

Abstract
Gene duplication is a substrate of evolution. However, the relative importance of positive selection versus relaxation of constraints in the functional divergence of gene copies is still under debate. Plant MADS-box genes encode transcriptional regulators key in various aspects of development and have undergone extensive duplications to form a large family. We recovered 104 MADS sequences from the Arabidopsis genome. Bayesian phylogenetic trees recover type II lineage as a monophyletic group and resolve a branching sequence of monophyletic groups within this lineage. The type I lineage is comprised of several divergent groups. However, contrasting gene structure and patterns of chromosomal distribution between type I and II sequences suggest that they had different evolutionary histories and support the placement of the root of the gene family between these two groups. Site-specific and site-branch analyses of positive Darwinian selection (PDS) suggest that different selection regimes could have affected the evolution of these lineages. We found evidence for PDS along the branch leading to flowering time genes that have a direct impact on plant fitness. Sites with high probabilities of having been under PDS were found in the MADS and K domains, suggesting that these played important roles in the acquisition of novel functions during MADS-box diversification. Detected sites are targets for further experimental analyses. We argue that adaptive changes in MADS-domain protein sequences have been important for their functional divergence, suggesting that changes within coding regions of transcriptional regulators have influenced phenotypic evolution of plants.
Keywords: positive Darwinian selection, duplication, functional divergence, Arabidopsis thaliana, development
Gene duplication provides a substrate for evolution, and understanding the fate of duplicates is fundamental to clarifying mechanisms of genetic redundancy and the link between gene family diversification and phenotypic evolution (1). Several empirical studies have evaluated the roles of duplication in adaptation and diversification (2), but the evolutionary forces at play during functional divergence of duplicates are still under debate (3, 4). Genomic studies are revealing that eukaryotes harbor large families of genes that have arisen during evolution through duplication and have persisted for longer periods of time than expected by classical models (5). Models that incorporate positive selection (6, 7) provide alternative explanations for the persistence of duplicates.
Empirical studies to test models on the fate of duplicates and the evolutionary forces driving their functional divergence will need complete and resolved gene family phylogenies. Several studies suggest that positive Darwinian selection (PDS) might have been important in protein evolution. However, most previous studies have involved few members of a gene family from various species (8, 9). In this article, we annotate, align, and analyze the complete MADS-box gene family of the plant model system Arabidopsis thaliana and provide resolved phylogenies as a basis to infer the role of PDS in protein evolution in this gene family.
The detection of an excess in the ratio of the rate of nonsynonymous (dN) over the synonymous (dS) substitutions (that is dN/dS > 1; dN/dS is also denoted ω) is a nonambiguous indicator of PDS at the coding sequence level. Early studies estimated this ratio as an average over all codon sites within complete or partial sequence stretches and over the entire evolutionary time that separates the sequences compared. This method appears to be conservative because many sites might be under purifying selection because of functional constraint (10). However, in adaptive evolution in developmental regulatory loci, such as the MADS (11) PDS most likely occurs along particular lineages and at specific sites. In such cases, average dN/dS ratios over time and sites might not be significantly greater than 1, even if PDS has occurred.
MADS-box genes are present in plants, animals, and fungi, and previous studies suggested the existence of two main monophyletic lineages (type I and II) among all eukaryotes that probably derived from at least one duplication event before the divergence of plants and animals (14). The trees presented here recover Arabidopsis type II genes as a strongly supported monophyletic lineage, and the type I genes seem to be monophyletic but comprise several divergent sublineages.
MADS-box genes encode transcriptional regulators with diverse functions that could have been key during important events of plant diversification (12, 13). Hence, phylogenetic analyses of MADS-box genes are useful guides for studying their roles in plant evolution. Plant MADS-box gene phylogeny resolution, especially at its basal nodes, has been hindered by incomplete data and by limitations of inference methods (14, 15). Here we show resolved gene phylogenies of the Arabidopsis MADS-box genes.
More than half of the Arabidopsis MADS-box sequences are type I and only share with type II the MADS-box (14). All but one (16) functionally characterized plant MADS-box genes are type II and encode the three floral homeotic functions of the flower development ABC model (17-19). They also encode regulators of flower initiation, flower meristem identity, and various aspects of ovule, fruit, leaf, and root development (11, 20-23). All characterized plant type II MADS-box genes encode proteins that share a stereotypical MIKC structure, with highly conserved MADS and K domains that are putative DNA-binding and protein-protein interaction domains, respectively, and less conserved I and COOH regions.
We show here that sequences of type I and II have contrasting gene structure and chromosome distribution, supporting the idea that these two lineages had different evolutionary histories with a contrasting role of PDS. These contrasting histories also support placing the root of the family tree between the two lineages. Indeed, we found a significant role of PDS at fixing specific residues within the MADS-domain after different duplications in the type I lineage, but in the type II lineage we found evidence of PDS only with branch-site models along specific lineages. We addressed whether PDS played a significant role during the evolution of genes that regulate the transition to flowering, a trait that is clearly linked to plant fitness. Our findings identify target proteins and residues for future functional analyses and suggest that changes in coding sequences of transcriptional regulators, and not only in their regulatory regions, played important roles during phenotypic evolution.
