

Abstract
Trichomonads are anaerobic flagellated protists that, based on analyses of ribosomal RNA sequences, represent one of the earliest branching lineages among the eukaryotes. The absence of mitochondria in these organisms coupled with their deep phylogenetic position has prompted several authors to suggest that trichomonads, along with other deeply-branching amitochondriate protist groups, diverged from the main eukaryotic lineage prior to the endosymbiotic origin of mitochondria. In this report we describe the presence of a gene in Trichomonas vaginalis specifically related to mitochondrial chaperonin 60 (cpn60). A recent study indicates that a protein immunologically related to cpn60 is located in trichomonad hydrogenosomes. Together, these data provide evidence that ancestors of trichomonads perhaps harbored the endosymbiotic progenitors of mitochondria, but that these evolved into hydrogenosomes early in trichomonad evolution.
Trichomonads are a protist group consisting mainly of parasitic flagellates. They lack mitochondria and diverge prior to all known mitochondrion-containing eukaryotes in phylogenetic trees based on small subunit ribosomal RNA (1, 2). While many authors suggest that this deep phylogenetic position indicates that trichomonads primitively lack mitochondria (3–5), others have argued that they may have secondarily lost mitochondrial functions (6).
The controversy hinges partly on differing interpretations of the origin of trichomonad hydrogenosomes, unusual energy-generating organelles found in these cells. Hydrogenosomes function in the metabolism of pyruvate produced by glycolysis, generating ATP by substrate level phosphorylation and evolving molecular hydrogen (7). Like mitochondria, they possess a double-membrane envelope and divide autonomously by fission (4). However, it is unclear whether trichomonad hydrogenosomes share a common ancestor with mitochondria or instead descend from a distinct endosymbiotic event: phylogenetic analyses of genes encoding hydrogenosomal proteins have so far failed to yield a strong link with the mitochondrial or any other specific eubacterial lineage (4, 8–10).
Here we report phylogenetic studies using a chaperonin 60 (cpn60) gene located in the nucleus of Trichomonas vaginalis. We undertook the search for this gene for two reasons. First, we reasoned that cpn60, which helps to refold proteins after their import into mitochondria and plastids in eukaryotes, might be used to perform a similar function in hydrogenosomes. Second, cpn60 sequences have proven the most reliable protein-coding genes for reconstructing evolutionary relationships within and between endosymbiosis-derived organelles and their eubacterial ancestors; the α-Proteobacteria and Cyanobacteria for mitochondria and plastids, respectively (11, 12).
MATERIALS AND METHODS
Organism Culture and DNA Extraction.
Cultures of axenic T. vaginalis flagellates, strain C-1:NIH (ATCC 30001), were grown in YI-S medium pH 6.0 with 10% bovine serum (13) and DNA was extracted according to the protocol described in ref. 14.
Cloning of the T. vaginalis cpn60 Gene.
Degenerate primers, HSP5.4 (5′-CCAAAARTTACWAAAGATGGAGTTACWGTT-3′) and TvHSP3.1 (5′-CCRACCTTGATRACAGCRACRCCRCC-3′), were designed based on an alignment of various cpn60 homologs (12). PCR amplification was performed using these primers and total genomic T. vaginalis DNA under standard conditions (12) producing a 1-kb fragment that was cloned and partially sequenced to confirm its homology with cpn60. This fragment was labeled with [α-32P]dATP and used as a hybridization probe to isolate cDNA clones from a λZAP II T. vaginalis cDNA library (15). Using the manufacturer’s protocols (Stratagene), plasmids containing the cDNA inserts were isolated from the λZAP II vector. The largest hybridizing cDNA clone was completely sequenced using a primer walking strategy. Attempts to obtain the missing N-terminal portion of the gene by several methods were unsuccessful.
Phylogenetic Analyses.
