

Abstract
An efficient diastereo- and enantioselective Ag-catalyzed method for additions of a commercially available siloxyfuran to α-ketoimine esters is disclosed. Catalytic transformations require an inexpensive metal salt (AgOAc) and an air stable chiral ligand that is prepared in three steps from commercially available materials in 42% overall yield. Aryl- as well as heterocyclic substituted ketoimines can be used effectively in the Ag-catalyzed process. Additionally, two examples regarding reactions of alkyl-substituted ketoimines are presented. An electronically modified N-aryl group is introduced that is responsible for high reaction efficiency (>98% conversion, 72−95% yields after purification), diastereo- (up to >98:2 dr) and enantioselectivity (up to 97:3 er or 94% ee). The new N-aryl unit is crucial for conversion of the asymmetric vinylogous Mannich (AVM) products to the unprotected amines in high yields. Spectroscopic and X-ray data are among the physical evidence provided that shed light on the identity of the Ag-based chiral catalysts and some of the mechanistic subtleties of this class of enantioselective C—C bond forming processes.
Introduction
Catalytic enantioselective additions of C-based nucleophiles to ketoimines offer direct access to non-racemic N-substituted quaternary carbon stereogenic centers, components of several biologically active molecules.1 Ketoimines, however, are relatively unreactive,2 often exist as a mixture of E and Z isomers, and contain a sterically congested C=N bond that carries difficult-to-differentiate substituents.3 There are only two reported studies of Mannich-type additions4 to ketoimines. One case corresponds to additions of aldehyde-derived enols to geometrically-constrained ketone-derived cyclic imines;5 another disclosure outlines reactions of simple ketene acetals with ketoimines that are mostly generated from methyl-substituted ketones.6 Herein, we disclose a method for catalytic asymmetric vinylogous Mannich (AVM) reactions of ketoimines; additions are performed in the presence of a chiral Ag complex, which can be easily prepared and stored in air for an extended period of time. Transformations are highly site-, diastereo- and enantioselective, require a commercially available siloxyfuran and readily accessible substrates, and furnish products that bear an α-quaternary amino ester.1 We have identified an electronically modified class of N-aryl ketoimines that readily undergo AVM, affording products that can be converted to the unprotected amines efficiently. Through the studies described below, we offer insight regarding the identity of this emerging class of Ag-based chiral catalysts for asymmetric Mannich reactions.7,8,9
Results and Discussion
I. Development of Ag-catalyzed AVM of α-ketoimine esters
a. Enhancement of reactivity through electronic alteration of the N-activating group
We began by investigating the ability of 1, a small-molecule ligand (molecular weight = 508.6 g/mol) used to promote Ag-catalyzed enolsilane additions to o-anisidyl aldimines,7,8 to initiate the reaction of 2 with α-ketoimine ester 3 (see Table 1).10 Although high diastereoselectivity is observed (>98:2 syn:anti; entries 1−3, Table 1), reactions are inefficient (20−68% conv, 15 h) and enantioselectivity is low (<35% ee); at lower temperatures (−78 °C), the desired products are not formed (entry 4).
Table 1.
Examination of Ketoimines Bearing Different N-aryl Groupsa
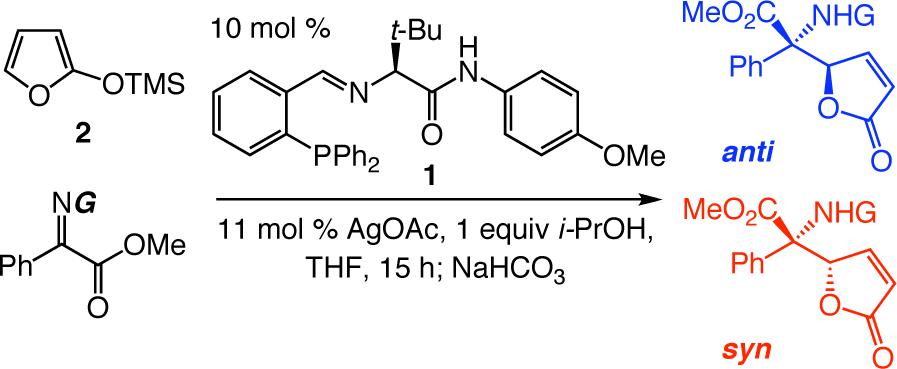 | |||||
|---|---|---|---|---|---|
| entry | G | temp (°C) | conv (%)b | anti:synb | er; ee (%)c |
| 1 | 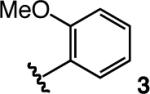 |
+4 | 68 | <2:>98 | 63.5:36.5; 27 |
| 2 | −15 | 45 | <2:>98 | 67:33; 34 | |
| 3 | −30 | 20 | <2:>98 | 64.5:35.5; 29 | |
| 4 | −78 | <2 | nd | nd | |
| 5 | 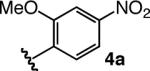 |
−78 | >98 | 93:7 | 95.5:4.5; 91 |
| 6 | 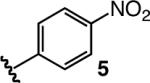 |
−78 | >98 | 80:20 | 89.5:10.5; 79 |
| 7 | 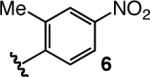 |
−78 | 43 | 8:92 | 77.5:22.5; 55 |
Reactions performed under N2 atmosphere.
Conversion levels and diastereomeric ratios were determined by analysis of 400 MHz 1H NMR spectra of unpurified products.
Enantiomeric ratio (er) values were determined by chiral HPLC analysis; see the Supporting Information for details.
nd = not determined.
