

Abstract
Myocardial infarction is the most common cause of cardiac injury and results in acute loss of a large number of myocardial cells. Because the heart has negligible regenerative capacity, cardiomyocyte death triggers a reparative response that ultimately results in formation of a scar and is associated with dilative remodeling of the ventricle. Cardiac injury activates innate immune mechanisms initiating an inflammatory reaction. Toll Like Receptor-mediated pathways, the complement cascade and reactive oxygen generation induce Nuclear Factor (NF)-κB activation and upregulate chemokine and cytokine synthesis in the infarcted heart. Chemokines stimulate the chemotactic recruitment of inflammatory leukocytes into the infarct, while cytokines promote adhesive interactions between leukocytes and endothelial cells, resulting in transmigration of inflammatory cells into the site of injury. Monocyte subsets play distinct roles in phagocytosis of dead cardiomyocytes and in granulation tissue formation through the release of growth factors. Clearance of dead cells and matrix debris may be essential for resolution of inflammation and transition into the reparative phase. Transforming Growth Factor (TGF)-β plays a crucial role in cardiac repair by suppressing inflammation while promoting myofibroblast phenotypic modulation and extracellular matrix deposition. Myofibroblast proliferation and angiogenesis result in formation of highly vascularized granulation tissue. As the healing infarct matures, fibroblasts become apoptotic and a collagen-based matrix is formed, while many infarct neovessels acquire a muscular coat and uncoated vessels regress. Timely resolution of the inflammatory infiltrate and spatial containment of the inflammatory and reparative response into the infarcted area are essential for optimal infarct healing. Targeting inflammatory pathways following infarction may reduce cardiomyocyte injury and attenuate adverse remodeling. In addition, understanding the role of the immune system in cardiac repair is necessary in order to design optimal strategies for cardiac regeneration.
Keywords: myocardial infarction, chemokine, cytokine, TGF-β, leukocyte
1. INTRODUCTION
Because the human heart has negligible regenerative capacity, cardiac injury poses a great challenge for the reparative mechanisms of the body, ultimately resulting in formation of a scar. Myocardial infarction is the most common and clinically significant form of acute cardiac injury and results in ischemic death of a large number of cardiomyocytes. Other injurious processes (such as chronic pressure or volume overload and chronic cardiomyopathic conditions) may cause more sporadic loss of cardiac muscle and activate hypertrophic and pro-fibrotic responses. Despite recent declines in the incidence of myocardial infarction, more than 1.5 million Americans suffer an acute infarct every year; approximately one fourth of all deaths are due to acute myocardial infarction [1]. Most myocardial infarcts result from coronary atherosclerosis, complicated by acute rupture of an atherosclerotic plaque and subsequent formation of a coronary thrombus. Sudden induction of ischemia by coronary artery occlusion triggers a series of events that culminates in the death of ischemic cardiomyocytes throughout the anatomic region supplied by this artery [2]. The necrotic muscle elicits an inflammatory cascade that serves to clear the infarct from dead cells and matrix debris, and ultimately results in healing and replacement of the damaged tissue with scar. Thus, cardiac repair following myocardial infarction is dependent on activation of inflammatory pathways. Infarct healing results in profound changes in ventricular architecture and geometry, also referred to as “ventricular remodeling” [3], [4]. The molecular and cellular changes associated with ventricular remodeling affect both the necrotic zone and the non-infarcted segments of the ventricle and manifest clinically as chamber dilation, cardiac hypertrophy, increased sphericity of the ventricle and worsened cardiac function [5]. Remodeling is linked to heart failure progression and is associated with poor prognosis following myocardial infarction. Ventricular dilation following myocardial infarction is an important predictor of mortality [6] and adverse cardiac events [7], including the development of heart failure and ventricular arrhythmias [8], [9]. The extent of adverse remodeling depends on the size of the infarct but is also directly affected by the pathologic and structural changes associated with infarct healing. In addition, defects in the healing process may be directly involved in the development of serious and potentially lethal complications, such as cardiac rupture and ventricular aneurysm formation. The cardiac rupture syndromes involve tearing of acutely infarcted tissue and result from mechanical weakening that occurs in the necrotic and inflamed myocardium. Rupture of the ventricular free wall results in cardiac tamponade and is usually fatal, whereas rupture of the ventricular septum leads to a left-to-right shunt and papillary muscle rupture results in acute mitral regurgitation. Formation of a ventricular aneurysm on the other hand is a late complication associated with a large transmural infarct that heals into a large region of thin scar tissue and paradoxically bulges during systole.
Although recent advances in cardiovascular therapy have resulted in the development of novel strategies that salvage myocardium and improve early mortality in patients with myocardial infarction through early reperfusion, approaches directly targeting the healing process are lacking. Because of the essential role of immunoinflammatory pathways in cardiac injury and repair, the inflammatory cascade may provide unique opportunities for interventions aimed at reducing cardiomyocyte injury, while optimizing the healing response and attenuating adverse remodeling. The current review examines the role of the immune system in activating reparative pathways following acute myocardial infarction and explores the inflammatory mechanisms of cardiac injury and repair.
2. THE PATHOLOGY OF MYOCARDIAL INFARCTION: AN OVERVIEW
Because the mammalian heart cannot produce enough energy under anaerobic conditions to maintain essential cellular processes, a constant supply of oxygen is indispensable to sustain cardiac function and viability. Ischemic myocardial injury results in decreased oxygen tension within the cell, subsequent loss of oxidative phosphorylation and decreased generation of high energy phosphates. Adenosine triphosphate (ATP) depletion leads to failure of the sodium pump, loss of potassium, influx of sodium and water, and cell swelling. Cessation of aerobic metabolism, ATP depletion and accumulation of products of anoxic metabolism (such as lactic acid) occur within ten seconds of occlusion. Striking loss of contractility occurs almost simultaneously and is evident within sixty seconds. Minutes after the onset of ischemia reversible ultrastructural cardiomyocyte changes appear, including cellular and mitochondrial swelling and glycogen depletion. Irreversible cardiomyocyte injury, evidenced by sarcolemmal disruption and the presence of small amorphous densities in the mitochondria, develops after 20–40 minutes of sustained severe ischemia [2]. The predominant mechanism of cardiomyocyte death in the infarcted heart is coagulation necrosis, although apoptosis is also likely to contribute to cardiomyocyte loss. Dying cells trigger an inflammatory reaction, activating reparative pathways that ultimately result in formation of a scar. Infarct healing can be divided into three overlapping phases: the inflammatory phase, the proliferative phase and the maturation phase [10] (Figure 1). During the inflammatory phase activation of chemokine and cytokine cascades results in recruitment of leukocytes into the infarcted area. Neutrophils and macrophages clear the wound from dead cells and matrix debris. Activated macrophages release cytokines and growth factors leading to formation of highly-vascularized granulation tissue. At this stage expression of pro-inflammatory mediators is suppressed, while fibroblasts and endothelial cells proliferate. During the proliferative phase of healing, activated myofibroblasts produce extracellular matrix proteins and an extensive microvascular network is formed. Maturation of the scar follows: fibroblasts and vascular cells undergo apoptosis and a collagen-based scar is formed. Several factors determine the time course of the cellular events associated with infarct healing. Reperfusion of the coronary vessel accentuates the inflammatory reaction and greatly accelerates the healing response. In addition, larger infarcts may not heal as readily as smaller ones. Finally, species-specific differences affect the time course of the inflammatory and reparative response following infarction. Rodents exhibit accelerated replacement of dead cardiomyocytes with granulation tissue in comparison to dogs and humans [11], [12], [13].
Figure 1. Infarct healing is closely intertwined with ventricular remodeling.

The healing response can be divided in three overlapping phases. During the inflammatory phase, chemokines and cytokines are induced in the infarct and marked leukocyte infiltration is noted. Neutrophils and macrophages clear the wound from dead cells and matrix debris. During the proliferative phase of healing, activated macrophages release cytokines and growth factors leading to formation of highly-vascularized granulation tissue. At this stage expression of pro-inflammatory mediators is suppressed, while fibroblasts and endothelial cells proliferate. Activated myofibroblasts produce extracellular matrix proteins and an extensive microvascular network is formed. Maturation of the scar follows: fibroblasts and vascular cells undergo apoptosis and a collagen-based scar is formed. As the infarct heals, dilative remodeling of the infarcted ventricle is noted (top panel). The time course of the cellular events presented in this figure is based on a reperfused model of infarction in the mouse. Large animals and humans exhibit delayed healing in comparison with rodents.
3. INITIATION OF THE POST-INFARCTION INFLAMMATORY RESPONSE. THE ROLE OF INNATE IMMUNITY
Cells dying by necrosis release their intracellular contents and initiate an intense inflammatory response by activating innate immune mechanisms. Cell surface receptors sense endogenous ligands released upon tissue injury as “danger signals” and activate inflammatory pathways. Evidence suggests that Toll-like receptor (TLR)-mediated pathways, the complement cascade and reactive oxygen species (ROS) play a significant role in triggering the post-infarction inflammatory response by activating the Nuclear factor (NF)-κB system.
TLR-mediated pathways
The TLRs represent a family of pattern recognition receptors that serve to recognize molecular patterns associated with pathogens and, upon binding of their ligands, induce activation of several kinases and NF-κB. Endogenous ligands from damaged tissues, including heat shock proteins, hyaluronan and fibronectin fragments serve as “danger signals” and are capable of activating TLRs [14]. Thus, even in the absence of microbial pathogens, alterations of the extracellular matrix due to tissue destruction can activate the innate immune system through TLR-mediated pathways [15]. To date, 12 members of the TLR family have been identified in mammals; however, their role in cardiac pathology remains poorly understood. TLR4 is expressed in the heart and is markedly induced in mouse and rat infarcts and in samples obtained from cardiomyopathic hearts [16]. Recent investigations demonstrated that TLR4 deficient mice have decreased infarct size and suppressed inflammation [17], and exhibit attenuated adverse remodeling following myocardial infarction [18], identifying TLR4 as a key component of the innate immune response in the infarcted heart. In contrast, TLR2 null animals had similar infarct size and comparable inflammatory leukocyte infiltration with their wildtype littermates, but exhibited decreased fibrosis in the non-infarcted area and attenuated post-infarction ventricular remodeling [19]. These findings suggested that TLR2 signaling may not critically affect the inflammatory response but may (directly or indirectly) modulate fibrous tissue deposition.
TLR engagement may also activate members of the pentraxin group, a superfamily of multimeric pattern-recognition proteins with an essential role in inflammatory processes. Pentraxins are divided into two groups: the short and long pentraxin families. C-reactive Protein (CRP) and Serum Amyloid P (SAP) Component are the prototypes of the short pentraxin family and are mainly produced by the liver as acute phase proteins in response to inflammatory signals [20]. It has been suggested that CRP deposition in the infarct may locally activate the complement cascade accentuating cardiomyocyte injury [21], [22]. On the other hand the prototypical long pentraxin PTX3 is a multifunctional protein at the crossroads between immunity and inflammation. PTX3 is induced in myocardial infarction and appears to play an important cardioprotective role possibly due to modulation of the complement cascade [23]. The diverse effects of the pentraxins in myocardial infarction highlight their complex role in the ischemic heart.
The complement cascade
The complement system is an important component of the innate immune response and a major effector in a variety of immunopathological diseases. The complement cascade is activated through three distinct mechanisms designated the classical, alternative and lectin pathways [24], [25]. Numerous studies have indicated that ischemic myocardial injury activates the complement cascade [26]. Hill and Ward [27] were the first to demonstrate that leukotactic activity in rat myocardial infarcts was in part due to C3 cleavage products. Subsequently Pinckard and colleagues [28] showed evidence of C1, C3 and C4 consumption in patients with acute myocardial infarction, suggesting that myocardial cell necrosis results in the release of subcellular membrane constituents capable of activating the complement cascade. Further studies [26, 29] have suggested that during myocardial ischemia, mitochondria, extruded through breaks in the sarcolemma, unfold and release membrane fragments rich in cardiolipin and protein. By binding C1 and supplying sites for the assembly of later acting complement components, these subcellular fragments provide the means to disseminate the complement-mediated inflammatory response to ischemic injury. mRNA and proteins for all the components of the classical complement pathway are upregulated in myocardial infarcts [30]. Complement activation may play an important role in mediating neutrophil and monocyte recruitment in the injured myocardium [31]. The contribution of complement activation in mononuclear cell recruitment appears to be particularly important during the first hour of reperfusion [32].
Blocking activation of the complement system can be achieved by consumptive depletion (such as with cobra venom factor injection), by antibody-induced inhibition of individual complement components (e.g. C5), or by infusion of modified native complement components that block complement activation, such as the soluble form of complement receptor type 1(sCR1) [33], [34]. Complement depletion using cobra venom factor injection at the time of experimental coronary artery occlusion has been shown to attenuate myocardial necrosis in a variety of animal models [35]. However, conclusions derived from studies with a focus on complement depletion overlook the prior systemic activation that may result in deactivation of neutrophils. Administration of C1-esterase inhibitor decreased infarct size in several experimental models of coronary occlusion and reperfusion [36, 37], [38–40]; however, high doses were detrimental, possibly due to pro-coagulant effects mediated through inactivation of the fibrinolytic system [38]. In addition, infusion of soluble human complement receptor type 1 (sCR1) significantly decreased infarct size in a rat model of myocardial ischemia and reperfusion [41]. These studies raised the possibility that interference with precisely targeted products of the complement system may reduce myocardial injury [42], [43].
Despite the promising experimental evidence, recent clinical trials testing the effects of complement inhibition in patients with acute myocardial infarction produced rather disappointing results. In the Complement Inhibition in Myocardial Infarction Treated with Angioplasty (COMMA) trial, administration of the humanized anti-C5 monoclonal antibody pexelizumab in patients with ST-elevation myocardial infarction undergoing primary angioplasty, did not affect infarct size, but decreased the ninety-day mortality rate when administered as a bolus plus infusion [44] (1.8% versus 5.9% with placebo); the bolus-only group had an intermediate mortality rate (4.2%). In contrast, when used in patients receiving thrombolytics, pexelizumab blocked complement activity but neither reduced infarct size, nor improved the clinical outcome [45]. In the Assessment of Pexelizumab in Acute Myocardial Infarction (APEX-AMI) trial, treatment with pexelizumab (bolus plus infusion) as an adjunct to percutaneous intervention had no effect on mortality and on the composite endpoint of death, shock and heart failure in patients with ST-elevation myocardial infarction [46].
Generation of reactive oxygen species (ROS) and the post-infarction inflammatory response
ROS are atoms or molecules with unpaired electrons in their outer orbit. They are highly reactive entities and can participate in a variety of biochemical reactions [47]. ROS react directly with cellular lipids, proteins and DNA, causing cell injury and death, and are critically involved in the oxidative burst reaction, which is essential for phagocyte function. In addition, ROS trigger cytokine and chemokine cascades through NF-κB activation [48], [49], [50]. Granger and colleagues [51] have provided evidence suggesting a role for reactive oxygen in leukocyte chemotaxis. Potential mechanisms through which ROS may generate a leukotactic stimulus include complement activation [52, 53], induction of P-selectin expression [54], chemokine upregulation [55–57] and increased ability of endothelial Intercellular Adhesion Molecule (ICAM)-1 to bind to neutrophils [58].
The normal heart possesses substantial ability to counterbalance the generation of ROS through enzymatic pathways (such as catalase, glutathione peroxidase and the superoxide dismutases) and through intracellular antioxidants. However, in the infarcted myocardium the antioxidant defenses are overwhelmed, resulting in generation of oxygen-related free radicals. ROS have been shown to exert a direct inhibitory effect on myocardial function in vivo and play a critical role in the pathogenesis of myocardial stunning [59, 60], [61]. Most of the evidence implicating ROS in the pathophysiology of myocardial infarction is derived from investigations using free radical scavengers. Jolly and coworkers [62] demonstrated that the combination of the antioxidant enzymes superoxide dismutase (SOD) and catalase significantly reduced infarct size in dogs undergoing experimental coronary occlusion and reperfusion, when the infusion started before ischemia or 15 min prior to reperfusion. In contrast, no effect was noted when antioxidant infusion was started 40 min after reperfusion, suggesting that free radical-mediated injury is an early event [62]. Other investigators found similar beneficial effects of antioxidant interventions in experimental models of myocardial infarction. However, there is a significant number of studies describing a failure of antioxidants to prevent injury, or demonstrating an early protective effect, that waned with increased duration of reperfusion [63, 64]. Transgenic mice that overexpress copper, zinc superoxide dismutase (SOD1) exhibited significant protection from postischemic injury [65]. In addition, mice overexpressing manganese SOD (MnSOD) demonstrated a significant decrease in infarct size in Langendorff-perfused hearts undergoing left coronary artery ligation [66]. Therapeutic strategies targeting free radical generation have not been successful in clinical practice: two small clinical studies using recombinant human SOD in patients with acute myocardial infarction undergoing thrombolysis [67] or balloon angioplasty [68] demonstrated no significant improvement in left ventricular function. Unfortunately, prolonged coronary occlusion (more than 2h) is usually present in the clinical setting of reperfused myocardial infarction and may cause extensive irreversible myocardial damage, leaving fewer myocytes to be affected by free radical mediated injury [69], [70]. Furthermore, ROS-mediated actions are not always harmful. Although at high concentrations ROS are detrimental to cells, increasing evidence suggests that at lower concentrations they exert beneficial effects and are involved in the protection afforded by ischemic preconditioning [71], [72].
Activation of the NF-κB system
Activation of the NF-κB system plays an essential role in induction of proinflammatory mediators in the ischemic myocardium [73]. NF-κB is activated by a large number of agents, including cytokines (such as Tumor Necrosis Factor (TNF)-α and Interleukin (IL)-1β) and ROS. The most prevalent activated form of NF-κB is a heterodimer consisting of a p50 or p52 subunit and p65, which contains transactivation domains necessary for gene induction [74]. The genes regulated by NF-κB are diverse and include those involved in the inflammatory response, cell adhesion and growth control [75]. In resting cells, NF-κB dimers reside in the cytoplasm in an inactive form, associated to regulatory proteins called inhibitors of κB (IκB), of which the most important are IκBα, IκBβ, and IκBε. Upon stimulation, IκB is phosphorylated, ubiquitinylated, and ultimately degraded by proteolytic cleavage by the proteasome system. Phosphorylation of IκB, a key step in NF-κB activation, is mediated by the IκB kinase (IKK). Release of the NF-κB dimers from the NF-κB–IκB complex results in activation of NF-κB, which translocates to the nucleus and binds to promoters or enhancer regions of target genes, initiating their transcription. NF-κB activation has been demonstrated in various models of experimental myocardial ischemia and reperfusion [76], [77]. In vivo transfer of NF-κB decoy oligodeoxynucleotides to bind the transcriptional factor blocking inflammatory gene activation, reduced the extent of myocardial infarction following reperfusion suggesting a crucial role for NF-κB in the regulation of the post-infarction inflammatory response [78]. In addition, treatment with an IKKβ inhibitor attenuated IκBα phosphorylation reduced infarct size, improved cardiac function and decreased pro-inflammatory cytokine levels in a model of reperfused infarction [79]. Furthermore, transgenic mice with cardiac-specific expression of a dominant-negative IκBα resulting in inhibition of cardiomyocyte NF-κB activation, exhibited significantly decreased infarct size in a model of reperfused infarction [80], and NF-κB p50 null mice had improved early survival and reduced left ventricular dilatation after non-reperfused infarction [81]. Although these studies demonstrated an injurious role of NF-κB activation in the infarcted heart, other investigations have suggested that the NF-κB pathway may also mediate cytoprotective responses [82]. Transgenic mice harboring cardiac-restricted expression of a mutated IκBα protein that prevents nuclear translocation of NF-κB in cardiac myocytes had larger infarcts and significantly enhanced myocyte apoptosis in a model of permanent coronary occlusion [83].
The contradictory findings on the role of NF-κB signaling in myocardial infarction reflect the diversity of cellular processes and molecular pathways affected by the NF-κB system. NF-κB is clearly one of the most important regulators of pro-inflammatory gene expression. However, it has also been suggested that NF-κB activation in leukocytes during the resolution phase of the inflammatory process results in upregulation of anti-inflammatory genes and induces leukocyte apoptosis [84] NF-κB activation is also involved in regulation of cell survival, mediates proliferative effects and modulates matrix metabolism by regulating Matrix Metalloproteinase (MMP) synthesis [85]. Thus, activation of the NF-κB signaling cascade in the infarcted heart involves various cell types and affects multiple parallel processes with an essential role in cardiac injury and repair.
4. THE CHEMOKINE FAMILY IN MYOCARDIAL INFARCTION
The chemokines comprise a family of small (8–14kDa) highly basic proteins with a strikingly similar tertiary structure [86]. They are divided into subfamilies on the basis of the number and sequential relationship of their conserved cysteine residues (CXC, CC, XC and CX3C subfamilies). Most chemokines contain at least four cysteines that form two disulfide bonds, one between the first and the third and one between the second and the fourth cysteine. In the CXC chemokine subfamily one aminoacid separates the first two cysteine residues, whereas in the CC chemokines the first two cysteines are adjacent to each other. Lymphotactin (XCL1) contains only two cysteines corresponding to the second and fourth cysteines of other classes and represents the XC subfamily. Fractalkine, on the other hand, has three aminoacids separating the first two cysteines (CX3C). CC chemokines are the largest and most diverse subfamily, including at least 28 ligands in humans. CXC chemokines are further classified according to the presence of the tripeptide motif glutamate-leucine-arginine (ELR) in the aminoterminal region [87].
Chemokines play a critical role in basal and inflammatory leukocyte locomotion and trafficking [88, 89] and their principal targets are bone marrow-derived cells. In addition to effects on cell locomotion, certain chemokines are capable of eliciting a variety of other responses affecting leukocyte activation, degranulation and apoptosis. Emerging evidence suggests that chemokines also exert important actions on many cell types beyond the immune system, including endothelial cells (resulting in angiogenic, or angiostatic effects) [90], smooth muscle cells, neurons and epithelial cells. Chemokines can be divided broadly into two categories: homeostatic chemokines are constitutively expressed in certain tissues and may be responsible for basal leukocyte trafficking and lymphoid organ formation, and inducible chemokines which are markedly upregulated by inflammatory or immune stimuli, actively participating in the inflammatory reactions by inducing leukocyte recruitment [88], [91, 92]. Although this approach is oversimplified, it offers valuable insight into the role of certain chemokines in pathological states. A wide variety of stimuli, such as ROS generation, activation of complement, TLR-mediated pathways, and activation of the NF-κB system can upregulate inducible chemokines, leading to a rapid increase in their local concentration followed by leukocyte infiltration and an inflammatory response.
Chemokine induction is a prominent feature of the post-infarction inflammatory response [93], [94], [95] (Figure 2). Recent investigations using experimental models of myocardial infarction demonstrated strong induction of several members of the chemokine family in the ischemic heart supporting their role in leukocyte recruitment [32], infarct angiogenesis and fibrous tissue deposition [96].
Figure 2. The role of the chemokines in cardiac injury.
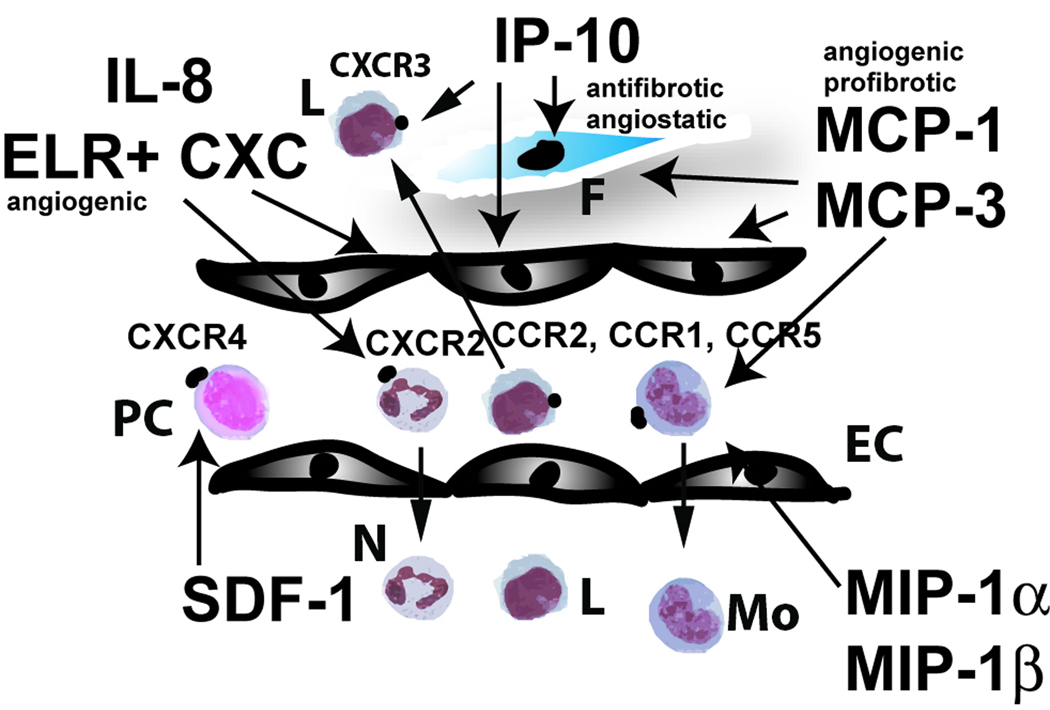
Both CC and CXC chemokines are induced in the infarcted myocardium. CXC chemokines that contain the ELR motif (such as IL-8) induce neutrophil infiltration and may exert angiogenic effects. In contrast, ELR-negative CXC chemokines (such as IP-10) have angiostatic and antifibrotic properties. Early IP-10 upregulation in the infarcted heart may inhibit granulation tissue formation until the wound is debrided from dead cells and a provisional matrix necessary to support fibroblast and endothelial cell migration is formed. In addition, IP-10 may play a role in recruitment of effector T cells. On the other hand, the CXC chemokine SDF-1 may induce infiltration of CXCR4+ progenitor cells enhancing tissue regeneration and angiogenesis. The best studied CC chemokine is MCP-1, a potent mononuclear cell chemoattractant with angiogenic and pro-fibrotic properties. MCP-1 deficiency is associated with attenuated adverse remodeling at the expense of delayed clearance of the infarct from dead cardiomyocytes. MCP-3, MIP-1α and MIP-1β are also induced in the infarct; however, their role in cardiac injury and repair remains unknown (Symbols: PC, immature progenitor cells; N, neutrophils; Mo, monocytes; L, lymphocytes; EC, endothelial cells; F, fibroblasts).