The partial amino acid sequence inferred from the T. vaginalis cpn60 gene (544 amino acids in length) was entered into an alignment of cpn60 homologs described previously (12). To improve the taxonomic representation of the dataset near the node of interest, additional sequences were added from the α-Proteobacteria: Cowdria ruminantium (GenBank accession no. U13638U13638), Bradyrhizobium japonicum groEL3 (GenBank accession no. Z22603Z22603), and Brucella abortus (GenBank accession no. L09273L09273). Distance and parsimony analyses were based on 519 positions of this alignment with regions of ambiguous alignment removed and gaps and missing regions of sequence scored as missing data.
The distance tree was generated using protdist employing the Dayhoff accepted point mutation correction and the neighbor program of the phylip package (16), version 3.57c. These programs, in addition to seqboot and consense, were used in bootstrap analysis of 500 resamplings of the dataset. Maximum parsimony trees were obtained by 50 random addition heuristic search replicates using paup, version 3.1.1 (17) and 500 bootstrap replicates were performed employing simple addition heuristic searches. Due to time constraints, 100 bootstrap resamplings were performed in the parsimony and distance analyses of datasets where various Rickettsiales species were excluded.
For most of the maximum likelihood analyses, shared missing data were removed from the dataset yielding 501 alignment positions for analysis. However, for the analysis where the partial Entamoeba histolytica sequence was included, all positions missing in this sequence were eliminated from the alignment, leaving a final dataset of 362 positions. Maximum likelihood trees were obtained by exhaustive tree-searching performed on a semiconstrained tree using the protml program version 2.2 (18), employing the Jones, Taylor, and Thornton frequencies (JTT-F) model of amino acid substitution. The semiconstrained tree was developed by using the distance tree to define the following subtrees: the spirochetes, the γ- and β-Proteobacteria, the non-Rickettsiales α-Protebacteria and a mitochondrial cpn60 subtree of animals, fungi, and plants. All possible topologies containing these subtrees were then examined and the trees of highest ln likelihood were determined for datasets with and without the E. histolytica sequence as well as for all of the combinations of Rickettsiales sequences. Bootstrap support for branches on these trees was estimated using the resampling estimated log-likelihood (RELL) method (19) and and bootstrap majority-rule trees were compiled using the mol2con (A. Stoltzfus, personal communication) and consense programs.
RESULTS
Our approach to finding a cpn60 gene in T. vaginalis was essentially the same as that we used in previous work on E. histolytica (12). Degenerate oligonucleotide primers were employed to amplify a 1.0-kb fragment of the gene with PCR, and this DNA fragment was used to screen a cDNA library.
The complete sequence of the largest cDNA clone was obtained. The restriction site used in the cDNA cloning was fused directly to the 5′ end of the coding region and no start codon was present in the sequence, indicating that the cDNA was truncated. The presence of a polyA tract 35 bp downstream of a UAA stop codon and the distinctive pattern of codon usage suggests that this cDNA was derived from T. vaginalis and not a bacterial contaminant. A Southern blot of T. vaginalis DNA digested with various restriction endonucleases, with this cDNA as a probe, revealed a single hybridizing band, implying that the protein is encoded by a single copy gene (data not shown) and confirming that T. vaginalis was its source.
The predicted T. vaginalis cpn60 protein, 544 amino acids in length, was entered into an alignment (12) containing the partial E. histolytica sequence, four eukaryotic mitochondrial homologs, 13 sequences from proteobacteria, two spirochetes, and one chlamydia. Preliminary phylogenetic analyses based on this full dataset using neighbor-joining distance (Fig. 1A), maximum parsimony and maximum likelihood methods were performed.
Figure 1.
