

Abstract
Animal research suggests that the consolidation of fear and extinction memories depends on N-methyl D-aspartate (NMDA)-type glutamate receptors. Using a fear conditioning and extinction paradigm in healthy normal volunteers, we show that postlearning administration of the NMDA partial agonist D-cycloserine (DCS) facilitates fear memory consolidation, evidenced behaviorally by enhanced skin conductance responses, relative to placebo, for presentations of a conditioned stimulus (CS) at a memory test performed 72 h later. DCS also enhanced CS-evoked neural responses in a posterior hippocampus/collateral sulcus region and in the medial prefrontal cortex at test. Our data suggest a role for NMDA receptors in regulating fear memory consolidation in humans.
Keywords: anxiety, conditioning, emotion, fMRI, hippocampus, MPFC
Introduction
Transforming a recently acquired memory into a durable memory trace is a critical component of adaptive behavior. Memory consolidation is also of considerable clinical interest in the treatment of both memory dysfunction and anxiety disorders such as posttraumatic stress disorder that may be based on maladaptive learning processes. Successful consolidation of many types of memories is believed to depend on early activation of NMDA-type glutamate receptors, setting into motion a cascade of molecular events that finally leads to durable changes in synaptic properties (Martin 2000; Izquierdo et al. 2006). The role of NMDA receptors in consolidation of both emotional and nonemotional memories has been extensively studied in rodents. In particular, there is evidence from various animal models of fear conditioning that NMDA receptor activation promotes fear memory consolidation (Stewart and McKay 2000; Rodrigues et al. 2001; Rubin et al. 2004). By contrast, there is a paucity of human data that address this issue. One study has found evidence suggesting that NMDA blockade impairs short-term consolidation of episodic memory (Parwani et al. 2005), whereas another study has failed to find evidence for a presumed positive modulatory role of the NMDA partial agonist D-cycloserine (DCS) on “extinction memory” consolidation (Guastella et al. 2007).
We investigated the contribution of NMDA receptors to consolidation of conditioned fear and extinction memory. In cued fear conditioning, an organism learns that an initially neutral stimulus (conditioned stimulus [CS]) predicts a noxious stimulus (unconditioned stimulus [UCS]), resulting in the formation of a conditioned response (CR) to presentation of the CS and a corresponding fear memory (CS–UCS association) (Pavlov 1927). To assess NMDA effects in fear memory consolidation, we first fear conditioned subjects and, postconditioning, administered either placebo or DCS (day 1). At a recall test 72 h later (day 2), we compared both groups for CS-evoked CRs. In addition to behavioral CR indices, we employed functional magnetic resonance imaging (fMRI) to measure correlated neural activity in predefined regions of interest (ROIs).
Fear memories can be extinguished by repeated unreinforced CS presentations, a process believed to involve formation of a new, inhibitory memory (CS–noUCS association) (Myers and Davis 2002; Bouton 2004; Delamater 2004). Successful recall of this extinction memory (i.e., CR inhibition) depends critically on the test context resembling the context in which extinction originally took place (Bouton 2004). To assess NMDA effects in extinction memory consolidation, we included extinction training and an extinction test in a context different from the context in which subjects were conditioned (see Fig. 1 for an overview of the design and methodology). Due to the contextualization of cued fear and extinction learning on day 1 in this paradigm, we expected context-dependent recall on day 2. That is, the fear memory should be preferentially recalled (an CR should be produced) when the CS was presented in the conditioning context, whereas the extinction memory should be preferentially recalled (a significantly smaller CR should be produced) when the CS was presented in the extinction context.
Figure 1.

Fear and extinction recall paradigm. On day 1, subjects (n = 31 healthy female volunteers) were fear conditioned to a CS+ (a face) through multiple pairings with an UCS (electric shock) in context A (conditioning context, block A1). Fear responses were then extinguished in context B (extinction context, block B1) through multiple CS+ presentations in the absence of the UCS. To ascertain retention of fear memories until a recall test 72 h later (see below), conditioning was repeated in a further block (A2). As a control for nonassociative effects, we also employed a nonpredictive CS− (a face of opposite gender) that was never paired with the UCS and presented intermixed with the CS+. Contexts were defined by screen color and auditory input. It was assumed that this procedure would create a CS-associated fear memory. Based on an earlier study by our group (Kalisch et al. 2006a), it was also assumed that the procedure would create an (extinction) context-dependent extinction memory. Learning was followed by administration of either placebo (n = 16) or 500-mg DCS (n = 15). On day 2 (72 h later), CSs were presented in both contexts A and B to test for CS-evoked fear and extinction memory recall, respectively. To this purpose, each context was presented 16 times in alternating order. Recall of fear memory in context A on day 2 was facilitated by additionally presenting 1 unpaired shock at the beginning of each context A block, thus again firmly associating context A with the UCS. The task was a speeded gender decision task in response to the face stimuli. Gender of faces and conditioning and extinction contexts were counterbalanced between groups. The design was randomized, double blind, and between subject. Flash denotes electric shock.
Based on the idea that consolidation involves NMDA activation, we predicted that DCS would enhance CRs in the conditioning context, relative to placebo. A secondary prediction was that such enhancement would be associated with increased CS-evoked activation in areas implicated across animal and human studies in recall of contextualized fear memories, namely posterior hippocampus and medial prefrontal cortex/anterior cingulate cortex (MPFC/ACC; Kim and Fanselow 1992; Maren and Fanselow 1997; Corcoran and Maren 2004; Frankland et al. 2004; Corcoran et al. 2005; Ji and Maren 2005; LaBar and Phelps 2005; Kalisch et al. 2006a). For CS presentations in the extinction context, we predicted further reduced CRs and concomitant neural activations in the DCS group.
Materials and Methods
The design consisted of Pavlovian fear conditioning in context A and extinction in context B on day 1. This was followed by testing CS-evoked responses in both the conditioning (A) and the extinction context (B) on a subsequent day (day 2, see Fig. 1 for an overview). To investigate the role of NMDA receptors in the consolidation of fear and extinction memory, subjects received either DCS or placebo after the experiment on day 1 in a randomized, double-blind, between-subject fashion. Drug effects on consolidation were then assessed by comparing the recall of fear and extinction memories on day 2 between the DCS and the placebo groups. In order to assure that the memory test was drug free (DCS has a plasma half-life of approximately 10–12 h), day 2 corresponded to 72 h after day 1.
Subjects
Thirty-two right-handed female volunteers, nonpregnant and nonbreastfeeding, participated in the study. In a face-to-face interview, subjects were preassessed by an experienced psychiatrist to exclude those currently under any medication or treatment, those with past or present mental or neurological illness, kidney impairment, heart condition, porphyria, porphyria among family members, and allergy to antibiotics. All subjects gave informed written consent, and the study was approved by the Joint Ethics Committee of the National Hospital for Neurology and Neurosurgery. One subject withdrew from the study 3 days after ingestion of DCS, complaining about headaches. The remaining 31 subjects had a mean age of 25 years (±1 year standard error of the mean). Subjects were randomly assigned to 1 of the 2 treatment groups, 1 receiving placebo (n = 16, mean age = 26 ± 2 years) and 1 receiving DCS (n = 15, mean age = 24 ± 1 years). Groups did not differ in terms of trait anxiety as assessed by their average scores on a trait anxiety inventory (STAI-T; Spielberger [1983]; 33.9 ± 2.4 (placebo) and 31.2 ± 1.7 [DCS], P = 0.252, 2-sample t-test, 2 tailed).
Stimuli
Unconditioned Stimulus
The UCS consisted of brief electric shocks to the right hand. Shocks were applied using a Digitimer DS7A electrical stimulator (Digitimer Ltd, Welwyn Garden City, UK) delivering electrical pulses of up to 20 mA and 1 or 2 ms duration through a silver chloride electrode. Stimulation parameters were individually adjusted prior to the experiment to achieve maximum tolerable pain. To this end, subjects were given a series of shocks, starting at a very low current level and slowly increasing in amplitude, until the subject indicated he or she did not want to receive any higher stimulation. The subject was explicitly asked whether the reached level was tolerable and could be used during the subsequent experiment. Note that fear-related areas show lateralized CS responses based on where the source of danger is located in space (Blair et al. 2005; Kalisch et al. 2005). To increase the probability of finding CS-evoked activation, we therefore applied the UCS to the same hand (right) in all subjects. This prohibits inference about lateralization of CS-evoked responses.