To enhance AVM efficiency, we chose to alter the N-aryl unit of the ketoimine by installing a nitro group para to the imine. Such a modification allows AVM of 2 with 4a (entry 5, Table 1) to proceed to >98% conversion at −78 °C, affording anti-7a in 91% ee (95.5:4.5 er). The sense of diastereoselectivity is reversed (vs 3), as the anti product isomer is now selectively generated (dr = 93:7). Pure anti-7a is obtained after purification in 72% yield (entry 1, Table 2).11 The finding in entry 6 (Table 1) indicates that the o-methoxy group is beneficial to diastereo- and enantioselectivity; it is the electron-withdrawing unit para to the N-aryl, however, that is largely responsible for the enhanced efficiency. The observation summarized in entry 7 of Table 1 suggests that the positive effect of the o-methoxy group is not simply due to steric effects. Reaction with a substrate that bears an o-methyl-substituted N-aryl group (6) proceeds to only 43% conversion and results in the formation of anti-7a in 55% ee (77.5:22.5 er); it should be noted, however, that such processes proceed with relatively high diastereoselectivity in favor of the syn product isomer (anti:syn = 8:92).
Table 2.
Ag-Catalyzed AVM of Aryl-Substituted α-Ketoimine Estersa
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| |
|
|
NaHC03 workup (−78 °C→22 °C) |
HOAc workup (−78 °C→22 °C) |
||||
| entry | Ar | substrate | 7:8b | yield 7 (%)c | 7 er; ee (%)d | 7:8b | yield 7 (%)c | 7 er; ee (%)d |
| 1 | C6H5 | 4a | 93:7 | 72 | 95.5:4.5; 91 | 95:5 | 88 | 96:4; 92 |
| 2 | m-OMeC6H4 | 4b | 80:20 | 76 | 93:7; 86 | 95:5 | 95 | 96.5:3.5; 93 |
| 3 | m-ClC6H4 | 4c | 75:25 | 51 | 90.5:9.5; 81 | 92:8 | 72 | 93.5:6.5; 87 |
| 4 | p-BrC6H4 | 4d | 88:12 | 78 | 96:4; 92 | 95:5 | 80 | 96:4; 92 |
| 5 | p-IC6H4 | 4e | 86:14 | 62 | 95:5; 90 | >98:<2 | 81 | 96.5:3.5; 93 |
| 6 | p-t-BuC6H4 | 4f | 75:25 | 68 | 93:7; 86 | 89:11 | 77 | 95:5; 90 |
| 7 | p-CF3C6H4 | 4g | 89:11 | 67 | 95.5: 4.5; 91 | 95:5 | 87 | 97:3; 94 |
| 8 | 2-naphthyl | 4h | 93:7 | 68 | 95:5; 90 | >98:<2 | 81 | 95.5:4.5; 91 |
| 9 | o-BrC6H4 | 4i | <2:>98 | 87e | 66:34; 32e | <2:>98 | 87e | 66:34; 32e |
Reactions performed under N2 atmosphere; >98% conversion in all cases (substrate consumption based on internal standard).
Diastereomeric ratios were determined by analysis of 400 MHz 1H NMR spectra of the unpurified reaction mixtures.
Yields are of the purified anti-7.
Enantiomer ratio (er) values were determined by chiral HPLC analysis; see the Supporting Information for details.
Data for syn-8i as the major isomer.
b. Range of aryl-substituted α-ketoimine esters
As the data summarized in Table 2 indicates, aryl-substituted α-ketoimine esters undergo AVM reactions to furnish anti-7a—h diastereoselectively (75:25−93:7 dr), in 81−92% ee (90.5−9.5 to 96:4 er) and 51−78% yield; minor diastereomers (syn-8a-h) are typically obtained in 40−60% ee. (Data involving NaHCO3 quench are shown in the left column; reactions with acid quench, the results of which are illustrated in the right hand columns, are discussed below).
As represented by the example in entry 9, ketoimines with an o-substituted aryl group are not transformed with high enantioselectivity (<40% ee); such processes are efficient (>98% conv), however, and occur diastereoselectively, although it is the syn isomer that is generated selectively (anti:syn = <2:>98). The outcome of the transformation shown in entry 9 of Table 2 is not as a result of electronic effects imposed by the presence of the halide substituent; catalytic AVM reactions of the corresponding α-ketoimine ester bearing an o-methylphenyl moiety exhibited a similar selectivity and reactivity profile. It is worth noting that a similar diastereoselectivity preference is observed when the N-aryl unit of the ketoimine bears an ortho substituent (see entry 7, Table 1).
c. Effect of workup conditions on AVM efficiency as well as diastereo- and enantioselectivity
We next considered whether the lower diastereomeric ratios (data in left columns, Table 2) and moderate yields of anti isomers could be improved by a better understanding of the origin of the selectivity reversal in reactions of 3 versus 4a (entries 1−3 and 5, Table 1). Such considerations led us to determine that when the reaction of 2 and 4a (−78 °C) is quenched with aqueous NaHCO3, there is 71% conversion after only 30 minutes; syn-8a, however, is formed predominantly (20:80 anti-7a:syn-8a vs 93:7 after 15 h) and enantioselectivity for both isomers is low (<25% ee). After six hours, there is >98% conversion and anti-7a is obtained in 90% ee as the major diastereomer of a 91:9 mixture.
A plausible interpretation of the above data is that the addition of aqueous NaHCO3 (Table 2) might not result in complete quenching of the reaction at −78 °C (i.e., excess 2 is notcompletely proto-desilylated);12 the syn-selective transformation might occur as the solution is allowed to warm up. When the reaction is quenched after extended periods of time (i.e., 10−15 h), anti-7a is generated predominantly, since most of the ketoimine has been consumed. Moreover, examination of the unpurified mixture pointed to the formation of a byproduct, derived from Michael addition of the unreacted enolsilane 2 with unsaturated lactones anti-7 and syn-8 (1H NMR and mass spectroscopic analysis).13
The above observations suggested that use of a quenching procedure that more efficiently removes unreacted enolsilane at −78 °C could enhance diastereo- and enantioselection (i.e., minimal Mannich addition at elevated temperatures) as well as afford higher yields (i.e., minimal Michael addition). Indeed, as illustrated in entries 1−8 of Table 2 (right column), when reaction mixtures are treated with HOAc in MeOH (vs aq. NaHCO3), anti-7a—h are formed in 89:11 to >98:2 dr (vs 75:25−93:7 dr), 93.5:6.5−97:3 er (vs 90.5:9.5−96:4 er) and 72−95% yield (vs 51−78% yield). As the data in entry 9 indicate, the modification in quenching conditions exerts little or no effect on the AVM of o-substituted α-ketoimine ester 4i.