Materials and Methods
Sequences and Alignment. To detect putative MADS-box genes in the Arabidopsis genome, two tblastn searches were performed on the complete Arabidopsis database (Table 3, which is published as supporting information on the PNAS web site). Sequences were assigned to either type I or II based on exon number and on careful comparisons of their MADS boxes (14, 24). Type I and II were then aligned separately with clustalw (25) launched from bioedit (26) and hand-corrected by using published alignments as guides (refs. 27 and 28; details can be found in Supporting Materials and Methods, which is published as supporting information on the PNAS web site).
Phylogenetic Reconstruction. Bayesian phylogenetic analyses were performed on mrbayes 2.01 (29). All searches were started from a random tree, on four different Markov chains for 2,500,000 generations and saving every 100th tree. At convergence (≈10,000 generations), the first 15,000 trees were discarded and a consensus was built. Posterior Bayesian probabilities were used to evaluate branch support. According to the recommendations of Foster (www.bioinf.org/molsys/data/like.pdf), we used the GTR model with a substitution rate that varies in an intracodon position-specific manner (GTR + SS).
Statistical Tests for Positive Selection. We applied the approach of Yang and coworkers (9, 30) to test for positive selection. First, we ran a test for the existence of sites with dN/dS ratios >1 by using a likelihood ratio test (LRT) to compare a model that does not allow for sites with dN/dS >1 to a model that does. If the LRT was statistically significant, then we identified the sites that were under positive selection. We calculated the posterior probability (PP) that a site was drawn from a given dN/dS class. Sites with PP > 0.5 are reported but we focus on those with a PP > 0.95. The program HYPHY 0.901b (S. L. Kosakovsky-Pond and S. V. Muse, www.hyphy.org) was used. Models tested were M3, M2, and M8 vs. null models M0, M1, and M7, respectively. We used the codon substitution model of Goldman and Yang (31) and 10 classes in the gamma distribution of M7 and M8. To avoid false positives, sites detected by models M3, M2, and M8 were considered as bona fide results only if the same sites were detected with at least two of these models and in both cases the LRT result was significant (32). To further minimize false positives, we performed all analyses on unambiguous and compact alignments (available on request). Thus, our conclusions on the role of PDS are based on very conservative analyses.
Additionally, we performed the branch-site analyses of Yang and Nielsen by using model MB (PAML 3.13, http://abacus.gene.ucl.ac.uk/software/paml.html; refs. 33 and 34) at the basal branch of two clades of flowering-time type II genes [FLC- and SVP-like genes (35-37)]. We hypothesized that functional change would be important precisely at the origin of these clades that evolved a distinct function (details of procedures can be found in refs. 31, 33, and 34). We also performed site-branch analyses for the branch that leads to type I PHERES1 gene.
Gene conversion and concerted evolution may violate the assumptions of site-specific positive selection models. We used geneconv 1.81 (www.math.wustl.edu/~sawyer/geneconv/index.html) and MEGA 2.1 (38) on the alignments tested for PDS.
Results
Annotation, Nomenclature, Gene Structure, and Duplications. The list of 104 MADS-box sequences found in the Arabidopsis genome database (www.ncbi.nlm.nih.gov/blast/Genome/ara.html), given agamous-like (AGL) number, accession number, chromosome location, intron-exon structure, and type of duplication are shown in Table 3.
Type I and II genes have contrasting gene structure and chromosome location. Type I genes have always one or two exons, whereas type II genes have more than five, and typically six to eight, suggesting that these two types of genes have different predisposition to gain or loss of introns or, alternatively, a difference in exon shuffling in the building of both types of genes, perhaps since their origin. All type II genes have a clear MIKC structure, except AGL33 with a very short transcript and AGL30-related sequences that do not have a clear IKC structure but share conserved motifs in their MADS-boxes with the rest of the type II genes. Also, a coiled-coil domain similar to the K domain is inferred for at least AGL104 (data not shown).
We used chromosome map locations to make qualitative inferences on past duplication events during MADS-box gene family evolution. The distribution of type I sequences among the five chromosomes is distinct to that of type II genes. Whereas the former are concentrated in chromosome I and V (χ2 = 26.77; P < 0.001 rejects uniform distribution among chromosomes correcting with chromosome size), type II are uniformly distributed (χ2 = 2.62; P > 0.1) among chromosomes. Also, most type I genes can be traced to intrachromosomal duplications, whereas approximately half of type II genes seem to have originated from interchromosomal duplications. Interestingly, Lynch and Conery (39) have found that recent duplications happened more frequently within than between two chromosomes, suggesting that most type I genes diverged more recently than type II genes (40). Moreover, a survey of the A. thaliana paralogous blocks database (http://wolfe.gen.tcd.ie/athal/dup) indicates that there are more duplicates from the type II group that seem to have persisted than those from type I. Eighteen out of 26 type II sequences that are found in a nonspurious duplicated region had a close paralog in a sister region, whereas among type I genes only 2 out of 22 did. Among type II genes found in nonspurious duplicated blocks, 14 are found in interchromosomal duplications and 12 in intrachromosomal duplications, whereas among type I genes the corresponding numbers are 15 and 7. But when we consider gene pairs of terminal clades of the trees, 13 out of 18 type I gene pairs involve intrachromosomal duplications, whereas in the type II we found this to be true of 6 out of 15 gene pairs (Fig. 1).
