

Abstract
The tightly packaged dsDNA genome in the mature particles of many tailed bacteriophages has been shown to form multiple concentric rings when reconstructed from cryo-electron micrographs. However, recent single-particle DNA packaging force measurements have suggested that incompletely packaged DNA (ipDNA) is less ordered when it is shorter than ∼25% of the full genome length. The study presented here initially achieves both the isolation and the ipDNA length-based fractionation of ipDNA-containing T3 phage capsids (ipDNA-capsids) produced by DNA packaging in vivo; some ipDNA has quantized lengths, as judged by high-resolution gel electrophoresis of expelled DNA. This is the first isolation of such particles among the tailed dsDNA bacteriophages. The ipDNA-capsids are a minor component (containing ∼10-4 of packaged DNA in all particles) and are initially detected by non-denaturing gel electrophoresis after partial purification by buoyant density centrifugation. The primary contaminants are aggregates of phage particles and empty capsids. This study then investigates ipDNA conformations by the first cryo-electron microscopy (cryo-EM) of ipDNA-capsids produced in vivo. The 3-D structures of DNA-free capsids, ipDNA-capsids with various lengths of ipDNA, and mature bacteriophage are reconstructed, which reveals the typical T=7l icosahedral shell of many tailed dsDNA bacteriophages. Though the icosahedral shell structures of these capsids are indistinguishable at the current resolution for the protein shell (∼15 Å), the conformations of the DNA inside the shell are drastically different. T3 ipDNA-capsids with 10.6 kb or shorter dsDNA (<28% of total genome) have an ipDNA conformation indistinguishable from random. However, T3 ipDNA-capsids with 22 kb DNA (58% of total genome) forms a single DNA ring next to the inner surface of the capsid shell. In contrast, dsDNA fully packaged (38.2 kb) in mature T3 phage particles forms multiple concentric rings like those seen in other tailed dsDNA bacteriophages. The distance between the icosahedral shell and the outermost DNA ring decreases in the mature, fully packaged phage structure. These results suggest that, in the early stage of DNA packaging, the dsDNA genome is randomly distributed inside the capsid, not preferentially packaged against the inner surface of the capsid shell, and that the multiple concentric dsDNA rings seen later are the results of pressure-driven close-packing.
Keywords: Agarose gel electrophoresis, Buoyant density centrifugation, Cryo-EM, 3-D reconstruction, Mass spectrometry, DNA packaging
Introduction
During morphogenesis, the DNA genome of tailed double-stranded DNA (dsDNA) bacteriophages is driven into a pre-assembled protein capsid by a DNA packaging motor protein complex that docks on a unique portal vertex and consumes ATP. The DNA packaging ATPase is gp19 in the case of the related bacteriophages, T3 and T7 1; 2; 3. Motor-driven DNA entry occurs at the portal vertex and begins while the capsid is in its initially assembled state (procapsid), both in vivo 4 and in vitro 5. The T3/T7 procapsid is sometimes also called capsid I. During packaging, the protein shell of capsid I undergoes several concerted changes, including thinning, enlarging and becoming more angular (Figure 1). After the conversion, the capsid is called capsid II in the case of T3/T7 1. For most bacteriophages, but not ϕ29, the DNA packaging ATPase has a C-terminal DNA cleaving activity (terminase) that cuts the encapsulated genome from a longer concatemer of genomes 3; 6; 7. The productive T3/T7 DNA packaging pathway is illustrated via the solid arrows in the middle section of Figure 1.
Figure 1.
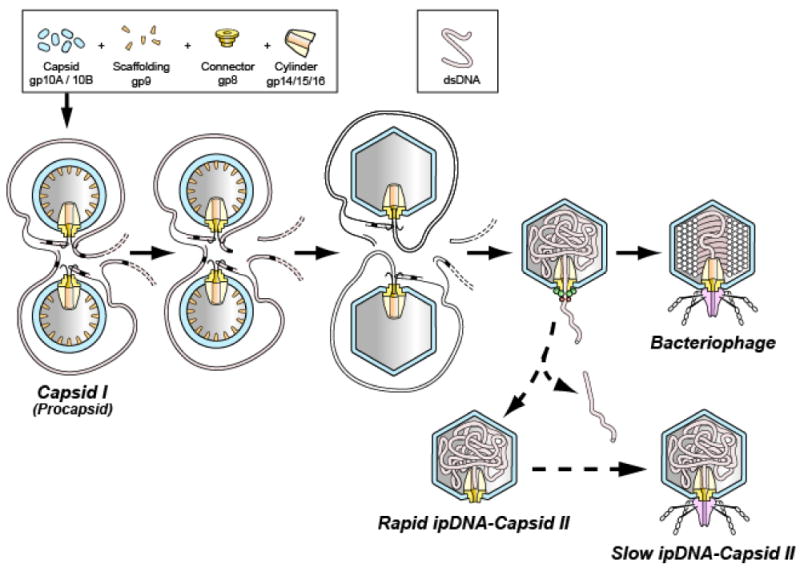
The DNA packaging pathway of the related bacteriophages, T3 and T7. The pathway indicated by solid arrows is the in vivo DNA packaging pathway for T7 1; 44. The in vivo pathway for T3 is the same, based on the intermediates produced in vivo 32. The pathway indicated by the dashed arrows is a proposed abortive branch that produces rapid and slow ipDNA-capsid II. A legend for protein components and DNA is at the top.
Complete analysis of dsDNA genome packaging includes analysis of the progression of the conformations that incompletely packaged DNA (ipDNA) assumes as a DNA molecule enters a capsid. The final conformation achieved by a fully packaged, mature genome produces concentric DNA rings when 3-D reconstruction is performed from cryo-EM images of the isolated particles preserved in a near-native state in vitreous ice. The concentric rings have been found for both ϕ29 and all studied concatemer-packaging bacteriophages 8; 9. The rings are co-axial with the tail for short-tail bacteriophages (podoviruses) P22 10; 11, T7 12; 13 and ε15 14. The usual, though not certain, interpretation of the rings is that packaging winds the DNA molecule uni-directionally around the axis of the portal vertex to form a DNA toroid 11; 13; 14; 15; 16; 17; 18. In any case, the DNA conformation has not been visualized at intermediate stages in packaging 15. Thus, neither the DNA conformations nor their evolution during packaging have been characterized.
Based on the assumption of a DNA toroid throughout packaging, the DNA condensation-derived force resisting packaging has been calculated as a function of the fraction of the length of DNA packaged 15; 17; 19. When external osmotic stress-based stabilization 20 is quantified as a function of DNA length 21, the results confirm the predicted DNA condensation-derived force for the high percentage (77.7 % and more) range of packaged bacteriophage λ DNA.
Based on the energetics of gradual bending (no kinks) and electrostatic attractive/repulsive DNA segment-segment interaction, the possibility has been proposed that a non-random conformation occurs even at the beginning of packaging 15; 16; 17. To perform a comparable test for lower percentages of packaged DNA, single-molecule, optical trap-derived force measurements have recently been performed while DNA was being packaged in vitro. When less than 25 % of the DNA was packaged in the case of bacteriophage ϕ29, this analysis revealed a DNA condensation-derived force that was over 5× higher than predicted via assumption of a DNA toroid, even when the theory was fitted to empirical force values measured at higher degrees of filling 22. In the case of bacteriophage λ, this discrepancy apparently did not occur 23. Rickgauer et al 22 present the hypothesis that the discrepancy for ϕ29 was caused by inaccuracy of the assumption of toroidal DNA conformation (inverted spool in this case) for incompletely packaged DNA (ipDNA). The results of molecular dynamics simulations yield basically the same “disordered ipDNA hypothesis” for bacteriophage DNA packaging in general 24; 25; 26; 27.
Thus, conflicts exist in models for dsDNA genome conformations during the dsDNA packaging process, especially in the early stages. Such conflicts can be directly resolved by cryo-EM imaging and 3-D reconstruction of the conformation of DNA in capsids with packaged ipDNA (to be called ipDNA-capsids). Ideally, imaging will be performed of ipDNA-capsids that are produced during packaging in vivo, to avoid perturbations that occur in vitro. For example, perturbations are potentially caused by the use of a non-concatemeric DNA as substrate for in vitro packaging in the case of well-studied bacteriophages other than ϕ29. Bacteriophages λ, P22, T4 and T3/T7, among others, package a concatemeric DNA in vivo 6, as illustrated for T3/T7 in Figure 1. However, isolation of the appropriate in vivo ipDNA-capsids has not been reported, to the knowledge of the authors, for any of the tailed dsDNA bacteriophages.
In the case of bacteriophage T7, we have shown the existence of ipDNA-capsids in vivo using a DNase protection assay 28. Some of the T7 ipDNA-capsids are produced with kinetics consistent with a DNA packaging intermediate. However, the T7 ipDNA-capsids were not purified. Subsequent experimentation (P. Serwer and E. T. Wright, unpublished data) revealed difficulty with the stability of the T7 ipDNA-capsids during purification. However, the present study first showed that the T7 close relative, T3 phage 29, produced apparently analogous in vivo ipDNA-capsids in stable form. We subsequently used cryo-EM to both image the ipDNA-capsids produced in vivo by bacteriophage T3 and visualize the conformations of the ipDNA at multiple intermediate stages of dsDNA packaging. These studies establish the foundation for more comprehensive future studies of the dynamics and mechanisms of DNA packaging by tailed dsDNA bacteriophages.
Results
Preparation of ipDNA-capsids
To prepare T3 ipDNA-capsids for analysis, particles in two simultaneous 6-liter lysates of wild-type T3-infected cells were pooled, concentrated by polyethylene glycol precipitation and digested with DNase I. The concentrated/DNase I-digested particles were centrifuged through a cesium chloride step gradient in an amount that was as large as possible without having a high chance of causing complete collapse of the fractionation. The sample concentration was high enough to produce some detectable overloading-induced band broadening. Observation of light scattering in the gradient revealed (1) a band of light scattering from bacteriophage particles (1.50 g/ml; labeled as ϕ in Figure 2a), (2) a zone of variable, less intense light scattering at the lower densities expected of ipDNA-capsids (1.26 – 1.50 g/ml; the lower part of this zone is indicated by a bracket in Figure 2a) and (3) a diffuse band that overlaps the bracketed zone of light scattering and is primarily from a mixture of capsid I, capsid II and fragments of the host cell envelope (horizontal arrow in Figure 2a).