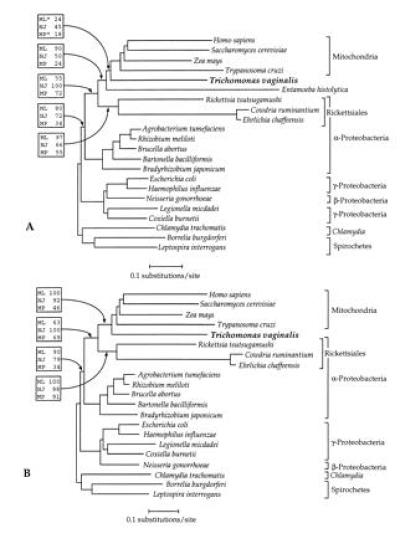
Phylogenies of cpn60 homologs. Sequences were selected from the database for organisms previously shown to branch in the region of the mitochondrial cpn60 clade (11, 12). The trees shown are derived from neighbor-joining analysis of a accepted point mutation corrected distance matrix. Percentage bootstrap support is shown above selected branches in boxes, from bootstrap analyses employing the protein maximum likelihood (ML), neighbor-joining distance (NJ), and maximum parsimony (MP) methods. (A) Cpn60 tree derived from the full alignment. The maximum likelihood tree (ln likelihood = −8933.6) differed from the neighbor-joining tree by the placement of the T. vaginalis and E. histolytica sequences as sister groups. Parsimony generated five trees of length = 2369 all of which differed principally from the tree shown by the placement of T. vaginalis as a sister group of the Rickettsiales species. Asterisks (∗) indicate that the method used did not recover this node in the majority of bootstrap replicates. (B) Cpn60 tree with the E. histolytica sequence excluded. Maximum likelihood yielded a tree of identical topology (ln likelihood = −12181.7), while parsimony generated three trees of length = 2209. Two of these differed from the neighbor-joining tree by the placement of the α-Protebacteria (excluding the Rickettsiales) as a sister group to the γ- and β-Proteobacteria. The third differed by placing T. vaginalis as an immediate relative to the Rickettsiales (see Results).
The three methods generated similar trees. Mitochondrial sequences were specifically related to the Rickettsiales group (comprised of Ehrlichia chaffeensis, C. ruminantium, and Rickettsia tsutsugamushi) of the α-Proteobacteria, similar to previously published phylogenies (11, 12). In both neighbor-joining distance and maximum likelihood analysis, the T. vaginalis sequence formed a clade with the mitochondrial and E. histolytica cpn60 homologs. By contrast, maximum parsimony yielded five trees of equal length all of which placed the T. vaginalis sequences as a specific sister group to the Rickettsiales sequences.
The bootstrap majority rule consensus trees from all three methods indicated that the T. vaginalis/E. histolytica/mitochondrial clade was the preferred topology in every case, including parsimony (Fig. 1A). For maximum likelihood, the support for this grouping was strong (90%) while distance and parsimony methods yielded significantly weaker support (50% and 24%, respectively).
Our previous analysis of the cpn60 gene from E. histolytica showed that the extremely divergent nature of this sequence sometimes resulted in an affinity for the rickettsia, Ehrlichia chaffeensis, an artifactual result likely due to the long branch attraction phenomenon (12, 20). We suspected, therefore, that the presence of the divergent E. histolytica sequence in the dataset may have been responsible for the poorly supported T. vaginalis/E. histolytica/mitochondria node in distance and parsimony analysis. To study the placement of the T. vaginalis sequence in the cpn60 tree without the confounding influence of the E. histolytica sequence, we chose to exclude the latter from the subsequent analysis.
Analysis of the dataset without the E. histolytica sequence using neighbor-joining distance and maximum likelihood analyses indicated that the T. vaginalis cpn60 sequence clustered with those of mitochondrial origin to the exclusion of all other sequences. As expected, the exclusion of E. histolytica caused bootstrap values for this relationship to increase for all three methods (Fig. 1B), with highly significant bootstrap values (>90%) for neighbor-joining and maximum likelihood analyses. However, maximum parsimony analysis still yielded relatively poor bootstrap support for this relationship. Moreover, three equally parsimonious trees were found, two of which displayed the T. vaginalis/mitochondria grouping whereas a third placed T. vaginalis as a specific sister group to the Rickettsiales (not shown).