Conditioned Stimuli
The 2 CSs (1 CS+ that was occasionally paired with the UCS and 1 CS− that was never paired) consisted of 1 male and 1 female face from the Ekman series (Ekman and Friesen 1976) whose hair was removed in view of the gender decision task (see below). Mildly (20%) angry faces were chosen based on earlier studies by our group showing successful conditioning and amygdala activation with mildly angry face CSs (Morris et al. 1998; Critchley et al. 2002). The 2 same faces were used for all subjects. In 15 of the 31 subjects (8 in the placebo and 7 in the DCS group), the CS+ was the male face; in the remaining 16 subjects (8 in the placebo and 8 in the DCS group), it was the female face.
Contexts
Conditioning and extinction occurred in 2 different contexts that were distinguished by background screen color and auditory input. The screen color was either black or rhythmically changing between red and orange. The fixation mark was a white cross in the black and a white dot in the red–orange context. There was no auditory input in the black context, whereas, in the red–orange context, subjects heard 2 sounds of different pitch, presented over headphones, which changed synchronously with the color of the screen. In 16 subjects (8 in the placebo and 8 in the DCS group), the black context was the context in which conditioning occurred (conditioning context or A) and the red–orange context was the context in which extinction occurred (extinction context or B). In the remaining 15 subjects (8 in the placebo and 7 in the DCS group), the red–orange context was the conditioning context and the black context was the extinction context.
Task
Subjects were told that the study would examine attentional performance under stress and its pharmacological manipulation and were only debriefed at the end of the study. The task was a speeded gender decision task for which subjects signaled the gender of the face by pressing the left (for female) or the right (for male) button on a keypad with their right hand's index or middle finger, respectively, as soon as they saw the face.
Drugs
DCS (King Pharamaceuticals, Leicester, UK; dose = 500 mg) was administered as 2 tablets of 250 mg. This dose has apparent (extinction) memory-enhancing effects in phobic patients treated with exposure therapy (Ressler et al. 2004). Subjects were asked not to eat 3 h before start of the experiment. Fasting facilitates DCS absorption (Zhu et al. 2001), and plasma concentrations peak within 2 h in sober subjects (van Berckel et al. 1998). This measure was intended to assure high DCS plasma levels during the theoretical critical time window for NMDA-dependent memory consolidation of 1- to 2-h postlearning, as determined in rats (Scavio et al. 1992). In addition, subjects were asked to refrain from alcohol or other drugs on the eve of, and during, the experimental days to limit potential drug effects on task performance as well as potential DCS–drug interactions—which can have amnestic effects in the case of ethanol (Trevisan et al. 2008). There were no restrictions with regard to nicotine consumption. Two 100-mg vitamin E tablets (Co-farmer, Hurts, UK) served as placebo.
Design
Day 1: Discrimination Learning and Drug Administration
Subjects were first habituated to the CSs and contexts by presenting each CS 3 times in each context prior to the actual experiment. Subjects then learned to discriminate the 2 CSs on the basis of how they predicted danger. In conditioning block A1, the CS+ and the CS− were each presented 15 times in a randomized order in the center of the screen. The duration of CSs ranged from 2 to 8 s with a mean duration of 5.7 s per CS type (2 CSs of 2 s, 1 of 3 s, 3 of 5, 6, 7, and 8 s each per CS type). The UCS was applied 250 ms before the offset of the CS+. Those 3 CS+ presentations that were shorter than 5 s were not coupled with an UCS, resulting in a reinforcement ratio of 80%. Varying delays between CS+ and UCS onset were meant to introduce additional uncertainty that would make conditioning somewhat more extinction resistant and therefore increase the likelihood of recall of fear memory on day 2, an effect that, according to our experience (Kalisch et al. 2006a), is difficult to obtain in humans and that we expected to be even more difficult to reproduce with a relatively long acquisition-test interval of 72 h. For the same reasons, the number of conditioning trials was increased compared with our earlier study where the acquisition-test interval was 24 h only (Kalisch et al. 2006a). A minimum delay of 5 s between CS+ and UCS onset allowed us to measure conditioned skin conductance responses (SCRs) to the CS+ without a confound from the subsequent unconditioned SCRs to the shock. CSs were separated by an interstimulus interval (ISI) of 9 s during which subjects saw a central fixation mark (low-level baseline). The length of the ISI was chosen to avoid complete masking of conditioned SCRs by preceding unconditioned SCRs to the shock.
In the following extinction block B1, which was different from the preceding conditioning block in terms of screen color and auditory input (see above), conditioned fear responses were extinguished by presenting the same 30 CSs in the same fashion but without shock. This was followed by another conditioning block (A2) in its corresponding context. This design allowed subjects to learn to discriminate between 2 different contexts on the basis of whether the CS+ was (conditioning context) or was not (extinction context) associated with the UCS. Each block lasted 6 min, and the corresponding context was already present at the beginning of each block 9 s before CS presentation started. Blocks were separated by a break of 30 s during which scanning continued, but subjects were allowed to close their eyes if they wanted.
Immediately (within 2 min) after the end of the experiment, subjects received either placebo or DCS. Following this, the subjects were asked whether they had noticed any relationship between the shock and the gender of the face and between the shock and the screen color. Twenty minutes after drug intake, subjects filled in a 7-item physical symptoms rating for dry mouth, dry skin, blurred vision, sedation, nausea, dizziness, and headache (rated not present, very mild, mild, moderate, moderately severe, severe, or extremely severe) and a 17-item mood rating scale using visual analog scales for pairs of words (alert–drowsy, tense–relaxed, etc.) as suggested by Bond and Lader (1974). Thirty minutes after drug intake, the subjects were allowed to leave the department.
Day 2: Memory Test 72 h Later
Subjects again completed the physical symptoms and mood rating scales. Then, each context was again presented 18 times in an alternating order (ABABAB…) for a duration of 29 s each, separated by 5-s breaks. At the beginning of each block, the context was present without any CS for 10 s. In 16 out of the 18 blocks of A and B each, 1 CS+ and 1 CS− were presented in random order for a duration of 5 s each, followed by an ISI of 4 s each. In those 16 A blocks, subjects received a shock 3 s after the beginning of the block that was, however, not paired with either the CS+ or the CS−. Associating context A with shock during recall theoretically promotes reinstatement, thus facilitating recall of the CS-associated fear memory in this context. This was another measure taken to increase the likelihood of fear memory recall on day 2 that should be additionally compromised by the ongoing extinction of CRs as a consequence of repeated unreinforced CS+ presentation at test (for detailed discussion, see Kalisch et al. [2006a]).
Efficiency of Blinding and Drug Side Effects
Before debriefing, subjects were asked whether they thought they had received drug or placebo. Out of 31 subjects 20 answered they did not know. Four placebo subjects correctly guessed that they had received placebo, whereas 2 placebo subjects incorrectly guessed that they had received drug. Four drug subjects correctly guessed that they had received drug, whereas 1 drug subject incorrectly guessed that she had received placebo. Correct guesses were most likely due to the absence/presence of side effects. In the mood rating scale completed 20 min after placebo/drug intake, drug subjects reported to feel more mentally slowed (compared with quick witted, mean = 5.3 ± 0.6) than placebo subjects (mean = 7.2 ± 0.4, P = 0.012, unpaired t-test, 2 tailed). Otherwise, the mood rating scale as well as the physical symptoms rating scale did not pick up any significant group differences, whether employed on the day of drug intake or at the beginning of the experiment on day 2. However, on day 2, 6 drug subjects but only 1 placebo subject told the experimenters of headaches typically starting a few hours after intake on day 1 and lasting a few hours up to a day. Additional side effects thus reported but not picked up by the rating scales were tiredness/lethargy (3 drug subjects), dizziness (3 drug subjects), nausea (2 drug subjects), and feeling hot/sweaty (1 drug subject). This suggests that a limited number of subjects may have been effectively unblinded. It is unlikely, though, that this influenced our measures of recall, given that these were incidental and subjects were not aware of the true purpose of the task.