CXC chemokines in healing infarction
Members of the CXC chemokine subfamily that contain the ELR motif, such as IL-8, Epithelial Neutrophil-Activating peptide (ENA)-78, Growth-Regulated Oncogene (GRO)-α, GRO-β, GRO-γ and Neutrophil-Activating Peptide (NAP)-2, induce neutrophil chemotaxis and activation [86, 97] and have angiogenic properties. In contrast with ELR-containing chemokines, the CXC chemokines lacking the ELR motif, (such as platelet factor 4 (PF4/CXCL4), Interferon-γ-inducible Protein (IP)-10/CXCL10, and monokine induced by γ-interferon (MIG/CXCL9)) do not induce neutrophil chemotaxis and not only fail to stimulate angiogenesis, but were found to be potent angiostatic factors in the presence of either ELR-CXC chemokines or the unrelated angiogenic factor, basic fibroblast growth factor (bFGF) [90, 98].
The prototypic CXC chemokine is IL-8/CXCL8, a critical regulator of neutrophil influx and activation [99, 100] with angiogenic properties [101]. IL-8 upregulation has been documented in canine [102] and rabbit [103] models of experimental myocardial infarction. Elucidation of the role of IL-8 in cardiac injury has been hampered by the absence of an IL-8 homologue in mice. IL-8, and possibly other neutrophil chemoattractant chemokines synthesized by microvascular endothelial cells, may play an important role in granulocyte recruitment in the infarcted myocardium. IL-8 induces the neutrophil respiratory burst and granule release, and enhances cellular adhesion, a β2 integrin-dependent event. IL-8-induced β2 integrin activation may be mediated through Mitogen-activated protein kinase (MAPK) and Protein Kinase C (PKC) signaling [104]. It appears that neutrophils may need to sample immobilized IL-8 molecules presented by the vessel wall before forming a sufficient number of high avidity β2 integrin bonds for firm adhesion [105]. In addition, IL-8 may also have effects beyond its neutrophil chemotactic properties [104]. IL-8 neutralization significantly reduces the degree of necrosis in a rabbit model of myocardial ischemia-reperfusion injury without affecting neutrophil infiltration [106].
Even less is known about the expression and potential role of other ELR-containing CXC chemokines in myocardial infarction. In rodents several ELR positive CXC chemokines, including GRO-α/KC, Macrophage Inflammatory Protein (MIP)-2 and Lipopolysaccharide-Induced CXC chemokine (LIX), induce neutrophil chemotaxis and activation through binding to their main receptor, CXCR2. Experiments in a rat model of infarction demonstrated that, although KC and MIP-2 are upregulated in the injured heart, neutrophil recruitment in reperfused rat infarcts appeared to be mainly due to expression of LIX [107]. Deficiency of CXCR2, the main receptor for the ELR-containing CXC chemokines, resulted in significantly decreased inflammatory leukocyte recruitment in murine infarcts, suggesting a crucial role for these chemokines in inflammatory cell infiltration [108]. However, experiments using a Langendorff preparation indicated protective effects of CXCR2 signaling on myocardial viability [108]. The molecular basis for the presumed direct effects of CXCR2 signaling on cardiomyocytes remains unclear.
The role of the ELR-negative CXC chemokines in the healing infarct is an area of active investigation. We have demonstrated a marked transient upregulation of IP-10 in reperfused canine myocardial infarcts [96]; whether MIG and I-TAC are also induced remains unknown. IP-10 mRNA expression is downregulated following 24 h of reperfusion, whereas IL-8 message levels remain high. The exact role of IP-10 upregulation in the infarcted myocardium remains unclear. IP-10 is critically involved in effector T cell trafficking [109], has angiostatic effects [110] and may exert direct antifibrotic actions [111], [112]. It is tempting to hypothesize that the early transient induction of IP-10 in the ischemic myocardium may serve to prevent premature wound angiogenesis and fibrous tissue deposition in the infarct, until the injured myocardium has been cleared from dead cells and debris by infiltrating phagocytes and a fibrin-rich provisional matrix is formed in order to support ingrowth of granulation tissue. Subsequent suppression of IP-10 through TGF-β-mediated actions may allow unopposed angiogenic and fibrogenic activity facilitating the reparative process. Thus, IP-10 may orchestrate infarct healing through its effects on angiogenesis and fibrous tissue deposition.
Stromal cell-Derived Factor (SDF)-1/CXCL12 is a non-ELR containing CXC chemokine with chemotactic effects for CD34+ progenitors [113] and primitive hematopoietic cells [114]. SDF-1α induction was recently reported in a rat model of non-reperfused myocardial infarction [115]; however, the role of this chemokine in regulating the post-infarction inflammatory response is unknown. SDF-1 may regulate the recruitment, maturation and function of CXCR4-expressing progenitor cells in ischemic tissues [116], [117]. Transplantation of cells engineered to express SDF-1 into the peri-infarct zone following coronary occlusion in the rat resulted in attenuated adverse remodeling [116]. The beneficial effects of SDF-1 may be due to therapeutic stem cell homing into the injured myocardium resulting in neovascularization and enhanced preservation of cardiomyocytes [118]. Local delivery of SDF-1 in the infarct may exert beneficial actions by augmenting vasculogenesis and by protecting ischemic cardiomyocytes from apoptosis without inducing cardiomyocyte regeneration. These findings suggest that enhanced recruitment of CXCR4+ blood-derived progenitors through activation of chemokine-mediated pathways may be a promising therapeutic approach in patients with myocardial infarction.
CC chemokines in myocardial infarction
CC chemokines are functionally diverse and their names more often reflect historical accidents of their cloning or isolation than their predominant functions [119]. One of the best-studied CC chemokines, Monocyte Chemoattractant Protein (MCP)-1/CCL2, has been implicated in a variety of diseases characterized by monocyte-rich infiltrates [120, 121]. In addition to its critical role in mononuclear cell recruitment, MCP-1 exerts important actions on non-hematopoietic cells, inducing angiogenic and arteriogenic effects [122] and modulating fibroblast phenotype and activity by increasing collagen expression and by regulating MMP synthesis [123]. MCP-1 upregulation has been demonstrated in a canine [124], a rat [125, 126] and a mouse model [127] of experimental myocardial infarction. Studies from our laboratory demonstrated that MCP-1 −/− mice had decreased and delayed macrophage infiltration in the healing infarct and exhibited delayed replacement of injured cardiomyocytes with granulation tissue. MCP-1 −/− infarcts had decreased mRNA expression of the cytokines TNF-α, IL-1β, Transforming Growth Factor (TGF)-β, and IL-10 and showed defective macrophage differentiation [128]. MCP-1 deficiency diminished myofibroblast accumulation but did not significantly affect infarct angiogenesis. Despite showing delayed phagocytotic removal of dead cardiomyocytes, MCP-1 −/− mice had attenuated left ventricular remodeling, but similar infarct size when compared with wildtype animals. MCP-1 antibody inhibition resulted in defects comparable with the pathological findings noted in infarcted MCP-1 −/− animals without an effect on macrophage recruitment [128].
Our findings indicated that MCP-1 has important effects on macrophage recruitment and activation, cytokine synthesis and myofibroblast accumulation in healing infarcts. The role of MCP-1 extends beyond its monocyte chemoattractant effects: MCP-1 inhibition with a neutralizing antibody results in defects comparable with the pathological findings noted in infarcted MCP-1 −/− animals in the absence of an impairment in monocyte recruitment. Absence of MCP-1 results in attenuated post-infarction left ventricular remodeling at the expense of a prolonged inflammatory phase and delayed replacement of injured cardiomyocytes with granulation tissue [129].
Thus, MCP-1 may mediate its effects on the healing infarct through several distinct pathways:
Recruitment of monocytes in the ischemic myocardium appears to be critically regulated by MCP-1. However, loss of MCP-1 may result in markedly decreased recruitment of specific subsets of monocytes. Peripheral blood monocytes are a heterogeneous population. In mice, two distinct subpopulations have been identified that circulate in approximately equal numbers [130]: a CCR2 positive subset with low expression of the fractalkine receptor CX3CR1 (CX3CR1 lo), preferentially recruited in inflammatory processes, and a CCR2 negative subpopulation comprised of cells with high level CX3CR1 expression (CX3CR1 hi), that home to normal tissues and become resident macrophages. In the absence of MCP-1, the “selection advantage” of the CCR2-expressing population may be lost, resulting in decreased and delayed infiltration of the infarct with both subsets of monocytes in equal numbers. CX3CR1 hi monocytes may exhibit decreased cytokine expression and/or phagocytic activity upon stimulation with pro-inflammatory mediators. Recent experiments demonstrated that infarcted mouse hearts sequentially recruit Ly-6C hi and Ly-6C lo monocytes through CCR2 and CX3CR1 respectively. The Ly-6C hi subset appears to exhibit phagocytic, proteolytic and pro-inflammatory functions, whereas Ly-6C lo cells promote healing and have profibrotic and angiogenic properties [131]. These concepts appear to be highly relevant in human pathobiology. Subsets of human peripheral blood monocytes with distinct chemokine receptor profiles have been identified. CD14+CD16+ monocytes express lower CCR2 but higher CCR5 levels; in contrast CD14++ monocytes exhibit high CCR2 and low CCR5 expression [132]. Polarized CCR2 expression is accompanied by differential chemotactic responsiveness to MCP-1 [132].
In addition to its chemotactic properties, MCP-1 may also directly modulate macrophage differentiation, phagocytic activity and cytokine expression. Previous investigations indicated that MCP-1 induces monocyte IL-1 [133] and IL-6 synthesis [134] and that it may be involved in differentiation of monocytes into foam macrophages [135].
The reduced myofibroblast density in MCP-1 null infarcts may result from decreased proliferative activity of resident fibroblasts or impaired recruitment of fibroblast progenitor cells, capable of differentiating into fibroblasts. The significance of these cells in the infarcted myocardium remains unknown.
The key role of MCP-1 signaling in the pathogenesis of post-infarction remodeling was also suggested by experiments using mice with genetic disruption of CCR2, the primary receptor for MCP-1 [136]. CCR2 null mice had decreased infiltration with macrophages and exhibited attenuated ventricular dilation following myocardial infarction. CCR2 absence was associated with markedly decreased MMP expression and lower gelatinolytic activity in the infarcted ventricle. Attenuated matrix degradation may explain, at least in part, the protection from the development of adverse remodeling noted in CCR2 null animals.
MCP-3, another potent mononuclear cell chemoattractant, is also transiently induced in mouse myocardial infarcts [137]. Transplantation of MCP-3-expressing cardiac fibroblasts into the infarct border zone one month after coronary ligation resulted in enhanced homing of injected mesenchymal stem cells in the infarcted myocardium [137]. However, the exact role of MCP-3-mediated interactions in ischemic tissues remains poorly understood. Macrophage Inflammatory Protein (MIP)-1α and MIP-1β are also mononuclear cell chemoattractants, although less efficient than MCP-1 [138]. A robust induction of MIP-1α and MIP-1β is noted in murine infarcts [11]; however, their importance in myocardial injury and repair has not been investigated. In addition, increased serum levels of RANTES (Regulated upon Activation, Normal T-cell Expressed and Secreted), a CC chemokine that induces chemotaxis of monocytes, eosinophils, and specific subsets of T-cells, were found in patients with acute myocardial infarction [139]. Whether these findings reflect upregulation of RANTES expression in the infarct is not known.
A recently published investigation demonstrated reduced functional impairment and attenuated structural remodeling after myocardial infarction in mice with genetic absence of CCR1, a promiscuous CC chemokine receptor primarily activated by MIP-1α and RANTES [140]. Protection of the infracted heart in CCR1 null mice was associated with abrogated early recruitment of neutrophils and improved infarct healing. Translation of these findings into human infarction is hampered by species differences in CCR1 biology. The CCR1 ligand MIP-1α is a potent chemoattractant for mouse, but not for human neutrophils [141].
5. PRO-INFLAMMATORY CYTOKINES IN MYOCARDIAL INFARCTION
Numerous studies have demonstrated activation of cytokine cascades in the infarcted myocardium [142], [143], [144]. Induction and release of the pro-inflammatory cytokines TNF-α, IL-1β and IL-6 is consistently found in experimental models of myocardial infarction [144], [145], [11]. Complement activation, ROS generation and NF-κB activation potently stimulate cytokine mRNA synthesis in both resident and blood-derived cells, resulting in marked cytokine upregulation in the infarcted area. One of the characteristic features of cytokines is their functional pleiotropy and redundancy: one cytokine exhibits a wide range of biological effects on various cell types and several cytokines exert similar and overlapping actions on the same cell type [146]. The multifunctional, overlapping and often contradictory effects of the cytokines have hampered understanding of their functional role in cardiac injury and repair.
TNF-α
TNF-α is capable of exerting diverse effects on all cell types involved in cardiac injury and repair. TNF-α suppresses cardiac contractility [147], [148] and enhances cardiomyocyte apoptosis [149]. In addition, TNF-α stimulates expression of proinflammatory cytokines, chemokines and adhesion molecules by leukocytes and endothelial cells and regulates extracellular matrix metabolism by reducing collagen synthesis and by enhancing MMP activity in cardiac fibroblasts [150]. Despite extensive research, the relative significance of these diverse effects in the infarcted myocardium remains poorly understood. Sun and co-workers demonstrated that TNF-α null mice were protected from cardiac rupture and chronic dysfunction following infarction, and exhibited suppressed inflammation and attenuated MMP activity in comparison with wildtype animals [151]. Several other investigations have suggested an essential role for TNF-α in mediating inflammatory injury following infarction [152], [145]. Inhibition studies have produced contradictory findings regarding the effects of TNF-α neutralization on the infarcted heart. Several studies demonstrated injurious effects of TNF-α signaling in mediating infarct expansion [153], and cardiac dysfunction [154]. On the other hand, other investigations indicated protective effects of TNF-α signaling in the infarcted myocardium. TNF-α inhibition using gene therapy with soluble TNF receptor had deleterious effects in a mouse infarction model promoting cardiac rupture and enhancing adverse remodeling [155]. In addition, TNFR1/TNFR2 double receptor knockout mice undergoing left coronary artery ligation had significantly higher infarct size and increased myocyte apoptosis when compared with wildtype controls [156]. These findings suggested that TNF-α may induce cytoprotective signals capable of preventing or delaying the development of myocyte apoptosis following myocardial infarction. It has been suggested that TNF-α may exert distinct biological effects through the TNFR1 and TNFR2 receptor. A recent investigation suggested that effects mediated through TNFR1 are deleterious, inducing cardiac dysfunction, whereas TNFR2-mediated actions may be protective by attenuating adverse remodeling [157]. The contradictory findings of the investigations exploring the role of TNF-α in myocardial infarction highlight the complex and pleiotropic actions of the cytokines in biological processes and may explain the unpredictable effects of cytokine-targeted therapeutic strategies in clinical trials.
The IL-1 family
The IL-1 gene family consists of three members: IL-1α, IL-1β and IL-1 receptor antagonist (IL-1Ra). IL-1α and IL-1β are agonists, whereas IL-1Ra is a specific receptor antagonist [158]. Both Il-1α and IL-1β are capable of inducing the expression of other cytokines, chemokines, growth factors, and adhesion molecules. Marked IL-1 upregulation has been reported in experimental models of myocardial infarction [142], [11]. In addition, a significant increase in IL-1β plasma levels has been documented in patients with acute myocardial infarction [159]. However, despite extensive descriptive evidence suggesting involvement of IL-1 in infarct healing, information regarding its biological role in myocardial infarction is limited. Cardiac transfection with human IL-1Ra significantly decreased infarct size, reduced apoptosis, and attenuated the inflammatory response in rat hearts undergoing ischemia/reperfusion protocols [160], suggesting an injurious role for IL-1 in the ischemic myocardium. In contrast, another investigation suggested a protective role for IL-1 demonstrating that IL-1β neutralization in the acute phase of myocardial infarction resulted in increased occurrence of cardiac rupture and enhanced adverse remodeling [161]. Because IL-1 signals exclusively through the IL-1 receptor type I (IL-1RI) we have recently examined the effects of disrupted IL-1 signaling on infarct healing and cardiac remodeling using IL-1RI −/− mice [162]. Following reperfused infarction IL-1RI null mice exhibited decreased infiltration of the infarcted myocardium with neutrophils and macrophages and reduced chemokine and cytokine expression. In the absence of IL-1 signaling, suppressed inflammation was followed by an attenuated fibrotic response. Infarcted IL-1RI −/− mice had attenuated fibrosis of the infarcted and remodeling myocardium. IL-1RI deficiency protected from the development of adverse remodeling; however, infarct size was comparable between groups suggesting that the beneficial effects of IL-1RI gene disruption were not due to decreased cardiomyocyte injury. Reduced chamber dilation in IL-1RI null animals was associated with decreased collagen deposition and attenuated MMP-2 and MMP-3 expression in the peri-infarct area, suggesting decreased fibrotic remodeling of the non-infarcted heart. IL-1β stimulated MMP mRNA synthesis in wildtype, but not in IL-1RI null cardiac fibroblasts. The findings suggested that IL-1 signaling is essential for activation of inflammatory and fibrogenic pathways in the healing infarct and may play an essential role in the pathogenesis of post-infarction remodeling [162].
IL-18 shares with IL-1β three-dimensional structural similarities, the same family of receptors and several identical signal transduction pathways [163]. It has potent pro-inflammatory properties inducing cytokine and chemokine synthesis and activating NF-κB. IL-18 upregulation was noted in the non-infarcted remodeling myocardium, while IL-18 infusion decreased left ventricular contractility in vivo [164]. However, the role of endogenous IL-18 in cardiac injury and repair has not been investigated.
The IL-6 family of cytokines
IL-6 is a member of a larger family of structurally related cytokines with overlapping biological effects. The family includes IL-11, Leukemia Inhibitory Factor (LIF), Oncostatin-M, Cardiotrophin-1 (CT-1), Ciliary Neurotrophic Factor (CNTF) and neurotrophin-1/B-cell stimulating factor-3 (NNT-1/BSF-3). All IL-6-related cytokines signal through multisubunit receptors that share the transmembrane glycoprotein (gp)130. Extensive experimental evidence demonstrated induction of members of the IL-6 family in healing infarcts [165]. IL-6 synthesis is rapidly induced in mononuclear cells and cardiomyocytes of the ischemic myocardium [145, 166, 167]. CT-1 is upregulated in fibroblasts and surviving cardiomyocytes and shows a prolonged time course of expression [165], [168], whereas LIF [11] and oncostatin-M [169] are induced during the inflammatory phase of healing. However, the functional role of these cytokines in infarct healing remains unknown. Members of the IL-6 family have profound effects on cardiac myocytes by promoting cardiac hypertrophy, but also by protecting them from apoptosis [170]. CT-1 administration resulted in decreased infarct size and reduced cardiomyocyte apoptosis in a rat model of myocardial ischemia and reperfusion [171]. However, CT-1 absence did not affect infarct size in a mouse model of reperfused infarction suggesting that endogenous CT-1 does not play an essential role in acute ischemic cardiac injury [172]. Prolonged CT-1 upregulation in the infracted myocardium may modulate the fibrotic response through effects on fibroblast proliferation [173]. Although the role of endogenous LIF expression in the infarct remains unknown, gene therapy with LIF cDNA prevented cardiomyocyte death and induced angiogenesis, enhancing recruitment of bone marrow-derived cells into the heart [174]. IL-6 is capable of modulating the phenotypic characteristics and gene expression of many cell types involved in infarct healing. IL-6 null mice demonstrated significantly delayed cutaneous wound healing suggesting a significant role for IL-6 in tissue repair [175]. However, Fuchs and co-workers [176] found that the absence of IL-6 did not affect infarct size, left ventricular function and post-infarction remodeling in non-reperfused infarcts. Although these findings do not preclude biologically significant actions of IL-6 in healing infarcts, it appears that in mice lacking IL-6, other mediators may act in a compensatory manner to activate the JAK/STAT pathway, thereby maintaining STAT3 phosphorylation, which is crucial for the cellular effects of IL-6-related cytokines.
6. THE CELLULAR BASIS OF THE POST-INFARCTION INFLAMMATORY RESPONSE
Activation of the inflammatory response in the infarcted myocardium is associated with induction of endothelial adhesion molecules and enhanced permeability of the microvasculature. Upregulation of chemokines and cytokines results in extravasation of activated blood-derived cells into the infarcted area. Platelets are the first cells recruited in the site of injury [177]. Subsequently various subsets of leukocytes infiltrate the myocardium and clear the wound from dead cells and matrix debris. Blood-derived cells play an important role in regulation of the reparative process following cardiac injury (Figure 3).
Figure 3. The cell biology of healing myocardial infarction.
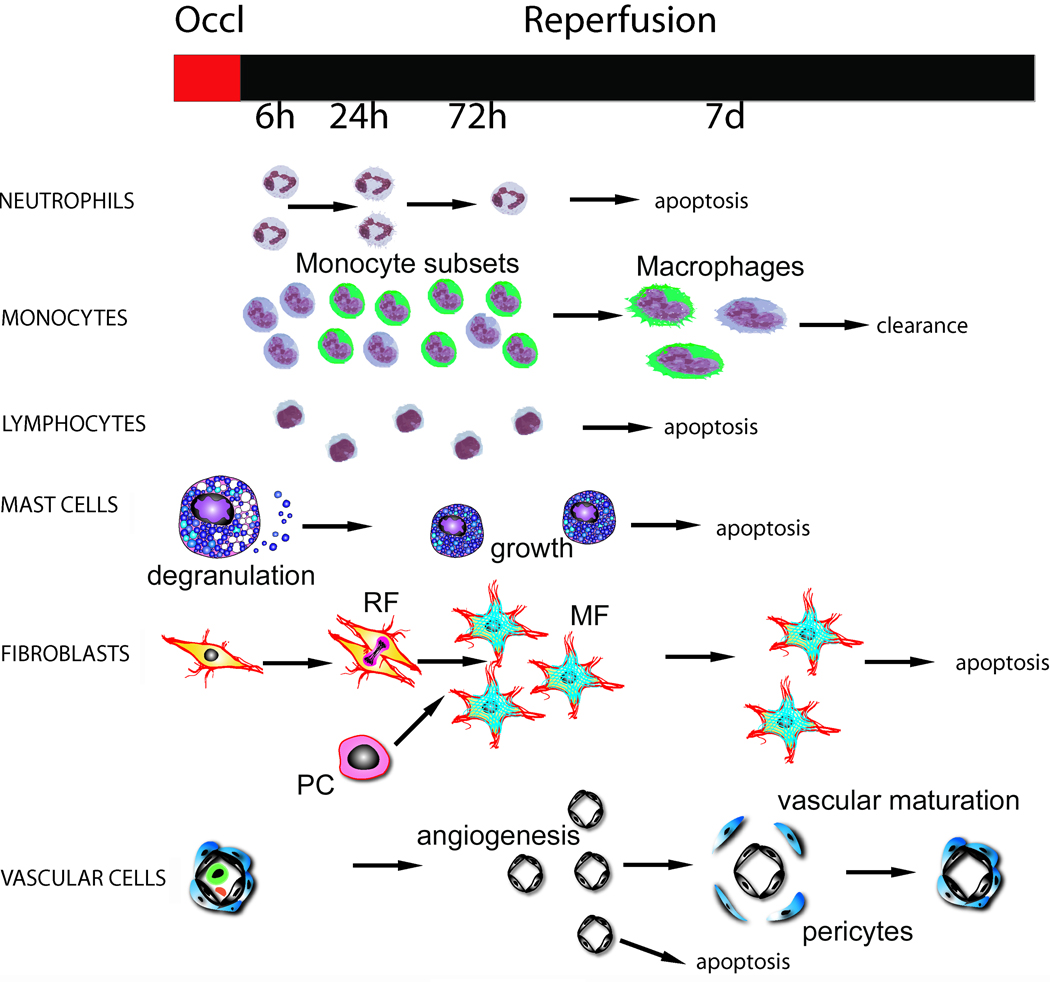
Infarct healing is dependent on the sequential infiltration of the injured myocardium with neutrophils, mononuclear cells, mast cells, fibroblasts and vascular cells (see text for more details). This is a dynamic and superbly orchestrated process: recruitment or proliferation of each cell type is followed by activation. Various cell populations have distinct but overlapping functions. Orchestration of the sequence of cellular events in the healing infarct is dependent on timely apoptosis of specific cell types. The time course presented here is based on reperfused mouse myocardial infarction; large mammals and humans exhibited a delayed inflammatory and reparative response (Symbols: RF, resident fibroblasts; PC, progenitor cells; MF, myofibroblasts).
Platelets
Beyond their eminent role in hemostasis and thrombosis, platelets modulate inflammatory responses and represent an important linkage between tissue injury and repair [178]. In the healing wound activated platelets may play multiple roles by aggregating in areas of injury, by contributing to the formation of a fibrin-based provisional matrix and by releasing a variety of chemokines (such as RANTES and PF4), cytokines (such as IL-1β) and growth factors (such as Platelet-Derived Growth Factor (PDGF) and TGF-β) [179]. In addition, platelets are capable of initiating complement activation and may play a role in localizing the inflammatory response into the area of injury [180]. Despite the ability of platelets to modulate the inflammatory and reparative response, thrombocytopenic mice did not exhibit defects in wound reepithelialization, angiogenesis and collagen deposition following cutaneous injury [181]. Whether activated platelets play an important role in inflammation and repair following cardiac injury has not been investigated.
The neutrophils
Neutrophils are recruited very early after cardiac injury. Neutrophil transmigration in the infarcted myocardium requires adhesive interactions with activated vascular endothelial cells and involves a cascade of molecular steps that correspond to the morphological changes that accompany adhesion (Figure 4). The selectin family of adhesion molecules mediates the initial capture of leukocytes from the rapidly flowing bloodstream to the blood vessel. Selectins promote leukocyte attachment and rolling at shear stresses characteristic of post-capillary venules. The selectin family consists of three closely related cell-surface molecules: L-selectin (CD62L), E-selectin (CD62E), and P-selectin (GMP-140, CD62P). All three selectins are involved in leukocyte entry into tissues.
Figure 4. Infiltration of the infarcted myocardium with leukocytes is dependent on a sequence of adhesive interactions.

