

Abstract
Much effort and interest have focused on assessing the importance of natural selection, particularly positive natural selection, in shaping the human genome. Although scans for positive selection have identified candidate loci that may be associated with positive selection in humans, such scans do not indicate whether adaptation is frequent in general in humans. Studies based on the reasoning of the MacDonald–Kreitman test, which, in principle, can be used to evaluate the extent of positive selection, suggested that adaptation is detectable in the human genome but that it is less common than in Drosophila or Escherichia coli. Both positive and purifying natural selection at functional sites should affect levels and patterns of polymorphism at linked nonfunctional sites. Here, we search for these effects by analyzing patterns of neutral polymorphism in humans in relation to the rates of recombination, functional density, and functional divergence with chimpanzees. We find that the levels of neutral polymorphism are lower in the regions of lower recombination and in the regions of higher functional density or divergence. These correlations persist after controlling for the variation in GC content, density of simple repeats, selective constraint, mutation rate, and depth of sequencing coverage. We argue that these results are most plausibly explained by the effects of natural selection at functional sites—either recurrent selective sweeps or background selection—on the levels of linked neutral polymorphism. Natural selection at both coding and regulatory sites appears to affect linked neutral polymorphism, reducing neutral polymorphism by 6% genome-wide and by 11% in the gene-rich half of the human genome. These findings suggest that the effects of natural selection at linked sites cannot be ignored in the study of neutral human polymorphism.
Author Summary
There is much reported evidence for positive selection at specific loci in the human genome. Additional papers based on comparisons between the genomes of humans and chimpanzees have also suggested that adaptive evolution may be quite common. At the same time, it has been surprisingly hard to find unambiguous evidence that either positive or negative (background) selection is affecting genome-wide patterns of variation at neutral sites. Here, we evaluate the prevalence of positive or background selection by using two genome-wide datasets of human polymorphism. We document that levels of neutral polymorphism are substantially lower in the regions of (i) higher density of genes and/or regulatory regions, (ii) higher protein or regulatory divergence, and (iii) lower recombination. These patterns are robust to a number of possible confounding factors and suggest that effects of selection at linked sites cannot be ignored in the study of the human genome.
Introduction
The neutral theory of molecular evolution [1] postulates that adaptive substitutions occur so rarely that they can be safely ignored in most studies in population genetics or molecular evolution. This view has dominated the field of molecular evolution for the past 40 years. However, the past 4–6 years have seen a strong challenge to this view. This challenge comes not only from numerous studies detailing specific cases of molecular adaptation in a number of organisms (for example, see [2]–[8]) but also, and most compellingly, from a number of studies that indicate that adaptation might be common on the genomic scale [9]–[17].
High rates of adaptation on the genomic scale have been inferred from the excess of substitutions in functional regions relative to neutral expectations. The neutral expectations are derived from the polymorphism data at functional and putatively neutral sites and the divergence at the neutral sites using the reasoning of the McDonald-Kreitman (MK) test [18]. The excess in the number of substitutions at functional sites over this expectation can be used to estimate the number of adaptive substitutions [10],[19]. McDonald-Kreitman approaches can be modified to account for the presence of deleterious polymorphisms in the sample and the effects of demographic processes on polymorphism [10],[20],[21]. The approach can also be extended to estimate rates of adaptation in regulatory regions [12],[22].
McDonald-Kreitman analysis indicates that adaptive evolution in functional regions might be common in a range of organisms. In Drosophila, it has been estimated that from 30 to 60% of amino acid substitutions and ∼20% of substitutions in non-coding regions are adaptive [10], [11], [16], [23]–[25]. The rate appears similarly high in E. coli (>56% of amino acid substitutions are adaptive) [26] but not in Arabidopsis (0–5% of amino acid substitutions are adaptive) [27] and yeast [28].
In humans, McDonald-Kreitman-based estimates have varied from zero to ∼35% of all amino acid substitutions being adaptive [15], [29]–[33]. A recent estimate by Boyko et al [21] used information from the allele spectra of nonsynonymous and synonymous SNPs in human genes and the divergence with chimpanzee orthologs to estimate that ∼10% of amino acid substitutions between humans and chimpanzees have been fixed by positive selection. Thus, some of these studies suggest that adaptation might be fairly common in humans, although probably substantially less common than in Drosophila or E. coli.
McDonald-Kreitman approaches are very powerful at detecting positive selection, however, they can be misleading for a variety of reasons [15],[34],[35]. For example, if the strength of purifying selection over the evolutionary period separating two species has been different than it is in the present, McDonald-Kreitman-based approaches can either over- or underestimate the rate of adaptive evolution. As these estimates do not provide consistent answers about the prevalence of adaptation in humans and because they can be misleading under plausible demographic scenarios, reaching more reliable conclusions about the importance of adaptations in humans requires the investigation of other signatures of positive selection.
An adaptive substitution reduces the level of polymorphism at neutral sites in its vicinity in a phenomenon known as a selective sweep [36]. The width of the region in which the polymorphism is reduced is inversely proportional to the local recombination rate and directly proportional to the selection coefficient associated with the adaptive substitution [37]–[39]. The reduction of polymorphism is transient and the levels of polymorphism are expected to recover within roughly Ne generations [40]. In addition to the reduction of the level of polymorphism, recurrent selective sweeps may also generate other signatures such as (i) an overabundance of low-frequency alleles [41],[42], (ii) a greater proportion of high-frequency derived alleles [43],[44], (iii) unusual haplotype structures [45],[46].
A number of these expectations have been used to define signatures of positive selection for genome-wide scans for recent adaptation in humans: i.e., the detection of candidate regions that are likely to be experiencing a selective sweep at present or that have experienced one recently. For example, Nielsen et al. [30] and Kelley et al [47] used the deviation of the allele frequency spectrum from its background characteristics to detect candidate regions that may have experienced a sweep; several other methods have used summaries of haplotype structure and their deviation from the background to detect candidate regions that are undergoing a selective sweep [45],[46].
Genomic scans for positive selection are primarily used to choose candidate regions for future investigation, but their application to the quantification of positive selection or even the establishment of its prevalence is problematic. To quantify the extent of positive selection based on the deviations of these signatures from the background requires a prior expectation about the likelihood of observing them under neutrality. These expectations, however, may be sensitive to the effects of non-equilibrium demography [44], [48]–[50]. As a result, it is difficult to generate robust a priori expectations for these statistics under neutrality. Therefore, scans for positive selection do not, by themselves, provide reliable quantification of the extent of positive selection in humans or establish that positive selection is prevalent in humans.
To evaluate whether selective sweeps are common in the human genome, we require signatures that are unlikely to be generated by demography alone. The effects of recurrent selective sweeps (RSS) should be stronger in the regions of lower recombination and in regions of more frequent and selectively consequential adaptation. In Drosophila, for example, the level of neutral polymorphism is positively correlated with the recombination rate [51]–[53] and negatively with the rate and number of nonsynonymous substitutions in a region [9],[13],[14]. These correlations, which are expected under models of RSS but should not be generated by demography alone, support the notion of high rates of adaptation in these taxa.
Despite several compelling examples of adaptations, clear genome-wide signatures of RSS have been difficult to detect in humans. A relationship of diversity and recombination has been reported, but was attributed primarily to an association between recombination and mutation processes rather than to the effects of selected at linked sites [54]–[57], with the possible exception of telomeric and centromeric regions [58]. In turn, the relationships between levels of polymorphism and functional divergence have not yet been examined.
If the recent MK estimates of the rate of adaptive evolution are correct and approximately 10% of amino acid substitutions are adaptive [21], we should expect to see a substantial number of recent selective sweeps in the polymorphism data. Indeed, ∼7×104 amino acid differences between human and chimpanzee proteins [31] have accumulated over the past ∼14 million years. If 10% of these have been adaptive, then we can estimate that ∼7×103 adaptive amino acid substitutions have taken place over ∼14 million years. Assuming a constant rate of adaptation, this translates into ∼100 adaptive amino acid substitutions that occurred during the past Ne generations (Ne = ∼2×105 years) [59]. Moreover, if regulatory adaptations are common as well, then hundreds of recent selective sweeps should be detectable in the human polymorphism data.
With these considerations in mind, we analyze genomic patterns of nucleotide polymorphism, recombination, functional density and functional divergence in humans using two independent, genome-wide SNP datasets. Consistent with the expectations of positive selection, we detect a positive correlation between levels of neutral polymorphism and recombination rate and a negative correlation between levels of nucleotide polymorphism and both functional density and functional divergence. These correlations remain intact after controlling for a number of possible covariates. The evidence is consistent with positive selection in both regulatory and protein-coding regions. We consider alternative explanations for these findings and argue that, in addition to recurrent selective sweep, only background selection (BS) (loss of neutral variants due to hitchhiking with linked deleterious mutations) can possibly generate most of these patterns. Hitchhiking of neutral polymorphisms with linked selected variants—either due to recurrent positive selection or background selection or possibly both—appears to be a substantial force determining levels of neutral polymorphism in the human genome.
Results
Neutral Variation in the Human Genome
To study the effects of RSS, we separate the genomic sequences into two mutually exclusive sets of sequences: “functional” (genic and regulatory) and “nonfunctional”. Both sets of sequences are taken only from the internal parts of autosomes; specifically, we remove all sequences located within 10 Mbp of a telomere or a centromere. We further remove all sequences that cannot be aligned with the chimpanzee genome [31]. The functional set is composed of several types of sequences (see Material and Methods). First, it contains all the genic regions, specifically those that (i) encode exons or are located within 1 kb of any predicted exon and (ii) are located within 5 kb from the starting and ending position of transcripts of protein-coding genes. Because many functional, noncoding sequences are located far from genes in the human genome [60]–[62], we also take all the sequences that can be aligned between primates and zebrafish; sequences that can be aligned over such large evolutionary distances are very unlikely to be unconstrained [63] (see Materials and Methods). The nonfunctional set contains all other sequences except for the repetitive sequences that are filtered out using RepeatMasker [64]. We remove repetitive regions because both alignment and SNP discovery are more problematic in such regions [65]. Hereafter, we will refer to the sequences in the primarily nonfunctional set (totaling ∼1,080 Mbp) as “neutral” sequences for brevity.