Autonomic Monitoring
Skin conductance measurements were acquired at a sampling rate of 1000 Hz from electrodes on the middle and ring finger of the left hand using an AT64 SCR apparatus (Autogenic Systems, Wood Dale, IL).
Imaging
Subjects were scanned on both days to maximize context identity across days. Only data from day 2 are reported here. A 3 Tesla MR head scanner (Magnetom Allegra, Siemens, Erlangen, Germany) was used to acquire gradient echo -weighted echo-planar images (EPI) with blood oxygen level–dependent contrast (time echo = 30 ms, time repetition = 1.43 s, flip angle = 70°, slice tilt = 30°, z-shim gradient prepulse = −1 mT/m ms). Each volume comprised 22 oblique axial slices of 2-mm thickness and 3 × 3 mm2 in-plane resolution with a slice gap of 1 mm. The slice package excluded the dorsal frontal, parietal, and occipital cortices. These parameters produced EPIs in which signal dropout due to susceptibility-induced field inhomogeneities was minimized for amygdala and orbitofrontal cortex (Deichmann et al. 2003). Subjects were placed in a light head restraint within the scanner to limit head movement during acquisition. A total of 1064 (day 1) and 899 (day 2) volumes were acquired continuously throughout the task, at 1.43-s intervals, starting 14.3 s before the onset of the experiment. As a result of the above timings, there was no systematic temporal relationship between the onsets of slices and stimuli, thus allowing for sampling, over the course of the experiment, the entire length of the stimulus-driven hemodynamic responses in each of the 22 slices. A T1-weighted structural image was also acquired (Deichmann et al. 2004).
Data Analysis
Behavioral Data
Skin conductance data were downsampled to 100 Hz, mean filtered, and then visually inspected for artifacts. Eight subjects did not show any apparent SCR to the UCS on day 1 and were excluded from further skin conductance analysis, reducing sample size (including for the imaging data analysis where SCR scores were used to model CR magnitude, see below) to n = 23 (n = 13 in the placebo and n = 10 in the DCS group). In the following, an SCR was defined as the maximum skin conductance in a time window of 5 s after CS onset minus skin conductance at the time of CS onset (Buchel et al. 1998). Data were z-transformed to account for interindividual differences in physiological reactivity (Buchel et al. 1998). In 2 subjects on day 1, reaction time (RT) recording failed for technical reasons, reducing sample size on day 1 to n = 29 (n = 15 in the placebo and n = 14 in the DCS group). As SCRs, RT data from the gender decision task were z-transformed.
Significance of behavioral effects was assessed using t-tests and analysis of variance (ANOVA). Because we had directed hypotheses for both SCR and RT effects, a 1-tailed threshold of P = 0.05 was used throughout, unless indicated otherwise. On day 1, where early and late experimental blocks were analyzed separately (LaBar et al. 1998; Phelps et al. 2004), this was done by subtracting responses to the first 8 CS− from the first 8 CS+ (early) and responses to the last 7 CS− from the last 7 CS+. Note that the order of CS+ and CS− was randomized. This means that some of the CSs classified as early may effectively have occurred in the last half of the block and some of the late CSs in the first half. However, such potential minor distortions should randomize out at the group level.
Response accuracy was 100% in all subjects (n = 29) recorded in all 4 conditions (CS+ in A, CS− in A, CS+ in B, and CS− in B) on day 1. On day 2, response accuracy ranged between 87.5% and 100% across subjects and conditions. The CS+ versus CS− difference scores were not significantly influenced by either context (A vs. B) or treatment (DCS vs. placebo) as assessed by 2-way repeated measures ANOVA.
Imaging Data
Imaging data were analyzed using SPM2 (www.fil.ion.ucl.ac.uk/spm; Ashburner et al. 2004). The 10 initial images were discarded to account for T1 equilibration. Scanner hardware problems caused occasional high–signal intensity artifacts (spikes) in 5 subjects. The affected slices were discarded and replaced by the mean of the preceding and the subsequent slice. To correct for motion artifacts, images were realigned to the 11th volume. Images were unwarped to correct for movement-by-distortion interactions, spatially normalized to a standard EPI template, spatially smoothed using a Gaussian kernel with a full width at half maximum (FWHM) of 4 mm, temporally high-pass filtered (cutoff = 128 s) and corrected for temporal autocorrelations using first-order autoregressive modeling. Statistical analysis was performed using a standard approach for fMRI, involving a general linear convolution model at the single-subject level and a random effects analysis at the group level (see Friston et al. 1994; Friston et al. 1995; Penny and Holmes 2004 for details). First, for each subject, condition-specific regressors were defined that modeled the time course of the experimental events and, after convolution with a canonical hemodynamic response function, served as predictors of the fMRI signal time courses at each voxel in the brain. CSs were modeled as a series of events (i.e., a series of delta functions, separately for CS+ in A, CS− in A, CS+ in B, and CS− in B). Additionally, these 4 categorical regressors were parametrically modulated using trial-by-trial z-transformed SCRs as an index of the magnitude of the evoked CR. Shocks were also modeled as events, whereas A and B blocks were modeled as 2 separate boxcar regressors (0 for off and 1 for on). As mentioned above, each regressor was convolved with a canonical hemodynamic response function. Using these regressors in a general linear model (multiple regression) of brain activation at each voxel yields parameter estimates of the contribution of each regressor to the fMRI signal measured in each voxel. Contrasts, that is, linear combinations of these parameter estimates, were then calculated voxel-wise to produce within-subject estimates of effects of interests (e.g., the contrast CS+ minus CS− in context A). Statistical inference was obtained using a t-statistics that takes into account the magnitude of the contrast value and its standard deviation. This yielded single-subject statistical parametric maps (SPM T maps) for each contrast of interest. For the random effects group analyses, the subject-specific contrast images were spatially smoothed (FWHM = 10 mm) to account for intersubject variation in the exact location of activations and compared across subjects. Within-group effects (i.e., effects within either the placebo or the DCS group) were tested for significance using voxel-wise 1-sample 1-tailed t-tests. Between-group effects (treatment effects) were tested using 2-sample unpaired t-tests. Averaged single-subject contrast estimates were used to illustrate group effect sizes in selected voxels (insets in the figures).
Correction for multiple comparisons following Gaussian random field theory was limited to predefined spherical ROIs (small volume correction [SVC]) and included all voxels active at an uncorrected threshold of P = 0.01. The bilateral amygdala (coordinates ±28, −2, −32) and posterior hippocampus (±38, −32, −12) ROIs for recall of fear memory were taken from Kalisch et al. (2006a) from their contrast (CS+ > CS−)A, RT-modulated (see their fig. 3). The bilateral MPFC/ACC ROI for anticipatory fear (coordinates ±2, 45, 27) was taken from Kalisch et al. (2005). The same coordinates have since successfully been used in 2 further studies (Kalisch et al. 2006b, 2006c) to identify MPFC/ACC activation during anticipatory fear. All ROIs had a radius of 6 mm around the seed point. Bilateral ROIs were chosen because the cited reference studies, albeit partly reporting unilateral coordinates only, did not test and were not designed to test lateralization (due to UCS application to the same hand in all subjects, as in the present study).
Structural images were coregistered onto functional images and spatially normalized using the nonlinear transformation estimated from the functional EPIs. Anatomical localization was carried out with reference to the atlas of Duvernoy (1999). Coordinates are described in the standard space defined by the Montreal Neurological Institute.