Fig. 1.

A. thaliana MADS-box gene family Bayesian phylogeny. Numbers above or below branches represent Bayesian posterior probabilities of finding a given clade. Branch lengths are proportional to number of nucleotide substitutions. AGL105 was excluded.
MADS-Box Gene Family Phylogeny. To corroborate the monophyletic origin of the two lineages that we had previously proposed (14), we obtained trees that included 103 of the MADS-box sequences found in the A. thaliana genome (Fig. 1). The global phylogeny recovers the two lineages (types I and II) of MADS-box sequences as two monophyletic groups with both alignments used (see supporting information) if the tree root is placed between type I and II genes. However, type I genes are more divergent among them than type II genes (Tables 4 and 5, which are published as supporting information on the PNAS web site). Nonetheless conserved motifs after the MADS suggest that type I sequences are not pseudogenes, an idea supported by the recent characterization of PHERES1 (16).
AGL30 had been incorrectly assigned to type I. However, this and related genes seem to be divergent type II. This is supported by their affiliation to type II-like moss genes that bear K domains as well as by their exon number (27, 41), and by conserved MADS-box motifs with respect to other type II genes. In our global tree (Fig. 1), these genes are resolved in a different position to that in the type II tree (Fig. 2b). Nonetheless, this remaining ambiguity does not affect PDS analyses. We ran PDS analyses for MIKC genes with an alternative type II topology similar to that in Fig. 1 and results recover the same sites with high PP as those obtained with the topology shown in Fig. 2b (data available on request).
Fig. 2.

Type I and type II A. thaliana MADS-box Bayesian phylogenies. (a) Type I tree polarized with type II sequences. (b) Type II tree polarized with type I sequences. Numbers above or below branches represent posterior probabilities. Branch lengths are proportional to the number of nucleotide substitutions. Boxed letters identify clades in which site-specific tests of positive selection yielded statistically significant LRT results for at least two model comparisons and in which at least one of the models detected sites under PDS with PP > 0.90. Branches underlined with a broken line identify cases in which the branch-site analyses yielded significant PDS results. #, Excluded from the site-specific analyses.
Given the ambiguity of the alignment of sequences after the MADS-boxes of both lineages, we resolved the internal phylogenetic relationships of each lineage separately. For the type I lineage tree, we used type II sequences as outgroups and vice versa for type II lineage. In both cases, tree topologies were very similar if regions downstream of the MADS-boxes of outgroup sequences were assumed to be homologous to those of ingroup sequences or were displaced. In contrast to previously published phylogenies, the trees shown here for both lineages resolve the branching sequence of the monophyletic groups (Fig. 2).
In the type I lineage, several strongly supported monophyletic clades are resolved and these are confirmed in trees with various outgroups (data not shown). It is noteworthy that no type I sequences bear the IKC region typical of lineage II and that several of the previously identified (14) privative amino acids within the putative MADS-domain of this lineage are found in most available sequences. Two main monophyletic groups are distinguished within the type I, suggesting at least one ancestral duplication within this lineage. DNA sequences beyond the MADS-box in the AGL23-like sublineage are conserved within each small monophyletic group resolved within this sublineage but are very divergent among groups (Fig. 2a and Table 4). Within the AGL26-like genes, two groups are resolved [AGL26-like genes themselves and PHERES1-like genes that includes the first type I gene functionally characterized (16)] and within each a very high degree of conservation is found in the putative domains beyond the MADS (Fig. 2a and Table 4). Sisters to these groups, AGL47 and AGL82, are resolved but have divergent putative domains beyond the MADS.
In the type II lineage, which includes all of the MIKC genes functionally characterized up to now, several clades are resolved and all are well supported (Fig. 2b). It is noteworthy that the AGAMOUS clade has AGL12, which had been reported as root-specific (42), as its sister gene. Another important finding is the strong association of AGL79, expressed in roots (data not shown), with AP1, CAL, and FUL, which are well characterized flower development genes (43). Therefore, this tree suggests that not all monophyletic groups resolved include genes with similar expression patterns and functions as previously thought (15, 21), but formal and robust inferences on evolution of MADS-box gene expression and function will have to await more experimental data and the inclusion of genes from additional taxa.
Positive Selection in MADS-Box Gene Evolution. We compared Models M0 and M3 to evaluate whether there had been dN/dS ratio variation among codon positions below each node of type I and II trees from Fig. 2 (Tables 1 and 2). We found rate variation at deep, intermediate, and recent duplications of both type I and II lineages (data available on request).
