

Summary
Theiler’s murine encephalomyelitis virus (TMEV or Theiler’s virus) is a neurotropic picornavirus that can persist lifelong in the central nervous system of infected mice, thereby causing a chronic inflammatory demyelinating disease. The leader (L) protein of the virus is an important determinant of viral persistence. It was shown to inhibit transcription of type I interferon (IFN) genes and to cause nucleocytoplasmic redistribution of host proteins. Here, we report that expression of L shuts-off the synthesis of green fluorescent protein and of firefly luciferase, suggesting that it induces a global shut-off of host protein expression. L inhibited neither the transcription nor the translation of the reporter genes but blocked cellular mRNA export from the nucleus. This activity correlated with the phosphorylation of nucleoporin 98 (Nup98) an essential component of the nuclear pore complex. On the other hand, our data confirm that L inhibits IFN expression at the transcriptional level and show that transcription of other chemokine or cytokine genes is affected by L. This transcriptional inhibition correlated with inhibition of interferon regulatory factor 3 (IRF-3) dimerization. Whether inhibition of IRF-3 dimerization and dysfunction of the nuclear pore complex are related phenomena remains an open question. In vivo, IFN antagonism appears to be an important role of L early during infection since a virus bearing a mutation in the zinc finger of L replicated as well as the wild-type virus in type I IFN receptor-deficient mice (IFNAR-KO) but had impaired fitness in IFN-competent mice.
Introduction
Theiler’s murine encephalomyelitis virus (TMEV or Theiler’s virus) is a neurotropic picornavirus which has a striking ability to cause persistent infections of the central nervous system (CNS) of the mouse (Theiler M, 1940) (reviewed in (Brahic et al., 2005)). The genome of TMEV is a single-stranded positive RNA molecule encoding a long polyprotein which is processed to yield the mature viral proteins. TMEV encodes two proteins, L and L*, that are not essential for replication of the virus in BHK-21 cells (Kong & Roos, 1991; Kong et al., 1994; Michiels et al., 1997), but that are required for persistent infection of the CNS in vivo (Ghadge et al., 1998; van Pesch et al., 2001; van Eyll & Michiels, 2002; Paul & Michiels, 2006). The L* protein is translated from an additional ORF, overlapping the ORF encoding the L, VP4 and VP2 regions of the viral polyprotein (Kong & Roos, 1991).
The leader (L) protein of TMEV is a 76 amino acid-long protein cleaved off from the N-terminal end of the viral polyprotein. It possesses a zinc finger which was shown to bind divalent cations (Chen et al., 1995) and to be crucial for L protein function in vitro and in vivo (van Pesch et al., 2001; Delhaye et al., 2004). It was shown that the leader protein could block the production of type I interferons (IFN) by infected cells and that this inhibition occurred at the transcriptional level (van Pesch et al., 2001; van Pesch & Michiels, 2003). The L protein of TMEV was also shown to interfere with the nucleocytoplasmic trafficking of cellular proteins in infected cells (Delhaye et al., 2004) and to trigger the subcellular redistribution of both nuclear and cytoplasmic cellular proteins, most notably interferon regulatory factor-3 (IRF-3).
Encephalomyocarditis virus (EMCV), a Cardiovirus related to TMEV, encodes a leader protein sharing 35% amino acid identity with that of TMEV. The EMCV and TMEV leaders share conserved Zn finger and negatively charged domains. However, the EMCV leader lacks a C-terminal Ser-Thr rich domain that is conserved in the leaders of TMEV strains. In spite of such sequence divergences, the EMCV leader shares with the leader protein of TMEV the antagonist activities directed against IFN production and nucleocytoplasmic trafficking (Zoll et al., 2002; Lidsky et al., 2006; Paul & Michiels, 2006; Porter et al., 2006; Hato et al., 2007). In the case of EMCV, IFN production inhibition was found to correlate with inhibition of NFκB activation (Zoll et al., 2002) and with suppression of IRF-3 dimerization (Hato et al., 2007). Perturbation of nucleocytoplasmic transport by the EMCV leader (Lidsky et al., 2006) correlated with the interaction of this protein with RAN GTPase, a crucial regulator of trafficking across the nuclear envelope (Porter et al., 2006). The blockade of macromolecule transport possibly explains the global shut-off of protein expression mediated by the EMCV leader (Zoll et al., 1996). Recent work suggests that the TMEV leader also mediates a global shut-off of host cell protein expression (Baida et al., 2008).
Here, we analyzed the possibility that global shut-off and inhibition of IFN gene transcription could result from a global inhibition of cellular transcription. Contrary to this hypothesis, our data show that the TMEV leader acts at two different levels in cells. On one hand, it blocks global protein expression at a post-transcriptional stage, by preventing nuclear export of cellular mRNAs. This activity correlated with Nup98 hyperphosphorylation. On the other hand, it acts at the transcriptional level on the expression of selected cytokine and chemokine genes, by inhibiting IRF-3 dimerization. In vivo, inhibition of the IFN pathway appears to be an important activity of the leader at early times following infection.
Materials and Methods
Cells and viruses
BHK-21, BALB/3T3 and L929 cells were cultured as described previously (van Pesch et al., 2001). TMEV derivatives were produced by electroporation of BHK-21 cells (Michiels et al., 1997) with genomic RNA transcribed in vitro from plasmids carrying the corresponding cDNAs: pTMDA1 carries the genome of virus DA1, a molecular clone of the Daniel’s strain of TMEV (Daniels et al., 1952; McAllister et al., 1989; Michiels et al., 1997). Virus TM598 is a DA1 derivative carrying mutations disrupting the zinc finger motif of the L protein (Lcys mutation) but which do not modify the amino acid sequence of the L* protein which is translated from an overlapping ORF (van Pesch et al., 2001). KJ6 is a DA1 derivative containing capsid mutations adapting the virus to infect L929 cells with high efficiency (Jnaoui & Michiels, 1998). The corresponding Lcys mutant was called TM659 (van Pesch et al., 2001). SB3 is another derivative of KJ6 which carries a deletion encompassing codons 6 to 67 of the L region (LΔ6-67). The pSB3 plasmid, carrying the genome of this virus, was obtained by replacing, in pTM564 (Michiels et al., 1997), a MscI-BamHI restriction fragment in the capsid-coding region, by the corresponding fragment of pKJ6.
