

Abstract
Motivation: Promoter-driven reporter genes, notably luciferase and green fluorescent protein, provide a tool for the generation of a vast array of time-course data sets from living cells and organisms. The aim of this study is to introduce a modeling framework based on stochastic differential equations (SDEs) and ordinary differential equations (ODEs) that addresses the problem of reconstructing transcription time-course profiles and associated degradation rates. The dynamical model is embedded into a Bayesian framework and inference is performed using Markov chain Monte Carlo algorithms.
Results: We present three case studies where the methodology is used to reconstruct unobserved transcription profiles and to estimate associated degradation rates. We discuss advantages and limits of fitting either SDEs ODEs and address the problem of parameter identifiability when model variables are unobserved. We also suggest functional forms, such as on/off switches and stimulus response functions to model transcriptional dynamics and present results of fitting these to experimental data.
Contact: b.f.finkenstadt@warwick.ac.uk
Supplementary Information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Imaging data from luciferase (LUC) and green fluorescent protein (GFP) reporters combined with fluorescent tagging of protein can provide very high quality data with good temporal resolution (Millar et al., 1995; Nelson et al., 2004). In this case the actual imaging time series is approximately proportional to the abundance of an artificial protein. The underlying transcriptional dynamics are unobserved and are masked by two degradation processes, namely of reporter mRNA and reporter protein. In this study, we address the problem of back-calculating from the observed protein activity to the hidden transcriptional dynamics where it is of interest to estimate the associated rates of degradation as part of the analysis. We formulate a probability model based on (stochastic) differential equations which provides the mechanistic rules for the back-calculation. In practise heterogeneous datasets may be available from different experiments which contain information about the transcription process and model parameters. Data sources may be of different quality and time resolution, as well as from single cells or an aggregated population of cells. Longitudinal measurements are discrete in time and can be irregularly spaced or on different time scales for different variables. Other realistic shortcomings of the data are that time-course measurements may not correspond to the same biological sample, or data on different variables may not be matched in time which would be preferable for fitting a multivariate dynamical model. As the quality and quantity of such datasets supports more or less complex modeling approaches, we consider both stochastic differential equations (SDEs) and ordinary differential equations (ODEs) with measurement noise. Information on rate constants may be incorporated through prior distributions in a Bayesian approach. We first describe the models and the statistical methods used for its inference. Then, we present three case studies each with the aim of reconstructing transcription and inferring any identifiable degradation rates from reporter gene data using available heterogeneous sources of data. These case studies serve to demonstrate the adaption of the methodology to different experimental scenarios.
2 MODELS AND INFERENCE
It is now well understood that, because of the stochastic nature of reaction events and the presence of internal noise due to the fluctuations in the molecular environment of the cell, regulatory and signalling systems are intrinsically stochastic. To develop a stochastic model one can attempt to model the individual stochastic events involved, such as binding of the transcription factors, the assembly and initiation of the polymerase and transcription. Although an exact simulation algorithm of the corresponding stochastic processes is provided by Gillespie (1977, 1992) such models are too detailed for there to be any hope of fitting to current data with its limitations. SDEs provide a good approximation of molecular population systems when one can assume that there is a macroscopic time scale for which (i) the event rates can be regarded as constant and (ii) there are many events of each type. An example of formulating and fitting an autoregulatory feedback system with transcriptional delay as a system of SDEs can be found in Heron et al. (2007). However, if the data are too sparsely sampled in time to reveal information about the volatility process, or if measurements are not realizations of the same continuous stochastic process in a cell, then the assumption of SDEs can be problematic in estimation. Simpler modeling approaches based on ODEs to represent the mean process with an additional stochastic error may provide a useful vehicle for estimation purposes at least in systems that have relatively regular and stable dynamics. The formulation of ODEs to model the dynamics of molecular population processes has become a widespread tool in systems biology [see, for example, systems studied in Goldbeter (2002), Jensen et al. (2003) and (Locke et al., 2005a, 2005b)], and early statistically less rigorous attempts in obtaining kinetic parameters from GFP reporter data can be found in Ronen et al. (2002) and Kalir and Alon (2004).