Table 1. Sites under PDS in the A. thaliana MADS-box gene family: “Site-specific analyses”.
| Site-specific analyses (type I) | n | dN/dS (ω) under M0 |
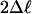 M3 vs. M0 (df LRT 3) M3 vs. M0 (df LRT 3) |
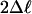 M2 vs. M1 (df 2) M2 vs. M1 (df 2) |
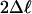 M8 vs. M7 (df 2) M8 vs. M7 (df 2) |
Parameter estimates (β and ω) under M8 β (p,q) | Positively selected sites under M3 | Positively selected sites under M2 | Positively selected sites under M8 |
|---|---|---|---|---|---|---|---|---|---|
| Node AG | 7 | 0.22 | 28.76*** | 10.10* | 10.57* | P1 = 0.98, ω = 8.44 β (0.87, 3,82) | 123 | No rate classes with dN/dS > 1 | 123 |
| Node AH | 7 | 0.26 | 59.09*** | 11.75** | 7.36* | P1 = 0.81, ω = 1.96 β (0.87, 3.48) | 73 7461 72 82 107 63 | 73 747261 82 63 107 | 73 72 7461 82 10763 48 58 81 110 |
| Node AL | 9 | 0.25 | 90.35*** | 18.39*** | 4.55 | P1 = 0.93, ω = 1.24 β (0.80, 2.28) | 61 72 73 82 107 110 48 74 8658 64 6663 65 109 | 72 73 827461 107 48 110 | 7273 8248 61 74 107 110 58 |
| Node AP | 12 | 0.25 | 130.47*** | 21.26*** | 7.59* | P1 = 0.75, ω = 1.18 β (0.56, 3.46) | 58 | 58 48 57 78 8972 12373 81 | 48 57 58 72 73 78 89 123 59 65 8147 6066 92 74 100 |
| Node AQ | 14 | 0.22 | 116.71*** | 14.97*** | 7.73* | P1 = 0.97, ω = 4.67 β (0.47, 1.52) | 5889 | 58 | 58 |
| Node AR | 20 | 0.15 | 102.65*** | 30.18*** | 8.22* | P1 = 0.96, ω = 1.12 β (1.64, 8.94) | No rate classes with dN/dS > 1. | 73 82 72 | 82 73 |
Each comparison has n sequences, dN/dS is average ratio over sites under a codon model with one ω. Proportion of the component of positively selected sites (P1) and parameters p and q of the beta distribution β(p,q) are given under M8. *, P < 0.5; **, P < 0.005; ***, P < 0.001; bold underlined, PP ≥ 0.99 of being under positive selection; bold, 0.99 > PP ≥ 0.95; italics, 0.95 > PP ≥ 0.90; underlined, 0.90 > PP ≥ 0.70; normal, 0.70 > PP ≥ 0.50.
Table 2. Sites under PDS in the A. thaliana MADS-box gene family: “Branch-site analyses”.
| Branch-site analyses | n |
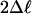 M3 (K = 2) vs. MB (df 2) M3 (K = 2) vs. MB (df 2) |
Parameter estimates under MB | Positively selected sites under MB |
|---|---|---|---|---|
| Type II | ||||
| Branch leading to the FLC-like genes | 39 | 20.50*** | P0 = 0.45, P1 = 0.38, (P2 + P3 = 0.17), ω0 = 0.06, ω1 = 0.36, ω2 = 4.47 | 42 56 59 154138 142 163 165 4 15 134 147 158 176 |
| Branch leading to the SVP-like genes | 39 | 13.13** | P0 = 0.44, P1 = 0.37, (P2 + P3 = 0.19), ω0 = 0.06, ω1 = 0.37, ω2 = 2.01 | 16 14426170 2 4 7 55 84 129 132 133 184 186 188 197 |
| Type I | ||||
| Branch leading to PHERESI (AGL37) | 48 | 10.10** | P0 = 0.35, P1 = 0.41, (P2 + P3 = 0.24), ω0 = 0.10, ω1 = 0.32, ω2 = 6.52 | 105 92 7212057 99 63 118 78 88 66 89 47 |
Each comparison has n sequences. Proportions of the component site classes 0 (P0), 1 (P1), and 2 + 3 (P2 + P3), as well as the values for the background ratios ω0 and ω1 and the foreground ratio ω2, are given under MB. **, P < 0.005; ***, P < 0.001; bold underlined, PP ≥ 0.99 of being under positive selection; bold, 0.99 > PP ≥ 0.95; italics, 0.95 > PP ≥ 0.90; underlined, 0.90 > PP ≥ 0.70; normal, 0.70 > PP ≥ 0.50.