To understand this result, we examined the impact of the inclusion and exclusion of Rickettsiales species on the bootstrap support for the T. vaginalis/mitochondria node and the alternative T. vaginalis/Rickettsiales node (Fig. 2). Deletion of the Rickettsia sequence causes bootstrap support for the T. vaginalis/mitochondria node (Fig. 2A) to increase in both neighbor-joining and parsimony analysis and support for the alternative T. vaginalis/Rickettsiales node (Fig. 2B) to decrease (the maximum likelihood bootstrap value was not strongly affected). Conversely, deletion of the highly similar Ehrlichia and Cowdria sequences causes bootstrap support for T. vaginalis/mitochondria to decrease for all methods with the alternative node receiving the majority of the remaining bootstrap support. It is clear from this that the affinity of the T. vaginalis sequence for the Rickettsiales is largely due to the presence of the R. tsutsugamushi sequence in the dataset. However, the effect is most apparent when maximum parsimony and neighbor-joining methods are used.
Figure 2.
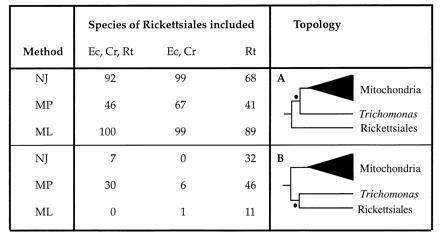
The impact of the sampling of Rickettsiales species on the bootstrap support for two alternative topologies of the cpn60 tree. The dataset excluding the E. histolytica sequence, was used to examine the bootstrap support for two alternative clades each indicated by • on the two trees. (A) The T. vaginalis/mitochondria clade found by neighbor-joining, maximum likelihood and two of the three maximum parsimony trees. (B) The T. vaginalis/Rickettsiales clade displayed by the third maximum parsimony tree (see Materials and Methods). Percentage bootstrap support for each clade is indicated to the left of the trees. Three different combinations of Rickettsiales species were used in the dataset. Species abbreviations are: Ec, Ehrlichia chaffeensis; Cr, C. ruminantium and Rt; R. tsutsugamushi. For each combination of species, bootstrap support for the clade was evaluated using the methods NJ (neighbor-joining distance), MP (maximum parsimony) and ML (protein maximum likelihood) as described in the Materials and Methods.
Maximum likelihood, by contrast, appears to be far less sensitive to this species sampling effect, in each case providing strong support for the T. vaginalis/mitochondria node. Since the maximum likelihood method has been shown to be more robust under conditions of substitution rate inequality between lineages (21, 22), we suggest that these conditions are the likely source of the T. vaginalis/Rickettsiales affinity observed in some of the parsimony and distance analyses. In any case, the T. vaginalis/mitochondria relationship is clearly preferred in eight out of nine of the phylogenetic analyses shown in Fig. 2, suggesting that this is likely the true gene phylogeny and that the alternative topology is artifactual.
DISCUSSION
Bozner (23) recently used heterologous antibodies to immunolocalise a cpn60 homolog in trichomonads of the genus Tritrichomonas, showing that the cellular distribution of the crossreacting protein is most consistent with a hydrogenosomal location. Since we detected no other homologs of cpn60 in T. vaginalis, the gene we report probably encodes a hydrogenosomal protein. Moreoever, in other eukaryotes cpn60 is known to function in the refolding of proteins following their transit across organellar membranes (24) suggesting that the T. vaginalis homolog may perform a similar function in the hydrogenosome. One other protein involved in protein refolding after organellar import is a specific isoform of the molecular chaperone hsp70. In the accompanying paper (25), Germot et al. report the existence of a gene encoding a mitochondrial isoform of hsp70 in T. vaginalis and also conclude that it likely has a hydrogenosomal location.
There are three possible origins, not mutually exclusive, for the T. vaginalis chaperonins. They could be derived from either the mitochondrial symbiont genome, the genome of the symbiont that gave rise to the hydrogenosome, or they could have been acquired by lateral transfer from another organism with which the ancestral trichomonad formed a transient symbiosis that did not result in the formation of an endosymbiotic organelle (26). Whichever of these possibilities is correct, the organism of origin for the chaperonin genes must have been very closely related, if not identical, to the mitochondrial endosymbiont.
