

Abstract
The iodothyronine deiodinases initiate or terminate thyroid hormone action and therefore are critical for the biological effects mediated by thyroid hormone. Over the years, research has focused on their role in preserving serum levels of the biologically active molecule T3 during iodine deficiency. More recently, a fascinating new role of these enzymes has been unveiled. The activating deiodinase (D2) and the inactivating deiodinase (D3) can locally increase or decrease thyroid hormone signaling in a tissue- and temporal-specific fashion, independent of changes in thyroid hormone serum concentrations. This mechanism is particularly relevant because deiodinase expression can be modulated by a wide variety of endogenous signaling molecules such as sonic hedgehog, nuclear factor-κB, growth factors, bile acids, hypoxia-inducible factor-1α, as well as a growing number of xenobiotic substances. In light of these findings, it seems clear that deiodinases play a much broader role than once thought, with great ramifications for the control of thyroid hormone signaling during vertebrate development and metamorphosis, as well as injury response, tissue repair, hypothalamic function, and energy homeostasis in adults.
I. Introduction
- II. Structure and Cell Biology of Deiodinases
- A. Subcellular localization and structure
- B. Selenoprotein synthesis
- C. Posttranslational regulation of deiodinases
- III. Cellular Control of Thyroid Hormone Signaling
- A. Transport across membranes
- B. Deiodinase-mediated mechanisms
- C. Deiodination in intact cells and cofactor availability
- D. Conjugation of iodothyronine and its effects on deiodination pathways
- E. Deiodinases and thyronamines
- IV. Deiodinase-Mediated Thyroid Hormone Signaling in Discrete Cell Systems
- A. Development and metamorphosis
- B. Energy homeostasis
- C. Deiodinases in the hypothalamus/pituitary systems
- D. Transgenic murine models of ectopic D2 expression in the heart
- V. Deiodination in Disease States
- A. Local impact of altered deiodinase activity on thyroid hormone signaling in disease states
- B. Systemic impact of abnormal deiodinase activity on thyroid hormone homeostasis
- C. Genetic basis of deiodinase-related diseases
VI. Conclusions
I. Introduction
ALTHOUGH T4 WAS ISOLATED as early as 1915 by Edward C. Kendall, T3 was only identified in humans in 1952 by Gross and Pitt-Rivers (1), sparking the idea that iodothyronine deiodination was a physiological pathway in vertebrates. However, another 18 yr passed until conclusive evidence was obtained when Braverman et al. (2) demonstrated that both stable T3 and 125I-T3 were found in the serum of athyrotic patients who were given mixtures of stable and 125I-labeled T4. Today, T4 is known to be the main secretory product of the thyroid gland in all vertebrates and is recognized as a long-lived minimally active molecule. Deiodination catalyzes the first step of thyroid hormone action by conferring biological activity to T4, a metabolic pathway that removes one iodine residue from the T4 molecule to produce T3, the short-lived most active form of thyroid hormone. Moreover, most circulating T3 is derived from extrathyroidal T4 deiodination. T3 modulates gene expression in virtually every cell through ligand-dependent transcription factors: the thyroid hormone receptors (TRs) (3,4,5).
T4 is a phenoxyphenyl polyiodinated molecule. To produce T3, deiodination must occur in the outer ring of the T4 molecule, a reaction catalyzed by the type 1 and type 2 iodothyronine deiodinases (D1 and D2, respectively). If deiodination occurs in the inner ring, a rT3 molecule is produced that lacks known TR-mediated biological activity. Inner ring deiodination is catalyzed by the type 3 deiodinase (D3), which can also deiodinate T3 and thereby terminate thyroid hormone action. Identification of the different deiodinase pathways took time (Fig. 1). Characterization of the D1 activity in liver and kidney homogenates (6,7,8) and a number of kinetic studies performed in animals and humans were the early experiments that attracted interest in this field and resulted in extensive progress (9,10,11,12,13). Today, the function of D1 in humans is poorly understood, aside from its role in producing high serum T3 concentrations in hyperthyroidism and susceptibility to inhibition by 6-n-propyl-2-thiouracil (PTU) and amiodarone (14,15,16,17,18). The discovery of the more widely expressed D2, which displays three orders of magnitude higher affinity for T4, not only revealed the predominant T3-generating pathway but also highlighted the new function of deiodinases in thyroid hormone homeostasis (19). For example, the finding that D2 is up-regulated by hypothyroidism and the opposite in hyperthyroidism illustrates that the changes in D2 expression and activity contribute to the constancy of T3 levels in the plasma as well as at the cellular level. The subsequent discovery of the inactivating deiodinase D3, which normally is expressed in the central nervous system and placenta, completed the picture (20,21,22). Combined, the roles played by D2 and D3 provided an elegant homeostatic mechanism by which coordinated reciprocal changes in their activities ensure adaptation to iodine availability in the environment.