Secondly, we applied the LRT to compare data fit to models M1 vs. M2 and M7 vs. M8 to address whether PDS promoted divergence of MADS-box genes below nodes and whether the action of selection has been heterogeneous among protein domains codified by these genes, using the trees of Fig. 2 (Tables 1 and 2 and Fig. 3). Below many of the deep nodes of type II tree, model M3 and at least one of M2 or M8 had significant LRT results (Fig. 2b). However, none of the sites with high PP were detected by more than one model comparison with significant LRT results (data available on request). In contrast, a similar analysis for type I lineage reveals several nodes below which specific sites appear to have been under PDS (Tables 1 and 2). For instance, in nodes AH and AL, positions 72-74 appear to have been under PDS with high PP, with position 72 showing five different amino acids for the seven sequences involved (Fig. 2a and Table 4). Positions 73 and 74 are part of the otherwise highly conserved “RQVTF” motif, and in the human serum response factor, position 72 has been shown to be involved in DNA contact (44). Below nodes AH and AR, position 82 was also found to be under PDS in two of the model pairs compared, although model M3 collapsed to only two rate classes. At this position, amino acid diversity is very high. For example, 10 different residues can be found for 20 sequences analyzed below node AR. In contrast, the homologous position of type II genes (position 26 in Table 4) shows only six different amino acids for 45 sequences.
Fig. 3.

Schematic representation of the distribution of sites under PDS in type I (a) and type II (b) sequences. MADS, I, K, and COOH domains are indicated. In a, the upper row corresponds to branch-site analysis along the branch leading to PHERES1, and the rest of the rows correspond, from top to bottom, to the site analyses performed below the nodes marked with the boxed letters AG, AH, AL, AP, AQ, and AR in Fig. 2a. In b, the upper row corresponds to branch-site analysis along the FLC branch and the lower row corresponds to branch-site analysis along the SVP branch. In a, the upper scale corresponds to amino acid position along the MADS domain and the lower scale corresponds to amino acid position along our alignment. In b, the scale corresponds to amino acid position along our alignment. Sites with PP > 0.70 are included. Shaded regions in b were excluded from PDS to avoid false positives. All sites are listed in Tables 1 and 2.
Strong evidence for positive selection in type I evolution was also found in less variable positions. This is the case for position 123 of node AG and position 58 of nodes AP and AQ. In position 123, there are only two different amino acids, although their distribution suggests that this site mutated twice during the history of descendants of node AG. In position 58, there are only five variable sites out of 14 sequences compared.
In the above analyses, PDS is detected at individual sites only if the average dN rate across lineages is higher than the average dS rate. This is observed mainly when recurrent positive selection occurs. However, PDS may change a few key residues of a protein but only at particular moments during its evolutionary history. In the latter case, detecting a significantly elevated dN would be hard if an average across-lineages estimate is considered. Thus, we applied the branch-site model of Yang and Nielsen (33) to test for PDS affecting individual sites along the branches leading to the flowering-time FLC- and SVP-like genes in type II lineage and along the branch leading to the only functionally characterized type I protein (PHERES1).
Parameter estimates suggest that in the basal branch of the FLC-like lineage, four residues were fixed by PDS with PP > 0.95 (Tables 1 and 2 and Fig. 3). Two of the residues were found within the MADS and correspond to amino acids that in other MADS-domain proteins participate in interactions between subunits (44-46). For example, site 42 is homologous to a site that in the myocyte enhancer factor-2 has been shown to intervene in subunit folding (45). Another site with high PP is 154. This site is within the K-box and has been shown to intervene in AP3/PI dimerization and determines functional specificity in AP1 and AG (47).
PDS seems to have been important also along the branch leading to the SVP-like genes, and we found two sites that appear to have been fixed by PDS (PP > 0.95). Site 16 is within the MADS-domain, and its homologous position in MEF2A plays a role in DNA-protein complex stabilization (46). Position 144 is found within the K-domain. SVP and AGL24 are the only type II proteins that have a lysine at that position.
Branch-site models also detected strong (PP > 0.95; see Tables 1 and 2) support for PDS along the branch leading to PHE1 at sites 92 and 72, which have been reported (44) to be important for α-β folding, and for position 105, which is involved in both dimerization and α-β folding.
Our analyses suggest that gene conversion or concerted evolution has not been prevalent during MADS-box genes evolution and does not bias PDS inferences. The overall modified Nei-Gojobori (48) means of synonymous-nonsynonymous differences were relatively high: 0.18 and 0.09 (transition/transversion ratio of 1.72 and 1.7, respectively) for types I and II, respectively (49). Gene conversion does not seem to have played significant roles in MADS-box evolution because only three conversion events were detected among type II genes, but none of these included genes for which we infer PDS and no conversion events were detected among type I sequences.
Discussion
We presented an annotated list of 104 MADS-box sequences from the complete A. thaliana genome database. Our phylogenetic analyses provide a resolved evolutionary hypothesis for the A. thaliana MADS-box gene family. This will be a useful reference for establishing orthology relationships, postulating functional hypotheses for uncharacterized MADS-box genes, and evaluating the role of MADS-box genes in plant morphological evolution.