During the inflammatory phase of infarct healing, chemoattractants for neutrophils, monocytes and lymphocytes are upregulated in the injured myocardium. Local release of inflammatory mediators results in endothelial cell activation and surface expression of adhesion molecules. The selectin family of adhesion molecules mediates the initial capture of leukocytes from the rapidly flowing bloodstream to the blood vessel. Selectins promote leukocyte attachment and rolling (1) at shear stresses characteristic of post-capillary venules. Although rolling is a prerequisite for eventual firm adherence to blood vessels under conditions of flow, selectin-dependent adhesion of leukocytes does not lead to firm adhesion (2) and transmigration (3) unless another set of adhesion molecules, the integrins, is engaged. The mechanisms responsible for leukocyte transmigration into the tissues are poorly understood. In vitro studies have demonstrated that infiltrating neutrophils are capable of inducing cytotoxic injury to cardiomyocytes (4); this process is dependent on adhesive interactions between activated neutrophils and cytokine-stimulated cardiomyocytes. Whether neutrophil-mediated injury significantly contributes to cardiomyocyte death following infarction remains controversial (Symbols: Ly, lymphocyte; Mo, monocyte; Ma, macrophage; N, neutrophil; EC, endothelial cell; CM, cardiomyocyte).
The role of the selectins in cardiac injury is not well defined at present. L-selectin is constitutively expressed in neutrophils in a highly specific distribution and its shedding upon activation may be important in regulating leukocyte rolling velocity and recruitment [182]. P-selectin surface expression occurs rapidly on endothelial cells under circumstances likely to be seen during cardiac injury. It is stored in the Weibel- Palade bodies and is rapidly translocated to the endothelial surface in response to thrombin and/or oxidative stress, both of which would be likely to be found in the ischemic myocardium, and to histamine, which is rapidly released in the injured myocardium by degranulating mast cells. Experimental studies have suggested that monoclonal antibodies against L-selectin and P-selectin [183, 184] reduced myocardial necrosis, preserving coronary endothelial function and attenuating neutrophil accumulation in the ischemic and reperfused feline myocardium. In addition, P-selectin deficient mice showed decreased infarct size after 30 min of coronary occlusion and 2 h of reperfusion [185]. In contrast, no difference in infarct size was noted after a 60 min ischemic period [185]. In addition, mice with a combined P-selectin and ICAM-1 deficiency demonstrated impaired neutrophil trafficking without a difference in infarct size due to myocardial ischemia and reperfusion [186]. Although current concepts suggest a role for the selectins in supporting leukocyte margination under shear stress, the effects of selectin-related interventions in experimental models of myocardial ischemia have been inconsistent [187, 188]. Peptide analogs of the soluble selectin ligand P-selectin glycoprotein ligand-1 (PSGL-1) have been constructed and may represent a promising approach in targeting selectin-dependent interactions. A recombinant analog of sPSGL-1 significantly reduced cardiac injury in a feline model of coronary occlusion and reperfusion [189].
Although rolling appears to be a prerequisite for eventual firm adherence to blood vessels under conditions of flow, selectin-dependent adhesion of leukocytes does not lead to firm adhesion and transmigration unless another set of adhesion molecules, the integrins, is engaged. Integrins are a family of heterodimeric membrane glycoproteins that consist of an α and a β subunit; these subunits are associated through noncovalent bonds and transported to the cell surface as a complex [190]. For neutrophils, firm adhesion requires activation of the β2 (CD18) integrins, which share the beta chain CD18 paired with CD11a (LFA-1), CD11b (Mac-1), or CD11c (p150,95). This results in binding to one of the intercellular adhesion molecules (ICAM) on the surface of endothelial cells. LFA-1, Mac-1 and p150,95 have different and yet overlapping roles in adhesion, in part due to their characteristics of expression on leukocytes. Stationary neutrophils adherent to the luminal endothelial surface frequently change shape and assume the characteristic bipolar configuration of motile cells [191, 192]. This event may result from interaction with surface-bound chemokines. Transendothelial migration follows and leads to neutrophil infiltration in the inflamed tissues. Antibodies that inhibit LFA-1 adhesion are effective in blocking transmigration [193] and LFA-1 deficient mice show dramatically decreased neutrophil extravasation at sites of inflammation [194]. In contrast, antibodies that block Mac-1 adhesion are marginally effective [193], and Mac-1 deficient mice demonstrate no deficit in neutrophil emigration [195]. These findings suggest that LFA-1 and not Mac-1 is critical for neutrophil extravasation in sites of inflammation.
Integrin-related strategies have been utilized to mitigate post-reperfusion inflammation in various experimental models. Inhibition of CD11/CD18 integrin resulted in significant reduction of infarct size in rat [196], feline [197], canine [198], and primate [199] models of experimental myocardial infarction. However, despite the promising results of the experimental studies, leukocyte integrin inhibition in clinical studies has led to disappointment [200]. A multicenter clinical trial [201] demonstrated that patients undergoing primary angioplasty for acute myocardial infarction had no difference in events and no reduction in infarct size upon administration of a humanized anti-CD11/CD18 antibody (Hu23F2G). It appears that the effectiveness of different anti-CD18 antibodies in preventing injury is highly dependent on the specific antibody employed [202]. Experiments using genetically targeted animals have contributed to our understanding of the role of integrins in experimental myocardial infarction. CD18 deficient mice demonstrated significant reduction in neutrophil accumulation following myocardial ischemia and reperfusion [203]. Furthermore, treatment with an antibody to Vascular Cell Adhesion Molecule (VCAM)-1 significantly attenuated neutrophil emigration in the infarcted myocardium of CD18-null mice, but did not diminish myocardial injury [204].
Integrin-mediated neutrophil adhesion is followed by infiltration of the leukocytes into the underlying tissue. However, very little is known regarding the mechanisms responsible for neutrophil diapedesis through the vessel wall. The major route for a leukocyte to pass the endothelial barrier is through the cleft between adjacent cells. Endothelial cells possess a number of junctions that maintain vascular integrity and regulate permeability. Endothelial cells are not just passive participants in diapedesis and besides expressing key adhesion molecules they actively downregulate their inter-endothelial junctions and contract to allow leukocyte transmigration. Junctional adhesion molecules (JAMs) of tight junctions are regulated by phosphorylation [177] and play an essential role in Transendothelial migration [205]. Corada and co-workers suggested an important role for the Junctional Adhesion Molecule (JAM)-A in regulating neutrophil infiltration into the ischemic myocardium [206]. JAM-A −/− mice exhibited impaired neutrophil diapedesis following myocardial ischemia/reperfusion. Microscopic examination of the heart microvasculature showed large numbers of neutrophils adherent on the endothelium, or entrapped between endothelial cells and the basement membrane. These defects were associated with enhanced cardiomyocyte injury in JAM-A null infarcts, perhaps due to impaired blood flow caused by prolonged entrapment of neutrophils in the microcirculation [206].
The concept of neutrophil-mediated cardiomyocyte injury
Infiltrating neutrophils generate free radicals and release enzymes contributing to the clearance of the infarct from dead cells and debris. In addition, they may express mediators capable of amplifying cell recruitment. Over the last twenty years, experimental evidence suggested that neutrophils may directly injure parenchymal cells through release of specific toxic products [207]. Obviously, neutrophils accumulating in the injured areas might release proteolytic enzymes, or reactive oxygen species to injure surrounding myocytes. Under conditions found in vivo these toxic products are almost exclusively secreted by adherent neutrophils. Thus, it appears that a ligand-specific adhesion of the neutrophils to the cardiac myocytes may be critical for the mediation of ischemia-induced myocyte injury. In vitro experiments suggested that the mechanism of neutrophil-cardiomyocyte adhesion is dependent on CD18 integrin activation on neutrophils and on expression of ICAM-1, one of the primary ligands for the CD18 integrins [208], by injured cardiomyocytes. However, the in vivo significance of neutrophil-induced cardiomyocyte injury remains unknown [209]. Neutrophil depletion in animals undergoing reperfused myocardial infarction led to a marked decrease in infarct size [210, 211] suggesting that a significant amount of myocardial injury induced by coronary artery occlusion followed by reperfusion may be neutrophil-dependent [212]. Although ICAM-1 deficient mice exhibited less myocardial injury at an early stage, they showed no significant difference in infarct size and scar formation after 1–3 weeks of reperfusion [213]. In addition, mice with a combined deficiency in both ICAM-1 and P-selectin showed no difference in infarct size due to myocardial ischemia and reperfusion despite exhibiting impaired neutrophil trafficking [186].
It has been suggested that, in addition to clearing the wound from dead cells and debris, the neutrophils may play a direct role in wound healing by secreting cytokines and growth factors. Although the contribution of the neutrophil to fibrous tissue deposition in the healing infarct cannot be excluded; it is unlikely that this cell type plays a critical role in the events leading to formation of a scar. In a study performed more than thirty years ago, Simpson and Ross demonstrated that depletion of circulating neutrophils in guinea pigs treated with anti-neutrophil serum had no effects on granulation tissue formation in a model of cutaneous healing, suggesting that the neutrophil response may not be essential in the repair process [214]. Neutrophils may contribute to the healing response through their apoptosis and subsequent clearance by macrophages; this process, as will be discussed later, releases TGF-β, resulting in resolution of inflammation and transition to fibrosis.
The mononuclear cells
Activation of inflammatory pathways rapidly triggers infiltration of the infarcted myocardium with mononuclear cells. As discussed previously, CCL2/MCP-1 plays an important role in monocyte recruitment in the infarcted myocardium. Other mediators including complement, TGF-β, free radicals and other CC chemokines may also play a role in regulating monocyte infiltration. Various monocyte chemoattractants are likely to play distinct roles through the recruitment of monocyte and lymphocyte subpopulations with distinct properties.
Recruitment of monocytes in the infarcted myocardium is followed by maturation and differentiation of these blood-derived cells into mature macrophages. This is a complex and poorly understood process that is likely to involve growth factors such as Macrophage-Colony Stimulating Factor (M-CSF) and Granulocyte Macrophage-Colony Stimulating Factor (GM-CSF). M-CSF is induced in healing canine infarcts and its expression is associated with macrophage accumulation and proliferation [215]. The effects of M-CSF on macrophage phenotype and activity are not fully understood; however, it appears to be crucial for survival of macrophages permitting them to respond to internal and external cues for their differentiation [216]. Differentiated macrophages play multiple roles in the healing infarct. First, they are responsible for phagocytosis of dead cells and debris and for clearance of apoptotic neutrophils and cardiomyocytes. Second, they serve as a source of cytokines and growth factors regulating fibroblast growth and angiogenesis. Third, they contribute to extracellular matrix remodeling by producing MMPs and their inhibitors. The gene expression profile of infiltrating macrophages may change during the healing process; these dynamic alterations may have a profound effect on cardiac repair.
The mast cells
Mast cells are multifunctional resident cells, capable of secreting a wide range of inflammatory and pro-fibrotic mediators. The mammalian heart contains a significant number of resident mast cells predominantly located in close proximity to vessels [217]. A possible role for mast cells in mediating cardiac injury was suggested in a porcine model of C5a-induced myocardial ischemia [218]. Our studies [145] indicated a role for mast cell-derived mediators in initiating the cytokine cascade in the reperfused canine myocardium. We found that resident cardiac mast cells rapidly degranulate following infarction releasing large amounts of histamine and TNF-α. Histamine may induce surface expression of P-selectin in endothelial cells, whereas mast cell-derived TNF-α played an important role in stimulating cytokine expression by infiltrating mononuclear cells. There is significant evidence that mast cells may also participate in the fibrotic process. We have demonstrated a striking accumulation of mast cells during the proliferative phase of healing, predominantly in areas of collagen deposition and cell proliferation [219]. The factors responsible for mast cell accumulation in areas of fibrosis remain to be defined. Stem Cell Factor (SCF) is a potent mast cell chemoattractant that stimulates directional motility of both mucosal-and connective tissue-type mast cells. Our studies demonstrated significant upregulation of SCF mRNA expression in the infarcted canine myocardium after 72h of reperfusion [219]. At the same time point, an increase in mast cell numbers is noted in the healing myocardium and immature mast cell progenitors are found in the infarcted area. Although the contribution of mast cell proliferation cannot be ruled out, chemotaxis of circulating mast cell precursors in the healing myocardium may be the predominant mechanism responsible for mast cell accumulation in the ischemic heart. The role of SCF in infarct healing may not be limited to its effects on mast cells. Experimental evidence suggested that SCF may promote recruitment and homing of primitive bone marrow-derived cells delivered into the infarct [220]. These progenitor cells may be capable of differentiating into cardiomyocytes and vascular cells [221]; thus, SCF may be a promising mediator for cardiac regeneration.
Mast cells can produce and release a wide variety of preformed and newly-synthesized mediators with pleiotropic actions. Mast cell-derived histamine may critically affect leukocyte-endothelial interactions by increasing endothelial P-selectin expression and by facilitating recruitment of rolling leukocytes [222]. Tryptase, the most abundant protease found in mast cell granules stimulates granulocyte recruitment [223], and upregulates cytokine and chemokine synthesis [224], [225]. Mast cell-derived cytokines, such as TNF-α may trigger the cytokine cascade in the infarct, regulating adhesion molecule expression and leukocyte recruitment. In addition, many mast cell-derived mediators may influence fibroblast growth and function. Histamine has been shown to stimulate fibroblast growth and collagen synthesis in vitro [226]. Tryptase induces fibroblast proliferation [227] and chemotaxis and upregulates type I collagen production [228]. Furthermore, mast cells are important sources of TGF-β [229], bFGF [230], and Vascular Endothelial Growth Factor (VEGF) [231], factors that can regulate fibroblast growth, modulate extracellular matrix metabolism and stimulate angiogenesis [232]. Mast cells may also influence healing and tissue remodeling by expressing gelatinases A and B [233–235] which are implicated in extracellular matrix metabolism. Finally, an important role for the chymase pathway in promoting angiotensin II generation and cardiac fibrosis has been suggested [236].
Although many mast cell-derived mediators are capable of modulating cellular events critical to the healing infarct, the role of mast cells and their secretory products in cardiac injury and repair remain poorly understood. Experiments in a canine model of reperfused infarction demonstrated that mast cell stabilization using lodoxamide significantly reduced infarct size [237]. However, the specific mediators responsible for the injurious effects of mast cell activation in the infarct were not investigated. In addition, elucidation of the role of mast cell-derived cytokines and growth factors in myocardial infarction is difficult because many of these secretory products are produced by other cell types involved in cardiac repair. On the other hand, the proteases chymase and tryptase are specific mast cell products may play unique roles in infarct healing. Chymase inhibition in a rat model of non-reperfused myocardial infarction attenuated left ventricular interstitial fibrosis and diastolic dysfunction without affecting the dilative pattern of cardiac remodeling [238]
The myofibroblasts
Inflammatory leukocytes secrete a variety of cytokines and growth factors with angiogenic and fibrogenic properties. This environment promotes the proliferation and activation of fibroblasts, the main source of extracellular matrix protein synthesis in the infarct, and stimulates the formation of neovessels, which are necessary to carry oxygen and nutrients in order to sustain cell metabolism. Willems and colleagues [239] have identified and characterized the interstitial nonvascular α-smooth muscle actin (α-SMA) positive cells, which were present in human myocardial scars 4–6 days after an infarction. These phenotypically modulated fibroblasts, termed myofibroblasts [240–242] develop ultrastructural and phenotypic characteristics of smooth muscle cells and possess a contractile apparatus that contains bundles of actin myofilaments with associated contractile proteins, such as non-muscle myosin [243]. Myofibroblasts are the predominant source of collagen mRNA in healing myocardial infarcts [244–246], [247]. They transiently appear during granulation tissue formation and become apoptotic when the scar matures [248].
The origin of the fibroblasts infiltrating the healing wound is one of the oldest and most controversial concepts in cell biology and has been debated for over 100 years [249]. [250]. Early studies have documented differentiation of leukocytes into fibroblasts and have cultured connective tissue-producing cells from the buffy coat [251], [252]. Although it is widely recognized that fibroblasts proliferate in the healing wound; the extent to which connective tissue is the result of an ingrowth of adjacent mesenchymal or fibroblast-like cells versus the hematogenous entry of circulating fibroblast precursors is debated. Early studies of connective tissue repair in implanted polyvinyl sponges and granulating wounds supported the local origin of fibroblasts [250]. More recently, Bucala and co-workers described and characterized a circulating population of leukocytes that traffic to fibrotic tissues and sites of injury and differentiate to fibroblasts [253]. The recruitment of blood borne fibroblast precursors is mediated through chemokine-dependent mechanisms and appears to be important in the development of pulmonary fibrosis [254]. The origin of fibroblasts in healing infarcts remains unclear. Although numerous studies have documented active proliferation of resident fibroblasts [255], [219], [256] the possibility that at least a subset of infarct fibroblasts may be derived from blood-borne precursors should be considered.
Fibroblasts infiltrating the infarct undergo myofibroblast differentiation, developing a microfilamentous apparatus that expresses α-SMA. They are predominantly localized in the border zone area, and exhibit intense proliferative activity [257], [255]. The mechanisms responsible for myofibroblast differentiation in the infarcted heart remain poorly understood; however, the modulation of a fibroblast toward myofibroblast likely results from the combined action of several distinct factors:
TGF-β is critically involved in myofibroblast differentiation in healing wounds by regulating α-SMA expression [258], and is likely to play an important role in modulating fibroblast phenotype in infarcts.
Alterations in the composition of the extracellular matrix may dictate phenotypic changes in infarct fibroblasts. The splice variant ED-A of cellular fibronectin is crucial for myofibroblastic phenotype induction by TGF-β [259].
Mechanical tension induces myofibroblast differentiation but also modulates fibroblast behavior by activating various signal transduction pathways [260].
In reperfused infarcts, the return to normoxic pO2 after a hypoxic period is perceived as “relative hyperoxia”. Perceived hyperoxia induces fibroblast alterations that resemble TGF-β1-induced morphological changes, including myofibroblast differentiation [261], [262].
Fibroblast gene expression is modulated by microenvironmental factors. In normal hearts resident fibroblasts are responsible for homeostatic maintenance of the extracellular matrix network. Although their contribution in the inflammatory phase of healing is unclear, fibroblasts are capable of producing large amounts of chemokines and cytokines upon stimulation with inflammatory mediators [263]. The cytokine milieu during the inflammatory phase of healing promotes a matrix-degrading fibroblast phenotype. Stimulation of cardiac fibroblasts with IL-1β and TNF-α, mediators induced and released in the early stages of infarct healing, results in decreased collagen expression and enhanced MMP activity [150]. In contrast, during the proliferative phase of healing, signals that promote extracellular matrix synthesis are activated. FGF-2 is released in the infarcted myocardium and may induce fibroblast proliferation [264]. TGF-β markedly increases extracellular matrix protein synthesis and enhances Tissue Inhibitor of Metalloproteinases (TIMP) expression promoting matrix preservation. Angiotensin II may play an important role in fibroblast proliferation and matrix synthesis, mediating effects transduced by AT1 receptors [265]. The effects of angiotensin II may be mediated, at least in part, through induction of fibrogenic growth factors, such as TGF-β.
During the maturation phase, infarct myofibroblasts undergo apoptosis and the highly cellular granulation tissue is replaced by a collagen-based scar. The mechanisms responsible for apoptotic death and clearance of the fibroblasts have not been investigated.
Vascular cells and pericytes
Angiogenesis is an integral part of wound healing. Neovessels are important components of granulation tissue and are required to provide oxygen and nutrients to the highly dynamic and metabolically active cells of the healing wound. Angiogenic growth factors, such as bFGF and VEGF, are induced and released during the first few hours following myocardial ischemia [266], [267] resulting in neovessel growth [96]. During the proliferative phase of healing, a rich network of capillaries is formed along with enlarged pericyte-poor “mother vessels” [268]. As the vasculature matures, some infarct neovessels are coated with pericytes, whereas uncoated vessels regress [269]. Although the time course of neovessel formation is well-described, the mechanisms responsible for infarct angiogenesis remain poorly understood. Hypoxia-Inducible Factor (HIF)-1α is exquisitely sensitive to hypoxic conditions making it one of the earliest effectors of the response to ischemia. HIF-1α activation results in VEGF induction and release, playing an important role in mediating endothelial sprouting in the healing infarct [270]. VEGF induces endothelial cell migration and proliferation resulting in the formation of hyperpermeable neovessels. VEGF acts in concert with the angiopoietins. Angiopoietin (Ang)-1 and Ang-2 are ligands for the Tie2 receptor tyrosine kinase, which is present on endothelial cells and endothelial progenitor cells. Ang-1 is widely expressed in the quiescent vasculature. Ang1-Tie2 signaling serves to inhibit endothelial cell activation and thus may impede the efficient initiation of the angiogenic response in the face of hypoxia or tissue injury. Ang-2 is an endogenous antagonist of Tie2 and inhibits Ang1–Tie2 signaling, facilitating endothelial activation in response to inducers of angiogenesis, such as VEGF [271], [272]. In healing infarcts Ang-2 is markedly induced in the early stages of healing, whereas Ang-1 expression is decreased [273]. Thus, Ang-2 may act in concert with VEGF as a permissive factor in the earliest stages of infarct angiogenesis by releasing endothelial cells from the inhibitory actions of Ang-1. Several other mediators, such as FGFs, TGF-β and the chemokines MCP-1, IL-8 and IP-10, are capable of modulating angiogenesis, and may participate in the complex process of neovessel formation following myocardial infarction. The composition of the extracellular matrix is also critical for neovascular growth and should be viewed as a dynamic player in the process.
During the maturation phase of infarct healing, granulation tissue is replaced by a collagen-rich scar, which exhibits a relatively low capillary density, but contains a large number of vessels coated with mural cells (Figure 5). Mature coated vessels show decreased angiogenic potential and are protected from regression, whereas uncoated endothelial cells undergo apoptosis. Acquisition of a muscular coat is a dynamic process that appears to involve PDGF-BB-PDGF-Rβ interactions [274]. Our recent experiments [275] identified perivascular cells expressing phosphorylated-PDGF-Rβ in healing mouse infarcts indicating activation of the PDGF-BB/PDGF-Rβ pathway. PDGF-Rβ blockade resulted in impaired maturation of the infarct vasculature, enhanced capillary density, and formation of a significant number of dilated uncoated vessels. Defective vascular maturation in antibody-treated mice was associated with increased and prolonged extravasation of red blood cells and monocyte/macrophages, suggesting increased permeability. These defects were associated with decreased collagen content in the healing infarct. In contrast, PDGFR-α inhibition did not affect vascular maturation, but significantly decreased collagen deposition in the infarct. These findings suggested that PDGF signaling plays a crucial role in regulating post-infarction repair. Both PDGFR-β and PDGFR-α-mediated pathways promote extracellular matrix deposition in the infarct. However, activation of the PDGF-B/PDGFR-β pathway is also involved in recruitment of mural cells by neovessels, regulating maturation of the infarct vasculature. Acquisition of a mural coat and maturation of the vasculature is an important step for suppression of granulation tissue formation following myocardial infarction, and promotes resolution of inflammation and stabilization of the scar [275].
Figure 5. Vascular maturation through acquisition of a pericyte coat regulates infarct angiogenesis.
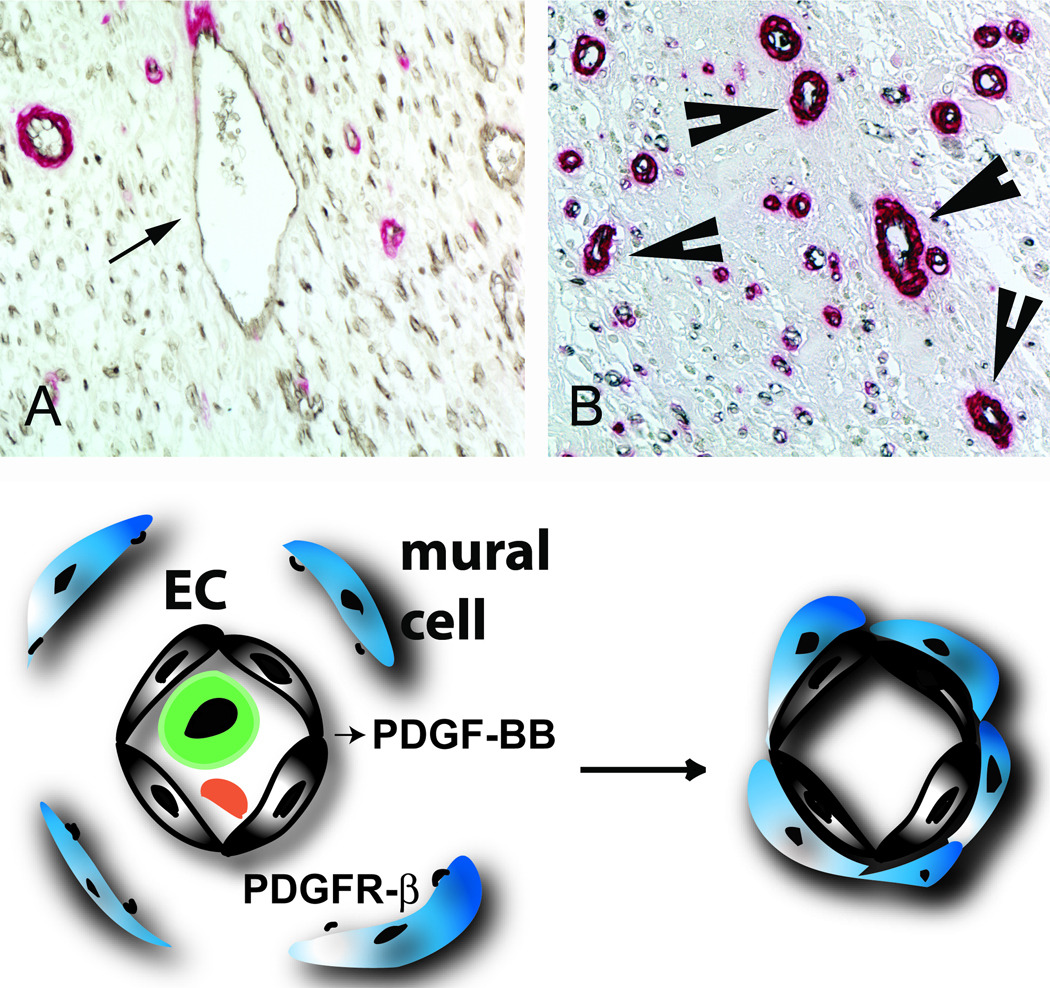
Panels A and B show dual immunohistochemical staining of reperfused canine infarcts for the endothelial cell marker CD31 (black) and α-smooth muscle actin (red). During the proliferative phase of healing (A) the infarct contains a large number of capillaries and several dilated pericyte-poor vessels (arrow - “mother vessels”). As the scar matures (B), some vessels acquire a mural cell coat (B- arrowheads), while uncoated vessels regress. PDGF-BB/PDGFR-β interactions are critically involved in vascular maturation following myocardial infarction.
