

Abstract
Signaling of transforming growth factor β (TGF-β) is mediated through a heteromeric complex of two types of transmembrane receptors and downstream intracellular proteins known as Smads. Alterations of TGF-β signaling underlie various forms of human cancer and developmental diseases. Human genetic studies have revealed both point mutations and deletions of Smad2 or Smad4 in several types of cancers. However, the role of Smad3 in tumorigenesis is not clear. Recent data indicate that Smad3 also functions as a tumor suppressor by inhibiting cell proliferation and promoting apoptosis. In addition, Smad3 is essential for TGF-β–mediated immune suppression, and it plays an important role in regulating transcriptional responses that are favorable to metastasis. Therefore, through regulating different transcriptional responses, Smad3 functions as both a negative and positive regulator of carcinogenesis depending on cell type and clinical stage of the tumor.
Keywords: tumor suppressor, growth inhibition, apoptosis, metastasis, immune suppression, transcription
I. INTRODUCTION
Transforming growth factor-β (TGF-β) is the prototype of a large family of secreted polypeptide growth factors that regulate a multitude of cellular processes affecting proliferation, differentiation, and apoptosis.1 In addition, TGF-β exerts a multifaceted influence on cancer pathogenesis. Among the six essential alterations of cell physiology that are required for malignant transformation,2 TGF-β is involved in the insensitivity to antigrowth signals, evasion of apoptosis, induction of tumor angiogenesis, and tendency to promote tissue invasion and metastasis. A large number of genetic or epigenetic changes affecting various components of the TGF-β pathway have been reported in a spectrum of human hyperproliferative disorders and various forms of cancers.1,3 Although it is a potent growth inhibitor in cultured cells, TGF-β is actually abundantly expressed in most human tumors, and high levels of TGF-β often bode poor prognosis.4,5 This dichotomy complicated early attempts at direct use of TGF-β in cancer treatment and stressed a need for a thorough understanding of its signaling mechanism. Following the identification of membrane-bound TGF-β receptors, a group of transcription factors known as Smads was discovered in the late 1990s to mediate transcriptional responses of TGF-β and its related factors.6,7 Among these, Smad2 and Smad3 are accredited with mediating TGF-β responses. Because of their close relatedness, these two proteins exhibit interchangeable functions in many signaling systems.8,9 In this review, we discuss recent advances in the understanding of how TGF-β suppresses tumor formation in some instances but promotes tumor growth in others. We will focus on roles and mechanisms of Smad3.
II. MECHANISM OF TGF-β SIGNALING THROUGH SMAD3
It is now generally accepted that the plethora of biologic activities of TGF-β is universally initiated by the binding of the ligand to a heteromeric complex of two types of transmembrane receptors: TGF-β receptor type I (TGFBR1) and TGFBR2, each equipped with an intrinsic serine/threonine kinase activity.7 Ligand occupancy causes an association between TGFBR2 and TGFBR1, which results in phosphorylation of TGFBR1 by the constitutively active TGFBR2. The phosphorylated TGFBR1 then triggers activation of Smad3 by phosphorylation at the C-terminal serine residues,6,7 forcing Smad3 to dissociate from the membrane-bound receptors and translocate into the nucleus. In the nucleus, it binds DNA at a preferential sequence of GTCT or the reverse complement AGAC, called Smad-binding element (SBE).10,11 However, the affinity of Smad3 for SBE does not support one on one binding in vivo; instead, Smad3 relies on cooperative binding with other transcription factors, including a common Smad4 that is essential for many Smad-mediated transcriptional responses. The Smad transcriptional complexes have the ability to either activate or repress transcription of a selected set of target genes depending on the nature of associated cofactors and the status of local chromatin structure in the context of signal-receiving cells. Now it is clear that this Smad-mediated signaling pathway is subject to control or function in conjunction with Smad-independent mechanisms, such as those governed by mitogen-activated protein kinases (MAPKs) or rho-like GTPases.12,13 The Smad-independent, noncanonic receptor-signaling conduits can provoke cellular response to ligands on their own right or modulate Smad activity to custom fit signaling output to a particular need, further lending complexity to the control of TGF-β signaling.
II. SMAD3 PLAYS A MAJOR ROLE IN TGF-β–MEDIATED GROWTH INHIBITION
TGF-β is a potent growth inhibitor of cells of epithelial, neuronal, and hematopoietic origins, causing them to arrest at the G1 phase of the cell cycle.3,14 Key to this inhibition are the down-regulation of protooncogene c-Myc15,16 and the induction of p15Ink4b and/or p21Cip1,17-19 which are inhibitors of G1-phase cyclin-dependent kinases (CDKs). TGF-β has long been recognized for an ability to rapidly downregulate c-Myc in various cell types,15 but it was not understood until recently that this downregulation occurs at the level of transcription through a cis-acting TGF-β inhibitory element (TIE) in the c-Myc promoter.20 TIE is composed of a repressive SBE (RSBE) and an overlapping E2F site, which are recognized by a tripartite complex involving Smad3/4, E2F4/5, and the transcriptional repressor p107.20,21 There is evidence to suggest that this multimeric Smad3 complex is preformed in the cytoplasm, and upon TGF-β induction, traverses into the nucleus to engage in the repression of c-Myc transcription (Fig. 1).21 RSBE is distinct from the more common SBE in both sequence and function,20 and loss of recognition of RSBE by the Smads–E2F4/5–p107 complex underlies the resistance of breast cancer cells to TGF-β–mediated growth inhibition.21
FIGURE 1.
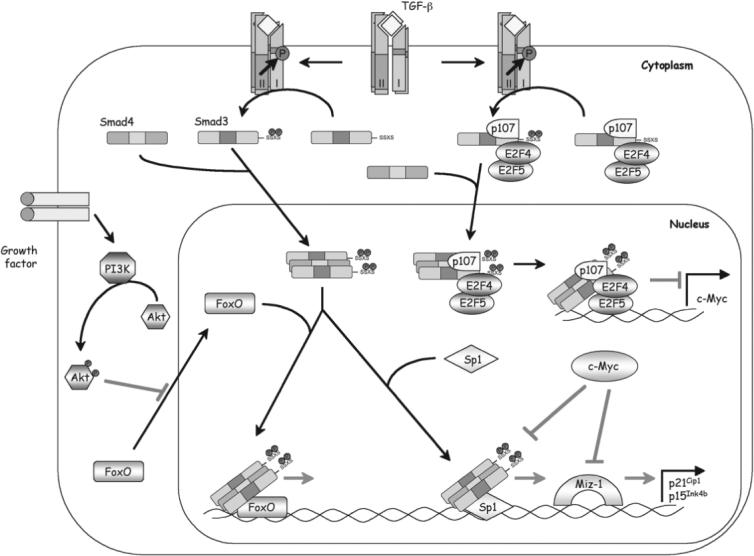
Mechanisms of Smad3 in TGF-β–induced antiproliferative transcriptional responses. In repressing the protooncogene c-Myc, a preexisting cytoplasmic complex containing Smad3, E2F4/5, and p107 likely begins to move into the nucleus in response to ligand stimulation and binds DNA at a RSBE/TIE site in the c-Myc promoter. Smad4 is recruited to this complex as well. In the induction of CDK inhibitors p15Ink4b and p21Cip1, a different Smad3/4 complex cooperates with FOXO at a distal promoter element to activate transcription. The PI3K/Akt pathway has the ability to antagonize TGF-β/Smad3 signaling by inhibiting FOXO nuclear localization. The Smad3/4 complex also stimulates transcription in conjunction with Sp1 at a proximal promoter element. Finally, transcription of p15Ink4b and p21Cip1 is directly repressed by c-Myc through its association with Miz-1 and/or Smad2/3 at the initiator site. Thus, attenuating c-Myc expression is a prerequisite for induction of p15Ink4b and p21Cip1.