Several distinct scenarios for the origin of the hydrogenosome are possible. (i) Hydrogenosomes might have evolved directly from mitochondria by the loss of mitochondrial DNA and the electron transport chain (6). If this is true, then proteins found in hydrogenosomes but lacking in mitochondria must have been secondarily acquired to complete the conversion. For hydrogenosomal enyzmes such as pyruvate:ferredoxin oxidoreductase, found in the cytosol of amitochondrial eukaryotes such as Giardia lamblia and E. histolytica (10), this may have only required the acquisition of a targeting peptide onto the N terminus of the protein. However, it is unclear how enzymes such as hydrogenase, unique to hydrogenosomes but lacking in mitochondria and the cytosol of other eukaryotes (10), were acquired by the ancestral trichomonad. The hydrogenosomal chaperonins in this case are derived from those of the mitochondrion. This scenario is supported by the fact that hydrogenosomes in other eukaryotes appear to have arisen by conversion of mitochondria. For instance, hydrogenosomes of some ciliates bear mitochondrial cristae-like structures (27) while those of Psalteriomonas lanterna are enveloped by a layer of endoplasmic reticulum (28, 29) in exactly the same arrangement as mitochondria are to be found associated with the endoplasmic reticulum in related heterolobosean amoeboflagellates (30).
(ii) A second view holds that hydrogenosomes and mitochondria are derived from a single endosymbiotic ancestor, which had all of the characteristics of both descendants (8). The lineage leading to trichomonads may have diverged from that leading to mitochondriate eukaryotes before the constituents of the present day mitochondrion became fixed, with the two lineages retaining different functions of their shared ancestral symbiont. This view is supported by the finding that most hydrogenosomal enzymes, where comparative data exist, tend to be more similar to their eubacterial than their archaebacterial homologs (10), consistent with an endosymbiotic origin. This scenario also implies that selection for aerobic metabolism need not have been the sole force driving the initial integration of the symbiont, as is often suggested for mitochondria (6).
(iii) A third possibility is that two independent endosymbioses involving closely related α-Proteobacteria occurred early after the divergence of trichomonads from the rest of the eukaryotes, giving rise (perhaps because of different selection pressures) to the hydrogenosome in the former case and mitochondria in the latter. In this scenario, the two organelles, and their chaperonins, share a pre-endosymbiosis common ancestry. However, the conversion from an endosymbiotic bacterium to an organelle likely requires many rare mutations to occur in succession (6). Since this scenario requires that two such conversions occurred independently from the same bacterial lineage, it seems less probable.
(iv) It is also possible that an ancestral trichomonad possessed both the hydrogenosome and the mitochondrion but the two organelles had quite distinct endosymbiotic origins (4). Mitochondria were subsequently lost and certain proteins, including cpn60 and hsp70, were coopted for use in the hydrogenosome. A distinct endosymbiotic origin for the hydrogenosome may explain the biochemical similarity noted between hydrogenosomes and some anaerobic bacteria (31).
(v) Finally, it is possible that the hydrogenosome is not of endosymbiotic origin and the chaperonin genes were derived from a lateral transfer event from a mitochondrion-containing eukaryote or an unknown proteobacterial endosymbiont. In this case, the chaperonin genes are not indicative of the origin of the hydrogenosome as a whole.
In our opininon, scenarios 1 and 2 are the most likely and we believe that trichomonad hydrogenosomes and mitochondria share a common endosymbiotic origin. Regardless of which scenario is true, however, the phylogenetic affinities of the cpn60 sequence in particular make it clear that an ancestor of trichomonads had an intimate relationship with an organism closely related to the mitochondrial symbiont that persisted long enough for gene transfer from its genome to the cell nucleus to take place.