Figure 2.
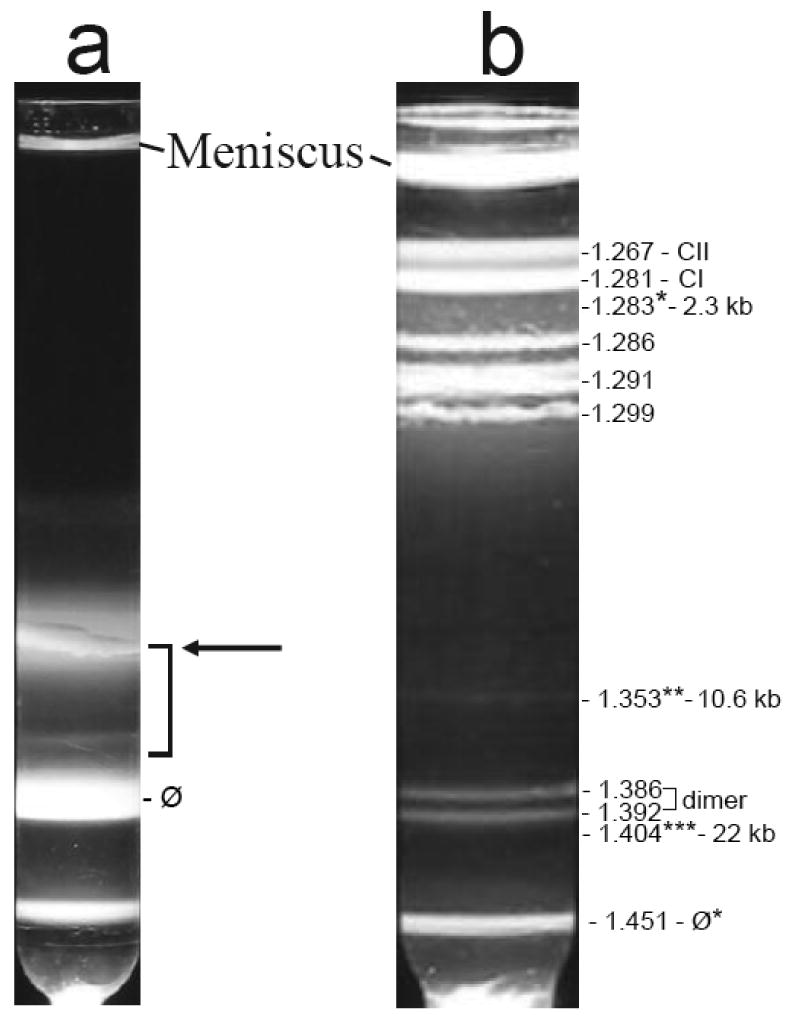
Preparation of ipDNA-capsids. Cesium chloride density gradient centrifugation in two stages was used to prepare ipDNA-capsids, as described in the Materials and Methods section. (a) Light scattering is shown from the step gradient. The arrow points to a diffuse band primarily from a mixture of capsid I, capsid II and fragments of host cell. (b) Light scattering is shown from the buoyant density gradient used to further fractionate the particles in the bracketed region of the gradient in (a). The symbol, ϕ*, indicates a band formed by bacteriophage particles. The symbol, CI, indicates capsid I; the symbol, CII, indicates capsid II. The three ipDNA-capsid fractions analyzed by cryo-EM are labeled with *, ** and *** and their corresponding density and ipDNA length.
The particles in the bracketed region of the gradient in Figure 2a were further purified by buoyant density centrifugation in a cesium chloride density gradient (Figure 2b). Several bands were observed with this second centrifugation. Bands formed by DNA-free capsid I (CI in Figure 2b) and capsid II (CII in Figure 2b) were at the top of the gradient of Figure 2b. Identification of these two band-forming particles is described in the next section.
Just below the CI and CII bands in Figure 2b were roughly four coarse grained, intensely light-scattering bands. The particles of these four bands are assumed to include host cell fragments that were aggregated, based on (1) the coarse grain and high light scattering intensity of the bands and (2) the non-entry of these particles into agarose gels during non-denaturing agarose gel electrophoresis (below). The distribution of light scattering from these aggregates differs from one preparation to another, unlike the other aspects of the light scattering profile.
Lower in the gradient of Figure 2b close to the phage band were two bands at densities of 1.386 g/ml and 1.392 g/ml (labeled as dimer). These two bands were produced by light scattered from bacteriophage/capsid dimers that are not further investigated here.
The densities of ipDNA-capsids are expected to be in the range of 1.28 g/ml to 1.49 g/ml, i.e., the range between the density of DNA-free capsids and the density of mature bacteriophage. Indeed, ipDNA-capsids were found and imaged by gel electrophoresis and cryo-EM (below) at 1.283 g/ml (Figure 2b, marked with single asterisk), 1.353 g/ml (Figure 2b, marked with double asterisk) and 1.404 g/ml (Figure 2b, marked with a triple asterisk). The ipDNA-capsids will be labeled either by density or by ipDNA length, the latter as determined below.
The ϕ* band of Figure 2b contained 1.1 % of the bacteriophage particles in the original step gradient of Figure 1a, as judged by optical density at 260 nm. The ipDNA-capsid bands marked with single, double and triple asterisks in Figure 2b each had less than 2% of the DNA of the ϕ* band, as judged by fluorometry of the DNA after the expulsion from capsids, fractionation and ethidium staining shown below (Figure 3). Thus, the ipDNA-capsids are a minor lysate component (less than one per cell) and were, at first, very difficult to detect.
Figure 3.
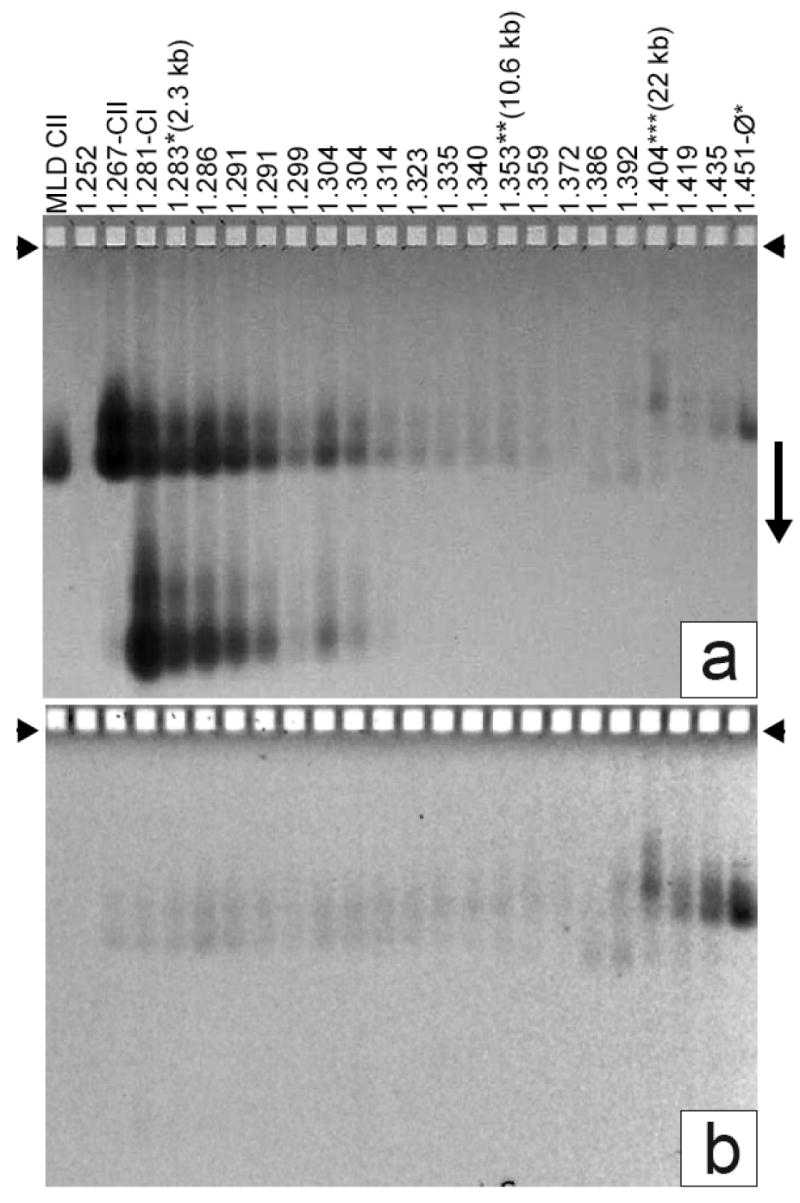
Detection of ipDNA-capsids by AGE. Fractions of a cesium chloride density gradient (Figure 2b) were analyzed by AGE. The gel was stained with (a) Coomassie blue after it had been stained with (b) ethidium. A number above a lane indicates the density of the sample (g/ml). MLD capsid II was also analyzed (leftmost lane). Capsid I and capsid II are labeled as CI and CII, respectively, on top of the lanes. The three samples (density gradient fractions) used to image ipDNA-capsids by cryo-EM are labeled at the top with their corresponding ipDNA lengths. Other density gradient fractions also had ipDNA-capsids, but these were not subjected to cryo-EM.
Detection of ipDNA-capsids: Non-denaturing agarose gel electrophoresis
By itself, the light scattering profile of Figure 2b does not provide sufficient information to determine whether ipDNA-capsids are present in the density gradient. To detect and identify the various capsids within the gradient of Figure 2b, analysis by non-denaturing agarose gel electrophoresis (AGE) was performed for fractions of this gradient (Figure 3a). AGE fractionates by both the average electrical surface charge density (σ) and radius R of a particle 30. Note that both σ and R are characteristics of the particle exterior and do not change as packaged ipDNA amount changes within a capsid, unless the structure of the capsid surface changes with ipDNA amount.