Here, we consider the following dynamic model as the mechanistic backbone for the reconstruction of transcription profiles from reporter protein data
| (1) |
where M denotes the abundance of mRNA molecules and P denotes the abundance of the corresponding protein. The first equation describes the dynamics of mRNA molecules where transcription is given by a non-negative function τ(t). The second equation states that the protein is synthesized at a rate proportional to the abundance of mRNA. The mRNA and the protein are degraded (or leave their molecular compartment otherwise) at time scales with mean 1/δm and 1/δp, respectively. The aim is to infer the transcription function τ(t) and possibly other rate constants of the system given time-series data proportional to one or both variables of the system. Suppose that we measure M,P proportionally to their population size, sMM(t) for the mRNA and sPP(t) for the reporter protein. Reparameterizing (1) gives a scaled model which is identical to (1) with scaled terms for α and τ (see Supplementary Material). However, degradation rates are not affected by scaling. Let Y={yi}i=1T={M(ti), P(ti)}i=1T denote experimental time-series data observed at discrete time points. In order to obtain a likelihood function that incorporates the mechanistic rules in (1) we consider two approaches. One is the SDE approach where (1) is formulated as an appropriate system of SDEs. This approach is rigorously modeling the volatility of the stochastic dynamics of the kinetic processes provided that the assumptions of the SDE approximation itself are valid. It is very challenging to incorporate additional measurement error unless its variance is known or assumed. The second is the mean ODE approach where we assume that a solution path to (1) represents the mean of a stochastic process whilst the modeler makes assumptions about the probability distribution of the residual process. This approach is less exact than the SDE approach in modeling the volatility of the underlying stochastic interaction between molecules. On the other hand, it naturally deals with measurement error and might also be useful for fitting to datasets which do not comply with the SDE assumption, for example, if data points are averages over replicates, come from different samples and/or represent a population of cells. We now introduce the two approaches and their likelihood derivation in more detail.
SDE approach: here, M and P are random variables of molecular population sizes and the rates of increase and decrease in model (1) are event probabilities of birth and death processes at the individual molecular level. One can derive the following Itô SDEs (see Supplementary Material)
| (2) |
where ζM(t, θ)=τ(t)−δmM(t), ζP(t, θ)=αM(t)−δPP(t), and σM(t)=sM1/2 (τ(t)+δmM(t))1/2, σP(t)=sP1/2 (αM(t)+δPP(t))1/2 are drift and volatility functions, respectively, and WM and WP are independent Wiener processes.1 Here and throughout the article, θ is used to denote a vector of model parameters. If M and P are indirect measurements of molecular populations in the sense that they are proportional to molecular abundance with factors sM, sP then these factors arise as additional parameters in the volatility functions and their estimation will be extremely useful allowing us to calibrate the model to the population level. Given data Y the likelihood function for the diffusion process is
| (3) |
where f(yi+1∣yi;θ) denotes the transition density of yi+1 given yi, that is the joint probability distribution of M(ti+1) and P(ti+1) given present values, under parameter vector θ. The exact transition density function for solutions of SDEs is rarely available in analytical form and usually approximations have to be considered. If the time-step Δ ti=ti+1−ti is small then a good approximation is given by assuming that, conditional on past values,
(*) Increments y(ti+1)−y(ti) are bivariate normal with mean vector ζ(ti)Δ ti and variance matrix Σ(ti)Δti where ζ(ti)=(ζM(ti), ζP(ti)), Σ(ti)=diag(σM2(ti), σP2(ti)) are the drift and volatility functions as defined above.
Thus, for sufficiently small sampling intervals Δti the likelihood function can be approximated by a product of the form
| (4) |
where Φ(x; μ, Σ) denotes the bivariate normal density function with mean vector μ and variance matrix Σ. Justifications for this approximation are given in Kloeden and Platen (1999).
Mean ODE approach: suppose there is a solution path μ(t; θ)=(M(t), P(t); θ) to the system in (1) from unknown initial conditions (M0, P0). Then a natural probabilistic model is to assume that Y has a joint distribution with mean function μ(t; θ) and a variance function σ2(t; θ). The distribution function and variance are specified according to assumptions that the modeler makes about the residual process and measurement error. If the error process is assumed independent then the likelihood in the mean ODE approach is
| (5) |
where θ now incorporates initial conditions (M0, P0) and g is a suitably chosen probability distribution.
Inference: by Bayes' theorem the posterior distribution is
| (6) |
where L is the likelihood function, derived for either the ODE or SDE approach, and π(θ) are prior densities of model parameters. Sampling from the posterior distribution is usually achieved using Markov chain Monte Carlo (MCMC), where each element of θ is updated by using an appropriately constructed Metropolis–Hastings acceptance/rejection scheme based on either random walk or independence proposals (Gamerman and Lopes, 2006). The reason for choosing a Bayesian approach combined with a MCMC algorithm is 2-fold: first, the Bayesian methodology is flexible allowing for portability of inference results between different experimental studies in a well-defined way and this is highly relevant to studies in systems biology. Second, the probabilistic imputation of missing data and/or unobserved variables can be implemented in a straightforward way as part of an MCMC sampler.
