MutS are very similar to one another (22). Despite this lack of differences, E. coli MutS-E38A is severely compromised for repair of base-base mismatches in vivo (21-23) and defective in the ATP-induced formation of the mobile clamp state in vitro (22). The latter finding led to the proposal that formation of a hydrogen bond between Glu and a mismatched base causes a post-recognition conformational change in the MutS mismatch complex that inhibits ATP hydrolysis and promotes formation of a mobile MutS clamp that signals repair. The AFM and ATPase data reported here suggest that the formation of the unbent URC is likely to be a precursor to the conformation that inhibits ATP hydrolysis and permits the formation of the mobile clamp state. Specifically, for a T-bulge, T. aquaticus MutS-E41A exhibits a DNA bend angle distribution and ATPase activity that are very similar to those of wtMutS (Figs. 2, i and c, and 3), indicating that MutS-E41A, like wtMutS, takes the next step in the repair reaction. In contrast, for a GT mismatch, the population of complexes that are “unbent” is shifted from an angle of 0° for wtMutS to an angle of ∼15° for MutS-E41A (Fig. 2, b versus h), and the burst of ATP hydrolysis by MutS-E41A is not suppressed by GT-DNA, indicating that MutS-E41A bound to a GT cannot attain the conformation necessary to signal repair. This slightly bent (15°) population appears to be trapped in a conformation with an intermediate bend angle, unable to attain the completely unbent conformation that appears to be necessary for repair. These results provide a plausible structural explanation for the loss of repair of base-base mismatches observed with the MutS-E38A mutant in E. coli (21, 22).
The observations that MutS-E41A induces the same conformations and exhibits the same ATPase activity as wild type protein on a T-bulge, but not on a GT mismatch, suggest that the Glu residue may be more important for repairing certain types of errors, such as base-base mismatches, than others, such as IDLs. Consistent with this hypothesis, mutation of the homologous residue in E. coli (E38A) or yeast (yMsh2-Msh6-E339A) to Ala results in different mutation rates for base-base mismatches and IDLs. Specifically, both E. coli MutS-E38A and yeast yMsh2-Msh6-E339A exhibit a significant increase in mutations resulting from base-base mismatches (21-23, 36), whereas E. coli MutS-E38A exhibits a lower mutation frequency for frameshift mutations than for base-base mismatches (indicating better repair of IDLs such as the T-bulge relative to mismatches) (23), and yMsh2-Msh6-E339A displays no mutator phenotype for frameshift mutations (20, 23, 36). The in vivo results are thus consistent with our prediction that a reduction in the population of the unbent state would lead to reduced repair and strongly support our proposal that formation of the unbent URC is essential for signaling DNA repair. Taken together, these results suggest that Glu plays a subtle but functionally critical role in the interaction between MutS and a mismatch.
The Conserved Glutamate Facilitates DNA Bending on Homoduplex DNA—For nonspecific complexes, the bend angle distribution for MutS-E41A is broader than that of wtMutS, with a significant percentage of complexes exhibiting lesser extents of DNA bending (Fig. 2, g versus a). This result suggests that Glu41 interacts with nonspecific DNA and helps induce DNA bending. Notably, Glu41 plays different roles in specific and nonspecific complexes, facilitating unbending in specific complexes but inducing bending in nonspecific complexes. These differences likely result from different types of interactions between Glu41 in specific versus nonspecific complexes. In specific complexes, Glu41 (Glu38 in E. coli) makes a hydrogen bond with one of the mismatched bases (11, 12, 22), whereas such an interaction is unlikely to occur between MutS and perfectly paired DNA (nonspecific complexes) (22). In complexes with homoduplex DNA, Glu41 probably facilitates DNA bending via nonspecific electrostatic interactions. Consistent with this suggestion, asymmetric distributions of both positively and negatively charged amino acids have been shown to promote protein-induced DNA bending, without significant changes in affinity (37), as seen for MutS-E41A (Table 1). In this scenario, Glu41 first forms nonspecific electrostatic interactions with homoduplex DNA, and upon interaction with a mismatch, this nonspecific interaction converts to a hydrogen bond with the mismatched base. Such changes from electrostatic interactions in nonspecific complexes to highly specific interactions of the same residue in a recognition complex have been observed previously for lac repressor (38).