Plasmids construction
Plasmids were constructed in the pcDNA3 vector backbone (Invitrogen), in which gene transcription is under the control of the cytomegalovirus (CMV) immediate-early promoter. A series of plasmids, pTM553, pTM592 and pTM641, were designed to express the L, Lcys and LΔ6-67 proteins, respectively. A second series of plasmids was designed to express fusion protein between the leader variants and eGFP (pCER01, pCER02, and pCER03). pCER03 encodes eGFP alone. In pCER01 (Lwt) and pCER02 (Lcys), the junction between L and eGFP encompasses codon 75-Pro, 76-Gln of L, a Val-Thr linker, and codons 4-Lys, 5-Gly of eGFP. A third series of plasmids was derived from pTM624. This vector contains a CMV promoter, a multicloning site, an IRES corresponding to nucleotides 392 to 1065 of TMEV (strain DA1), driving the translation of eGFP (unpublished). These plasmids co-express the L variants and eGFP. The matrix (M) protein coding region of Vesicular Stomatitis Virus (VSV) was PCR amplified with primers TM822 and TM823, from cDNA generated from VSV (strain Indiana)-infected L929 cells and cloned in the pTM624 bicistronic construct. Plasmid pCS41 is a pcDNA3 derivative expressing the firefly luciferase gene (Sommereyns et al., 2008). Table 1 summarizes the properties of the plasmids used in this study. Primers are presented in Table 2.
Table 1. Plasmids used.
| virus cDNA | Lwt | Lcys | LΔ6-67 |
|---|---|---|---|
| wild-type capsid | pTMDA1 | pTM598 | pTM564 |
| adapted to L929 cells | pKJ6 | pTM659 | pSB3 |
| L expression plasmids | Lwt | Lcys | LΔ6-67 |
|---|---|---|---|
| PCMV - L | pTM553 | pTM592 | pTM641 |
| PCMV - L-eGFP (fusion) | pCER01 | pCER02 | pCER03 * |
| PCMV - L-IRES-eGFP (bicistronic) | pTM625 | pTM626 | pTM624 * |
| Luc expression plasmid | |||
|---|---|---|---|
| PCMV - luciferase | pCS41 | ||
absence of L instead of LΔ6-67
Table 2. Primers used.
| Names * | Sequence (5′⇒ 3′) | Use |
|---|---|---|
| TM41 (s) | TCC GGA TCC GCC ACC ATG GCT TGC AAA CAT GGA TAC | mutagenesis |
| TM42 (as) | AAG ATC TAG ATC ACT GGG GTT CCA TGA CAA TAT C | mutagenesis |
| TM56 (as) | AAC GGC TGT GCG AAT AGT GCG CAC ATC TGG GT | mutagenesis |
| TM247 (s) | TAA TAC GAC TCA CTA TAG GGA | mutagenesis |
| TM580 (as) | CAT GGG TTA CCT GGG GTT CCA TGA CAA TA | mutagenesis |
| TM495 (s) | CAT GGT AAC CAA GGG CGA GGA GCT GTT | mutagenesis |
| TM496 (as) | AGT AAA ACC TCT ACA AAT GTG | mutagenesis |
| TM822 (s) | TCC GGA TCC GCC ACC ATG AGT TCC TTA AAG AAG AT | mutagenesis |
| TM823 (as) | AAG ATC TAG ATC ATT TGA AGT GGC TGA | mutagenesis |
| TM346 (s) | GCC GCT CTT CAC ACC CAT | qPCR virus |
| TM347 (as) | AGC AGG GCA GAA AGC ATC AC | qPCR virus |
| TM427 (s) | AAG AGT TGT GCA ATG GCA ATT CT | qPCR IL-6 |
| TM428 (as) | AAA TTT TCA ATA GGC AAA TTT CCT GAT | qPCR IL-6 |
| TM528 (s) | CTT CTG GGC CTG CTG TTC A | qPCR MCP-1 |
| TM529 (as) | CCA GCC TAC TCA TTG GGA TCA | qPCR MCP-1 |
| TM367 (s) | AGA CGG AAT ACA GGG CTT TCG | qPCR TGF-β |
| TM368 (as) | CAT GAG GAG CAG GAA GGG C | qPCR TGF-β |
| TM839 (s) | TCA AAG AGG CGA ACT GTG TG | qPCR luciferase |
| TM840 (as) | GGT GTT GGA GCA AGA TGG AT | qPCR luciferase |
s = sense; as = antisense
Capped RNA transcript synthesis
Capped luciferase mRNA was transcribed in vitro from plasmid pCS41, using T7 RNA polymerase (Roche) and Ribo m7G cap analog 0,5mM (Promega), in the conditions recommended by the latter supplier. To eliminate template plasmid DNA, the transcription reaction products were treated with DNase I as described (Shaw-Jackson & Michiels, 1999). We checked that residual contaminating DNA amounts, measured by real-time PCR, were not sufficient to generate detectable luciferase activity (data not shown).
DNA and RNA transfection
Plasmid DNA was transfected with TransIT-LT1 Transfection Reagent (Mirus) according to the manufacturer’s recommendations. 3 μl of transfection reagent were used with 1 μg of DNA to transfect about 300 000 cells grown in a well of a 24-well plate.
RNA/DNA mixtures were transfected with the TransIT-mRNA Transfection Kit (Mirus) in BALB/3T3 cells, at ratios of 1:9 or 1:39 (100 ng of the capped RNA transcript and 900 ng of plasmid DNA or 25 ng of the capped RNA transcript and 975 ng of plasmid DNA). 1 μl of boost reagent was mixed with 1,5 μl of transfection reagent and 1 μg of nucleic acid in 50 μl of DMEM. After 15 min of incubation at room temperature, the mixture was added on sub-confluent cells grown in 24 well-plates.
Oligo(dT) In Situ Hybridization (ISH)
BALB/3T3 were cultured on 13mm coverslips. At 50% of confluence, the cells were transfected with plasmid DNA expressing the L constructs. At chosen time after transfection, the cells were fixed for 8 min with paraformaldehyde 4% in phosphate buffered saline (PBS) and washed 3 times in PBS. The ISH protocol was from B. Fontoura (Chakraborty et al., 2006). The probe consisted in a 45-mer oligo(dT), biotinylated at its 3′ extremity and purified by reverse phase chromatography (Eurogentec). Streptavidine-Cy3 1/100 (Sigma) was used for detection. Coverslips were mounted in Mowiol (Mowiol 4-88 (Calbiochem; ref. 475904), 25% (m/v) glycerol, 0,1% (w/v) diazabicyclo-octane (Sigma; ref D2522) in 100 mM Tris-HCl, pH 8,5). Data were analyzed by conventional (Leica DM IRB) or confocal (Zeiss Axiovert 135M equipped with the Bio-Rad MRC1024 confocal device) fluorescent microscopy.