It should be noted that ketoimine substrates are required for adventitious syn-selective AVM to occur at higher temperatures during work-up. Control experiments (internal reference) illustrate that after 15 hours, >98% of the substrate is converted to AVM products. It is therefore likely that retro-Mannich reactions take place to a limited extent during the basic NaHCO3 quench, generating the small amount of ketoimine.
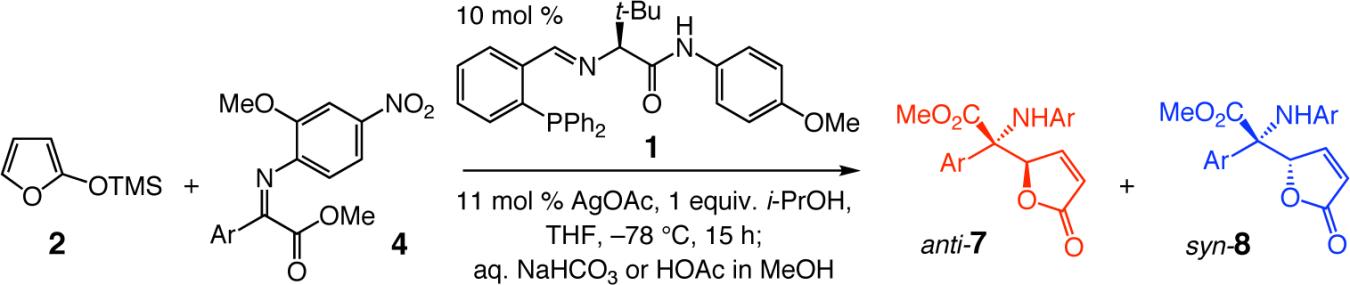
As illustrated by the enantioselective synthesis of amino esters 7j and 7k, respectively, transformations can be performed with heteroaromatic α-ketoimine esters. The lower selectivities obtained for 7j versus 7k may be due to competitive chelation of the Lewis basic furyl oxygen (vs OMe of the N-aryl) with the Lewis acidic Ag-based complex.
d. Issues of practicality and removal of the N-activating group
The Ag-catalyzed process is performed in the presence of an air stable chiral ligand that can be prepared, on gram scale, in three simple steps and 42% overall yield from commercially available starting materials; the amino acid-based phosphine ligand can be purified by a single crystallization. Moreover, the requisite AgOAc and siloxyfuran are commercially available.
Ag-catalyzed AVM reactions can be easily performed on significant amounts of α-ketoimine esters. As an example, reaction with 2.0 grams of 4a affords 2.24 grams of purified anti-7a (88% yield) with high selectivity (96:4 er). Furthermore, although t-Leu-containing ligand 1 is optimal, the corresponding phosphine 9, bearing the less expensive valine moiety, promotes AVM efficiently and in lower — but useful — selectivity; the example in eq (1) is illustrative.

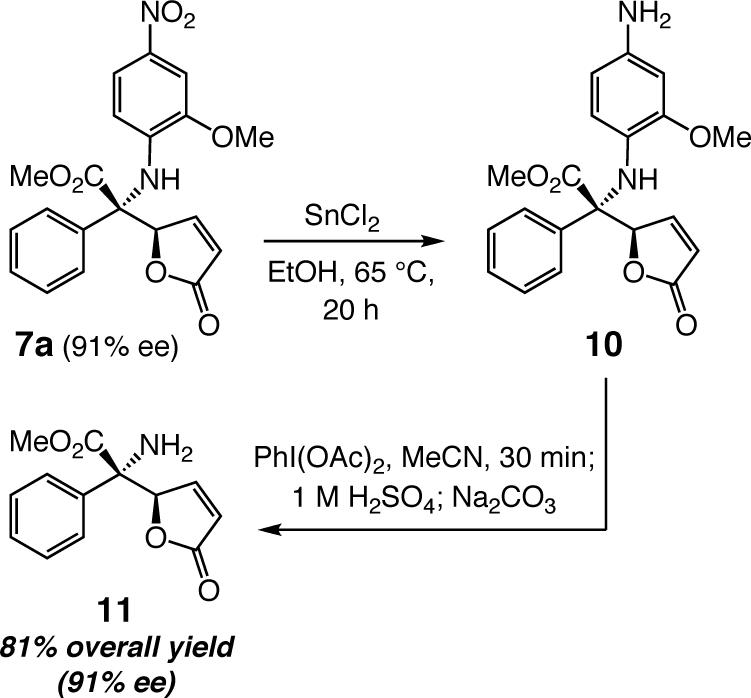
The N-aryl group can be removed by a two-step procedure that affords the desired amines in >80% yield; an example is presented in Scheme 1. Several points regarding deprotection of the N-aryl unit are worth noting: (a) Removal of the o-anisidyl group in a corresponding AVM product (e.g., 3, entry 1, Table 1) is substantially less efficient (<25% yield). That is, the sterically unencumbered amine of the N-aryl unit, generated from reduction of the NO2 group, likely to react more readily with PhI(OAc)2 than the more sterically encumbered heteroatom of the quaternary carbon. (b) Attempts to unmask the amine through oxidative procedures involving DDQ (e.g., 1.5 equiv oxidant, 20:1 CH2Cl2:H2O, 22 °C, 2 h) resulted in <2% conversion. (c) When ceric ammonium nitrate (CAN) is used, unprotected 11 can be obtained in one step but only in 45% yield.14 (c) Subjection of 7a to catalytic hydrogenation conditions (1 atm. H2, 10% w/w Pd(C), EtOAc, 22 °C, 4 h) leads to isolation of the saturated lactone bearing the p-nitroaryl moiety in 79% yield along with 10% of the saturated aniline.