A complex scenario is at play in the induction of CDK inhibitors triggered by TGF-β. Initial studies suggested that Smad3 and Smad2 activate the transcription of p15Ink4b in concert with Sp1 at a compound SBE–Sp1 site in the proximal promoter region22 and attributed the inhibition of p15Ink4b transcription by c-Myc to the interference brought in by the interaction between c-Myc and the MAD homology 2 domain of Smad3 in the Smad/Sp1 complex.23 Although later studies confirmed the contribution of Sp1, the TGF-β inducibility conferred by this proximal promoter region was nevertheless accredited to the sequences surrounding the transcription initiation site, which is recognized by the Myc-interacting zinc-finger protein Miz-1.24 Recruiting c-Myc by Miz-1 to the p15Ink4b promoter interferes with activation of the p15Ink4b gene by TGF-β.24,25 A similar mechanism also applies to the induction of p21Cip1 by TGF-β.26-29 Downregulation of c-Myc is prerequisite for induction of p15Ink4b and p21Cip1 but is not sufficient.30,31 A transactivation complex containing Smad3, Smad4, and the forkhead box O (FOXO) family of transcription factors is also required.32,33 Smads and FOXO bind cooperatively to a distal promoter region in both p15Ink4b and p21Cip1 promoters containing multiple copies of SBE that are flanked by the forkhead-binding element. Thus, formation of the Smad/FOXO complex in response to TGF-β stimulation delivers the final activation signal for induction of transcription of p15Ink4b and p21Cip1 genes. Because FOXO proteins are targets of the phosphoinositide 3 kinase (PI3K)/Akt survival pathway, Akt can inhibit nuclear localization of FOXO proteins by phosphorylating them and thus barring them from target genes. This finding offered a mechanistic account for the antagonizing interaction between the TGF-β pathway and the PI3K/Akt pathway (Fig. 1).
Consistent with the essential role of Smad3 in the above transcriptional responses, depletion of Smad3 but not Smad2 by RNA interference sufficiently blocked TGF-β–mediated cell cycle arrest and growth inhibition.34 Similar results were also obtained using primary T cells or hepatocytes isolated from Smad3-deficient mice.35,36
III. SMAD3 MUTATIONS IN HUMAN CANCER
The role of TGF-β signaling as a tumor suppressor pathway is best illustrated by the presence of mutations in genes encoding components of the TGF-β signaling pathway.3,14 Mutations in TGFBR2 are frequently found in colon cancer,37-39 gastric cancer,39-42 glioma,43 and non–small cell lung carcinoma,44 whereas mutations in Smad4 have been found in more than 50% of pancreatic cancers45 and in a subgroup of patients with juvenile polyposis syndromes.46 Although less common, mutations in TGFBR1 have been observed in ovarian cancer,47,48 metastatic breast cancer,49 pancreatic cancer,50 and T-cell lymphoma,51 whereas mutations in Smad2 have been detected in a small portion of colorectal and lung cancers.52-54 However, it is only most recently that a heterozygous missense mutation (R373H) in Smad3 was reported in colorectal cancer cell line SNU-769A, originated from a metastatic site in a lymph node.55 This mutation is localized to the C-terminal domain of Smad3 that is involved in the interaction with TGFBR1, formation of Smad homomeric or heteromeric complex, and transcriptional activation. Although this mutation could functionally disable the Smad3 protein, its role in tumorigenesis is not clear because the same mutation is not present in the SNU-769B cell line, originated from a primary tumor site in the same patient. Moreover, a homozygous mutation in the TGFBR2 gene is already present in both SNU-769A and SNU-769B cell lines.55
Despite the lack of inactivating mutations in human cancers, direct evidence supporting a tumor-suppressing role of Smad3 comes from the observation of the absence of Smad3 protein in T-cell acute lymphoblastic leukemia (ALL).35 Interestingly, the loss of Smad3 protein in T-cell ALL is not due to mutations in the Smad3 gene or alterations in the level of Smad3 mRNA expression. Nevertheless, these results suggest that the level of Smad3 protein can be an important determinant in the suppression of tumorigenesis.
IV. TUMOR DEVELOPMENT IN SMAD3-DEFICIENT MOUSE MODELS
Smad3-deficient mice have been generated in three different laboratories with different strategies.56-58 One of these lines, Smad3exo2/exo2, was originally reported to develop colon carcinomas, including adenocarcinomas and metastatic carcinomas.58 However, this phenotype was not seen in two other independently generated lines despite all of the lines being reported to have impaired TGF-β signaling.56,57 This discrepancy could be due to differences in genetic background and/or in environmental factors associated with animal holding. For example, on the 129/Sv background, 100% of Smad3exo2/exo2 mice had tumors by age 6 months,58 whereas on the hybrid background of 129/Sv and C57BL/6, a less aggressive tumor phenotype was observed: tumor onset was delayed to 10 months with only 30% of mice developing tumors.58 Moreover, a follow-up study indicated that, when maintained in a Helicobacter-free environment for up to 9 months, previously reported colon cancer phenotypes were not developed in Smad3exo2/exo2 mice on the 129/Sv background; infection of the same mice with Helicobacter triggered colon cancer in 50% to 66% of the animals.59 These results suggested that the original Smad3exo2/exo2 mice may have been infected with Helicobacter or other colitis-inducing organisms.58 Bacterial infection, including Helicobacter infection, can induce chronic inflammation, during which reactive oxygen species may arise,59 ultimately resulting in DNA damage, genomic instability, and transformation.60 Impairment of TGF-β–mediated immunosuppression has been documented in all three lines of Smad3-deficient mice,56,57,59 which could contribute to increased inflammatory responses that predispose the animals to cancer formation.
Interestingly, although infection with Helicobacter restored the colon cancer phenotype in Smad3-deficient mice, it alone had no effect on another rodent intestinal cancer model, the Apcmin/+ mouse.59 In contrast, Smad3 deficiency promotes tumorigenesis in the distal colon of Apcmin/+ mouse.61 A mixture of adenomas and invasive carcinomas were observed in Apcmin/+ Smad3exo2/exo2 mice at 2 months, exclusively in the distal colon, closely mimicking the familial adenomatous polyposis disease.61 Transcriptional profiling revealed higher expression of several TGF-β activators in the normal distal mucosa than in the proximal mucosa, suggesting a stronger reliance on TGF-β–mediated growth control in the distal than in the proximal colon. Therefore, the absence of Smad3 could also predispose the colon epithelium toward neoplasia in the context of reduction of TGF-β–mediated antiproliferative signals.61
Furthermore, the studies using Smad3-deficient mice indicated that the loss of Smad3 alone is insufficient to initiate tumorigenesis.59,61 Instead, a reduction in Smad3 increased the risk or tendency of tumorigenesis when associated with alterations in other factors that control cellular proliferation and apoptosis. This is best illustrated by the studies of Wolfraim et al.,35 which showed that loss of one allele of Smad3 in mice impairs the inhibitory effect of TGF-β on the proliferation of normal T cells and works in tandem with the homozygous inactivation of p27Kip1, a CDK inhibitor whose gene is frequently altered in human T-cell ALL, to promote T-cell ALL.