We use two SNP datasets: (i) ∼1.2 million Perlegen [66] “A” SNPs discovered using Perlegen chip technology [67] in a panel of 71 individuals of mixed ancestry [68] and (ii) ∼2.0 million SNPs discovered in the diploid sequence of James Watson [69] (see Materials and Methods). In the remainder of the paper, we show the results derived from the analysis of the Perlegen dataset. The results derived from the analysis of the Watson SNPs are shown in the Supplementary Materials. All of the conclusions in the paper are supported by the analysis of either dataset.
We measure the level of neutral nucleotide variation in a genomic window using the number of SNPs within the neutral regions divided by the total number of neutral sites (θneu) in a window (see Materials and Methods). This measure is proportional to the conventional Watterson's θ [70]. In the remainder of the paper, all measurements are carried out over 400 kb windows. We have also carried out all of the analyses with two other window sizes, 200 and 600 kb; none of the conclusions change depending on the window size (Table S1, S2 and S3, Figures S7, S8, S9, and S10).
The level of neutral polymorphism (θneu)
depends both on the average time to coalescence within a particular genomic
region and on the local constraint and mutation rate. For the purposes of
detecting signatures of RSS, variation in constraint and mutation rate generates
noise. We assess variability in constraint and mutation rate by measuring
divergence per neutral site (dneu) within the
neutral regions between the human and chimpanzee genomes (see Materials and Methods). We detect a positive
correlation between dneu and
θneu (Table 1), confirming that, as expected,
constraint and/or mutation rate vary across the human genome. We control for the
variation in neutral mutation rate either by carrying out partial correlations
with dneu or by using a normalized measure of
neutral variation, 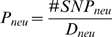 , where #SNPneu stands for the number of SNPs found
in the neutral regions and Dneu stands for the
number of divergent sites within neutral regions between humans and chimpanzee
genomes. Pneu and
θneu also correlate significantly with
repeat density (RD) and GC content (GC) (Table 1, Table S1).
Finally, in the case of the Watson data, we further carry out controls for the
depth of sequence coverage (Table S4).
, where #SNPneu stands for the number of SNPs found
in the neutral regions and Dneu stands for the
number of divergent sites within neutral regions between humans and chimpanzee
genomes. Pneu and
θneu also correlate significantly with
repeat density (RD) and GC content (GC) (Table 1, Table S1).
Finally, in the case of the Watson data, we further carry out controls for the
depth of sequence coverage (Table S4).
Table 1. Correlation coefficients among the studied variables: the level of neutral polymorphism (θneu), the level of normalized neutral polymorphism (Pneu = θneu/dneu), recombination rate (RR), GC content (GC), the density of simple repeats (RD), the divergence at coding sites (Dn), the divergence at conserved noncoding region (Dx), the number of codons (FDn), the number of conserved noncoding sites (FDx), and the level of neutral divergence (dneu).
| θneu | Pneu | RR | GC | RD | Dn | Dx | FDn | FDx | dneu | |
| θneu | — | 0.9364** | 0.2187** | −0.2747** | −0.1046** | −0.2939** | −0.1655** | −0.3210** | −0.3094** | 0.2868** |
| Pneu | 0.7880** | — | 0.1309** | −0.2460** | −0.1306** | −0.2467** | −0.1552** | −0.2363** | −0.2161** | −0.0166NS |
| (1.27e-2) | ||||||||||
| RR | 0.1486** | 0.0886** | — | 0.3535** | −0.2769** | 0.0480* | −0.0454* | 0.0267* | −0.0243** | 0.2934** |
| (5.36e-13) | (8.58e-12) | (6.04e-5) | (2.57e-4) | |||||||
| GC | −0.1837** | −0.1630** | 0.2421** | — | −0.0617** | 0.5694** | 0.1899** | 0.6100** | 0.5096** | −0.1322** |
| RD | −0.0703** | −0.0878** | −0.1876** | −0.0412* | — | 0.0226* | 0.0617** | −0.0248* | −0.0356* | 0.0539* |
| (1.63e-20) | (6.81e-4) | (1.93e-4) | (8.86e-8) | (5.55e-16) | ||||||
| Dn | −0.2079** | −0.1733** | 0.0337* | 0.4141** | 0.0166* | — | 0.3027** | 0.8941** | 0.6772** | −0.1727** |
| (2.81e-13) | (3.17e-4) | |||||||||
| Dx | −0.1150** | −0.1080** | −0.0313* | 0.1296** | 0.0425* | 0.2204** | — | 0.3008** | 0.4965** | −0.0444* |
| (8.27e-12) | (1.81e-20) | (2.53e-11) | ||||||||
| FDn | −0.2213** | −0.1606** | 0.0188* | 0.4397** | −0.0163* | 0.7379** | 0.2119** | — | 0.8260** | −0.3022** |
| (3.03e-5) | (3.08e-4) | |||||||||
| FDx | −0.2096** | −0.1446** | −0.0157* | 0.3535** | −0.0238* | 0.5045** | 0.3524** | 0.6493** | — | −0.3242** |
| (4.00e-4) | (7.89e-8) | |||||||||
| dneu | 0.2011** | −0.0109NS | 0.2011** | −0.0917** | 0.0365* | −0.1264** | −0.0320* | −0.2115** | −0.2226** | — |
| (1.38e-2) | (2.12e-16) | (2.96e-12) |
**: P<1e-20.
*: 1e-20≤P<1e-3.
P> = 1e-3.
Spearman's ρ and Kendall's τ are given at the upper and lower diagonal parts of the table, respectively. P-values are given in parentheses for marginally significant (1e-20≤P<1e-3) and nonsignificant (NS, P> = 1e-3) values.
Positive Correlation between Levels of Neutral Polymorphism and Recombination Rate
The overall effect of RSS on the regional levels of neutral polymorphism should depend on (i) the regional rate of recombination, (ii) the number of recent sweeps (the rate of RSS), and (iii) the strength of positive natural selection associated with a typical adaptive substitution (the strength of RSS). The levels of neutral polymorphism across the genome should correlate positively with the rate of recombination and negatively with the rate and the strength of RSS.
We take estimates of recombination rate from Myers et al. [71], who used a statistical approach to infer recombination rates from linkage disequilibrium data in humans; these rates have been shown to be highly reliable by comparison to pedigree data [72]. The levels of neutral polymorphism measured by both θneu and Pneu increase with the recombination rate (Figures 1, S1, and S2). The correlation remains when we control for possible confounders such GC content (GC), repeat density (RD), and divergence at neutral sites (dneu) separately (Table 2S) or together (Pearson r (θneu, RR|GC, RD, dneu) = 0.254, Pearson r (Pneu, RR|GC, RD) = 0.209, P<0.001 in both cases).
Figure 1. Correlations between recombination rate and neutral divergence rate and neutral polymorphism.

Scatter plots display values of two variables in orange dots for (A) recombination rate and the level of neutral divergence rate (dneu), (B) recombination rate and the level of neutral polymorphism (θneu), and (C) recombination rate and the level of normalized neutral polymorphism (Pneu = θneu/dneu). Black circles are average values for orange dots pooled in 100 bins each containing 1% of the data points.
Table 2. Spearman rank correlation and partials correlation coefficients between the number of codons (FDn) and the levels of neutral polymorphism (θneu) or the normalized neutral polymorphism (Pneu = θneu/dneu), and between the number of conserved noncoding sites (FDx) and the levels of neutral polymorphism (θneu) or normalized neutral polymorphism (Pneu = θneu/dneu).
| FDn vs θneu | FDn vs Pneu | FDx vs θneu | FDx vs Pneu | RR, GC, RD | Dn, Dx | FDn (FDx§) | dneu |
| −0.321 ** | −0.236 ** | −0.309 ** | −0.216 ** | ○ | ○ | ○ | ○ |
| −0.126 ** | −0.060 ** | −0.130 ** | −0.059 ** | • | ○ | ○ | ○ |
| −0.096 ** | —† | −0.099 ** | —† | • | ○ | ○ | • |
| −0.036* | −0.025* † | −0.042* | −0.021† * | • | ○ | •§ | •(○)† |
| (6.67e-8) | (1.94e-4) | (2.34e-10) | (1.35e-3) | ||||
| 0.010NS | 0.027† * | −0.025* | 0.007† NS | • | • | •§ | •(○)† |
| (1.51e-1) | (4.80e-5) | (1.42e-4) | (3.16e-1) |
§: Correlation coefficients for FDn versus θneu or Pneu was calculated here controlling for FDx and the correlation coefficients for FDx versus θneu or Pneu was calculated here controlling for FDn.
†: Correlation coefficients for FDn or FDx versus Pneu were not calculated or were calculated without controlling for dneu, as Pneu ( = θneu /dneu) is not independent from dneu.
**: P<1e-10.
*: 1e-10≤P<1e-3.
P> = 1e-3.
Closed circles (•) indicate the controlled variables. Highly significant values (P<1e-10) are in bold. P-values are given in parentheses for marginally significant (1e-10≤P<1e-3) and nonsignificant (NS, P> = 1e-3) values.
Lower Levels of Neutral Polymorphism in the Functionally Dense Regions
Under a model of RSS regions experiencing more frequent or stronger selective sweeps should show lower levels of neutral polymorphism. Because positive selection should be more prevalent in regions of greater functional density, RSS is expected to generate a negative correlation between the degree of functional density and the level of neutral polymorphism. We measure functional density in two complementary ways. First, in each 400 kb window, we count the number of protein-coding codons (FDn) as a proxy of protein-coding density. In addition, we count the number of nongenic sites that can be aligned between primates and zebrafish (FDx) as a proxy of the number of conserved noncoding sites (CNRs) (see Materials and Methods for details).
Consistent with the predictions under RSS, there are strongly negative correlations between either measure of functional density (FDn,FDx) and measures of neutral variability (Figures 2, S3, S4, and Tables 2, S3). After controlling for GC content (GC), recombination rate (RR), repeat density (RD), and divergence at putatively neutral sites (dneu) (in the case of θneu) the correlations become substantially weaker but do remain statistically significant (Tables 2, 3S). The correlations between FDn and both θneu and Pneu remain significant after we control for FDx; and similarly, the correlations between FDx and both θneu and Pneu are still significant when we control for FDn (Tables 2, 3S).
Figure 2. Relationships among the levels of functional density and neutral polymorphism.