Results
Day 1: Fear Conditioning and Extinction
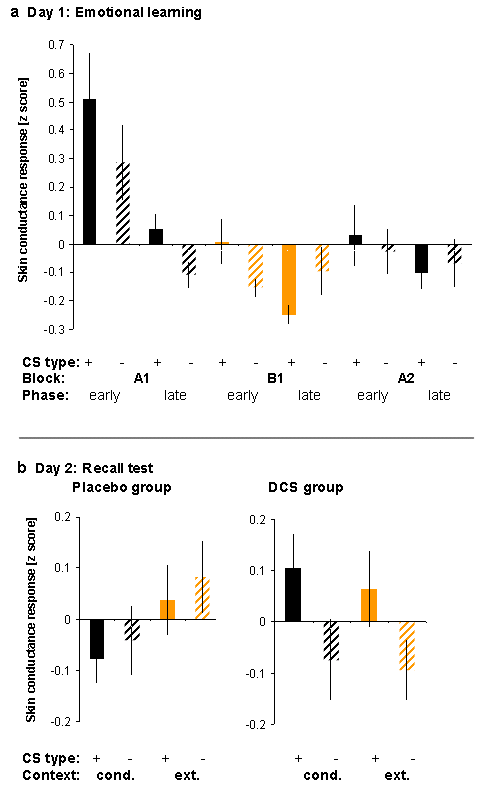
Out of 31 subjects 23 (11 in the placebo group and 12 in the DCS group) reported awareness of the CS+–UCS contingency and 28 out of 31 subjects (15 in the placebo group and 13 in the DCS group) reported awareness of the context A–UCS contingency when interviewed after the experiment on day 1, suggesting successful conditioning and context discrimination. Conditioning was confirmed by analysis of CS-evoked SCRs. Figure 2a shows larger SCRs to the CS+ than to the CS− in the first conditioning block (A1). The effect reached significance in the second half of the first conditioning block (A1 late: t(22) = 2.19, P = 0.02, paired t-test, 1 tailed), that is, after an initial learning phase (A1 early). (For a separate depiction of SCRs evoked by CS+ and CS−, see Supplementary Fig. 1.) This differential SCR effect extended into the first half of the subsequent extinction block (B1 early: t(22) = 1.82, P = 0.041). The effect was not observed in the second conditioning block (A2) (but compare the change relative to the preceding (late) extinction block).
Figure 2.

SCRs showing facilitated fear memory consolidation by DCS. (a) Day 1: Significantly larger SCRs to the CS+ than to the CS− during late fear conditioning in context A indicate learning of the CS+−UCS contingency (fear memory) in context A on day 1. A reversal of the conditioning effect during late extinction in context B indicates learning of the CS+−noUCS contingency (extinction memory) in context B. (b) Day 2: SCRs to the CS+ in the conditioning context were again significantly larger than to the CS− on day 2, but only in those subjects receiving DCS after learning on day 1. A similar effect was apparent in the extinction context, that is, there was no evidence for extinction memory recall. Scale: z scores (unit: standard deviations [SDs]). *P < 0.05, (*)P < 0.1, 1-tailed t-test versus 0.
The SCR data also showed evidence of successful extinction in context B. During the second half of the extinction block (B1 late), SCRs to the CS+ tended to become smaller than to the CS− (t(22) = −1.67, P = 0.054). That is, extinction learning not only abolished but also reversed the conditioning effect. The context discrimination effect ((CS+ > CS−)A > (CS+ > CS−)B) was significant when comparing A1 late to B1 late (t(22) = 2.4, P = 0.013). There were no significant differences between those subjects who were subsequently treated with placebo versus those treated with DCS (ingested immediately following learning). We observed no significant RT effects in the gender decision task.
Day 2: Recall of Fear Memory as a Function of Postlearning Drug Treatment
Placebo
In the placebo group, test at 72 h showed no differential (CS+ > CS−) SCRs in either context nor was there any contextual modulation of SCR differences (Fig. 2b). The data indicate an absence of fear memory recall 72 h after learning. Given the apparent decay of fear memory, no conclusions about extinction memory recall (which can only be deduced from a reduction of CRs) can be drawn. Absence of CRs in the placebo group highlights the difficulty to obtain fear memory recall in human subjects (where the UCS does not evoke substantial threat and the conditioning experience can be cognitively reappraised) despite a relatively extended conditioning procedure on day 1 and presentation of unpaired UCSs, which theoretically promotes reinstatement of fear, in the conditioning context on day 2 (see Materials and Methods).
D-Cycloserine
By contrast, the DCS group showed significant differential SCRs in both contexts, indicating recall of fear memory independent of context (A: t(9) = 1.91, P = 0.044; B: t(9) = 2.33, P = 0.022; Fig. 2b). SCR differences reached levels similar to block A1 late on day 1 (compare Fig. 2a). This effect did not differ between contexts ((CS+ > CS−)A > (CS+ > CS−)B: t(9) = 0.17, P = 0.435); that is, there was no evidence for recall of extinction memory (comparatively reduced CRs) in the extinction context. As on day 1, there were no RT effects in either group (see Supplementary Fig. 2).
Placebo versus DCS
The group comparison showed that postlearning drug treatment had a significant influence on our SCR measure of fear memory recall. A 2-way repeated measures ANOVA with treatment (placebo, DCS) as between-subject factor, context (A, B) as within-subject factor, and the SCR CS+ > CS− difference score as the independent variable revealed a significant effect of treatment (F1,21 = 5.7, P = 0.026) but no effect of context (F1,21 = 0.03, P = 0.86) and no treatment by context interaction (F1,21 = 0.0, P = 0.948). Planned post hoc t-tests showed that in the conditioning context A, where we predicted stronger recall of fear following DCS treatment, the DCS subjects had increased differential SCRs compared with the placebo subjects (t(21) = 1.99, P = 0.03, unpaired t-test, 1 tailed). This suggests that DCS treatment facilitated fear memory recall at test. The DCS subjects also had larger differential SCRs in the extinction context B, but the effect did not reach significance (t(21) = 1.57, P = 0.131, 2 tailed). The data thus allow no inferences on the role of DCS in extinction memory consolidation within the context of our experimental design.
Day 2: Neural Activity Associated with Fear Recall Facilitation by DCS
Our behavioral data showed a treatment effect on differential responses to CS+ versus CS− stimuli 72 h after treatment. To capture this effect in our analysis of the fMRI data from day 2, we modeled CS-evoked responses as events. Moreover, categorical effects were parametrically modulated by trial-by-trial SCRs as an index of CR magnitude (Carter et al. 2006). That is, the statistical model included 1 categorical regressor per trial type (CS+A, CS−A, CS+B, CS−B) that predicted equivalent hemodynamic activation for each trial of a trial type. In addition, there was 1 parametric regressor per trial type that predicted activation varying with SCRs over trials and hence expressed the behaviorally determined magnitude of the associated CR (see Materials and Methods). The behavioral results led us to focus our analysis on differential (DCS > placebo) parametric activations in areas related to fear recall. Bilateral hippocampal ROIs were defined based on an earlier study by our group (Kalisch et al. 2006a) where fear recall was tested 24 h after conditioning, yielding CS+-evoked activation in posterior hippocampus. Because the same study had also produced amygdala activation, albeit at lower threshold, and because of the documented role of the amygdala in cued fear recall in rats (Schafe et al. 2005), we also included bilateral amygdala ROIs (see Materials and Methods for coordinates). Bilateral MPFC/ACC ROIs were defined based on studies of anticipatory fear for impending pain that used explicit instructional learning to fear condition subjects and tested for CRs at variable delays after conditioning (see Materials and Methods).
As indicated above, the behavioral results suggested that the data could be collapsed across contexts, thereby maximizing statistical power in the fMRI analysis. To ensure that this was justified, we first tested the placebo and the DCS data separately for differences in the contrast (CS+ > CS−) as a function of context ((CS+ > CS−)A vs. (CS+ > CS−)B). As expected, there were only minor differences (some voxels active at a threshold of P ≤ 0.001 uncorrected for multiple comparisons) between contexts in either group, irrespective of whether the categorical or the parametric effects were examined; no differences were observed in the ROIs (data not shown). The following analysis therefore reports collapsed data only.
We observed a significant parametric interaction of the type treatment (DCS > placebo) by stimulus (CS+ > CS−) in the left hippocampal and right MPFC/ACC ROIs. Both activations survived correction for multiple comparisons within the respective search volume (SVC; see Materials and Methods) at a threshold of P ≤ 0.05 (left posterior hippocampus: coordinates x = −34, y = −32, z = −16, P = 0.022 SVC; bilateral MPFC/ACC: x = 2, y = 46, z = 34, P = 0.009 SVC, x = −2, y = 46, z = 34, P = 0.011 SVC, x = −2, y = 48, z = 28, P = 0.012 SVC; unpaired 2-sample t-test, 1 tailed; see Supplementary Table 1 for details). For the activation peak in the hippocampal ROI, our anatomical precision was not sufficient to segregate between a localization in hippocampus versus collateral sulcus; the MPFC/ACC peak was consistent with Brodmann area 9 (Fig. 3). By analogy to our earlier study (Kalisch et al. 2006a), a corresponding amygdala activation was only observed at trend-level significance (left amygdala: x = −30, y = 2, z = −36, P = 0.082 SVC; right amygdala: x = 32, y = −4, z = −28, P = 0.086 SVC). Inspection of averaged single-subject contrast estimates in hippocampus/collateral sulcus and MPFC/ACC (Fig. 3) confirmed differential (CS+ > CS−) activation of both areas in the DCS, but not placebo, group across contexts. This suggests that enhanced fear memory consolidation with DCS administration, as indexed in behavior, was also expressed in enhanced neural activity at recall within the posterior hippocampus/collateral sulcus and the MPFC/ACC. No significant effects were observed in the corresponding categorical contrast. Supplementary Table 1 gives results from the entire scan volume and from the within-group contrasts; Supplementary Table 2 gives results from context A only.