Figure 1.

Schematic of deiodinase-mediated metabolism of the major iodothyronines. [Figure was modified with permission from Bianco and Kim: J Clin Invest 116:2571–2579, 2006 (33). ©American Society for Clinical Investigation.]
Departing from the concept that deiodinase function is to maintain thyroid hormone homeostasis, it has become increasingly clear that these enzymes also locally control thyroid hormone action. This concept has evolved from the original observation that D2 activation increases intracellular T3 concentration in the brown adipose tissue (BAT) of cold exposed rats (23) and that this saturates local TR (24,25,26). Most importantly, D2-mediated TR saturation increased thyroid hormone signaling at a local level, causing significant effects on the transcription of T3-responsive genes in D2-expressing cells (24,25). Analogous to the studies involving D2 expression, ectopic expression of D3 has been shown to promote cell-specific hypothyroidism and to decrease T3-dependent gene transcription, relevant in development and tissue injury (27,28). Today, this concept has gained tremendous support from the discovery of a number of cellular systems in which thyroid hormone signaling is regulated by the deiodinases (29,30,31,32). Thus, a well-accepted paradigm has emerged in which deiodinases control thyroid hormone action at the cellular level, relatively independent of thyroid hormone serum concentrations (33). This review will focus on the most recent progress achieved in this field, particularly the cellular and molecular aspects of local control of thyroid hormone signaling provided by deiodinases. For more broad reviews on thyroid hormone deiodination, readers are referred to a series of articles that have been published in the recent years (5,34,35,36,37), including the features of deiodinase-deficient transgenic mice summarized in Table 1 (33,35).
Table 1.
Characteristics of deiodinase-deficient adult mice compared with wild-type
| Knockout models | D1−/− | D2−/− | D3−/− | D1/D2−/− |
|---|---|---|---|---|
| Serum T4 | ↑ | ↑ | ↓ | ↑ |
| Serum T3 | Normal | Normal | ↓ | ↓ |
| Serum rT3 | ↑ | NR | NR | ↑ |
| TSH | Normal | ↑ | Normal or modest ↑ | ↑ |
| Systemic phenotype | Euthyroid | Euthyroid | Hypothyroid | NR |
| Specific features | Increased fecal excretion of iodothyronines | Mild cold intolerance, hearing impairment | Central hypothyroidism, growth retardation, neonatal brain thyrotoxicosis | NR |
| Reproduction | Normal | Normal | Impaired | NR |
II. Structure and Cell Biology of Deiodinases
Iodothyronine deiodinases constitute a group of three dimeric thioredoxin (TRX) fold-containing selenoproteins that catalyze the reductive dehalogenation of iodothyronines. The subcellular localization of the deiodinases varies according to the subtype, and this affects their systemic vs. cellular contributions to thyroid hormone homeostasis and action (38,39). Importantly, these enzymes are distinct from the iodotyrosine deiodinase 1 (DEHAL 1) that is expressed in the thyroid and acts on the mono- and diiodotyrosines released during the hydrolysis of thyroglobulin to liberate and recycle iodide (40).