After AGE and Coomassie blue staining (protein specific), stained particles in the agarose gel had the following distribution:
Particles collected from the center of the uppermost light-scattering band of Figure 2b (1.267 g/ml lane, also labeled CII) formed two agarose gel bands (lane labeled as CII in Figure 3a); the more origin-distal band was at the position of MLD capsid II (MLD CII lane in Figure 3a). MLD capsid II is a subfraction of the capsid II in Figure 2b and was previously found to have no tail and no DNA and to have the angular, thin-walled appearance diagnostic for all capsid II particles 31. The band-forming, more slowly migrating capsid II will be called slow capsid II; the band-forming, more rapidly migrating capsid II will be called rapid capsid II. The cause of the difference between rapid and slow capsid II will be analyzed below. In summary, these two types of capsid II particles have indistinguishable density (1.267 g/ml), but different electrophoretic mobility.
Most particles collected from the center of the next light-scattering band (1.281 g/ml, labeled as CI) of Figure 2b migrated in an agarose gel band more distant from the origin than both slow and rapid capsid II, though some 1.281 g/ml particles did form the two capsid II bands (Figure 3a). The primary 1.281 g/ml agarose gel band is at the position expected of capsid I (procapsid; labeled CI in Figure 3a) 32. Capsid I migrated more rapidly than capsid II because of both smaller R and larger magnitude of σ32.
Particles collected from the lowermost band (labeled ϕ*) co-migrated with T3 mature phage particles purified from the ϕ band of Figure 2a (not shown). However, the ϕ* particles were less dense for reasons that have not been investigated.
As density became higher than the density of particles at the capsid I peak in Figure 2b, the intensity of the CI bands dropped below detection in the agarose gel of Figure 3a. The intensity of the CII bands also dropped as density increased in Figure 3a. But, in contrast to the result for the CI bands, the intensity of the CII bands did not become too low to detect. The intensities of the two CII bands would have dropped below detection level, as did the CI band, unless some of the CII particles were denser than DNA-free capsid II, as ipDNA-capsids would be.
This observation raises the possibility that the denser capsid II particles are ipDNA-capsids, i.e., capsids with protein shell similar to that of DNA-free capsid II but with ipDNA inside the protein shell. To determine whether either these or any other particles in Figure 3a had DNA, the agarose gel had been stained with ethidium (DNA stain) before it was stained with Coomassie blue.
For the 1.267 g/ml particles (CII lane) in Figure 3, an ethidium fluorescence (DNA) signal was observed (Figure 3b) from rapid capsid II; this signal was weak in comparison to the Coomassie (protein) signal. As the density increased, the average DNA/protein signal ratio increased by 2-3 orders of magnitude; the DNA signal also shifted from rapid capsid II to slow capsid II. The presence of the DNA signal is an indication of the presence of a DNA-containing, denser form of capsid II particles, possibly either ipDNA-capsid II or a capsid-bacteriophage aggregate that happened to have a similar mobility in the agarose gel. To distinguish these possibilities, the DNA was expelled from the capsid and stained again after completion of both the electrophoresis and the first ethidium staining (Materials and Methods Section). The second ethidium staining did not increase the ethidium signal for the slow capsid II and rapid capsid II bands, though the second ethidium-staining of T3 bacteriophage particles did (data not shown). This observation implies that both slow and rapid capsid II are less full of DNA than bacteriophage particles 30. That is to say, they are ipDNA-capsids. Determination of effective radius by two-dimensional non-denaturing agarose gel electrophoresis (2d-AGE) 30 of particles in one of these fractions (1.349 g/ml) also showed that the particles migrating as either fast or slow capsid II were not aggregates (data not shown). However, some denser particles at 1.386-1.404 g/ml in Figure 3 had somewhat different mobilities and were found below to be aggregates.
The ethidium signal from the intact 1.335-1.372 g/ml ipDNA-capsids of Figure 3b was higher for slow ipDNA-capsid II than it was for rapid ipDNA-capsid II; the opposite was true for the Coomassie signal from the same particles (Figure 3a). Assuming that all ipDNA in any given fraction had the same length, most of the capsid II particles are rapid capsid II particles that did not have ipDNA; the slow capsid II particles had most of the ipDNA. The DNA-free rapid capsid II particles would not have entered this region of the density gradient unless originally either (1) reversibly aggregated with other particles (bacteriophage particles, for example) and subsequently disaggregated or (2) partially filled with ipDNA that was subsequently expelled. Aggregation is favored by the stability of ipDNA-capsids, as shown below. That is to say, the 1.335-1.372 g/ml ipDNA-capsids are contaminated with bacteriophage/empty capsid II aggregates.
Length of ipDNA: High resolution gel electrophoresis of expelled DNA
To measure ipDNA length, high-resolution gel electrophoresis (Figure 4a) was performed of DNA expelled from particles in the density gradient of Figure 2b. The length standards are shown in the rightmost four lanes of Figure 4a. The resolution of the DNA fractionation was high enough in the 5-40 kb region so that the DNA co-migrating with mature DNA is not more than 2% shorter or longer and may well be exactly the length of mature T3 DNA. The high resolution at the position of mature T3 DNA is appreciated by comparing the separation of mature T3 DNA (T3 lane of Figure 4a) from mature T7 DNA (T7 lane of Figure 4a) which is 4.5% longer.
Figure 4.
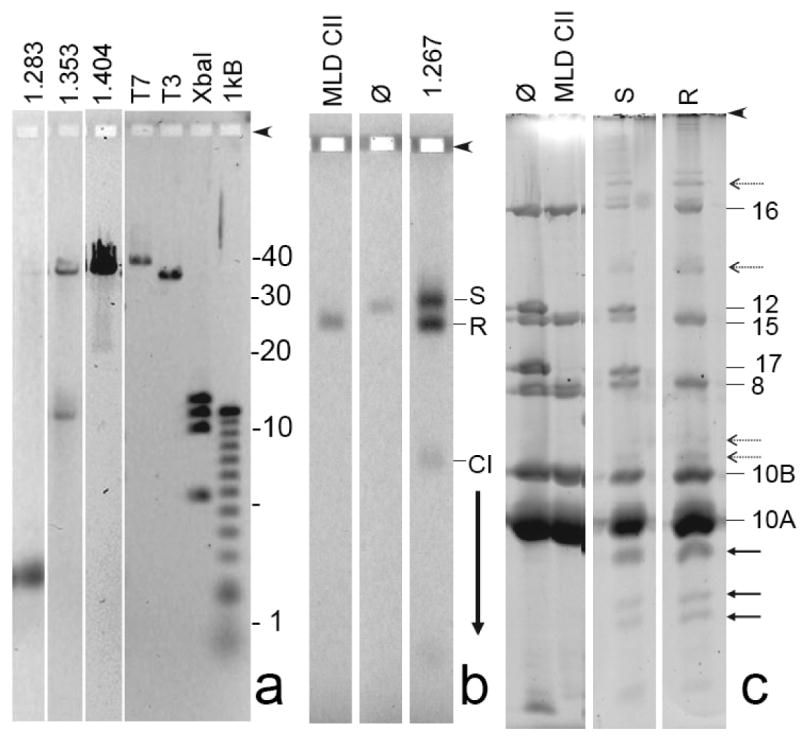
Preliminary characterization of ipDNA-capsids. (a) DNA from samples of 1.283, 1.353, and 1.404 g/ml ipDNA-capsids was expelled from capsids and subjected to high-resolution agarose gel electrophoresis. The following standards are in the rightmost four lanes: T7, mature T7 DNA (39.937 kb); T3, mature T3 DNA (38.208 kb); XbaI, a T7 XbaI digest (5.64; 10.10; 11.37; 12.83 kb), and 1 kB, a 1 kb ladder obtained from Invitrogen. (b) A sample of 1.267 g/ml ipDNA-capsids was removed from a cesium chloride density gradient (Figure 2b), stained with Alexa 488 and then subjected to non-denaturing agarose gel electrophoresis, as in Figure 3. (c) Gel fragments with particles of both the slow and rapid capsid II bands were separately sliced from the agarose gel in (b) and subjected to SDS-PAGE and then stained with SYPRO Ruby (S and R lanes, respectively). Additional lanes during SDS-PAGE have the following samples that had not been subjected to agarose gel electrophoresis (lane marking, followed by sample): ϕ, bacteriophage T3; MLD CII, MLD capsid II. Proteins from the polyacrylamide gel in Figure 4c were eluted, digested with trypsin and analyzed by mass spectrometry, as described in the Materials and Methods Section. Proteins identities (numbers) are marked on the right side.
For the 1.283 g/ml fraction, the result (1.283 g/ml lane in Figure 4a) was finding of a single major ipDNA band of 2.3 kb ipDNA. A trace amount of mature length DNA, presumably from capsid II/bacteriophage aggregates, was also present. For the 1.353 g/ml fraction, the result (1.353 g/ml lane in Figure 4a) for the ipDNA component was finding of a predominant, sharp ipDNA band of 10.6 kb ipDNA and also some more non-uniform length DNA between 10.6 and ∼15 kb. In comparison to the 1.283 g/ml fraction, an increased amount of mature length DNA was also present. For the ipDNA component of the 1.404 g/cm fraction, a band was observed at 22 kb and also some background of longer ipDNAs. In addition, the amount of mature length DNA increased in this fraction to the point that it was the dominant component. The increase in ipDNA length with increasing particle density is expected from the density of DNA, 1.70 g/ml, which is higher than the density of T3 capsid protein.
In Figure 4a, the amount of contaminating mature length DNA increased as the density increased. The presence of this mature length DNA is explained by the presence in the ipDNA-capsid fractions of contaminating aggregates that (1) did not, in some cases, form bands during agarose gel electrophoresis and (2) contained mature bacteriophage particles. By this hypothesis, the aggregation shifted the density of bacteriophage particles to the density of the fractions analyzed for ipDNA. In support, bacteriophage particles and DNA-free capsid II particles were observed in the ipDNA-capsid fractions by cryo-EM (below), though most were monomeric and, therefore, are assumed to have dissociated after centrifugation.