Scatter plots display values of two variables in orange dots for (A) the number of codons (FDn) and the level of neutral polymorphism (θneu), (B) the number of conserved noncoding sites (FDx) and the level of neutral polymorphism (θneu), (C) the number of codons (FDn) and the level of normalized neutral polymorphism (Pneu = θneu/dneu), and (D) the number of conserved noncoding sites (FDx) and the level of normalized neutral polymorphism (Pneu = θneu/dneu). Black circles are average values for orange dots pooled in 100 bins each containing 1% of the data points.
Table 3. Spearman rank correlation and partials correlation coefficients between the divergence at coding sites (Dn) and the levels of neutral polymorphism (θneu) or normalized neutral polymorphism (Pneu = θneu/dneu) and between the divergence at conserved noncoding region (Dx) and the levels of neutral polymorphism (θneu) or normalized neutral polymorphism (Pneu = θneu/dneu).
| Dn vs θneu | Dn vs Pneu | Dx vs θneu | Dx vs Pneu | RR, GC, RD | FDn, FDx | Dn (Dx§) | dneu |
| −0.294 ** | −0.247 ** | −0.165 ** | −0.155 ** | ○ | ○ | ○ | ○ |
| −0.113 ** | −0.089 ** | −0.082 ** | −0.084 ** | • | ○ | ○ | ○ |
| −0.105 ** | —† | −0.160 ** | —† | • | ○ | ○ | • |
| −0.089 ** | −0.073 ** † | −0.064 ** | −0.066 ** † | • | ○ | •§ | •(○)† |
| −0.047 ** | −0.065 ** † | −0.042* | −0.056 ** † | • | • | •§ | •(○)† |
| (2.06e-10) |
§: Correlation coefficients for Dn versus θneu or Pneu were calculated controlling for Dx and the correlation coefficients for Dx versus θneu or Pneu were calculated controlling for Dn.
†: Correlation coefficients for Dn or Dx versus Pneu were not calculated or were calculated without controlling for dneu, as Pneu ( = θneu /dneu) is not independent of dneu.
**: P<1e-10.
*: 1e-10≤P<1e-3.
P> = 1e-3.
Closed circles (•) indicate the controlled variables. Highly significant values (P<1e-10) are in bold. P-values are given in parentheses for marginally significant (1e-10≤P<1e-3) value.
Lower Levels of Neutral Polymorphism in the Regions of Higher Functional Divergence
The number of differences between humans and chimpanzee genomes at functional regions is likely to be a more direct proxy of the rate of positive selection than the functional density. Consistent with the expectations of RSS, we detect lower levels of θneu (Pneu) in regions of higher Dn (the count of divergent amino acid coding sites) or Dx (the count of divergent sites within conserved noncoding regions) (Tables 3, S3, Figures 3, S5, S6). These correlations remain significant when we control for GC content (GC), recombination rate (RR), repeat density (RD), and functional density (FDn, FDx, or both) (Tables 3, S4). The correlations between either Dn or Dx and either of the two measures of neutral variation (θneu or Pneu) remain statistically significant when we control/correct for the other measure of functional divergence (i.e. control for Dn in the case of correlations of neutral diversity with Dx and, similarly, control for Dx in the case of correlations of neutral diversity with Dn) (Tables 3, S4).
Figure 3. Relationships among the levels of functional divergence and neutral polymorphism.

Scatter plots display values of two variables in orange dots for (A) the divergence at coding sites (Dn) and the level of neutral polymorphism (θneu), (B) the divergence at conserved noncoding region (Dx) and the level of neutral polymorphism (θneu), (C) the divergence at coding sites (Dn) and the level of normalized neutral polymorphism (Pneu = θneu/dneu), and (D) the divergence at conserved noncoding region (Dx) and the level of normalized neutral polymorphism (Pneu = θneu/dneu). Black circles are average values for orange dots pooled in 100 bins each containing 1% of the data points.
Discussion
The genome-wide patterns of nucleotide polymorphism in the human genome contain much information about the historical patterns of mutation, recombination, natural selection and population histories of modern humans. Here we search for traces of recurrent positive selection in the patterns of diversity at (mostly) neutral sites across the human genome. A number of studies argued that positive selection is reasonably common in humans [15],[30],[31],[33], although substantially less common than in Drosophila [10], [11], [16], [23]–[25],[29] and E. coli [26]. A recent study estimated that ∼10% of all amino acid substitutions between humans and chimpanzees have been driven by positive selection [21]. If true, then signatures of hundreds of recent selective sweeps should still be detectable in the pattern of neutral variation in the human genome.
Because recurrent adaptive substitutions leave local (on the order of 0.1 s/ρ) and transient (on the order of Ne generations) dips in neutral polymorphism, persistent adaptation should lead to lower levels of neutral polymorphism in regions of lower recombination and regions where selective sweeps are more frequent and/or stronger on average. Here we have confirmed these predictions by showing that levels of SNP density are lower in the regions of lower recombination and in the regions of higher functional density and functional divergence.
In addition to RSS, a number of other evolutionary forces can generate heterogeneous patterns of polymorphism: (i) variation in mutation rates and selective constraint, (ii) demographic events such as population structure, bottlenecks, and fast recent population growth, and (iii) hitchhiking of neutral variants with recurrent deleterious mutations (background selection (BS)). In addition, uneven ascertainment of SNPs across the genome could generate spurious variability in SNP density. Below we discuss the evidence in relation to these alternative possibilities and argue that hitchhiking—due to selective sweeps or background selection—needs to be invoked to explain the detected patterns.
Ascertainment Biases
All SNP datasets suffer from ascertainment biases during the SNP discovery phase that can systematically under- or overestimate numbers of SNPs in particular genomic regions or at particular types of sites. We address this concern by using two very different SNP datasets that are likely to have different ascertainment biases: (i) the high quality (type A) SNPs from the Perlegen dataset [66] and (ii) SNPs discovered in the sequenced diploid genome of James Watson [69]. The type A SNPs were discovered using Perlegen oligo hybridization chip technology in a panel of 71 individuals of mixed ancestry [66]. This set is biased against SNPs located in repetitive regions, given that it is difficult to design uniquely hybridizing oligonucleotides in such repetitive regions [66]. The diploid genome of James Watson was sequenced using the 454 technology and does not suffer from the same technological problems as the Perlegen oligonucleotide chip hybridization technology.
We obtain very similar results using both datasets, which argues that it is unlikely that specific ascertainment biases are responsible for the observed patterns. In addition, we also used the density of the repeats, GC content and functional density as variables in our statistical analyses and showed that all of the signatures of genetic hitchhiking in our data are robust to statistical controls for these variables. The depth of coverage in the Watson sequencing data also does not noticeably affect any of the detected correlations (Table S4).
Noise in Polymorphism due to Demographic Phenomena
The demographic history of human populations in general, and specifically of the populations that have been used for SNP discovery and SNP typing in the Perlegen data, is very complex. Bottlenecks, quick population growth and complex patterns of admixture (for example in the African–American population) are expected to perturb levels of neutral polymorphism across the genome. Collectively, we will denote these forces as “demography”.
The effects of recent demography undoubtedly generate much variation in neutral polymorphism; however, the correlations that we observe are likely to be weakened and unlikely to be generated by the demographic processes alone. For instance, the lower levels of neutral polymorphism in the regions that have large numbers of the protein-coding (Dn) and functional noncoding (Dx) differences are hard to explain by demography; demographic events cannot easily affect the longer-term rates of functional divergence that have been accumulating for ∼10–14 million years between chimpanzees and humans [73]. On the other hand, it is clear that demography needs to be taken into account in order to use the detected signatures to evaluate the strength of hitchhiking in the human genome.
Variation in the Rate of Mutation and Selective Constraint
Some of the variation in levels of polymorphism in the sequences that we use to
measure levels of neutral polymorphism could be due to the variability in the
rates of mutation and levels of selective constraint. We measure levels of
neutral variation in the sequences that are less likely to be under selective
constraint: they are noncoding, located far from exons, and cannot be aligned
with distantly related species such as zebrafish. Nevertheless, some residual
variation in constraint is likely to remain. Indeed, the positive correlation
between our measures of the levels of neutral polymorphism
(θneu) and divergence
(dneu) (Table 1 and 1S)
suggests that mutation rates and/or levels of constraint vary systematically in
these regions. It is therefore important to control for the variation in the
levels of selective constraint and mutation rate; we do so by using the levels
of divergence (dneu) as a variable in partial
correlation analyses or by using the measure Pneu (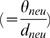 ).The levels of neutral variation correlate strongly with
recombination rate, functional density and functional divergence after
controlling for neutral divergence suggesting that these correlations are not
due to the variation in mutation rate or constraint
).The levels of neutral variation correlate strongly with
recombination rate, functional density and functional divergence after
controlling for neutral divergence suggesting that these correlations are not
due to the variation in mutation rate or constraint
Partial correlations may not remove all of the effects of the variation in mutation rate and constraint, however. The variation in selective constraint among neutral regions should have a stronger effect on the levels of neutral divergence (dneu) than on the levels of neutral polymorphism (θneu) because deleterious mutations have a greater chance of segregating in the population than to become fixed. This implies that if the negative correlation between θneu and levels of functional density were entirely due to the variation in selective constraint (specifically higher remaining constraint in regions of higher functional density), then controlling for divergence (dneu) should make the partial correlation between neutral polymorphism (θneu) and functional density positive. Yet we see the opposite: the correlations between Pneu and functional density and the partial correlation between θneu and functional density with respect to dneu both remain strongly negative. This suggests that the variation in selective constraint is unlikely to generate the correlations between levels of neutral variation and recombination rate, functional density and functional divergence that we see in this study.
On the other hand, variability in mutation rates might contribute to some of the observed patterns. Specifically, the positive correlation between neutral diversity and rates of recombination could be due to the mutagenic effects of recombination. Because rates of recombination at local scales (although not necessarily at the 200–600 kb scales relevant to this study) evolve fast [55], [56], [74]–[77], mutagenic effects of recombination should have more pronounced effects on the levels of polymorphism than on the levels of divergence. If so, controlling for neutral divergence (dneu) may not entirely account for the higher mutation rates produced by recent recombination [55].