Figure 3.

Effects of postlearning DCS on fMRI correlates of recall of fear memory on day 2. Activations associated with recall of fear memory on day 2 in left posterior hippocampus/collateral sulcus (x = −34, y = −32, z = −16) (a) and right MPFC/ACC (x = 2, y = 46, z = 34) (b) were larger in the DCS than in the placebo group. Images show the parametric contrast (CS+ > CS−)DCS > (CS+ > CS−)placebo, display threshold P ≤ 0.01. Hair cross denotes activation peak surviving SVC at P ≤ 0.05. Activations are superimposed on the mean structural image. The bar graphs show average contrast estimates for the parametric CS+ > CS− contrasts in both groups and contexts.
Discussion
We provide behavioral and neuronal data indicating that DCS administration following acquisition of cued conditioned fear facilitates recall of the conditioned memory at test in humans. DCS administration postlearning led to significantly enhanced SCRs to CS+ probes presented 72 h later, at a time where placebo subjects showed no CS+-evoked SCRs. This was accompanied by CS+-evoked activation, in the DCS group, within the posterior hippocampus and/or collateral sulcus and the MPFC/ACC. On this basis, we suggest that NMDA receptor activation is involved in fear memory consolidation in humans.
A large animal literature has demonstrated the importance of NMDA receptor activation in fear memory consolidation. NMDA antagonists administered postlearning impair the later recall of fear in cued (Rodrigues et al. 2001; Rubin et al. 2004) and context fear conditioning (Stewart and McKay 2000; Rodrigues et al. 2001; Rubin et al. 2004) and in avoidance learning paradigms (reviewed in Izquierdo et al. [2006]). In a minority of studies (Scavio et al. 1992; Gould et al. 2002), NMDA antagonists showed a facilitatory effect. Our results support the former findings, though we acknowledge that a partial agonist like DCS can show antagonistic behavior when endogenous ligand levels are very high. This might be the case during severe stress, a potential corollary of the testing and scanning procedures in this experiment. In particular, DCS is only a weak partial agonist at NMDA receptors that bear the NR2A and NR2B subunits (e.g., Sheinin et al. 2001) and that are likely to be involved in conditioning- and extinction-related synaptic plasticity (e.g., Rodrigues et al. 2001; Bauer et al. 2002; Sotres-Bayon et al. 2007). In any case, our results strongly suggest an involvement of NMDA receptors in fear memory consolidation in humans.
The only human study indicating a role of NMDA receptors in memory consolidation reported a deleterious effect of the NMDA antagonist ketamine on episodic memory recall when given immediately after learning (Parwani et al. 2005). However, drug administration in this study was continued throughout recall. The findings are also hard to reconcile with other episodic memory studies where ketamine impaired recall when given during learning but not when given immediately after learning (Hetem et al. 2000; Honey et al. 2005). Such inconsistent effects of NMDA receptor manipulations may best be explained by the short interval of less than 60 min between learning and recall in these experiments. Rat data show that fear memory recall 24 h and 7 days, but not 3 h, after learning is affected by NMDA agonists and antagonists (Lee et al. 2006), in agreement with findings from other molecular pathways generally showing that different systems support the consolidation of long- versus short-term memory (Izquierdo et al. 2006). The existing human data thus appear to mirror the time frame of effects seen in animals.
In the present study, we were able to replicate findings from Kalisch et al. (2006a) of hippocampal and (at lower threshold) amygdalar activations during recall of fear memory. Because we here tested for fear recall 72 h after conditioning (as opposed to 24 h in Kalisch et al. [2006a]), we also expected medial prefrontal/anterior cingulate activations. This hypothesis was based on rodent studies of trace and context fear conditioning that have shown a contribution of the MPFC/ACC to the recall of remote fear memories (Takehara et al. 2003, Frankland et al. 2004; Runyan et al. 2004). The rodent literature did not allow us to define a circumscribed medial prefrontal ROI. Using a conservative analytical approach, we therefore took advantage of a large human fMRI literature on anticipatory fear for impending pain, a paradigm where subjects have to recall a cue-pain contingency to prepare for the signaled application of a painful stimulus. Consistent with a crucial role for the MPFC/ACC in fear recall, an earlier review of the literature (see Materials and Methods in Kalisch et al. [2005]) identified the MPFC/ACC (mean coordinates −2, 45, 27) as the area most consistently activated across anticipatory fear studies. Subsequent experiments have successfully employed these coordinates to identify anticipatory activation in the MPFC/ACC (Kalisch et al. 2005, 2006b, 2006c). MPFC/ACC activation in these studies appeared as large clusters spanning both cingulate and medial prefrontal areas, with activation peaks in Brodmann areas 9 and 32. Areas 9 and 32 are connected with medial temporal and cingulate areas (Amaral and Price 1984; Carmichael and Price 1995) and thus in a position to process emotional information. We have been able to demonstrate engagement of this region during emotional appraisal of stimuli, a process that can involve recall of fearful associations (Kalisch et al. 2006b). The functional significance of MPFC/ACC for fear recall is further substantiated by the fact that MPFC/ACC responses in the present study scaled with the magnitude of the CS+-evoked CR, as modeled using trial-by-trial SCRs as a parametric modulator of the neural CS+-evoked response. Thus, our finding of prior DCS treatment enhancing conditioned responding of a functionally defined subregion of the medial frontal wall is in agreement with the previous literature and points to a key role for this specific region in fear recall in humans. Given the indirect derivation of our MPFC/ACC ROI, we cannot exclude a contribution of other medial prefrontal areas identified in the same contrast, notably the bilateral ventral MPFC and the right rostral ACC (see Supplementary Table 1). Activation patterns in the latter foci were similar to the one shown for the MPFC/ACC in Figure 3b (see Supplementary Fig. 3).
We note that DCS effects in the hippocampus and the MPFC/ACC were driven in part by a smaller response to CS− compared with CS+ in the placebo group. Comparison of CS+ and CS− responses between groups (i.e., CS+DCS vs. CS+placebo and CS−DCS vs. CS−placebo) suggested that this effect was mainly due to group differences in CS+ responses rather than in CS− responses (data not shown). Speculatively, this might reflect some degree of inhibition of CS+ responses in the placebo group, perhaps related to online extinction in the absence of negative reinforcement on day 2. Such CS+ < CS− differences have also been observed by Phelps et al. (2004) during extinction. If this interpretation is correct, one could conclude that there must be at least some fear memory recall in the placebo group on day 2, as otherwise there could be no extinction.