Capsid proteins: SDS-PAGE and mass spectrometry
To determine the reason for the electrophoretic mobility difference between slow and rapid ipDNA-capsid II, 1.267 g/ml capsid II particles were stained with Alexa 488 for detection and then preparatively fractionated by non-denaturing agarose gel electrophoresis for subsequent protein analysis. Fluorescence detection revealed two bands in the preparative agarose gel (1.267 g/ml lane in Figure 4b), as expected.
To analyze the proteins in both slow and rapid capsid II, particles from a band-forming region of the agarose gel of Figure 4b were removed by slicing the gel and further analyzed by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) of a boiled gel slice. SDS-PAGE often is sufficient to identify proteins based on their mass. However, in this case, proteins extracted from the polyacrylamide gel used for SDS-PAGE were further identified by mass spectrometry (MS) because at least one agarose-extracted protein suffered significant damage during AGE, as described below, and agarose present in the samples for SDS-PAGE significantly changed the positions of bands during SDS-PAGE.
The following were the results of the SDS-PAGE/MS.
Rapid capsid II was missing two major tail proteins, gp12 and gp17. The absence of gp12 and gp17 is observed in Figure 4c by visually comparing the SDS-PAGE of rapid capsid II (lane marked R) with the SDS-PAGE of mature bacteriophage T3 (lane marked ϕ) and was confirmed by MS. Thus, rapid capsid II is missing the T3 tail.
In contrast, Figure 4c revealed that slow capsid II (lane marked S) had proteins that migrated close to the distance migrated by tail proteins, gp12 and gp17, in amount comparable to what is present in the bacteriophage T3 particle (lane marked ϕ). MS revealed that these two proteins were, indeed, gp12 and gp17; the SDS-PAGE mobility of these two proteins was slightly changed by the presence of agarose. Thus, slow capsid II has a tail.
Both rapid and slow capsid II had the major outer shell protein, gp10A, and its minor variant gp10B (Figure 4c). No difference in relative amount of gp10A and gp10B between the slow and rapid capsid II particles was observed.
The scaffolding protein (gp9), found in capsid I (data not shown), was not found in either the rapid or the slow capsid II. This result depended heavily on the mass spectrometry because gp9 migrates close to and sometimes co-migrates with gp10A.
Some bands were present in the SDS-PAGE profile of capsids fractionated by the agarose gel electrophoresis, but not of capsids that were obtained directly from the density gradient fractions, before agarose gel electrophoresis. Some of these bands are from cleavage products of gp10B (solid arrows in Figure 4c) that are missing their N-terminus. MLD capsid II also formed these cleavage products after agarose gel electrophoresis, followed by SDS-PAGE (not shown). In addition, several bands formed by gp10 were found closer to the origin than either gp10A or gp10B (dashed arrows in Figure 4d). The A/B designation is not known for the proteins that form these latter bands because the mass spectrometry analysis did not reveal a C-terminal peptide. The working assumption is made that the slow migration of these proteins is caused by damage (possibly cross-links) during agarose gel electrophoresis. Further details have not been investigated.
In summary, the difference in tail proteins was the only significant difference detected between slow ipDNA-capsid II and rapid ipDNA-capsid II. The conclusions are that (1) rapid ipDNA-capsid II has no tail and (2) slow ipDNA-capsid II has a tail, independent of ipDNA length and including the case of DNA length = 0.
Stabilizing ipDNA-capsids and reducing contamination with aggregates
The ipDNA-capsids were stable in cesium chloride solution as judged by repeatedly performing the analysis of Figure 4a on the same density gradient fractions, stored at 4 °C, without removal of the cesium chloride. The profile of ipDNA did not vary for up to 6 months (not shown); longer periods have not been tested. However, dialysis against T/M buffer and storage for 2 days at 4 °C caused loss of about 50% of the ipDNA and also caused loss of sharpness of the ipDNA bands (not shown). Presumably, after dialysis, DNA leaked out of the ipDNA-capsids and was digested. Because of their stability in cesium chloride solutions, the ipDNA-capsids were transported for cryo-electron microscopy without dialysis.
The stability of ipDNA-capsids in cesium chloride solution was purposely used during purification to reduce contamination of ipDNA-capsids with particles that contained mature-length T3 DNA (Figure 4a). The fractions of the step gradient of Figure 2a were left at 4 °C for a week before the second buoyant density centrifugation (Methods Section). This procedure reduced the amount of contaminating mature length DNA and caused aggregation of cell envelope components (1.286-1.299 g/ml bands in Figure 2a), but did not reduce the amount of ipDNA-capsids, as judged by agarose gel electrophoresis of expelled DNA. The reduction in mature length DNA-containing particles was presumably caused by changes in, possibly dissociation of bacteriophage particle-containing aggregates during prolonged incubation in the cesium chloride solution.
Cryo-electron microscopy: 2-D images
Because of the stability of the ipDNA-capsids in cesium chloride solution, cryo-EM was performed on samples of ipDNA-capsids that were dialyzed just before plunge-freezing the specimen onto EM grids. Cryo-electron micrographs of 10.6 kb ipDNA-capsids revealed particles of the following three types, all observed in the field shown in Figure 5b: (1) bacteriophage particles indicated by the symbol, ϕ, (2) capsid II without any detectable DNA indicated by CII, and (3) capsid II with internal material, indicated by ipDNA-CII. The internal material has been confirmed to be ipDNA as detailed in the next paragraph. Capsids of all three of these types were found in micrographs of the entire dataset, taken with defocus values in the range of 1.3 to 3.5 μm (Figure 5b and Supplementary Figure 1). Though only about ∼30% of the ipDNA-capsids had a visible tail as shown in Figure 5b (indicated by the arrows), the actual fraction of tailed particles should be significantly higher since the tails in some views are expected to be masked by the capsid shell. Similar to the image of the 10.6 kb ipDNA-capsid fraction, cryo-EM images of the 22 kb ipDNA-capsid fraction revealed particles of three types, phage, 22 kb ipDNA-capsid, and capsid II (Supplementary Figure 2a). The observation of particles of these three types in the same ipDNA-capsid fraction is consistent with the data shown in Figure 3 and Figure 4. In contrast, cryo-EM of the phage fraction revealed only phage particles, not particles of other types (Supplementary Figure 2b).
Figure 5.
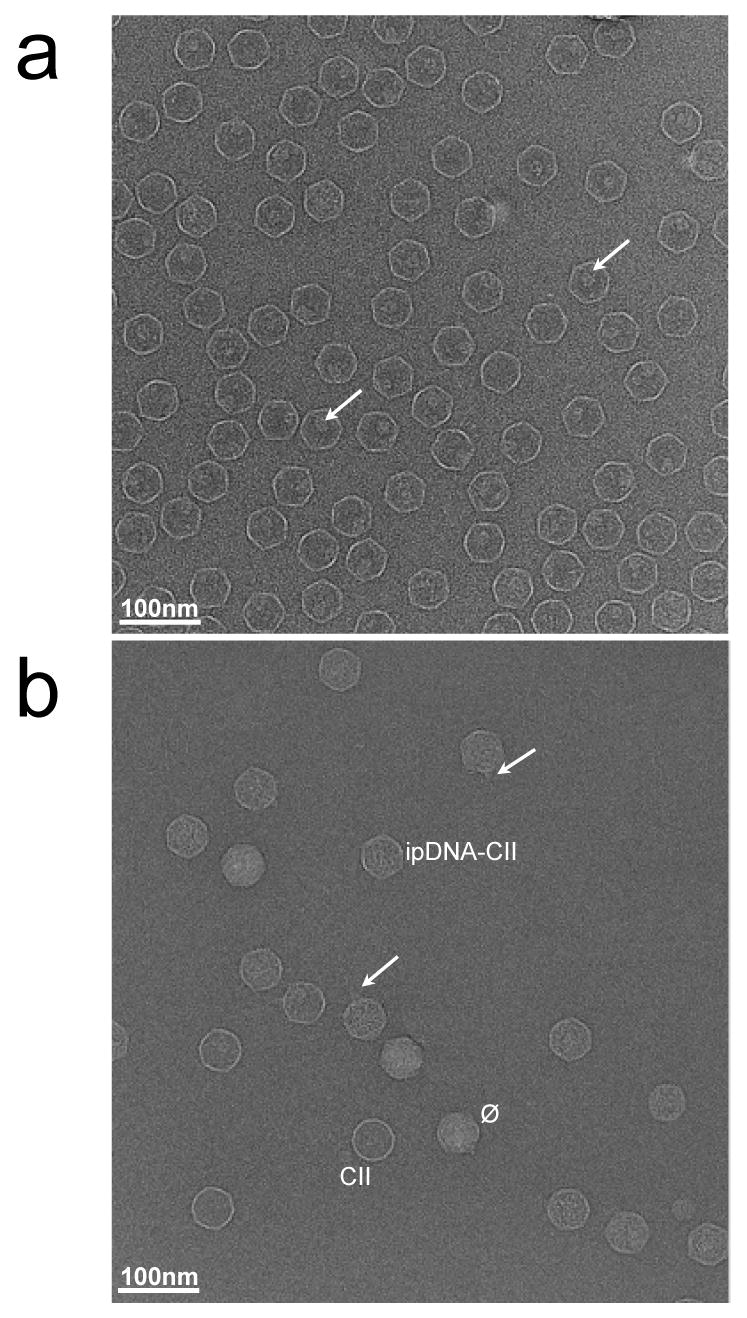
Cryo-electron microscopy of T3 capsids. Fields are shown of (a) MLD capsid II at 3.4 μm defocus and (b) 10.6 kb ipDNA-capsids at 2.5 μm defocus, as described in the Materials and Methods section. The length of the bar is 100 nm. The arrows in (a) point to the internal cylindrical shaped density in the capsid. The labels in (b) mark particles representative of the different types of capsids. The arrows in (b) point to tails on the ipDNA-capsids.
As a reference for a capsid without DNA, a purified capsid II without detectable DNA and tail, MLD capsid II, was imaged here for the first time by cryo-EM (Figure 5a). MLD capsid II preparations were not detectably contaminated by particles of other type, by the criteria of the cryo-EM imaging (Figure 5a), 2d-AGE and SDS-PAGE (not shown). Note that, although DNA-free, MLD capsid II does have an internal protein cylinder clearly visible in some particles (indicated by the arrows in Figure 5a) as previously described 31. This cylinder is also present in the mature bacteriophage particle (see Figure 1), though more difficult to see in cryo-EM images of phage particles due to the interference of packaged DNA.