Immunoblotting (IRF-3 dimerization and Nup98 phosphorylation)
To analyze IRF-3 dimerization, L929 cells were infected for 9 hours with KJ6, TM659 or SB3, or were mock-infected. Proteins were extracted from the cells in non denaturing conditions and were separated on Native PAGE gels as described (Hato et al., 2007). For analysis of Nup98, BHK-21 cells were infected for 12 hours with KJ6 or TM659 or were mock-infected. Proteins were extracted using Tx lysis buffer as described previously (Park et al., 2008). Where indicated, 40 μg of protein was treated with 4U of calf alkaline phosphatase (Promega) in the manufacturer’s recommended buffer for 1 hour at 37 °C. Samples were analyzed by SDS PAGE followed by blotting with a rat monoclonal anti-nup98 antibody (2H10, SigmaAldrich).
Mice and infections
129/Sv IFNAR -/- mice (Muller et al., 1994) were bred at the animal facility of the University of Louvain. 129/Sv mice were purchased from the same source or from Charles River Laboratories. Handling of mice and experimental procedures were conducted in accordance with national and institutional guidelines for animal care and use. Groups of 4 (1 and 4 dpi) or 6 (5 dpi), 3 to 4 week-old mice were infected by intracranial injection of 40 μl of serum-free medium containing 105 PFU of the virus (129/Sv mice) or 103 PFU of virus (IFNAR -/- mice). Control mice were injected with 40 μl of serum-free culture medium. Mice were anesthetized with xylazine and ketamine before sacrifice for organ harvest.
Real-time quantitative RT-PCR
Total RNA preparations, reverse transcription and real-time PCR reactions were performed as previously described (Paul & Michiels, 2006). Real-time PCR standards consisted of 10-fold dilutions of known concentrations of plasmids carrying the sequence to be amplified: pcDNA3-IFN-β (van Pesch et al., 2004) or genomic DNA for IFN-β, pTM410 for TMEV, pTM849 for MCP-1, pTM793 for β-actin, pTM799 for RANTES, pTM844 for IL-6, pTM842 for TGF-β and pCS41 for luciferase. Plasmids pTM793, pTM799, pTM842, pTM844, pTM849 are derivatives of pCR4-ToPo (Invitrogen) in which the corresponding PCR fragments were cloned. Sense (s) and antisense (as) primers used for IFN-β and MCP-1 amplification were from Petro et al. (Petro, 2005) and Overbergh et al. (Overbergh et al., 2003), respectively. Primer sequences are given in Table 2.
Results
The leader protein shuts off the synthesis of cellular proteins
We first confirmed L-mediated shut-off of protein expression by testing reporter protein levels in BALB/3T3 cells transfected with plasmids encoding either wild-type or mutant L-eGFP protein fusions or with plasmids encoding bicistronic L-IRES-eGFP constructs. 24 hours after transfection, cells expressing the Lwt-eGFP fusion showed significantly less fluorescence than cells expressing Lcys-eGFP (5-fold) or LΔ-eGFP (16-fold), in a representative triplicate experiment. Mean fluorescence of cells transfected with the bicistronic plasmid encoding Lwt was also more than 10 times lower than that of cells that received bicistronic constructs expressing Lcys or LΔ, confirming inhibition of eGFP expression by the leader protein (Fig. 1A).
Figure 1. Post-transcriptional shut-off of cellular protein synthesis by the TMEV leader protein.
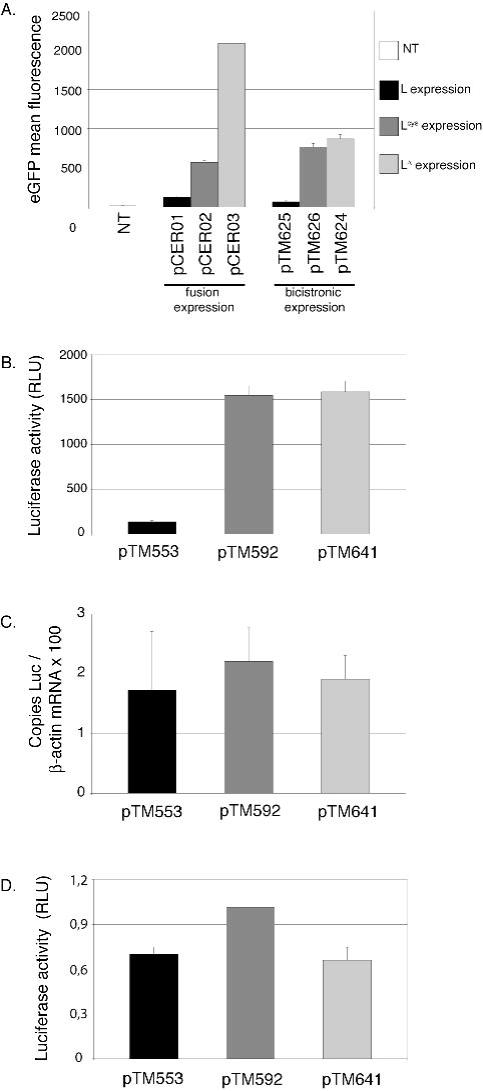
BALB/3T3 cells were transfected with constructs encoding L, Lcys and LΔ. Histograms show the means and Standard Deviation of one representative experiment performed in triplicate. A. eGFP mean fluorescence measured by FACS, 24 hours after transfection of the plasmids encoding the L-eGFP fusions or the L-IRES-eGFP constructs. (NT: non transfected cells) B. Luciferase activity (relative luciferase units) measured 7 hours after co-transfection of cells with a plasmid expressing luciferase (pCS41) and either L (pTM553), Lcys (pTM592) or LΔ6-67 (pTM641). C. Amount of luciferase mRNA, measured by real-time RT-PCR in cells co-transfected as described in B. Values were normalized to β-actin. Note that no effect of L on β-actin mRNA levels was detected at that time point. D. Leader does not affect translation of cytoplasmic mRNA. BALB/3T3 cells were co-transfected with luciferase capped RNA and with plasmid DNA (ratio 1/10) coding for L (pTM533), Lcys (pTM592) and LΔ6-67 (pTM641). Histograms show luciferase activity, measured 14 hours after co-transfection.
To detect shut-off at earlier time points after transfection, we used the more sensitive luciferase reporter system. We co-transfected the firefly luciferase expression vector (pCS41) and either the L, Lcys or LΔ6-67 expression vectors into BALB/3T3 cells and assayed luciferase activity (Fig. 1B). Luciferase activity measured 7 hours after transfection was 12-fold lower in cells expressing L than in cells expressing Lcys or LΔ6-67 (Fig. 1B). Interestingly, luciferase mRNA levels, measured by real-time RT-PCR, varied less than 1.3-fold between cells expressing mutated or wild-type leader proteins (Fig. 1C). We concluded that shut-off of luciferase expression by the L protein occurred at a post-transcriptional level.