Discrete data and unobserved variables: molecular time-series data are discretely measured and it cannot be guaranteed that the sampling interval is small enough for the approximation (*) to work well. A remedy suggested in econometric applications of SDEs (Durham and Gallant, 2002; Elerian et al., 2001) is to augment the observed data by introducing a number of latent or unobserved data points, called a bridge, in-between the measurements with the aim of creating a virtual fine discrete time grid for which the assumption in (*) is valid. The bridges are treated as missing or latent data. Let Y* denote the collection of all latent data. We wish to sample from the joint posterior f(θ, Y*∣Y) of the parameters θ and the latent variables Y* given the data Y, using the fact that, by Bayes' theorem,
| (7) |
where L(Y*, Y∣θ) is the approximated augmented likelihood. This is achieved by sampling in turn from the full-conditional densities of θ∣Y*, Y and Y*∣θ, Y (Tanner and Wong, 1987). Thus, in the framework of an MCMC, one can generate proposal bridge processes and accept these with an appropriately constructed acceptance probability. In practise, we have used (Heron et al., 2007) a bridging method based on an independence sampler suggested by Elerian et al. (2001) (see Supplementary Material). The treatment of other forms of missing data, such as unobserved variables as part of the inference algorithm is theoretically the same. In practise, this is challenging as the dimension of the posterior density in (7) can become very large. We present applications of bridge building and stochastic reconstruction of unobserved processes in our case studies. One also needs to decide upon the size of a virtual sampling interval for which one can safely assume that (*) holds. Since there are no analytical results we base our choice on Monte Carlo studies of simulated systems.
3 CASE STUDIES
3.1 Case study 1: red light pulse experiment
The Arabidopsis thaliana gene Chlorophyll A/B binding Protein 2 CAB2 is regulated by light and the circadian clock (Millar and Kay, 1996). The aim here is to estimate degradation rate of CAB2 mRNA and to reconstruct the transcriptional dynamics of the CAB2:LUC reporter gene as a result of a 20 min red-light induction. At subjective dawn on the 6th day of the experiment (see Supplementary Material for a description of experiment), the grown Arabidopsis seedlings were given a 20 min red light pulse to induce CAB2 expression. Samples were harvested at the indicated time points and total RNA and total protein was extracted. Steady-state levels of LUC mRNA were measured by quantitative PCR (Q-PCR) and an in vitro LUC assay was used to measure LUC activity in the protein samples. Concurrently, red light-pulsed seedlings were also imaged for LUC activity using light sensitive cameras (Millar et al., 1995). This allows the measurement of LUC activity within the same seedlings throughout the entire experiment, whereas the in vitro LUC assays and Q-PCR experiments necessarily sacrificed different samples for each time point. All data are probes from whole leaves (plots of all time series in Supplementary Material) representing cell populations and the activity of the clock gene can be assumed to be synchronized between cells by the light pulse. There are three replicates of each measurement variable sampled every half hour for a length of 7 h. Matching control replicates that have not been subject to light induction were sampled for the same time length albeit more sparsely for the Q-PCR and in vitro assay data.
Assuming that molecular populations all scale differently with the Q-PCR, in vitro and in vivo imaging data, we use (1) to describe the dynamics of mRNA and imaged LUC protein and add a third equation
| (8) |
which represents the protein dynamics measured by the in vitro LUC protein assays (see Supplementary Material for full model statement). The two protein equations are identical except for differently scaled translation rates αP and αPv. Furthermore, a constant cP is added to the imaging data to represent some threshold level at which the camera is able to detect a signal. To specify a form for the transcription τ(t) consider an indicator function L(t)=1 for the time of the red light pulse, and L(t)=0 otherwise [L(t)=0 for all control experiments]. The response of mRNA transcription to the stimulus can then be modeled as a convolution of L(t) and d(u) which is a probability density for the waiting time u between the pulse and the initiation of transcription, i.e.
| (9) |
where  represents a baseline transcription. We take d(u) to be a Gamma density with mean μΓ and SD σΓ to be estimated. The specification in (9) is motivated by the fact that it successfully reproduced the qualitative features observed in the data in preliminary model simulations and because d is flexible. Since data are from aggregated cell populations, the imaged protein data are very smooth and successive data points of the Q-PCR and in vitro time series come from different samples of cell populations, we choose to fit the model using the mean ODE approach with independent error. To ensure all variables are strictly non-negative, we used an independent Gamma distribution for g in the likelihood (9) for each of the three variables where parameters were specified to have mean process equal to an ODE solution and time constant variance σM2, σP2, σPv2. Applying (5) the likelihood of replicate r=1,2,3 is
represents a baseline transcription. We take d(u) to be a Gamma density with mean μΓ and SD σΓ to be estimated. The specification in (9) is motivated by the fact that it successfully reproduced the qualitative features observed in the data in preliminary model simulations and because d is flexible. Since data are from aggregated cell populations, the imaged protein data are very smooth and successive data points of the Q-PCR and in vitro time series come from different samples of cell populations, we choose to fit the model using the mean ODE approach with independent error. To ensure all variables are strictly non-negative, we used an independent Gamma distribution for g in the likelihood (9) for each of the three variables where parameters were specified to have mean process equal to an ODE solution and time constant variance σM2, σP2, σPv2. Applying (5) the likelihood of replicate r=1,2,3 is
| (10) |
where yir,R is the vector of observed data points i=1,…,TR for variables M, P, Pv for replicate r under the red light experiment, yjr,C denotes observed data points j=1,…,TC for the corresponding control experiment and g is a product of Gamma densities. The ODE model was fitted to each of the replicates r=1,2,3 and to the average of the replicates where prior distributions for all parameters were chosen to be uninformative. Results of posterior estimates are summarized in Table 1 and the model fit can be seen in Figure 1. The mean delay time between light induction and transcription is about 2 h with almost all transcription happening between 0.8 h and 3.2 h after the pulse. Convergence of the Markov chains for parameters associated with the Gamma delay is relatively quick and precise. Chains for αM and δM are correlated and convergence for these is slower. The half life of LUC mRNA is estimated to be around 0.5 h with some small variation between replicates. In contrast, the chains for δL converged quickly due to the abundance and smoothness of the imaging data. Protein half life was estimated to be around 2–2.5 h. Although the control data do not seem very dynamic they are useful in inferring the base rates of transcription and translation. If the control series are omitted from the analysis these rates were estimated with considerably less precision and slower convergence due to correlations.