Mutagenic effects of recombination are expected to affect levels of polymorphism proportionately to the rate of recombination in the area, whereas hitchhiking (RSS or BS) is expected to affect levels polymorphism in regions of very low recombination much more substantially [78]. We observe a mostly linear effect of recombination on divergence (dneu) suggestive of the mutagenic effect of recombination and further arguing that the regional recombination rates at the level of our analysis (200 to 600 kb) do not evolve as fast as the location of recombination hotspots. In contrast, the effect of recombination on the levels of polymorphism (θneu and Pneu) is curvilinear, with most of the effect limited to the regions of the lowest recombination rates (Figure 1 and S1). Indeed, when we split the data by the median value of recombination rate (RR = 1.040 cM/MBp), the correlation between the levels of neutral divergence (dneu) and recombination rate (RR) for the two halves of the data are of similar strength (r (dneu, RR|RR<1.040) = 0.197 and r (dneu, RR|RR>1.040) = 0.220). However, the correlations between recombination rate and levels of polymorphism (θneu or Pneu) are much stronger in the low recombination regions than in the high recombination regions ((r (θneu, RR|RR<1.040) = 0.249 versus r (θneu, RR|RR>1.040) = 0.045; r (Pneu, RR|RR<1.040) = 0.194 versus r (Pneu, RR|RR>1.040) = −0.0241). These considerations suggest that most of the positive correlation between recombination rates and levels of neutral polymorphism, and especially the reduction at lower recombination rates, is caused by some form of hitchhiking. These results are consistent with the findings of Hellman et al [58] who detected lower levels of polymorphism in the areas of low recombination close to centromeres and telomeres. Note that in our study we explicitly excluded telomeric and centromeric regions (see Materials and Methods), making our findings complementary to those of Hellman et al [58].
Effects due to Background Selection
Background selection (BS) is the process of hitchhiking of neutral or weakly deleterious polymorphism with linked strongly deleterious polymorphisms [79]–[82]. BS should be more efficacious and lead to lower levels of neutral polymorphism in regions of lower recombination. It is thus quite possible that the positive correlation between neutral polymorphism and recombination rate is due in part to BS. In addition, BS should be stronger in the more constrained genomic regions because such regions should experience higher rates of deleterious mutation (e.g. [58]). Therefore BS is likely to contribute to the negative correlation between levels of neutral polymorphism and functional density as well. Because regions of higher functional density also exhibit higher rates of functional divergence (Tables 1 and S1), BS could contribute to the negative correlation between levels of neutral polymorphism and functional divergence as well.
It is less clear whether BS could generate the negative correlation between the levels of neutral polymorphism and functional divergence after controlling for levels of functional density (Tables 3, S4, Figure S6). Two regions of equal functional density can differ in the strength of BS if they differ in the rate of deleterious mutations in the functional sequences. The higher level of deleterious mutations should lead to stronger BS and therefore lower levels of polymorphism in the linked neutral sequences. At the same time, the higher rate of deleterious mutations is likely to come at the expense of neutral mutations at functional sites and thus should lead to lower levels of protein and regulatory divergence. The reduction of neutral mutation rate in the regions of higher deleterious mutation should lead to a positive correlation between levels of neutral polymorphism and functional divergence after controlling for functional density—the opposite of what is seen. On the other hand, the increase in the rate of fixation of weakly deleterious mutations, also expected in the regions of stronger BS, counteracts the reduction of the rate of functional divergence due to the reduction of neutral mutation rate. The combined effect is difficult to estimate given that we do not have information about the distribution of the rates of mutations of different selective effects along the genome.
There is another pattern we observed that is not naturally predicted by BS. The correlations between functional density (FDn or FDx) and neutral polymorphism weaken very substantially and in some cases become nonsignificant when we control for functional divergence at replacement (Dn) and conserved noncoding sites (Dx) (Table 2). Functional density is likely to be a better of proxy of regional constraint than functional divergence. If BS is indeed the dominant force in the generation of the observed patterns, we might have expected correlations between neutral polymorphisms with FDn and/or FDx to be the most robust.
Without a better understanding of the distribution of selective effects and rates of new mutations, we cannot reject the possibility that BS contributes substantially to all of the detected patterns. It appears, however, that only specific distributions of selective effects of new mutations would generate all of the observed patterns. Whether such a distribution exists in principle and whether the distribution of selective effects of human mutation satisfies these requirements in fact remains to be determined.
The Nature and the Effect of Natural Selection at Linked Sites
The arguments above suggest strongly that some form of hitchhiking, either BS or RSS, needs to be invoked to explain the results presented in this paper. These results also suggest that natural selection at both coding and regulatory sites affect linked neutral polymorphism. This is because the measures of the rate of functional evolution at coding and regulatory sites appear to influence levels of neutral polymorphism independently of each other. Specifically, divergence at coding sites and divergence at regulatory sites correlate negatively with the levels of neutral polymorphism after controlling for each other and for the variation in levels of functional divergence (Table 3, S3). To the extent that this is due to recurrent adaptation selection at both coding and regulatory sites, this would echo results of McDonald-Kreitman analyses of adaptation in Drosophila [12].
Levels of neutral polymorphism correlate stronger with divergence at coding than at non-coding regions, possibly implying that either a higher proportion of nonsynonymous changes are adaptive compared to changes in regulatory regions or that the nonsynonymous adaptations have higher selective coefficients. It is also possible and even likely that Dx is a noisier measure than Dn due to greater difficulties in identification of regulatory regions and the noise in estimating Dx due to misalignments. This pattern may also be due to different rates or distributions of the selective effects of deleterious mutations located in coding and regulatory regions, leading to varying effects of BS on linked neutral polymorphism and functional divergence.
These results can also be used to assess the importance of hitchhiking (either RSS or BS) in affecting patterns of neutral polymorphism. The levels of neutral polymorphism appear to be ∼50% lower in the regions of high Dn or Dx (Figures 3, S5) relative to the regions of zero functional divergence (Dn or Dx = 0). If we assume that this effect is entirely due to hitchhiking, then by using the observed correlation between θneu and Dn, we estimate that the levels of polymorphism genome-wide are reduced by 6% genome-wide (Materials and Methods). This reduction is much more pronounced in the more gene-rich regions. For instance, in the 50% of the most gene-rich regions (regions that have greater than the median density of codons (FDn)), the neutral polymorphism is reduced by 11%, while in the regions that contain 50% of the genes (regions that have greater than the mean density of codons (FDn)), the neutral polymorphism is reduced by 13%.
It is clear that hitchhiking has left a significant imprint on the patterns and levels of neutral variability in the human genome and that the effects of natural selection at linked sites cannot be ignored in the analysis of polymorphism data in humans. The challenge for the future is to use these signatures to answer a number of outstanding questions. What are the selective effects and genomic distributions of adaptive and deleterious changes responsible for RSS and BS? What is the biological nature of these changes? What is the relative importance of RSS and BS? Can we estimate parameters of adaptive evolution in the presence of BS? The availability of whole genome sequences in a large number of humans may provide the necessary data to answer these questions. What is needed now are the models and tools to harness these data to provide a cogent picture of the effects of natural selection on human genome and human evolution.
Materials and Methods
SNP Datasets
All analyses have been carried out using two SNP data sets—Perlegen data [66] and Watson data [69]. Perlegen data were downloaded from http://genome.perlegen.com. These data were annotated based on the NCBI build 35 of the human genome sequence. We updated all the genomic positions of the SNPs to match the latest NCBI build 36, according to the rs number of SNPs in the dbSNP build 127. During the processing, 1,361 SNPs were discarded because they could not be uniquely mapped to the human genome. Perlegen data contain three classes of SNPs: (A) array-based genomic resequencing, (B) reliable external SNP collections, and (C) unvalidated, lower confidence sources (see Supplementary text of [66]). We excluded class B and C SNPs and retained 1,235,057 class A SNPs located on autosomes for our analysis. The Watson data were downloaded from http://jimwatsonsequence.cshl.edu/. The genome of James Watson was sequenced at 6× coverage using 454 Life Sciences Technology [69] and matched to the human genome project's published reference sequence [83]. In the Watson DNA sequence, heterozygous sites, in which each site was sequenced multiple times and both forms of the base were found in the diploid genome, were ascertained as SNPs. Homozygous sites of Watson's DNA sequence that have been sequenced multiple times and that differ from the reference sequence of the human genome were also ascertained as SNPs. In total our Watson dataset consisted of 2,020,767 SNPs.
Neutral Genomic Regions
Whole-genome alignments of human (H), chimpanzee (C), and zebrafish (Z) sequences were obtained from the Ensembl compara database [84] through the Ensembl Application Program Interfaces (APIs). We defined the “neutral” genomic regions of the human genome if the regions were: (1) H-C aligned, (2) not H-C-Z aligned, (3) located at least 5 kb away from the starting and ending position of transcripts of protein-coding genes and at least 1 kb away from any exons, (4) located on autosomes at least 10 Mbp away from the boundaries of centromeres and the ends of telomeres, (5) not located in the simple repetitive regions of the human genome. The chromosomal coordinates of exons, transcripts and simple repeats were obtained from the finished and annotated human chromosome sequence from the Ensembl database (build 36).
Neutral Divergence and Polymorphism
Neutral divergence was assessed from H-C alignments. The accuracy of estimation
of neutral divergence may be influenced by the misaligned sequences. Indeed, we
discovered some short (2 kb on average) neutral genomic regions having extremely
high levels of divergent sites, which may result from misalignments (data not
shown). To minimize the possible influence of misalignments, we only counted
“isolated” substitutions that are flanked by two monomorphic
positions on each side (i.e. no substitutions
or SNPs were mapped to these sites). We denoted the number of isolated
substitutions between human and chimpanzee sequences as
Dneu, and the number of isolated substitutions per
neutral site, dneu. To measure neutral polymorphism,
we counted the number of SNPs in neutral regions and denoted the number of SNPs
per site as θneu. Alternatively, we
measured neutral polymorphism with 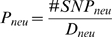 . Data manipulation was done using Matlab functions based on
PGEToolbox [85] and MBEToolbox [86].
. Data manipulation was done using Matlab functions based on
PGEToolbox [85] and MBEToolbox [86].