Scheme 1.
Removal of the N-Aryl Unit
II. Structures of Chiral Ag-based Complexes and Related Mechanistic Issues
a. Dependence of selectivity on temperature
We verified the strong dependence of diastereoselectivity on reaction temperature through systematic investigations; the results of these studies are summarized in Table 3. Such findings have notable mechanistic implications. The reversal of selectivity (see entries 1 and 5, Table 3) implies a change in the energetics of the catalytic cycle; alterations in the structure of the chiral catalyst may also be responsible for different stereochemical outcomes. To gain additional insight, we explored the identity of the active chiral complex.
Table 3.
Dependence of Ag-catalyzed AVM selectivity on temperaturea
 | |||||
|---|---|---|---|---|---|
| entry | temp (°C) | 7a:8ab | yield (%)c | 7a er; ee (%)d | 8a er; ee (%)d |
| 1 | −78 | 93:7 | 88 | 95.5:4.5; 91 | 82.5:17.5; 65 |
| 2 | −50 | 33:66 | 76 | 85:15; 70 | 85:15; 70 |
| 3 | −30 | 14:86 | 58 | 58:42; 16 | 81:19; 62 |
| 4 | −15 | 10:90 | 32 | 63:37; 26 | 79.5:20.5; 59 |
| 5 | +4 | 8:92 | 41 | <55:45; <10 | 77:23; 54 |
Reactions performed under N2 atmosphere; all reactions were quenched at the specified temperature and kept at that temperature for 3 hours before allowing to warm to 22 °C. All conversions = >98% (substrate consumption based on internal standard).
Diastereomeric ratios were determined by 400 MHz 1H NMR analyses of product mixtures prior to purification.
Yields are of purified products; with the exception of entry 1 (yield of only anti-7a), total yields of isomeric mixtures are shown.
Enantiomer ratio (er) values were determined by chiral HPLC analysis; see the Supporting Information for details.
b. Identity and catalytic activity of Ag-based chiral complexes
Treatment of 1 with AgOAc (1.0 equiv, 22 °C, THF) for five minutes followed by filtration (to remove uncomplexed AgOAc, as determined by IR analysis) and solvent removal results in isolation of an air stable white powder that can serve as an effective AVM catalyst. The transformations in entries 1−2 and 7 of Table 2 are initiated by 10 mol % (based on ligand 1; −78 °C, 15 h) of a one-year old sample of the powder, affording anti-7a-b and 7g with nearly identical reactivity and selectivity. For example, in the presence of the aforementioned powder sample, 7g is isolated in 95:5 dr, 97:3 er and 89% yield. When a solution of the powder (d8-THF) is allowed to cool to −78 °C, there is a notable change in the 1H NMR spectrum (see spectra a—e, Figure 1): while the signal for the amide proton shifts downfield from δ 10.09 ppm (spectrum a, Figure 1, 22 °C), it splits into two peaks of approximately equal intensity at δ 10.87 and δ 10.78 ppm at −78 °C (spectrum e, Figure 1).15 Signals re-coalesce and return to their original chemical shifts when the sample is allowed to warm to 22 °C.15 These data indicate that the powder consists of two complexes that rapidly inter-convert at ambient temperature.
Figure 1.
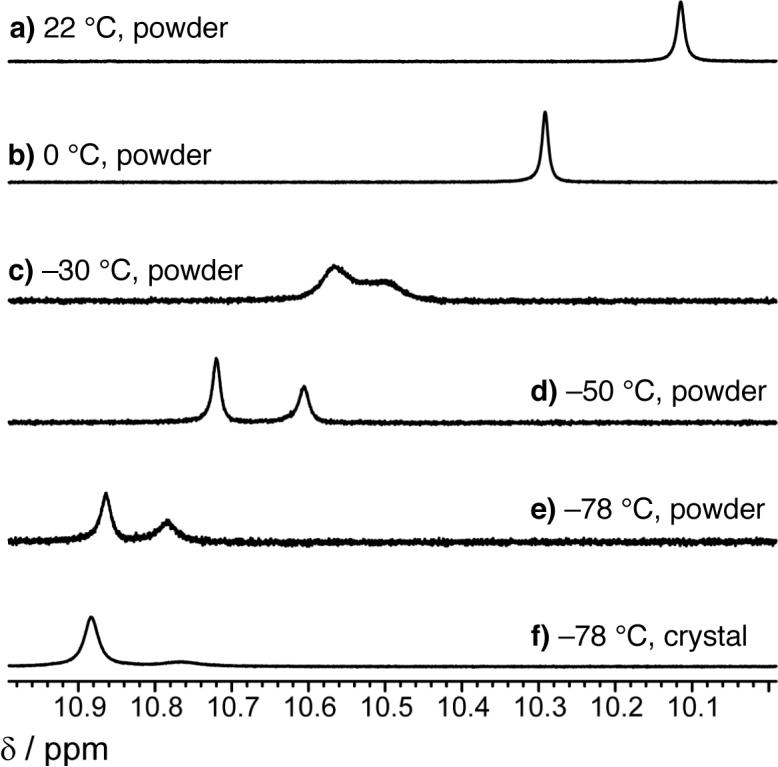
1H NMR spectra (400 MHz, d8-THF) of chiral Ag-based complexes of 1.