Table 1.
Case 1: posterior results for selected parameters
| Parameter | Average | r1 | r2 | r3 |
|---|---|---|---|---|
| δM | 1.542 (0.019) | 1.726 (0.044) | 1.417 (0.121) | 3.526 (0.315) |
| half-life | 0.45 h | 0.4 h | 0.49 h | 0.2 h |
| μΓ | 2.008 (0.011) | 2.101 (0.014) | 1.902 (0.045) | 2.362 (0.0289) |
| σΓ | 0.631 (0.013) | 0.692 (0.014) | 0.686 (0.039) | 0.723 (0.0217) |
 |
0.012 (0.001) | 0.014 (0.001) | 0.014 (0.002) | 0.013 (0.002) |
| δP | 0.305 (0.0045) | 0.286 (0.0040) | 0.272 (0.010) | 0.365 (0.0093) |
| half-life | 2.27 h | 2.42 h | 2.5 h | 1.9 h |
Posterior means and SDs of selected estimated parameters (See Supplementary Material for all parameters), where the red light pulse model was fitted to average data and to single replicate datasets denoted by r1, r2, r3. Estimated rates are per hour. Degradation rates are translated into half-lives as follows: half-life (in hours)=ln(2)/degradation rate (per hour).
Fig. 1.

This figure shows mean ODE fit for average data (data points given by big dots) of red light pulse experiment. LUC mRNA (top left), LUC activity in vitro (bottom left) and imaging the luminescence from LUC protein (top right) under two experimental conditions: with and without red light pulse. Solid lines give the mean ODE fit using mean posterior estimates for the parameters. The 95% credible intervals (dashed lines) are shown for the control experiments. The reconstructed transcription profile τ(t) is shown in the bottom right panel (the area between dashed lines gives 95% central values of the transcription profile for 10 000 iterations of the Markov chain).
3.2 Case study 2: a switch model for CCA1
The Circadian Clock associated 1 (CCA1) gene in A.thaliana has been identified as one of the core genes of the circadian clock (Wang and Tobin, 1998). In this case study, we show results for the reconstruction of an ON/OFF switching transcription profile from the following two experimental datasets:
Native mRNA Q-PCR data: Q-PCR measurements were taken at 2 h intervals over 72 h on CCA1 mRNA entrained under a photoperiod of 18 h before being released into constant light. The data used are an average of concentrations relative to the start of two biological replicates.
Protein imaging: high resolution imaging data for a different experiment with identical conditions as for data (1) were sampled at 1.5 h intervals over a length of 91.5 h on LUC protein activity resulting from LUC reporter constructs fused to the CCA1 promoter. Similar to case study 1, all data come from whole leaves and thus represent a population of cells where the activity of the clock gene is synchronized between cells during the exposure to dark, light cycles during the entrainment period (see Supplementary Material for further details of experiment). The data used are an average of concentrations relative to the start of 20 replicates.2
No data were available for the CCA1:LUC mRNA. However, if we assume that CCA1:LUC and CCA1 mRNA have the same transcriptional dynamics, then the available two time series are connected in a dynamic model with three variables where LUC mRNA and LUC protein dynamics are described by (1) and a further equation
| (11) |
is added for the native CCA1 mRNA. We assume that observed variables are proportional to Mg and P populations with scaling factors sMg and sP, while M is unobserved. To describe the oscillatory nature of the data, we consider an ON/OFF switching function for the transcription τ(t)=τon if transcription is active at time t, and τ(t)=τoff if transcription is inactive. This function has the advantage of being interpretable and parsimonious. If it produces realistic oscillations then its simple structure makes it an interesting ingredient to models of larger networks. Let Sw=(s1,…,sR) where s1 < s2 < ··· < sR are the times at which a switching between an ON and OFF state occurs. They are estimated as part of the MCMC algorithm where we assume that the number of switches and the initial state are known.3 To set the phase of the clock both the data series experienced a light–dark (LD) cycle of 18 h of L and 6 h of D at the beginning of the sampling period and this seems to generate a higher amplitude. We allow for this by setting the transcription on-rate to pdτon during the first 35 h (allowing also for some delayed effect of the dark period). For purpose of estimation, the mean ODE approach will be appropriate for similar reasons as case study 1. However, an SDE approach is a superior theoretical model that should be considered even if data do not (yet) strictly comply with its underlying assumptions. We use this case study to show the application of both approaches.