To further test the conclusion that the internal material of particles marked “ipDNA-CII” in Figure 5b is indeed ipDNA, ten randomly selected 10.6 kb ipDNA-capsid particles (Figure 6b) were visually compared to ten randomly selected MLD capsid II particles (Figure 6a) and also ten randomly selected bacteriophage particles (Figure 6d). By inspection, the presumed 10.6 kb ipDNA-capsids in Figure 6b had more non-cylinder, spaghetti-like internal content than the MLD capsid II particles in Figure 6a and less than the bacteriophage particles in Figure 6d. The analogous presumed 22 kb ipDNA-capsids (Figure 6c) had more internal non-cylinder, spaghetti-like content than the 10.6 kb ipDNA-capsids (Figure 6b), but less than the bacteriophage particles (Figure 6d). In the cryo-EM 2-D images of the 2.3 kb ipDNA-capsids (data not shown), the capsid had less internal content than the 10.6 kb ipDNA-capsids, though more than MLD capsid II. The correlation of cesium chloride buoyant density, agarose gel electrophoresis-determined DNA length and cryo-EM appearance is taken as strong evidence that particles marked as “ipDNA-CII” are ipDNA-capsid II, even though the ipDNA-capsids were not completely purified.
Figure 6.
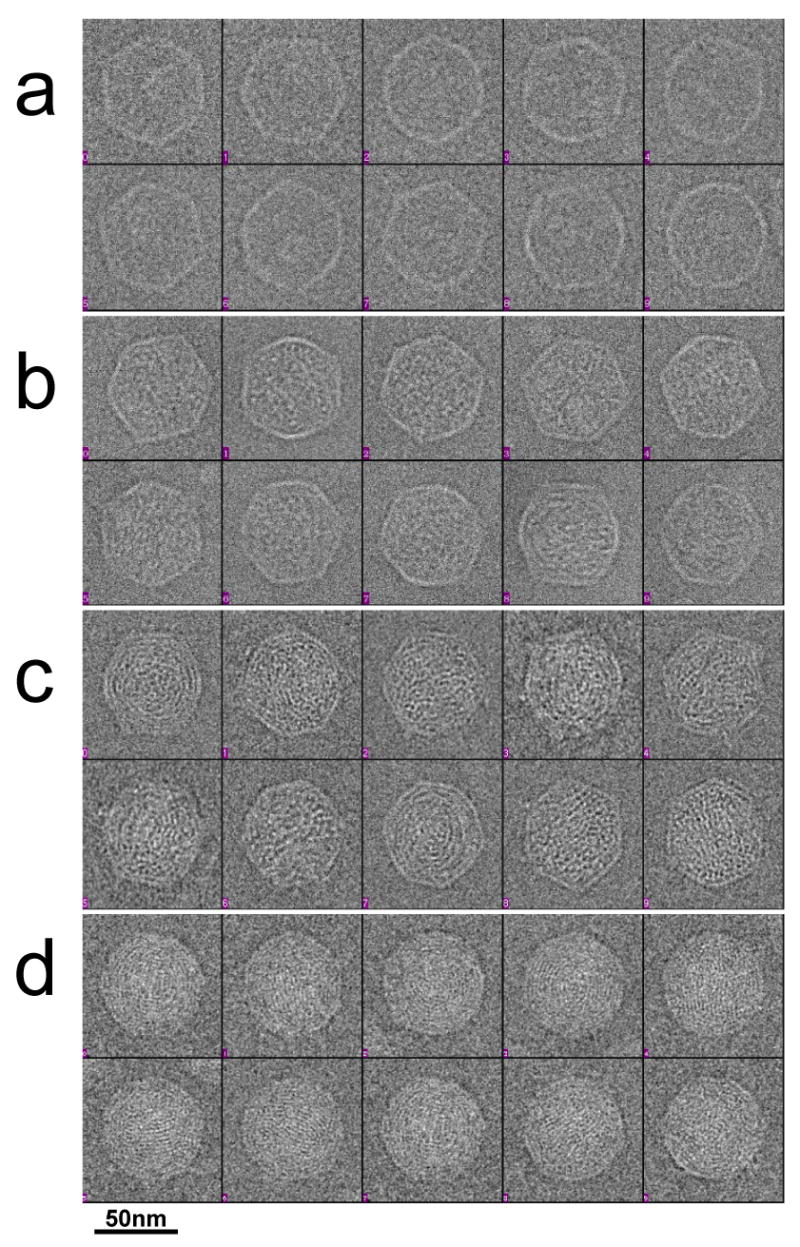
Cryo-electron microscopy of representative particles. Images are shown of ten randomly selected particles of: (a) MLD capsid II, (b) 10.6 kb ipDNA-capsids, (c) 22 kb ipDNA-capsids, and (d) bacteriophage T3. The length of the bar is 50 nm.
Cryo-electron microscopy: 3-D reconstructions
To examine differences among the capsid components of ipDNA-capsids and phage, icosahedral 3-D reconstruction was performed for the particles of Figure 6 and also for the 2.3 kb ipDNA-capsid. The resolution of the reconstructions was about 20 Å, except for the 22 kb ipDNA-capsid which was reconstructed at lower resolution (35 Å) due to a smaller number of particles used in reconstruction. No significant differences among these 3-D reconstructions were seen in capsid surface features among MLD capsid II (Figure 7a), 2.3 kb ipDNA-capsid (not shown), 10.6 kb ipDNA-capsid (Figure 7b), 22 kb ipDNA-capsid (Figure 7c) and the mature bacteriophage (Figure 7d). All these T3 capsids were forms of capsid II with gp10 arranged in a T=7l icosahedral lattice that exhibited a nondistorted six-fold symmetry of hexamers. This type of capsid shell structure is typical among many tailed dsDNA bacteriophages 2; 10; 11; 14; 33.
Figure 7.

3-D icosahedral reconstructions of particles observed by cryo-electron microscopy. The 3-fold surface views are shown for the 3-D reconstruction of (a) MLD capsid II; (b) 10.6 kb ipDNA-capsid; (c) 22 kb ipDNA-capsid; (d) bacteriophage T3. The central cross sections perpendicular to the 5-fold axis are shown for the 3-D reconstructions of (e) MLD capsid II; (f) 10.6 kb ipDNA-capsid; (g) 22 kb ipDNA-capsid; (h) bacteriophage T3. The average distance between the center of the protein shell and that of the first DNA ring is indicated for the 22 kb ipDNA-capsid (g) and bacteriophage T3 (h). The radial density profile of 22 kb ipDNA-capsid and mature phage is also shown in (g) and (h) respectively to measure the distance between the peaks for the protein shell and the outmost DNA ring.
However, significant differences among these 3-D reconstructions were seen in the DNA-containing cavity, as shown in the central cross section of the density maps (Figure 7e-h). In the reconstructions of MLD II capsid and the 2.3 and 10.6 kb ipDNA-capsids, uniform density was seen, except for noise (Figure 7f for 10.6 kb ipDNA-capsid; Figure 7e for MLD capsid II). The simplest interpretation of these results is that the short ipDNA (10.6 kb or shorter) packaged in a capsid has organization indistinguishable from random, in contrast to organization that produces the concentric rings seen in bacteriophage particles. However, two potential limitations of this interpretation exist.
A first potential limitation of any interpretation of ipDNA reconstruction is either introduction of a non-existent (artifactual) ipDNA ring or artifactual removal of a real ipDNA ring by the icosahedral averaging during reconstruction. To test for these potential artifacts, we icosahedrally averaged simulated ipDNA of different conformations inside the capsid. The results of the simulations confirmed that (1) a random conformation for 10.6 kb ipDNA produces a uniform reconstructed ipDNA density, without preferential distribution near the protein shell inner surface and (2) icosahedral averaging enhances an ipDNA ring, if the ring exists, but does not artificially produce a ring-like feature if the ipDNA is randomly distributed (Supplementary Figure 3). Thus, this first potential limitation of interpretation is not a significant limitation.
A second potential limitation of interpretation is the potential missing of a 10.6 kb ipDNA ring because of incomplete ring occupancy caused by short ipDNA length. The amount of ipDNA needed to fill the outermost ring can be estimated based on the number of observed rings and the mean distance of each ring from the particle center (radius) in the icosahedral reconstruction of the T3 phage (Figure 7h). Assuming uniform packing density, the DNA length needed for each ring is proportional to the area of each ring, which in turn is proportional to the square of the radius of the ring. By partitioning the total genome length of T3 phage (38.2kb) in proportion to ring area, it was found that ∼24% or ∼9.2 kb ipDNA fills the outermost ring of the mature phage; 9.0 kb fills the smaller ring observed below for the 22 kb ipDNA. The 10.6 kb ipDNA is thus sufficiently long to fill the outermost ring and will be visible in the icosahedral reconstruction if it is indeed preferentially located near the shell inner surface. The absence of evidence of such a 10.6 kb ring indicates the lack of such preferred localization for the shorter ipDNAs, at least up to 10.6 kb. Thus, the second potential limitation of interpretation is, also not a significant limitation.
In contrast to the result with the 10.6 kb ipDNA-capsid, in the 3-D reconstruction of 22 kb ipDNA-capsid, a ring was seen next to the inner surface of the capsid shell (Figure 7g). The ring is interpreted as the ordered fraction of the 22kb ipDNA. This assignment is supported by the absence of such a ring in the control reconstruction of the contaminating DNA-free capsid II particles from the same set of images (Supplementary Figure 4). The average distance between the mean radius of the protein shell and the mean radius of this ipDNA ring is 34 Å (Figure 7g). In addition, the 3-D reconstruction of the mature bacteriophage T3 particles (Figure 7h) exhibited multiple concentric DNA rings like those of other bacteriophages. Interestingly, the average distance between the mean radius of the capsid shell and the mean radius of the first ring of DNA in the mature phage is slightly smaller (31 Å) (Figure 7h). From these observations, we conclude that the ipDNA is neither preferentially packed next to the inner surface of the capsid shell nor ordered in any other way. Instead, the DNA assumes random organization inside the capsid in the early stages of DNA packaging when the ipDNA length is short (10.6 kb or shorter, compared to the full genome length of 38.2 kb). Concentric DNA rings begin to form when the ipDNA length is longer, 22 kb ipDNA. The ordering of ipDNA begins next to the inner surface of the capsid shell and propagates inward. Thus our cryo-EM observations corroborate the hypothesis (see Introduction) that the DNA order observed in mature bacteriophage particles is not present at the early stages of DNA packaging. Further indication of ipDNA disordering during packaging is the observation that ordering encompasses only one ring in the case of the 22kb ipDNA-capsid, even though the 22kb ipDNA is sufficiently long for more than two rings. Similarly, the outermost DNA rings in the mature phage exhibit better order than those in the center region of the capsid.