When petroleum ether is added to a THF solution of the powder, a crystalline solid is formed, which, as confirmed by X-ray analysis, is the 2:1 complex 12 (Figure 2). In contrast to the powder form, 12 is not an effective catalyst at −78 °C (10 mol %, 15 h: 48% conv) and delivers products with low selectivity (anti-7a:syn-8a = 33:67 dr; eranti = 74:26; ersyn = 83:17).
Figure 2.

X-ray structure of Ag-based complex 12 (Ar = p-OMeC6H4).
At 22 °C, the chemical shifts of the signals in the 1H NMR spectrum of the crystalline form are identical to those observed for the powder.15 Upon cooling, an identical downfield shift of the amide proton is observed; as shown in spectrum f in Figure 1, however, one of the singlets (δ 10.87 ppm at −78 °C) now predominates (2:1 by peak integration; see below for more details). The above observations imply that the powder consists of a mixture of 12 and another complex (NH at δ 10.78 ppm at −78 °C), which likely promotes reactions more readily and with higher selectivity at −78 °C. Thus, in the crystalline sample, the more catalytically active Ag-based complex is the minor constituent.16
The findings summarized below indicate that the active catalyst is likely the less sterically encumbered 1:1 ligand:Ag complex (component represented by the more upfield signal in spectra e—f, Figure 1):17
First, as illustrated in Figure 3 (spectrum b), analysis of the 31P NMR of a sample of the powder form indicates two distinct sets of doublets, with coupling constants consistent with complexes bearing a P—Ag—P (2:1 complex; JP—Ag = 413 and 477 Hz)18 and a P—Ag (1:1 complex; JP—Ag = 622 and 762 Hz) unit.19 In agreement with the 1H NMR data discussed above (spectrum e, Figure 1), the 1:1 and 2:1 complexes exist in a 2:1 ratio in the powder form,20 as judged by the 31P NMR spectrum b (Figure 3). Analysis of the 31P NMR spectrum derived from 12 (spectrum c, Figure 3) indicates a 2:1 mixture with the 1:1 complex being the minor component,20 a finding that is also consistent with 1H NMR spectrum f in Figure 1.
Figure 3.

31P NMR spectra (162 MHz, d8-THF) of the powder and crystal forms of the chiral Ag-based complexes of 1 at various temperatures.
Second, peaks corresponding to the cationic 1:1 (calculated m/z for 1•Ag+: 615.1331, found: 615.1327) and the cationic 2:1 complex (calculated m/z for 12•Ag+: 1123.3610, found: 1123.3625) are detected in the high-resolution mass spectra of the powder as well as of the crystal.
Third, addition of excess AgOAc21 to a THF solution of 2:1 complex 12 causes an increase in the amount of the catalytically more active system (1H NMR analysis);[15] nearly identical results are obtained from this sample as observed with the aforementioned powder.15 Similarly, addition of excess 1 to a solution of the crystalline form (see spectrum f, Figure 1 and spectrum c, Figure 3) affords a sample that is >98% 12 (<2% 1:1 complex).
The availability of a sample of 12 uncontaminated by the 1:1 complex has allowed us to substantiate that the former is a relatively ineffective catalyst at −78 °C (∼25% conv, 15 h, 95:5 syn:anti, 76:24 er in favor of the syn diastereomer), and that at −50 °C, the 2:1 complex promotes a highly syn-selective AVM with improved efficiency (∼70% conv, 15 h, 95:5 syn:anti, 71:29 er). These observations indicate that the turnover in diastereoselectivity as a function of temperature (see Tables 1 and 3) is due to higher activity of the syn-selective 2:1 complex (12) at higher temperatures as well as a change in the stereochemical preference in the reaction promoted by the 1:1 complex (e.g., due to a change in the identity of the stereochemistry-determining step). At −78 °C, the 1:1 complex preferably catalyzes the formation of the anti diastereomer but favors more of the syn adduct at elevated temperatures.
c. Transition state working models
The above mechanistic considerations support the scenarios put forth previously for reactions of aldimines, involving a 1:1 Ag:ligand complex.8a A model (endo mode of approach I, G = H, Figure 4), wherein the substrate binds in a manner to minimize interaction with the bulky amino acid substituent (R in I) and ketoimine's sterically hindered aryl substituent is situated trans to the Si-based nucleophile, may be proposed. Another noteworthy feature is the Lewis base activation22 of the siloxyfuran by the amide terminus. We have previously demonstrated the importance of the chiral phosphine's Lewis basic C-terminus to the facility of this class of transformations; for example, we have shown8a that replacement of the p-methoxyphenyl amide with a p-trifluoromethylphenyl amide results in significant diminution in AVM efficiency.
Figure 4.
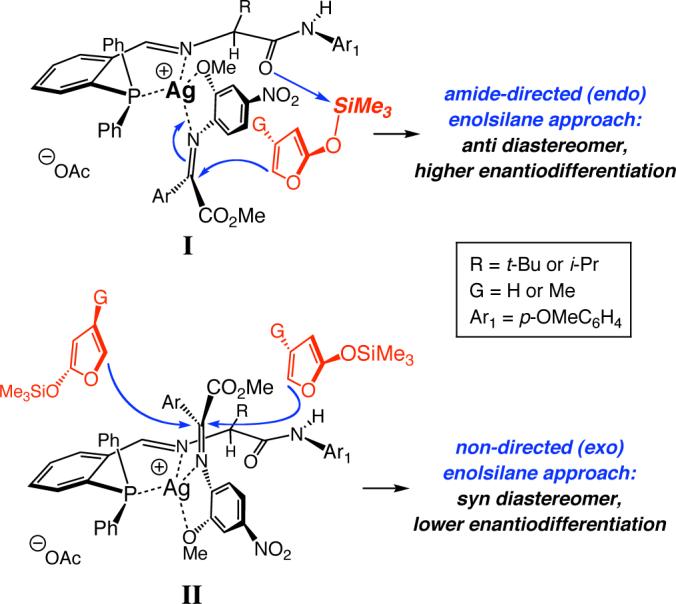
Proposed working models for Ag-catalyzed AVM reactions of aryl-substituted α-ketoimine esters, including Lewis acid activation of the substrate and Lewis base activation of the enolsilane nucleophile.