MutS Searches for and Recognizes DNA Mismatches via a Multi-step Pathway—Taken together, the results for wtMutS and the two mutants in the Phe-Xaa-Glu motif present a detailed picture of how MutS searches for and recognizes DNA mismatches and suggest how this recognition may signal repair (Fig. 4). In searching for a DNA mismatch, MutS binds to nonspecific DNA and bends it, with the energy to bend the DNA being derived from the extensive nonspecific protein-DNA interactions. In the nonspecific complexes, the negative charge on the conserved Glu facilitates DNA bending (39-41), whereas the conserved Phe does not appear to make any significant interactions with homoduplex DNA. As discussed previously (9), the DNA in nonspecific complexes is likely to be smoothly bent instead of kinked, because in the crystal structures (4, 5, 11-17), the majority of the nonspecific protein-DNA contacts are flanking the mismatch but not immediately adjacent to it. Consequently, in the absence of a mismatch, it is probably more energetically favorable to spread the bend over several base pairs rather than localizing it to a kink (9). Because Glu41 appears to be interacting electrostatically with nonspecific DNA, it seems likely that it would be poised to make a hydrogen bond with a DNA base if it were slightly extruded from the helix. Consequently, we suggest that the first step in mismatch recognition is a conformational change in the DNA from a smooth bend to a local kink, because of the local flexibility of the mismatch, as has been suggested previously (3, 8, 12, 16, 42, 43). We further propose that this kink, in turn, allows the mismatched base to form a hydrogen bond with the Glu residue and that the Phe then stacks with the mismatched base to form the kinked IRC (Fig. 4). As such, the conserved Glu may serve as a “scanning head” facilitating bending and searching for possible hydrogen-bonding interactions with the DNA bases. Such a mechanism allows an efficient search of the DNA for potential mismatches by checking for specific interactions only at positions where the DNA has a propensity to form a kink. In addition, by using nonspecific interactions to bend or kink the DNA instead of using the conserved Phe, which is essential for specific site recognition, all of the interaction energy between the Phe and the mismatched base can be used to stabilize the specific complex over nonspecific ones (44). Once the kinked IRC is formed, both Phe and Glu and the energy stored in the bent DNA (9) facilitate the formation of the unbent URC, which can then undergo the ATP-induced conformational change to the mobile clamp state that leads to repair. Because the ATP-induced formation of the mobile clamp state at a mismatch is essentially irreversible on the time scale of repair, the URC only needs to be modestly populated to efficiently signal repair (9).
FIGURE 4.

Model for MutS searching for and finding a mismatch and then signaling repair. MutS is depicted as the blue and green theta-shaped structure. Phe39 and Glu41 are shown in stick notation. The DNA is yellow and lavender except the mismatched base, which is red. In the kinked IRC and in the unbent URC, conformational changes in the protein are indicated by shape changes in the two monomers. See text for discussion of the model.
We have previously suggested that the mismatched base may be extruded from the DNA helix in the URC (3, 9). Although there is no direct evidence for this proposed mechanism, our observation that MutS-Glu41 appears to be important for forming the URC at GT mismatch but not at a T-bulge is consistent with this suggestion. Specifically, to flip out a base at a GT mismatch requires more energy than to flip out an unpaired T because hydrogen bonds between the G and T must be broken, whereas no hydrogen bonds are required to be broken for an unpaired T. Consequently, the additional energy gained from the hydrogen bond between the Glu and DNA may be more important for mismatches than for IDLs. The observation that Phe39 is absolutely essential for formation of the unbent URC is also consistent with the suggestion of base flipping because Phe is commonly used by proteins that flip out bases (32, 33, 35, 45, 46). For instance, for N-6 adenine DNA methyltransferase M-TaqI, Phe196, which makes an edge-to-face π interaction with the extruded adenine, has been shown to significantly stabilize the base in the extrahelical position (32, 46). Flipping of the mismatched base in the URC by MutS is consistent with both in vitro and in vivo studies of DNA MMR (3). It is also possible that the unbent MutS-DNA complexes are in some alternative conformation that does not involve base flipping; however, it is clear that both the conserved Phe and Glu are involved in promoting the unbent state and that the loss of this conformation correlates with impaired DNA repair.