Our design, involving postlearning drug administration and a delayed recall test, minimizes potential confounds. First, because the drug was given after learning, the observed facilitation of recall on day 2 cannot be attributed to a potential DCS effect on learning on day 1. And second, the fact that we measured recall 72 h after drug administration makes it highly unlikely that DCS affected recall itself. DCS has a plasma half-life of approximately 10–12 h, that is, the recall test occurred after 6–7 half-lives. With an estimated peak plasma concentration of 125 μM at 1.5 h after intake (compare van Berckel et al. [1998]), plasma levels at test would have dropped to 2 μM or less. We do not know corresponding brain concentrations and their dose–response relationships for DCS and thus cannot absolutely exclude that residual DCS at test affected fear recall. Yet, it is noteworthy that DCS present at test does not affect or may decrease, rather than enhance, fear responses in human and rat models of conditioned or unconditioned fear (Anthony and Nevins 1993; Karcz-Kubicha et al. 1997; Klodzinska and Chojnacka-Wojcik 2000; Heresco-Levy et al. 2002; Walker et al. 2002; Ressler et al. 2004; Mao et al. 2006; Yang et al. 2007). In the light of a pronounced enhancing effect on fear recall in our study, we suggest that the most likely explanation for our results is a facilitatory effect of DCS on fear memory consolidation, but not on recall itself. A potential confound that our design is not able to exclude is a possible influence of adverse drug side effects, which were reported by a majority of subjects in the DCS group (see Materials and Methods), on memory consolidation. Adverse side effects may induce stress and a concomitant rise in stress hormone levels, factors known to influence emotional memory (McGaugh 2004). However, a facilitatory role of endogenous cortisol on fear memory consolidation is observed only in male subjects and in subjects with high cortisol levels (Zorawski et al. 2006), suggesting a rather minor impact of this factor in our (random female) sample. Furthermore, subjects usually reported onset of side effects several hours after the experiment, that is, after the theoretical critical time window for NMDA-dependent memory consolidation of 1–2 h postlearning, as determined in rats (Scavio et al. 1992). A more plausible explanation for the observed effects therefore is DCS action on the NMDA receptor.
In rats, DCS facilitates extinction learning (Walker et al. 2002). The effect is due to the enhancement of extinction memory consolidation as the drug does not improve within-session extinction learning but recall of the extinction memory at a later test, an effect that is also observed when the drug is given immediately after extinction training (Ledgerwood et al. 2003; for recent reviews, see Davis et al. [2006] and Vervliet [2007]). DCS has been successfully employed to augment extinction-based (i.e., exposure) therapy of height phobia (Ressler et al. 2004), social phobia (Hofmann et al. 2006), and obsessive-compulsive disorder (Kushner et al. 2007). However, so far, there is no proof that, in humans, DCS acts by facilitating extinction memory consolidation. One experimental conditioning study failed to show any beneficial effect of DCS on skin conductance and shock expectancy measures of extinction memory (Guastella et al. 2007), possibly due to a floor effect related to very rapid and near-complete fear extinction already in the placebo group. From our data, we are unable to draw any conclusions about potential DCS effects on extinction memory consolidation as there was no evidence of recall of extinction memory on day 2 in the first place. In the placebo group, subjects showed no behavioral signs of recall of fear memory in the conditioning context on day 2, a necessary comparison condition against which to measure loss of fear (i.e., extinction) in the extinction context. In the DCS group, although there was apparent fear memory recall in the conditioning context, very similar CRs were observed in the extinction context as well. There are at least 2 potential reasons for this absence of extinction effects on day 2 that are in contrast to our previous study that showed reduced CRs in an extinction context at test (Kalisch et al. 2006a). First, to avoid a potential floor effect, we used only 1 extinction block bracketed by 2 conditioning blocks (A1, A2) on day 1, whereas in the previous study, the last conditioning block (A2) had been followed by another extinction block. Thus, despite evidence for extinction learning on day 1, this may have been insufficient to create a (stable) extinction memory manifesting on day 2. Second, the acquisition-test interval was 72 h in this study compared with 24 h in the previous study, a modification we introduced to guarantee a drug-free state at test. However, this may effectively have contributed to a further decline of any remaining extinction memory trace (and also of any remaining fear memory trace in the placebo group). Other potential reasons for an absence of extinction memory recall in the DCS group are interactions of DCS with menstrual cycle or subjects’ potential alcohol consumption (Trevisan et al. 2008) that might theoretically affect (impair) extinction memory consolidation more strongly than fear memory consolidation. Finally, it cannot be excluded that, particularly in humans, the NMDA receptor subtypes involved in fear versus extinction memory consolidation differ in terms of their subunit composition and, hence, in their DCS response. DCS might thus preferentially enhance fear memory consolidation in humans, although the above clinical results would argue against this interpretation. As a result of these shortcomings of the study, the question whether and how DCS interacts with extinction learning in humans awaits further experimental clarification.
Given a high intraindividual trial-by-trial variability in CR magnitude, as is commonly observed in human fear conditioning studies, we modeled CS-evoked neural responses as proportional to trial-by-trial CR magnitude, determined using our skin conductance index of conditioning (see Materials and Methods). This method should theoretically be more sensitive in detecting CR-related brain activity than a simple categorical analysis that ignores the possibility of fluctuations in CR magnitude over time. The method should therefore reduce the likelihood of a type II error while not compromising on type I error probability. Similar approaches, modeling an exponential (Buchel et al. 1998) or stepwise (LaBar et al. 1998) decline in CR magnitude over the course of the experiment on purely theoretical grounds, have allowed for identifying the amygdala as a major component of the human fear conditioning circuitry in the first fMRI studies on fear conditioning. More recently, researchers have tended to use behavioral data-driven approaches (e.g., Seymour et al. 2004; Carter et al. 2006; Kalisch et al. 2006a) as these generate biologically more plausible predictions of activity time courses.
We have presented evidence suggesting involvement of NMDA receptors in the consolidation of human fear memory. Our findings will require corroboration from experiments with an NMDA antagonist such as ketamine, in particular because DCS, although safe, well tolerable, and therefore highly suitable for human experiments, is only a partial agonist. Likewise, future experiments should use a range of doses of DCS to establish dose dependency. An important topic for future research will be how the NMDA system interacts with other neurotransmitter and hormone systems, such as the stress hormone system, known to be crucial for memory consolidation (McGaugh 2004). Our research opens the possibility of using psychopharmacological manipulations in conjunction with fMRI to provide insights into therapeutic strategies for treating mental conditions associated with memory impairment or pathologically enhanced memories.
Supplementary Material
Supplementary material can be found at http://www.cercor.oxfordjournals.org/.
Funding
Wellcome Trust Programme Grant to R.J.D.; European Union Presencia Programme Grant to R.J.D. and R.K.
Supplementary Material
Acknowledgments
We thank O. Josephs, B. Pleger, B. Draganski, R. Weil, M. Gray, N. Weiskopf, P. Aston, and E. Featherstone for help with experiments and F. Eippert and T. Sommer-Blöchl for comments on an earlier draft of the manuscript. Conflict of Interest: None declared.