A. Subcellular localization and structure
The molecular weight of the deiodinases varies between 29 and 33 kDa, and their sequence identity is lower than approximately 50%; nevertheless, they all share an approximately 15 amino acid-long conserved selenocysteine-containing active center, and all have one transmembrane domain (5). Protein modeling using hydrophobic cluster analysis (41,42) was helpful in developing a general three-dimensional (3D) model for the deiodinase family and also in providing insights into structural aspects of deiodinase dimerization and D2 ubiquitination (43,44). These studies have provided insight into the mechanisms responsible for the complementary and divergent abilities of the deiodinases to modulate thyroid hormone signaling.
1. Subcellular localization.
The cellular location of the individual deiodinases is an important feature of these enzymes, given that thyroid hormone is supplied from and eventually equilibrates with the plasma compartment, whereas thyroid hormone action is mediated inside the nucleus of the cell. Studies using cell models where the deiodinases were either transiently or endogenously expressed indicate that D1 and D3 are located in the plasma membrane, whereas D2 is an endoplasmic reticulum (ER) resident protein (45,46,47,48,49,50) (Fig. 2). The presence of D1 in the plasma membrane explains previous findings that T3 generated in the liver and kidney via D1 rapidly equilibrates with plasma T3, and the poor contribution of locally generated T3 to the overall TR occupancy in such tissues (51).
Figure 2.

Subcellular localization of deiodinases by immunofluorescence. HEK-293 cells were transiently transfected with FLAG-tagged D1, D2, or D3 (left). D1 (green) is located in the plasma membrane region and does not colocalize with the ER marker GRP78/BIP (red), in contrast to D2 (green), which is located in the ER. D3 (green) was colocalized to the plasma membrane with Na/K ATPase (red). Immunofluorescence staining of endogenously expressed deiodinases is shown in the right panel. D1 in porcine thyrocytes is found in the plasma membrane (image kindly provided by Dr. Peter Arvan, Ann Arbor, MI), whereas endogenously expressed D2 in MSTO-211H cells colocalized with GRP78/BIP in the ER. Endogenous D3 protein was detected in the plasma membrane of NCLP-6E cells. Scale bar, 10 μm. [Reprinted with permission from Baqui et al.: Endocrinology 141:4309–4312, 2000 (48), ©The Endocrine Society. Reproduced with permission of the Company of Biologists from Prabakaran et al.: J Cell Sci 112:1247–1256, 1999 (47); Curcio et al.: J Biol Chem 276:30183–30187, 2001 (49), ©ASBMB, Inc.; and Baqui et al.: J Biol Chem 278:1206–1211, 2003 (50), ©ASBMB, Inc.]
D2 is subjected to static retention in the ER and does not reach the Golgi compartment during its trafficking (52). Therefore, D2 generates T3 in the cytosol with potential ready access to the nucleus by virtue of the physical proximity between the ER and the nuclear compartment. This would explain the much longer residence time of the D2-generated T3 (hours) when compared with T3 entering the cell directly from the plasma (minutes) (53). In tissues such as brain, however, a more complex mechanism regulating T3 access to the nucleus is likely to exist, given that D2 is expressed in glial cells that are not the ultimate destination of T3 (neurons) (see Section IV.C). This could also include the existence of a stereospecific transporter system capable of generating a higher nuclear/cytoplasm T3 ratio as previously proposed (54).
Although the mechanism of ER retention remains to be fully elucidated, in an experimental setting D2 ER-retention could be overridden by fusion of a transmembrane-less D2 molecule to the long-lived plasma membrane protein sodium-iodine symporter (NIS) (52). The finding that the D2-NIS fusion protein is targeted to the plasma membrane suggests that the signal determining D2 ER retention is located in the transmembrane domain, although it is also possible that the D2 retention mechanism might be weaker than the plasma membrane determinant of NIS. On the other hand, using similar strategies, a transmembrane-less D1 could be directed to the ER by fusion to the ER resident protein Sec62 (52).
Like many other proteins anchored in the plasma membrane, D3 is internalized and becomes part of the endosomal vesicles (50). Using immunofluorescence, D3 was colocalized with Na,K-ATPase α in the plasma membrane and intracellularly with the early endosomal marker EEA-1 and clathrin, but not with two ER-resident proteins (50). These studies also indicated that there is constant internalization of D3 that is blocked by sucrose or methyl-β-cyclodextrin-containing medium. Additionally, exposing cells to a weak base such as primaquine increases the pool of internalized D3, suggesting that D3 is recycled between plasma membrane and early endosomes (50). However, the signal(s) controlling the partition of D3 between these two pools are currently unknown.