Proxies of the Rate of Adaptive Evolution
We used four metrics as proxies of the rate of adaptive evolution for a given region in the human genome. Functional density was measured using FDn, the number of codons, and FDx, the number of aligned bases in the H-C-Z three-way alignments. Functional divergence was measured using Dn, the number of codons involved in nonsynonymous substitutions between H-C orthologous gene pairs, and Dx, the number of H-C substitutions in H-C-Z alignments that are located in noncoding human genomic regions. For each pair of genes, the amino-acid sequences were extracted and aligned using CLUSTALW [87] with the default parameters. The corresponding nucleotide sequence alignments were derived by substituting the respective coding sequences from the protein sequences. The synonymous substitution rate (Ks) was then estimated by the maximum-likelihood method implemented in the CODEML program of PAML [88]. Insertions and deletions within alignments were discarded. Poorly aligned orthologous pairs, as indicated by Ks>5, were excluded. The codons containing nonsynonymous substitutions were mapped back onto the human genome and positions were recorded. For simplicity we counted the numbers of codons causing amino-acid changes instead of the numbers of single nucleotide replacement substitutions. In calculation of Dx, we excluded “tri-allelic” sites where the bases of H, C and Z all differ from each other.
Correlation Analysis
We used 400 kb (as well as 200 and 600 kb) sliding window with a step of 100 kb to scan along the human genome. For each window, two measures of neutral polymorphism (θneu and Pneu) and four proxies of the rate adaptive evolution (FDn, FDx, Dn, and Dx) were estimated. To reduce noise arising from small sample size, we also discarded the windows with Dneu<500 and the ones with the total amount of “neutral” sequence less than 2 kb. 22,553 400 kb windows have been used for the correlation analysis. Spearman rank correlation or Kendall's correlation coefficients have been calculated in all cases. To visualize correlations between variables, we used scatter plots with regression lines superimposed. We also pooled the data points of neutral polymorphism by the values of the proxy of adaptation under consideration (e.g. Dn). To do this, we ranked all the data points of the neutral polymorphism by the values of the proxy and then pooled them into 100 bins such that each bin had equal size (i.e., 1%) of the data points. We then computed average values of the proxies of adaptation and the average value of neutral polymorphism for each bin, and superimposed them onto the scatter plots.
To control for confounding variables, we calculated Spearman partial correlation coefficients between variables X and Y controlling for Z, using the function partialcorr in the Matlab statistic toolbox. Recombination rate estimated by using the coalescent method of [89] were downloaded from http://hapmap.org/downloads/recombination/. The density of simple repeats was computed as the proportion of bases of simple repeats in the given region. Chromosomal coordinates of simple repeats in the human genome, identified by RepeatMasker [64], were obtained from the UCSC genome browser [90].
We also calculated the partial correlation coefficients between variables X and Y by calculating the correlation between the two sets of residuals formed by two linear models X∼Z and Y∼Z (see also [91]) where Z stands for either one or a series of variables. The distribution of Dn, Dx, FDn, and FDx values is approximately exponential, which is a problem in a least squares linear model framework in controlling for a third variable, Z. The linear model used to regress out Z is sensitive to the highly non-normal distribution of variables, and the residuals will be highly non-normal, making the results difficult to interpret. Therefore, we quantile-normalized values, replacing the original estimates with their theoretical quantiles based on a normal distribution. Then, we fitted linear models, using as the response variable quantile-normalized Dn, Dx, FDn, or FDx, and using as the predictor variables various combinations of recombination rate, GC content, and the density of simple repeats.
Estimation of the effect of hitchhiking on the level of neutral polymorphism was calculated using the regression between θneu on Dn, using the formula
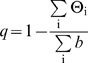 |
where q is the reduction of polymorphism due to hitchhiking, i is a window count for the subsets of windows used in the analysis (e.g. FDn>median (FDn)), b is the intercept of the regression of θneu on Dn.
Supporting Information
Correlations between recombination rate and neutral divergence rate and neutral polymorphism.
Scatter plots display values of two variables in gray dots for (A) recombination rate (RR) and the level of neutral divergence rate (dneu), (B) recombination rate (RR) and the level of neutral polymorphism (θneu), and (c) recombination rate (RR) and the level of normalized neutral polymorphism (Pneu = θneu/dneu). Red circles are average values for the pooled gray dots in 100 bins each containing 1% of the data points. The solid green line shows the fit of a linear model. Spearman's correlation coefficients for (A) to (C) are 0.302, 0.316, and 0.210, respectively. These coefficients are significantly different from zero (P<0.001). The values of θneu and Pneu here are based on the Watson data. The results derived from the Perlegen data are given in Figure 1.
(0.1 MB PDF)
Residual-residual plot between recombination rate (RR) and neutral polymorphism [i.e., the level of neutral polymorphism (θneu) or the level of normalized neutral polymorphism (Pneu = θneu/dneu)] after statistically removing the effects of GC content (GC), repeat density (RD), functional divergences [i.e., the divergence at coding sites (Dn) and the divergence at conserved noncoding region (Dx)], and functional constraints [i.e., the number of codons (FDn) and the number of conserved noncoding sites (FDx)].
e(Y|X) is the difference between the observed value of the response variable, Y, and the value suggested by the regression model of Y on several predictor variables X = {GC, RD, Dn, Dx, FDn, FDx}. The values of θneu and Pneu here are based on the Perlegen data are in (A) and based on the Watson data are in (B).
(0.08 MB PDF)
Relationships among the levels of functional density [i.e., the number of codons (FDn) or the number of conserved noncoding sites (FDx)] and neutral polymorphism [i.e., the level of neutral polymorphism (θneu) or the level of normalized neutral polymorphism (Pneu = θneu/dneu)].
Scatter plots display values of two variables in gray dots for (A) FDn and θneu, (B) FDx and θneu, (C) FDn and Pneu, and (D) FDx and Pneu. Red circles are average values for the pooled gray dots in 100 bins each containing 1% of the data points. The solid, green line shows the fit of a linear model. The values of θneu and Pneu here are based on the Watson data. The results derived from the Perlegen data are given in Figure 2.
(0.1 MB PDF)
Residual-residual plots between functional density [i.e., the number of codons (FDn) or the number of conserved noncoding sites (FDx)] and neutral polymorphism [i.e., the level of neutral polymorphism (θneu) or the level of normalized neutral polymorphism (Pneu = θneu/dneu)], after both have been adjusted for effects of GC content (GC), repeat density (RD), functional divergences [i.e., the divergence at coding sites (Dn) and the divergence at conserved noncoding region (Dx)], and functional density (FDn or FDx, excluding the response variable under test).
e(Y|X) is the difference between the observed value of the response variable, Y, and the value suggested by the regression model of Y on several predictor variables X = {GC, RD, Dn, Dx, FDn, FDx}. The values of θneu and Pneu here are based on the Perlegen data are in (A) and based on the Watson data are in (B).
(0.08 MB PDF)
Relationships among the levels of functional divergence [i.e., the divergence at coding sites (Dn) or the divergence at conserved noncoding region (Dx)] and neutral polymorphism [i.e., the level of neutral polymorphism (θneu) or the level of normalized neutral polymorphism (Pneu = θneu/dneu)].
Scatter plots display values of two variables in gray dots for (A) Dn and θneu, (B) Dx and θneu, (C) Dn and Pneu, and (D) Dx and Pneu. Red circles are average values for the pooled gray dots in 100 bins each containing 1% of the data points. The solid, green line shows the fit of a linear model. The values of θneu and Pneu here are based on the Watson data. The results derived from the Perlegen data are given in Figure 3.
(0.1 MB PDF)
Residual-residual plots between functional divergence [i.e., the divergence at coding sites (Dn) or the divergence at conserved noncoding region (Dx)] and neutral polymorphism [i.e., the level of neutral polymorphism (θneu) and the level of normalized neutral polymorphism (Pneu = θneu/dneu)], after both have been adjusted for effects of GC content (GC), repeat density (RD), functional constraints [i.e., the number of codons (FDn) and the number of conserved noncoding sites (FDx)], and functional divergence (Dn or Dx, excluding the response variable under test).
e(Y|X) is the difference between the observed value of the response variable, Y, and the value suggested by the regression model of Y on several predictor variables X = {GC, RD, Dn, Dx, FDn, FDx}. The values of θneu and Pneu here are based on the Perlegen data are in (A) and based on the Watson data are in (B).
(0.08 MB PDF)
Results derived from the sliding windows of 200 kb. Correlations between functional density [i.e., the number of codons (FDn) or the number of conserved noncoding sites (FDx)], and divergence [i.e., the divergence at coding sites (Dn) or the divergence at conserved noncoding region (Dx)] and neutral polymorphism [i.e., the level of neutral polymorphism (θneu) and the level of normalized neutral polymorphism (Pneu = θneu/dneu)] are given.
The results are based on the Perlegen data.
(0.1 MB PDF)
Results derived from the sliding windows of 200 kb.
Correlations between functional density [i.e., the number of codons (FDn) or the number of conserved noncoding sites (FDx)], and divergence [i.e., the divergence at coding sites (Dn) or the divergence at conserved noncoding region (Dx)] and neutral polymorphism [i.e. the level of neutral polymorphism (θneu) or the level of normalized neutral polymorphism (Pneu = θneu/dneu)] are given. The results are based on the Watson data.
(0.1 MB PDF)
Results derived from the sliding windows of 600 kb.
Correlations between functional density [i.e. the number of codons (FDn) or the number of conserved noncoding sites (FDx)], and divergence [i.e. the divergence at coding sites (Dn) or the divergence at conserved noncoding region (Dx)] and neutral polymorphism [i.e., the level of neutral polymorphism (θneu) or the level of normalized neutral polymorphism (Pneu = θneu/dneu)] are given. The results are based on the Perlegen data.
(0.1 MB PDF)
Results derived from the sliding windows of 600 kb.
Correlations between functional density [i.e., the number of codons (FDn) or the number of conserved noncoding sites (FDx)], and divergence [i.e., the divergence at coding sites (Dn) or the divergence at conserved noncoding region (Dx)] and neutral polymorphism [i.e., the level of neutral polymorphism (θneu) and the level of normalized neutral polymorphism (Pneu = θneu/dneu)] are given. The results are based on the Watson data.
(0.1 MB PDF)
Correlation coefficients among the studied variables: the level of neutral polymorphism (θneu), he level of normalized neutral polymorphism (Pneu = θneu/dneu), recombination rate (RR), GC content (GC), the density of simple repeats (RD), the depth of sequencing coverage (SC), the divergence at coding sites (Dn), the divergence at conserved noncoding region (Dx), the number of codons (FDn), the number of conserved noncoding sites (FDx), and the level of neutral divergence (dneu).
Spearman's ρ and Kendall's τ are given at the upper and lower diagonal parts of the table, respectively. The values of θneu and Pneu are based on the Watson data.