The monophyly of the type II lineage is strongly supported in the present analyses, and type I comprises several sublineages with divergent putative domains after the MADS. However, previous analyses (14), as well as contrasting exon-intron structure and chromosomal distribution between type I and II sequences, still support the placement of the root between the type I and II genes in the tree of the complete gene family (Fig. 1). This tree hence resolves type I and II sequences in two monophyletic lineages. Nonetheless, genes from other plant, animal, and fungal species should be included in future analyses to trace MADS-box gene duplications with respect to taxa divergence and to specifically reevaluate the number of MADS-box gene duplications that occurred before the divergence of plants and animals (14). Such analyses will provide further evidence to reevaluate the monophyly of type I and II lineages.
Gene family structure has to be understood in the context of extensive gene duplications that have occurred in the evolutionary history of A. thaliana (40). Duplications leading to the chromosome stretches identified in the Arabidopsis Genome Initiative occurred 65 million years ago or before (refs. 50 and 51, but see ref. 52). Most retained groups within these stretches belong to type II genes, and duplications among type II seem to have been more ancient than those among type I, as suggested by their differential distribution among chromosomes. Retention due to a balance between genetic drift and mutation (5, 53) would depend on population characteristics (mainly effective population size) and hence would affect sequences of type I and II equally. But contrasting roles of selection between these two lineages could underlie the contrasting retention rates observed between them. The different evolutionary histories could have been determined by the fact that genes from these two lineages were recruited for different functions.
Interestingly, although duplications of type I seem to have occurred more recently than those leading to the type II lineage, type I sequences are more divergent among them in comparison to type II. This finding would suggest that whereas type II genes have been affected by sporadic PDS at the origin of new functions, followed by strong functional constraint, type I genes have been subject to recurrent events of PDS. In turn, this could be an indication that the functional roles of type I genes are overall distinct to those of type II genes. This pattern also allows us to put forward the hypothesis that type I orthologues from other taxa are less conserved than most type II orthologues.
Indeed, PDS analyses presented here suggest that type I and II lineages have been subject to overall contrasting selection regimes. We found that recurrent positive selection could have played a role in fixing specific amino acids after several duplication events during the evolution of type I genes. In contrast, analyses of site models did not provide strong evidence for PDS selection among type II genes. We had to use site-branch models to detect a role for PDS among type II genes. Indeed, we found evidence for PDS along the branches leading to the groups of genes that control flowering time that evolved a new function with respect to most other genes characterized up to now that are involved in cell- or organ-type specification. Indeed, probably by their control of life-history traits, flowering-time genes may have directly impacted plant fitness, and this could also explain the prevalence of positive selection during protein evolution among them.
Sites with high PP of having been fixed by natural selection in both lineages were found mainly in the MADS and K domains. However, our analyses are biased toward these domains because we only focused on conserved stretches that may be unambiguously aligned and excluded most variable domains. Future studies focusing on particular closely related genes for several species will be useful to address the role of PDS within COOH and other divergent domains. Indeed, a recent study showed that regions within the C-terminal domain determine functional specificity in AP3 and PI and may be relevant for floral organ evolution (54). The localization of the sites with high PP identified here suggest a role for PDS in MADS-domain protein diversification through interactions with protein partners and changes in affinity to binding motifs (46, 47, 55). Sites and genes identified here to have been under PDS become interesting targets for functional evaluations.
Evaluations of assumptions and predictions made by models of gene duplication and persistence will require phylogenetically driven analyses of functional and population level data for related genes in different species. The MADS-box gene family might become a good “model family” for such a purpose. For example, our site model analyses did not find that PDS played a role in the divergence of redundant AP1, CAL, and FUL (56). However, population-level data do suggest a role for positive selection in the divergence of these genes (57). More powerful analyses should be performed to rule out false negatives in our analyses due to low gene number (32). Moreover, our conclusions were based on conservative analyses and unambiguously aligned sequences to avoid false positives. Additional tests (data available on request; Fig. 2b) suggest that the role of PDS in MADS-box gene evolution might be more widespread. Other approaches (58) and the inclusion of sequences for additional taxa should be considered when further investigating the role of PDS in MADS evolution.
Our results suggest a role for positive selection during MADS-box evolution in plants. Previous studies have emphasized the role of changes in cis-regulatory regions of transcriptional regulators during plant evolution (59). Fewer recent studies, however, have also demonstrated that the evolution of transcriptional regulators' cDNA sequences played important roles in plant evolution (54). The detection of positive selection in MADS protein sequences that are developmentally important also indicates that changes in cDNA, and not only in the regulatory regions of these genes, have played a role in the evolution of plant body plans.