7. RESOLUTION OF THE POST-INFARCTION INFLAMMATORY RESPONSE
Induction of pro-inflammatory mediators and leukocyte infiltration play a crucial role in phagocytotic removal of dead cells and matrix debris from the infarcted myocardium. However, this acute localized inflammatory response is transient, and is followed by resolution of the inflammatory infiltrate and fibrous tissue deposition [11]. A crucial commitment is made during the late stages of the inflammatory phase to convert the response from phagocytosis and clearance of dead cells and debris, to a mode that promotes tissue repair and scar formation [276]. Inhibition of chemokine and cytokine synthesis after a dramatic early peak is crucial for the repair process, preventing prolonged expression of inflammatory mediators in the healing infarct, and suppressing continuous leukocyte recruitment and injury. Thus, optimal healing requires mechanisms inhibiting chemokine and cytokine synthesis resulting in resolution of the inflammatory infiltrate and transition to fibrous tissue deposition. These mechanisms involve: a) clearance of the neutrophilic infiltrate and removal of matrix debris, b) inhibition of cytokine and chemokine synthesis, c) removal of the fibrin-based provisional matrix, and d) activation of fibroblasts and collagen deposition. Although very few studies have dealt with the process of resolution of inflammation in the healing infarct, understanding these concepts is crucial for planning strategies targeting the reparative response.
Clearance of apoptotic neutrophils and removal of matrix debris
Clearance of the granulocytic infiltrate by professional phagocytes is a prerequisite for resolution of the inflammatory process. Apoptosis is the predominant mechanism that determines the functional longevity of neutrophils in inflamed and infarcted tissues [277]. Ingestion of apoptotic cells by macrophages results in powerful anti-inflammatory and immunosuppressive effects [278]. TGF-β released by macrophages that are ingesting apoptotic cells exerts potent anti-inflammatory effects in the inflamed peritoneum and the endotoxin-stimulated lung [279]. In contrast, cellular necrosis is a pro-inflammatory stimulus in most tissues, perhaps because dying cells disintegrate releasing contents before the debris is ingested by phagocytes [280]. Recognition and phagocytosis of apoptotic cells by macrophages is a complex process that involves several distinct molecular pathways [278]. The importance of apoptotic cell removal in myocardial infarction has not been investigated. However, timely clearance of the leukocytic infiltrate may play an essential role in suppression of the post-infarction inflammatory response.
Removal of matrix debris may also be essential for resolution of the post-infarction inflammatory response. Increased turnover of extracellular matrix is a hallmark of tissue injury and results in generation of degradation products. Hyaluronan, a ubiquitously present constituent of the extracellular matrix [281], exists as a high molecular weight polymer under physiologic conditions, but undergoes degradation resulting in accumulation of lower molecular weight species after tissue injury. Hyaluronan fragments induce the expression of a variety of inflammatory genes by endothelial cells and macrophages [282], including chemokines and cytokines, and may play an important role in regulating inflammatory processes. Furthermore, clearance of hyaluronan fragments from the injured tissue is crucial for resolution of chronic inflammation [283]. Hyaluronan modulates the inflammatory response through interactions with the transmembrane adhesion molecule CD44 [284], a ubiquitously distributed glycoprotein that mediates a wide variety of cell-cell and cell-matrix interactions. A growing body of evidence suggests that CD44 serves as a key factor in resolution of inflammation through removal of matrix breakdown products and clearance of apoptotic neutrophils [283]. CD44 also exerts fibrogenic actions mediating fibroblast migration and invasion in the wound provisional matrix [285]. Recent experiments from our laboratory tested the hypothesis that CD44 may play an essential role in infarct healing by regulating the inflammatory and fibrotic response [286]. We found that CD44 null mice exhibit enhanced and prolonged inflammation in the infarcted heart followed by reduced myofibroblast infiltration. In CD44 null infarcts the enhanced inflammatory phase was followed by decreased fibroblast infiltration, reduced collagen deposition and diminished proliferative activity. Isolated CD44 null cardiac fibroblasts had reduced proliferation upon stimulation with serum and decreased collagen synthesis in response to TGF-β in comparison to wildtype fibroblasts. The healing defects in CD44 −/− mice were associated with enhanced dilative remodeling of the infarcted ventricle, without affecting the size of the infarct. Thus, the role of CD44 in the injured myocardium extends beyond its significance in resolution of inflammation and involves regulation of fibrogenic responses.
The potential role of IL-10 in resolution of inflammation
IL-10, a cytokine predominantly expressed by activated Th2 lymphocytes and stimulated monocytes, possesses potent anti-inflammatory properties [287, 288]. Among the different cell types affected by IL-10, monocyte-macrophages appear to be particularly modified in regard to their function, morphology and phenotype. IL-10 inhibits the production of IL1α, IL1β, TNF-α, IL-6 and IL-8 by lipopolysaccharide-activated monocytes, suppressing the inflammatory response. Furthermore, IL-10 may play a significant role in extracellular matrix remodeling by promoting TIMP-1 synthesis, leading to stabilization of the matrix [289].
We have previously demonstrated IL-10 mRNA and protein upregulation in reperfused canine infarction. IL-10 expression was first detected at 5 hours and peaked following 96–120 hours of reperfusion. In contrast, IL-12, a Th1 related cytokine associated with macrophage activation, was not detected in the infarct. IL-10 induction was associated with decreased synthesis of the proinflammatory cytokine IL-6. In vitro experiments demonstrated that late postischemic cardiac lymph induced TIMP-1 mRNA expression by isolated canine mononuclear cells. This effect was inhibited when the incubation contained a neutralizing anti-IL-10 antibody [290]. The findings suggested that IL-10 may regulate extracellular matrix metabolism following myocardial infarction.
The role of IL-10 in resolution of post-infarction inflammation remains controversial. Yang and co-workers suggested that IL-10 −/− mice had markedly increased mortality and exhibited an enhanced inflammatory response following reperfused myocardial infarction showing accentuated neutrophil recruitment, elevated plasma levels of TNF-α and higher tissue expression of ICAM-1 [291]. The study did not examine the effects of IL-10 deficiency on resolution of the inflammatory response and on remodeling of the infarcted heart. In contrast, our recent investigation exploring the role of IL-10 in cardiac repair following reperfused infarction showed only subtle differences between IL-10 null and wildtype animals [292]. Infarcted IL-10 −/− mice exhibited comparable mortality rates with their wildtype littermates. Although IL-10 −/− mice had higher peak TNF-α and MCP-1 mRNA levels in the infracted heart than wildtype animals, both groups demonstrated timely repression of pro-inflammatory cytokine and chemokine mRNA synthesis after 24h of reperfusion and exhibited a similar time course of resolution of the neutrophil infiltrate. IL-10 gene disruption did not alter fibrous tissue deposition and dilative remodeling of the infarcted heart [292]. These findings suggested that IL-10 signaling plays a non-critical role in suppression of inflammatory mediators, resolution of the inflammatory response and fibrous tissue deposition following myocardial infarction. This may be due to the relative selectivity of IL-10-mediated anti-inflammatory actions, with respect to cell type and stimulus. Resolution of post-infarction inflammation is likely to involve multiple overlapping regulatory mechanisms controlling various proinflammatory pathways activated in the infarcted myocardium.
8. TGF-β: A KEY REGULATOR IN CARDIAC REPAIR
The biology of TGF-β
The TGF-βs are pleiotropic, multifunctional cytokines with a wide range of biological effects regulating cell proliferation, differentiation, and apoptosis and modulating the immune response. The complexity and diversity of TGF-β–mediated effects is demonstrated by its multiple roles in immune system suppression, wound healing, and fibrosis. In mammalian species TGF-β is found in three isoforms (TGF-β1, 2 and 3), encoded by three distinct genes. [293]. These isoforms signal through the same cell surface receptors and have similar cellular targets, although each isoform is expressed in a distinct pattern under control of a unique promoter [294]. TGF-β1 is the prevalent isoform and is found almost ubiquitously, whereas the other isoforms are expressed in a more limited spectrum of cells and tissues. Because of its anti-inflammatory and fibrogenic properties TGF-β may be an essential mediator for cardiac repair by mediating the transition from inflammation to fibrosis.
TGF-β is produced by many cell types as a latent complex, unable to associate with its receptors. The extracellular concentration of TGF-β activity is primarily regulated by conversion of latent TGF-β to active TGF-β. Most tissues contain significant amounts of latent TGF-β; activation of only a small fraction of this latent TGF-β generates maximal cellular response [295]. Mature TGF-β is secreted as a homodimer of two 12.5 kD polypeptides joined by a disulfide bond. This latent dimeric complex contains the C-terminal mature TGF-β and its N-terminal pro-domain, LAP (TGF-β Latency Associated Peptide) [296]. Upon secretion it is covalently bound to one of the four Latent TGF-β Binding Proteins (LTBPs). LTBP is covalently associated with the extracellular matrix and serves to localize the complex in specific areas [297].
As part of the large latent complex (LLC), TGF-β cannot interact with its receptors, because the LAP functions as an inhibitor due a non-covalent high affinity association with TGF-β. After proteolytic cleavage of TGF-β from its propeptide, the LAP propeptide dimer remains associated with the TGF-β dimer by noncovalent interactions forming the small latent TGF-β; this LAP:TGF-β interaction is sufficient to inhibit TGF-β bioactivity. Activation of TGF-β involves liberation of active TGF-β from the activation-competent LAP:TGF-β complex.
A variety of molecules have been described as TGF-β activators. Proteases including plasmin, MMP-2 and MMP-9 are capable of activating TGF-β, coupling matrix degradation with activation of a molecule that has a primary role in maintaining matrix integrity and stability [295], [298], [299]. The matricellular protein Thrombospondin (TSP)-1 is a key TGF-β activator which acts by disrupting the non-covalent interactions between LAP and the TGF-β molecule [300]. Binding of TSP-1 to the sequence LSKL in the LAP alters the conformation of TGF-β making it accessible to its receptor. The αvβ6 integrin appears to be important in epithelial TGF-β activation through a direct interaction with the RGD sequence of the LAP molecule [301]. ROS generation [302] and a mildly acidic environment [303] are also capable of inducing TGF-β activation.
TGF-β induction and activation in cardiac injury
TGF-β is markedly upregulated in experimental models of myocardial infarction [304]. TGF-β isoforms demonstrate distinct patterns of expression in the infarct: TGF-β1 and -β2 are induced early, whereas TGF-β3 shows delayed and prolonged upregulation [305], [11]. TGF-β expression in the infarcted heart is attenuated by angiotensin-converting enzyme inhibitors and angiotensin receptor blockers [306], [307], [308], suggesting that angiotensin II signaling plays an important role in stimulating TGF-β synthesis in the infarct [309]. TGF-β expression is predominantly localized in the infarct border zone, associated with expression of Smad2, 3 and 4 [310] and phosphorylation of Smad1 and Smad2 [311]. Although evidence suggests that bioactive TGF-β is released in the cardiac extracellular fluids 3–5h following reperfused infarction [32], the mechanisms responsible for TGF-β activation in the infarcted heart are poorly understood. Our recent experiments suggested that TSP-1 induction in the infarct border zone may play an important role in activation of TGF-β signaling pathways in mouse and canine infarcts [312].
The role of TGF-β in myocardial infarction
Through its pleiotropic effects, TGF-β is ideally suited as a key mediator in the transition from inflammation to fibrosis [313] (Figure 6). TGF-β suppresses cytokine and chemokine expression by stimulated mononuclear and endothelial cells. TGF-β also inhibits proliferation of most cells, and modulates fibroblast behavior by stimulating the synthesis of various extracellular matrix proteins including collagens, fibronectin, tenascin and proteoglycans [314], and by suppressing matrix degradation through decreased expression of proteinases, such as plasminogen activators and collagenases, and increased synthesis of proteinase inhibitors, such as Plasminogen Activator Inhibitor (PAI)-1 and TIMP-1 [315], [316]. In the healing infarct TGF-β may play a dual role suppressing cytokine and chemokine synthesis by endothelial cells and leukocytes, while promoting extracellular matrix deposition. Unfortunately, the complex biology of TGF-β activation and its pleiotropic actions have hampered our efforts to understand its role in infarct healing.
Figure 6. The complex role of TGF-β signaling in myocardial infarction.
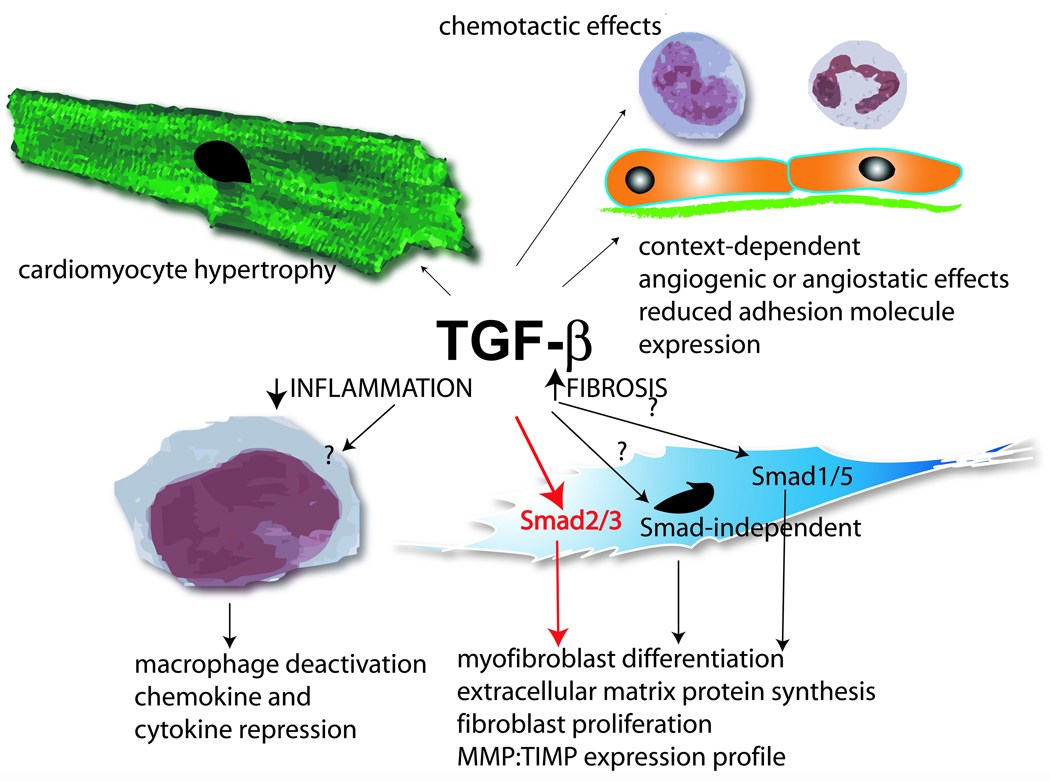
Evidence suggests that TGF-β plays a dual role in cardiac injury, contributing to resolution of inflammation through deactivation of macrophages, while exerting fibrogenic actions. Thus TGF-β may be the “master switch” responsible for transition from inflammation to fibrosis. TGF-β is a highly pleiotropic mediator capable of modulating the phenotype of all cells involved in cardiac repair. Evidence suggests that TGF-β induces cardiomyocyte hypertrophy, has complex, context-dependent angiogenic and angiostatic effects and stimulates leukocyte chemotaxis, while suppressing endothelial cell adhesion molecule synthesis. Because of the complex and multifunctional role of TGF-β, dissection of the signaling pathways responsible for specific actions is critical in order to identify therapeutic targets. It appears that the fibrogenic effects of TGF-β in the healing infarct are, at least in part, dependent on Smad3 signaling. The role of the Smad1/5 and Smad-independent pathways remains unknown.
TGF-β injection during the inflammatory phase of healing significantly reduced ischemic myocardial injury, presumably by attenuating the deleterious effects of pro-inflammatory cytokines such as TNF-α [317]. Two recent studies showed that inhibition of TGF-β signaling by injection of an adenovirus harboring soluble TGF-β type II receptor in the hindlimb muscles resulted in attenuated left ventricular remodeling by modulating cardiac fibrosis [318], [319]. However, early TGF-β inhibition significantly increased mortality and exacerbated left ventricular dilation enhancing cytokine synthesis, suggesting that during the phase of resolution of the inflammatory response, TGF-β signaling plays an important role in suppression of inflammatory mediator synthesis [318].
TGF-β signaling pathways in cardiac injury
Active TGF-β binds to the constitutively active type II receptor (TβRII) at the cell surface. The complex subsequently interacts with, and transphosphorylates the cytoplasmic domain of the type I receptor (TβRI). Phosphorylation of the TβRI propagates downstream intracellular signals, through the Smad proteins, essential components of the TGF-β signaling pathway [320]. There are eight vertebrate Smads: Smad1 to Smad8. The receptor-activated Smads (R-Smads, Smad1, Smad2, Smad3, Smad5, Smad8) are released from the receptor complex to form a heterotrimeric complex of two R-Smads and the common Smad, Smad4, and translocate to the nucleus where they regulate gene transcription together with nuclear transcription factors, co-activators, or co-repressors. In contrast, the structurally divergent Smad6 and Smad7 inhibit TGF-β signaling (inhibitory Smads). Recently, it has become increasingly apparent that TGF-β not only activates Smads, but also signals through Smad-independent pathways [321] that may involve c-Abl [322], p21-activated kinase-2 (PAK2) [323], TGF-β Activated Kinase (TAK)-1 [324], and p38 MAPK [325].
Because of the pleiotropic effects of TGF-β, dissection of the signaling pathways responsible for its actions in the infarcted myocardium is essential for identification of therapeutic targets. Extensive evidence suggests that Smad3 signaling plays an essential role in fibrotic processes [326]. Furthermore, a growing body of evidence suggests that TGF-β-mediated suppression of inflammatory mediator synthesis may be, at least in part, Smad3-dependent [327] Accordingly, we examined the effects of Smad3 gene disruption on infarct healing and the pathogenesis of cardiac remodeling. Smad3 null animals had suppressed peak chemokine expression and decreased neutrophil recruitment in the infarcted myocardium but showed timely repression of inflammatory gene synthesis and resolution of the inflammatory infiltrate. Although myofibroblast density was higher in Smad3 null infarcts, interstitial deposition of collagen and the matricellular protein tenascin-C, a marker of active interstitial remodeling [328], in the peri-infarct zone and the non-infarcted myocardium was markedly reduced. TGF-β-mediated induction of procollagen type III and tenascin-C in isolated cardiac fibroblasts was dependent on Smad3, suggesting that decreased fibrotic remodeling in infarcted Smad3 null hearts may be due to abrogation of the pro-fibrotic TGF-β responses. On the other hand the increased myofibroblast density in Smad3 −/− infarcts may reflect a partial resistance of Smad3 null fibroblasts to the antiproliferative actions of TGF-β [329]. Thus, in the absence of Smad3 signaling the infarct was filled with a large number of dysfunctional fibroblasts. Attenuated fibrosis on the non-infarcted heart in Smad3 −/− mice was associated with reduced dilative remodeling and improved diastolic function; however infarct size was comparable between groups [311]. Our findings suggested that Smad3 loss does not alter the time course of resolution of inflammation in healing infarcts, but prevents interstitial fibrosis in the non-infarcted myocardium and attenuates cardiac remodeling. Thus, the Smad3 cascade may be a promising therapeutic target for the treatment of myocardial infarction [330]. The role of the Smad1/5 and Smad-independent pathways in cardiac repair is an area of active investigation.
9. THE INFARCT BORDER ZONE AS A BARRIER PREVENTING EXPANSION OF THE INFLAMMATORY REACTION
Optimal cardiac repair requires containment of the inflammatory response into the infarcted area. Extension of the inflammatory reaction into the non-infarcted area could result in expansion of the leukocyte infiltrate leading to matrix degradation and worse adverse remodeling. Thus, it is tempting to hypothesize that endogenous protective mechanisms may be activated in the infarct border zone and the peri-infarct area, preventing expansion of the inflammatory reaction into the viable myocardium. Recent evidence from our laboratory suggested that, through its unique extracellular matrix composition, the infarct border zone may serve as a “barrier” that protects the non-infarcted area from expansion of the inflammatory reaction (Figure 7) [312]. Using a canine model of reperfused infarction we demonstrated a strikingly selective localization of TSP-1, a matricellular protein with potent angiostatic properties and a crucial role in TGF-β activation [331], in the extracellular matrix and microvascular endothelium of the ischemic border zone. Murine infarcts also had marked TSP-1 deposition in the border zone. Infarcted TSP-1 knockout mice exhibited sustained upregulation of the chemokines MCP-1, MIP-1α and IP-10 and the cytokines IL-1β, IL-6 and TGF-β, suggesting an enhanced and prolonged post-infarction inflammatory response. In addition, TSP-1 knockouts had markedly increased macrophage and myofibroblast density in infarcts and in remodeling non-infarcted myocardial areas neighboring the myocardial scar, suggesting expansion of granulation tissue formation into the non-infarcted territory. TSP-1 deficient animals had more extensive post-infarction remodeling than wildtype mice, although infarct size was similar in both groups [312]. Our findings suggested that the selective endogenous expression of TSP-1 in the infarct border zone may protect the non-infarcted myocardium from inflammatory cell infiltration and fibrotic remodeling. Several distinct TSP-1-mediated actions may be responsible for its protective effects [332]. First, enhanced TGF-β activation in the border zone may locally suppress inflammatory mediator synthesis. When compared with wildtype animals, TSP-1 null mice exhibited a trend towards reduced Smad2 phosphorylation in the infarcted heart, suggesting that impaired TGF-β activation may explain, at least in part, the healing defects observed in the absence of TSP-1. Second, reduced angiogenesis may prevent extension of the inflammatory reaction. Third, direct anti-inflammatory actions of the TSP-1 molecule, mediated through CD47 [333], may suppress the inflammatory response.
Figure 7. Through its unique composition, the infarct border zone may protect the non-infarcted myocardium from expansion of inflammatory injury.

Immunohistochemical staining of canine myocardial infarction demonstrates a strikingly selective deposition of the matricellular protein Thrombospondin (TSP)-1 (arrows) in the infarct border zone (BZ). TSP-1 is a key TGF-β activator, has direct anti-inflammatory effects mediated through CD47 and exerts potent angiostatic effects. Thus, TSP-1 induction in the border zone may serve as a “barrier” preventing expansion of the inflammatory reaction into the non-infarcted myocardium (C). (Symbol: I, infarct)
10. TARGETING THE IMMUNE RESPONSE TO OPTIMIZE CARDIAC REPAIR
A vast body of evidence suggests an essential role for the immune response in the reparative process following myocardial infarction [334]. Although no specific immunomodulatory approach is currently part of our therapeutic armamentarium for patients with myocardial infarction, established therapeutic strategies, such as β-adrenergic blockade and ACE inhibition, may exert their beneficial effects in part by interfering with the inflammatory cascade. β-adrenergic blockade attenuated TNF-α and IL-1β expression in failing hearts [335] and decreased IL-1 expression levels in the infarcted myocardium [336]. Several studies have demonstrated that ACE inhibition and angiotensin receptor blockade result in decreased TGF-β expression in healing infarcts [306], [308], [309], [307]. However, the contribution of TGF-β signaling inhibition in mediating the salutary effects of ACE inhibitors remains unknown.
Over the past thirty years numerous experimental studies have shown dramatic reduction in infarct size, or attenuation of adverse remodeling, with the use of specific strategies that modulate the inflammatory response. Initially the main concept guiding these interventions was the notion that inflammatory cells and mediators may induce injury of viable cardiomyocytes. As the role of the inflammatory cascade in cardiac repair was recognized, attempts to optimize the healing response were introduced, aimed at attenuating adverse remodeling. Recently, the concept of cell-based cardiac repair has evolved and strategies enhancing the regenerative capacity of the heart have been tested in both experimental animal models and in clinical settings. In order to identify suitable therapeutic targets for patients with myocardial infarction several important questions need to be answered:
Do anti-inflammatory approaches reduce cardiomyocyte injury?
Although extensive experimental evidence suggested that infiltrating leukocytes and inflammatory mediators may induce injurious effects on viable cardiomyocytes in the infarcted heart, attempts to mitigate inflammatory injury in clinical practice have been in general unsuccessful. The catastrophic experience of the methylprednisolone trial [337] emphasized the need for better understanding of the cellular and molecular events associated with the inflammatory response in order to achieve effective suppression of injurious processes without interfering with healing and cardiac repair. Recently, the disappointing results of the anti-CD18 trials led to criticism regarding the usefulness of strategies targeting the inflammatory cascade in myocardial infarction [201]. It has been suggested that these failures may represent the inherent risk of using animal models, which may have fundamental differences from the respective human disease process. Although species-specific effects may be in some cases significant, the most important lesson we have learned from studying experimental myocardial infarction is that a sound understanding of the biology is necessary before a specific intervention is pursued on a therapeutic basis. So, why did anti-inflammatory strategies fail in reducing cardiac injury?
First, the significance of inflammatory cardiomyocyte injury may have been overstated. Although many published experimental studies demonstrated an impressive reduction in infarct size upon inhibition of specific inflammatory mediators, these findings may not adequately represent our collective experience. In contrast to clinical trials, experimental studies may not be published when the data do not reveal an effect of the intervention. This bias against “negative” findings may have prevented publication of reports that showed no significant effects of anti-inflammatory strategies.
Second, one has to consider that the inflammatory cascade is based on a complex network of molecular mediators with pleiotropic effects, dictated by critical cellular, spatial and temporal variables. Typical properties of cytokines in networks are redundancy, pleiotropic, synergistic activity and antagonistic effects upon each other. Thus, cytokines and other inflammatory mediators that may appear reasonable therapeutic targets considering their injurious role in the early stages of the inflammatory response may also be essential regulators of cardiac repair. Interventions attenuating early inflammatory injury may also result in impaired healing, leading to accentuated adverse remodeling through alterations in the qualitative characteristics of the scar. For many anti-inflammatory interventions, the possible benefit from reduction of inflammatory cardiomyocyte injury may not outweigh the detrimental effects on the reparative response.