Although immunocytochemistry places the D3 globular domain in the extracellular space (50), it was suggested that catalysis is an intracellular event because coexpression of the monocarboxylate transporter 8 (MCT8), a thyroid hormone transporter, increases D3-mediated deiodination (55). Although this could be explained by the active center of D3 being cytosolic, it might also indicate that D3 catalysis takes place in endosomal vesicles, with MCT8 necessary for T3 entry into these vesicles.
2. Deiodinases are TRX-fold containing proteins.
Despite their different substrate preferences and cellular localization, the deiodinases have a similar protein structure. Structural analyses of the deiodinases have been hindered by their integral membrane nature and the inherent inefficiency of selenoprotein synthesis (see Section II.B). Nevertheless, insights into their structures have been obtained through in silico protein modeling. Based on hydrophobic cluster analysis, it has become clear that the three deiodinases share a common general structure composed of a single transmembrane segment in the N termini of D1, D2, and D3, and several clusters typical of α-helices or β-strands that correspond to core secondary structures composing the deiodinase globular domains (43). A striking common feature is the presence of the TRX fold, defined by βαβ and ββα motifs. This finding that deiodinases are TRX-fold family proteins is not completely unexpected because glutathione peroxidase (GPX) and other selenocysteine-containing thiol-interacting oxidoreductases are also members of this group (56). Interestingly, the relationship between the βαβ and ββα motifs within the canonical TRX fold is locally interrupted by distinct secondary structure elements, a feature also observed in other proteins of the TRX-fold family (56). However, a unique structural aspect of the deiodinases is that they contain intervening elements within their TRX-fold structure that are highly similar (47% identity with D1 and D3, 60% with D2) to α-L-iduronidase (IDUA), a lysosomal enzyme that cleaves α-linked iduronic acid residues from the nonreducing end of glycosaminoglycans (57).
3. Deiodinase dimerization.
It has become increasingly clear that the protein structure of the deiodinases leads to higher order interactions that influence their activity. In early studies, gel filtration and gradient centrifugation of solubilized kidney or liver microsomal membranes were used in an attempt to purify D1 and D2 (58,59). These studies detected deiodinase activity in higher molecular weight forms than predicted from their respective deduced amino acid sequences (29–33 kDa) (58,59). However, it was unclear whether these high molecular weight bands represented association of deiodinases with other cellular proteins or dimerization of the deiodinases themselves (60,61,62,63).
Expression of the inactive rat D1 Sec126Ser protein in porcine LLC-PK1 cells with endogenous D1, followed by the immunoprecipitation of D1 activity using a rat D1-specific antibody, indicated that D1 homodimers can be found in cells (64). However, such dimers or other deiodinase-containing high molecular weight complexes could not be visually identified in cells endogenously expressing deiodinases (49,65,66,67). Subsequent studies using three independent approaches in a cellular model transiently expressing the deiodinases demonstrated the existence of D1, D2, and D3 dimers (68). Evidence for dimer formation includes: 1) identification of higher molecular weight bands of appropriate size for a putative dimeric enzyme by Western blot analysis; 2) immunoprecipitation of deiodinase (D1 or D2) activity from cells coexpressing inactive FLAG-tagged deiodinase and the respective untagged active enzymes (D1 or D2) using anti-FLAG antibodies; and 3) immunoprecipitation of 75Se-deiodinase (D1, D2, or D3) from cells coexpressing unlabeled FLAG-tagged deiodinases and the respective selenocysteine-containing enzymes using anti-FLAG antibodies.
Deletion/truncation analysis coupled with immunodepletion assays suggested that a conserved C-terminal region of D1 corresponding to rat amino acids 148–163 serves as a deiodinase dimerization domain responsible for posttranslational assembly of deiodinases (69). Further characterization of this domain using alanine scanning mutagenesis identified 152IYI154 as critical residues required for D1 dimer assembly (70). Use of a similar strategy identified five residues (153FLIVY157) at the beginning and three residues (164SDG166) at the end of a homologous region of D2 as required for dimerization (70).