Supplementary Material
Acknowledgments
We shared unpublished results and agreed on AGL numbers with Lucia Colombo and her collaborators. We thank Lorenzo Segovia and Julio Collado for computer time. Discussions with Francisco Vergara-Silva were insightful, and Alejandra Vásquez-Lobo, Rodolfo Salas, and Lev Jardón made comments and helped with the figures. Gary Ditta and Marty Yanofsky provided a preliminary list of MADS-like sequences. The comments of three anonymous reviewers improved this paper. Elizabeth Núñez helped with logistical tasks. This work was supported by Ph.D. fellowships to L.P.M.-C. from Consejo Nacional de Ciencia y Tecnología (CONACYT) and Dirección General de Estudios de Posgrado (DGEP) at the National Autonomous University of Mexico, and by grants from CONACYT, Programa de Apoyo para Proyectos de Investigación e Innovación Tecnológica, the Human Frontiers Science Program, and the University of California-Mexico (to E.R.A.-B.).
Abbreviations: PDS, positive Darwinian selection; LRT, likelihood ratio test; AGL, agamous-like.
References
- 1.Ohno, S. (1970) Evolution by Gene Duplication (Springer, Heidelberg, Germany).
- 2.Zhang, J., Zhang, Y.-P. & Rosenberg, H. F. (2002) Nat. Genet. 30, 411-415. [DOI] [PubMed] [Google Scholar]
- 3.Ohta, T. (2000) Gene 259, 45-52. [DOI] [PubMed] [Google Scholar]
- 4.Hughes, A. L. (2002) Trends Genet. 18, 433-434. [DOI] [PubMed] [Google Scholar]
- 5.Lynch, M., O'Hely, M., Walsh, B. & Force, A. (2001) Genetics 159, 1789-1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark, A. G. (1994) Proc. Natl. Acad. Sci. USA 91, 2950-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner, A. (1999) J. Evol. Biol. 12, 1-16. [Google Scholar]
- 8.Messier, W. & Stewart, C.-B. (1997) Nature 385, 151-154. [DOI] [PubMed] [Google Scholar]
- 9.Swanson, W. J., Yang, Z., Wolfner, M. F. & Aquadro, C. F. (2001) Proc. Natl. Acad. Sci. USA 98, 2509-2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang, Z. & Bielawski, J. P. (2000) Trends Ecol. Evol. 15, 496-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jack, T., Brockmann, L. L. & Meyerowitz, E. M. (1992) Cell 68, 683-697. [DOI] [PubMed] [Google Scholar]
- 12.Doyle, J. (1994) Syst. Biol. 43, 307-328. [Google Scholar]
- 13.Purugganan, M. D. (1998) BioEssays 20, 700-711. [DOI] [PubMed] [Google Scholar]
- 14.Alvarez-Buylla, E. R., Pelaz, S., Liljegren, S. J., Gold, S. E., Burgeff, C., Ditta, G. S., Ribas de Pouplana, L., Martínez-Castilla, L. & Yanofsky, M. F. (2000) Proc. Natl. Acad. Sci. USA 97, 5328-5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Purugganan, M. D. (1997) J. Mol. Evol. 45, 392-396. [DOI] [PubMed] [Google Scholar]
- 16.Kohler, C., Hennig, L., Spillane, C., Pien, S., Gruissem, W. & Grossniklaus, U. (2003) Genes Dev. 17, 1540-1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwarz-Sommer, Z., Huijser, P., Nacken, W., Saedler, H. & Sommer, H. (1990) Science 250, 931-936. [DOI] [PubMed] [Google Scholar]
- 18.Bowman, J. L., Smyth, D. R. & Meyerowitz, E. M. (1991) Development (Cambridge, U.K.) 112, 1-20. [DOI] [PubMed] [Google Scholar]
- 19.Carpenter, R. & Coen, E. S. (1990) Genes Dev. 4, 1483-1493. [DOI] [PubMed] [Google Scholar]
- 20.Goto, K. & Meyerowitz, E. M. (1994) Genes Dev. 8, 1548-1560. [DOI] [PubMed] [Google Scholar]
- 21.Alvarez-Buylla, E. R., Liljegren, S. J., Pelaz, S., Gold, S. E., Burgeff, C., Ditta, G. S., Vergara-Silva, F. & Yanofsky, M. F. (2000) Plant J. 24, 457-466. [DOI] [PubMed] [Google Scholar]
- 22.Liljegren, S. J., Ferrándiz, C., Alvarez-Buylla, E. R., Pelaz, S. & Yanofsky, M. F. (1998) Flowering Newslett. 25, 9-19. [Google Scholar]
- 23.Zhang, H. & Forde, B. G. (1998) Science 279, 407-409. [DOI] [PubMed] [Google Scholar]
- 24.Johansen, B., Pedersen, L. B., Skipper, M. & Frederiksen, S. (2002) Mol. Phylogenet. Evol. 23, 458-480. [DOI] [PubMed] [Google Scholar]
- 25.Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994) Nucleic Acids Res. 22, 4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hall, T. A. (1999) Nucleic Acids Symp. Ser. 41, 95-98. [Google Scholar]