From this example it is clear that the lack of mitochondrial functions coupled with a deeply branching position in phylogenetic trees are not sufficient evidence to conclude that an organism evolved prior to the endosymbiotic origin of mitochondria. Two other amitochondrial protist groups occupy the deepest branches in small subunit ribosomal RNA trees: diplomonads and microsporidia (1, 2, 32). A 60-kDa protein that crossreacts with anti-mitochondrial cpn60 antibodies has already been described in the diplomonad G. lamblia. (33). In addition, it has been suggested that typical eukaryotic cytosolic glyceraldehyde-3-phosphate dehydrogenase genes may derive from the mitochondrial endosymbiont (26). If this is correct, then the existence of eukaryotic cytosolic glyceraldehyde-3-phosphate dehydrogenase genes in G. lamblia (26), other diplomonads (34) and Microsporidia (A.J.R., unpublished data) may also betray the secondary loss of mitochondria in these groups. However, further evidence is needed and a concentrated search for endosymbiotically-derived genes in these deeply-branching amitochondrial groups may help to decide which groups, if any, truly never had mitochondria.
Note.
After this work was accepted for publication, Horner et al. (35) and Bui et al. (36) reported similar results.
Acknowledgments
Thanks are due to Dr. Miklós Müller for useful discussions regarding hydrogenosomal origins. We also thank Dr. Patricia Johnson for providing a sample of the T. vaginalis cDNA library and Dr. Arlin Stoltzfus for providing his mol2con program used in the maximum likelihood bootstrap analysis. This work was supported by a grant awarded to W.F.D. by the Medical Research Council of Canada. A.J.R. was supported by a 1967 Science and Engineering grant from the Natural Sciences and Engineering Research Council of Canada. Part of this work was performed while C.G.C. was at the Laboratory of Parasitic Diseases, National Institutes of Health, Bethesda.
Footnotes
References
- 1.Gunderson J, Hinkle G, Leipe D L, Morrison H G, Stickel S K, Odelson D A, Breznak J A, Nerad T A, Müller M, Sogin M L. J Eukaryotic Microbiol. 1995;42:411–415. doi: 10.1111/j.1550-7408.1995.tb01604.x. [DOI] [PubMed] [Google Scholar]
- 2.Sogin M L. Curr Opin Genet Dev. 1991;1:457–463. doi: 10.1016/s0959-437x(05)80192-3. [DOI] [PubMed] [Google Scholar]
- 3.Patterson D J, Sogin M L. In: The Origin and Evolution of Prokaryotic and Eukaryotic Cells. Hartman H, Matsuno K, editors. Singapore: World Scientific; 1992. pp. 13–46. [Google Scholar]
- 4.Müller M. J Gen Microbiol. 1993;139:2879–2889. doi: 10.1099/00221287-139-12-2879. [DOI] [PubMed] [Google Scholar]
- 5.Margulis L. Proc Natl Acad Sci USA. 1996;93:1071–1076. doi: 10.1073/pnas.93.3.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cavalier-Smith T. Ann NY Acad Sci. 1987;503:55–71. doi: 10.1111/j.1749-6632.1987.tb40597.x. [DOI] [PubMed] [Google Scholar]
- 7.Steinbuchel A, Müller M. Mol Biochem Parasitol. 1986;20:57–65. doi: 10.1016/0166-6851(86)90142-8. [DOI] [PubMed] [Google Scholar]
- 8.Johnson P J, Lahti C J, Bradley P J. J Parasitol. 1993;79:664–670. [PubMed] [Google Scholar]
- 9.Hrdy I, Müller M. J Eukaryotic Microbiol. 1995;42:593–603. doi: 10.1111/j.1550-7408.1995.tb05913.x. [DOI] [PubMed] [Google Scholar]
- 10.Müller M. In: Christian Gottfried Ehrenberg-Festschrift. Schlegel M, Hausmann K, editors. Leipzig: Leipziger Universitaetverlag; 1996. pp. 63–76. [Google Scholar]
- 11.Viale A M, Arakaki A K. FEBS Lett. 1994;341:146–151. doi: 10.1016/0014-5793(94)80446-x. [DOI] [PubMed] [Google Scholar]
- 12.Clark C G, Roger A J. Proc Natl Acad Sci USA. 1995;92:6518–6521. doi: 10.1073/pnas.92.14.6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diamond L S, Clark C G, Cunnick C C. J Eukaryotic Microbiol. 1995;42:277–278. doi: 10.1111/j.1550-7408.1995.tb01579.x. [DOI] [PubMed] [Google Scholar]
- 14.Clark C G. In: Protocols in Protozoology. Lee J J, Soldo A T, editors. Vol. 1. Lawrence, KS: Allen; 1992. pp. D3.1–D3.2. [Google Scholar]
- 15.Lahti C J, Bradley P J, Johnson P J. Mol Biochem Parasitol. 1994;66:309–318. doi: 10.1016/0166-6851(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 16.Felsenstein, J. (1995) phylip: Phylogeny Inference Package (Univ. of Washington, Seattle), Version 3.57c.