References
- Amaral DG, Price JL. Amygdalo-cortical projections in the monkey (Macaca fascicularis) J Comp Neurol. 1984;230:465–496. doi: 10.1002/cne.902300402. [DOI] [PubMed] [Google Scholar]
- Anthony EW, Nevins ME. Anxiolytic-like effects of N-methyl-D-aspartate-associated glycine receptor ligands in the rat potentiated startle test. Eur J Pharmacol. 1993;250:317–324. doi: 10.1016/0014-2999(93)90397-z. [DOI] [PubMed] [Google Scholar]
- Ashburner J, Friston K, Penny W. Imaging neuroscience—theory and analysis. In: Frackowiak RS, Friston KJ, Frith C, Dolan RJ, Price CJ, editors. Human brain function. 2nd ed. San Diego (CA): Academic Press; 2004. pp. 599–1104. [Google Scholar]
- Bauer EP, Schafe GE, LeDoux JE. NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci. 2002;22:5239–5249. doi: 10.1523/JNEUROSCI.22-12-05239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair HT, Huynh VK, Vaz VT, Van J, Patel RR, Hiteshi AK, Lee JE, Tarpley JW. Unilateral storage of fear memories by the amygdala. J Neurosci. 2005;25:4198–4205. doi: 10.1523/JNEUROSCI.0674-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond A, Lader M. The use of analogue scales in rating subjective feelings. Br J Med Psychol. 1974;47:211–218. [Google Scholar]
- Bouton ME. Context and behavioral processes in extinction. Learn Mem. 2004;11:485–494. doi: 10.1101/lm.78804. [DOI] [PubMed] [Google Scholar]
- Buchel C, Morris J, Dolan RJ, Friston KJ. Brain systems mediating aversive conditioning: an event-related fMRI study. Neuron. 1998;20:947–957. doi: 10.1016/s0896-6273(00)80476-6. [DOI] [PubMed] [Google Scholar]
- Carmichael ST, Price JL. Limbic connections of the orbital and medial prefrontal cortex in macaque monkeys. J Comp Neurol. 1995;363:615–641. doi: 10.1002/cne.903630408. [DOI] [PubMed] [Google Scholar]
- Carter RM, O'Doherty JP, Seymour B, Koch C, Dolan RJ. Contingency awareness in human aversive conditioning involves the middle frontal gyrus. Neuroimage. 2006;29:1007–1012. doi: 10.1016/j.neuroimage.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Corcoran KA, Desmond TJ, Frey KA, Maren S. Hippocampal inactivation disrupts the acquisition and contextual encoding of fear extinction. J Neurosci. 2005;25:8978–8987. doi: 10.1523/JNEUROSCI.2246-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran KA, Maren S. Factors regulating the effects of hippocampal inactivation on renewal of conditional fear after extinction. Learn Mem. 2004;11:598–603. doi: 10.1101/lm.78704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchley HD, Mathias CJ, Dolan RJ. Fear conditioning in humans: the influence of awareness and autonomic arousal on functional neuroanatomy. Neuron. 2002;33:653–663. doi: 10.1016/s0896-6273(02)00588-3. [DOI] [PubMed] [Google Scholar]
- Davis M, Ressler K, Rothbaum BO, Richardson R. Effects of D-cycloserine on extinction: translation from preclinical to clinical work. Biol Psychiatry. 2006;60:369–375. doi: 10.1016/j.biopsych.2006.03.084. [DOI] [PubMed] [Google Scholar]
- Deichmann R, Gottfried JA, Hutton C, Turner R. Optimized EPI for fMRI studies of the orbitofrontal cortex. Neuroimage. 2003;19:430–441. doi: 10.1016/s1053-8119(03)00073-9. [DOI] [PubMed] [Google Scholar]
- Deichmann R, Schwarzbauer C, Turner R. Optimisation of the 3D MDEFT sequence for anatomical brain imaging: technical implications at 1.5 and 3 T. Neuroimage. 2004;21:757–767. doi: 10.1016/j.neuroimage.2003.09.062. [DOI] [PubMed] [Google Scholar]
- Delamater AR. Experimental extinction in Pavlovian conditioning: behavioural and neuroscience perspectives. Q J Exp Psychol B. 2004;57:97–132. doi: 10.1080/02724990344000097. [DOI] [PubMed] [Google Scholar]
- Duvernoy HM. The human brain: surface, blood supply, and three-dimensional sectional anatomy. Wien (Austria): Springer; 1999. [Google Scholar]
- Ekman P, Friesen WV. Pictures of facial affect. Palo Alto (CA): Consulting Psychologists; 1976. [Google Scholar]
- Frankland PW, Bontempi B, Talton LE, Kaczmarek L, Silva AJ. The involvement of the anterior cingulate cortex in remote contextual fear memory. Science. 2004;304:881–883. doi: 10.1126/science.1094804. [DOI] [PubMed] [Google Scholar]
- Friston KJ, Holmes AP, Worsley KJ, Poline JB, Frith CD, Frackowiak RSJ. Statistical parametric maps in functional imaging: a general linear approach. Hum Brain Mapp. 1995;2:189–210. [Google Scholar]
- Friston KJ, Jezzard P, Turner R. Analysis of functional MRI timeseries. Hum Brain Mapp. 1994;1:153–171. [Google Scholar]
- Gould TJ, McCarthy MM, Keith RA. MK-801 disrupts acquisition of contextual fear conditioning but enhances memory consolidation of cued fear conditioning. Behav Pharmacol. 2002;13:287–294. doi: 10.1097/00008877-200207000-00005. [DOI] [PubMed] [Google Scholar]
- Guastella AJ, Lovibond PF, Dadds MR, Mitchell P, Richardson R. A randomized controlled trial of the effect of D-cycloserine on extinction and fear conditioning in humans. Behav Res Ther. 2007;45:663–672. doi: 10.1016/j.brat.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Heresco-Levy U, Kremer I, Javitt DC, Goichman R, Reshef A, Blanaru M, Cohen T. Pilot-controlled trial of D-cycloserine for the treatment of post-traumatic stress disorder. Int J Neuropsychopharmacol. 2002;5:301–307. doi: 10.1017/S1461145702003061. [DOI] [PubMed] [Google Scholar]
- Hetem LA, Danion JM, Diemunsch P, Brandt C. Effect of a subanesthetic dose of ketamine on memory and conscious awareness in healthy volunteers. Psychopharmacology (Berl) 2000;152:283–288. doi: 10.1007/s002130000511. [DOI] [PubMed] [Google Scholar]
- Hofmann SG, Meuret AE, Smits JAJ, Simons NM, Pollack MH, Eisenmenger K, Shiekh M, Otto MW. Augmentation of exposure therapy for social anxiety disorder with D-Cycloserine. Arch Gen Psychiatry. 2006;63:298–304. doi: 10.1001/archpsyc.63.3.298. [DOI] [PubMed] [Google Scholar]
- Honey GD, Honey RA, Sharar SR, Turner DC, Pomarol-Clotet E, Kumaran D, Simons JS, Hu X, Rugg MD, Bullmore ET, et al. Impairment of specific episodic memory processes by sub-psychotic doses of ketamine: the effects of levels of processing at encoding and of the subsequent retrieval task. Psychopharmacology (Berl) 2005;181:445–457. doi: 10.1007/s00213-005-0001-z. [DOI] [PubMed] [Google Scholar]
- Izquierdo I, Bevilaqua LR, Rossato JI, Bonini JS, Medina JH, Cammarota M. Different molecular cascades in different sites of the brain control memory consolidation. Trends Neurosci. 2006;29:496–505. doi: 10.1016/j.tins.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Ji J, Maren S. Electrolytic lesions of the dorsal hippocampus disrupt renewal of conditional fear after extinction. Learn Mem. 2005;12:270–276. doi: 10.1101/lm.91705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalisch R, Korenfeld E, Stephan KE, Weiskopf N, Seymour B, Dolan RJ. Context-dependent human extinction memory is mediated by a ventromedial prefrontal and hippocampal network. J Neurosci. 2006a;26:9503–9511. doi: 10.1523/JNEUROSCI.2021-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalisch R, Wiech K, Critchley HD, Dolan RJ. Levels of appraisal: a medial prefrontal role in high-level appraisal of emotional material. Neuroimage. 2006b;30:1458–1466. doi: 10.1016/j.neuroimage.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Kalisch R, Wiech K, Critchley HD, Seymour B, O'Doherty JP, Oakley DA, Allen P, Dolan RJ. Anxiety reduction through detachment: subjective, physiological, and neural effects. J Cogn Neurosci. 2005;17:874–883. doi: 10.1162/0898929054021184. [DOI] [PubMed] [Google Scholar]
- Kalisch R, Wiech K, Herrmann K, Dolan RJ. Neural correlates of self-distraction from anxiety and a process model of cognitive emotion regulation. J Cogn Neurosci. 2006c;18:1266–1276. doi: 10.1162/jocn.2006.18.8.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karcz-Kubicha M, Jessa M, Nazar M, Plaznik A, Hartmann S, Parsons CG, Danysz W. Anxiolytic activity of glycine-B antagonists and partial agonists—no relation to intrinsic activity in the patch clamp. Neuropharmacology. 1997;36:1355–1367. doi: 10.1016/s0028-3908(97)00130-5. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- Klodzinska A, Chojnacka-Wojcik E. Anticonflict effect of the glycineB receptor partial agonist, D-cycloserine, in rats. Pharmacological analysis. Psychopharmacology (Berl) 2000;152:224–228. doi: 10.1007/s002130000547. [DOI] [PubMed] [Google Scholar]