The leader protein does not interfere with cytoplasmic luciferase mRNA translation
To determine if L acted at the translational level, capped luciferase mRNA was synthesized in vitro and co-transfected into BALB/3T3 cells along with plasmids encoding the Lwt, Lcys or LΔ6-67 proteins. To ensure that cells receiving luciferase mRNA also received the L expression plasmid, luciferase mRNA and plasmid DNA were transfected at ratios of 1:9 or 1:39. Luciferase activity was measured at 7, 12 or 14 hours post-transfection (Fig. 1D and data not shown). No clear inhibition of luciferase expression by the L protein was observed at any time point in any of four independent experiments involving two productions of luciferase capped mRNA and three different plasmid DNA preparations. In addition, no luciferase activity inhibition occurred in cells that were co-transfected with a 1:9 ratio of mRNA encoding luciferase and L, respectively (data not shown). Thus, L protein of TMEV acted on luciferase expression at a post-transcriptional stage, but upstream of mRNA translation.
The leader protein interferes with cellular mRNA export and promotes Nup98 phosphorylation
Porter et al. proposed that the L protein of EMCV inhibited mRNA export from the nucleus (Porter et al., 2006).To examine the effect of TMEV L on mRNA export in a direct manner, we used in situ hybridisation (ISH) with an oligo(dT) probe to localise the main pool of polyA+ RNA in cells transfected with plasmids expressing the Lwt, Lcys or LΔ - proteins or with the bicistronic plasmids co-expressing these proteins and eGFP (Fig. 2A). As a control, we transfected pCER26, a bicistronic construct that encodes the matrix (M) protein of vesicular stomatitis virus (VSV) which has been shown to block mRNA export (Her et al., 1997). Non-transfected cells rarely showed nuclear retention of polyA+ RNA. In contrast, 17,3% of the cell population transfected with the VSV-M expressing plasmid exhibited a clear nuclear mRNA accumulation. Similarly, 14% and 9,3% of cells transfected with the monocistronic (pTM533) and bicistronic (pTM625) plasmids expressing the leader protein, respectively, showed nuclear retention of polyA+ RNA (Fig 2A). In contrast, less than 2% of cells transfected with plasmids expressing the corresponding leader deletion mutants (pTM641 and pTM624) exhibited a nuclear signal. The cell population transfected with plasmids expressing the Lcys mutant (pTM592 and pTM626) exhibited a variable and intermediate phenotype.
Figure 2. The leader promotes nuclear retention of cellular polyA+ RNA and triggers phosphorylation of Nup98.
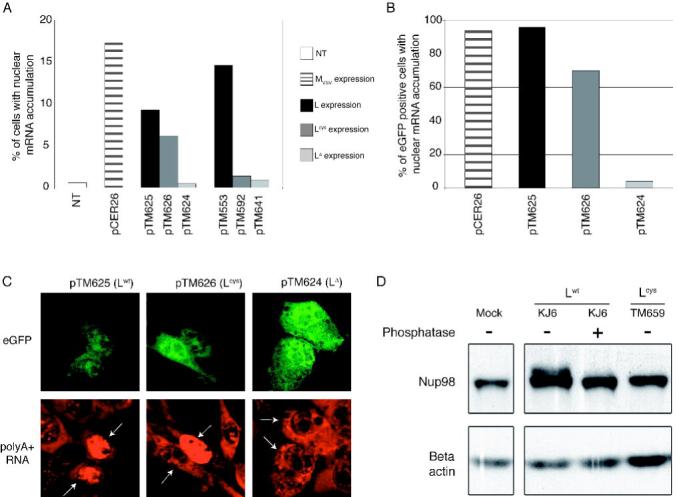
A. BALB/3T3 cells were transfected with L, Lcys or LΔ6-67 expression plasmids (pTM553, pTM592 and pTM641, respectively) or with the bicistronic constructs co-expressing these proteins and eGFP (pTM625, pTM626 and pTM624, respectively). The bicistronic construct expressing VSV M protein (pCER26) was used as control. PolyA+ RNA was detected by fluorescent in situ hybridization using an oligo(dT) probe. The percentage of cells showing higher fluorescent intensity in the nucleus than in the cytoplasm at 16 hours after transfection is shown. (NT: non transfected cells). B. Percentage of cells showing higher fluorescent intensity in the nucleus, among 50 eGFP positive cells. C. Representative ISH results in cells transfected with the bicistronic constructs, showing eGFP fluorescence (above) and detection of polyA+ RNA by ISH (below). In the latter panel, white arrows point to eGFP positive cells. D. Phosphorylation of Nup98 in cells infected with TMEV. BHK-21 cells were infected for 12 hours with either KJ6 or TM659 or were mock-infected. Lysates were prepared and analyzed by SDS PAGE and immunoblotting to detect Nup98. Phosphatase indicates whether protein extracts were treated with calf alkaline phosphatase.
Although the leader protein and, to a lesser extent, the Lcys mutant inhibited eGFP expression (Fig. 1A), using confocal microscopy, it was possible to detect eGFP-positive cells and to evaluate the percentage of cells displaying nuclear polyA+ RNA retention among cells that expressed the bicistronic construct (Fig. 2B-2C). Cells expressing LΔ (pTM624) displayed a mostly cytoplasmic polyA+ RNA staining, as typically observed in non-transfected cells. In contrast, nearly 100% of the cells expressing the TMEV leader (pTM625) displayed nuclear accumulation of cellular polyA+ RNA, very similar to the results of cells expressing the VSV M protein (pCER26). Surprisingly, 70% of cells expressing the Lcys mutant (pTM626) showed nuclear accumulation of cellular polyA+ RNA, in spite of the disruption of the Zn finger in this mutant protein (Fig. 2B). This experiment shows that the L protein of TMEV provokes a nuclear retention of cellular poly(A)-mRNA. L-mediated nuclear accumulation of mRNA became detectable from 8 hours post-transfection and increased with time (data not shown).
The VSV M protein inhibits mRNA export by forming a complex with the transport factor Rae1 and with Nup98 (Faria et al., 2005), a component of the nuclear pore complex that is thought to play a key role in mRNA export (Powers et al., 1997). To determine if TMEV L might also target Nup98 to inhibit mRNA export, we examined the status of Nup98 in cells infected with TMEV encoding wild-type L or the Lcys mutant. Figure 2D shows that 12 hours following infection with wild-type TMEV (KJ6) a slower migrating form of Nup98 was apparent in cell lysates. This result is likely due to phosphorylation as treatment with alkaline phosphatase converted Nup98 to the single faster migrating form. Phosphorylation of Nup98 required a functional L protein, as infection with the Lcys mutant (TM659) had no effect on the migration of Nup98. These results suggest that inhibition of mRNA export by L may be mediated by phosphorylation of Nup98.