(0.07 MB PDF)
Correlation coefficients and partials correlation coefficients between recombination rate (RR) and levels of neutral polymorphism [i.e., the level of neutral polymorphism (θneu) and the level of normalized neutral polymorphism (Pneu = θneu/dneu)].
Spearman's partial correlation coefficients were calculated by controlling for all possible combinations of potentially confounding variables. The results of representative combinations are given here. Closed circles (•) indicate the controlled variables. These variables are GC content (GC), the density of simple repeats (RD), the divergence at coding sites (Dn), the divergence at conserved noncoding region (Dx), the number of codons (FDn), the number of conserved noncoding sites (FDx), and the level of neutral divergence rate (dneu). Open circles (○) indicate the variables that were not controlled in a particular analysis. (P) indicates the results are based on the Perlegen data, (W) indicates the results are based on the Watson data. P-values are given in parentheses.
(0.1 MB PDF)
Spearman rank correlation coefficients and partials correlation coefficients between functional divergence [i.e., the divergence at coding sites (Dn) or the divergence at conserved noncoding region (Dx)] and neutral polymorphism [i.e., the level of neutral polymorphism (θneu) and the level of normalized neutral polymorphism (Pneu = θneu/dneu)], and between functional constraints [i.e., the number of codons (FDn) and the number of conserved noncoding sites (FDx)] and neutral polymorphism (θneu and Pneu).
Spearman's partial correlation coefficients were calculated by controlling for all possible combinations of potentially confounding variables. The results of representative combinations are given here. Closed circles (•) indicate the controlled variables. These variables are GC content (GC), the density of simple repeats (RD), the divergence at coding sites (Dn), the divergence at conserved noncoding region (Dx), the number of codons (FDn), the number of conserved noncoding sites (FDx), and the level of neutral divergence rate (dneu). Open circles (○) indicate the variables that were not controlled in a particular analysis. (P) indicates the results are based on the Perlegen data, (W) indicates the results are based on the Watson data. P-values are given in parentheses.
(0.2 MB PDF)
Partial Spearman rank correlation coefficients controlling for the depth of sequencing coverage (SC) of Watson data.
Partial Spearman rank correlation coefficients between functional divergence [i.e. the divergence at coding sites (Dn) or the divergence at conserved noncoding region (Dx)] and neutral polymorphism [i.e.,the level of neutral polymorphism (θneu) and the level of normalized neutral polymorphism (Pneu = θneu/dneu)], and between functional constraints [i.e.,the number of codons (FDn) and the number of conserved noncoding sites (FDx)] and neutral polymorphism (θneu and Pneu) are given. Spearman's partial correlation coefficients were calculated by controlling for all possible combinations of potentially confounding variables. The results of representative combinations are given here. Closed circles (•) indicate the controlled variables. These variables are GC content (GC), the density of simple repeats (RD), the depth of sequencing coverage (SC), the divergence at coding sites (Dn), the divergence at conserved noncoding region (Dx), the number of codons (FDn), the number of conserved noncoding sites (FDx), and the level of neutral divergence rate (dneu). Open circles (○) indicate the variables that were not controlled in a particular analysis. (W) indicates the results are based on the Watson data. P-values are given in parentheses.
(0.2 MB PDF)
Footnotes
The authors have declared that no competing interests exist.
This research was supported by the grant from the National Institutes of Health (GM077368) and the National Science Foundation (0317171) to DAP and a Flegg Fellowship and by the Israel Science Foundation (grant no. 1435/07) to GS.
References
- 1.Kimura M. The neutral theory of molecular evolution. Cambridge [Cambridgeshire]; New York: Cambridge University Press; 1983. xv367 [Google Scholar]
- 2.Kelley JL, Swanson WJ. Dietary change and adaptive evolution of enamelin in humans and among primates. Genetics. 2008;178:1595–1603. doi: 10.1534/genetics.107.077123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tishkoff SA, Reed FA, Ranciaro A, Voight BF, Babbitt CC, et al. Convergent adaptation of human lactase persistence in Africa and Europe. Nat Genet. 2007;39:31–40. doi: 10.1038/ng1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoekstra HE, Hirschmann RJ, Bundey RA, Insel PA, Crossland JP. A single amino acid mutation contributes to adaptive beach mouse color pattern. Science. 2006;313:101–104. doi: 10.1126/science.1126121. [DOI] [PubMed] [Google Scholar]
- 5.Aminetzach YT, Macpherson JM, Petrov DA. Pesticide resistance via transposition-mediated adaptive gene truncation in Drosophila. Science. 2005;309:764–767. doi: 10.1126/science.1112699. [DOI] [PubMed] [Google Scholar]
- 6.Shapiro MD, Marks ME, Peichel CL, Blackman BK, Nereng KS, et al. Genetic and developmental basis of evolutionary pelvic reduction in threespine sticklebacks. Nature. 2004;428:717–723. doi: 10.1038/nature02415. [DOI] [PubMed] [Google Scholar]
- 7.Colosimo PF, Peichel CL, Nereng K, Blackman BK, Shapiro MD, et al. The genetic architecture of parallel armor plate reduction in threespine sticklebacks. PLoS Biol. 2004;2:E109. doi: 10.1371/journal.pbio.0020109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daborn PJ, Yen JL, Bogwitz MR, Le Goff G, Feil E, et al. A single p450 allele associated with insecticide resistance in Drosophila. Science. 2002;297:2253–2256. doi: 10.1126/science.1074170. [DOI] [PubMed] [Google Scholar]
- 9.Begun DJ, Holloway AK, Stevens K, Hillier LW, Poh YP, et al. Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol. 2007;5:e310. doi: 10.1371/journal.pbio.0050310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fay JC, Wyckoff GJ, Wu CI. Testing the neutral theory of molecular evolution with genomic data from Drosophila. Nature. 2002;415:1024–1026. doi: 10.1038/4151024a. [DOI] [PubMed] [Google Scholar]
- 11.Smith NG, Eyre-Walker A. Adaptive protein evolution in Drosophila. Nature. 2002;415:1022–1024. doi: 10.1038/4151022a. [DOI] [PubMed] [Google Scholar]
- 12.Andolfatto P. Adaptive evolution of non-coding DNA in Drosophila. Nature. 2005;437:1149–1152. doi: 10.1038/nature04107. [DOI] [PubMed] [Google Scholar]
- 13.Andolfatto P. Hitchhiking effects of recurrent beneficial amino acid substitutions in the Drosophila melanogaster genome. Genome Res. 2007;17:1755–1762. doi: 10.1101/gr.6691007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Macpherson JM, Sella G, Davis JC, Petrov DA. Genomewide spatial correspondence between nonsynonymous divergence and neutral polymorphism reveals extensive adaptation in Drosophila. Genetics. 2007;177:2083–2099. doi: 10.1534/genetics.107.080226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eyre-Walker A. The genomic rate of adaptive evolution. Trends Ecol Evol. 2006;21:569–575. doi: 10.1016/j.tree.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 16.Shapiro JA, Huang W, Zhang C, Hubisz MJ, Lu J, et al. Adaptive genic evolution in the Drosophila genomes. Proc Natl Acad Sci U S A. 2007;104:2271–2276. doi: 10.1073/pnas.0610385104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williamson SH, Hubisz MJ, Clark AG, Payseur BA, Bustamante CD, et al. Localizing recent adaptive evolution in the human genome. PLoS Genet. 2007;3:e90. doi: 10.1371/journal.pgen.0030090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- 19.Charlesworth B. The effect of background selection against deleterious mutations on weakly selected, linked variants. Genet Res. 1994;63:213–227. doi: 10.1017/s0016672300032365. [DOI] [PubMed] [Google Scholar]
- 20.Charlesworth J, Eyre-Walker A. The McDonald-Kreitman Test and Slightly Deleterious Mutations. Mol Biol Evol. 2008 doi: 10.1093/molbev/msn005. [DOI] [PubMed] [Google Scholar]
- 21.Boyko AR, Williamson SH, Indap AR, Degenhardt JD, Hernandez RD, et al. Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet. 2008;4:e1000083. doi: 10.1371/journal.pgen.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kohn MH, Fang S, Wu CI. Inference of positive and negative selection on the 5′ regulatory regions of Drosophila genes. Mol Biol Evol. 2004;21:374–383. doi: 10.1093/molbev/msh026. [DOI] [PubMed] [Google Scholar]
- 23.Sawyer SA, Kulathinal RJ, Bustamante CD, Hartl DL. Bayesian analysis suggests that most amino acid replacements in Drosophila are driven by positive selection. J Mol Evol. 2003;57(Suppl 1):S154–164. doi: 10.1007/s00239-003-0022-3. [DOI] [PubMed] [Google Scholar]
- 24.Bierne N, Eyre-Walker A. The genomic rate of adaptive amino acid substitution in Drosophila. Mol Biol Evol. 2004;21:1350–1360. doi: 10.1093/molbev/msh134. [DOI] [PubMed] [Google Scholar]
- 25.Welch JJ. Estimating the genomewide rate of adaptive protein evolution in Drosophila. Genetics. 2006;173:821–837. doi: 10.1534/genetics.106.056911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charlesworth J, Eyre-Walker A. The rate of adaptive evolution in enteric bacteria. Mol Biol Evol. 2006;23:1348–1356. doi: 10.1093/molbev/msk025. [DOI] [PubMed] [Google Scholar]
- 27.Bustamante CD, Nielsen R, Sawyer SA, Olsen KM, Purugganan MD, et al. The cost of inbreeding in Arabidopsis. Nature. 2002;416:531–534. doi: 10.1038/416531a. [DOI] [PubMed] [Google Scholar]
- 28.Doniger SW, Kim HS, Swain D, Corcuera D, Williams M, et al. A catalog of neutral and deleterious polymorphism in yeast. PLoS Genet. 2008;4:e1000183. doi: 10.1371/journal.pgen.1000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fay JC, Wyckoff GJ, Wu CI. Positive and negative selection on the human genome. Genetics. 2001;158:1227–1234. doi: 10.1093/genetics/158.3.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nielsen R, Williamson S, Kim Y, Hubisz MJ, Clark AG, et al. Genomic scans for selective sweeps using SNP data. Genome Res. 2005;15:1566–1575. doi: 10.1101/gr.4252305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chimpanzee-Sequencing-and-Analysis-Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- 32.Zhang L, Li WH. Human SNPs reveal no evidence of frequent positive selection. Mol Biol Evol. 2005;22:2504–2507. doi: 10.1093/molbev/msi240. [DOI] [PubMed] [Google Scholar]