- 27.De Bodt, S., Raes, J., Florquin, K., Rombauts, S., Rouze, P., Theissen, G. & Van De Peer, Y. (2003) J. Mol. Evol. 56, 573-586. [DOI] [PubMed] [Google Scholar]
- 28.Kramer, E. M., Dorit, R. L. & Irish, V. F. (1998) Genetics 149, 765-783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huelsenbeck, J. P. & Ronquist, F. (2001) Bioinformatics 17, 754-755. [DOI] [PubMed] [Google Scholar]
- 30.Yang, Z., Nielsen, R., Goldman, N. & Krabbe-Pedersen, A.-M. (2000) Genetics 155, 431-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldman, N. & Yang, Z. (1994) Mol. Biol. Evol. 11, 725-736. [DOI] [PubMed] [Google Scholar]
- 32.Anisimova, M., Bielawski, J. P. & Yang, Z. (2001) Mol. Biol. Evol. 18, 1585-1592. [DOI] [PubMed] [Google Scholar]
- 33.Yang, Z. & Nielsen, R. (2002) Mol. Biol. Evol. 19, 908-917. [DOI] [PubMed] [Google Scholar]
- 34.Yang, Z. (1997) Comput. Appl. Biosci. 13, 555-556. [DOI] [PubMed] [Google Scholar]
- 35.Michaels, S. D., Ditta, G., Gustafson-Brown, C., Pelaz, S., Yanofsky, M. & Amasino, R. M. (2003) Plant J. 33, 867-874. [DOI] [PubMed] [Google Scholar]
- 36.Hartmann, U., Hohmann, S., Nettesheim, K., Wisman, E., Saedler, H. & Huijser, P. (2000) Plant J. 21, 351-360. [DOI] [PubMed] [Google Scholar]
- 37.Ratcliffe, O. J., Kumimoto, R. W., Wong, B. J. & Riechmann, J. L. (2003) Plant Cell 15, 1159-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar, S., Tamura, K., Jakobsen, I. B. & Nei, M. (2001) Bioinformatics 17, 1244-1245. [DOI] [PubMed] [Google Scholar]
- 39.Lynch, M. & Conery, J. S. (2000) Science 290, 1151-1155. [DOI] [PubMed] [Google Scholar]
- 40.The Arabidopsis Initiative (2000) Nature 408, 796-815. [DOI] [PubMed] [Google Scholar]
- 41.Henschel, K., Kofuji, R., Hasebe, M., Saedler, H., Munster, T. & Theissen, G. (2002) Mol. Biol. Evol. 19, 801-814. [DOI] [PubMed] [Google Scholar]
- 42.Rounsley, S. D., Ditta, G. S. & Yanofsky, M. F. (1995) Plant Cell 7, 1259-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Riechmann, J. L. & Meyerowitz, E. M. (1997) J. Biol. Chem. 378, 1079-1101. [PubMed] [Google Scholar]
- 44.Pellegrini, L., Tan, S. & Richmond, T. J. (1995) Nature 376, 490-498. [DOI] [PubMed] [Google Scholar]
- 45.Santelli, E. & Richmond, T. J. (2000) J. Mol. Biol. 297, 437-449. [DOI] [PubMed] [Google Scholar]
- 46.Huang, K., Louis, J. M., Donaldson, L., Lim, F.-L., Sharrocks, A. D. & Clore, G. M. (2000) EMBO J. 19, 2615-2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krizek, B. A. & Meyerowitz, E. M. (1996) Proc. Natl. Acad. Sci. USA 93, 4063-4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang, J., Rosenberg, H. F. & Nei, M. (1998) Proc. Natl. Acad. Sci. USA 95, 3708-3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nei, M., Rogozin, I. B. & Piontkivska, H. (2000) Proc. Natl. Acad. Sci. USA 97, 10866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vision, T., Brown, D. G. & Tanskley, S. D. (2000) Science 290, 2114-2117. [DOI] [PubMed] [Google Scholar]
- 51.Simillion, C., Vandepoele, K., Van Montagu, M. C., Zabeau, M. & Van de Peer, Y. (2002) Proc. Natl. Acad. Sci. USA 99, 13627-13632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blanc, G., Hokamp, K. & Wolfe, K. H. (2003) Genome Res. 13, 137-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Force, A., Lynch, M., Pickett, F. B., Amores, A., Yan, Y.-l. & Postlethwait, J. (1999) Genetics 151, 1531-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lamb, R. S. & Irish, V. F. (2003) Proc. Natl. Acad. Sci. USA 100, 6558-6563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Riechmann, J., Wang, M. & Meyerowitz, E. (1996) Nucleic Acids Res. 24, 3134-3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferrándiz, C., Gu, Q., Martienssen, R. & Yanofsky, M. F. (2000) Development (Cambridge, U.K.) 127, 725-734. [DOI] [PubMed] [Google Scholar]
- 57.Purugganan, M. D. & Suddith, J. I. (1998) Proc. Natl. Acad. Sci. USA 95, 8130-8134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suzuki, Y. & Nei, M. (2002) Mol. Biol. Evol. 19, 1865-1869. [DOI] [PubMed] [Google Scholar]
- 59.Doebley, J. & Lukens, L. (1998) Plant Cell 10, 1075-1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.