Third, the complexity of the clinical scenario cannot be adequately simulated in experimental studies. Variables such as age, gender, the presence of comorbid conditions such as diabetes, obesity and hyperlipidemia, the timing of reperfusion and genetic variations between individuals greatly complicate prediction of the potential effects of a therapeutic intervention. Our recent investigation on the effects of aging on infarct healing provides insight into the challenges of predicting the effects of a therapeutic intervention on the basis of experimental animal studies [338]. We demonstrated that enhanced post-infarction remodeling in senescent mice is associated with suppressed inflammation, delayed granulation tissue formation and markedly reduced collagen deposition, in part due to blunted responses of senescent fibroblasts to growth factors. In contrast, young animals exhibit a robust post-infarction inflammatory response and form dense collagenous scars. Animal studies are almost always performed in young adult animals and, although they provide valuable insight into the mechanisms involved in cardiac injury and repair, they may not accurately reflect the pathology of myocardial infarction in middle aged or elderly human populations. Thus, the injurious potential of inflammatory mediators in patients with myocardial infarction may have been overstated due to extrapolation from young animals to human patients. In addition, senescent ischemic hearts show impairment of important cardioprotective pathways involving TNF-α [339] and PDGF-AB [340] and have a decreased anti-apoptotic response to administration of Granulocyte-Colony Stimulating Factor (G-CSF) and SCF [341] in comparison with young animals.
Does modulation of the inflammatory response attenuate adverse remodeling?
Even if interventions targeting the inflammatory response do not reduce cardiomyocyte death, modulation of the reparative process in order to optimize the mechanical and functional properties of the infarcted heart remains an interesting direction. Experimental evidence suggests that inhibition of chemokine signaling, modulation of extracellular matrix remodeling, and enhancement of the endogenous protective mechanisms that contribute to resolution and containment of the inflammatory response may be promising therapeutic strategies [334]. Timing and topography are key determinants of the success of a specific approach. Effective healing is dependent on a well-orchestrated cellular response and on timely induction and suppression of specific mediators in a locally restricted manner. Thus, interventions targeting inflammatory mediators should take into account both topographic and temporal parameters. An approach that may have favorable effects if locally applied in the center of the infarct may result in deleterious changes in the border zone or the non-infarcted remodeling myocardium. For example, a strategy that decreases the collagen content in the infarcted heart may attenuate interstitial fibrosis in the non-infarcted areas, but may also result in enhanced remodeling by suppressing collagen deposition in the center of the infarct, leading to decreased tensile strength and subsequent left ventricular dilation.
Is cardiac regeneration a realistic therapeutic target?
Cardiac myocytes are thought to be terminally differentiated cells unable to regenerate and replace damaged myocardium. This concept was challenged by recent evidence suggesting that a fraction of cardiomyocytes may be able to re-enter the cell cycle, and that limited cardiac regeneration may occur through recruitment of resident and circulating stem cells [342, 343]. Although the issue of cardiomyocyte regeneration remains highly controversial [344], [345], [346] these concepts led to basic and clinical studies exploring the effects of cell-therapy strategies in cardiac repair following infarction [345], [346]. Cell-based therapy began with the transplantation of autologous skeletal myoblasts; these cells did not appear to transdifferentiate into cardiomyocytes after cardiac grafting [347] and remained functionally isolated from their host [348]. Several groups have independently studied the potential of bone marrow-derived cells to promote cardiac regeneration. Orlic and co-workers suggested that hematopoietic stem cells can transdifferentiate into cardiomyocytes when injected into the infarcted myocardium resulting in extensive cardiac regeneration and improved function [221]. Unfortunately several other groups could not reproduce these findings and concluded that there was no significant cardiac differentiation of bone marrow-derived cells [349, 350], although endogenously derived circulating cells were noted to fuse with host myocytes in the infarct border zone. Thus, the ability of injected bone marrow-derived cells to give rise to cardiac myocytes after myocardial infarction remains controversial [345], [346]. Mesenchymal stem cells have recently attracted significant interest as a promising cell-based therapeutic strategy for patients with myocardial infarction [351]. Enthusiasm from the early experimental findings fueled numerous small clinical studies examining the effects of cell therapy in patients with acute myocardial infarction [342, 352, 353]. Although some of these studies have reported beneficial effects of progenitor cell transfer in the infarcted heart [354], [355], the mechanisms responsible for these actions remain poorly understood [356]. Carefully conducted clinical trials will clarify the role of cell therapy in the treatment of patients with myocardial infarction.
The discovery of the beneficial effects of marrow-derived progenitors in cardiac repair led to attempts to mobilize these cells using cytokines and growth factors. Inflammatory mediators play an important role in mobilizing progenitor cells and may regulate their homing into the infarcted myocardium [357]. Several studies demonstrated that treatment with SCF and G-CSF results in improved function and attenuated left ventricular remodeling following myocardial infarction [220, 342]. Whether these beneficial effects are mediated through enhanced recruitment of progenitor cells remains unclear. Growth factors are pleiotropic and multifunctional and induce a diverse range of effects on a variety of cell types involved in infarct healing. In addition to its actions on progenitor cell recruitment, G-CSF may prevent post-infarction remodeling by exerting direct anti-apoptotic effects on cardiomyocytes [358]. Thus, the potential beneficial actions of growth factor therapy in the infarcted myocardium may not be mediated through stem cell mobilization.
The field of cell-based therapy in myocardial infarction is rapidly evolving. Although the initial ambitious goal to rebuild the heart from its component parts remains elusive, extensive experimental evidence suggests beneficial effects from therapeutic strategies injecting a wide variety of cell types. The benefit is usually not associated with cardiomyocyte regeneration, but may be mediated through paracrine effects of the injected cells, inducing enhanced repair and attenuating dilative remodeling of the infracted ventricle. Many challenging questions remain to be answered. What is the mechanism responsible for functional improvement in the absence of cardiomyocyte transdifferentiation? Are the benefits from cell therapy simply due to alterations in the mechanical properties of the infarcted heart? Considering the significant species differences in the biology of progenitor cells and tissue regeneration, are the conclusions derived from studies in rodents applicable to human patients? How can we ensure optimal vascularization of the grafted cells? Do the injected cells serve as a vehicle for continuous delivery of specific soluble mediators that exert protective effects? Which combination of cytokines and growth factors would provide optimal enhancement of the reparative response? These and other critical issues need to be addressed in order to fulfill the visionary goal of cardiac regeneration.
ACKNOWLEDGMENTS
Research in Dr Frangogiannis’ laboratory is supported by grants from the National Institutes of Health (R01 HL-76246 and R01 HL-85440).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Tavazzi L. Clinical epidemiology of acute myocardial infarction. Am Heart J. 1999;138:S48–S54. doi: 10.1016/s0002-8703(99)70320-0. [DOI] [PubMed] [Google Scholar]
- 2.Jennings RB, Murry CE, Steenbergen C, Jr, Reimer KA. Development of cell injury in sustained acute ischemia. Circulation. 1990;82:II2–II12. [PubMed] [Google Scholar]
- 3.Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81:1161–1172. doi: 10.1161/01.cir.81.4.1161. [DOI] [PubMed] [Google Scholar]
- 4.Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA. Controversies in ventricular remodelling. Lancet. 2006;367:356–367. doi: 10.1016/S0140-6736(06)68074-4. [DOI] [PubMed] [Google Scholar]
- 5.Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling--concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. 2000;35:569–582. doi: 10.1016/s0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- 6.White HD, Norris RM, Brown MA, Brandt PW, Whitlock RM, Wild CJ. Left ventricular end-systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation. 1987;76:44–51. doi: 10.1161/01.cir.76.1.44. [DOI] [PubMed] [Google Scholar]
- 7.St John Sutton M, Pfeffer MA, Plappert T, Rouleau JL, Moye LA, Dagenais GR, Lamas GA, Klein M, Sussex B, Goldman S, et al. Quantitative two-dimensional echocardiographic measurements are major predictors of adverse cardiovascular events after acute myocardial infarction The protective effects of captopril. Circulation. 1994;89:68–75. doi: 10.1161/01.cir.89.1.68. [DOI] [PubMed] [Google Scholar]
- 8.Sogaard P, Gotzsche CO, Ravkilde J, Norgaard A, Thygesen K. Ventricular arrhythmias in the acute and chronic phases after acute myocardial infarction. Effect of intervention with captopril. Circulation. 1994;90:101–107. doi: 10.1161/01.cir.90.1.101. [DOI] [PubMed] [Google Scholar]
- 9.St John Sutton M, Lee D, Rouleau JL, Goldman S, Plappert T, Braunwald E, Pfeffer MA. Left ventricular remodeling and ventricular arrhythmias after myocardial infarction. Circulation. 2003;107:2577–2582. doi: 10.1161/01.CIR.0000070420.51787.A8. [DOI] [PubMed] [Google Scholar]
- 10.Frangogiannis NG. The mechanistic basis of infarct healing. Antioxid Redox Signal. 2006;8:1907–1939. doi: 10.1089/ars.2006.8.1907. [DOI] [PubMed] [Google Scholar]
- 11.Dewald O, Ren G, Duerr GD, Zoerlein M, Klemm C, Gersch C, Tincey S, Michael LH, Entman ML, Frangogiannis NG. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol. 2004;164:665–677. doi: 10.1016/S0002-9440(10)63154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dobaczewski M, Bujak M, Zymek P, Ren G, Entman ML, Frangogiannis NG. Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell Tissue Res. 2006;324:475–488. doi: 10.1007/s00441-005-0144-6. [DOI] [PubMed] [Google Scholar]
- 13.Jugdutt BI, Joljart MJ, Khan MI. Rate of collagen deposition during healing and ventricular remodeling after myocardial infarction in rat and dog models. Circulation. 1996;94:94–101. doi: 10.1161/01.cir.94.1.94. [DOI] [PubMed] [Google Scholar]
- 14.Beg AA. Endogenous ligands of Toll-like receptors: implications for regulating inflammatory and immune responses. Trends Immunol. 2002;23:509–512. doi: 10.1016/s1471-4906(02)02317-7. [DOI] [PubMed] [Google Scholar]
- 15.Frantz S, Ertl G, Bauersachs J. Mechanisms of disease: Toll-like receptors in cardiovascular disease. Nat Clin Pract Cardiovasc Med. 2007;4:444–454. doi: 10.1038/ncpcardio0938. [DOI] [PubMed] [Google Scholar]
- 16.Frantz S, Kobzik L, Kim YD, Fukazawa R, Medzhitov R, Lee RT, Kelly RA. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J Clin Invest. 1999;104:271–280. doi: 10.1172/JCI6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oyama J, Blais C, Jr, Liu X, Pu M, Kobzik L, Kelly RA, Bourcier T. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109:784–789. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 18.Riad A, Jager S, Sobirey M, Escher F, Yaulema-Riss A, Westermann D, Karatas A, Heimesaat MM, Bereswill S, Dragun D, et al. Toll-like receptor-4 modulates survival by induction of left ventricular remodeling after myocardial infarction in mice. J Immunol. 2008;180:6954–6961. doi: 10.4049/jimmunol.180.10.6954. [DOI] [PubMed] [Google Scholar]
- 19.Shishido T, Nozaki N, Yamaguchi S, Shibata Y, Nitobe J, Miyamoto T, Takahashi H, Arimoto T, Maeda K, Yamakawa M, et al. Toll-like receptor-2 modulates ventricular remodeling after myocardial infarction. Circulation. 2003;108:2905–2910. doi: 10.1161/01.CIR.0000101921.93016.1C. [DOI] [PubMed] [Google Scholar]
- 20.Mantovani A, Garlanda C, Doni A, Bottazzi B. Pentraxins in innate immunity: from C-reactive protein to the long pentraxin PTX3. J Clin Immunol. 2008;28:1–13. doi: 10.1007/s10875-007-9126-7. [DOI] [PubMed] [Google Scholar]
- 21.Pepys MB, Hirschfield GM, Tennent GA, Gallimore JR, Kahan MC, Bellotti V, Hawkins PN, Myers RM, Smith MD, Polara A, et al. Targeting C-reactive protein for the treatment of cardiovascular disease. Nature. 2006;440:1217–1221. doi: 10.1038/nature04672. [DOI] [PubMed] [Google Scholar]
- 22.Griselli M, Herbert J, Hutchinson WL, Taylor KM, Sohail M, Krausz T, Pepys MB. C-reactive protein and complement are important mediators of tissue damage in acute myocardial infarction. J Exp Med. 1999;190:1733–1740. doi: 10.1084/jem.190.12.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salio M, Chimenti S, De Angelis N, Molla F, Maina V, Nebuloni M, Pasqualini F, Latini R, Garlanda C, Mantovani A. Cardioprotective function of the long pentraxin PTX3 in acute myocardial infarction. Circulation. 2008;117:1055–1064. doi: 10.1161/CIRCULATIONAHA.107.749234. [DOI] [PubMed] [Google Scholar]
- 24.Nijmeijer R, Lagrand WK, Visser CA, Meijer CJ, Niessen HW, Hack CE. CRP, a major culprit in complement-mediated tissue damage in acute myocardial infarction? Int Immunopharmacol. 2001;1:403–414. doi: 10.1016/s1567-5769(00)00044-8. [DOI] [PubMed] [Google Scholar]
- 25.Fujita T. Evolution of the lectin-complement pathway and its role in innate immunity. Nat Rev Immunol. 2002;2:346–353. doi: 10.1038/nri800. [DOI] [PubMed] [Google Scholar]
- 26.Rossen RD, Michael LH, Kagiyama A, Savage HE, Hanson G, Reisberg MA, Moake JN, Kim SH, Self D, Weakley S. Mechanism of complement activation after coronary artery occlusion: evidence that myocardial ischemia in dogs causes release of constituents of myocardial subcellular origin that complex with human C1q in vivo. Circ Res. 1988;62:572–584. doi: 10.1161/01.res.62.3.572. [DOI] [PubMed] [Google Scholar]
- 27.Hill JH, Ward PA. The phlogistic role of C3 leukotactic fragments in myocardial infarcts of rats. J Exp Med. 1971;133:885–900. doi: 10.1084/jem.133.4.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinckard RN, Olson MS, Giclas PC, Terry R, Boyer JT, O'Rourke RA. Consumption of classical complement components by heart subcellular membranes in vitro and in patients after acute myocardial infarction. J Clin Invest. 1975;56:740–750. doi: 10.1172/JCI108145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rossen RD, Michael LH, Hawkins HK, Youker K, Dreyer WJ, Baughn RE, Entman ML. Cardiolipin-protein complexes and initiation of complement activation after coronary artery occlusion. Circ Res. 1994;75:546–555. doi: 10.1161/01.res.75.3.546. [DOI] [PubMed] [Google Scholar]
- 30.Yasojima K, Schwab C, McGeer EG, McGeer PL. Human heart generates complement proteins that are upregulated and activated after myocardial infarction. Circ Res. 1998;83:860–869. doi: 10.1161/01.res.83.8.860. [DOI] [PubMed] [Google Scholar]
- 31.Dreyer WJ, Michael LH, Nguyen T, Smith CW, Anderson DC, Entman ML, Rossen RD. Kinetics of C5a release in cardiac lymph of dogs experiencing coronary artery ischemia-reperfusion injury. Circ Res. 1992;71:1518–1524. doi: 10.1161/01.res.71.6.1518. [DOI] [PubMed] [Google Scholar]
- 32.Birdsall HH, Green DM, Trial J, Youker KA, Burns AR, MacKay CR, LaRosa GJ, Hawkins HK, Smith CW, Michael LH, et al. Complement C5a, TGF-beta 1, and MCP-1, in sequence, induce migration of monocytes into ischemic canine myocardium within the first one to five hours after reperfusion. Circulation. 1997;95:684–692. doi: 10.1161/01.cir.95.3.684. [DOI] [PubMed] [Google Scholar]
- 33.Makrides SC. Therapeutic inhibition of the complement system. Pharmacol Rev. 1998;50:59–87. [PubMed] [Google Scholar]
- 34.Czermak BJ, Lentsch AB, Bless NM, Schmal H, Friedl HP, Ward PA. Role of complement in in vitro and in vivo lung inflammatory reactions. J Leukoc Biol. 1998;64:40–48. doi: 10.1002/jlb.64.1.40. [DOI] [PubMed] [Google Scholar]
- 35.Maroko PR, Carpenter CB, Chiariello M, Fishbein MC, Radvany P, Knostman JD, Hale SL. Reduction by cobra venom factor of myocardial necrosis after coronary artery occlusion. J Clin Invest. 1978;61:661–670. doi: 10.1172/JCI108978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buerke M, Prufer D, Dahm M, Oelert H, Meyer J, Darius H. Blocking of classical complement pathway inhibits endothelial adhesion molecule expression and preserves ischemic myocardium from reperfusion injury. J Pharmacol Exp Ther. 1998;286:429–438. [PubMed] [Google Scholar]
- 37.Buerke M, Murohara T, Lefer AM. Cardioprotective effects of a C1 esterase inhibitor in myocardial ischemia and reperfusion. Circulation. 1995;91:393–402. doi: 10.1161/01.cir.91.2.393. [DOI] [PubMed] [Google Scholar]
- 38.Horstick G, Kempf T, Lauterbach M, Bhakdi S, Kopacz L, Heimann A, Malzahn M, Horstick M, Meyer J, Kempski O. C1-esterase-inhibitor treatment at early reperfusion of hemorrhagic shock reduces mesentry leukocyte adhesion and rolling. Microcirculation. 2001;8:427–433. doi: 10.1038/sj/mn/7800115. [DOI] [PubMed] [Google Scholar]
- 39.Horstick G, Heimann A, Gotze O, Hafner G, Berg O, Boehmer P, Becker P, Darius H, Rupprecht HJ, Loos M, et al. Intracoronary application of C1 esterase inhibitor improves cardiac function and reduces myocardial necrosis in an experimental model of ischemia and reperfusion. Circulation. 1997;95:701–708. doi: 10.1161/01.cir.95.3.701. [DOI] [PubMed] [Google Scholar]
- 40.Horstick G. C1-esterase inhibitor in ischemia and reperfusion. Immunobiology. 2002;205:552–562. doi: 10.1078/0171-2985-00154. [DOI] [PubMed] [Google Scholar]
- 41.Weisman HF, Bartow T, Leppo MK, Marsh HC, Jr, Carson GR, Concino MF, Boyle MP, Roux KH, Weisfeldt ML, Fearon DT. Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science. 1990;249:146–151. doi: 10.1126/science.2371562. [DOI] [PubMed] [Google Scholar]
- 42.Lucchesi BR, Kilgore KS. Complement inhibitors in myocardial ischemia/reperfusion injury. Immunopharmacology. 1997;38:27–42. doi: 10.1016/s0162-3109(97)00060-x. [DOI] [PubMed] [Google Scholar]
- 43.Kirschfink M. Targeting complement in therapy. Immunol Rev. 2001;180:177–189. doi: 10.1034/j.1600-065x.2001.1800116.x. [DOI] [PubMed] [Google Scholar]
- 44.Granger CB, Mahaffey KW, Weaver WD, Theroux P, Hochman JS, Filloon TG, Rollins S, Todaro TG, Nicolau JC, Ruzyllo W, et al. Pexelizumab, an anti-C5 complement antibody, as adjunctive therapy to primary percutaneous coronary intervention in acute myocardial infarction: the COMplement inhibition in Myocardial infarction treated with Angioplasty (COMMA) trial. Circulation. 2003;108:1184–1190. doi: 10.1161/01.CIR.0000087447.12918.85. [DOI] [PubMed] [Google Scholar]
- 45.Mahaffey KW, Granger CB, Nicolau JC, Ruzyllo W, Weaver WD, Theroux P, Hochman JS, Filloon TG, Mojcik CF, Todaro TG, et al. Effect of pexelizumab, an anti-C5 complement antibody, as adjunctive therapy to fibrinolysis in acute myocardial infarction: the COMPlement inhibition in myocardial infarction treated with thromboLYtics (COMPLY) trial. Circulation. 2003;108:1176–1183. doi: 10.1161/01.CIR.0000087404.53661.F8. [DOI] [PubMed] [Google Scholar]
- 46.Armstrong PW, Granger CB, Adams PX, Hamm C, Holmes D, Jr, O'Neill WW, Todaro TG, Vahanian A, Van de Werf F. Pexelizumab for acute ST-elevation myocardial infarction in patients undergoing primary percutaneous coronary intervention: a randomized controlled trial. Jama. 2007;297:43–51. doi: 10.1001/jama.297.1.43. [DOI] [PubMed] [Google Scholar]
- 47.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hensley K, Robinson KA, Gabbita SP, Salsman S, Floyd RA. Reactive oxygen species, cell signaling, and cell injury. Free Radic Biol Med. 2000;28:1456–1462. doi: 10.1016/s0891-5849(00)00252-5. [DOI] [PubMed] [Google Scholar]
- 49.Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: Part I: basic mechanisms and in vivo monitoring of ROS. Circulation. 2003;108:1912–1916. doi: 10.1161/01.CIR.0000093660.86242.BB. [DOI] [PubMed] [Google Scholar]
- 50.Sen CK, Packer L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996;10:709–720. doi: 10.1096/fasebj.10.7.8635688. [DOI] [PubMed] [Google Scholar]
- 51.Granger DN. Role of xanthine oxidase and granulocytes in ischemia-reperfusion injury. Am J Physiol. 1988;255:H1269–H1275. doi: 10.1152/ajpheart.1988.255.6.H1269. [DOI] [PubMed] [Google Scholar]
- 52.Shingu M, Nonaka S, Nishimukai H, Nobunaga M, Kitamura H, Tomo-Oka K. Activation of complement in normal serum by hydrogen peroxide and hydrogen peroxide-related oxygen radicals produced by activated neutrophils. Clin Exp Immunol. 1992;90:72–78. doi: 10.1111/j.1365-2249.1992.tb05834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shingu M, Nobunaga M. Chemotactic activity generated in human serum from the fifth component of complement by hydrogen peroxide. Am J Pathol. 1984;117:201–206. [PMC free article] [PubMed] [Google Scholar]
- 54.Patel KD, Zimmerman GA, Prescott SM, McEver RP, McIntyre TM. Oxygen radicals induce human endothelial cells to express GMP-140 and bind neutrophils. J Cell Biol. 1991;112:749–759. doi: 10.1083/jcb.112.4.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lakshminarayanan V, Beno DW, Costa RH, Roebuck KA. Differential regulation of interleukin-8 and intercellular adhesion molecule-1 by H2O2 and tumor necrosis factor-alpha in endothelial and epithelial cells. J Biol Chem. 1997;272:32910–32918. doi: 10.1074/jbc.272.52.32910. [DOI] [PubMed] [Google Scholar]
- 56.Lakshminarayanan V, Drab-Weiss EA, Roebuck KA. H2O2 and tumor necrosis factor-alpha induce differential binding of the redox-responsive transcription factors AP-1 and NF-kappaB to the interleukin-8 promoter in endothelial and epithelial cells. J Biol Chem. 1998;273:32670–32678. doi: 10.1074/jbc.273.49.32670. [DOI] [PubMed] [Google Scholar]
- 57.Lakshminarayanan V, Lewallen M, Frangogiannis NG, Evans AJ, Wedin KE, Michael LH, Entman ML. Reactive oxygen intermediates induce monocyte chemotactic protein-1 in vascular endothelium after brief ischemia. Am J Pathol. 2001;159:1301–1311. doi: 10.1016/S0002-9440(10)62517-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sellak H, Franzini E, Hakim J, Pasquier C. Reactive oxygen species rapidly increase endothelial ICAM-1 ability to bind neutrophils without detectable upregulation. Blood. 1994;83:2669–2677. [PubMed] [Google Scholar]
- 59.Kloner RA, Jennings RB. Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 2. Circulation. 2001;104:3158–3167. doi: 10.1161/hc5001.100039. [DOI] [PubMed] [Google Scholar]
- 60.Kloner RA, Jennings RB. Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 1. Circulation. 2001;104:2981–2989. doi: 10.1161/hc4801.100038. [DOI] [PubMed] [Google Scholar]
- 61.Bolli R, Marban E. Molecular and cellular mechanisms of myocardial stunning. Physiol Rev. 1999;79:609–634. doi: 10.1152/physrev.1999.79.2.609. [DOI] [PubMed] [Google Scholar]