- Kushner MG, Kim SW, Donahue C, Thuras P, Adson D, Kotlyar M, McCabe J, Peterson J, Foa EB. D-cycloserine augmented exposure therapy for obsessive-compulsive disorder. Biol Psychiatry. 2007;62:835–838. doi: 10.1016/j.biopsych.2006.12.020. [DOI] [PubMed] [Google Scholar]
- LaBar KS, Gatenby JC, Gore JC, LeDoux JE, Phelps EA. Human amygdala activation during conditioned fear acquisition and extinction: a mixed-trial fMRI study. Neuron. 1998;20:937–945. doi: 10.1016/s0896-6273(00)80475-4. [DOI] [PubMed] [Google Scholar]
- LaBar KS, Phelps EA. Reinstatement of conditioned fear in humans is context dependent and impaired in amnesia. Behav Neurosci. 2005;119:677–686. doi: 10.1037/0735-7044.119.3.677. [DOI] [PubMed] [Google Scholar]
- Ledgerwood L, Richardson R, Cranney J. Effects of D-cycloserine on extinction of conditioned freezing. Behav Neurosci. 2003;117:341–349. doi: 10.1037/0735-7044.117.2.341. [DOI] [PubMed] [Google Scholar]
- Lee JL, Milton AL, Everitt BJ. Reconsolidation and extinction of conditioned fear: inhibition and potentiation. J Neurosci. 2006;26:10051–10056. doi: 10.1523/JNEUROSCI.2466-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao SC, Hsiao YH, Gean PW. Extinction training in conjunction with a partial agonist of the glycine site on the NMDA receptor erases memory trace. J Neurosci. 2006;26:8892–8899. doi: 10.1523/JNEUROSCI.0365-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Fanselow MS. Electrolytic lesions of the fimbria/fornix, dorsal hippocampus, or entorhinal cortex produce anterograde deficits in contextual fear conditioning in rats. Neurobiol Learn Mem. 1997;67:142–149. doi: 10.1006/nlme.1996.3752. [DOI] [PubMed] [Google Scholar]
- Martin SJ. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu Rev Neurosci. 2004;27:1–28. doi: 10.1146/annurev.neuro.27.070203.144157. [DOI] [PubMed] [Google Scholar]
- Morris JS, Ohman A, Dolan RJ. Conscious and unconscious emotional learning in the human amygdala. Nature. 1998;393:467–470. doi: 10.1038/30976. [DOI] [PubMed] [Google Scholar]
- Myers KM, Davis M. Behavioral and neural analysis of extinction. Neuron. 2002;36:567–584. doi: 10.1016/s0896-6273(02)01064-4. [DOI] [PubMed] [Google Scholar]
- Parwani A, Weiler MA, Blaxton TA, Warfel D, Hardin M, Frey K, Lahti AC. The effects of a subanesthetic dose of ketamine on verbal memory in normal volunteers. Psychopharmacology (Berl) 2005;183:265–274. doi: 10.1007/s00213-005-0177-2. [DOI] [PubMed] [Google Scholar]
- Pavlov IP. Conditioned reflexes: an investigation of the physiological activity of the cerebral cortex. Oxford (United Kingdom): Oxford University Press; 1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penny W, Holmes AP. Random effects analysis. In: Frackowiak RS, Friston KJ, Frith C, Dolan RJ, Price CJ, editors. Human brain function. 2nd ed. San Diego (CA): Academic Press; 2004. pp. 843–850. [Google Scholar]
- Phelps EA, Delgado MR, Nearing KI, LeDoux JE. Extinction learning in humans: role of the amygdala and vmPFC. Neuron. 2004;43:897–905. doi: 10.1016/j.neuron.2004.08.042. [DOI] [PubMed] [Google Scholar]
- Ressler KJ, Rothbaum BO, Tannenbaum L, Anderson P, Graap K, Zimand E, Hodges L, Davis M. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61:1136–1144. doi: 10.1001/archpsyc.61.11.1136. [DOI] [PubMed] [Google Scholar]
- Rodrigues SM, Schafe GE, LeDoux JE. Intra-amygdala blockade of the NR2B subunit of the NMDA receptor disrupts the acquisition but not the expression of fear conditioning. J Neurosci. 2001;21:6889–6896. doi: 10.1523/JNEUROSCI.21-17-06889.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin MA, Berlese DB, Stiegemeier JA, Volkweis MA, Oliveira DM, dos Santos TL, Fenili AC, Mello CF. Intra-amygdala administration of polyamines modulates fear conditioning in rats. J Neurosci. 2004;24:2328–2334. doi: 10.1523/JNEUROSCI.1622-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runyan JD, Moore AN, Dash PK. A role for prefrontal cortex in memory storage for trace fear conditioning. J Neurosci. 2004;24:288–1295. doi: 10.1523/JNEUROSCI.4880-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scavio MJ, Clift PS, Wills JC. Posttraining effects of amphetamine, chlorpromazine, ketamine, and scopolamine on the acquisition and extinction of the rabbit's conditioned nictitating membrane response. Behav Neurosci. 1992;106:900–908. doi: 10.1037//0735-7044.106.6.900. [DOI] [PubMed] [Google Scholar]
- Schafe GE, Doyere V, LeDoux JE. Tracking the fear engram: the lateral amygdala is an essential locus of fear memory storage. J Neurosci. 2005;25:10010–10014. doi: 10.1523/JNEUROSCI.3307-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielberger CD. State-trait anxiety inventory (form Y) Redwood City (CA): Mind Garden; 1983. [Google Scholar]
- Seymour B, O'Doherty JP, Dayan P, Koltzenburg M, Jones AK, Dolan RJ, Friston KJ, Frackowiak RS. Temporal difference models describe higher-order learning in humans. Nature. 2004;429:664–667. doi: 10.1038/nature02581. [DOI] [PubMed] [Google Scholar]
- Sheinin A, Shavit S, Benveniste M. Subunit specificity and mechanism of action of NMDA partial agonist D-cycloserine. Neuropharmacology. 2001;41:151–158. doi: 10.1016/s0028-3908(01)00073-9. [DOI] [PubMed] [Google Scholar]
- Sotres-Bayon F, Bush DE, LeDoux JE. Acquisition of fear extinction requires activation of NR2B-containing NMDA receptors in the lateral amygdala. Neuropsychopharmacology. 2007;32:1929–1940. doi: 10.1038/sj.npp.1301316. [DOI] [PubMed] [Google Scholar]
- Stewart LS, McKay BE. Acquisition deficit and time-dependent retrograde amnesia for contextual fear conditioning in agmatine-treated rats. Behav Pharmacol. 2000;11:93–97. doi: 10.1097/00008877-200002000-00011. [DOI] [PubMed] [Google Scholar]
- Takehara K, Kawahara S, Kirino Y. Time-dependent reorganization of the brain components underlying memory retention in trace eyeblink conditioning. J Neurosci. 2003;23:9897–9905. doi: 10.1523/JNEUROSCI.23-30-09897.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevisan L, Petrakis IL, Pittman B, Gueorguieva R, D'Souza DC, Perry E, Limoncelli D, Krystal JH. Absence of significant interactive effects of high-dose D-cycloserine and ethanol in healthy human subjects: preliminary insights into ethanol actions at the glycine B site of NMDA glutamate receptors. Alcohol Clin Exp Res. 2008;32:36–42. doi: 10.1111/j.1530-0277.2007.00543.x. [DOI] [PubMed] [Google Scholar]
- van Berckel BN, Lipsch C, Gispen-de Wied C, Wynne HJ, Blankenstein MA, van Ree JM, Kahn RS. The partial NMDA agonist D-cycloserine stimulates LH secretion in healthy volunteers. Psychopharmacology (Berl) 1998;138:190–197. doi: 10.1007/s002130050662. [DOI] [PubMed] [Google Scholar]
- Vervliet B. Learning and memory in conditioned fear extinction: effects of D-cycloserine. Acta Psychol. 2007;127:601–613. doi: 10.1016/j.actpsy.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Walker DL, Ressler KJ, Lu KT, Davis M. Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of D-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci. 2002;22:2343–2351. doi: 10.1523/JNEUROSCI.22-06-02343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YL, Chao PK, Ro LS, Wo YY, Lu KT. Glutamate NMDA receptors within the amygdala participate in the modulatory effect of glucocorticoids on extinction of conditioned fear in rats. Neuropsychopharmocology. 2007;32:1042–1051. doi: 10.1038/sj.npp.1301215. [DOI] [PubMed] [Google Scholar]
- Zhu M, Nix DE, Adam RD, Childs JM, Peloquin CA. Pharmacokinetics of cycloserine under fasting conditions and with high-fat meal, orange juice, and antacids. Pharmacotherapy. 2001;21:891–897. doi: 10.1592/phco.21.11.891.34524. [DOI] [PubMed] [Google Scholar]
- Zorawski M, Blanding NQ, Kuhn CM, LaBar KS. Effects of stress and sex on acquisition and consolidation of human fear conditioning. Learn Mem. 2006;13:441–450. doi: 10.1101/lm.189106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}