Discussion
Isolation and stabilization of ipDNA-capsids
The following two questions arise from the data obtained here. (1) Was the T3 ipDNA generated during packaging or was it generated some other way, perhaps during partial exit/cleavage of DNA that had been completely packaged? (2) What mechanism generated band-forming discrete-length DNA's, in contrast to fragments that vary continuously in length? Several observations support the hypothesis that these ipDNA-capsids were generated during in vivo DNA packaging from a DNA packaging intermediate (pathway indicated by dashed arrows in Figure 1).
First, the presence of a tail on most ipDNA-capsids implies that the capsid had received a signal for tail assembly. This signal is normally preceded by terminase-generated DNA cleavage. After cleaving the packaged DNA from a concatemer of several genomes, the terminase complex dissociates from the portal vertex of the capsid and subsequently the tail is attached to the same portal vertex 3; 6; 7 (Figure 1). By this reasoning, either capsid-associated terminase did not make the cleavages or it made them during packaging, not during exit of the DNA from the capsid. Second, the following consideration supports the idea that capsid-associated T3 terminase made the cleavages as DNA was being packaged, i.e., the cleavages occurred the normal way, but prematurely (pathway indicated by dashed arrows in Figure 1): The presence of discrete bands indicates non-random cleavage that is unlikely if the DNA was cleaved by enzymes that just happened to be present when the DNA exited the capsid. Third, the kinetics of appearance follows that of a DNA packaging intermediate in the case of some ipDNA-capsids of T7, a phage closely related to T3 28. Fourth, the ipDNA-capsids are stable for at least six months in cesium chloride solution used during the purification and storage.
Both the isolation and the 3-D reconstruction of ipDNA-capsid intermediates produced in vivo are achieved here for the first time for any of the numerous tailed dsDNA phages that have been studied. The yield of the ipDNA-capsids is rather low which has limited our isolation procedure to partial purification. The low yield is most likely caused by intrinsically low amount of the ipDNA-capsids in the infected cells since the ipDNA-capsids appear to be DNA packaging intermediates that do not accumulate with time.
Stabilizing ipDNA-capsids was critical for the analysis performed here and was achieved by a surprisingly simple procedure: leaving the ipDNA-capsids in the cesium chloride solution in which they were isolated by buoyant density centrifugation. This procedure does not work well with bacteriophage T7 and the extent to which it works with other bacteriophages is not known.
DNA packing order
Studies of packaged DNA/capsid crosslinking have previously led to the conclusion that no significant inside/outside ordering exists in the case of fully packaged bacteriophage λ DNA 34 and only partial inside/outside ordering exists in the case of bacteriophage T7 (last end packaged preferentially outside) 35. In agreement, molecular dynamics modeling 27 indicates that significant thermal motion-induced inside/outside disordering should exist for all mature packaged DNAs. Molecular dynamics simulations also predict that steric effects of the internal cylinder found in T7, but not in λ, are sufficient to explain the above difference in packaged DNA ordering 23; 26. Simulations have also shown that spooling is not necessary to produce the observed DNA rings in the cryo-EM reconstructions of mature bacteriophages 25.
The data presented here demonstrate that T3 ipDNA shorter than 10.6 kb is packaged without any of the DNA ordering observed in mature bacteriophage particles and without any other ordering detectable at the level of cryo-EM 2-D images and icosahedral 3-D reconstruction. These results confirm the hypothesis that the packaged DNA assumes no overall order in the initial stages of packaging. However, incompletely packaged DNA does exhibit ring-like ordering with increased length packaged. The observed ordering starts next to the inner surface of the capsid shell with a single ring in the 22 kb ipDNA (∼58% of full length) and presumably propagates inward to produce multiple rings as a DNA molecule becomes fully packaged.
The spacing between the capsid shell and the first DNA ring decreases with increased amount of packaged DNA. This observation and the absence of any ordering for shorter ipDNAs suggest that no strong interaction occurs between the inner surface of the capsid shell and the packaged DNA. Such interaction, if it exists, would have caused ordering even at the initial stage of packaging when ipDNA is short (2.3 kb and 10.6 kb) and would have produced a fixed DNA-capsid spacing. Thus, ordering is most likely induced by gradually increasing internal pressure as more DNA is packaged. The disorder-to-order transition begins when the ipDNA length is between 10.6 kb and 22 kb (i.e. ∼28% to ∼58% of full DNA genome).
In the current study, icosahedral symmetry was assumed in the 3-D reconstruction which necessarily prevents visualization of the densities contributed by the non-icosahedral components such as the connector and tail. We cannot rule out that a small portion of the ipDNA might assume ordered packing in the neighborhood of the portal vertex, in analogy to what was seen in the asymmetric reconstructions of mature ε15 14, P22 10; 11 and T7 12 phages. Due to the low amount of ipDNA-capsid particles, asymmetric 3-D reconstructions of the T3 capsids were not performed in this study. Work is in progress to develop procedures for obtaining larger datasets in studies of this type.
If one assumes that the fraction of DNA packaged is a proxy variable for time, analysis of the state of the capsid as a function of the ipDNA length is a procedure conceptually equivalent to single-molecule procedures 22; 23; 36 for observing the cycle of a DNA packaging motor 37. The ipDNA-capsids with increasing lengths of ipDNA, as shown here, will be useful for studies of the dynamics of DNA packaging and its associated capsid transformations.
Materials and Methods
Production and purification of bacteriophage T3 particles
Bacteriophage T3 was originally received from F. W. Studier 38. The host for T3 was Escherichia coli BB/1. The growth medium was 2xLB broth: 20 g Bacto tryptone, 10 g yeast extract, 5 g NaCl in 1 liter of water. To produce a lysate of T3-infected cells, an overnight culture of the host was diluted 200× into either one or two 6-liter amounts of 2xLB broth. The culture was grown with aeration-via-bubbler at 37 °C to 3 × 108 per ml, as judged by counting in a Petroff-Hauser chamber.
The culture was then infected with T3 at a multiplicity of 0.01. Incubation with vigorous aeration was continued until visible lysis, typically at ∼80 min. after infection. After addition of sterile NaCl to a concentration of 0.5 M, debris was immediately removed from the lysed culture by pelleting in six 1.0 liter bottles at 4,000 rpm, for 15 min. in a Beckman J-6B centrifuge (JS4.2 rotor). The lysate was then brought to 9% polyethylene glycol 8,000 by the addition of solid polyethylene glycol 8,000. The capsids and bacteriophage particles were precipitated by leaving this mixture at 4 °C for 2- 4 days.
The supernatant was then decanted and the precipitate was collected by centrifugation at 4,000 rpm for 45 min in a Beckman JS4.2 rotor. Precipitated particles were resuspended in 7 ml per liter of lysate of the following buffer: 0.5 M NaCl, 0.01 M Tris-Cl, pH 7.4, 0.001 M MgCl2. The resuspended lysate was clarified by centrifugation for 10 min. at 10,000 rpm, 4 °C in a Beckman Avanti J-25 centrifuge (JLA16.250 rotor). The supernatant and two washes of the pellet were combined and brought to a volume of 160 ml. Then, the resuspended particles were again brought to 9% polyethylene glycol 8,000 and precipitated at 4 °C, this time for ∼ 16 hr.
After resuspension (final volume = ∼17 ml) and clarification, concentrated particles were then digested with DNase I (2.5 μg/ml) mg/ml, final concentration) for 60 min at 30 °C. The DNase-digested mixture was layered on a cesium chloride density gradient formed by layering of cesium chloride solutions of increasing density (step gradient). The buffer in the cesium chloride solutions was 0.01 M Tris-Cl, pH 7.4, 0.001 M MgCl2. The steps of the gradient were the following (volume in ml, followed by density in g/ml,): 1.50, 1.226; 1.00, 1.349; 1.50, 1.446; 1.00, 1.510; 1.00, 1.728. A 5.6 ml amount of DNase-digested, concentrated particles was then layered on the gradient solution and centrifuged at 33,000 rpm, for 3.0 hr, at 18 °C, in a Beckman SW41 rotor.
After removal of the centrifuge tube from the rotor, light scattering from particles in a density gradient was photographed (Figure 2a) and the gradient was fractionated by pipeting from the top. In comparison to tube puncture, pipeting from the top reduces contamination of ipDNA-capsids with bacteriophage particles and minimizes problems with non-uniform flow across the tube cross-section. The latter problem always exists and becomes aggravated by the viscosity of some regions of the density gradient which, in this case, was mildly overloaded for the purpose of concentrating ipDNA-capsids. Pipeting from the top is also more reliable than tube puncture for controlling the contents of fractions.
The ipDNA-capsids were further purified by, first, diluting their step gradient fraction (the bracketed region in Figure 2a) with an equal amount of gradient buffer and clarifying by centrifugation at 10,000 rpm, for 10 min., at 4 °C in a Beckman JLA16.250 rotor. The supernatant was then brought to a density of 1.340 g/ml with solid cesium chloride and subjected to buoyant density centrifugation for 20 hr., at 42,000 rpm in a Beckman SW50.1 rotor, LE80 ultracentrifuge. Identical fractions of three step gradients were pooled for the buoyant density centrifugation of Figure 2b. Light scattering from particles in the density gradient was photographed (Figure 2b) and the gradient was collected by pipeting from the top. To prepare MLD capsid II, the particles in the CII band (Figure 2b) were subjected to buoyant density centrifugation in a Metrizamide density gradient 31.