- 62.Jolly SR, Kane WJ, Bailie MB, Abrams GD, Lucchesi BR. Canine myocardial reperfusion injury. Its reduction by the combined administration of superoxide dismutase and catalase. Circ Res. 1984;54:277–285. doi: 10.1161/01.res.54.3.277. [DOI] [PubMed] [Google Scholar]
- 63.Uraizee A, Reimer KA, Murry CE, Jennings RB. Failure of superoxide dismutase to limit size of myocardial infarction after 40 minutes of ischemia and 4 days of reperfusion in dogs. Circulation. 1987;75:1237–1248. doi: 10.1161/01.cir.75.6.1237. [DOI] [PubMed] [Google Scholar]
- 64.Richard VJ, Murry CE, Jennings RB, Reimer KA. Therapy to reduce free radicals during early reperfusion does not limit the size of myocardial infarcts caused by 90 minutes of ischemia in dogs. Circulation. 1988;78:473–480. doi: 10.1161/01.cir.78.2.473. [DOI] [PubMed] [Google Scholar]
- 65.Wang P, Chen H, Qin H, Sankarapandi S, Becher MW, Wong PC, Zweier JL. Overexpression of human copper, zinc-superoxide dismutase (SOD1) prevents postischemic injury. Proc Natl Acad Sci U S A. 1998;95:4556–4560. doi: 10.1073/pnas.95.8.4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen Z, Siu B, Ho YS, Vincent R, Chua CC, Hamdy RC, Chua BH. Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. J Mol Cell Cardiol. 1998;30:2281–2289. doi: 10.1006/jmcc.1998.0789. [DOI] [PubMed] [Google Scholar]
- 67.Murohara Y, Yui Y, Hattori R, Kawai C. Effects of superoxide dismutase on reperfusion arrhythmias and left ventricular function in patients undergoing thrombolysis for anterior wall acute myocardial infarction. Am J Cardiol. 1991;67:765–767. doi: 10.1016/0002-9149(91)90538-v. [DOI] [PubMed] [Google Scholar]
- 68.Flaherty JT, Pitt B, Gruber JW, Heuser RR, Rothbaum DA, Burwell LR, George BS, Kereiakes DJ, Deitchman D, Gustafson N, et al. Recombinant human superoxide dismutase (h-SOD) fails to improve recovery of ventricular function in patients undergoing coronary angioplasty for acute myocardial infarction. Circulation. 1994;89:1982–1991. doi: 10.1161/01.cir.89.5.1982. [DOI] [PubMed] [Google Scholar]
- 69.Lefer DJ, Granger DN. Oxidative stress and cardiac disease. Am J Med. 2000;109:315–323. doi: 10.1016/s0002-9343(00)00467-8. [DOI] [PubMed] [Google Scholar]
- 70.Maxwell SR, Lip GY. Reperfusion injury: a review of the pathophysiology, clinical manifestations and therapeutic options. Int J Cardiol. 1997;58:95–117. doi: 10.1016/s0167-5273(96)02854-9. [DOI] [PubMed] [Google Scholar]
- 71.Tritto I, Ambrosio G. Role of oxidants in the signaling pathway of preconditioning. Antioxid Redox Signal. 2001;3:3–10. doi: 10.1089/152308601750100425. [DOI] [PubMed] [Google Scholar]
- 72.Kabir AM, Clark JE, Tanno M, Cao X, Hothersall JS, Dashnyam S, Gorog DA, Bellahcene M, Shattock MJ, Marber MS. Cardioprotection initiated by reactive oxygen species is dependent on activation of PKCepsilon. Am J Physiol Heart Circ Physiol. 2006;291:H1893–H1899. doi: 10.1152/ajpheart.00798.2005. [DOI] [PubMed] [Google Scholar]
- 73.Lenardo MJ, Baltimore D. NF-kappa B: a pleiotropic mediator of inducible and tissue-specific gene control. Cell. 1989;58:227–229. doi: 10.1016/0092-8674(89)90833-7. [DOI] [PubMed] [Google Scholar]
- 74.Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stancovski I, Baltimore D. NF-kappaB activation: the I kappaB kinase revealed? Cell. 1997;91:299–302. doi: 10.1016/s0092-8674(00)80413-4. [DOI] [PubMed] [Google Scholar]
- 76.Kupatt C, Habazettl H, Goedecke A, Wolf DA, Zahler S, Boekstegers P, Kelly RA, Becker BF. Tumor necrosis factor-alpha contributes to ischemia- and reperfusion-induced endothelial activation in isolated hearts. Circ Res. 1999;84:392–400. doi: 10.1161/01.res.84.4.392. [DOI] [PubMed] [Google Scholar]
- 77.Chandrasekar B, Freeman GL. Induction of nuclear factor kappaB and activation protein 1 in postischemic myocardium. FEBS Lett. 1997;401:30–34. doi: 10.1016/s0014-5793(96)01426-3. [DOI] [PubMed] [Google Scholar]
- 78.Morishita R, Sugimoto T, Aoki M, Kida I, Tomita N, Moriguchi A, Maeda K, Sawa Y, Kaneda Y, Higaki J, et al. In vivo transfection of cis element "ecoy"against nuclear factor-kappaB binding site prevents myocardial infarction. Nat Med. 1997;3:894–899. doi: 10.1038/nm0897-894. [DOI] [PubMed] [Google Scholar]
- 79.Moss NC, Stansfield WE, Willis MS, Tang RH, Selzman CH. IKKbeta inhibition attenuates myocardial injury and dysfunction following acute ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2007;293:H2248–H2253. doi: 10.1152/ajpheart.00776.2007. [DOI] [PubMed] [Google Scholar]
- 80.Brown M, McGuinness M, Wright T, Ren X, Wang Y, Boivin GP, Hahn H, Feldman AM, Jones WK. Cardiac-specific blockade of NF-kappaB in cardiac pathophysiology: differences between acute and chronic stimuli in vivo. Am J Physiol Heart Circ Physiol. 2005;289:H466–H476. doi: 10.1152/ajpheart.00170.2004. [DOI] [PubMed] [Google Scholar]
- 81.Frantz S, Hu K, Bayer B, Gerondakis S, Strotmann J, Adamek A, Ertl G, Bauersachs J. Absence of NF-kappaB subunit p50 improves heart failure after myocardial infarction. Faseb J. 2006;20:1918–1920. doi: 10.1096/fj.05-5133fje. [DOI] [PubMed] [Google Scholar]
- 82.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 83.Misra A, Haudek SB, Knuefermann P, Vallejo JG, Chen ZJ, Michael LH, Sivasubramanian N, Olson EN, Entman ML, Mann DL. Nuclear factor-kappaB protects the adult cardiac myocyte against ischemia-induced apoptosis in a murine model of acute myocardial infarction. Circulation. 2003;108:3075–3078. doi: 10.1161/01.CIR.0000108929.93074.0B. [DOI] [PubMed] [Google Scholar]
- 84.Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA. Possible new role for NF-kappaB in the resolution of inflammation. Nat Med. 2001;7:1291–1297. doi: 10.1038/nm1201-1291. [DOI] [PubMed] [Google Scholar]
- 85.Bond M, Baker AH, Newby AC. Nuclear factor kappaB activity is essential for matrix metalloproteinase-1 and -3 upregulation in rabbit dermal fibroblasts. Biochem Biophys Res Commun. 1999;264:561–567. doi: 10.1006/bbrc.1999.1551. [DOI] [PubMed] [Google Scholar]
- 86.Clark-Lewis I, Kim KS, Rajarathnam K, Gong JH, Dewald B, Moser B, Baggiolini M, Sykes BD. Structure-activity relationships of chemokines. J Leukoc Biol. 1995;57:703–711. doi: 10.1002/jlb.57.5.703. [DOI] [PubMed] [Google Scholar]
- 87.Luster AD. Chemokines--chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 88.Gerard C, Rollins BJ. Chemokines and disease. Nat Immunol. 2001;2:108–115. doi: 10.1038/84209. [DOI] [PubMed] [Google Scholar]
- 89.Moser B, Loetscher P. Lymphocyte traffic control by chemokines. Nat Immunol. 2001;2:123–128. doi: 10.1038/84219. [DOI] [PubMed] [Google Scholar]
- 90.Strieter RM, Polverini PJ, Arenberg DA, Walz A, Opdenakker G, Van Damme J, Kunkel SL. Role of C-X-C chemokines as regulators of angiogenesis in lung cancer. J Leukoc Biol. 1995;57:752–762. doi: 10.1002/jlb.57.5.752. [DOI] [PubMed] [Google Scholar]
- 91.Zlotnik A, Morales J, Hedrick JA. Recent advances in chemokines and chemokine receptors. Crit Rev Immunol. 1999;19:1–47. [PubMed] [Google Scholar]
- 92.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- 93.Frangogiannis NG. Chemokines in ischemia and reperfusion. Thromb Haemost. 2007;97:738–747. [PubMed] [Google Scholar]
- 94.Frangogiannis NG. Chemokines in the ischemic myocardium: from inflammation to fibrosis. Inflamm Res. 2004;53:585–595. doi: 10.1007/s00011-004-1298-5. [DOI] [PubMed] [Google Scholar]
- 95.Frangogiannis NG. The role of the chemokines in myocardial ischemia and reperfusion. Curr Vasc Pharmacol. 2004;2:163–174. doi: 10.2174/1570161043476375. [DOI] [PubMed] [Google Scholar]
- 96.Frangogiannis NG, Mendoza LH, Lewallen M, Michael LH, Smith CW, Entman ML. Induction and suppression of interferon-inducible protein 10 in reperfused myocardial infarcts may regulate angiogenesis. FASEB J. 2001;15:1428–1430. doi: 10.1096/fj.00-0745fje. [DOI] [PubMed] [Google Scholar]
- 97.Clark-Lewis I, Schumacher C, Baggiolini M, Moser B. Structure-activity relationships of interleukin-8 determined using chemically synthesized analogs. Critical role of NH2-terminal residues and evidence for uncoupling of neutrophil chemotaxis, exocytosis, and receptor binding activities. J Biol Chem. 1991;266:23128–23134. [PubMed] [Google Scholar]
- 98.Strieter RM, Polverini PJ, Kunkel SL, Arenberg DA, Burdick MD, Kasper J, Dzuiba J, Van Damme J, Walz A, Marriott D, et al. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem. 1995;270:27348–27357. doi: 10.1074/jbc.270.45.27348. [DOI] [PubMed] [Google Scholar]
- 99.Mukaida N. Interleukin-8: an expanding universe beyond neutrophil chemotaxis and activation. Int J Hematol. 2000;72:391–398. [PubMed] [Google Scholar]
- 100.Zeilhofer HU, Schorr W. Role of interleukin-8 in neutrophil signaling. Curr Opin Hematol. 2000;7:178–182. doi: 10.1097/00062752-200005000-00009. [DOI] [PubMed] [Google Scholar]
- 101.Koch AE, Polverini PJ, Kunkel SL, Harlow LA, DiPietro LA, Elner VM, Elner SG, Strieter RM. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science. 1992;258:1798–1801. doi: 10.1126/science.1281554. [DOI] [PubMed] [Google Scholar]
- 102.Kukielka GL, Smith CW, LaRosa GJ, Manning AM, Mendoza LH, Daly TJ, Hughes BJ, Youker KA, Hawkins HK, Michael LH, et al. Interleukin-8 gene induction in the myocardium after ischemia and reperfusion in vivo. J Clin Invest. 1995;95:89–103. doi: 10.1172/JCI117680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ivey CL, Williams FM, Collins PD, Jose PJ, Williams TJ. Neutrophil chemoattractants generated in two phases during reperfusion of ischemic myocardium in the rabbit. Evidence for a role for C5a and interleukin-8. J Clin Invest. 1995;95:2720–2728. doi: 10.1172/JCI117974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Takami M, Terry V, Petruzzelli L. Signaling pathways involved in IL-8-dependent activation of adhesion through Mac-1. J Immunol. 2002;168:4559–4566. doi: 10.4049/jimmunol.168.9.4559. [DOI] [PubMed] [Google Scholar]
- 105.DiVietro JA, Smith MJ, Smith BR, Petruzzelli L, Larson RS, Lawrence MB. Immobilized IL-8 triggers progressive activation of neutrophils rolling in vitro on P-selectin and intercellular adhesion molecule-1. J Immunol. 2001;167:4017–4025. doi: 10.4049/jimmunol.167.7.4017. [DOI] [PubMed] [Google Scholar]
- 106.Boyle EM, Jr, Kovacich JC, Hebert CA, Canty TG, Jr, Chi E, Morgan EN, Pohlman TH, Verrier ED. Inhibition of interleukin-8 blocks myocardial ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 1998;116:114–121. doi: 10.1016/S0022-5223(98)70249-1. [DOI] [PubMed] [Google Scholar]
- 107.Chandrasekar B, Smith JB, Freeman GL. Ischemia-reperfusion of rat myocardium activates nuclear factor-KappaB and induces neutrophil infiltration via lipopolysaccharide-induced CXC chemokine. Circulation. 2001;103:2296–2302. doi: 10.1161/01.cir.103.18.2296. [DOI] [PubMed] [Google Scholar]
- 108.Tarzami ST, Miao W, Mani K, Lopez L, Factor SM, Berman JW, Kitsis RN. Opposing effects mediated by the chemokine receptor CXCR2 on myocardial ischemia-reperfusion injury: recruitment of potentially damaging neutrophils and direct myocardial protection. Circulation. 2003;108:2387–2392. doi: 10.1161/01.CIR.0000093192.72099.9A. [DOI] [PubMed] [Google Scholar]
- 109.Khan IA, MacLean JA, Lee FS, Casciotti L, DeHaan E, Schwartzman JD, Luster AD. IP-10 is critical for effector T cell trafficking and host survival in Toxoplasma gondii infection. Immunity. 2000;12:483–494. doi: 10.1016/s1074-7613(00)80200-9. [DOI] [PubMed] [Google Scholar]
- 110.Strieter RM, Polverini PJ, Arenberg DA, Kunkel SL. The role of CXC chemokines as regulators of angiogenesis. Shock. 1995;4:155–160. doi: 10.1097/00024382-199509000-00001. [DOI] [PubMed] [Google Scholar]
- 111.Shiraha H, Glading A, Gupta K, Wells A. IP-10 inhibits epidermal growth factor-induced motility by decreasing epidermal growth factor receptor-mediated calpain activity. J Cell Biol. 1999;146:243–254. doi: 10.1083/jcb.146.1.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tager AM, Kradin RL, LaCamera P, Bercury SD, Campanella GS, Leary CP, Polosukhin V, Zhao LH, Sakamoto H, Blackwell TS, et al. Inhibition of pulmonary fibrosis by the chemokine IP-10/CXCL10. Am J Respir Cell Mol Biol. 2004;31:395–404. doi: 10.1165/rcmb.2004-0175OC. [DOI] [PubMed] [Google Scholar]
- 113.Aiuti A, Webb IJ, Bleul C, Springer T, Gutierrez-Ramos JC. The chemokine SDF-1 is a chemoattractant for human CD34+ hematopoietic progenitor cells and provides a new mechanism to explain the mobilization of CD34+ progenitors to peripheral blood. J Exp Med. 1997;185:111–120. doi: 10.1084/jem.185.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jo DY, Rafii S, Hamada T, Moore MA. Chemotaxis of primitive hematopoietic cells in response to stromal cell-derived factor-1. J Clin Invest. 2000;105:101–111. doi: 10.1172/JCI7954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pillarisetti K, Gupta SK. Cloning and relative expression analysis of rat stromal cell derived factor-1 (SDF-1)1: SDF-1 alpha mRNA is selectively induced in rat model of myocardial infarction. Inflammation. 2001;25:293–300. doi: 10.1023/a:1012808525370. [DOI] [PubMed] [Google Scholar]
- 116.Askari AT, Unzek S, Popovic ZB, Goldman CK, Forudi F, Kiedrowski M, Rovner A, Ellis SG, Thomas JD, DiCorleto PE, et al. Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet. 2003;362:697–703. doi: 10.1016/S0140-6736(03)14232-8. [DOI] [PubMed] [Google Scholar]
- 117.Togel F, Isaac J, Hu Z, Weiss K, Westenfelder C. Renal SDF-1 signals mobilization and homing of CXCR4-positive cells to the kidney after ischemic injury. Kidney Int. 2005;67:1772–1784. doi: 10.1111/j.1523-1755.2005.00275.x. [DOI] [PubMed] [Google Scholar]
- 118.Zhang M, Mal N, Kiedrowski M, Chacko M, Askari AT, Popovic ZB, Koc ON, Penn MS. SDF-1 expression by mesenchymal stem cells results in trophic support of cardiac myocytes after myocardial infarction. Faseb J. 2007;21:3197–3207. doi: 10.1096/fj.06-6558com. [DOI] [PubMed] [Google Scholar]
- 119.Rollins BJ. Chemokines. Blood. 1997;90:909–928. [PubMed] [Google Scholar]
- 120.Rollins BJ. Monocyte chemoattractant protein 1: a potential regulator of monocyte recruitment in inflammatory disease. Mol Med Today. 1996;2:198–204. doi: 10.1016/1357-4310(96)88772-7. [DOI] [PubMed] [Google Scholar]
- 121.Gu L, Tseng SC, Rollins BJ. Monocyte chemoattractant protein-1. Chem Immunol. 1999;72:7–29. doi: 10.1159/000058723. [DOI] [PubMed] [Google Scholar]
- 122.Salcedo R, Ponce ML, Young HA, Wasserman K, Ward JM, Kleinman HK, Oppenheim JJ, Murphy WJ. Human endothelial cells express CCR2 and respond to MCP-1: direct role of MCP-1 in angiogenesis and tumor progression. Blood. 2000;96:34–40. [PubMed] [Google Scholar]
- 123.Gharaee-Kermani M, Denholm EM, Phan SH. Costimulation of fibroblast collagen and transforming growth factor beta1 gene expression by monocyte chemoattractant protein-1 via specific receptors. J Biol Chem. 1996;271:17779–17784. doi: 10.1074/jbc.271.30.17779. [DOI] [PubMed] [Google Scholar]
- 124.Kumar AG, Ballantyne CM, Michael LH, Kukielka GL, Youker KA, Lindsey ML, Hawkins HK, Birdsall HH, MacKay CR, LaRosa GJ, et al. Induction of monocyte chemoattractant protein-1 in the small veins of the ischemic and reperfused canine myocardium. Circulation. 1997;95:693–700. doi: 10.1161/01.cir.95.3.693. [DOI] [PubMed] [Google Scholar]
- 125.Ono K, Matsumori A, Furukawa Y, Igata H, Shioi T, Matsushima K, Sasayama S. Prevention of myocardial reperfusion injury in rats by an antibody against monocyte chemotactic and activating factor/monocyte chemoattractant protein-1. Lab Invest. 1999;79:195–203. [PubMed] [Google Scholar]
- 126.Kakio T, Matsumori A, Ono K, Ito H, Matsushima K, Sasayama S. Roles and relationship of macrophages and monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 in the ischemic and reperfused rat heart. Lab Invest. 2000;80:1127–1136. doi: 10.1038/labinvest.3780119. [DOI] [PubMed] [Google Scholar]
- 127.Tarzami ST, Cheng R, Miao W, Kitsis RN, Berman JW. Chemokine expression in myocardial ischemia: MIP-2 dependent MCP-1 expression protects cardiomyocytes from cell death. J Mol Cell Cardiol. 2002;34:209–221. doi: 10.1006/jmcc.2001.1503. [DOI] [PubMed] [Google Scholar]
- 128.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, Michael LH, Rollins BJ, Entman ML, Frangogiannis NG. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–889. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 129.Xia Y, Frangogiannis NG. MCP-1/CCL2 as a therapeutic target in myocardial infarction and ischemic cardiomyopathy. Inflamm Allergy Drug Targets. 2007;6:101–107. doi: 10.2174/187152807780832265. [DOI] [PubMed] [Google Scholar]
- 130.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 131.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Weber C, Belge KU, von Hundelshausen P, Draude G, Steppich B, Mack M, Frankenberger M, Weber KS, Ziegler-Heitbrock HW. Differential chemokine receptor expression and function in human monocyte subpopulations. J Leukoc Biol. 2000;67:699–704. doi: 10.1002/jlb.67.5.699. [DOI] [PubMed] [Google Scholar]
- 133.Gavrilin MA, Deucher MF, Boeckman F, Kolattukudy PE. Monocyte chemotactic protein 1 upregulates IL-1beta expression in human monocytes. Biochem Biophys Res Commun. 2000;277:37–42. doi: 10.1006/bbrc.2000.3619. [DOI] [PubMed] [Google Scholar]
- 134.Jiang Y, Beller DI, Frendl G, Graves DT. Monocyte chemoattractant protein-1 regulates adhesion molecule expression and cytokine production in human monocytes. J Immunol. 1992;148:2423–2428. [PubMed] [Google Scholar]
- 135.Tabata T, Mine S, Kawahara C, Okada Y, Tanaka Y. Monocyte chemoattractant protein-1 induces scavenger receptor expression and monocyte differentiation into foam cells. Biochem Biophys Res Commun. 2003;305:380–385. doi: 10.1016/s0006-291x(03)00771-x. [DOI] [PubMed] [Google Scholar]
- 136.Kaikita K, Hayasaki T, Okuma T, Kuziel WA, Ogawa H, Takeya M. Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am J Pathol. 2004;165:439–447. doi: 10.1016/S0002-9440(10)63309-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schenk S, Mal N, Finan A, Zhang M, Kiedrowski M, Popovic Z, McCarthy PM, Penn MS. Monocyte chemotactic protein-3 is a myocardial mesenchymal stem cell homing factor. Stem Cells. 2007;25:245–251. doi: 10.1634/stemcells.2006-0293. [DOI] [PubMed] [Google Scholar]
- 138.Uguccioni M, D'Apuzzo M, Loetscher M, Dewald B, Baggiolini M. Actions of the chemotactic cytokines MCP-1, MCP-2, MCP-3, RANTES, MIP-1 alpha and MIP-1 beta on human monocytes. Eur J Immunol. 1995;25:64–68. doi: 10.1002/eji.1830250113. [DOI] [PubMed] [Google Scholar]
- 139.Parissis JT, Adamopoulos S, Venetsanou KF, Mentzikof DG, Karas SM, Kremastinos DT. Serum profiles of C-C chemokines in acute myocardial infarction: possible implication in postinfarction left ventricular remodeling. J Interferon Cytokine Res. 2002;22:223–229. doi: 10.1089/107999002753536194. [DOI] [PubMed] [Google Scholar]
- 140.Liehn EA, Merx MW, Postea O, Becher S, Djalali-Talab Y, Shagdarsuren E, Kelm M, Zernecke A, Weber C. Ccr1 deficiency reduces inflammatory remodelling and preserves left ventricular function after myocardial infarction. J Cell Mol Med. 2008;12:496–506. doi: 10.1111/j.1582-4934.2007.00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gao JL, Wynn TA, Chang Y, Lee EJ, Broxmeyer HE, Cooper S, Tiffany HL, Westphal H, Kwon-Chung J, Murphy PM. Impaired host defense, hematopoiesis, granulomatous inflammation and type 1-type 2 cytokine balance in mice lacking CC chemokine receptor 1. J Exp Med. 1997;185:1959–1968. doi: 10.1084/jem.185.11.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Herskowitz A, Choi S, Ansari AA, Wesselingh S. Cytokine mRNA expression in postischemic/reperfused myocardium. Am J Pathol. 1995;146:419–428. [PMC free article] [PubMed] [Google Scholar]
- 143.Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. 2004;94:1543–1553. doi: 10.1161/01.RES.0000130526.20854.fa. [DOI] [PubMed] [Google Scholar]
- 144.Frangogiannis NG, Youker KA, Rossen RD, Gwechenberger M, Lindsey MH, Mendoza LH, Michael LH, Ballantyne CM, Smith CW, Entman ML. Cytokines and the microcirculation in ischemia and reperfusion. J Mol Cell Cardiol. 1998;30:2567–2576. doi: 10.1006/jmcc.1998.0829. [DOI] [PubMed] [Google Scholar]
- 145.Frangogiannis NG, Lindsey ML, Michael LH, Youker KA, Bressler RB, Mendoza LH, Spengler RN, Smith CW, Entman ML. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation. 1998;98:699–710. doi: 10.1161/01.cir.98.7.699. [DOI] [PubMed] [Google Scholar]
- 146.Kishimoto T, Akira S, Narazaki M, Taga T. Interleukin-6 family of cytokines and gp130. Blood. 1995;86:1243–1254. [PubMed] [Google Scholar]
- 147.Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, Simmons RL. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science. 1992;257:387–389. doi: 10.1126/science.1631560. [DOI] [PubMed] [Google Scholar]
- 148.Yokoyama T, Vaca L, Rossen RD, Durante W, Hazarika P, Mann DL. Cellular basis for the negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian heart. J Clin Invest. 1993;92:2303–2312. doi: 10.1172/JCI116834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Engel D, Peshock R, Armstong RC, Sivasubramanian N, Mann DL. Cardiac myocyte apoptosis provokes adverse cardiac remodeling in transgenic mice with targeted TNF overexpression. Am J Physiol Heart Circ Physiol. 2004;287:H1303–H1311. doi: 10.1152/ajpheart.00053.2004. [DOI] [PubMed] [Google Scholar]
- 150.Siwik DA, Chang DL, Colucci WS. Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res. 2000;86:1259–1265. doi: 10.1161/01.res.86.12.1259. [DOI] [PubMed] [Google Scholar]
- 151.Sun M, Dawood F, Wen WH, Chen M, Dixon I, Kirshenbaum LA, Liu PP. Excessive tumor necrosis factor activation after infarction contributes to susceptibility of myocardial rupture and left ventricular dysfunction. Circulation. 2004;110:3221–3228. doi: 10.1161/01.CIR.0000147233.10318.23. [DOI] [PubMed] [Google Scholar]
- 152.Maekawa N, Wada H, Kanda T, Niwa T, Yamada Y, Saito K, Fujiwara H, Sekikawa K, Seishima M. Improved myocardial ischemia/reperfusion injury in mice lacking tumor necrosis factor-alpha. J Am Coll Cardiol. 2002;39:1229–1235. doi: 10.1016/s0735-1097(02)01738-2. [DOI] [PubMed] [Google Scholar]
- 153.Berthonneche C, Sulpice T, Boucher F, Gouraud L, de Leiris J, O'Connor SE, Herbert JM, Janiak P. New insights into the pathological role of TNF-alpha in early cardiac dysfunction and subsequent heart failure after infarction in rats. Am J Physiol Heart Circ Physiol. 2004;287:H340–H350. doi: 10.1152/ajpheart.01210.2003. [DOI] [PubMed] [Google Scholar]
- 154.Sugano M, Tsuchida K, Hata T, Makino N. In vivo transfer of soluble TNF-alpha receptor 1 gene improves cardiac function and reduces infarct size after myocardial infarction in rats. Faseb J. 2004;18:911–913. doi: 10.1096/fj.03-1148fje. [DOI] [PubMed] [Google Scholar]
- 155.Monden Y, Kubota T, Tsutsumi T, Inoue T, Kawano S, Kawamura N, Ide T, Egashira K, Tsutsui H, Sunagawa K. Soluble TNF receptors prevent apoptosis in infiltrating cells and promote ventricular rupture and remodeling after myocardial infarction. Cardiovasc Res. 2007;73:794–805. doi: 10.1016/j.cardiores.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 156.Kurrelmeyer KM, Michael LH, Baumgarten G, Taffet GE, Peschon JJ, Sivasubramanian N, Entman ML, Mann DL. Endogenous tumor necrosis factor protects the adult cardiac myocyte against ischemic-induced apoptosis in a murine model of acute myocardial infarction. Proc Natl Acad Sci U S A. 2000;97:5456–5461. doi: 10.1073/pnas.070036297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Monden Y, Kubota T, Inoue T, Tsutsumi T, Kawano S, Ide T, Tsutsui H, Sunagawa K. Tumor necrosis factor-alpha is toxic via receptor 1 and protective via receptor 2 in a murine model of myocardial infarction. Am J Physiol Heart Circ Physiol. 2007;293:H743–H753. doi: 10.1152/ajpheart.00166.2007. [DOI] [PubMed] [Google Scholar]