V. TUMOR-SUPPRESSING ROLE OF SMAD3 IN CHEMICALLY HEPATOCELLULAR CARCINOMA (HCC) MODELS
Perhaps more convincing evidence for a tumorsuppressing role of Smad3 stems from a recent study of chemically induced HCC in mice expressing Smad3 transgenes specifically in the liver.62 This study showed that enhancing Smad3 function through forced expression of wild-type or constitutively active Smad3 protected mouse livers from diethyl nitrosamine/phenobarbital-induced carcinogenic insult, whereas disrupting Smad3 function by dominant-negative mutant Smad3 aggravated the tumor load in the affected livers. It was also found that while the liver tumors lost response to the growth inhibition of TGF-β, they still succumbed to a high rate of apoptosis, particularly when the function of Smad3 was elevated. These results suggest that Smad3 is a tumor suppressor of mouse liver cancers and it does so by promoting TGF-β–mediated apoptosis.
Induction of apoptosis is a well-recognized mechanism of TGF-β to exert its tumor suppression function, but a detailed understanding of the chain of events from ligand occupying the receptor leading to the activation of apoptotic machinery is lacking. Several studies have indicated that overexpression of Smad3, but not Smad2, promotes TGF-β–induced apoptosis, whereas interfering with Smad3 function by dominant-negative mutants, RNA interference, or inactivation of the Smad3 gene locus, protects against apoptosis,36, 62-64 suggesting that Smad3 is a physiologic mediator in this signaling process. One target of Smad3 in inducing apoptosis is Bcl-2, a key antiapoptotic inhibitor.62 Upregulation of Bcl-2 is one of the traits widely acquired by cancer cells to evade apoptosis,65 and the significance of Bcl-2 as an anticancer target is reflected by the sheer number of clinical trials designed for various strategies of inactivating Bcl-2.66,67 Smad3 can bind directly to a GC-rich element in the Bcl-2 promoter, and the interaction between Smad3 and the Bcl-2 promoter correlates with a repressed state of transcription of Bcl-2 in vivo.62 Thus, by attenuating the level of the major apoptosis inhibitor Bcl-2, TGF-β/Smad3 signaling facilitates a permissive cellular context that is conducive to apoptosis. Besides Bcl-2, several other proteins, including death-associated protein kinase (DAPK),68 growth arrest and DNA damage–inducible β (GADD45B),69,70 and BH3-only protein Bim,71 have also been identified as Smad3's targets in mediating the proapoptotic response of TGF-β (Fig. 2).
FIGURE 2.
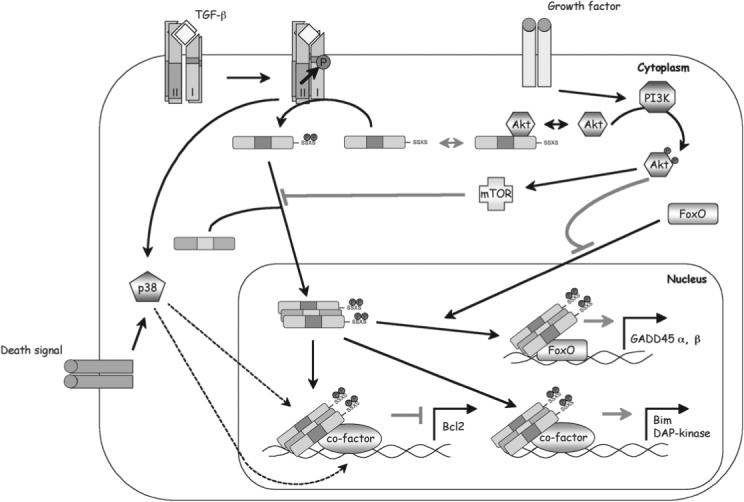
Mechanisms of Smad3 in TGF-β–mediated proapoptotic transcriptional responses. An activated Smad3/4 complex is required in the nucleus to activate the transcription of GADD45 (along with FOXO) and Bim or DAPK and to repress Bcl-2. p38 MAPK is likely involved in the repression of Bcl-2, but the mechanism is not clear. Three mechanisms have been reported for the PI3K/Akt pathway to dampen the proapoptotic signaling of Smad3: Akt can sequester Smad3 in the cytoplasm by the binding of Smad3, activity of Smad3 can be inhibited by mTOR, or Akt may inhibit FOXO by sequestering it from participating in Smad3-mediated transcription of GADD45.
Interestingly, the mouse livers that ectopically expressed constitutively active mutant Smad3SD exhibited no phenotype even though it was clearly shown that Smad3SD was localized to the nucleus. This argues that C-terminal phosphorylation and nuclear translocation alone are probably not sufficient to render Smad3 fully active in inducing apoptosis.62 The proapoptotic activity of Smad3 may require additional activation of other cytoplasmic kinase pathways in vivo. One of the candidates is p38 MAPK, which has been reported to phosphorylate Smad3 at the linker region.72,73 More importantly, activation of p38 MAPK was shown to be essential to TGF-β–induced apoptosis in hepatocytes, mammary epithelial cells, and B cells.62,74-77 Another kinase, Akt (protein kinase B), which can be activated in response to insulin and serum, has been shown to associate directly with Smad3. The interaction between Akt and Smad3 prevents Smad3 from being activated by TGF-β and translocating into the nucleus.78,79 As such, Akt can decrease the activity of Smad3 and protect cells against TGF-β–induced apoptosis. In addition, like in the case of p15Ink4b and p21Cip1 promoters, Akt could negatively regulate Smad3-dependent target genes that act in apoptotic response through FOXO (e.g., GADD45A and GADD45B).33 A third protein, mTOR, which is a key mediator downstream from Akt, was also recognized to have an ability to suppress Smad3 activation and inhibit the apoptosis induced by TGF-β.80
VI. ROLES OF SMAD3 IN PROMETASTATIC TRANSFORMATION
Although TGF-β has long been regarded as a tumor suppressor, it also promotes tumor progression during the late stages of cancer by suppressing immune surveillance, inducing epithelium to mesenchyme transition (EMT), and enhancing cell migration and transcription of factors favorable to metastasis (Fig. 3).81-84 Again, Smad3 plays an indispensable role in these multifaceted pathway activities and thus can function as a tumor promoter in certain instances.
FIGURE 3.
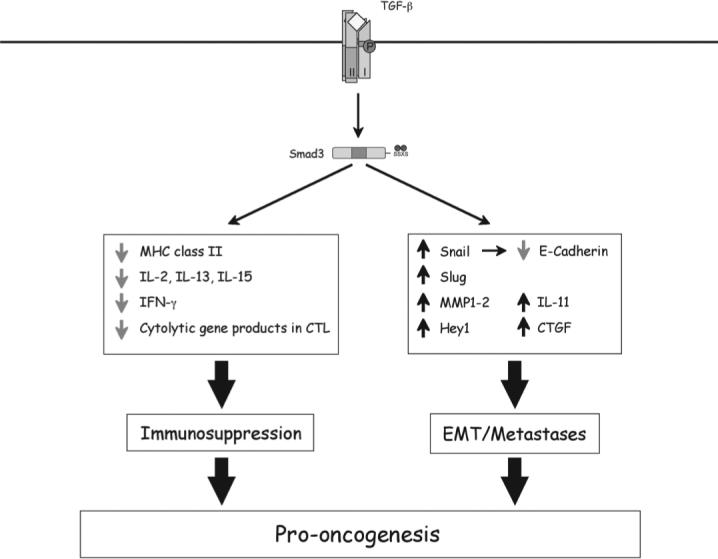
Mechanisms underlying prooncogenic activities of Smad3 during the late stages of carcinogenesis. Smad3 is essential for TGF-β–mediated immune suppression, and it regulates transcriptional responses that are favorable to EMT and metastasis. The induced genes are labeled with upward arrows and repressed ones with downward arrows.