The leader protein inhibits cytokine and chemokine gene transcription
The lack of transcriptional inhibition of luciferase mRNA reported here contrasts with the transcriptional inhibition of type I IFN genes reported previously (van Pesch et al., 2001; van Pesch & Michiels, 2003). To confirm IFN gene transcription inhibition and to test whether transcriptional inhibition might involve other genes, we compared the mRNA levels of selected cytokine and chemokine genes, in L929 cells infected with viruses expressing wild-type or mutated L proteins. We studied the transcription of two chemokine genes: RANTES (=Regulated on Activation Normal T cell Expressed and Secreted or CCL5) and MCP-1 (=Monocyte chemoattractant protein-1 or CCL2), and of two cytokine genes, IL-6 (=Interleukin 6) which is pro-inflammatory and TGF-β =Transforming Growth Factor-β) which is anti-inflammatory. IFN-β was tested as a control to confirm previous data (Fig. 3). In the conditions used, the viruses infected more than 95% of the cells as measured by immunohistochemistry (data not shown). 9 hours after infection, gene transcription of IFN-β RANTES, IL-6, MCP-1 and TGF-β was upregulated in cells infected with the Lcys and LΔ6-67 mutant viruses but not in cells infected with virus KJ6 expressing Lwt (Fig. 3). However, induction levels differed strongly between the genes analyzed, being in the range of 3000 and 300-fold for IFN-β and RANTES respectively, and in the range of 30 and 10-fold for IL-6 and MCP-1 mRNA. Upregulation of TGF-β transcription was minimal.
Figure 3. Influence of the TMEV leader on the transcription of cytokine and chemokine genes.
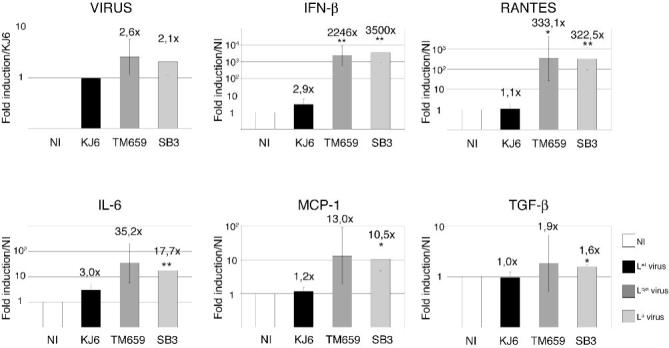
A. L929 cells were infected for 9 hours with 2 PFU per cell of either KJ6 (Lwt) (black), TM659 (Lcys) (medium grey), or SB3 (LΔ6-67) (light grey), or were mock-infected (NI) (white). mRNA levels, detected by comparative real-time RT-PCR, were normalized to the amounts of β-actin for each sample. Histograms show the means and SD of relative gene expressions. Ratios to KJ6 infected samples are shown in the case of viral RNA. Ratios to mock-infected cells are shown for other genes. * and ** indicate significant differences with the corresponding KJ6-infected sample (p values < 0.05 and <0.01 respectively, determined in an unpaired t-test).
These data suggest that, in addition to the post-transcriptional effect of the L protein on mRNA nuclear export, the L protein acts at the transcriptional level on the expression of cytokine and chemokine genes that are typically activated in response to viral infection. The effect was particularly prominent in the case of IFN-β and RANTES genes whose transcription strongly depends on the IRF-3 transcription factor.
The leader protein inhibits IRF-3 dimerization
We next analyzed whether L protein-mediated transcriptional inhibition of cytokine and chemokine genes could result from inhibition of IRF-3 activation. IRF-3 dimerization was examined in cells infected with TMEV encoding wild-type and mutant L proteins. As shown on Fig. 4, IRF-3 dimers were detected in cells infected with the L-mutant viruses (Lcys or LΔ6-67). In contrast, IRF-3 was almost entirely monomeric in non-infected cells or in cells infected with the virus expressing Lwt. Thus, viral infection can trigger IRF-3 dimerization but this dimerization is impaired by the L protein.
Figure 4. Inhibition of IRF-3 dimerization.
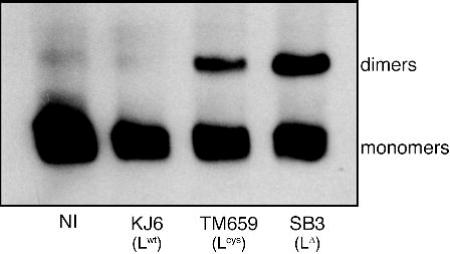
L929 cells were infected for 9 hours with either KJ6 (L), TM659 (Lcys), or SB3 (LΔ6-67), or were mock-infected (NI). Protein extracts were separated by acrylamide gel electrophoresis under non-denaturing conditions and IRF-3 dimer formation was detected by immunoblotting.
Influence of the leader protein on mouse infection
To determine the biological relevance of L protein functions, and in particular, the importance of IFN production inhibition by the leader, we compared the replication of the wild-type DA1 virus and of the Lcys TM598 mutant in the CNS of wild-type and IFNAR -/- 129/Sv mice (Fig. 5). As reported previously IFNAR -/- mice were extremely susceptible to TMEV infection (Fiette et al., 1995). After inoculation of 103 PFU of DA1 or TM598, IFNAR -/- mice rarely survived more than 4 days post-infection. At this time point, viral load analyzed by RT-PCR in the brain of these mice, was between 50 and 100-fold higher than in the brain of wild-type mice despite the fact that the latter mice had been inoculated with 100-fold higher virus doses (Fig. 5). In IFNAR -/- mice, the Lcys mutant virus replicated as well as the wild-type virus. In contrast, in interferon-competent mice, mutation of the leader protein led to a 3,75-fold decrease of viral replication at day 4 post-inoculation (Fig. 5A central panel). This replication difference between the Lcys mutant virus and the WT virus increased with time and became significant at 5 days post-inoculation (Fig. 5A, right panel). The fact that the Lcys mutation affected viral yield in WT mice but not in IFNAR-KO mice suggests that, at least during the first days of the infection, the leader protein acts to counteract the IFN-α β response.
Figure 5. Comparison of viral load and IFN-β and RANTES gene expression in brains of infected mice.