- 33.Bustamante CD, Fledel-Alon A, Williamson S, Nielsen R, Hubisz MT, et al. Natural selection on protein-coding genes in the human genome. Nature. 2005;437:1153–1157. doi: 10.1038/nature04240. [DOI] [PubMed] [Google Scholar]
- 34.Andolfatto P. Controlling Type-I Error of the McDonald-Kreitman Test in Genome-wide Scans for Selection on Non-coding DNA. Genetics. 2008 doi: 10.1534/genetics.108.091850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sella G, Petrov D, Przeworski M, Andolfatto P (in review) Pervasive natural selection in the Drosophila genome? PLoS Genet. doi: 10.1371/journal.pgen.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maynard Smith J, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res. 1974;23:23–35. [PubMed] [Google Scholar]
- 37.Kaplan NL, Hudson RR, Langley CH. The “hitchhiking effect” revisited. Genetics. 1989;123:887–899. doi: 10.1093/genetics/123.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim Y, Stephan W. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics. 2002;160:765–777. doi: 10.1093/genetics/160.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gillespie JH. Genetic drift in an infinite population. The pseudohitchhiking model. Genetics. 2000;155:909–919. doi: 10.1093/genetics/155.2.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Przeworski M. Estimating the time since the fixation of a beneficial allele. Genetics. 2003;164:1667–1676. doi: 10.1093/genetics/164.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Braverman JM, Hudson RR, Kaplan NL, Langley CH, Stephan W. The hitchhiking effect on the site frequency spectrum of DNA polymorphisms. Genetics. 1995;140:783–796. doi: 10.1093/genetics/140.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fay JC, Wu CI. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Przeworski M. The signature of positive selection at randomly chosen loci. Genetics. 2002;160:1179–1189. doi: 10.1093/genetics/160.3.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sabeti PC, Reich DE, Higgins JM, Levine HZ, Richter DJ, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- 46.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kelley JL, Madeoy J, Calhoun JC, Swanson W, Akey JM. Genomic signatures of positive selection in humans and the limits of outlier approaches. Genome Res. 2006;16:980–989. doi: 10.1101/gr.5157306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jensen JD, Kim Y, DuMont VB, Aquadro CF, Bustamante CD. Distinguishing between selective sweeps and demography using DNA polymorphism data. Genetics. 2005;170:1401–1410. doi: 10.1534/genetics.104.038224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nielsen R. Statistical tests of selective neutrality in the age of genomics. Heredity. 2001;86:641–647. doi: 10.1046/j.1365-2540.2001.00895.x. [DOI] [PubMed] [Google Scholar]
- 50.Macpherson JM, Gonzalez J, Witten DM, Davis JC, Rosenberg NA, et al. Nonadaptive explanations for signatures of partial selective sweeps in Drosophila. Mol Biol Evol. 2008;25:1025–1042. doi: 10.1093/molbev/msn007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Begun DJ, Aquadro CF. Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature. 1992;356:519–520. doi: 10.1038/356519a0. [DOI] [PubMed] [Google Scholar]
- 52.Kulathinal RJ, Bennett SM, Fitzpatrick CL, Noor MA. Fine-scale mapping of recombination rate in Drosophila refines its correlation to diversity and divergence. Proc Natl Acad Sci U S A. 2008;105:10051–10056. doi: 10.1073/pnas.0801848105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Berry AJ, Ajioka JW, Kreitman M. Lack of polymorphism on the Drosophila fourth chromosome resulting from selection. Genetics. 1991;129:1111–1117. doi: 10.1093/genetics/129.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hellmann I, Ebersberger I, Ptak SE, Paabo S, Przeworski M. A neutral explanation for the correlation of diversity with recombination rates in humans. Am J Hum Genet. 2003;72:1527–1535. doi: 10.1086/375657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hellmann I, Prufer K, Ji H, Zody MC, Paabo S, et al. Why do human diversity levels vary at a megabase scale? Genome Res. 2005;15:1222–1231. doi: 10.1101/gr.3461105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spencer CC, Deloukas P, Hunt S, Mullikin J, Myers S, et al. The influence of recombination on human genetic diversity. PLoS Genet. 2006;2:e148. doi: 10.1371/journal.pgen.0020148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nachman MW. Single nucleotide polymorphisms and recombination rate in humans. Trends Genet. 2001;17:481–485. doi: 10.1016/s0168-9525(01)02409-x. [DOI] [PubMed] [Google Scholar]
- 58.Hellmann I, Mang Y, Gu Z, Li P, de la Vega FM, et al. Population genetic analysis of shotgun assemblies of genomic sequences from multiple individuals. Genome Res. 2008;18:1020–1029. doi: 10.1101/gr.074187.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takahata N. Allelic genealogy and human evolution. Mol Biol Evol. 1993;10:2–22. doi: 10.1093/oxfordjournals.molbev.a039995. [DOI] [PubMed] [Google Scholar]
- 60.Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, et al. Ultraconserved elements in the human genome. Science. 2004;304:1321–1325. doi: 10.1126/science.1098119. [DOI] [PubMed] [Google Scholar]
- 61.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ahituv N, Rubin EM, Nobrega MA. Exploiting human–fish genome comparisons for deciphering gene regulation. Hum Mol Genet. 2004;13 Spec No 2:R261–266. doi: 10.1093/hmg/ddh229. [DOI] [PubMed] [Google Scholar]
- 63.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 64.Smit AFA, Hubley R, Green P. 1996–2004 RepeatMasker Open-3.0 ( http://www.repeatmasker.org) [Google Scholar]
- 65.Gajer P, Schatz M, Salzberg SL. Automated correction of genome sequence errors. Nucleic Acids Res. 2004;32:562–569. doi: 10.1093/nar/gkh216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hinds DA, Stuve LL, Nilsen GB, Halperin E, Eskin E, et al. Whole-genome patterns of common DNA variation in three human populations. Science. 2005;307:1072–1079. doi: 10.1126/science.1105436. [DOI] [PubMed] [Google Scholar]
- 67.Hinds DA, Stokowski RP, Patil N, Konvicka K, Kershenobich D, et al. Matching strategies for genetic association studies in structured populations. Am J Hum Genet. 2004;74:317–325. doi: 10.1086/381716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Coriell-Cell-Repositories http://locus.umdnj.edu/ccr/ [Google Scholar]
- 69.Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 70.Watterson GA. On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- 71.Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–324. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- 72.Coop G, Wen X, Ober C, Pritchard JK, Przeworski M. High-resolution mapping of crossovers reveals extensive variation in fine-scale recombination patterns among humans. Science. 2008;319:1395–1398. doi: 10.1126/science.1151851. [DOI] [PubMed] [Google Scholar]
- 73.Goodman M. The genomic record of Humankind's evolutionary roots. Am J Hum Genet. 1999;64:31–39. doi: 10.1086/302218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Przeworski M, Wall JD. Why is there so little intragenic linkage disequilibrium in humans? Genet Res. 2001;77:143–151. doi: 10.1017/s0016672301004967. [DOI] [PubMed] [Google Scholar]
- 75.Winckler W, Myers SR, Richter DJ, Onofrio RC, McDonald GJ, et al. Comparison of fine-scale recombination rates in humans and chimpanzees. Science. 2005;308:107–111. doi: 10.1126/science.1105322. [DOI] [PubMed] [Google Scholar]
- 76.Jeffreys AJ, Neumann R, Panayi M, Myers S, Donnelly P. Human recombination hot spots hidden in regions of strong marker association. Nat Genet. 2005;37:601–606. doi: 10.1038/ng1565. [DOI] [PubMed] [Google Scholar]
- 77.Ptak SE, Hinds DA, Koehler K, Nickel B, Patil N, et al. Fine-scale recombination patterns differ between chimpanzees and humans. Nat Genet. 2005;37:429–434. doi: 10.1038/ng1529. [DOI] [PubMed] [Google Scholar]
- 78.Innan H, Stephan W. Distinguishing the hitchhiking and background selection models. Genetics. 2003;165:2307–2312. doi: 10.1093/genetics/165.4.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Charlesworth B, Morgan MT, Charlesworth D. The effect of deleterious mutations on neutral molecular variation. Genetics. 1993;134:1289–1303. doi: 10.1093/genetics/134.4.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Charlesworth D, Charlesworth B, Morgan MT. The pattern of neutral molecular variation under the background selection model. Genetics. 1995;141:1619–1632. doi: 10.1093/genetics/141.4.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McVean GA, Charlesworth B. The effects of Hill-Robertson interference between weakly selected mutations on patterns of molecular evolution and variation. Genetics. 2000;155:929–944. doi: 10.1093/genetics/155.2.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gordo I, Charlesworth B. Genetic linkage and molecular evolution. Curr Biol. 2001;11:R684–686. doi: 10.1016/s0960-9822(01)00408-0. [DOI] [PubMed] [Google Scholar]
- 83.International-Human-Genome-Sequencing-Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 84.Hubbard TJ, Aken BL, Beal K, Ballester B, Caccamo M, et al. Ensembl 2007. Nucleic Acids Res. 2007;35:D610–617. doi: 10.1093/nar/gkl996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cai JJ. PGEToolbox: A Matlab toolbox for population genetics and evolution. J Hered. 2008;99:438–440. doi: 10.1093/jhered/esm127. [DOI] [PubMed] [Google Scholar]
- 86.Cai JJ, Smith DK, Xia X, Yuen KY. MBEToolbox: a MATLAB toolbox for sequence data analysis in molecular biology and evolution. BMC Bioinformatics. 2005;6:64. doi: 10.1186/1471-2105-6-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 89.McVean GA, Myers SR, Hunt S, Deloukas P, Bentley DR, et al. The fine-scale structure of recombination rate variation in the human genome. Science. 2004;304:581–584. doi: 10.1126/science.1092500. [DOI] [PubMed] [Google Scholar]
- 90.Karolchik D, Kuhn RM, Baertsch R, Barber GP, Clawson H, et al. The UCSC Genome Browser Database: 2008 update. Nucleic Acids Res. 2008;36:D773–779. doi: 10.1093/nar/gkm966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bullaughey K, Przeworski M, Coop G. No effect of recombination on the efficacy of natural selection in primates. Genome Res. 2008 doi: 10.1101/gr.071548.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Correlations between recombination rate and neutral divergence rate and neutral polymorphism.