A. Smad3 is Essential for TGF-β–Mediated Immune Suppression
TGF-β–mediated immunosuppression may contribute to its tumor-suppressing role by inhibiting inflammatory responses upon bacterial infections that predispose animals to cancer formation as documented in Smad3-deficient mice59; it also stimulates tumor development and progression by escaping immune surveillance during the late stage of carcinogenesis.85,86 It has been shown that secretion and expression of TGF-β by tumor cells downregulates major histocompatibility complex class II antigens and renders the tumor cell surface less immunogenic.87,88 Moreover, TGF-β produced by a wide variety of noncancerous cells present at tumor sites also contributes to the local suppression of immune function and the escape of tumor cells from immune surveillance. The inhibitory role of TGF-β–mediated immune suppression in tumor eradication has been demonstrated in various mouse tumor models. When challenged with TGF-β–producing tumors, mice that express a dominant-negative TGF-β receptor transgene in T cells were able to generate an immune response capable of eliminating tumors.89 In another study, systemic neutralization of TGF-β activity in the EL4 thymoma cells by knocking down TGF-β expression through RNA interference or inhibiting TGF-β receptor activity restored T-cell cytotoxicity that was responsible for the antigen-specific tumor clearance in vivo.84
Analysis of Smad3-deficient mice revealed an essential role of Smad3 in mediating the immune-suppressing activity of TGF-β. This notion was supported by the finding that T cells in these mutant mice are constitutively activated and resistant to TGF-β–mediated growth inhibition.56,57,59 Consistent with this observation, TGF-β–mediated inhibition in the production of several cytokines, such as interleukin 2 (IL-2), IL-13, IL-15, inter-feron γ (IFN-γ), and expression of CDK4 was lost in the Smad3-deficient primary T-cell cultures.35,56,90,91 Furthermore, TGF-β has been shown to directly inhibit expression of five cytolytic gene products in cytotoxic T cells (CTLs): perforin, granzyme A, granzyme B, Fas ligand, and IFN-γ, which are collectively responsible for CTL-mediated tumor cytotoxicity.84 In each of the above cases, Smad3 was shown to be crucial in mediating these TGF-β–mediated transcriptional responses.84
B. Smad3 is Directly Involved In EMT
EMT is a process of delaminating mesenchymal cells from epithelium. It often associates with weaker cell–cell interactions and acquisition of motile and invasive properties of the cells that are prerequisite for progressing to advanced metastatic tumors.92 TGF-β is a major player in EMT in vivo. The tumor-promoting role of TGF-β by inducing EMT at late stages of carcinogenesis is best exemplified in a study based on transgenic mice overexpressing TGF-β1 specifically in epidermis.93,94 When subjected to a standard 7,12-dimethylbenz-α-anthracene–initiated and 12-O-tetradecanoylphorbol 13-acetate–promoted skin tumor induction protocol, these mice developed fewer benign tumors than controls, consistent with the potent growth inhibitory action of TGF-β. Nevertheless, despite fewer benign papillomas, they developed more carcinomas, and the carcinomas were more aggressive, with a noticeable increase in the incidence of overt fibroblastoid spindle tumors. Recent experiments with conditional transgenic mice have provided further support for the requirement of TGF-β activity in inducing tumor cell EMT in vivo.94 Mice that overexpressed TGF-β1 in keratinocytes developed spindle carcinoma. In contrast, such tumor types were rare in mice that harbored an empty control vector, a dominant-negative TGFBR2, or TGF-β1 plus a dominant-negative TGFBR2.
Requirements of the Smad pathway in TGF-β–induced EMT of malignant keratinocytes and normal mammary epithelial cells were first demonstrated in experiments with constitutively active or dominant-negative forms of Smad2 and Smad3 in cultured cells.95,96 Most prominently, a mutant TGFBR1 defective in Smad binding was shown to lack the ability to induce EMT in mammary epithelial cells,76 indicating that Smad-dependent signaling is required for TGF-β–mediated EMT response. Direct evidence for a role of Smad3 stems from observations that the loss of Smad3 in mice blocked EMT in response to the injury of the lens, retina, and kidney in vivo or by exposure to exogenous TGF-β in organ culture.97-99 Furthermore, TGF-β fails to induce EMT in primary tubular epithelial cells derived from kidneys of Smad3-deficient mice.100
At the molecular level, EMT is characterized by a transcriptional program shift marking the transition from epithelium to mesenchyme.95,96,101,102 One of the transcriptional changes is the downregulation of E-cadherin.102,103 Snail, a zinc-finger transcription factor known to act as a potent repressor of the E-cadherin gene,104 is one of the immediate Smad3 target genes.105 In Smad3-deficient cells, expression of Snail was ablated during TGF-β–mediated EMT initiated by injury of resident epithelial cells. In addition, Smad3 can directly or indirectly regulate transcription of a number of other genes involved in the EMT process, including epithelial and mesenchymal markers, extracellular matrix/cytoskeleton proteins, inhibitors of differentiation, and components of the Notch signaling pathway (including Krt1−19, Slug, N-cadherin, α smooth muscle actin, MMP1−2, Id1−3, and Hey1).96,100,106
C. Smad3-Mediated Transcriptional Response Fosters Metastasis in Humans
The increased expression and activation of TGF-β also affects the tumor microenvironment that permits tumor growth, invasion, and angiogenesis.103,107-109 There is substantial evidence supporting the notion that excessive TGF-β production is associated with poor prognosis.4,5 In mouse breast cancer models, TGF-β signaling promoted bone and lung metastasis.110-112 Both Smad-independent and Smad–dependent signaling pathways contributed to the prometastatic role of TGF-β. In the case of the Smad pathway, it was observed that reduction in the signaling flux through the Smad2/3 pathway is sufficient to block metastases of oncogenically transformed or tumor-derived cells, whereas overexpression of Smad3 in these same cells increased the number and size of lung metastases.113 It appears that Smad3 signaling is required for cells to either extravasate into the lung or proliferate in that tissue and induce an angiogenic response. Using the MDA-MB-231 human breast cancer cell line as a model system, IL-11 and connective tissue growth factor (CTGF) have been identified among a group of 48 genes with expression elevated in MDA-MB-231 subpopulations selected in vivo for high bone metastatic activity.114 Enforced expression of IL-11 and CTGF increases the osteolytic bone metastatic activity in MDA-MB-231 xenografts. Both IL-11 and CTGF are direct targets of TGF-β. TGF-β stimulation induces binding of Smad3 (and/or Smad2) and Smad4 to the relevant regions of the IL-11 and CTGF promoters.114 In the IL-11 promoter, Smads also cooperate with AP1 transcription factors to enhance transcription.82 Therefore, these Smad-dependent transcriptional responses could provide an advantage to cancer cells for osteolytic metastasis in a TGF-β–rich bone microenvironment.