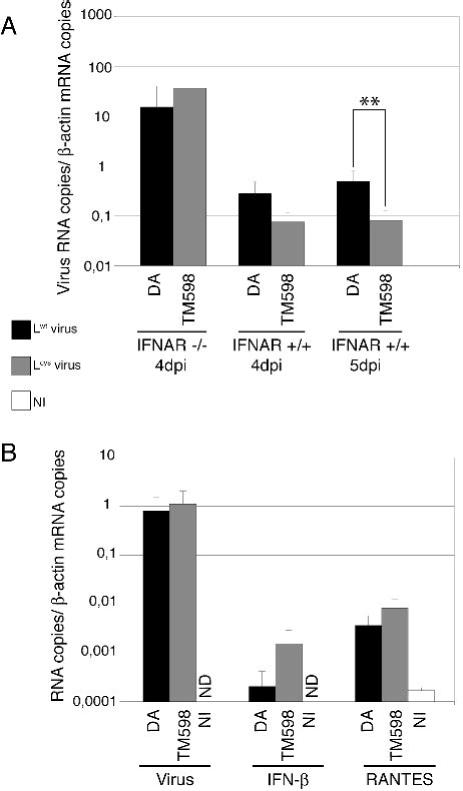
Real-time quantitative PCR was used to measure viral RNA or IFN-β and RANTES mRNA in total RNA extracted from the brain of infected mice. A. Comparison of Lwt (DA1) and Lcys (TM598) viral RNA amounts (mean and SD) in the brains of IFNAR -/- or IFNAR +/+ mice, 4 and 5 days post-infection. B. Viral genomes, RANTES mRNA and IFN-β mRNA levels measured in the brain of IFNAR -/- mice infected with Lwt (DA1) and Lcys (TM598) viruses, or mock-infected (NI), 1 day post-inoculation. ND indicates that the cDNA copy number was under the detection threshold (<10-4 copies per copy of β-actin cDNA). **: t test, p < 0.01.
We next compared the abundance of IFN-β and RANTES transcripts in the CNS of IFNAR-/- mice, 1 day after infection with the DA1 and TM598 viruses. IFNAR -/- mice were chosen for this analysis to minimize the effect of cell priming by IFN and because the Lcys mutant and WT viruses replicated to similar levels in these mice.
IFN-β and RANTES transcription was induced in brains of mice infected by both DA1 and TM598 viruses. As discussed below, IFN-β and RANTES transcripts were only slightly more abundant in mice infected by the Lcys mutant than in mice infected by the wild-type virus (6.5 and 2.1 fold)(Fig. 5B). Thus, the inhibition of cytokine and chemokine gene transcription observed in vitro was less extensive in vivo.
Discussion
The leader protein of TMEV can trigger a global shut-off of protein expression. Interestingly, this global shut-off, assessed by inhibition of eGFP and luciferase expression, turned out to be neither transcriptional nor translational. Instead, using ISH with an oligo(dT) probe, we showed in a direct manner that the TMEV leader inhibited export of cellular polyA+ RNA from the nucleus. Such nuclear retention of mRNA could result from L-mediated inhibition of RAN GTPase activity (Porter et al., 2006). Although RAN is not directly involved in mRNA export, RAN inhibition might affect shuttling of proteins such as Dbp5p or TAP/NXF1 (export receptor) or Aly/REF (export adaptor) (Rodriguez et al., 2004), that mediate mRNA export from the nucleus. However, our data reveal that the TMEV leader also triggers phosphorylation of Nup98. This nucleoporin notably interacts with TAP/NXF1 and RAE1, factors involved in mRNA export (Pritchard et al., 1999; Blevins et al., 2003). Thus, our observation that the TMEV L protein mediates phosphorylation of Nup98, suggests another possible mechanism whereby L could inhibit mRNA export by modulating the activity of components of the nuclear pore complex.
We previously reported that type I IFN and RANTES gene expression were inhibited by the TMEV leader (van Pesch et al., 2001; Paul & Michiels, 2006). Here, we show that expression of other cytokine and chemokine genes including MCP-1 and IL-6 is also inhibited by L. In contrast to the post-transcriptional shut-off described here-above, L-mediated inhibition of cytokine and chemokine gene expression occurred at the transcriptional level. Transcription of RANTES and IFN-β, which was the most severely affected by the L protein, is known to be upregulated by IRF-3 (Schafer et al., 1998; Lin et al., 1999a). Upon viral infection, IRF-3 is phosphorylated by TBK-1 and IKK-ε kinases. This induces IRF-3 dimerization and translocation into the nucleus (Yoneyama et al., 1998; Lin et al., 1999a; Lin et al., 1999b; Fitzgerald et al., 2003; Sharma et al., 2003) where IRF-3 associates with other transcription factors and CBP/p300 on specific promoter motifs to activate cytokine and chemokine gene transcription (Panne et al., 2007). As recently reported in the case of the EMCV leader (Hato et al., 2007), TMEV leader inhibited IRF-3 dimerization in infected cells (Fig 4). Intriguingly, IRF-3 dimerization is believed to take place in the cell cytoplasm and to precede nuclear translocation of this factor. Thus, whether inhibition of IRF-3 dimerization can be consequent to nucleocytoplasmic trafficking alteration deserves future investigations.
Mutation of the zinc-finger of the leader affected all the tested activities of this protein. A possible explanation is that disruption of the Zn finger modified the global fold of L, thus impacting the different functions of this protein. An alternative explanation is that the various functions of the leader might be coupled.
It is also interesting to note that not all functions of L were equally affected by the Zn finger disruption. This mutation had a high impact on cytokine gene transcription inhibition. It also almost fully prevented Nup98 phosphorylation. Yet, it only partially inhibited polyA+ RNA export from the nucleus (Fig 2). This raises the possibility that full inhibition of mRNA export by L is due to disruption of multiple factors in the mRNA export pathway. Alternatively, residual activity of the Lcys mutant might go undetected or become apparent according to the experimental settings.
In vivo, IFN-α/β are key components in the defence against viruses. IFNAR KO mice, that are unable to respond to type-I IFN, are extremely sensitive to a large panel of viruses and notably to TMEV (Fiette et al., 1995). Here, the importance of the IFN system in the CNS of mice was confirmed by the fact that survival of IFNAR KO mice was compromised as soon as 4 days after IC inoculation and that viral load at this time was impressive (more than 10 copies of viral RNA per copy of β-actin mRNA in the entire brain). We also tested the importance of the L protein in the antagonism of the IFN response in vivo. In IFN-competent mice, a Lcys mutant virus exhibited less fitness than the WT virus. In contrast, in the IFNAR KO mice, the mutant virus replicated at least as well as the WT virus (Fig 5A). The fact that the Lcys mutation affected viral yield in WT but not in IFNAR-KO mice suggests that, in vivo, antagonism of the IFN system is an important function of the L protein, at least during the first days of the infection. However, in contrast to the in vitro situation, inhibition of IFN-β mRNA expression by the leader protein was quite modest. This difference between the in vitro and the in vivo situations might be accounted for by the fact that, in vivo, only a small percentage of the cells are infected. On one hand, this allows priming of some cells by IFN or by other cytokines before their infection by virus. Such priming was shown to override the ability of L to block IFN production (van Pesch & Michiels, 2003). On the other hand, it was shown previously that, in the CNS of mice infected with TMEV or LaCrosse virus, a substantial part of IFN-α β producing cells were viral antigen-negative (Delhaye et al., 2006). Recent work notably showed that uptake of picornavirus-infected cells by dendritic cells allowed the activation of the IFN system (Kramer et al., 2008). Thus, non-infected cells likely detect viral replication products released by infected cells, possibly through TLR recognition. As L is not expressed in these cells, no inhibition of IFN production would be expected. Yet, even if IFN production inhibition in vivo is low, the importance of the leader as an IFN antagonist in vivo is underlined by its influence on viral replication in wild-type compared to IFNAR-KO mice. One might also consider that a small overall reduction in IFN production could hide more extensive local effects that can be physiologically relevant.