Different DNA Repair Enzymes Employ Dramatically Different Searching Mechanisms—This mechanism of MutS searching and specific site recognition (Fig. 4) differs markedly from those recently proposed for two other DNA damage recognition enzymes: the 8-oxoguanine (oxoG) DNA glycosylase, MutM, (47) and the uracil DNA glycosylase, UNG (48). Although there is no homology between MutS and these glycosylases, they are faced with similar tasks of finding their specific site among the vast amount of normal DNA; MutM and UNG must find oxoG and uracil bases, respectively, whereas MutS must find mismatched bases and IDLs. Analysis of several crystal structures of MutM bound to nonspecific DNA revealed that MutM inserted a Phe into DNA in all of the complexes, inducing significant DNA distortion. These observations led to the proposal that MutM searches for oxoG-C base pairs by invading the DNA double helix at most positions along the DNA and that this intercalation preferentially causes oxoG to flip out of the helix to interact specifically with MutM because oxoG-C is slightly less stable than a GC base pair (47). In contrast to this active participation of MutM in interrogating the DNA for the presence of its specific site, results from crystallographic and kinetic studies on wild type and mutant UNG proteins have shown that although UNG also employs DNA intercalation coupled with base flipping, it does not distinguish between its target base uracil and correct DNA bases within the context of the DNA helix (48). Instead, UNG passively searches the DNA and recognizes its target (uracil) by trapping the transient extrahelical state of the specific target base using specific interactions with uracil, but not normal DNA bases, within its binding pocket (48). Our results present another very different picture for MutS. MutS binds DNA nonspecifically and bends it without any Phe intercalation or interaction. Upon reaching a mismatch, the DNA bending becomes localized to a kink at the mismatch, causing one of the mismatched bases to be rotated slightly out of the helix and allowing interaction with the conserved Glu and Phe to form a kinked IRC, which then undergoes a conformational change to the final unbent URC (Fig. 4). MutS thus uses a partially passive mechanism, bending the nonspecific DNA and only interrogating the DNA when it has a propensity to kink. The differences in these mechanisms undoubtedly result from the fact that the glycosylases MutM and UNG have specific recognition pockets for their substrates and can therefore discriminate between their targets and other bases after the base has been flipped into the pocket. In contrast, MutS must discriminate based solely on differences in noncovalent interactions between bases, and the formation of the unbent state is the final check before signaling repair. As such, MutS has apparently evolved a delicate balance between locating a mismatch and forming the kinked IRC and the unbent URC, such that the most common mismatches, which are also the least destabilizing, are repaired while avoiding signaling excessive futile repair. Taken together, these results suggest that the mechanism of searching for a specific site in the DNA may be as varied as the many different types of specific recognition mechanisms.
In closing, by using AFM to investigate the binding affinities and conformational properties of wild type and mutant MutS-DNA complexes at the level of individual molecules, we have been able to tease apart the mechanism by which MutS searches for, finds, and recognizes a mismatch. Our results show different roles for the conserved Phe-Xaa-Glu motif of MutS in binding to specific and nonspecific sites. The conserved glutamate facilitates MutS-induced DNA bending at nonspecific sites, but it promotes the formation of the unbent state at specific sites. Surprisingly, the conserved Phe is not responsible for inducing DNA bending at either specific or nonspecific sites but does appear to be necessary for the formation of the unbent conformation at the specific sites. Finally, we observe a direct correlation between the ability of MutS to adopt an unbent DNA conformation at a mismatch or IDL and the ability of the MutS-DNA complex to progress to the next step in the MMR reaction. This combination of AFM and ATPase studies yields insight into the structural properties of MutS-DNA complexes that are essential for signaling repair and provides a structural framework to explain the in vivo phenotypes of MutS mutants.
Supplementary Material
Acknowledgments
We thank Shannon Holmes and Thomas Kunkel for critical reading of this manuscript and for sharing data on the in vivo phenotypes of MutS mutants prior to publication.
This work was supported, in whole or in part, by National Institutes of Health Grant GM079480 (to D. A. E.). This work was also supported by Grant RSG-03-047-04-GMC from the American Cancer Society (to D. A. E.), National Science Foundation Grant MCB 0448379 (to M. M. H.), and the Intramural Research Program of NIDDK (to P. H. and C. D.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
Footnotes
The abbreviations used are: MMR, mismatch repair; IDL, insertions or deletions; AFM, atomic force microscopy; IRC, initial recognition complex; URC, ultimate recognition complex; wt, wild type; MDCC-PBP, N-[2-(1-maleimidyl)ethyl]-7-(diethylamino)coumarin-3-carboxamide; oxoG, 8-oxoguanine.