V. CONCLUDING REMARKS
In the past few years, considerable progress has been made toward the understanding of the signaling networks and biologic function of TGF-β and Smad3 during carcinogenesis. It has been greatly appreciated that, like TGF-β, Smad3 could function as both a tumor suppressor and prometastatic factor. However, it is still not known how Smad3 switches from a tumor suppressor to a prometastatic factor during carcinogenesis. It is presumed that TGF-β/Smad3–mediated prometastatic responses can emerge once the pathway becomes uncoupled from the tumor suppressor effect. Finally, as we continue to advance our understanding of the function of Smad3 and how subtle perturbation of Smad3 function can result in pathologic situations, consideration should be paid to the development of practical and clinical approaches for pharmacologic interventions that target Smad3 to enhance or retain its growth-inhibiting and apoptosis-inducing effects but also inhibit its prometastatic activities.
ACKNOWLEDGMENT
The research in Y. E. Zhang's group was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
REFERENCES
- 1.Massague J, Blain SW, Lo RS. TGFβ signaling in growth control, cancer, and heritable disorders. Cell. 2000;103(2):295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 3.Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumor suppression and cancer progression. Nat Genet. 2001;29(2):117–29. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 4.Gold LI. The role for transforming growth factor β (TGF-β) in human cancer. Crit Rev Oncog. 1999;10:303–60. [PubMed] [Google Scholar]
- 5.Watanabe T, Wu TT, Catalano PJ, Ueki T, Satriano R, Haller DG, Benson AB, III, Hamilton SR. Molecular predictors of survival after adjuvant chemo-therapy for colon cancer. N Engl J Med. 2001;344(16):1196–206. doi: 10.1056/NEJM200104193441603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19(23):2783–810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 7.Feng XH, Derynck R. Specificity and versatility in TGF-β signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–93. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 8.Dunn NR, Koonce CH, Anderson DC, Islam A, Bikoff EK, Robertson EJ. Mice exclusively expressing the short isoform of Smad2 develop normally and are viable and fertile. Genes Dev. 2005;19(1):152–63. doi: 10.1101/gad.1243205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunn NR, Vincent SD, Oxburgh L, Robertson EJ, Bikoff EK. Combinatorial activities of Smad2 and Smad3 regulate mesoderm formation and patterning in the mouse embryo. Development. 2004;131(8):1717–28. doi: 10.1242/dev.01072. [DOI] [PubMed] [Google Scholar]
- 10.Shi Y, Wang YF, Jayaraman L, Yang H, Massague J, Pavletich NP. Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-β signaling. Cell. 1998;94(5):585–94. doi: 10.1016/s0092-8674(00)81600-1. [DOI] [PubMed] [Google Scholar]
- 11.Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B, Kern SE. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998;1(4):611–7. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]
- 12.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. 2003;425(6958):577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 13.Moustakas A, Heldin CH. Non-Smad TGF-β signals. J Cell Sci. 2005;118(Pt 16):3573–84. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- 14.Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-β in homeostasis and cancer. Nat Rev Cancer. 2003;3(11):807–21. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 15.Alexandrow MG, Kawabata M, Aakre M, Moses HL. Overexpression of the c-Myc oncoprotein blocks the growth-inhibitory response but is required for the mitogenic effects of transforming growth factor β1. Proc Natl Acad Sci U S A. 1995;92(8):3239–43. doi: 10.1073/pnas.92.8.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen CR, Kang Y, Massague J. Defective repression of c-myc in breast cancer cells: a loss at the core of the transforming growth factor β growth arrest program. Proc Natl Acad Sci U S A. 2001;98(3):992–9. doi: 10.1073/pnas.98.3.992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-β-induced cell cycle arrest. Nature. 1994;371(6494):257–61. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 18.Reynisdottir I, Polyak K, Iavarone A, Massague J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-β. Genes Dev. 1995;9(15):1831–45. doi: 10.1101/gad.9.15.1831. [DOI] [PubMed] [Google Scholar]
- 19.Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y, Wang XF. Transforming growth factor β induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc Natl Acad Sci U S A. 1995;92(12):5545–9. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frederick JP, Liberati NT, Waddell DS, Shi Y, Wang XF. Transforming growth factor β-mediated transcriptional repression of c-myc is dependent on direct binding of Smad3 to a novel repressive Smad binding element. Mol Cell Biol. 2004;24(6):2546–59. doi: 10.1128/MCB.24.6.2546-2559.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen CR, Kang Y, Siegel PM, Massague J. E2F4/5 and p107 as Smad cofactors linking the TGFβ receptor to c-myc repression. Cell. 2002;110(1):19–32. doi: 10.1016/s0092-8674(02)00801-2. [DOI] [PubMed] [Google Scholar]
- 22.Feng XH, Lin X, Derynck R. Smad2, Smad3 and Smad4 cooperate with Sp1 to induce p15Ink4B transcription in response to TGF-β. EMBO J. 2000;19(19):5178–93. doi: 10.1093/emboj/19.19.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng XH, Liang YY, Liang M, Zhai W, Lin X. Direct interaction of c-Myc with Smad2 and Smad3 to inhibit TGF-β-mediated induction of the CDK inhibitor p15Ink4B. Mol Cell. 2002;9(1):133–43. doi: 10.1016/s1097-2765(01)00430-0. [DOI] [PubMed] [Google Scholar]
- 24.Seoane J, Pouponnot C, Staller P, Schader M, Eilers M, Massague J. TGFβ influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat Cell Biol. 2001;3(4):400–8. doi: 10.1038/35070086. [DOI] [PubMed] [Google Scholar]
- 25.Staller P, Peukert K, Kiermaier A, Seoane J, Lukas J, Karsunky H, Moroy T, Bartek J, Massague J, Hanel F, Eilers M. Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol. 2001;3(4):392–9. doi: 10.1038/35070076. [DOI] [PubMed] [Google Scholar]
- 26.Pardali K, Kurisaki A, Moren A, ten Dijke P, Kardassis D, Moustakas A. Role of Smad proteins and transcription factor Sp1 in p21Waf1/Cip1 regulation by transforming growth factor-β. J Biol Chem. 2000;275(38):29244–56. doi: 10.1074/jbc.M909467199. [DOI] [PubMed] [Google Scholar]
- 27.Pardali K, Kowanetz M, Heldin CH, Moustakas A. Smad pathway-specific transcriptional regulation of the cell cycle inhibitor p21WAF1/Cip1. J Cell Physiol. 2005;204(1):260–72. doi: 10.1002/jcp.20304. [DOI] [PubMed] [Google Scholar]
- 28.Seoane J, Le HV, Massague J. Myc suppression of the p21Cip1 Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature. 2002;419(6908):729–34. doi: 10.1038/nature01119. [DOI] [PubMed] [Google Scholar]