The o-methoxy group of the N-activating unit is required for the anti-diastereomer to be strongly favored as well as high enantioselectivity to be achieved (see entries 5−7, Table 1), since the bidentate mode of coordination enhances transition structure organization within the catalyst-substrate complex. Thus, in spite of unfavorable steric repulsion caused by the presence of the o-methoxy unit, it elevates catalytic activity (compare entries 5−7, Table 1).
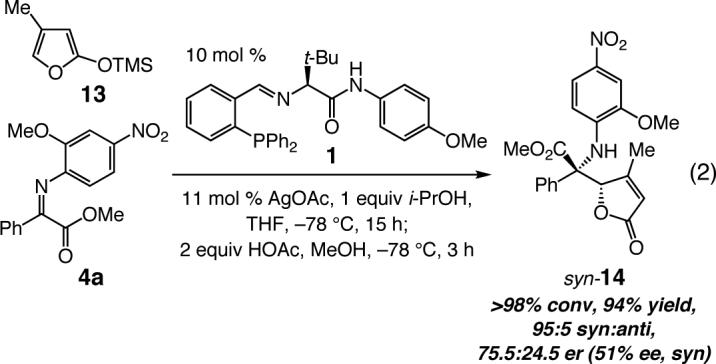
The relatively lower asymmetric induction observed with substrates bearing an o-substituted aryl unit (e.g., entry 9, Table 2) could be attributed to the steric interactions caused by such a structural modification, leading to a preference for AVM to proceed through the exo catalyst-substrate complex II (G = H, Figure 4). The suggested scenario accounts for the reversal of diastereoselectivity such cases as well (preferential formation of syn isomers). The diminished enantioselectivity might be attributed to the geometric constraints associated with interaction of the Lewis basic amide terminus with the Si atom of the siloxyfuran. Enantiofacial control may thus be largely governed by the steric differentiation between the two modes of attack, as illustrated in II (Figure 4); while the phenylphosphine partially hinders approach from one face of the ketoimine in II, the sterically hindered t-Leu might block siloxyfuran from the opposite enantiotopic face of C=N bond. Such a model implies that increasing the size of the Si-based nucleophile should have a deleterious effect on enantioselectivity, giving rise to preferable generation of the syn diastereomer. As shown by the example in eq 2, Ag-catalyzed AVM of α-ketoimine ester 4a (G = Me, Figure 4) with 4-methyl-substituted siloxyfuran 13 results in near exclusive formation of syn-14 (95:5 syn:anti) in 75.5:24.5 er (51% ee).23
It is plausible that the difference in the stereochemical preferences of the two types of substrates discussed above lies in the identity of the turnover-limiting (stereochemistry-determining) steps. With ketoimines carrying a p-nitroaryl group (e.g., 4a-h, anti selective), substrate-catalyst association may be relatively slow, since the electrophile is a ketoimine of diminished Lewis basicity (more reluctant to associate with the chiral Lewis acidic Ag). Thus, once I which may be kinetically favored is generated, it might readily undergo AVM to afford the anti diastereomer in high enantioselectivity. In contrast, with substrates bearing an unactivated N-aryl moiety (e.g., 3 in Table 1 and 15 in Scheme 2), association with the Ag-based catalyst might be relatively facile and reversible, while the addition step being turnover-limiting. The above mechanistic scheme is consistent with the proposal that reactions involving sterically bulky imines or siloxyfurans proceeds through complex II, allowing the nucleophile to have less obstructed access to the catalyst-bound ketoimine.
Scheme 2.
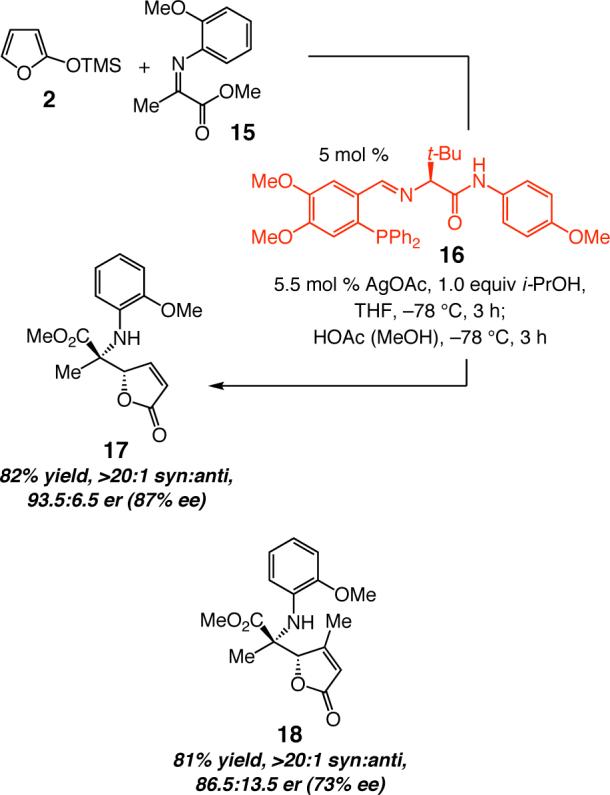
Ag-Catalyzed AVM Reactions of Methyl-Substituted α-Ketoimine Esters and Electronically Unactivated N-Aryl Groups
Initial studies regarding Ag-catalyzed reactions of alkyl-substituted ketoimines indicate that caution must be exercised in extending the mechanistic conclusions described above to other classes of related processes. As the preliminary data summarized in Scheme 224 regarding transformations of alkyl-substituted α-ketoimine esters suggest, AVM reactions of varying types of electrophiles can be governed by different mechanistic regimes and energetic nuances. With the smaller methyl substituent in 15 (vs aryl groups in 4a-i), the presence of an N-activating group bearing a p-nitro unit is no longer required; this is a critical difference that, as indicated by the data in Table 1, leads to the exclusive formation of the syn isomer (>98:<2 syn:anti for 1725 and 18).26 Studies to develop efficient catalytic AVM of other alkyl-substituted AVM reactions and delineation of the aforementioned mechanistic differences are subjects of ongoing studies.