SDE approach: consider a system of SDEs formulated analogously to (2). Since M is unobserved it can be imputed stochastically as realizations of the SDE but the cost of computation is high. Simulation studies suggested that the more practicable way of imputing M as solution to an ODE from an initial condition M0 to be estimated had no discernable impact on our inference results here. In order to fit an SDE model to discrete data points for Mg and P, we augment the coarse grid to a virtually fine grid [for which assumption (*) is valid] by imputing auxiliary data in the form of bridges. Let θ=(Sw, τon, τoff, δMg, M0, δM, SMg, α, δP, SP) denote the vector of unknown parameters and let Mg* and P* be the auxiliary data for Mg and P, respectively. Then according to (7) the posterior distribution for the unknown (Θ, Mg*, P* is given by
where we approximate L(Mg, P, Mg*, P*∣θ) with the augmented likelihood in (4) for small sampling intervals for all observed and auxiliary data, i.e. y=(Mg, P, Mg*, P*). More details of the SDE inference algorithm are provided in the Supplementary Material.
Mean ODE approach: here the likelihood is given by (5) where the unobserved variable M is reconstructed as a solution of an ODE from an initial condition M0 to be estimated. The density g was specified to be the product of two independent normal distributions with mean equal to the joint ODE solutions for Mg and P and with variance parameters σMg2 and σP2. We have set τoff=0 for the off-time as initial estimations showed that it was not different from zero.4 As the variables are concentrations relative to initial conditions the ODE solutions are assumed to start at one. Thus, the parameter vector for the mean ODE approach is θ=(Sw, τon, τoff, δMg, M0, δM, α, δP, σMg, σP).
To ensure identifiability in both estimation approaches the prior distribution for CCA1:LUC mRNA degradation δM has to be informative. We hence used a Gamma distribution with mean 1.542 and SD 0.019, corresponding to the results in Table 1. All other priors were taken independently uniform in an attempt to estimate all remaining parameters only from the experimental data at hand. Posterior estimates are given in Table 2. Figure 2 shows the transcription profiles and model fits for both approaches. The plots suggest that the switch model is remarkably able at reproducing the observed oscillations. The main feature of the reconstructed profiles is that the inactive times (around 15–18 h) are at least twice as long as the active times (around 7 h) and this produces the pronounced asymmetric cycles in the protein and mRNA time series. The estimates also suggest that there is a shorter but larger burst of transcription during the dark period. Both approaches deliver similar posterior rates for degradation. Our results for CCA1 mRNA degradation are in remarkable agreement with the analysis in Yakir et al. (2007) whose estimates correspond to 0.23 in darkness to 0.46 in light for δMg. Both approaches reliably estimate the half-life of the LUC protein to be around 9.5 h. This is surprisingly long and is probably due to a lack in provision of luciferin. The most notable difference between the two approaches lies in the variance estimation. The SDE approach has to deal with the estimation of the two scaling parameters, sP and sMg. We find that their identification from the experimental data is problematic as convergence could not achieved although this did not affect convergence of all other parameters. The two scaling parameters were thus sampled within some chosen bounded region of parameter space. In particular, in order for the bridge sampling to remain numerically stable for low values of the mRNA series, the sampling of sMg had to be bounded to artificially low values. The identifiability problem of the scaling parameters leads to problems in realistically quantifying the volatility. The estimated intervals in Figure 2 illustrate this for the mRNA series. For the mean ODE approach variability is measured by the posterior standard error of the fit similar to a regression and the graph shows that predictions can be made more precisely about the protein dynamics than about the native mRNA. This is reflecting the fact that the protein data are more aggregated and smoother time series than the mRNA series.
Table 2.
Case 2: posterior results for selected parameters
| (δMg) | (δM) | (δP) | |
|---|---|---|---|
| SDE | 0.426 (0.0043) | 1.54 (0.019) | 0.072 (0.0057) |
| ODE | 0.313 (0.0273) | 1.42 (0.101) | 0.075 (0.0018) |
Posterior mean and standard error estimates of selected parameters of model in case 2 using the SDE and mean ODE approach. All rates are per hour. Estimates for all parameters and switch times are provided in Supplementary Material.
Fig. 2.

Results of fitting SDEs (left) and ODEs (right) in case study 2. Top panel shows the mean reconstructed transcription profile τ(t) using the switch approximation. Middle panel shows results for Mg. Bottom panel gives results for P. Big dots are experimental data for Mg (middle panel) and P (bottom panel). The variation is shown as follows: for SDE approach (left): solid lines in middle and bottom panel give the 5%, mean and 95% values computed from 10 000 simulations of the SDE (using mean posterior parameter estimates). For ODE approach (right): solid lines corresponds to the mean ODE fit (using mean posterior parameter estimates) plus/minus twice the mean posterior standard error.