D2 dimerization is the best understood among the deiodinases. Live-cell fluorescence resonance energy transfer (FRET) and bioluminescence resonance energy transfer were used to study the interaction between D2 domains in the D2:D2 homodimer (44) (Fig. 3). D2 has a typical transmembrane helix containing potentially charged residues (D, E, K, R, H) that can be stabilized in the hydrophobic membrane environment by dimerization. In this way, charged residues could be neutralized via an intermolecular interaction of residue couples such as D29/K35 as well as direct contacts between polar residues such as H36-K35′, which is compatible with modeling of the two D2 transmembrane segments in an αα dimeric architecture. In support of this, a truncated D2 molecule missing the first 42 amino acids is a cytosolic protein that does not homodimerize. In addition to this critical role played by the transmembrane domain, interaction at the globular domain has also been demonstrated because a full-length D2 molecule dimerizes with the truncated D2 monomer lacking the transmembrane domain (44).
Figure 3.

Deiodinases are dimers. FRET measures the transfer of energy from an excited CFP-tagged molecule to a YFP-tagged acceptor molecule in close proximity. The FRET between D2-CFP and D2-YFP fusion proteins in transfected cells is shown. The location of the CFP and YFP chromophores relative to the D2 protein is indicated by N (amino) or C (carboxyl), respectively, and results are expressed as a percentage of the FRET of a positive control YFP-CFP fusion protein indicated by a plus symbol. CFP + YFP is a negative control for both proteins expressed alone. Notably, D2 with an N-terminal fusion of CFP has no FRET with D2 a C-terminal fusion of YFP, whereas FRET is observed between an N-terminal fusion of CFP to D2 and a N-terminal fusion of YFP to D2, or between a C-terminal fusion of CFP to D2 and C-terminal fusion of YFP to D2 (left panel, last 3 columns). For the bioluminescence resonance energy transfer studies (right panel), YFP fused to the N or C terminus of D2 and Renilla luciferase fused to the C terminus of D2 were expressed in transfected cells. Luminesence produced by the luciferase molecule can then excite a YFP molecule in close proximity, and the resulting YFP emission is measured. YFP, Yellow fluorescent protein; CFP, cyan fluorescent protein. [Reprinted with permission from Sagar et al.: Mol Cell Biol 27:4774–4783, 2007 (44). ©The American Society for Microbiology.]
Further characterization of the deiodinase globular dimerization interface was performed by taking advantage of the finding that deiodinases are TRX fold-containing proteins (43). The deiodinase globular interacting interface was tentatively modeled by comparison to the human TRX fold (44) (Fig. 4A) and shows a clear propensity to dimerize via a large area formed by the alignment of two β strands that constitute a small β sheet (71). Due to the high sequence identity around the canonical TRX β1-α1-β2 motif, the deiodinase globular dimerization model could be fitted on the crystal structure of dimeric oxidized TRX with no evident conflict. These studies indicate that the native deiodinase dimer is formed by interactions at both the transmembrane and the globular cytosolic domains (Fig. 4B). The fact that dimerization is critical for catalytic activity indicates that proper conformation for each active center can only be achieved in the dimeric state. All three deiodinases contain a region of negative electrostatic potential around their active sites, whereas the rest of the molecule is positively charged (44) (Fig. 4C).
Figure 4.

Globular interfaces mediate D2 dimerization and are critical for catalytic activity. A, Two orthogonal views of the modeled D2 dimer on the template of the crystal structure of human TRX dimer. At the top, the twofold axis is vertical, and at the bottom, it is perpendicular to the figure. Secondary structures are colored. The putative structure of the iduronidase-like active site insertion has been modeled as a ββ secondary structure (βd1 and βd2) lying between β2 and αB. βd1 is green/light purple, βd2 is dark purple, and, at the bottom of the dimer, the two symmetrical small βT in pink are the counterparts of the canonical TRX pairing. B, 3D model of the D2–D2 homodimer. N′ter and Cter indicate the N terminus of the TRX fold head domain and the C terminus of D2, respectively. A single large cavity is created upon D2 dimerization at the level of the active site (Sec133). C, Visualization of the Russian-doll-shaped electrostatic field around the D2 dimer (the −1.8-kT/e gradient limit is red and the +1.8-kT/e gradient limit is blue). [Reprinted with permission from Sagar et al.: Mol Cell Biol 27:4774–4783, 2007 (44). ©The American Society for Microbiology.]