Conclusions
We have developed a Ag-catalyzed process for AVM of α-ketoimine esters that involves a commercially available metal salt (AgOAc) and nucleophile (siloxyfuran 2) and an easily prepared chiral ligand (amino acid-based phosphines 1 or 9). The products, generated in high diastereo- and enantioselectivity, bear an N-substituted all-carbon quaternary stereogenic center as well as an unsaturated lactone moiety that is amenable to various functionalization procedures.8a These investigations highlight one of the important aspects of N-aryl units as general imine activating groups. The reactivity and, in certain instances, the stereoselectivity of the corresponding catalytic reactions can be controlled effectively by electronic modification of such moieties. The p-nitro unit introduced in ketomine substrates discussed above is not only crucial to obtaining high efficiency in addition to diastereo- and enantioselectivity, the resulting aniline (after reduction of the NO2 unit) is critical to the facility with which the N-aryl group is removed (see the example in eq 2).
The studies detailed above demonstrate that the active chiral Ag complex in catalytic Mannich reactions of this particular class of substrates is a 1:1 complex that exhibits different stereochemical preferences at different temperatures. We illustrate that, depending on the reaction conditions, a competing 2:1 complex can prove competitive and, in at least certain cases, cause diminution of stereoselectivity. The investigations described herein should therefore prove crucial to future investigations in catalyst design, as they suggest that structurally modified chiral ligands that are less prone to generate the less active 2:1 complexes might prove to be more effective catalysts in at least certain types of Ag-catalyzed asymmetric Mannich reactions.
Studies directed towards more efficient chiral catalysts for enantioselective Mannich reactions, and design of other sterically and/or electronically modified N-aryl groups for reactions of aldimines and ketoimines are in progress. Future investigations will include development of catalytic AVM processes for a wider range of substrates and additional Agcatalyzed asymmetric processes with ketoimines.
Supplementary Material
Acknowledgments
Financial support was provided by the NIH (GM-57212) and Novartis (fellowship to L. C. W.). We are grateful to Dr. H. Mandai (Boston College) for valuable discussions. We are indebted to Mr. S. J. Malcolmson (Boston College), Ms. K. Mandai (Boston College), Dr. B. Li (Boston College), Dr. P. Müller (MIT) and Dr. R. Staples (Harvard University) for their generous assistance in securing X-ray structures. We thank Mr. A. T. Colucci (Boston College) for experimental assistance. Mass spectrometry facilities at Boston College are supported by the NSF (DBI-0619576).
Footnotes
Supporting Information Available. Experimental procedures and spectral data for substrates and products (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
Footnotes and References
- 1.For stereoselective synthesis of N-substituted quaternary carbon stereogenic centers and quaternary α-amino acids, see: Cativiela C, Díaz-de-Villegas MD. Tetrahedron: Asymmetry. 2007;18:569–623.
- 2.For catalytic enantioselective additions of C-based nucleophiles to ketoimines, see: (CN additions) Funabashi K, Ratni H, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2001;123:10784–10785. doi: 10.1021/ja016935c.Vachal P, Jacobsen EN. J. Am. Chem. Soc. 2002;124:10012–10014. doi: 10.1021/ja027246j.Masumoto S, Usuda H, Suzuki M, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2003;125:5634–5635. doi: 10.1021/ja034980+.Wang J, Hu X, Jiang J, Gou S, Huang X, Liu X, Feng X. Angew. Chem., Int. Ed. 2007;46:8468–8470. doi: 10.1002/anie.200703188.Rueping M, Sugiono E, Moreth SA. Adv. Synth. Catal. 2007;349:759–764. (alkylmetal additions) Lauzon C, Charette AB. Org. Lett. 2006;8:2743–2745. doi: 10.1021/ol0607847.Fu P, Snapper ML, Hoveyda AH. J. Am. Chem. Soc. 2008;130:5530–5541. doi: 10.1021/ja8001343. (allyl addition) Wada R, Shibuguchi T, Makino S, Oisaki K, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2006;128:7687–7691. doi: 10.1021/ja061510h.
- 3.For a review on catalytic asymmetric reactions of ketoimines and ketones, see: Riant O, Hannedouche J. Org. Biomol. Chem. 2007;5:873–888. doi: 10.1039/b617746h.
- 4.For a recent review of Mannich reactions of aldimines, see: Córdova A. Acc. Chem. Res. 2004;37:102–112. doi: 10.1021/ar030231l. For select recent reports of catalytic asymmetric Mannich reactions (not AVM) involving aldimines, see: Hamada T, Manabe K, Kobayashi S. J. Am. Chem. Soc. 2004;126:7768–7769. doi: 10.1021/ja048607t.Matsunaga S, Yoshida T, Morimoto H, Kumagai N, Shibasaki M. J. Am. Chem. Soc. 2004;126:8777–8785. doi: 10.1021/ja0482435.Hamashima Y, Sasamoto N, Hotta D, Somei H, Umebayashi N, Sodeoka M. Angew. Chem., Int. Ed. 2005;44:1525–1529. doi: 10.1002/anie.200462202.Uraguchi D, Sorimachi K, Terada M. J. Am. Chem. Soc. 2004;126:11804–11805. doi: 10.1021/ja046185h.Cozzi PG, Rivalta E. Angew. Chem., Int. Ed. 2005;44:3600–3603. doi: 10.1002/anie.200462757.Song J, Wang Y, Deng L. J. Am. Chem. Soc. 2006;128:6048–6049. doi: 10.1021/ja060716f.Chi Y, Gellman SH. J. Am. Chem. Soc. 2006;128:6804–6805. doi: 10.1021/ja061731n.Chen Z, Yakura K, Matsunaga S, Shibasaki M. Org. Lett. 2008;10:3239–3242. doi: 10.1021/ol800965t.