Nondenaturing agarose gel electrophoresis
To detect ipDNA-capsids in a density gradient, fractions were added to a 0.35× amount of the following buffer (electrophoresis buffer), without dialysis: 0.09 M Tris-acetate, pH 8.4, 0.001 M MgCl2. A 0.11× amount of 50% sucrose in electrophoresis buffer was then added. The fractions were then layered in the wells of a horizontal, submerged agarose slab gel. The gel had been cast in and submerged under electrophoresis buffer. The layered fractions were allowed to remain in place for 60 min. pre-electrophoresis, in order to dialyze enough cesium chloride out of the sample so that increased ionic strength at the origin did not interfere with the initial movement of particles during electrophoresis. Electrophoresis was then conducted at 2.0 V/cm, 25 °C for 10 hr. After electrophoresis, the gel was stained for DNA with 1 μg/ml ethidium bromide (final concentration) in electrophoresis buffer both before and after bursting capsids by soaking for 1.5 hr. in 10% acetic acid and then neutralizing in electrophoresis buffer. The gel was then stained for protein with Coomassie blue by the procedure of reference 39.
Gel electrophoresis of DNA
To expel DNA from capsids for analysis, the following was sequentially added to 10 μl of a fraction from a density gradient: 10 μl of 0.1 M NaCl, 0.01 M Tris-Cl, pH 7.4, 0.001 M EDTA and then 5 μl of 30% sucrose, 0.6 M NaCl, 0.06 M Tris-Cl, pH 7.4, 0.06 M EDTA, 6% Sarkosyl. Next, the temperature was raised to 85 °C for 10 min. The DNA was then subjected to electrophoresis through a 0.25 % horizontal, agarose gel (Seakem Gold agarose; Lonza/Cambrex) cast in and submerged under the following buffer: 0.05 M sodium phosphate, pH 7.4, 0.001 M EDTA. The electrophoresis was conducted at 0.34 V/cm for 26 hr at 20 °C. These conditions were chosen for high resolution in the 5 - 40 kb range.
SDS-PAGE and Mass spectrometry
To identify capsid-associated proteins, capsids fractionated by nondenaturing agarose gel electrophoresis were obtained by slicing of the agarose gel. To visually detect capsids in an agarose gel, the particles had previously been stained by addition of Alexa 488 (100 μg/ml final concentration) and incubation for 2.0 hr. at 20 °C in the cesium chloride solution in which they had been fractionated. The agarose gel was observed while illuminated by an ultraviolet trans-illuminator. Proteins in an agarose gel slice were denatured for SDS-PAGE by soaking the slice in the following buffer (three changes) for 1.5 hr. and, then, boiling for 3 min: 20.5% glycerol, 4.8% SDS, 0.23 M Tris-Cl, pH 6.8, 0.008 M EDTA, 4.8% β-mercaptoethanol. The boiled mixture was kept at 70 °C to avoid re-gelation of the agarose and was loaded for SDS-PAGE with pipets pre-warmed to 37 °C. SDS-PAGE was performed in a 10% polyacrylamide gel by use of procedures described in reference 32.
After SDS-PAGE, Coomassie-stained gel bands were manually excised and digested in situ with trypsin (Promega modified) in 40 mM NH4HCO3 at 37 °C for 4 hr. The digests were analyzed by mass spectrometry without further purification. Capillary HPLC-electrospray ionization tandem mass spectra (HPLC-ESI-MS/MS) were acquired on a Thermo Fisher LTQ linear ion trap mass spectrometer fitted with a New Objective PicoView 550 nanospray interface. On-line HPLC separation of the digests was accomplished with an Eksigent NanoLC micro HPLC: column, PicoFrit™ (New Objective; 75 μm i.d.) packed to 10 cm with C18 adsorbent (Vydac; 218MS 5 μm, 300 Å); mobile phase A, 0.5% acetic acid (HAc)/0.005% trifluoroacetic acid (TFA); mobile phase B, 90% acetonitrile/0.5% HAc/0.005% TFA; gradient 2 to 42% B in 30 min; flow rate, 0.4 μl/min. MS conditions were: ESI voltage, 2.9 kV; isolation window for MS/MS, 3; relative collision energy, 35%; scan strategy, survey scan followed by acquisition of data dependent collision-induced dissociation (CID) spectra of the seven most intense ions in the survey scan above a set threshold. The uninterpreted CID spectra were searched against the NCBI nr database using Mascot (Matrix Science; London, UK). Methionine oxidation was considered as a variable modification for all searches. Cross correlation of the Mascot results with X! Tandem and determination of protein identity probabilities were accomplished by Scaffold (Proteome Software).
Cryo-electron microscopy
T3 ipDNA-capsid samples were stored and shipped from San Antonio to West Lafayette in the cesium chloride gradient solution used for the last step of isolation (see Figure 2b). Other samples were shipped after dialysis against the following buffer: 0.2 M NaCl, 0.01 M Tris-Cl, pH 7.4, 0.001 M MgCl2 (T/M buffer). The ipDNA-capsid samples were dialyzed against the same buffer immediately before cryo-EM sample grid preparation. An ∼4 μl aliquot of sample was (1) pipetted onto Quantifoil grids with 2 μm holes, (2) blotted, and (3) plunge frozen in liquid ethane cooled by liquid nitrogen. The T3 MLD capsid II and ipDNA-capsids were imaged using a CM200 electron cryo-microscope with field emission gun (FEG) operated at 200 kV. The T3 bacteriophage particles were imaged using a CM300 electron cryo-microscope operated at 300 kV. All the images were recorded on Kodak SO163 films at a nominal magnification of ×50,000 with varying underfocus values in the range of 1.3 to 3.5 μm and at low dose (∼20 e/Å2) conditions. Micrographs without ice contamination were selected and digitized using a Nikon Super Coolscan 9000ED scanner with step size of 6.35 μm/pixel. The scanned images were binned, resulting in final calibrated pixel sizes of 2.49 Å/pixel for T3 capsid II and ipDNA-capsids and 2.7 Å/pixel for T3 bacteriophage.
Single particle image processing and 3-D reconstruction
Particles were manually selected from the scanned micrograph images using the graphic program boxer in the single particle image processing software package EMAN 39. Due to the low concentration of the ipDNA-capsids, only a small number of ipDNA-capsids (several to teens) could be selected from one micrograph and the entire dataset consists of many micrographs of varying defocuses. The total number of particles selected were as follows: MLD capsid II, 684; 2.3 kb ipDNA-capsid, 721; 10.6 kb ipDNA-capsid, 692; 22 kb ipDNA-capsid, 76 and mature bacteriophage, 677. The fields of 22 kb ipDNA-capsids were contaminated enough by (presumably aggregation-derived) DNA-free capsid II and mature phage particles (see, for example, the 1.404 g/ml lane in Figure 4a) that a significantly smaller number of usable ipDNA-capsid particles was available.
The contrast transfer function (CTF) parameters, including defocus, experimental B-factor and background, of these particles were determined using EMAN ctfit program 40. The CTF-caused phase inversions of particle images were corrected before subsequent image processing. The initial icosahedral models for each dataset of these particles were independently built de novo using randomly selected raw particles with the starticos program in EMAN 39. Cross-common line algorithm was then used to determine the center and orientation of raw particles 41. The EMAN make3d program was used to reconstruct the 3-D maps from the 2-D images. Icosahedral symmetry was imposed during reconstruction for all the samples. During the reconstruction, CTF amplitude correction was performed which includes Wiener filtration and SNR weighting of different particles at different defocuses 39. The orientation/center determination and 3-D reconstruction were iterated until convergence, defined by the point at which the resolution of the 3-D reconstructions stopped improving. The final resolutions of the reconstructions were determined using the Fourier shell correlation (0.5 criterion) of two reconstructions of half datasets 42. For fair comparison with the structure of 22 kb ipDNA-capsid built from a comparatively small number (76) of particles, independent de novo reconstructions were repeated for the other four datasets with 76 randomly selected particles of the full datasets. These control reconstructions exhibited similar structures of the capsid shell and internal DNA, albeit at lower resolutions (data not shown). The full dataset was used for the final higher resolution structures shown in Figure 7. To test the possibility that the DNA ring seen in the 22 kb ipDNA-capsid reconstruction (Figure 7g) was due to residual artefacts of CTF, independent de novo reconstruction was repeated for the same number (76) of empty capsid II particles from the same set of micrographs (Supplementary Figure 4). The final resolution of these 3-D reconstructions, is 21.5 Å, MLD capsid II; 19.9 Å, 2.3 kb ipDNA–capsid; 20.1 Å, 10.6 kb ipDNA-capsid; 35.6 Å, 22 kb ipDNA-capsid and 19.6 Å, mature phage. These resolutions were calculated for the entire density map including the external background and the internal DNA without additional masking (as shown in right column of Figure 7). The resolutions for the icosahedral shell region were slightly better at ∼15 Å if only the density regions of the icosahedral protein shell were included in the resolution evaluation by removing the internal DNA and external background with soft masks. The final 3-D maps were visualized using UCSF Chimera software 43.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health to P.S. (GM24365) and W.J. (AI072035) and from the Welch Foundation to P.S. (AQ-764). The cryo-EM images were taken in the Purdue Biological Electron Microscopy Facility and the Purdue Rosen Center for Advanced Computing (RCAC) provided the computational resource for the 3-D reconstructions. Protein identifications were conducted in the UTHSCSA Institutional Mass Spectrometry Laboratory.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Serwer P. T3/T7 DNA packaging. In: Catalano CE, editor. Viral genome packaging machines: genetics, structure, and mechanism. Landes Publishing; Georgetown, TX: 2004. pp. 59–79. [Google Scholar]