FRET was also used to study the dimerization of D1 and D3 in living cells (72). Similar to D2, both enzymes also form homodimers in their native state via dimerization surfaces located in the transmembrane and globular domains. As with D2, dimerization is required for the catalytic activity of both enzymes. Minor heterodimerization was also detected between D3:D1 or D3:D2, but the significance of this observation is currently unknown (72).
4. The selenocysteine-containing active center.
The structure and dimerization of the deiodinases leads to a general 3D model that predicts an active center defined by the β1-α1-β2 motifs of the TRX fold and one of the IDUA intervening elements (43). In this pocket is the rare amino acid selenocysteine (Sec), initially found in D1 (73,74,75), that is critical for the deiodination reaction catalyzed by all three deiodinases (55). Replacement of the selenocysteine with cysteine decreases by two or three orders of magnitude the affinity of D2 for T4 and that of D3 for T3, whereas D1 affinity for rT3 is less affected (5). Mutagenesis studies demonstrated the functional relevance of this model by revealing that other amino acid mutations in the TRX-IDUA active center change important kinetic properties of the deiodinases (43). For example, D1 is normally sensitive to PTU and follows ping-pong kinetics with binding alternating between its two substrates, the reducing cofactor (e.g., dithiothreitol) and the iodothyronine. However, Ser128Pro modification of this enzyme resulted in resistance to PTU and the loss of ping-pong kinetics (43). A corresponding substitution of Ser for Pro in the equivalent position of D2 (135 a.a.) altered D2 such that its kinetics were now D1-like, being sensitive to PTU and displaying ping-pong kinetics and an increased Michaelis-Menten constant (Km) for T4 (43). In D3, substitution of Ser for Pro in the equivalent position (146 a.a.) also resulted in a more D1-like enzyme with a high sensitivity to inhibition by PTU and an increased Km for T3 (43).
B. Selenoprotein synthesis
Selenoproteins are unique in that they contain one or more residues of the rare amino acid selenocysteine in their structure. To date, less than 30 selenoproteins have been identified, and their function is not always well characterized. D1 was only the second eukaryotic selenoprotein to be identified, the cloning of which also led to the discovery of the mechanisms leading to selenocysteine incorporation in higher organisms (73,76).
1. Deiodinases must recode UGA from a signal for stop.
D1, D2, and D3 all contain selenocysteine at their active center, making them part of the 25-member selenoprotein family (77). UGA encodes for the insertion of selenocysteine during translation, yet in most cases of protein synthesis this codon is normally read as a signal for termination. In order for UGA to specify selenocysteine incorporation, additional components are required to facilitate translation, previously detailed elsewhere (5,78). In brief, selenoprotein synthesis requires an in-frame UGA codon and a downstream stem loop structure termed the selenocysteine insertion sequence (SECIS) that binds SECIS binding protein-2 (SBP-2). SBP-2 in turn interacts with an elongation factor, EFsec, promoting the insertion of selenocysteine from a specific tRNA (Sec-tRNASec) by the ribosome at the UGA codon (Fig. 5). Selenocysteine incorporation is not very efficient, and if the UGA codon in D1 is replaced with a cysteine codon (CysD1), up to 400-fold more D1 protein is produced (79). All the constraints that apply to selenoprotein production will also apply to deiodinase synthesis. This concept has been further validated with the discovery of patients with impaired deiodinase activity due to a mutation in SBP-2 (80) (see Section V.C.3).
Figure 5.