In conclusion, as other viral proteins (NS1 of influenza, M of VSV, 2A of polio, leader of FMDV...), the leader protein may be a multifunctional protein devoted to antagonize the cell and the host innate defences and in particular the type I IFN response. Two activities have now been identified for the Cardiovirus L protein. First, the L protein represses transcription of virus-induced genes by preventing IRF-3 dimerization. Second, by interacting with RAN GTPase (as shown for the EMCV leader) and/or by promoting phosphorylation of nuclear pore complex proteins (as shown for the TMEV leader), leader proteins would affect nucleocytoplasmic trafficking of proteins and mRNA export, thereby triggering a late global shut-off of protein expression. Whether these two activities of the leader proteins indeed represent different functions or whether ther are connected remains an open question that clearly deserves future investigations.
Acknowledgements
We thank Muriel Minet for expert technical assistance. We are grateful to André Tonon for help in FACS analysis, to Guy Warnier for help in animal care, and to Sofia Buonocore for pSB3 construction. We thank Beatriz Fontoura for communication of detailed ISH protocols. C.R. and B.M. were fellows of the Belgian FNRS (National Fund for Scientific Research). S.D. was the recipient of a FRIA fellowship. S.V.H. is supported by a mosaic grant from the Netherlands Organisation for Scientific Research (NWO-017.002.025). Work in the lab of T.M. was supported by the National Fund for Medical Scientific Research (FRSM), by Crédits aux chercheurs of the FNRS, by the FSR of the University of Louvain, by the Actions de Recherche Concertées, Communauté Française de Belgique. Work in the lab of F.J.M.v.K. was partly supported by grants from the NWO (NWO-VIDI-917.46.305) and the Beijerink Virology Fund from the Royal Netherlands Academy of Sciences. Work in the lab of K.E.G. was supported by ACS RSG109705, NIH AI059467 and AI064432.
Footnotes
Publisher's Disclaimer: This is an author manuscript that has been accepted for publication in Journal of General Virology, copyright Society for General Microbiology, but has not been copy-edited, formatted or proofed. Cite this article as appearing in Journal of General Virology. This version of the manuscript may not be duplicated or reproduced, other than for personal use or within the rule of ‘Fair Use of Copyrighted Materials’ (section 17, Title 17, US Code), without permission from the copyright owner, Society for General Microbiology. The Society for General Microbiology disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by any other parties. The final copy-edited, published article, which is the version of record, can be found at http://vir.sgmjournals.org, and is freely available without a subscription.
References
- Baida G, Popko B, Wollmann RL, Stavrou S, Lin W, Tretiakova M, Krausz TN, Roos RP. A subgenomic segment of Theiler’s murine encephalomyelitis virus RNA causes demyelination. J Virol. 2008;82:5879–5886. doi: 10.1128/JVI.02432-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blevins MB, Smith AM, Phillips EM, Powers MA. Complex formation among the RNA export proteins Nup98, Rae1/Gle2, and TAP. J Biol Chem. 2003;278:20979–20988. doi: 10.1074/jbc.M302061200. [DOI] [PubMed] [Google Scholar]
- Brahic M, Bureau JF, Michiels T. The Genetics of the Persistent Infection and Demyelinating Disease Caused by Theiler’s Virus. Annu Rev Microbiol. 2005 doi: 10.1146/annurev.micro.59.030804.121242. [DOI] [PubMed] [Google Scholar]
- Chakraborty P, Satterly N, Fontoura BM. Nuclear export assays for poly(A) RNAs. Methods. 2006;39:363–369. doi: 10.1016/j.ymeth.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Chen HH, Kong WP, Roos RP. The leader peptide of Theiler’s murine encephalomyelitis virus is a zinc-binding protein. J Virol. 1995;69:8076–8078. doi: 10.1128/jvi.69.12.8076-8078.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels JB, Pappenheimer AM, Richardson S. Observations on encephalomyelitis of mice (DA strain) J Exp Med. 1952;96:517–530. doi: 10.1084/jem.96.6.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhaye S, van Pesch V, Michiels T. The leader protein of Theiler’s virus interferes with nucleocytoplasmic trafficking of cellular proteins. J Virol. 2004;78:4357–4362. doi: 10.1128/JVI.78.8.4357-4362.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhaye S, Paul S, Blakqori G, Minet M, Weber F, Staeheli P, Michiels T. Neurons produce type I interferon during viral encephalitis. Proc Natl Acad Sci U S A. 2006;103:7835–7840. doi: 10.1073/pnas.0602460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria PA, Chakraborty P, Levay A, Barber GN, Ezelle HJ, Enninga J, Arana C, van Deursen J, Fontoura BM. VSV disrupts the Rae1/mrnp41 mRNA nuclear export pathway. Molecular cell. 2005;17:93–102. doi: 10.1016/j.molcel.2004.11.023. [DOI] [PubMed] [Google Scholar]
- Fiette L, Aubert C, Muller U, Huang S, Aguet M, Brahic M, Bureau JF. Theiler’s virus infection of 129Sv mice that lack the interferon alpha/beta or interferon gamma receptors. J Exp Med. 1995;181:2069–2076. doi: 10.1084/jem.181.6.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- Ghadge GD, Ma L, Sato S, Kim J, Roos RP. A protein critical for a Theiler’s virus-induced immune system-mediated demyelinating disease has a cell type-specific antiapoptotic effect and a key role in virus persistence. J Virol. 1998;72:8605–8612. doi: 10.1128/jvi.72.11.8605-8612.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hato SV, Ricour C, Schulte BM, Lanke KH, de Bruijni M, Zoll J, Melchers WJ, Michiels T, van Kuppeveld FJ. The mengovirus leader protein blocks interferon-alpha/beta gene transcription and inhibits activation of interferon regulatory factor 3. Cell Microbiol. 2007;9:2921–2930. doi: 10.1111/j.1462-5822.2007.01006.x. [DOI] [PubMed] [Google Scholar]
- Her LS, Lund E, Dahlberg JE. Inhibition of Ran guanosine triphosphatase-dependent nuclear transport by the matrix protein of vesicular stomatitis virus. Science. 1997;276:1845–1848. doi: 10.1126/science.276.5320.1845. [DOI] [PubMed] [Google Scholar]
- Jnaoui K, Michiels T. Adaptation of Theiler’s virus to L929 cells: mutations in the putative receptor binding site on the capsid map to neutralization sites and modulate viral persistence. Virology. 1998;244:397–404. doi: 10.1006/viro.1998.9134. [DOI] [PubMed] [Google Scholar]