Scatter plots display values of two variables in gray dots for (A) recombination rate (RR) and the level of neutral divergence rate (dneu), (B) recombination rate (RR) and the level of neutral polymorphism (θneu), and (c) recombination rate (RR) and the level of normalized neutral polymorphism (Pneu = θneu/dneu). Red circles are average values for the pooled gray dots in 100 bins each containing 1% of the data points. The solid green line shows the fit of a linear model. Spearman's correlation coefficients for (A) to (C) are 0.302, 0.316, and 0.210, respectively. These coefficients are significantly different from zero (P<0.001). The values of θneu and Pneu here are based on the Watson data. The results derived from the Perlegen data are given in Figure 1.
(0.1 MB PDF)
Residual-residual plot between recombination rate (RR) and neutral polymorphism [i.e., the level of neutral polymorphism (θneu) or the level of normalized neutral polymorphism (Pneu = θneu/dneu)] after statistically removing the effects of GC content (GC), repeat density (RD), functional divergences [i.e., the divergence at coding sites (Dn) and the divergence at conserved noncoding region (Dx)], and functional constraints [i.e., the number of codons (FDn) and the number of conserved noncoding sites (FDx)].
e(Y|X) is the difference between the observed value of the response variable, Y, and the value suggested by the regression model of Y on several predictor variables X = {GC, RD, Dn, Dx, FDn, FDx}. The values of θneu and Pneu here are based on the Perlegen data are in (A) and based on the Watson data are in (B).
(0.08 MB PDF)
Relationships among the levels of functional density [i.e., the number of codons (FDn) or the number of conserved noncoding sites (FDx)] and neutral polymorphism [i.e., the level of neutral polymorphism (θneu) or the level of normalized neutral polymorphism (Pneu = θneu/dneu)].
Scatter plots display values of two variables in gray dots for (A) FDn and θneu, (B) FDx and θneu, (C) FDn and Pneu, and (D) FDx and Pneu. Red circles are average values for the pooled gray dots in 100 bins each containing 1% of the data points. The solid, green line shows the fit of a linear model. The values of θneu and Pneu here are based on the Watson data. The results derived from the Perlegen data are given in Figure 2.
(0.1 MB PDF)
Residual-residual plots between functional density [i.e., the number of codons (FDn) or the number of conserved noncoding sites (FDx)] and neutral polymorphism [i.e., the level of neutral polymorphism (θneu) or the level of normalized neutral polymorphism (Pneu = θneu/dneu)], after both have been adjusted for effects of GC content (GC), repeat density (RD), functional divergences [i.e., the divergence at coding sites (Dn) and the divergence at conserved noncoding region (Dx)], and functional density (FDn or FDx, excluding the response variable under test).
e(Y|X) is the difference between the observed value of the response variable, Y, and the value suggested by the regression model of Y on several predictor variables X = {GC, RD, Dn, Dx, FDn, FDx}. The values of θneu and Pneu here are based on the Perlegen data are in (A) and based on the Watson data are in (B).
(0.08 MB PDF)
Relationships among the levels of functional divergence [i.e., the divergence at coding sites (Dn) or the divergence at conserved noncoding region (Dx)] and neutral polymorphism [i.e., the level of neutral polymorphism (θneu) or the level of normalized neutral polymorphism (Pneu = θneu/dneu)].
Scatter plots display values of two variables in gray dots for (A) Dn and θneu, (B) Dx and θneu, (C) Dn and Pneu, and (D) Dx and Pneu. Red circles are average values for the pooled gray dots in 100 bins each containing 1% of the data points. The solid, green line shows the fit of a linear model. The values of θneu and Pneu here are based on the Watson data. The results derived from the Perlegen data are given in Figure 3.
(0.1 MB PDF)
Residual-residual plots between functional divergence [i.e., the divergence at coding sites (Dn) or the divergence at conserved noncoding region (Dx)] and neutral polymorphism [i.e., the level of neutral polymorphism (θneu) and the level of normalized neutral polymorphism (Pneu = θneu/dneu)], after both have been adjusted for effects of GC content (GC), repeat density (RD), functional constraints [i.e., the number of codons (FDn) and the number of conserved noncoding sites (FDx)], and functional divergence (Dn or Dx, excluding the response variable under test).
e(Y|X) is the difference between the observed value of the response variable, Y, and the value suggested by the regression model of Y on several predictor variables X = {GC, RD, Dn, Dx, FDn, FDx}. The values of θneu and Pneu here are based on the Perlegen data are in (A) and based on the Watson data are in (B).
(0.08 MB PDF)
Results derived from the sliding windows of 200 kb. Correlations between functional density [i.e., the number of codons (FDn) or the number of conserved noncoding sites (FDx)], and divergence [i.e., the divergence at coding sites (Dn) or the divergence at conserved noncoding region (Dx)] and neutral polymorphism [i.e., the level of neutral polymorphism (θneu) and the level of normalized neutral polymorphism (Pneu = θneu/dneu)] are given.
The results are based on the Perlegen data.
(0.1 MB PDF)
Results derived from the sliding windows of 200 kb.
Correlations between functional density [i.e., the number of codons (FDn) or the number of conserved noncoding sites (FDx)], and divergence [i.e., the divergence at coding sites (Dn) or the divergence at conserved noncoding region (Dx)] and neutral polymorphism [i.e. the level of neutral polymorphism (θneu) or the level of normalized neutral polymorphism (Pneu = θneu/dneu)] are given. The results are based on the Watson data.
(0.1 MB PDF)
Results derived from the sliding windows of 600 kb.
Correlations between functional density [i.e. the number of codons (FDn) or the number of conserved noncoding sites (FDx)], and divergence [i.e. the divergence at coding sites (Dn) or the divergence at conserved noncoding region (Dx)] and neutral polymorphism [i.e., the level of neutral polymorphism (θneu) or the level of normalized neutral polymorphism (Pneu = θneu/dneu)] are given. The results are based on the Perlegen data.
(0.1 MB PDF)
Results derived from the sliding windows of 600 kb.
Correlations between functional density [i.e., the number of codons (FDn) or the number of conserved noncoding sites (FDx)], and divergence [i.e., the divergence at coding sites (Dn) or the divergence at conserved noncoding region (Dx)] and neutral polymorphism [i.e., the level of neutral polymorphism (θneu) and the level of normalized neutral polymorphism (Pneu = θneu/dneu)] are given. The results are based on the Watson data.
(0.1 MB PDF)
Correlation coefficients among the studied variables: the level of neutral polymorphism (θneu), he level of normalized neutral polymorphism (Pneu = θneu/dneu), recombination rate (RR), GC content (GC), the density of simple repeats (RD), the depth of sequencing coverage (SC), the divergence at coding sites (Dn), the divergence at conserved noncoding region (Dx), the number of codons (FDn), the number of conserved noncoding sites (FDx), and the level of neutral divergence (dneu).
Spearman's ρ and Kendall's τ are given at the upper and lower diagonal parts of the table, respectively. The values of θneu and Pneu are based on the Watson data.
(0.07 MB PDF)
Correlation coefficients and partials correlation coefficients between recombination rate (RR) and levels of neutral polymorphism [i.e., the level of neutral polymorphism (θneu) and the level of normalized neutral polymorphism (Pneu = θneu/dneu)].
Spearman's partial correlation coefficients were calculated by controlling for all possible combinations of potentially confounding variables. The results of representative combinations are given here. Closed circles (•) indicate the controlled variables. These variables are GC content (GC), the density of simple repeats (RD), the divergence at coding sites (Dn), the divergence at conserved noncoding region (Dx), the number of codons (FDn), the number of conserved noncoding sites (FDx), and the level of neutral divergence rate (dneu). Open circles (○) indicate the variables that were not controlled in a particular analysis. (P) indicates the results are based on the Perlegen data, (W) indicates the results are based on the Watson data. P-values are given in parentheses.
(0.1 MB PDF)
Spearman rank correlation coefficients and partials correlation coefficients between functional divergence [i.e., the divergence at coding sites (Dn) or the divergence at conserved noncoding region (Dx)] and neutral polymorphism [i.e., the level of neutral polymorphism (θneu) and the level of normalized neutral polymorphism (Pneu = θneu/dneu)], and between functional constraints [i.e., the number of codons (FDn) and the number of conserved noncoding sites (FDx)] and neutral polymorphism (θneu and Pneu).
Spearman's partial correlation coefficients were calculated by controlling for all possible combinations of potentially confounding variables. The results of representative combinations are given here. Closed circles (•) indicate the controlled variables. These variables are GC content (GC), the density of simple repeats (RD), the divergence at coding sites (Dn), the divergence at conserved noncoding region (Dx), the number of codons (FDn), the number of conserved noncoding sites (FDx), and the level of neutral divergence rate (dneu). Open circles (○) indicate the variables that were not controlled in a particular analysis. (P) indicates the results are based on the Perlegen data, (W) indicates the results are based on the Watson data. P-values are given in parentheses.
(0.2 MB PDF)
Partial Spearman rank correlation coefficients controlling for the depth of sequencing coverage (SC) of Watson data.
Partial Spearman rank correlation coefficients between functional divergence [i.e. the divergence at coding sites (Dn) or the divergence at conserved noncoding region (Dx)] and neutral polymorphism [i.e.,the level of neutral polymorphism (θneu) and the level of normalized neutral polymorphism (Pneu = θneu/dneu)], and between functional constraints [i.e.,the number of codons (FDn) and the number of conserved noncoding sites (FDx)] and neutral polymorphism (θneu and Pneu) are given. Spearman's partial correlation coefficients were calculated by controlling for all possible combinations of potentially confounding variables. The results of representative combinations are given here. Closed circles (•) indicate the controlled variables. These variables are GC content (GC), the density of simple repeats (RD), the depth of sequencing coverage (SC), the divergence at coding sites (Dn), the divergence at conserved noncoding region (Dx), the number of codons (FDn), the number of conserved noncoding sites (FDx), and the level of neutral divergence rate (dneu). Open circles (○) indicate the variables that were not controlled in a particular analysis. (W) indicates the results are based on the Watson data. P-values are given in parentheses.
(0.2 MB PDF)