3.3 Case study 3: Stochastic transcription for single cell data
In this experiment protein activity was imaged from GH3 rat pituitary cells stably transfected with a construct comprising a 5 kb human prolactin gene promoter fragment linked to a destabilized EGFP reporter gene (hPRL-d2EGFP) (see Supplementary Material for details of experiment). Images were taken 108 times in 15 min intervals giving a total of 27 h of data for a single cell (Fig. 3). We assume that the dynamics are described by the SDE model in (2). Since M is not observed we cannot identify the degradation rates (δM, δP) and a strongly informative prior density is needed. Here, we assume that each of them have an independent Gamma distribution with mean 0.4 for δM and 0.5 for δP.5 The prior variance was arbitrarily chosen to be small at 0.02 for both parameters. Since M is unobserved we can arbitrarily fix sM=1. Given the particular form of an experiment, where transcription is induced and afterwards comes back to its initial level, we have specified τ(t) as follows
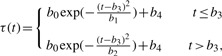 |
(12) |
where the parameters bi are to be estimated. Priors for parameters different than degradation rates were intended to be uninformative. Here, we used exponential priors with means given in Table 3. The challenge for inference here is to integrate over a fully unobserved process M whilst sampling bridges to augment the discretely observed P. Let P* denote the vector of bridges augmenting the P process and M* denote the latent M variable [we chose a grid size of 1 min for which we assume that (*) holds]. The vector of unknown parameters is θ=(δM, δP, α, sP, b0, b1, b2, b3, b4). The posterior distribution takes the form
| (13) |
where we approximate L(M*, P*, P∣Θ) with the likelihood (4) for the augmented data case, i.e. y=(M*, P*, P). In practise, this is a challenging sampling problem as the dimension of the posterior is very large and traces were highly autocorrelated. Faster convergence is achieved by reparameterizing the model (details of this and the algorithm are given in the Supplementary Material). The algorithm was first tested on simulated data from the SDE model with chosen parameters (Table 3). Artificial data are simulated on a fine scale of 15/51 min and coarse data are extracted for P at 15 min intervals. The simulated and observed time series, and the reconstructed τ(t) are shown in Figure 3. Posterior inference results are given in Table 3. Note that since M is not scaled the transcription profile corresponds to molecular population sizes which here are about 150 mRNA molecules per hour. This case study demonstrates that for high frequency single cell data the SDE approach can be extremely powerful as it allows estimation of absolute transcription rates in terms of molecule numbers and since sP can be estimated it is possible to calculate back to molecular levels of protein and translation rate. The need for precise prior information about degradation rates is irrespective of either SDE or ODE approach. The problem of non-identifiability of these parameters is due to not observing M as one can infer both degradation rates in either approach if both M and P are observed.
Fig. 3.
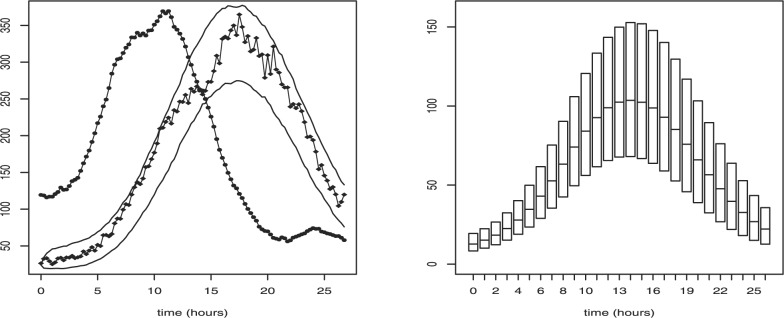
Left: Time series of fluorescence intensity used in case study 3. Solid and dashed lines represent experimental and simulated data, respectively. The variation of the SDE fit to the real data is shown by the 5% and 95% values computed from 1000 simulations of the SDE (using mean posterior parameter estimates). Right: Box-plot representing transcription profile in molecules per hour inferred from experimental data presented in the top figure. Each box represents 50% credibility interval and median of posterior distribution of the reconstructed transcription rate at particular time point.
Table 3.
Case 3: posterior inference results
| Value | Prior | Simulation | Experiment | |
|---|---|---|---|---|
| δM | 0.44 | Γ(0.44,0.02) | 0.56 (0.36–0.92) | 0.45 (0.26–0.82) |
| δP | 0.52 | Γ(0.52,0.02) | 0.59 (0.38–0.89) | 0.71 (0.45–1.09) |
| α | 20 | Exp(100) | 16.97 (6.54–78.98) | 0.46 (0.14–1.51) |
| sP | 0.2 | Exp(1) | 0.17 (0.09–0.3) | 2.11 (1.24–3.56) |
Priors, posterior medians and 95% credibility intervals inferred from both simulated and experimental data. Rates are per hour. Γ(μ, σ2) denotes Gamma distribution with mean μ and variance σ2. Full list of all parameter estimates is provided in Supplementary Material.