- 2.Cerritelli ME, Conway JF, Cheng N, Trus BL, Steven AC. Molecular mechanisms in bacteriophage T7 procapsid assembly, maturation, and DNA containment. Adv Protein Chem. 2003;64:301–23. doi: 10.1016/s0065-3233(03)01008-8. [DOI] [PubMed] [Google Scholar]
- 3.Fujisawa H, Morita M. Phage DNA packaging. Genes Cells. 1997;2:537–45. doi: 10.1046/j.1365-2443.1997.1450343.x. [DOI] [PubMed] [Google Scholar]
- 4.Son M, Watson RH, Serwer P. The direction and rate of bacteriophage T7 DNA packaging in vitro. Virology. 1993;196:282–9. doi: 10.1006/viro.1993.1476. [DOI] [PubMed] [Google Scholar]
- 5.Shibata H, Fujisawa H, Minagawa T. Characterization of the bacteriophage T3 DNA packaging reaction in vitro in a defined system. J Mol Biol. 1987;196:845–51. doi: 10.1016/0022-2836(87)90409-8. [DOI] [PubMed] [Google Scholar]
- 6.Catalano CE. Viral genome packaging machines: an overview. In: Catalano CE, editor. Viral Genome Packaging Machines: Genetics, Structure and Mechanism. Landes Bioscience; Georgetown, TX: 2005. pp. 1–4. [Google Scholar]
- 7.Catalano CE. The terminase enzyme from bacteriophage lambda: a DNA-packaging machine. Cell Mol Life Sci. 2000;57:128–48. doi: 10.1007/s000180050503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson JE, Chiu W. DNA packaging and delivery machines in tailed bacteriophages. Curr Opin Struct Biol. 2007;17:237–43. doi: 10.1016/j.sbi.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 9.Grimes S, Jardine PJ, Anderson D. Bacteriophage phi 29 DNA packaging. Adv Virus Res. 2002;58:255–94. doi: 10.1016/s0065-3527(02)58007-6. [DOI] [PubMed] [Google Scholar]
- 10.Chang J, Weigele P, King J, Chiu W, Jiang W. Cryo-EM asymmetric reconstruction of bacteriophage P22 reveals organization of its DNA packaging and infecting machinery. Structure. 2006;14:1073–82. doi: 10.1016/j.str.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 11.Lander GC, Tang L, Casjens SR, Gilcrease EB, Prevelige P, Poliakov A, Potter CS, Carragher B, Johnson JE. The structure of an infectious P22 virion shows the signal for headful DNA packaging. Science. 2006;312:1791–1795. doi: 10.1126/science.1127981. [DOI] [PubMed] [Google Scholar]
- 12.Agirrezabala X, Martin-Benito J, Caston JR, Miranda R, Valpuesta JM, Carrascosa JL. Maturation of phage T7 involves structural modification of both shell and inner core components. EMBO J. 2005;24:3820–3829. doi: 10.1038/sj.emboj.7600840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cerritelli ME, Cheng N, Rosenberg AH, McPherson CE, Booy FP, Steven AC. Encapsidated conformation of bacteriophage T7 DNA. Cell. 1997;91:271–80. doi: 10.1016/s0092-8674(00)80409-2. [DOI] [PubMed] [Google Scholar]
- 14.Jiang W, Chang J, Jakana J, Weigele P, King J, Chiu W. Structure of epsilon15 bacteriophage reveals genome organization and DNA packaging/injection apparatus. Nature. 2006;439:612–6. doi: 10.1038/nature04487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Purohit PK, Inamdar MM, Grayson PD, Squires TM, Kondev J, Phillips R. Forces during bacteriophage DNA packaging and ejection. Biophys J. 2005;88:851–66. doi: 10.1529/biophysj.104.047134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tzlil S, Kindt JT, Gelbart WM, Ben-Shaul A. Forces and pressures in DNA packaging and release from viral capsids. Biophys J. 2003;84:1616–27. doi: 10.1016/S0006-3495(03)74971-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kindt J, Tzlil S, Ben-Shaul A, Gelbart WM. DNA packaging and ejection forces in bacteriophage. Proc Natl Acad Sci U S A. 2001;98:13671–4. doi: 10.1073/pnas.241486298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Odijk T. Hexagonally packed DNA within bacteriophage T7 stabilized by curvature stress. Biophys J. 1998;75:1223–7. doi: 10.1016/S0006-3495(98)74041-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purohit PK, Kondev J, Phillips R. Mechanics of DNA packaging in viruses. Proc Natl Acad Sci USA. 2003;100:3173–3178. doi: 10.1073/pnas.0737893100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Serwer P, Masker WE, Allen JL. Stability and in vitro DNA packaging of bacteriophages: effects of dextrans, sugars, and polyols. J Virol. 1983;45:665–71. doi: 10.1128/jvi.45.2.665-671.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grayson P, Evilevitch A, Inamdar MM, Purohit PK, Gelbart WM, Knobler CM, Phillips R. The effect of genome length on ejection forces in bacteriophage lambda. Virology. 2006;348:430–6. doi: 10.1016/j.virol.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rickgauer JP, Fuller DN, Grimes S, Jardine PJ, Anderson DL, Smith DE. Portal motor velocity and internal force resisting viral DNA packaging in bacteriophage phi29. Biophys J. 2008;94:159–67. doi: 10.1529/biophysj.107.104612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fuller DN, Raymer DM, Rickgauer JP, Robertson RM, Catalano CE, Anderson DL, Grimes S, Smith DE. Measurements of single DNA molecule packaging dynamics in bacteriophage lambda reveal high forces, high motor processivity, and capsid transformations. J Mol Biol. 2007;373:1113–22. doi: 10.1016/j.jmb.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Z, Wu J, Wang ZG. Osmotic pressure and packaging structure of caged DNA. Biophys J. 2008;94:737–46. doi: 10.1529/biophysj.107.112508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petrov AS, Harvey SC. Structural and thermodynamic principles of viral packaging. Structure. 2007;15:21–7. doi: 10.1016/j.str.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 26.Forrey C, Muthukumar M. Langevin dynamics simulations of genome packing in bacteriophage. Biophys J. 2006;91:25–41. doi: 10.1529/biophysj.105.073429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spakowitz AJ, Wang ZG. DNA packaging in bacteriophage: is twist important? Biophys J. 2005;88:3912–23. doi: 10.1529/biophysj.104.052738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khan SA, Hayes SJ, Watson RH, Serwer P. Specific, nonproductive cleavage of packaged bacteriophage T7 DNA in vivo. Virology. 1995;210:409–20. doi: 10.1006/viro.1995.1357. [DOI] [PubMed] [Google Scholar]
- 29.Pajunen MI, Elizondo MR, Skurnik M, Kieleczawa J, Molineux IJ. Complete nucleotide sequence and likely recombinatorial origin of bacteriophage T3. J Mol Biol. 2002;319:1115–32. doi: 10.1016/S0022-2836(02)00384-4. [DOI] [PubMed] [Google Scholar]
- 30.Serwer P, Khan SA, Griess GA. Non-denaturing gel electrophoresis of biological nanoparticles: viruses. J Chromatogr A. 1995;698:251–61. doi: 10.1016/0021-9673(94)01259-h. [DOI] [PubMed] [Google Scholar]
- 31.Serwer P. A metrizamide-impermeable capsid in the DNA packaging pathway of bacteriophage T7. J Mol Biol. 1980;138:65–91. doi: 10.1016/s0022-2836(80)80005-2. [DOI] [PubMed] [Google Scholar]
- 32.Serwer P, Watson RH, Hayes SJ, Allen JL. Comparison of the physical properties and assembly pathways of the related bacteriophages T7, T3 and phi II. J Mol Biol. 1983;170:447–69. doi: 10.1016/s0022-2836(83)80157-0. [DOI] [PubMed] [Google Scholar]
- 33.Jiang W, Li Z, Zhang Z, Baker ML, Prevelige PE, Jr, Chiu W. Coat protein fold and maturation transition of bacteriophage P22 seen at subnanometer resolutions. Nat Struct Biol. 2003;10:131–5. doi: 10.1038/nsb891. [DOI] [PubMed] [Google Scholar]
- 34.Widom J, Baldwin RL. Tests of spool models for DNA packaging in phage lambda. J Mol Biol. 1983;171:419–37. doi: 10.1016/0022-2836(83)90038-4. [DOI] [PubMed] [Google Scholar]
- 35.Serwer P, Hayes SJ, Watson RH. Conformation of DNA packaged in bacteriophage T7. Analysis by use of ultraviolet light-induced DNA-capsid cross-linking. J Mol Biol. 1992;223:999–1011. doi: 10.1016/0022-2836(92)90258-l. [DOI] [PubMed] [Google Scholar]
- 36.Hugel T, Michaelis J, Hetherington CL, Jardine PJ, Grimes S, Walter JM, Falk W, Anderson DL, Bustamante C. Experimental test of connector rotation during DNA packaging into bacteriophage phi29 capsids. PLoS Biol. 2007;5:e59. doi: 10.1371/journal.pbio.0050059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Serwer P. Analysis of biological motors via multidimensional fractionation: a strategy. Electrophoresis. 2005;26:494–9. doi: 10.1002/elps.200406166. [DOI] [PubMed] [Google Scholar]
- 38.Studier FW. Relationships among different strains of T7 and among T7-related bacteriophages. Virology. 1979;95:70–84. doi: 10.1016/0042-6822(79)90402-1. [DOI] [PubMed] [Google Scholar]
- 39.Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 40.Saad A, Ludtke SJ, Jakana J, Rixon FJ, Tsuruta H, Chiu W. Fourier amplitude decay of electron cryomicroscopic images of single particles and effects on structure determination. J Struct Biol. 2001;133:32–42. doi: 10.1006/jsbi.2001.4330. [DOI] [PubMed] [Google Scholar]
- 41.Liu X, Jiang W, Jakana J, Chiu W. Averaging tens to hundreds of icosahedral particle images to resolve protein secondary structure elements using a Multi-Path Simulated Annealing optimization algorithm. J Struct Biol. 2007;160:11–27. doi: 10.1016/j.jsb.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harauz G, van Heel M. Exact filters for general geometry three dimensional reconstruction. Optik. 1986;73:146–156. [Google Scholar]
- 43.Goddard TD, Huang CC, Ferrin TE. Visualizing density maps with UCSF Chimera. J Struct Biol. 2007;157:281–7. doi: 10.1016/j.jsb.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 44.Sun M, Louie D, Serwer P. Single-event analysis of the packaging of bacteriophage T7 DNA concatemers in vitro. Biophys J. 1999;77:1627–37. doi: 10.1016/S0006-3495(99)77011-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.