- 29.Wu S, Cetinkaya C, Munoz-Alonso MJ, von der Lehr N, Bahram F, Beuger V, Eilers M, Leon J, Larsson LG. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene. 2003;22(3):351–60. doi: 10.1038/sj.onc.1206145. [DOI] [PubMed] [Google Scholar]
- 30.Warner BJ, Blain SW, Seoane J, Massague J. Myc downregulation by transforming growth factor β required for activation of the p15Ink4b G1 arrest pathway. Mol Cell Biol. 1999;19(9):5913–22. doi: 10.1128/mcb.19.9.5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Claassen GF, Hann SR. A role for transcriptional repression of p21CIP1 by c-Myc in overcoming transforming growth factor β-induced cell-cycle arrest. Proc Natl Acad Sci U S A. 2000;97(17):9498–503. doi: 10.1073/pnas.150006697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seoane J, Le HV, Shen L, Anderson SA, Massague J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117(2):211–23. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- 33.Gomis RR, Alarcon C, He W, Wang Q, Seoane J, Lash A, Massague J. A FoxO-Smad synexpression group in human keratinocytes. Proc Natl Acad Sci U S A. 2006;103(34):12747–52. doi: 10.1073/pnas.0605333103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim SG, Kim HA, Jong HS, Park JH, Kim NK, Hong SH, Kim TY, Bang YJ. The endogenous ratio of Smad2 and Smad3 influences the cytostatic function of Smad3. Mol Biol Cell. 2005;16(10):4672–83. doi: 10.1091/mbc.E05-01-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolfraim LA, Fernandez TM, Mamura M, Fuller WL, Kumar R, Cole DE, Byfield S, Felici A, Flanders KC, Walz TM, Roberts AB, Aplan PD, Balis FM, Letterio JJ. Loss of Smad3 in acute T-cell lymphoblastic leukemia. N Engl J Med. 2004;351(6):552–9. doi: 10.1056/NEJMoa031197. [DOI] [PubMed] [Google Scholar]
- 36.Ju W, Ogawa A, Heyer J, Nierhof D, Yu L, Kucherlapati R, Shafritz DA, Bottinger EP. Deletion of Smad2 in mouse liver reveals novel functions in hepatocyte growth and differentiation. Mol Cell Biol. 2006;26(2):654–67. doi: 10.1128/MCB.26.2.654-667.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu SL, Zhang WC, Akiyama Y, Nomizu T, Yuasa Y. Genomic structure of the transforming growth factor beta type II receptor gene and its mutations in hereditary nonpolyposis colorectal cancers. Cancer Res. 1996;56(20):4595–8. [PubMed] [Google Scholar]
- 38.Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B, Brattain M, Willson JKV. Inactivation of the type II TGF-β receptor in colon cancer cells with microsatellite instability. Science. 1995;268(5215):1336–8. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 39.Myeroff LL, Parsons R, Kim SJ, Hedrick L, Cho KR, Orth K, Mathis Michael, Kinzler KW, Lutterbaugh J, Park K, Bang Y-J, Lee HY, Park J-G, Lynch HT, Roberts AB, Vogeistein B, Markowitz SD. A transforming growth factor β receptor type II gene mutation common in colon and gastric but rare in endometrial cancers with microsatellite instability. Cancer Res. 1995;55(23):5545–7. [PubMed] [Google Scholar]
- 40.Chung YJ, Song JM, Lee JY, Jung YT, Seo EJ, Choi SW, Rhyu MG. Microsatellite instability-associated mutations associate preferentially with the intestinal type of primary gastric carcinomas in a high-risk population. Cancer Res. 1996;56(20):4662–5. [PubMed] [Google Scholar]
- 41.Ohue M, Tomita N, Monden T, Miyoshi Y, Ohnishi T, Izawa H, Kawabata Y, Sasaki M, Sekimoto M, Nishisho I, Shiozaki H, Monden M. Mutations of the transforming growth factor β type II receptor gene and microsatellite instability in gastric cancer. Int J Cancer. 1996;68(2):203–6. doi: 10.1002/(SICI)1097-0215(19961009)68:2<203::AID-IJC11>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 42.Park K, Kim SJ, Bang YJ, Park JG, Kim NK, Roberts AB, Sporn MB. Genetic changes in the transforming growth factor β (TGF-β) type II receptor gene in human gastric cancer cells: correlation with sensitivity to growth inhibition by TGF-β. Proc Natl Acad Sci U S A. 1994;91(19):8772–6. doi: 10.1073/pnas.91.19.8772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Izumoto S, Arita N, Ohnishi T, Hiraga S, Taki T, Tomita N, Ohue M, Hayakawa T. Microsatellite instability and mutated type II transforming growth factor-β receptor gene in gliomas. Cancer Lett. 1997;112(2):251–6. doi: 10.1016/s0304-3835(96)04583-1. [DOI] [PubMed] [Google Scholar]
- 44.Wang JC, Su CC, Xu JB, Chen LZ, Hu XH, Wang GY, Bao Y, Huang Q, Fu SB, Li P, Lu CQ, Zhang RM, Luo ZW. Novel microdeletion in the transforming growth factor β type II receptor gene is associated with giant and large cell variants of nonsmall cell lung carcinoma. Genes Chromosomes Cancer. 2007;46(2):192–201. doi: 10.1002/gcc.20400. [DOI] [PubMed] [Google Scholar]
- 45.Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271(5247):350–3. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- 46.Howe JR, Roth S, Ringold JC, Summers RW, Jarvinen HJ, Sistonen P, Tomlinson IP, Houlston RS, Bevan S, Mitros FA, Stone EM, Aaltonen LA. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998;280:1086–8. doi: 10.1126/science.280.5366.1086. [DOI] [PubMed] [Google Scholar]
- 47.Wang D, Kanuma T, Mizunuma H, Takama F, Ibuki Y, Wake N, Mogi A, Shitara Y, Takenoshita S. Analysis of specific gene mutations in the transforming growth factor-β signal transduction pathway in human ovarian cancer. Cancer Res. 2000;60(16):4507–12. [PubMed] [Google Scholar]
- 48.Chen T, Triplett J, Dehner B, Hurst B, Colligan B, Pemberton J, Graff JR, Carter JH. Transforming growth factor-β receptor type I gene is frequently mutated in ovarian carcinomas. Cancer Res. 2001;61(12):4679–82. [PubMed] [Google Scholar]
- 49.Chen T, Carter D, Garrigue-Antar L, Reiss M. Transforming growth factor β type I receptor kinase mutant associated with metastatic breast cancer. Cancer Res. 1998;58:4805–10. [PubMed] [Google Scholar]
- 50.Goggins M, Shekher M, Turnacioglu K, Yeo CJ, Hruban RH, Kern SE. Genetic alterations of the transforming growth factor β receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res. 1998;58(23):5329–32. [PubMed] [Google Scholar]
- 51.Schiemann WP, Pfeifer WM, Levi E, Kadin ME, Lodish HF. A deletion in the gene for transforming growth factor β type I receptor abolishes growth regulation by transforming growth factor β in a cutaneous T-cell lymphoma. Blood. 1999;94(8):2854–61. [PubMed] [Google Scholar]
- 52.Eppert K, Scherer SW, Ozcelik H, Pirone R, Hoodless P, Kim H, Tsui LC, Bapat B, Gallinger S, Andrulis IL, Thomsen GH, Wrana JL, Attisano L. MADR2 maps to 18q21 and encodes a TGFβ-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell. 1996;86(4):543–52. doi: 10.1016/s0092-8674(00)80128-2. [DOI] [PubMed] [Google Scholar]
- 53.Takagi Y, Koumura H, Futamura M, Aoki S, Ymaguchi K, Kida H, Tanemura H, Shimokawa K, Saji S. Somatic alterations of the SMAD-2 gene in human colorectal cancers. Br J Cancer. 1998;78(9):1152–5. doi: 10.1038/bjc.1998.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yanagisawa K, Uchida K, Nagatake M, Masuda A, Sugiyama M, Saito T, Yamaki K, Takahashi T, Osada H. Heterogeneities in the biological and biochemical functions of Smad2 and Smad4 mutants naturally occurring in human lung cancers. Oncogene. 2000;19(19):2305–11. doi: 10.1038/sj.onc.1203591. [DOI] [PubMed] [Google Scholar]