4 DISCUSSION
In this study, we suggest a dynamical model relating protein and corresponding mRNA dynamics via transcription and translation and suggest methods for model fitting. The applications here were motivated by the availability of gene reporter data but the model and methodology apply to many other scenarios where it is of interest to link protein and mRNA dynamics. While a stochastic model, such as (2) applies to single cell data, caution needs to be exercised in formulating an ODE model, such as (1) for multi-cell data. In order to reasonably assume such a joint mechanistic model it is essential that the individual cell activities are synchronized with respect to the gene of interest. Rate constants associated with processes of degradation, transcription and translation arise as model parameters and it is an important question whether these can be identified. In addition to a functional kind of non-identifiability of parameters in complex dynamic models as considered in Hengl et al. (2007) here, we find that practical or statistical non-identifiability of model parameters may result from unobserved variables. Case study 1 demonstrates that one can estimate all rate constants in systems of equations of the type given in (1) if all model variables—albeit coarse—are observed over time. Inference precision increases with the frequency at which the processes are sampled. In contrast, Cases 2 and 3 have latent variables and model inference is only feasible with informative prior knowledge of some parameters. Simulation studies of the model (using artificial parameters) help in identifying which sets of parameters need to be informed from other experiments. In case 3, prior knowledge of both degradation rates was needed as with M unobserved, parameters can trade-off giving rise to protein dynamics that is virtually indistinguishable via likelihood from the observed protein process. The specification of the functional form for the transcription profile also plays a role in practical identification. Even if M is observed the parameter estimates associated with transcription and degradation are correlated for obvious reasons. Such correlations affect precision of estimates and convergence of the Markov chain but can be alleviated by sampling more frequently, choosing a parsimonious functional form for transcription, and by technical aids such as the construction of independence samplers and reparameterization of the model. We believe that the functional specifications for τ(t) suggested in our case studies are useful in conjunction with gene transcription. A theoretical application of the switch function in clock modeling can be found in Aase and Ruoff (2008). Although the estimation of the switch model seems too high dimensional for datasets with many switches, this could be overcome by assigning probability distributions to the on- and off-times in the framework of a Bayesian hierarchical model.
Our results demonstrate that MCMC methods for ODEs and SDEs provide practical algorithms for reconstruction transcription profiles whilst estimating some of the rate parameters involved. As the real population dynamics are naturally stochastic SDEs provide the superior theoretical model. However, the mean ODE approach can be useful as a vehicle for estimation when the data are not fully compatible with the SDE assumptions. Whilst they usually describe the same model in the mean, their difference lies in the specification of the variance. The SDE model provides a rigid description of the volatility process which is rigorously derived for the stochastic dynamics of the molecular processes. In theory, it is straightforward to allow for additive measurement error [see Heron et al. (2007) for estimation of SDEs with measurement error]. However, identification of an unknown measurement error variance is difficult and—to our knowledge—is not possible when the data are coarse and indirectly measured with unknown scaling factors. The variance process of the mean ODE approach is not rigorously derived and can be specified by the modeler in an attempt to capture anything known about the residual process and measurement error. Estimation algorithms for the mean ODE approach are straightforward to implement although for higher dimensional or less stable systems more difficulties may occur. The algorithm for SDE estimation can be challenging to implement due to bridge sampling and is computationally expensive. Case 2 shows a problem that we have also encountered in Heron et al. (2007), namely if molecular populations are measured indirectly then the estimation of unknown scaling parameters can be difficult in practise. This may happen as a consequence of observing data that are too coarse, in the sense that too little information about the volatility process is revealed, or that are otherwise not directly compatible with the SDE assumption. However, drawbacks of the SDE approach are associated with the current quality, quantity and availability of the data. Case study 3 exemplifies that SDE estimation constitutes a very informative approach in calibrating all processes back to the molecular population levels as the scaling parameters can be identified. Under suitable assumptions the SDE model provides a theoretically well-founded modeling approach for describing the dynamics of molecular populations in a single cell. Estimation of SDEs is well studied and feasible and is highly informative when relatively frequent and clean (i.e. with little measurement error) single cell data are available on all model variables.
Funding: This research was funded by BBSRC and EPSRC (Inter-disciplinary Programme for Cellular Regulation GR/S29256/01) and EU (BIOSIM Network Contract 005137). The Millar group's work (K.E. and A.J.M.) was funded by BBSRC via award E015263 and CSBE (Centre for Integrative and Systems Biology funded by BBSRC and EPSRC). D.A.R. is funded by EPSRC Senior Research Fellowship EP/C544587/1 and M.K. by studentship, Department of Statistics, University of Warwick. C.V.H. was funded by Wellcome Trust Programme Grant (067252, to J.R.E.D. and M.R.H.W.) and now is recipient of The Prof. John Glover Memorial Postdoctoral Fellowship.
Conflict of Interest: none declared.