References
- 1.Iyer, R. R., Pluciennik, A., Burdett, V., and Modrich, P. L. (2006) Chem. Rev. 106 302-323 [DOI] [PubMed] [Google Scholar]
- 2.Schofield, M. J., and Hsieh, P. (2003) Annu. Rev. Microbiol. 57 579-608 [DOI] [PubMed] [Google Scholar]
- 3.Kunkel, T. A., and Erie, D. A. (2005) Annu. Rev. Biochem. 74 681-710 [DOI] [PubMed] [Google Scholar]
- 4.Yang, W., Junop, M. S., Ban, C., Obmolova, G., and Hsieh, P. (2000) Cold Spring Harbor Symp. Quant. Biol. 65 225-232 [DOI] [PubMed] [Google Scholar]
- 5.Junop, M. S., Obmolova, G., Rausch, K., Hsieh, P., and Yang, W. (2001) Mol. Cell 7 1-12 [DOI] [PubMed] [Google Scholar]
- 6.Allen, D. J., Makhov, A., Grilley, M., Taylor, J., Thresher, R., Modrich, P., and Griffith, J. D. (1997) EMBO J. 16 4467-4476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gradia, S., Subramanian, D., Wilson, T., Acharya, S., Makhov, A., Griffith, J., and Fishel, R. (1999) Mol. Cell 3 255-261 [DOI] [PubMed] [Google Scholar]
- 8.Isaacs, R. J., Rayens, W. S., and Spielmann, H. P. (2002) J. Mol. Biol. 319 191-207 [DOI] [PubMed] [Google Scholar]
- 9.Wang, H., Yang, Y., Schofield, M. J., Du, C., Fridman, Y., Lee, S. D., Larson, E. D., Drummond, J. T., Alani, E., Hsieh, P., and Erie, D. A. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 14822-14827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peyret, N., Seneviratne, P. A., Allawi, H. T., and SantaLucia, J., Jr. (1999) Biochemistry 38 3468-3477 [DOI] [PubMed] [Google Scholar]
- 11.Lamers, M. H., Perrakis, A., Enzlin, J. H., Winterwerp, H. H., de Wind, N., and Sixma, T. K. (2000) Nature 407 711-717 [DOI] [PubMed] [Google Scholar]
- 12.Obmolova, G., Ban, C., Hsieh, P., and Yang, W. (2000) Nature 407 703-710 [DOI] [PubMed] [Google Scholar]
- 13.Sixma, T. K. (2001) Curr. Opin. Struct. Biol. 11 47-52 [DOI] [PubMed] [Google Scholar]
- 14.Alani, E., Lee, J. Y., Schofield, M. J., Kijas, A. W., Hsieh, P., and Yang, W. (2003) J. Biol. Chem. 278 16088-16094 [DOI] [PubMed] [Google Scholar]
- 15.Lamers, M. H., Winterwerp, H. H., and Sixma, T. K. (2003) EMBO J. 22 746-756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Natrajan, G., Lamers, M. H., Enzlin, J. H., Winterwerp, H. H., Perrakis, A., and Sixma, T. K. (2003) Nucleic Acids Res. 31 4814-4821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamers, M. H., Georgijevic, D., Lebbink, J. H., Winterwerp, H. H., Agianian, B., de Wind, N., and Sixma, T. K. (2004) J. Biol. Chem. 279 43879-43885 [DOI] [PubMed] [Google Scholar]
- 18.Bowers, J., Sokolsky, T., Quach, T., and Alani, E. (1999) J. Biol. Chem. 274 16115-16125 [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto, A., Schofield, M. J., Biswas, I., and Hsieh, P. (2000) Nucleic Acids Res. 28 3564-3569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drotschmann, K., Yang, W., Brownewell, F. E., Kool, E. T., and Kunkel, T. A. (2001) J. Biol. Chem. 276 46225-46229 [DOI] [PubMed] [Google Scholar]
- 21.Schofield, M. J., Brownewell, F. E., Nayak, S., Du, C., Kool, E. T., and Hsieh, P. (2001) J. Biol. Chem. 276 45505-45508 [DOI] [PubMed] [Google Scholar]
- 22.Lebbink, J. H., Georgijevic, D., Natrajan, G., Fish, A., Winterwerp, H. H., Sixma, T. K., and de Wind, N. (2006) EMBO J. 25 409-419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmes, S. F., Scarpinato, K. D., McCulloch, S. D., Schaaper, R. M., and Kunkel, T. A. (2007) DNA Repair 6 293-303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang, Y., Sass, L. E., Du, C., Hsieh, P., and Erie, D. A. (2005) Nucleic Acids Res. 33 4322-4334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biswas, I., Ban, C., Fleming, K. G., Qin, J., Lary, J. W., Yphantis, D. A., Yang, W., and Hsieh, P. (1999) J. Biol. Chem. 274 23673-23678 [DOI] [PubMed] [Google Scholar]