Schematic of selenoprotein synthesis. As reviewed in Section II.B, in order for UGA to encode for selenocysteine insertion and not translational termination, the selenoprotein mRNAs require a downstream stem loop structure, the SECIS. The SECIS element binds SBP-2, which in turn interacts with a selenocysteine-specific elongation factor, EFsec. EFsec also binds the selenocysteine tRNA (Sec-tRNASec) and promotes selenocysteine incorporation in the elongating protein by the ribosome at the UGA codon. An additional SECIS binding protein, L30, can displace SBP-2 and anchor the loaded SECIS complex to the ribosome. The role of SECp43 remains to be defined; however this protein has been shown either to interact directly with or facilitate the interaction between many components needed for selenoprotein synthesis. The recently defined mammalian pathway of selenocysteine synthesis is also illustrated in the lower part of the figure, with the tRNASec initially being misacylated with serine, which is then phosphorylated by phosphoseryl-tRNA[Ser]Sec kinase (PSTK). SLA then dephosphorylates this serine, which is then followed by acceptance of active selenium generated via SPS-2.
2. Refining the model of selenoprotein synthesis.
In the last 5 yr, the model of how selenoproteins such as the deiodinases are translated has been significantly refined by numerous molecular studies. Recent areas of progress include the elucidation of the components needed for eukaryotic selenocysteine-tRNASec synthesis, a further definition of the role of SBP-2, and the identification of an additional SECIS binding protein, L30. Furthermore, several studies have provided insight into the interactions between the components needed to facilitate selenocysteine incorporation. A summary of these recent findings is detailed below.
In prokaryotes, selenocysteine synthesis occurs on its tRNA with selenocysteine-tRNASec initially being misacylated with serine, followed by dehydroxylation by the selenocysteine synthetase (SelA), and then addition of an activated selenium generated by the selenophosphate synthetase (SelD) (81). In eukaryotes, two SelD homologs, selenophosphate synthetase 1 and 2 (SPS-1 and SPS-2), had been identified, yet a functional SelA candidate remained elusive (82,83). The first piece of this puzzle was solved with the discovery of phosphoseryl-tRNA[Ser]Sec kinase, a kinase that specifically phosphorylates seryl-tRNASec to form O-phosphoseryl-tRNASec (Fig. 5) (84). Subsequently, soluble liver antigen (SLA), a selenocysteine-tRNASec binding protein of previously unknown function, was shown to dephosphorylate O-phosphoseryl-tRNASec in a pyridoxal phosphate-dependent manner (85,86). After dephosphorylation, activated selenium generated by SPS-2 can then be accepted to form selenocysteine in eukaryotes (85).
Recent studies have further defined the role of SBP-2 in promoting UGA read-through and provided insight into its role in the “hierarchy” of selenoprotein synthesis because some SECIS elements have a higher affinity for SBP-2 than others (87,88,89). A greater affinity for SBP-2 would enable more relative translation of a particular mRNA in conditions of limiting SBP-2, while also facilitating the pioneer round of translation, thereby rendering that mRNA potentially more resistant to nonsense-mediated decay. With this in mind, when SBP-2 was knocked down in a cell line with endogenous D2 activity, D2 message was unchanged, whereas other selenoprotein mRNA levels decreased (89). This suggests that the D2 mRNA has a higher affinity for SBP-2 and is more resistant to nonsense-mediated decay than other selenoprotein mRNAs such as selenoprotein H, GPX types 1 and 4, or the TRX reductases.
Recent work has identified the ribosomal protein L30 as an additional factor that binds SECIS elements and enhances selenocysteine incorporation (Fig. 5) (90). These studies determined that SECIS elements could exist in either an open or kinked conformation with SBP-2 preferring to bind to the open form, whereas L30 binds to both conformations. L30 also competes with SBP-2 for SECIS binding, leading to a model where SBP-2 initially recruits EFsec and selenocysteine-tRNASec to the translating selenoprotein mRNA, and a subsequent conformational shift in the SECIS leads to displacement of SBP-2 and binding of L30, anchoring the loaded SECIS complex to the ribosome.