- 55.Ku JL, Park SH, Yoon KA, Shin YK, Kim KH, Choi JS, Kang HC, Kim IJ, Han IO, Park JG. Genetic alterations of the TGF-β signaling pathway in colorectal cancer cell lines: a novel mutation in Smad3 associated with the inactivation of TGF-β-induced transcriptional activation. Cancer Lett. 2007;247(2):283–92. doi: 10.1016/j.canlet.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 56.Datto MB, Frederick JP, Pan L, Borton AJ, Zhuang Y, Wang XF. Targeted disruption of Smad3 reveals an essential role in transforming growth factor β-mediated signal transduction. Mol Cell Biol. 1999;19(4):2495–504. doi: 10.1128/mcb.19.4.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, Roberts AB, Deng C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J. 1999;18(5):1280–91. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu Y, Richardson JA, Parada LF, Graff JM. Smad3 mutant mice develop metastatic colorectal cancer. Cell. 1998;94(6):703–14. doi: 10.1016/s0092-8674(00)81730-4. [DOI] [PubMed] [Google Scholar]
- 59.Maggio-Price L, Treuting P, Zeng W, Tsang M, Bielefeldt-Ohmann H, Iritani BM. Helicobacter infection is required for inflammation and colon cancer in SMAD3-deficient mice. Cancer Res. 2006;66(2):828–38. doi: 10.1158/0008-5472.CAN-05-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bartsch H, Nair J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: role of lipid peroxidation, DNA damage, and repair. Langenbecks Arch Surg. 2006;391(5):499–510. doi: 10.1007/s00423-006-0073-1. [DOI] [PubMed] [Google Scholar]
- 61.Sodir NM, Chen X, Park R, Nickel AE, Conti PS, Moats R, Bading JR, Shibata D, Laird PW. Smad3 deficiency promotes tumorigenesis in the distal colon of ApcMin/+ mice. Cancer Res. 2006;66(17):8430–8. doi: 10.1158/0008-5472.CAN-06-1437. [DOI] [PubMed] [Google Scholar]
- 62.Yang YA, Zhang GM, Feigenbaum L, Zhang YE. Smad3 reduces susceptibility to hepatocarcinoma by sensitizing hepatocytes to apoptosis through down-regulation of Bcl-2. Cancer Cell. 2006;9(6):445–57. doi: 10.1016/j.ccr.2006.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yamamura Y, Hua X, Bergelson S, Lodish HF. Critical role of Smads and AP-1 complex in transforming growth factor-β-dependent apoptosis. J Biol Chem. 2000;275(46):36295–302. doi: 10.1074/jbc.M006023200. [DOI] [PubMed] [Google Scholar]
- 64.Kim BC, Mamura M, Choi KS, Calabretta B, Kim SJ. Transforming growth factor β1 induces apoptosis through cleavage of BAD in a Smad3-dependent mechanism in FaO hepatoma cells. Mol Cell Biol. 2002;22(5):1369–78. doi: 10.1128/mcb.22.5.1369-1378.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Graninger WB, Seto M, Boutain B, Goldman P, Korsmeyer SJ. Expression of Bcl-2 and Bcl-2-Ig fusion transcripts in normal and neoplastic cells. J Clin Invest. 1987;80(5):1512–5. doi: 10.1172/JCI113235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O'Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 67.Reed JC. Apoptosis-based therapies. Nat Rev Drug Discov. 2002;1(2):111–21. doi: 10.1038/nrd726. [DOI] [PubMed] [Google Scholar]
- 68.Jang CW, Chen CH, Chen CC, Chen JY, Su YH, Chen RH. TGF-β induces apoptosis through Smad-mediated expression of DAP-kinase. Nat Cell Biol. 2002;4(1):51–8. doi: 10.1038/ncb731. [DOI] [PubMed] [Google Scholar]
- 69.Takekawa M, Tatebayashi K, Itoh F, Adachi M, Imai K, Saito H. Smad-dependent GADD45β expression mediates delayed activation of p38 MAP kinase by TGF-β. EMBO J. 2002;21(23):6473–82. doi: 10.1093/emboj/cdf643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yoo J, Ghiassi M, Jirmanova L, Balliet AG, Hoffman B, Fornace AJ, Jr, Liebermann DA, Bottinger EP, Roberts AB. Transforming growth factor-β-induced apoptosis is mediated by Smad-dependent expression of GADD45β through p38 activation. J Biol Chem. 2003;278(44):43001–7. doi: 10.1074/jbc.M307869200. [DOI] [PubMed] [Google Scholar]
- 71.Wildey GM, Patil S, Howe PH. Smad3 potentiates transforming growth factor β (TGFβ)-induced apoptosis and expression of the BH3-only protein Bim in WEHI 231 B lymphocytes. J Biol Chem. 2003;278(20):18069–77. doi: 10.1074/jbc.M211958200. [DOI] [PubMed] [Google Scholar]
- 72.Furukawa F, Matsuzaki K, Mori S, Tahashi Y, Yoshida K, Sugano Y, Yamagata H, Matsushita M, Seki T, Inagaki Y, Nishizawa M, Fujisawa J, Inoue K. p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology. 2003;38(4):879–89. doi: 10.1053/jhep.2003.50384. [DOI] [PubMed] [Google Scholar]
- 73.Mori S, Matsuzaki K, Yoshida K, Furukawa F, Tahashi Y, Yamagata H, Sekimoto G, Seki T, Matsui H, Nishizawa M, Fujisawa J, Okazaki K. TGF-β and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker regions. Oncogene. 2004;23(44):7416–29. doi: 10.1038/sj.onc.1207981. [DOI] [PubMed] [Google Scholar]
- 74.Liao JH, Chen JS, Chai MQ, Zhao S, Song JG. The involvement of p38 MAPK in transforming growth factor β1-induced apoptosis in murine hepatocytes. Cell Res. 2001;11(2):89–94. doi: 10.1038/sj.cr.7290072. [DOI] [PubMed] [Google Scholar]
- 75.Schrantz N, Bourgeade MF, Mouhamad S, Leca G, Sharma S, Vazquez A. p38-mediated regulation of an Fas-associated death domain protein-independent pathway leading to caspase-8 activation during TGFβ-induced apoptosis in human Burkitt lymphoma B cells BL41. Mol Biol Cell. 2001;12(10):3139–51. doi: 10.1091/mbc.12.10.3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu L, Hebert MC, Zhang YE. TGF-β receptor-activated p38 MAP kinase mediates Smad-independent TGF-β responses. EMBO J. 2002;21(14):3749–59. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim KY, Kim BC, Xu Z, Kim SJ. Mixed lineage kinase 3 (MLK3)-activated p38 MAP kinase mediates transforming growth factor-β-induced apoptosis in hepatoma cells. J Biol Chem. 2004;279(28):29478–84. doi: 10.1074/jbc.M313947200. [DOI] [PubMed] [Google Scholar]
- 78.Conery AR, Cao Y, Thompson EA, Townsend CM, Jr, Ko TC, Luo K. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-β induced apoptosis. Nat Cell Biol. 2004;6(4):366–72. doi: 10.1038/ncb1117. [DOI] [PubMed] [Google Scholar]
- 79.Remy I, Montmarquette A, Michnick SW. PKB/Akt modulates TGF-β signalling through a direct interaction with Smad3. Nat Cell Biol. 2004;6(4):358–65. doi: 10.1038/ncb1113. [DOI] [PubMed] [Google Scholar]
- 80.Song K, Wang H, Krebs TL, Danielpour D. Novel roles of Akt and mTOR in suppressing TGF-β/ ALK5-mediated Smad3 activation. EMBO J. 2006;25(1):58–69. doi: 10.1038/sj.emboj.7600917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Akhurst RJ, Derynck R. TGF-β signaling in cancer—a double-edged sword. Trends Cell Biol. 2001;11(11):S44–51. doi: 10.1016/s0962-8924(01)02130-4. [DOI] [PubMed] [Google Scholar]
- 82.Kang Y, He W, Tulley S, Gupta GP, Serganova I, Chen CR, Manova-Todorova K, Blasberg R, Gerald WL, Massague J. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci U S A. 2005;102(39):13909–14. doi: 10.1073/pnas.0506517102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wakefield LM, Roberts AB. TGF-β signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev. 2002;12(1):22–9. doi: 10.1016/s0959-437x(01)00259-3. [DOI] [PubMed] [Google Scholar]