- 158.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- 159.Guillen I, Blanes M, Gomez-Lechon MJ, Castell JV. Cytokine signaling during myocardial infarction: sequential appearance of IL-1 beta and IL-6. Am J Physiol. 1995;269:R229–R235. doi: 10.1152/ajpregu.1995.269.2.R229. [DOI] [PubMed] [Google Scholar]
- 160.Suzuki K, Murtuza B, Smolenski RT, Sammut IA, Suzuki N, Kaneda Y, Yacoub MH. Overexpression of interleukin-1 receptor antagonist provides cardioprotection against ischemia-reperfusion injury associated with reduction in apoptosis. Circulation. 2001;104:I308–I303. doi: 10.1161/hc37t1.094871. [DOI] [PubMed] [Google Scholar]
- 161.Hwang MW, Matsumori A, Furukawa Y, Ono K, Okada M, Iwasaki A, Hara M, Miyamoto T, Touma M, Sasayama S. Neutralization of interleukin-1beta in the acute phase of myocardial infarction promotes the progression of left ventricular remodeling. J Am Coll Cardiol. 2001;38:1546–1553. doi: 10.1016/s0735-1097(01)01591-1. [DOI] [PubMed] [Google Scholar]
- 162.Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Na L, Reddy A, Frangogiannis NG. Interleukin-1 Receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. 2008 doi: 10.2353/ajpath.2008.070974. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Dinarello CA. Interleukin-18, a proinflammatory cytokine. Eur Cytokine Netw. 2000;11:483–486. [PubMed] [Google Scholar]
- 164.Woldbaek PR, Tonnessen T, Henriksen UL, Florholmen G, Lunde PK, Lyberg T, Christensen G. Increased cardiac IL-18 mRNA, pro-IL-18 and plasma IL-18 after myocardial infarction in the mouse; a potential role in cardiac dysfunction. Cardiovasc Res. 2003;59:122–131. doi: 10.1016/s0008-6363(03)00339-0. [DOI] [PubMed] [Google Scholar]
- 165.Aoyama T, Takimoto Y, Pennica D, Inoue R, Shinoda E, Hattori R, Yui Y, Sasayama S. Augmented expression of cardiotrophin-1 and its receptor component, gp130, in both left and right ventricles after myocardial infarction in the rat. J Mol Cell Cardiol. 2000;32:1821–1830. doi: 10.1006/jmcc.2000.1218. [DOI] [PubMed] [Google Scholar]
- 166.Gwechenberger M, Mendoza LH, Youker KA, Frangogiannis NG, Smith CW, Michael LH, Entman ML. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation. 1999;99:546–551. doi: 10.1161/01.cir.99.4.546. [DOI] [PubMed] [Google Scholar]
- 167.Kukielka GL, Smith CW, Manning AM, Youker KA, Michael LH, Entman ML. Induction of interleukin-6 synthesis in the myocardium. Potential role in postreperfusion inflammatory injury. Circulation. 1995;92:1866–1875. doi: 10.1161/01.cir.92.7.1866. [DOI] [PubMed] [Google Scholar]
- 168.Freed DH, Moon MC, Borowiec AM, Jones SC, Zahradka P, Dixon M. Cardiotrophin-1: expression in experimental myocardial infarction and potential role in post-MI wound healing. Mol Cell Biochem. 2003;254:247–256. doi: 10.1023/a:1027332504861. [DOI] [PubMed] [Google Scholar]
- 169.Gwechenberger M, Moertl D, Pacher R, Huelsmann M. Oncostatin-M in myocardial ischemia/reperfusion injury may regulate tissue repair. Croat Med J. 2004;45:149–157. [PubMed] [Google Scholar]
- 170.Wollert KC, Drexler H. The role of interleukin-6 in the failing heart. Heart Fail Rev. 2001;6:95–103. doi: 10.1023/a:1011401825680. [DOI] [PubMed] [Google Scholar]
- 171.Liao Z, Brar BK, Cai Q, Stephanou A, O'Leary RM, Pennica D, Yellon DM, Latchman DS. Cardiotrophin-1 (CT-1) can protect the adult heart from injury when added both prior to ischaemia and at reperfusion. Cardiovasc Res. 2002;53:902–910. doi: 10.1016/s0008-6363(01)00531-4. [DOI] [PubMed] [Google Scholar]
- 172.Gritman K, Van Winkle DM, Lorentz CU, Pennica D, Habecker BA. The lack of cardiotrophin-1 alters expression of interleukin-6 and leukemia inhibitory factor mRNA but does not impair cardiac injury response. Cytokine. 2006;36:9–16. doi: 10.1016/j.cyto.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Freed DH, Cunnington RH, Dangerfield AL, Sutton JS, Dixon IM. Emerging evidence for the role of cardiotrophin-1 in cardiac repair in the infarcted heart. Cardiovasc Res. 2005;65:782–792. doi: 10.1016/j.cardiores.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 174.Zou Y, Takano H, Mizukami M, Akazawa H, Qin Y, Toko H, Sakamoto M, Minamino T, Nagai T, Komuro I. Leukemia inhibitory factor enhances survival of cardiomyocytes and induces regeneration of myocardium after myocardial infarction. Circulation. 2003;108:748–753. doi: 10.1161/01.CIR.0000081773.76337.44. [DOI] [PubMed] [Google Scholar]
- 175.Gallucci RM, Simeonova PP, Matheson JM, Kommineni C, Guriel JL, Sugawara T, Luster MI. Impaired cutaneous wound healing in interleukin-6-deficient and immunosuppressed mice. Faseb J. 2000;14:2525–2531. doi: 10.1096/fj.00-0073com. [DOI] [PubMed] [Google Scholar]
- 176.Fuchs M, Hilfiker A, Kaminski K, Hilfiker-Kleiner D, Guener Z, Klein G, Podewski E, Schieffer B, Rose-John S, Drexler H. Role of interleukin-6 for LV remodeling and survival after experimental myocardial infarction. Faseb J. 2003;17:2118–2120. doi: 10.1096/fj.03-0331fje. [DOI] [PubMed] [Google Scholar]
- 177.Martin P, Leibovich SJ. Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol. 2005;15:599–607. doi: 10.1016/j.tcb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 178.von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res. 2007;100:27–40. doi: 10.1161/01.RES.0000252802.25497.b7. [DOI] [PubMed] [Google Scholar]
- 179.Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. 2005;115:3378–3384. doi: 10.1172/JCI27196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Del Conde I, Cruz MA, Zhang H, Lopez JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med. 2005;201:871–879. doi: 10.1084/jem.20041497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Szpaderska AM, Egozi EI, Gamelli RL, DiPietro LA. The effect of thrombocytopenia on dermal wound healing. J Invest Dermatol. 2003;120:1130–1137. doi: 10.1046/j.1523-1747.2003.12253.x. [DOI] [PubMed] [Google Scholar]
- 182.Hafezi-Moghadam A, Thomas KL, Prorock AJ, Huo Y, Ley K. L-selectin shedding regulates leukocyte recruitment. J Exp Med. 2001;193:863–872. doi: 10.1084/jem.193.7.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ma XL, Weyrich AS, Lefer DJ, Buerke M, Albertine KH, Kishimoto TK, Lefer AM. Monoclonal antibody to L-selectin attenuates neutrophil accumulation and protects ischemic reperfused cat myocardium. Circulation. 1993;88:649–658. doi: 10.1161/01.cir.88.2.649. [DOI] [PubMed] [Google Scholar]
- 184.Weyrich AS, Ma XY, Lefer DJ, Albertine KH, Lefer AM. In vivo neutralization of P-selectin protects feline heart and endothelium in myocardial ischemia and reperfusion injury. J Clin Invest. 1993;91:2620–2629. doi: 10.1172/JCI116501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Palazzo AJ, Jones SP, Anderson DC, Granger DN, Lefer DJ. Coronary endothelial P-selectin in pathogenesis of myocardial ischemia-reperfusion injury. Am J Physiol. 1998;275:H1865–H1872. doi: 10.1152/ajpheart.1998.275.5.H1865. [DOI] [PubMed] [Google Scholar]
- 186.Briaud SA, Ding ZM, Michael LH, Entman ML, Daniel S, Ballantyne CM. Leukocyte trafficking and myocardial reperfusion injury in ICAM-1/P-selectin-knockout mice. Am J Physiol Heart Circ Physiol. 2001;280:H60–H67. doi: 10.1152/ajpheart.2001.280.1.H60. [DOI] [PubMed] [Google Scholar]
- 187.Jones SP, Girod WG, Granger DN, Palazzo AJ, Lefer DJ. Reperfusion injury is not affected by blockade of P-selectin in the diabetic mouse heart. Am J Physiol. 1999;277:H763–H769. doi: 10.1152/ajpheart.1999.277.2.H763. [DOI] [PubMed] [Google Scholar]
- 188.Birnbaum Y, Patterson M, Kloner RA. The effect of CY1503, a sialyl Lewisx analog blocker of the selectin adhesion molecules, on infarct size and "no-reflow" in the rabbit model of acute myocardial infarction/reperfusion. J Mol Cell Cardiol. 1997;29:2013–2025. doi: 10.1006/jmcc.1997.0393. [DOI] [PubMed] [Google Scholar]
- 189.Hayward R, Campbell B, Shin YK, Scalia R, Lefer AM. Recombinant soluble P-selectin glycoprotein ligand-1 protects against myocardial ischemic reperfusion injury in cats. Cardiovasc Res. 1999;41:65–76. doi: 10.1016/s0008-6363(98)00266-1. [DOI] [PubMed] [Google Scholar]
- 190.Luscinskas FW, Lawler J. Integrins as dynamic regulators of vascular function. Faseb J. 1994;8:929–938. doi: 10.1096/fasebj.8.12.7522194. [DOI] [PubMed] [Google Scholar]
- 191.Smith CW. Introduction: functional polarity of motile neutrophils. Blood. 2000;95:2459–2461. [PubMed] [Google Scholar]
- 192.Smith CW, Marlin SD, Rothlein R, Toman C, Anderson DC. Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J Clin Invest. 1989;83:2008–2017. doi: 10.1172/JCI114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Furie MB, Tancinco MC, Smith CW. Monoclonal antibodies to leukocyte integrins CD11a/CD18 and CD11b/CD18 or intercellular adhesion molecule-1 inhibit chemoattractant-stimulated neutrophil transendothelial migration in vitro. Blood. 1991;78:2089–2097. [PubMed] [Google Scholar]
- 194.Ding ZM, Babensee JE, Simon SI, Lu H, Perrard JL, Bullard DC, Dai XY, Bromley SK, Dustin ML, Entman ML, et al. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol. 1999;163:5029–5038. [PubMed] [Google Scholar]
- 195.Lu H, Smith CW, Perrard J, Bullard D, Tang L, Shappell SB, Entman ML, Beaudet AL, Ballantyne CM. LFA-1 is sufficient in mediating neutrophil emigration in Mac-1-deficient mice. J Clin Invest. 1997;99:1340–1350. doi: 10.1172/JCI119293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lefer DJ, Shandelya SM, Serrano CV, Jr, Becker LC, Kuppusamy P, Zweier JL. Cardioprotective actions of a monoclonal antibody against CD-18 in myocardial ischemia-reperfusion injury. Circulation. 1993;88:1779–1787. doi: 10.1161/01.cir.88.4.1779. [DOI] [PubMed] [Google Scholar]
- 197.Ma XL, Tsao PS, Lefer AM. Antibody to CD-18 exerts endothelial and cardiac protective effects in myocardial ischemia and reperfusion. J Clin Invest. 1991;88:1237–1243. doi: 10.1172/JCI115427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Arai M, Lefer DJ, So T, DiPaula A, Aversano T, Becker LC. An anti-CD18 antibody limits infarct size and preserves left ventricular function in dogs with ischemia and 48-hour reperfusion. J Am Coll Cardiol. 1996;27:1278–1285. doi: 10.1016/0735-1097(95)00578-1. [DOI] [PubMed] [Google Scholar]
- 199.Aversano T, Zhou W, Nedelman M, Nakada M, Weisman H. A chimeric IgG4 monoclonal antibody directed against CD18 reduces infarct size in a primate model of myocardial ischemia and reperfusion. J Am Coll Cardiol. 1995;25:781–788. doi: 10.1016/0735-1097(94)00443-T. [DOI] [PubMed] [Google Scholar]
- 200.Dove A. CD18 trials disappoint again. Nat Biotechnol. 2000;18:817–818. doi: 10.1038/78412. [DOI] [PubMed] [Google Scholar]
- 201.Faxon DP, Gibbons RJ, Chronos NA, Gurbel PA, Sheehan F. The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: the results of the HALT-MI study. J Am Coll Cardiol. 2002;40:1199–1204. doi: 10.1016/s0735-1097(02)02136-8. [DOI] [PubMed] [Google Scholar]
- 202.Perez RG, Arai M, Richardson C, DiPaula A, Siu C, Matsumoto N, Hildreth JE, Mariscalco MM, Smith CW, Becker LC. Factors modifying protective effect of anti-CD18 antibodies on myocardial reperfusion injury in dogs. Am J Physiol. 1996;270:H53–H64. doi: 10.1152/ajpheart.1996.270.1.H53. [DOI] [PubMed] [Google Scholar]
- 203.Palazzo AJ, Jones SP, Girod WG, Anderson DC, Granger DN, Lefer DJ. Myocardial ischemia-reperfusion injury in CD18- and ICAM-1-deficient mice. Am J Physiol. 1998;275:H2300–H2307. doi: 10.1152/ajpheart.1998.275.6.H2300. [DOI] [PubMed] [Google Scholar]
- 204.Bowden RA, Ding ZM, Donnachie EM, Petersen TK, Michael LH, Ballantyne CM, Burns AR. Role of alpha4 integrin and VCAM-1 in CD18-independent neutrophil migration across mouse cardiac endothelium. Circ Res. 2002;90:562–569. doi: 10.1161/01.res.0000013835.53611.97. [DOI] [PubMed] [Google Scholar]
- 205.Chavakis T, Preissner KT, Santoso S. Leukocyte trans-endothelial migration: JAMs add new pieces to the puzzle. Thromb Haemost. 2003;89:13–17. [PubMed] [Google Scholar]
- 206.Corada M, Chimenti S, Cera MR, Vinci M, Salio M, Fiordaliso F, De Angelis N, Villa A, Bossi M, Staszewsky LI, et al. Junctional adhesion molecule-A-deficient polymorphonuclear cells show reduced diapedesis in peritonitis and heart ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2005;102:10634–10639. doi: 10.1073/pnas.0500147102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Jaeschke H, Smith CW. Mechanisms of neutrophil-induced parenchymal cell injury. J Leukoc Biol. 1997;61:647–653. doi: 10.1002/jlb.61.6.647. [DOI] [PubMed] [Google Scholar]
- 208.Albelda SM, Smith CW, Ward PA. Adhesion molecules and inflammatory injury. Faseb J. 1994;8:504–512. [PubMed] [Google Scholar]
- 209.Vinten-Johansen J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc Res. 2004;61:481–497. doi: 10.1016/j.cardiores.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 210.Romson JL, Hook BG, Kunkel SL, Abrams GD, Schork MA, Lucchesi BR. Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation. 1983;67:1016–1023. doi: 10.1161/01.cir.67.5.1016. [DOI] [PubMed] [Google Scholar]
- 211.Litt MR, Jeremy RW, Weisman HF, Winkelstein JA, Becker LC. Neutrophil depletion limited to reperfusion reduces myocardial infarct size after 90 minutes of ischemia. Evidence for neutrophil-mediated reperfusion injury. Circulation. 1989;80:1816–1827. doi: 10.1161/01.cir.80.6.1816. [DOI] [PubMed] [Google Scholar]
- 212.Jordan JE, Zhao ZQ, Vinten-Johansen J. The role of neutrophils in myocardial ischemia-reperfusion injury. Cardiovasc Res. 1999;43:860–878. doi: 10.1016/s0008-6363(99)00187-x. [DOI] [PubMed] [Google Scholar]
- 213.Metzler B, Mair J, Lercher A, Schaber C, Hintringer F, Pachinger O, Xu Q. Mouse model of myocardial remodelling after ischemia: role of intercellular adhesion molecule-1. Cardiovasc Res. 2001;49:399–407. doi: 10.1016/s0008-6363(00)00261-3. [DOI] [PubMed] [Google Scholar]
- 214.Simpson DM, Ross R. The neutrophilic leukocyte in wound repair a study with antineutrophil serum. J Clin Invest. 1972;51:2009–2023. doi: 10.1172/JCI107007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Frangogiannis NG, Mendoza LH, Ren G, Akrivakis S, Jackson PL, Michael LH, Smith CW, Entman ML. MCSF expression is induced in healing myocardial infarcts and may regulate monocyte and endothelial cell phenotype. Am J Physiol Heart Circ Physiol. 2003;285:H483–H492. doi: 10.1152/ajpheart.01016.2002. [DOI] [PubMed] [Google Scholar]
- 216.Lagasse E, Weissman IL. Enforced expression of Bcl-2 in monocytes rescues macrophages and partially reverses osteopetrosis in op/op mice. Cell. 1997;89:1021–1031. doi: 10.1016/s0092-8674(00)80290-1. [DOI] [PubMed] [Google Scholar]
- 217.Frangogiannis NG, Burns AR, Michael LH, Entman ML. Histochemical and morphological characteristics of canine cardiac mast cells. Histochem J. 1999;31:221–229. doi: 10.1023/a:1003541332070. [DOI] [PubMed] [Google Scholar]
- 218.Ito BR, Engler RL, del Balzo U. Role of cardiac mast cells in complement C5a-induced myocardial ischemia. Am J Physiol. 1993;264:H1346–H1354. doi: 10.1152/ajpheart.1993.264.5.H1346. [DOI] [PubMed] [Google Scholar]
- 219.Frangogiannis NG, Perrard JL, Mendoza LH, Burns AR, Lindsey ML, Ballantyne CM, Michael LH, Smith CW, Entman ML. Stem cell factor induction is associated with mast cell accumulation after canine myocardial ischemia and reperfusion. Circulation. 1998;98:687–698. doi: 10.1161/01.cir.98.7.687. [DOI] [PubMed] [Google Scholar]
- 220.Orlic D, Kajstura J, Chimenti S, Limana F, Jakoniuk I, Quaini F, Nadal-Ginard B, Bodine v, Leri A, Anversa P. Mobilized bone marrow cells repair the infarcted heart, improving function and survival. Proc Natl Acad Sci U S A. 2001;98:10344–10349. doi: 10.1073/pnas.181177898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, Pickel J, McKay R, Nadal-Ginard B, Bodine DM, et al. Bone marrow cells regenerate infarcted myocardium. Nature. 2001;410:701–705. doi: 10.1038/35070587. [DOI] [PubMed] [Google Scholar]
- 222.Asako H, Kurose I, Wolf R, DeFrees S, Zheng ZL, Phillips ML, Paulson JC, Granger DN. Role of H1 receptors and P-selectin in histamine-induced leukocyte rolling and adhesion in postcapillary venules. J Clin Invest. 1994;93:1508–1515. doi: 10.1172/JCI117129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.He S, Peng Q, Walls AF. Potent induction of a neutrophil and eosinophil-rich infiltrate in vivo by human mast cell tryptase: selective enhancement of eosinophil recruitment by histamine. J Immunol. 1997;159:6216–6225. [PubMed] [Google Scholar]
- 224.Compton SJ, Cairns JA, Holgate ST, Walls AF. The role of mast cell tryptase in regulating endothelial cell proliferation, cytokine release, and adhesion molecule expression: tryptase induces expression of mRNA for IL-1 beta and IL-8 and stimulates the selective release of IL-8 from human umbilical vein endothelial cells. J Immunol. 1998;161:1939–1946. [PubMed] [Google Scholar]
- 225.Somasundaram P, Ren G, Nagar H, Kraemer D, Mendoza L, Michael LH, Caughey GH, Entman ML, Frangogiannis NG. Mast cell tryptase may modulate endothelial cell phenotype in healing myocardial infarcts. J Pathol. 2005;205:102–111. doi: 10.1002/path.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Hatamochi A, Fujiwara K, Ueki H. Effects of histamine on collagen synthesis by cultured fibroblasts derived from guinea pig skin. Arch Dermatol Res. 1985;277:60–64. doi: 10.1007/BF00406482. [DOI] [PubMed] [Google Scholar]
- 227.Ruoss SJ, Hartmann T, Caughey GH. Mast cell tryptase is a mitogen for cultured fibroblasts. J Clin Invest. 1991;88:493–499. doi: 10.1172/JCI115330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Gruber BL, Kew RR, Jelaska A, Marchese MJ, Garlick J, Ren S, Schwartz LB, Korn JH. Human mast cells activate fibroblasts: tryptase is a fibrogenic factor stimulating collagen messenger ribonucleic acid synthesis and fibroblast chemotaxis. J Immunol. 1997;158:2310–2317. [PubMed] [Google Scholar]
- 229.Pennington DW, Lopez AR, Thomas PS, Peck C, Gold WM. Dog mastocytoma cells produce transforming growth factor beta 1. J Clin Invest. 1992;90:35–41. doi: 10.1172/JCI115853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Qu Z, Liebler JM, Powers MR, Galey T, Ahmadi P, Huang XN, Ansel JC, Butterfield JH, Planck SR, Rosenbaum JT. Mast cells are a major source of basic fibroblast growth factor in chronic inflammation and cutaneous hemangioma. Am J Pathol. 1995;147:564–573. [PMC free article] [PubMed] [Google Scholar]
- 231.Boesiger J, Tsai M, Maurer M, Yamaguchi M, Brown LF, Claffey KP, Dvorak HF, Galli SJ. Mast cells can secrete vascular permeability factor/ vascular endothelial cell growth factor and exhibit enhanced release after immunoglobulin E-dependent upregulation of fc epsilon receptor I expression. J Exp Med. 1998;188:1135–1145. doi: 10.1084/jem.188.6.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Shiota N, Rysa J, Kovanen PT, Ruskoaho H, Kokkonen JO, Lindstedt KA. A role for cardiac mast cells in the pathogenesis of hypertensive heart disease. J Hypertens. 2003;21:1935–1944. doi: 10.1097/00004872-200310000-00022. [DOI] [PubMed] [Google Scholar]
- 233.Brower GL, Chancey AL, Thanigaraj S, Matsubara BB, Janicki JS. Cause and effect relationship between myocardial mast cell number and matrix metalloproteinase activity. Am J Physiol Heart Circ Physiol. 2002;283:H518–H525. doi: 10.1152/ajpheart.00218.2000. [DOI] [PubMed] [Google Scholar]
- 234.Chancey AL, Brower GL, Janicki JS. Cardiac mast cell-mediated activation of gelatinase and alteration of ventricular diastolic function. Am J Physiol Heart Circ Physiol. 2002;282:H2152–H2158. doi: 10.1152/ajpheart.00777.2001. [DOI] [PubMed] [Google Scholar]
- 235.Fang KC, Wolters PJ, Steinhoff M, Bidgol A, Blount JL, Caughey GH. Mast cell expression of gelatinases A and B is regulated by kit ligand and TGF-beta. J Immunol. 1999;162:5528–5535. [PubMed] [Google Scholar]
- 236.Matsumoto T, Wada A, Tsutamoto T, Ohnishi M, Isono T, Kinoshita M. Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure. Circulation. 2003;107:2555–2558. doi: 10.1161/01.CIR.0000074041.81728.79. [DOI] [PubMed] [Google Scholar]
- 237.Jolly SR, Abrams GD, Romson JL, Bailie MB, Lucchesi BR. Effects of lodoxamide on ischemic reperfused myocardium. J Cardiovasc Pharmacol. 1982;4:441–448. doi: 10.1097/00005344-198205000-00015. [DOI] [PubMed] [Google Scholar]
- 238.Kanemitsu H, Takai S, Tsuneyoshi H, Nishina T, Yoshikawa K, Miyazaki M, Ikeda T, Komeda M. Chymase inhibition prevents cardiac fibrosis and dysfunction after myocardial infarction in rats. Hypertens Res. 2006;29:57–64. doi: 10.1291/hypres.29.57. [DOI] [PubMed] [Google Scholar]
- 239.Willems IE, Havenith MG, De Mey JG, Daemen MJ. The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am J Pathol. 1994;145:868–875. [PMC free article] [PubMed] [Google Scholar]
- 240.Gabbiani G. The cellular derivation and the life span of the myofibroblast. Pathol Res Pract. 1996;192:708–711. doi: 10.1016/S0344-0338(96)80092-6. [DOI] [PubMed] [Google Scholar]
- 241.Gabbiani G, Le Lous M, Bailey AJ, Bazin S, Delaunay A. Collagen and myofibroblasts of granulation tissue. A chemical, ultrastructural and immunologic study. Virchows Arch B Cell Pathol. 1976;21:133–145. doi: 10.1007/BF02899150. [DOI] [PubMed] [Google Scholar]
- 242.Gabbiani G. The role of contractile proteins in wound healing and fibrocontractive diseases. Methods Achiev Exp Pathol. 1979;9:187–206. [PubMed] [Google Scholar]
- 243.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 244.Weber KT, Sun Y, Katwa LC, Cleutjens JP, Zhou G. Connective tissue and repair in the heart. Potential regulatory mechanisms. Ann N Y Acad Sci. 1995;752:286–299. doi: 10.1111/j.1749-6632.1995.tb17438.x. [DOI] [PubMed] [Google Scholar]
- 245.Cleutjens JP, Kandala JC, Guarda E, Guntaka RV, Weber KT. Regulation of collagen degradation in the rat myocardium after infarction. J Mol Cell Cardiol. 1995;27:1281–1292. doi: 10.1016/s0022-2828(05)82390-9. [DOI] [PubMed] [Google Scholar]
- 246.Weber KT, Sun Y, Cleutjens JP. Structural remodeling of the infarcted rat heart. Exs. 1996;76:489–499. doi: 10.1007/978-3-0348-8988-9_30. [DOI] [PubMed] [Google Scholar]
- 247.Squires CE, Escobar GP, Payne JF, Leonardi RA, Goshorn DK, Sheats NJ, Mains IM, Mingoia JT, Flack EC, Lindsey ML. Altered fibroblast function following myocardial infarction. J Mol Cell Cardiol. 2005;39:699–707. doi: 10.1016/j.yjmcc.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 248.Desmouliere A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol. 1995;146:56–66. [PMC free article] [PubMed] [Google Scholar]
- 249.Quan TE, Cowper S, Wu SP, Bockenstedt LK, Bucala R. Circulating fibrocytes: collagen-secreting cells of the peripheral blood. Int J Biochem Cell Biol. 2004;36:598–606. doi: 10.1016/j.biocel.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 250.Edwards LC, Dunphy JE. Wound healing. I. Injury and normal repair. N Engl J Med. 1958;259:224–233. doi: 10.1056/NEJM195807312590506. [DOI] [PubMed] [Google Scholar]
- 251.Petrakis NL, Davis M, Lucia SP. The in vivo differentiation of human leukocytes into histiocytes, fibroblasts and fat cells in subcutaneous diffusion chambers. Blood. 1961;17:109–118. [PubMed] [Google Scholar]
- 252.Stirling GA, Kakkar VV. Cells in the circulating blood capable of producing connective tissue. Br J Exp Pathol. 1969;50:51–55. [PMC free article] [PubMed] [Google Scholar]
- 253.Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1:71–81. [PMC free article] [PubMed] [Google Scholar]