- 5.a Saaby S, Nakama K, Alstrup Lie M, Hazell RG, Jørgensen KA. Chem. Eur. J. 2003;9:6145–6154. doi: 10.1002/chem.200305302. [DOI] [PubMed] [Google Scholar]; b Zhuang W, Saaby S, Jørgensen KA. Angew. Chem., Int. Ed. 2004;43:4476–4478. doi: 10.1002/anie.200460158. [DOI] [PubMed] [Google Scholar]
- 6.Suto Y, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2007;129:500–501. doi: 10.1021/ja068226a. [DOI] [PubMed] [Google Scholar]
- 7.For Ag-catalyzed Mannich-type reactions to aldimines (not AVM) in the presence of 1, see: Josephsohn NS, Snapper ML, Hoveyda AH. J. Am. Chem. Soc. 2003;125:4018–4019. doi: 10.1021/ja030033p.Josephsohn NS, Carswell EL, Snapper ML, Hoveyda AH. Org. Lett. 2005;7:2711–2713. doi: 10.1021/ol050910r.
- 8.For Ag-catalyzed AVM reactions to aldimines in the presence of ligand 1, see: Carswell EL, Snapper ML, Hoveyda AH. Angew. Chem., Int. Ed. 2006;45:7230–7233. doi: 10.1002/anie.200603496. For related investigations, see: Uraguchi D, Sorimachi K, Terada M. J. Am. Chem. Soc. 2004;126:11804–11805. doi: 10.1021/ja046185h.Yamaguchi A, Matsunaga S, Shibasaki M. Org. Lett. 2008;10:2319–2322. doi: 10.1021/ol800756r.Akiyama T, Honma Y, Itoh J, Fuchibe K. Adv. Synth. Catal. 2008;350:399–402.
- 9.For a review regarding Ag-catalyzed enantioselective reactions, see: Naodovic M, Yamamoto H. Chem. Rev. 2008;108 doi: 10.1021/cr068413r., ASAP.
- 10.Ketoimine substrates exist predominantly as an E isomer (2.5−20:1 E:Z); there appears to be no correlation between diastereo- or enantioselectivity and such ratios. For example, 4b (93% ee) is a 5:1 E:Z ratio whereas 4h (94% ee) is nearly exclusively E (95:5). Spectroscopic studies indicate that the ketoimine isomers can readily interconvert at 22 °C.
- 11.The absolute stereochemistry of anti product diastereomers in Table 2 is based on an X-ray crystal structure analysis. See the Supporting Information for details.
- 12.Use of two equivalents of enolsilane is required for efficient AVM (to achieve >98% conv).
- 13.Control experiments indicate that Michael addition is Ag-catalyzed but does not stereoselectively consume one product diastereomer. Thus, selective catalytic Michael reaction is likely not the cause of the difference in dr values observed with different quenching procedures.
- 14.Removal of the N-aryl group in 7a in the presence of CAN was performed in the following manner: 3 equiv CAN, 1:1 MeCN:H2O, 0 °C, 10 min; 10 equiv H2SO4, 0 °C, 10 min; Na2CO3 (pH = 10), wash with CH2Cl2.
- 15.For further details, see the Supporting Information.
- 16.The lower selectivity in reactions with 12, containing a small amount of the more active 1:1 complex, is consistent with the observations that at low catalyst loadings, AgOAc and 1 deliver inferior results.
- 17.The minor amount of the 1:1 complex in the solution derived from crystalline 12 is likely due to a small degree of phosphine dissociation. Accordingly, addition of excess 1 leads to complete disappearance of the 1:1 complex (NH at δ 10.78 ppm).
- 18.For P–Ag–P coupling constants, see: Ohkouchi M, Masui D, Yamaguchi M, Yamagishi T. J. Mol. Catal. A. 2001;170:1–15.
- 19.For P–Ag coupling constants, see: Goel RG, Pilon P. Inorg. Chem. 1978;17:2876–2879.
- 20.The integration value obtained by analysis of the 1H and 31P NMR spectra must be corrected since one complex bears two amide protons and two P atoms, respectively.
- 21.The precise amount of excess AgOAc cannot be measured due to the salt's incomplete solubility in the reaction solvent.
- 22.For a review regarding Lewis base activation of Lewis acids, see: Denmark SE, Beutner GL. Angew. Chem., Int. Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943.
- 23.The stereochemical identity of AVM product syn-14 is based on 1H NMR analysis (comparison with other related syn and anti diastereomers, the structures of which have been determined by X-ray analysis).
- 24.The chiral ligand related to 16 but lacking the two methoxy substituents at the N-terminus is only slightly less effective (>98% conv under identical conditions; 91.5:8.5 er, 83% ee, 66% yield for 16).
- 25.The relative and absolute stereochemistry of 17 has been established by X-ray crystal structure analysis. See the Supporting Information for details.
- 26.Attempts to prepare and study Ag-catalyzed AVM reactions of the methyl-substituted ketoimine ester that bears a p-nitro-o-methoxyphenylimine led to complex mixtures of products, presumably as a result of enamine formation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.