- 26.Schofield, M. J., Nayak, S., Scott, T. H., Du, C., and Hsieh, P. (2001) J. Biol. Chem. 276 28291-28299 [DOI] [PubMed] [Google Scholar]
- 27.Antony, E., and Hingorani, M. M. (2004) Biochemistry 43 13115-13128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malkov, V. A., Biswas, I., Camerini-Otero, R. D., and Hsieh, P. (1997) J. Biol. Chem. 272 23811-23817 [DOI] [PubMed] [Google Scholar]
- 29.Jacobs-Palmer, E., and Hingorani, M. M. (2007) J. Mol. Biol. 366 1087-1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Antony, E., and Hingorani, M. M. (2003) Biochemistry 42 7682-7693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazur, D. J., Mendillo, M. L., and Kolodner, R. D. (2006) Mol. Cell 22 39-49 [DOI] [PubMed] [Google Scholar]
- 32.Horton, J. R., Zhang, X., Maunus, R., Yang, Z., Wilson, G. G., Roberts, R. J., and Cheng, X. (2006) Nucleic Acids Res. 34 939-948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allain, F. H., Yen, Y. M., Masse, J. E., Schultze, P., Dieckmann, T., Johnson, R. C., and Feigon, J. (1999) EMBO J. 18 2563-2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andera, L., Schneider, G. J., and Geiduschek, E. P. (1994) Biochimie (Paris) 76 933-940 [DOI] [PubMed] [Google Scholar]
- 35.Kim, J. L., Nikolov, D. B., and Burley, S. K. (1993) Nature 365 520-527 [DOI] [PubMed] [Google Scholar]
- 36.Salsbury, F. R., Jr., Clodfelter, J. E., Gentry, M. B., Hollis, T., and Scarpinato, K. D. (2006) Nucleic Acids Res. 34 2173-2185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hardwidge, P. R., Wu, J., Williams, S. L., Parkhurst, K. M., Parkhurst, L. J., and Maher, L. J., III (2002) Biochemistry 41 7732-7742 [DOI] [PubMed] [Google Scholar]
- 38.Kalodimos, C. G., Biris, N., Bonvin, A. M., Levandoski, M. M., Guennuegues, M., Boelens, R., and Kaptein, R. (2004) Science 305 386-389 [DOI] [PubMed] [Google Scholar]
- 39.Strauss-Soukup, J. K., and Maher, L. J., III (1998) Biochemistry 37 1060-1066 [DOI] [PubMed] [Google Scholar]
- 40.Hardwidge, P. R., Kahn, J. D., and Maher, L. J., III (2002) Biochemistry 41 8277-8288 [DOI] [PubMed] [Google Scholar]
- 41.Leonard, D. A., Rajaram, N., and Kerppola, T. K. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 4913-4918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rajski, S. R., Jackson, B. A., and Barton, J. K. (2000) Mutat. Res. 447 49-72 [DOI] [PubMed] [Google Scholar]
- 43.Isaacs, R. J., and Spielmann, H. P. (2004) DNA Repair 3 455-464 [DOI] [PubMed] [Google Scholar]
- 44.Erie, D. A., Yang, G., Schultz, H. C., and Bustamante, C. (1994) Science 266 1562-1566 [DOI] [PubMed] [Google Scholar]
- 45.Huffman, J. L., Sundheim, O., and Tainer, J. A. (2005) Mutat. Res. 577 55-76 [DOI] [PubMed] [Google Scholar]
- 46.Pues, H., Bleimling, N., Holz, B., Wolcke, J., and Weinhold, E. (1999) Biochemistry 38 1426-1434 [DOI] [PubMed] [Google Scholar]
- 47.Banerjee, A., Santos, W. L., and Verdine, G. L. (2006) Science 311 1153-1157 [DOI] [PubMed] [Google Scholar]
- 48.Parker, J. B., Bianchet, M. A., Krosky, D. J., Friedman, J. I., Amzel, L. M., and Stivers, J. T. (2007) Nature 449 433-438 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.