- 254.Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, Belperio JA, Keane MP, Strieter RM. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114:438–446. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Virag JI, Murry CE. Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair. Am J Pathol. 2003;163:2433–2440. doi: 10.1016/S0002-9440(10)63598-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Yano T, Miura T, Ikeda Y, Matsuda E, Saito K, Miki T, Kobayashi H, Nishino Y, Ohtani S, Shimamoto K. Intracardiac fibroblasts, but not bone marrow derived cells, are the origin of myofibroblasts in myocardial infarct repair. Cardiovasc Pathol. 2005;14:241–246. doi: 10.1016/j.carpath.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 257.Frangogiannis NG, Michael LH, Entman ML. Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb) Cardiovasc Res. 2000;48:89–100. doi: 10.1016/s0008-6363(00)00158-9. [DOI] [PubMed] [Google Scholar]
- 258.Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993;122:103–111. doi: 10.1083/jcb.122.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, Gabbiani G. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol. 1998;142:873–881. doi: 10.1083/jcb.142.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.MacKenna D, Summerour SR, Villarreal FJ. Role of mechanical factors in modulating cardiac fibroblast function and extracellular matrix synthesis. Cardiovasc Res. 2000;46:257–263. doi: 10.1016/s0008-6363(00)00030-4. [DOI] [PubMed] [Google Scholar]
- 261.Roy S, Khanna S, Bickerstaff AA, Subramanian SV, Atalay M, Bierl M, Pendyala S, Levy D, Sharma N, Venojarvi M, et al. Oxygen sensing by primary cardiac fibroblasts: a key role of p21(Waf1/Cip1/Sdi1) Circ Res. 2003;92:264–271. doi: 10.1161/01.res.0000056770.30922.e6. [DOI] [PubMed] [Google Scholar]
- 262.Roy S, Khanna S, Wallace WA, Lappalainen J, Rink C, Cardounel AJ, Zweier JL, Sen CK. Characterization of perceived hyperoxia in isolated primary cardiac fibroblasts and in the reoxygenated heart. J Biol Chem. 2003;278:47129–47135. doi: 10.1074/jbc.M308703200. [DOI] [PubMed] [Google Scholar]
- 263.Lafontant PJ, Burns AR, Donnachie E, Haudek SB, Smith CW, Entman ML. Oncostatin M Differentially Regulates Cxc Chemokines In Mouse Cardiac Fibroblasts. Am J Physiol Cell Physiol. 2006 doi: 10.1152/ajpcell.00322.2005. [DOI] [PubMed] [Google Scholar]
- 264.Detillieux KA, Sheikh F, Kardami E, Cattini PA. Biological activities of fibroblast growth factor-2 in the adult myocardium. Cardiovasc Res. 2003;57:8–19. doi: 10.1016/s0008-6363(02)00708-3. [DOI] [PubMed] [Google Scholar]
- 265.Sun Y, Weber KT. Infarct scar: a dynamic tissue. Cardiovasc Res. 2000;46:250–256. doi: 10.1016/s0008-6363(00)00032-8. [DOI] [PubMed] [Google Scholar]
- 266.Lee SH, Wolf PL, Escudero R, Deutsch R, Jamieson SW, Thistlethwaite PA. Early expression of angiogenesis factors in acute myocardial ischemia and infarction. N Engl J Med. 2000;342:626–633. doi: 10.1056/NEJM200003023420904. [DOI] [PubMed] [Google Scholar]
- 267.Li J, Brown LF, Hibberd MG, Grossman JD, Morgan JP, Simons M. VEGF, flk-1, and flt-1 expression in a rat myocardial infarction model of angiogenesis. Am J Physiol. 1996;270:H1803–H1811. doi: 10.1152/ajpheart.1996.270.5.H1803. [DOI] [PubMed] [Google Scholar]
- 268.Ren G, Michael LH, Entman ML, Frangogiannis NG. Morphological characteristics of the microvasculature in healing myocardial infarcts. J Histochem Cytochem. 2002;50:71–79. doi: 10.1177/002215540205000108. [DOI] [PubMed] [Google Scholar]
- 269.Dobaczewski M, Akrivakis S, Nasser K, Michael LH, Entman ML, Frangogiannis NG. Vascular mural cells in healing canine myocardial infarcts. J Histochem Cytochem. 2004;52:1019–1029. doi: 10.1369/jhc.3A6210.2004. [DOI] [PubMed] [Google Scholar]
- 270.Fukuda S, Kaga S, Sasaki H, Zhan L, Zhu L, Otani H, Kalfin R, Das DK, Maulik N. Angiogenic signal triggered by ischemic stress induces myocardial repair in rat during chronic infarction. J Mol Cell Cardiol. 2004;36:547–559. doi: 10.1016/j.yjmcc.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 271.Conway EM, Collen D, Carmeliet P. Molecular mechanisms of blood vessel growth. Cardiovasc Res. 2001;49:507–521. doi: 10.1016/s0008-6363(00)00281-9. [DOI] [PubMed] [Google Scholar]
- 272.Fam NP, Verma S, Kutryk M, Stewart DJ. Clinician guide to angiogenesis. Circulation. 2003;108:2613–2618. doi: 10.1161/01.CIR.0000102939.04279.75. [DOI] [PubMed] [Google Scholar]
- 273.Sandhu R, Teichert-Kuliszewska K, Nag S, Proteau G, Robb MJ, Campbell AI, Kuliszewski MA, Kutryk MJ, Stewart DJ. Reciprocal regulation of angiopoietin-1 and angiopoietin-2 following myocardial infarction in the rat. Cardiovasc Res. 2004;64:115–124. doi: 10.1016/j.cardiores.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 274.Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–3055. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- 275.Zymek P, Bujak M, Chatila K, Cieslak A, Thakker G, Entman ML, Frangogiannis NG. The role of platelet-derived growth factor signaling in healing myocardial infarcts. J Am Coll Cardiol. 2006;48:2315–2323. doi: 10.1016/j.jacc.2006.07.060. [DOI] [PubMed] [Google Scholar]
- 276.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 277.Haslett C. Granulocyte apoptosis and its role in the resolution and control of lung inflammation. Am J Respir Crit Care Med. 1999;160:S5–S11. doi: 10.1164/ajrccm.160.supplement_1.4. [DOI] [PubMed] [Google Scholar]
- 278.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 279.Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Cichy J, Pure E. The liberation of CD44. J Cell Biol. 2003;161:839–843. doi: 10.1083/jcb.200302098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004;279:17079–17084. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- 283.Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Pure E, Henson PM, Noble PW. Resolution of lung inflammation by CD44. Science. 2002;296:155–158. doi: 10.1126/science.1069659. [DOI] [PubMed] [Google Scholar]
- 284.Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. 2003;4:33–45. doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- 285.Svee K, White J, Vaillant P, Jessurun J, Roongta U, Krumwiede M, Johnson D, Henke C. Acute lung injury fibroblast migration and invasion of a fibrin matrix is mediated by CD44. J Clin Invest. 1996;98:1713–1727. doi: 10.1172/JCI118970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Huebener P, Abou-Khamis T, Zymek P, Bujak M, Ying X, Chatila K, Haudek S, Thakker G, Frangogiannis NG. CD44 Is Critically Involved in Infarct Healing by Regulating the Inflammatory and Fibrotic Response. J Immunol. 2008;180:2625–2633. doi: 10.4049/jimmunol.180.4.2625. [DOI] [PubMed] [Google Scholar]
- 287.Mosmann TR. Properties and functions of interleukin-10. Adv Immunol. 1994;56:1–26. [PubMed] [Google Scholar]
- 288.Moore KW, O'Garra A, de Waal Malefyt R, Vieira P, Mosmann TR. Interleukin-10. Annu Rev Immunol. 1993;11:165–190. doi: 10.1146/annurev.iy.11.040193.001121. [DOI] [PubMed] [Google Scholar]
- 289.Lacraz S, Nicod LP, Chicheportiche R, Welgus HG, Dayer JM. IL-10 inhibits metalloproteinase and stimulates TIMP-1 production in human mononuclear phagocytes. J Clin Invest. 1995;96:2304–2310. doi: 10.1172/JCI118286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Frangogiannis NG, Mendoza LH, Lindsey ML, Ballantyne CM, Michael LH, Smith CW, Entman ML. IL-10 is induced in the reperfused myocardium and may modulate the reaction to injury. J Immunol. 2000;165:2798–2808. doi: 10.4049/jimmunol.165.5.2798. [DOI] [PubMed] [Google Scholar]
- 291.Yang Z, Zingarelli B, Szabo C. Crucial role of endogenous interleukin-10 production in myocardial ischemia/reperfusion injury. Circulation. 2000;101:1019–1026. doi: 10.1161/01.cir.101.9.1019. [DOI] [PubMed] [Google Scholar]
- 292.Zymek P, Nah DY, Bujak M, Ren G, Koerting A, Leucker T, Huebener P, Taffet G, Entman M, Frangogiannis NG. Interleukin-10 is not a critical regulator of infarct healing and left ventricular remodeling. Cardiovasc Res. 2007;74:313–322. doi: 10.1016/j.cardiores.2006.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Schiller M, Javelaud D, Mauviel A. TGF-beta-induced SMAD signaling and gene regulation: consequenc es for extracellular matrix remodeling and wound healing. J Dermatol Sci. 2004;35:83–92. doi: 10.1016/j.jdermsci.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 294.Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- 295.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 296.Koli K, Saharinen J, Hyytiainen M, Penttinen C, Keski-Oja J. Latency, activation, and binding proteins of TGF-beta. Microsc Res Tech. 2001;52:354–362. doi: 10.1002/1097-0029(20010215)52:4<354::AID-JEMT1020>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 297.Hyytiainen M, Penttinen C, Keski-Oja J. Latent TGF-beta binding proteins: extracellular matrix association and roles in TGF-beta activation. Crit Rev Clin Lab Sci. 2004;41:233–264. doi: 10.1080/10408360490460933. [DOI] [PubMed] [Google Scholar]
- 298.Ignotz RA, Massague J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986;261:4337–4345. [PubMed] [Google Scholar]
- 299.Rifkin DB, Mazzieri R, Munger JS, Noguera I, Sung J. Proteolytic control of growth factor availability. APMIS. 1999;107:80–85. doi: 10.1111/j.1699-0463.1999.tb01529.x. [DOI] [PubMed] [Google Scholar]
- 300.Murphy-Ullrich JE, Poczatek M. Activation of latent TGF-beta by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev. 2000;11:59–69. doi: 10.1016/s1359-6101(99)00029-5. [DOI] [PubMed] [Google Scholar]
- 301.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 302.Barcellos-Hoff MH, Derynck R, Tsang ML, Weatherbee JA. Transforming growth factor-beta activation in irradiated murine mammary gland. J Clin Invest. 1994;93:892–899. doi: 10.1172/JCI117045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Lyons RM, Keski-Oja J, Moses HL. Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J Cell Biol. 1988;106:1659–1665. doi: 10.1083/jcb.106.5.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Thompson NL, Bazoberry F, Speir EH, Casscells W, Ferrans VJ, Flanders KC, Kondaiah P, Geiser AG, Sporn MB. Transforming growth factor beta-1 in acute myocardial infarction in rats. Growth Factors. 1988;1:91–99. doi: 10.3109/08977198809000251. [DOI] [PubMed] [Google Scholar]
- 305.Deten A, Holzl A, Leicht M, Barth W, Zimmer HG. Changes in extracellular matrix and in transforming growth factor beta isoforms after coronary artery ligation in rats. J Mol Cell Cardiol. 2001;33:1191–1207. doi: 10.1006/jmcc.2001.1383. [DOI] [PubMed] [Google Scholar]
- 306.Youn TJ, Kim HS, Oh BH. Ventricular remodeling and transforming growth factor-beta 1 mRNA expression after nontransmural myocardial infarction in rats: effects of angiotensin converting enzyme inhibition and angiotensin II type 1 receptor blockade. Basic Res Cardiol. 1999;94:246–253. doi: 10.1007/s003950050149. [DOI] [PubMed] [Google Scholar]
- 307.Yu CM, Tipoe GL, Wing-Hon Lai K, Lau CP. Effects of combination of angiotensin-converting enzyme inhibitor and angiotensin receptor antagonist on inflammatory cellular infiltration and myocardial interstitial fibrosis after acute myocardial infarction. J Am Coll Cardiol. 2001;38:1207–1215. doi: 10.1016/s0735-1097(01)01518-2. [DOI] [PubMed] [Google Scholar]
- 308.Sun Y, Zhang JQ, Zhang J, Ramires FJ. Angiotensin II, transforming growth factor-beta1 and repair in the infarcted heart. J Mol Cell Cardiol. 1998;30:1559–1569. doi: 10.1006/jmcc.1998.0721. [DOI] [PubMed] [Google Scholar]
- 309.Hao J, Wang B, Jones SC, Jassal DS, Dixon IM. Interaction between angiotensin II and Smad proteins in fibroblasts in failing heart and in vitro. Am J Physiol Heart Circ Physiol. 2000;279:H3020–H3030. doi: 10.1152/ajpheart.2000.279.6.H3020. [DOI] [PubMed] [Google Scholar]
- 310.Dean RG, Balding LC, Candido R, Burns WC, Cao Z, Twigg SM, Burrell LM. Connective tissue growth factor and cardiac fibrosis after myocardial infarction. J Histochem Cytochem. 2005;53:1245–1256. doi: 10.1369/jhc.4A6560.2005. [DOI] [PubMed] [Google Scholar]
- 311.Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, Wang XF, Frangogiannis NG. Essential Role of Smad3 in Infarct Healing and in the Pathogenesis of Cardiac Remodeling. Circulation. 2007;116:2127–2138. doi: 10.1161/CIRCULATIONAHA.107.704197. [DOI] [PubMed] [Google Scholar]
- 312.Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, Winkelmann K, Michael LH, Lawler J, Entman ML. The critical role of endogenous Thrombospondin (TSP)-1 in preventing expansion of healing myocardial infarcts. Circulation. 2005;111:2935–2942. doi: 10.1161/CIRCULATIONAHA.104.510354. [DOI] [PubMed] [Google Scholar]
- 313.Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184–195. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Bassols A, Massague J. Transforming growth factor beta regulates the expression and structure of extracellular matrix chondroitin/dermatan sulfate proteoglycans. J Biol Chem. 1988;263:3039–3045. [PubMed] [Google Scholar]
- 315.Laiho M, Saksela O, Andreasen PA, Keski-Oja J. Enhanced production and extracellular deposition of the endothelial-type plasminogen activator inhibitor in cultured human lung fibroblasts by transforming growth factor-beta. J Cell Biol. 1986;103:2403–2410. doi: 10.1083/jcb.103.6.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Edwards DR, Murphy G, Reynolds JJ, Whitham SE, Docherty AJ, Angel P, Heath JK. Transforming growth factor beta modulates the expression of collagenase and metalloproteinase inhibitor. Embo J. 1987;6:1899–1904. doi: 10.1002/j.1460-2075.1987.tb02449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Lefer AM, Tsao P, Aoki N, Palladino MA., Jr Mediation of cardioprotection by transforming growth factor-beta. Science. 1990;249:61–64. doi: 10.1126/science.2164258. [DOI] [PubMed] [Google Scholar]
- 318.Ikeuchi M, Tsutsui H, Shiomi T, Matsusaka H, Matsushima S, Wen J, Kubota T, Takeshita A. Inhibition of TGF-beta signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc Res. 2004;64:526–535. doi: 10.1016/j.cardiores.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 319.Okada H, Takemura G, Kosai K, Li Y, Takahashi T, Esaki M, Yuge K, Miyata S, Maruyama R, Mikami A, et al. Postinfarction gene therapy against transforming growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation. 2005;111:2430–2437. doi: 10.1161/01.CIR.0000165066.71481.8E. [DOI] [PubMed] [Google Scholar]
- 320.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 321.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 322.Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, Leof EB. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J Clin Invest. 2004;114:1308–1316. doi: 10.1172/JCI19603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Wang S, Wilkes MC, Leof EB, Hirschberg R. Imatinib mesylate blocks a non-Smad TGF-beta pathway and reduces renal fibrogenesis in vivo. Faseb J. 2005;19:1–11. doi: 10.1096/fj.04-2370com. [DOI] [PubMed] [Google Scholar]
- 324.Zhang D, Gaussin V, Taffet GE, Belaguli NS, Yamada M, Schwartz RJ, Michael LH, Overbeek PA, Schneider MD. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat Med. 2000;6:556–563. doi: 10.1038/75037. [DOI] [PubMed] [Google Scholar]
- 325.Tsukada S, Westwick JK, Ikejima K, Sato N, Rippe RA. SMAD and p38 MAPK signaling pathways independently regulate alpha1(I) collagen gene expression in unstimulated and transforming growth factor-beta-stimulated hepatic stellate cells. J Biol Chem. 2005;280:10055–10064. doi: 10.1074/jbc.M409381200. [DOI] [PubMed] [Google Scholar]
- 326.Flanders KC. Smad3 as a mediator of the fibrotic response. Int J Exp Pathol. 2004;85:47–64. doi: 10.1111/j.0959-9673.2004.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Feinberg MW, Shimizu K, Lebedeva M, Haspel R, Takayama K, Chen Z, Frederick JP, Wang XF, Simon DI, Libby P, et al. Essential role for Smad3 in regulating MCP-1 expression and vascular inflammation. Circ Res. 2004;94:601–608. doi: 10.1161/01.RES.0000119170.70818.4F. [DOI] [PubMed] [Google Scholar]
- 328.Frangogiannis NG, Shimoni S, Chang SM, Ren G, Dewald O, Gersch C, Shan K, Aggeli C, Reardon M, Letsou GV, et al. Active interstitial remodeling: an important process in the hibernating human myocardium. J Am Coll Cardiol. 2002;39:1468–1474. doi: 10.1016/s0735-1097(02)01792-8. [DOI] [PubMed] [Google Scholar]
- 329.Datto MB, Frederick JP, Pan L, Borton AJ, Zhuang Y, Wang XF. Targeted disruption of Smad3 reveals an essential role in transforming growth factor beta-mediated signal transduction. Mol Cell Biol. 1999;19:2495–2504. doi: 10.1128/mcb.19.4.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Seo D, Hare JM. The transforming growth factor-beta/Smad3 pathway: coming of age as a key participant in cardiac remodeling. Circulation. 2007;116:2096–2098. doi: 10.1161/CIRCULATIONAHA.107.735381. [DOI] [PubMed] [Google Scholar]
- 331.Lawler J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J Cell Mol Med. 2002;6:1–12. doi: 10.1111/j.1582-4934.2002.tb00307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Chatila K, Ren G, Xia Y, Huebener P, Bujak M, Frangogiannis NG. The role of the thrombospondins in healing myocardial infarcts. Cardiovasc Hematol Agents Med Chem. 2007;5:21–27. doi: 10.2174/187152507779315813. [DOI] [PubMed] [Google Scholar]
- 333.Doyen V, Rubio M, Braun D, Nakajima T, Abe J, Saito H, Delespesse G, Sarfati M. Thrombospondin 1 is an autocrine negative regulator of human dendritic cell activation. J Exp Med. 2003;198:1277–1283. doi: 10.1084/jem.20030705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Frangogiannis NG. Targeting the inflammatory response in healing myocardial infarcts. Curr Med Chem. 2006;13:1877–1893. doi: 10.2174/092986706777585086. [DOI] [PubMed] [Google Scholar]
- 335.Prabhu SD, Chandrasekar B, Murray DR, Freeman GL. beta-adrenergic blockade in developing heart failure: effects on myocardial inflammatory cytokines, nitric oxide, and remodeling. Circulation. 2000;101:2103–2109. doi: 10.1161/01.cir.101.17.2103. [DOI] [PubMed] [Google Scholar]
- 336.Deten A, Volz HC, Holzl A, Briest W, Zimmer HG. Effect of propranolol on cardiac cytokine expression after myocardial infarction in rats. Mol Cell Biochem. 2003;251:127–137. [PubMed] [Google Scholar]
- 337.Roberts R, DeMello V, Sobel BE. Deleterious effects of methylprednisolone in patients with myocardial infarction. Circulation. 1976;53:I204–I206. [PubMed] [Google Scholar]
- 338.Bujak M, Kweon HJ, Chatila K, Li N, Taffet G, Frangogiannis NG. Aging-related defects are associated with adverse cardiac remodeling in a mouse model of reperfused myocardial infarction. J Am Coll Cardiol. 2008;51:1384–1392. doi: 10.1016/j.jacc.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Cai D, Xaymardan M, Holm JM, Zheng J, Kizer JR, Edelberg JM. Age-associated impairment in TNF-alpha cardioprotection from myocardial infarction. Am J Physiol Heart Circ Physiol. 2003;285:H463–H469. doi: 10.1152/ajpheart.00144.2003. [DOI] [PubMed] [Google Scholar]
- 340.Xaymardan M, Zheng J, Duignan I, Chin A, Holm JM, Ballard VL, Edelberg JM. Senescent impairment in synergistic cytokine pathways that provide rapid cardioprotection in the rat heart. J Exp Med. 2004;199:797–804. doi: 10.1084/jem.20031639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Lehrke S, Mazhari R, Durand DJ, Zheng M, Bedja D, Zimmet JM, Schuleri KH, Chi AS, Gabrielson KL, Hare JM. Aging impairs the beneficial effect of granulocyte colony-stimulating factor and stem cell factor on post-myocardial infarction remodeling. Circ Res. 2006;99:553–560. doi: 10.1161/01.RES.0000238375.88582.d8. [DOI] [PubMed] [Google Scholar]
- 342.Wollert KC, Drexler H. Clinical applications of stem cells for the heart. Circ Res. 2005;96:151–163. doi: 10.1161/01.RES.0000155333.69009.63. [DOI] [PubMed] [Google Scholar]
- 343.Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Myocyte death, growth, and regeneration in cardiac hypertrophy and failure. Circ Res. 2003;92:139–150. doi: 10.1161/01.res.0000053618.86362.df. [DOI] [PubMed] [Google Scholar]
- 344.Anversa P, Nadal-Ginard B. Myocyte renewal and ventricular remodelling. Nature. 2002;415:240–243. doi: 10.1038/415240a. [DOI] [PubMed] [Google Scholar]
- 345.Anversa P, Leri A, Rota M, Hosoda T, Bearzi C, Urbanek K, Kajstura J, Bolli R. Concise review: stem cells, myocardial regeneration, and methodological artifacts. Stem Cells. 2007;25:589–601. doi: 10.1634/stemcells.2006-0623. [DOI] [PubMed] [Google Scholar]
- 346.Laflamme MA, Murry CE. Regenerating the heart. Nat Biotechnol. 2005;23:845–856. doi: 10.1038/nbt1117. [DOI] [PubMed] [Google Scholar]
- 347.Reinecke H, Poppa V, Murry CE. Skeletal muscle stem cells do not transdifferentiate into cardiomyocytes after cardiac grafting. J Mol Cell Cardiol. 2002;34:241–249. doi: 10.1006/jmcc.2001.1507. [DOI] [PubMed] [Google Scholar]
- 348.Leobon B, Garcin I, Menasche P, Vilquin JT, Audinat E, Charpak S. Myoblasts transplanted into rat infarcted myocardium are functionally isolated from their host. Proc Natl Acad Sci U S A. 2003;100:7808–7811. doi: 10.1073/pnas.1232447100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Murry CE, Soonpaa MH, Reinecke H, Nakajima H, Nakajima HO, Rubart M, Pasumarthi KB, Ismail Virag J, Bartelmez SH, Poppa V, et al. Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature. 2004 doi: 10.1038/nature02446. [DOI] [PubMed] [Google Scholar]
- 350.Balsam LB, Wagers AJ, Christensen JL, Kofidis T, Weissman IL, Robbins RC. Haematopoietic stem cells adopt mature haematopoietic fates in ischaemic myocardium. Nature. 2004 doi: 10.1038/nature02460. [DOI] [PubMed] [Google Scholar]
- 351.Mazhari R, Hare JM. Mechanisms of action of mesenchymal stem cells in cardiac repair: potential influences on the cardiac stem cell niche. Nat Clin Pract Cardiovasc Med. 2007;4 Suppl 1:S21–S26. doi: 10.1038/ncpcardio0770. [DOI] [PubMed] [Google Scholar]
- 352.Janssens S, Theunissen K, Boogaerts M, de Werf FV. Bone marrow cell transfer in acute myocardial infarction. Nat Clin Pract Cardiovasc Med. 2006;3 Suppl 1:S69–S72. doi: 10.1038/ncpcardio0440. [DOI] [PubMed] [Google Scholar]
- 353.Dawn B, Bolli R. Cardiac progenitor cells: the revolution continues. Circ Res. 2005;97:1080–1082. doi: 10.1161/01.RES.0000195610.71671.c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Janssens S, Dubois C, Bogaert J, Theunissen K, Deroose C, Desmet W, Kalantzi M, Herbots L, Sinnaeve P, Dens J, et al. Autologous bone marrow-derived stem-cell transfer in patients with ST-segment elevation myocardial infarction: double-blind, randomised controlled trial. Lancet. 2006;367:113–121. doi: 10.1016/S0140-6736(05)67861-0. [DOI] [PubMed] [Google Scholar]
- 355.Wollert KC, Meyer GP, Lotz J, Ringes-Lichtenberg S, Lippolt P, Breidenbach C, Fichtner S, Korte T, Hornig B, Messinger D, et al. Intracoronary autologous bone-marrow cell transfer after myocardial infarction: the BOOST randomised controlled clinical trial. Lancet. 2004;364:141–148. doi: 10.1016/S0140-6736(04)16626-9. [DOI] [PubMed] [Google Scholar]
- 356.Dimmeler S, Zeiher AM, Schneider MD. Unchain my heart: the scientific foundations of cardiac repair. J Clin Invest. 2005;115:572–583. doi: 10.1172/JCI24283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Vandervelde S, van Luyn MJ, Tio RA, Harmsen MC. Signaling factors in stem cell-mediated repair of infarcted myocardium. J Mol Cell Cardiol. 2005;39:363–376. doi: 10.1016/j.yjmcc.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 358.Harada M, Qin Y, Takano H, Minamino T, Zou Y, Toko H, Ohtsuka M, Matsuura K, Sano M, Nishi J, et al. G-CSF prevents cardiac remodeling after myocardial infarction by activating the Jak-Stat pathway in cardiomyocytes. Nat Med. 2005;11:305–311. doi: 10.1038/nm1199. [DOI] [PubMed] [Google Scholar]