- 84.Thomas DA, Massague J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8(5):369–80. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 85.Letterio JJ, Roberts AB. Regulation of immune responses by TGF-β. Annu Rev Immunol. 1998;16:137–61. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- 86.Gorelik L, Flavell RA. Transforming growth factor-β in T-cell biology. Nat Rev Immunol. 2002;2(1):46–53. doi: 10.1038/nri704. [DOI] [PubMed] [Google Scholar]
- 87.Geiser AG, Letterio JJ, Kulkarni AB, Karlsson S, Roberts AB, Sporn MB. Transforming growth factor β1 (TGF-β1) controls expression of major histocompatibility genes in the postnatal mouse: aberrant histocompatibility antigen expression in the pathogenesis of the TGF-β1 null mouse phenotype. Proc Natl Acad Sci U S A. 1993;90(21):9944–8. doi: 10.1073/pnas.90.21.9944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Letterio JJ, Geiser AG, Kulkarni AB, Dang H, Kong L, Nakabayashi T, Mackall CL, Gress RE, Roberts AB. Autoimmunity associated with TGF-β1-deficiency in mice is dependent on MHC class II antigen expression. J Clin Invest. 1996;98(9):2109–19. doi: 10.1172/JCI119017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-β signaling in T cells. Nat Med. 2001;7(10):1118–22. doi: 10.1038/nm1001-1118. [DOI] [PubMed] [Google Scholar]
- 90.Letterio JJ. TGF-β signaling in T cells: roles in lymphoid and epithelial neoplasia. Oncogene. 2005;24(37):5701–12. doi: 10.1038/sj.onc.1208922. [DOI] [PubMed] [Google Scholar]
- 91.McKarns SC, Schwartz RH, Kaminski NE. Smad3 is essential for TGF-β1 to suppress IL-2 production and TCR-induced proliferation, but not IL-2-induced proliferation. J Immunol. 2004;172(7):4275–84. doi: 10.4049/jimmunol.172.7.4275. [DOI] [PubMed] [Google Scholar]
- 92.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2(6):442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 93.Cui W, Fowlis DJ, Bryson S, Duffie E, Ireland H, Balmain A, Akhurst RJ. TGFβ1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell. 1996;86(4):531–42. doi: 10.1016/s0092-8674(00)80127-0. [DOI] [PubMed] [Google Scholar]
- 94.Han G, Lu SL, Li AG, He W, Corless CL, Kulesz-Martin M, Wang XJ. Distinct mechanisms of TGF-β1-mediated epithelial-to-mesenchymal transition and metastasis during skin carcinogenesis. J Clin Invest. 2005;115(7):1714–23. doi: 10.1172/JCI24399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Piek E, Moustakas A, Kurisaki A, Heldin CH, ten Dijke P. TGF-β type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal trans-differentiation in NMuMG breast epithelial cells. J Cell Sci. 1999;112(Pt 24):4557–68. doi: 10.1242/jcs.112.24.4557. [DOI] [PubMed] [Google Scholar]
- 96.Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A. TGF-β and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell. 2005;16(4):1987–2002. doi: 10.1091/mbc.E04-08-0658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Saika S, Kono-Saika S, Tanaka T, Yamanaka O, Ohnishi Y, Sato M, Muragaki Y, Ooshima A, Yoo J, Flanders KC, Roberts AB. Smad3 is required for dedifferentiation of retinal pigment epithelium following retinal detachment in mice. Lab Invest. 2004;84(10):1245–58. doi: 10.1038/labinvest.3700156. [DOI] [PubMed] [Google Scholar]
- 98.Saika S, Kono-Saika S, Ohnishi Y, Sato M, Muragaki Y, Ooshima A, Flanders KC, Yoo J, Anzano M, Liu CY, Kao WW, Roberts AB. Smad3 signaling is required for epithelial-mesenchymal transition of lens epithelium after injury. Am J Pathol. 2004;164(2):651–63. doi: 10.1016/S0002-9440(10)63153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-β1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest. 2003;112(10):1486–94. doi: 10.1172/JCI19270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of TGF-β/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004;23(5):1155–65. doi: 10.1038/sj.emboj.7600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, Arteaga CL, Moses HL. Transforming growth factor-β1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. 2001;12(1):27–36. doi: 10.1091/mbc.12.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-β induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. 1994;127(6 Pt 2):2021–36. doi: 10.1083/jcb.127.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Oft M, Peli J, Rudaz C, Schwarz H, Beug H, Reichmann E. TGF-β1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 1996;10(19):2462–77. doi: 10.1101/gad.10.19.2462. [DOI] [PubMed] [Google Scholar]
- 104.Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2(2):84–9. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 105.Shirai K, Saika S, Tanaka T, Okada Y, Flanders KC, Ooshima A, Ohnishi Y. A new model of anterior subcapsular cataract: involvement of TGFβ/Smad signaling. Mol Vis. 2006;12:681–91. [PubMed] [Google Scholar]
- 106.Zavadil J, Bitzer M, Liang D, Yang YC, Massimi A, Kneitz S, Piek E, Bottinger EP. Genetic programs of epithelial cell plasticity directed by transforming growth factor-β. Proc Natl Acad Sci U S A. 2001;98(12):6686–91. doi: 10.1073/pnas.111614398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ueki N, Nakazato M, Ohkawa T, Ikeda T, Amuro Y, Hada T, Higashino K. Excessive production of transforming growth-factor β1 can play an important role in the development of tumorigenesis by its action for angiogenesis: validity of neutralizing antibodies to block tumor growth. Biochim Biophys Acta. 1992;1137(2):189–96. doi: 10.1016/0167-4889(92)90201-l. [DOI] [PubMed] [Google Scholar]
- 108.Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J, Beug H, Grunert S. Ras and TGFβ cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol. 2002;156(2):299–313. doi: 10.1083/jcb.200109037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Oft M, Heider KH, Beug H. TGFβ signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol. 1998;8(23):1243–52. doi: 10.1016/s0960-9822(07)00533-7. [DOI] [PubMed] [Google Scholar]
- 110.Siegel PM, Shu W, Cardiff RD, Muller WJ, Massague J. Transforming growth factor β signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci U S A. 2003;100(14):8430–5. doi: 10.1073/pnas.0932636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Muraoka-Cook RS, Dumont N, Arteaga CL. Dual role of transforming growth factor β in mammary tumorigenesis and metastatic progression. Clin Cancer Res. 2005;11(2 Pt 2):937s–43s. [PubMed] [Google Scholar]
- 112.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2(8):584–93. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 113.Tian F, DaCosta Byfield S, Parks WT, Yoo S, Felici A, Tang B, Piek E, Wakefield LM, Roberts AB. Reduction in Smad2/3 signaling enhances tumorigenesis but suppresses metastasis of breast cancer cell lines. Cancer Res. 2003;63(23):8284–92. [PubMed] [Google Scholar]
- 114.Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3(6):537–49. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]