- 17.Swofford, D. L. (1993) paup: Phylogenetic Analysis Using Parsimony (Illinois Nat. Hist. Survey, Champaign), Version 3.1.1.
- 18.Adachi J, Hasegawa M. Computer Science Monographs. Tokyo: Inst. Stat. Math.; 1992. Publ. No. 27. [Google Scholar]
- 19.Kishino H, Miyata T, Hasegawa M. J Mol Evol. 1990;30:151–160. doi: 10.1007/BF02109497. [DOI] [PubMed] [Google Scholar]
- 20.Felsenstein J. Syst Zool. 1978;27:401–410. [Google Scholar]
- 21.Hasegawa M, Fujiwara M. Mol Phylogenet Evol. 1993;2:1–5. doi: 10.1006/mpev.1993.1001. [DOI] [PubMed] [Google Scholar]
- 22.Hillis D M, Huelsenbeck J P, Cunningham C W. Science. 1994;264:671–677. doi: 10.1126/science.8171318. [DOI] [PubMed] [Google Scholar]
- 23.Bozner P. J Parasitol. 1996;82:103–111. [PubMed] [Google Scholar]
- 24.Stuart R A, Cyr D M, Craig E A, Neupert W. Trends Biochem Sci. 1994;19:87–92. doi: 10.1016/0968-0004(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 25.Germot A, Philippe H, Le Guyader H. Proc Natl Acad Sci USA. 1996;93:14614–14617. doi: 10.1073/pnas.93.25.14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henze K, Badr A, Wettern M, Cerff R, Martin W. Proc Natl Acad Sci USA. 1995;92:9122–9126. doi: 10.1073/pnas.92.20.9122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finlay B J, Fenchel T. FEMS Microbiol Lett. 1989;65:311–314. [Google Scholar]
- 28.Broers C A M, Stumm C K, Vogels G D, Brugerolle G. Eur J Protistol. 1990;25:369–380. doi: 10.1016/S0932-4739(11)80130-6. [DOI] [PubMed] [Google Scholar]
- 29.Brul S, Veltman R H, Lombardo M C P, Vogels G D. Biochim Biophys Acta. 1994;1183:544–546. doi: 10.1016/0005-2728(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 30.Page F C, Blanton R L. Protistologica. 1985;21:121–132. [Google Scholar]
- 31.Müller M. Symp Soc Gen Microbiol. 1980;31:127–142. [Google Scholar]
- 32.Cavalier-Smith T. Nature (London) 1989;339:100–101. doi: 10.1038/339100a0. [DOI] [PubMed] [Google Scholar]
- 33.Soltys B J, Gupta R S. J Parasitol. 1994;80:580–590. [PubMed] [Google Scholar]
- 34.Rozario C, Morin L, Roger A J, Smith M W, Müller M. J Eukaryotic Microbiol. 1996;43:330–340. doi: 10.1111/j.1550-7408.1996.tb03997.x. [DOI] [PubMed] [Google Scholar]
- 35.Horner D S, Hirt R P, Kilvington S, Lloyd D, Embley T M. Proc R Soc London B. 1996;263:1053–1059. doi: 10.1098/rspb.1996.0155. [DOI] [PubMed] [Google Scholar]
- 36.Bui E T N, Bradley P J, Johnson P J. Proc Natl Acad Sci USA. 1996;93:9651–9656. doi: 10.1073/pnas.93.18.9651. [DOI] [PMC free article] [PubMed] [Google Scholar]