- Kong WP, Roos RP. Alternative translation initiation site in the DA strain of Theiler’s murine encephalomyelitis virus. J Virol. 1991;65:3395–3399. doi: 10.1128/jvi.65.6.3395-3399.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong WP, Ghadge GD, Roos RP. Involvement of cardiovirus leader in host cell-restricted virus expression. Proc Natl Acad Sci U S A. 1994;91:1796–1800. doi: 10.1073/pnas.91.5.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer M, Schulte BM, Toonen LW, Barral PM, Fisher PB, Lanke KH, Galama JM, van Kuppeveld FJ, Adema GJ. Phagocytosis of picornavirus-infected cells induces an RNA-dependent antiviral state in human dendritic cells. J Virol. 2008;82:2930–2937. doi: 10.1128/JVI.02376-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidsky PV, Hato S, Bardina MV, Aminev AG, Palmenberg AC, Sheval EV, Polyakov VY, van Kuppeveld FJ, Agol VI. Nucleocytoplasmic traffic disorder induced by cardioviruses. J Virol. 2006;80:2705–2717. doi: 10.1128/JVI.80.6.2705-2717.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Heylbroeck C, Genin P, Pitha PM, Hiscott J. Essential role of interferon regulatory factor 3 in direct activation of RANTES chemokine transcription. Mol Cell Biol. 1999a;19:959–966. doi: 10.1128/mcb.19.2.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Mamane Y, Hiscott J. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol Cell Biol. 1999b;19:2465–2474. doi: 10.1128/mcb.19.4.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister A, Tangy F, Aubert C, Brahic M. Molecular cloning of the complete genome of Theiler’s virus, strain DA, and production of infectious transcripts. Microb Pathog. 1989;7:381–388. doi: 10.1016/0882-4010(89)90041-7. [DOI] [PubMed] [Google Scholar]
- Michiels T, Dejong V, Rodrigus R, Shaw-Jackson C. Protein 2A is not required for Theiler’s virus replication. J Virol. 1997;71:9549–9556. doi: 10.1128/jvi.71.12.9549-9556.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Overbergh L, Giulietti A, Valckx D, Decallonne R, Bouillon R, Mathieu C. The use of real-time reverse transcriptase PCR for the quantification of cytokine gene expression. J Biomol Tech. 2003;14:33–43. [PMC free article] [PubMed] [Google Scholar]
- Panne D, Maniatis T, Harrison SC. An atomic model of the interferon-beta enhanceosome. Cell. 2007;129:1111–1123. doi: 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park N, Katikaneni P, Skern T, Gustin KE. Differential targeting of nuclear pore complex proteins in poliovirus-infected cells. J Virol. 2008;82:1647–1655. doi: 10.1128/JVI.01670-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Michiels T. Cardiovirus leader proteins are functionally interchangeable and have evolved to adapt to virus replication fitness. J Gen Virol. 2006;87:1237–1246. doi: 10.1099/vir.0.81642-0. [DOI] [PubMed] [Google Scholar]
- Petro TM. ERK-MAP-kinases differentially regulate expression of IL-23 p19 compared with p40 and IFN-beta in Theiler’s virus-infected RAW264.7 cells. Immunol Lett. 2005;97:47–53. doi: 10.1016/j.imlet.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Porter FW, Bochkov YA, Albee AJ, Wiese C, Palmenberg AC. A picornavirus protein interacts with Ran-GTPase and disrupts nucleocytoplasmic transport. Proc Natl Acad Sci U S A. 2006;103:12417–12422. doi: 10.1073/pnas.0605375103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers MA, Forbes DJ, Dahlberg JE, Lund E. The vertebrate GLFG nucleoporin, Nup98, is an essential component of multiple RNA export pathways. The Journal of cell biology. 1997;136:241–250. doi: 10.1083/jcb.136.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard CE, Fornerod M, Kasper LH, van Deursen JM. RAE1 is a shuttling mRNA export factor that binds to a GLEBS-like NUP98 motif at the nuclear pore complex through multiple domains. The Journal of cell biology. 1999;145:237–254. doi: 10.1083/jcb.145.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez MS, Dargemont C, Stutz F. Nuclear export of RNA. Biol Cell. 2004;96:639–655. doi: 10.1016/j.biolcel.2004.04.014. [DOI] [PubMed] [Google Scholar]
- Schafer SL, Lin R, Moore PA, Hiscott J, Pitha PM. Regulation of type I interferon gene expression by interferon regulatory factor-3. J Biol Chem. 1998;273:2714–2720. doi: 10.1074/jbc.273.5.2714. [DOI] [PubMed] [Google Scholar]
- Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- Shaw-Jackson C, Michiels T. Absence of internal ribosome entry site-mediated tissue specificity in the translation of a bicistronic transgene. J Virol. 1999;73:2729–2738. doi: 10.1128/jvi.73.4.2729-2738.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS pathogens. 2008;4 doi: 10.1371/journal.ppat.1000017. e1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theiler M GS. Encephalomyelitis of mice. I. Characteristics and Pathogenesis of the Virus. J Exp Med. 1940;72:49–67. doi: 10.1084/jem.72.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eyll O, Michiels T. Non-AUG-initiated internal translation of the L* protein of Theiler’s virus and importance of this protein for viral persistence. J Virol. 2002;76:10665–10673. doi: 10.1128/JVI.76.21.10665-10673.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Pesch V, van Eyll O, Michiels T. The leader protein of Theiler’s virus inhibits immediate-early alpha/beta interferon production. J Virol. 2001;75:7811–7817. doi: 10.1128/JVI.75.17.7811-7817.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Pesch V, Michiels T. Characterization of interferon-alpha 13, a novel constitutive murine interferon-alpha subtype. J Biol Chem. 2003;278:46321–46328. doi: 10.1074/jbc.M302554200. [DOI] [PubMed] [Google Scholar]
- van Pesch V, Lanaya H, Renauld JC, Michiels T. Characterization of the murine alpha interferon gene family. J Virol. 2004;78:8219–8228. doi: 10.1128/JVI.78.15.8219-8228.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. Embo J. 1998;17:1087–1095. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoll J, Galama JM, van Kuppeveld FJ, Melchers WJ. Mengovirus leader is involved in the inhibition of host cell protein synthesis. J Virol. 1996;70:4948–4952. doi: 10.1128/jvi.70.8.4948-4952.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoll J, Melchers WJ, Galama JM, van Kuppeveld FJ. The mengovirus leader protein suppresses alpha/beta interferon production by inhibition of the iron/ferritin-mediated activation of NF-kappa B. J Virol. 2002;76:9664–9672. doi: 10.1128/JVI.76.19.9664-9672.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]