Supplementary Material
Footnotes
1The Wiener process, or Brownian motion, is a continuous time stochastic process that has independent normally distributed increments.
2For computational precision, we amplified the mRNA concentrations by factor 105 and the protein concentrations by 104.
3The number of switches and initial state are fairly obvious here. The inference algorithm can, however, be generalized to allow for an arbitrary number of switches and where the initial state is estimated. We will describe work on this elsewhere.
4We could not set τoff=0 in the SDE case for the practical problem that the bridge building algorithm becomes numerically unstable for values of the mRNA too close to zero.
5These rates were motivated by preliminary estimation using a small dataset from other experiments. They are used here only to demonstrate the case as their estimates may change if more data were available.
REFERENCES
- Aase SO, Ruoff P. Semi-algebraic optimization of temperature compensation in a general switch-type negative feedback model of circadian clocks. J. Math. Biol. 2008;56:279–292. doi: 10.1007/s00285-007-0115-5. [DOI] [PubMed] [Google Scholar]
- Durham GB, Gallant AR. J. Bus. Econ. Stat. 2002;20:297–316. [Google Scholar]
- Elerian O, et al. Likelihood inference for discretely observed non-linear diffusions. Econometrica. 2001;69:959–993. [Google Scholar]
- Gamerman D, Lopes HF. Markov Chain Monte Carlo Stochastic Simulation for Bayesian Inference. 2nd edn. Boca Raton, London, New York: Chapman & Hall/CRC; 2006. [Google Scholar]
- Gillespie DT. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977;81:2340–2361. [Google Scholar]
- Gillespie DT. A rigorous derivation of the chemical master equation. Physica A. 1992;188:404–425. [Google Scholar]
- Goldbeter A. Computational approaches to cellular rhythms. Nature. 2002;420:238–245. doi: 10.1038/nature01259. [DOI] [PubMed] [Google Scholar]
- Hengl S, et al. Data-based identifiability analysis of non-linear dynamical models. Bioinformatics. 2007;23:2612–2618. doi: 10.1093/bioinformatics/btm382. [DOI] [PubMed] [Google Scholar]
- Heron EA, et al. Bayesian inference for dynamic transcriptional regulation; the hes1 system as a case study. Bioinformatics. 2007;23:2589–2595. doi: 10.1093/bioinformatics/btm367. [DOI] [PubMed] [Google Scholar]
- Jensen MH, et al. Sustained oscillations and time delays in gene expression of proteinHes1. Febs Lett. 2003;541:176–177. doi: 10.1016/s0014-5793(03)00279-5. [DOI] [PubMed] [Google Scholar]
- Kalir S, Alon U. Using a quantitative blueprint to reprogram the dynamics of the flagella gene network. Cell. 2004;117:713–720. doi: 10.1016/j.cell.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Kloeden PE, Platen E. Numerical Solution of Stochastic Differential Equations. 3rd edn. Berlin, New York: Springer-Verlag; 1999. [Google Scholar]
- Locke JCW, et al. Modelling genetic networks with noisy and varied experimental data: the circadian clock in Arabidopsis thaliana. J. Theor. Biol. 2005a;234:383–393. doi: 10.1016/j.jtbi.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Locke JCW, et al. Extension of a genetic network model by iterative experimentation and mathematical analysis. Mol. Syst. Biol. 2005b;1:E1–E9. doi: 10.1038/msb4100018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar AJ, et al. Circadian clock mutants in Arabidopsis identified by luciferase imaging. Science. 1995;267:1161–1163. doi: 10.1126/science.7855595. [DOI] [PubMed] [Google Scholar]
- Millar AJ, Kay SA. Integration of circadian and phototransduction pathways in the network controlling cab gene transcription in Arabidopsis. Proc. Natl Acad. Sci. USA. 1996;93:15491–15496. doi: 10.1073/pnas.93.26.15491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DE, et al. Oscillations in protect NF-kappa B signaling control the dynamics of gene expression. Science. 2004;306:704–708. doi: 10.1126/science.1099962. [DOI] [PubMed] [Google Scholar]
- Ronen M, et al. Assigning numbers to the arrows: parameterizing a gene regulatory network by using accurate expression kinetics. Proc. Natl Acad. Sci. USA. 2002;99:10555–10560. doi: 10.1073/pnas.152046799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner MA, Wong WH. The calculation of posterior distributions by data augmentation. J. Am. Stat. Assoc. USA. 1987;82:528–540. [Google Scholar]
- Wang Z, Tobin EM. Constitutive expression of the circadian clock associated 1 (cca1) gene disrupts circadian rhythms and suppresses its own expression. Cell. 1998;93:1207–1217. doi: 10.1016/s0092-8674(00)81464-6. [DOI] [PubMed] [Google Scholar]
- Yakir E, et al. Circadian clock associated1 transcript stability and the entrainment of the circadian clock in arabidopsis. Plant Physiol. 2007;145:925–932. doi: 10.1104/pp.107.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.