Much of the current work on selenoprotein synthesis has additionally addressed the role of subcellular localization and intermolecular interactions in selenocysteine incorporation. SPB-2 has several nuclear localization sequences and shuttles between the cytoplasm and nucleus with EFsec, and this can be modulated by cellular oxidation state (91,92). SBP-2 is found to associate with the ribosome but is unable to simultaneously interact with the ribosome and SECIS element, lending further support to a model where SBP-2 binding to the SECIS is displaced by L30 (90,93). Other work indicates that a selenocysteine-tRNASec interacting protein of previously unknown function, SECp43, may coordinate multiple steps leading to selenoprotein synthesis (Fig. 5) (94). SECp43 has been found to interact with SLA and SPS1, and this interaction shifts their localization to the nucleus. SECp43 also promotes interactions between SBP-2 and EFsec and increases selenocysteine incorporation and selenoprotein mRNA levels (94,95).
C. Posttranslational regulation of deiodinases
1. Regulation of D2 activity half-life.
D2 is considered the critical homeostatic T3-generating deiodinase due to its substantial physiological plasticity. A number of transcriptional and posttranscriptional mechanisms have evolved to ensure limited expression and tight control of D2 protein levels, which is critical for its homeostatic function (96,97,98). D2 activity/mRNA ratios are variable, indicating that there is significant posttranslational regulation of D2 expression (99,100,101). In fact, the decisive biochemical property that characterizes the homeostatic behavior of D2 is its short half-life (∼40 min) (102), which can be further reduced by exposure to physiological concentrations of its substrate, T4, and in experimental situations, rT3 or even high concentrations of T3 (102,103,104,105,106). This down-regulation of D2 activity by substrate is a rapid and potent regulatory feedback loop that efficiently controls T3 production and intracellular T3 concentration based on availability of T4 (102,103,105,106,107,108).
The first indication that D2 undergoes selective degradation in the proteasome originated from studies in rat pituitary tumor cells (GH4C1) that endogenously express D2. Treatment of these cells with the proteasomal inhibitor carbobenzoxy-L-leucyl-L-leucyl-L-leucinal stabilized D2 activity and prevented the loss of D2 activity after cells were exposed to T4 or rT3 or when protein synthesis was inhibited with cycloheximide (109). Although these data indicate that the proteasomal system is involved in the regulation of D2 activity, they do not provide a mechanistic explanation for the findings. Later, D2 protein levels were quantified under similar conditions after labeling with 75Se and immunoprecipitation with anti-D2 antisera (110). These studies revealed that the loss of D2 activity is due to a decrease in D2 protein levels, indicating that proteolysis of D2 is a critical step in D2 regulation. Additionally, if the selenocysteine in D2 was replaced with Cys (CysD2), an approximately 1000-fold higher T4 concentration was required for substrate induced degradation, proportional to the higher Km of CysD2. At the same time, a catalytically inactive AlaD2 mutant was not affected by exposure to substrate (110). This indicates that the T4-induced degradation of D2 requires a direct interaction of T4 with the catalytic center of the enzyme.
Selective proteasomal degradation of many short-lived proteins is initiated by conjugation to ubiquitin (Ub), a conserved approximately 8-kDa protein that is covalently bound to lysine side chains in the target protein (111). After Ub is activated by the Ub activating enzyme (E1), the combined actions of Ub conjugation enzymes (E2) and Ub ligases (E3) mediate Ub conjugation to the specific substrate. Ubiquitinated proteins can then be either degraded in the proteasome complex or deubiquitinated to its unconjugated form (111,112). In contrast to a single E1, about two dozen E2 proteins have been identified, and all share a conserved core domain of 150 amino acids (113). E2s have limited substrate specificity and are involved in the ubiquitination of different general classes of substrates (114). In contrast, E3 ligases have no overt sequence homology, and this large group of proteins is primarily responsible for the target specificity of the ubiquitination process (111,113).
2. Ubiquitination regulates D2 activity.
Direct evidence for D2 ubiquitination has been obtained using CHO-ts20 cells where E1-mediated activation of Ub can be blocked at restrictive higher temperatures. Under these conditions, E1 inactivation decreased the amount of ubiquitinated D2 (115). In addition, high molecular mass (100–300 kDa) ubiquitinated forms of D2 have been identified in HEK293 cells transiently expressing D2, and the amount of Ub-D2 increased as expected upon substrate exposure or by treatment with carbobenzoxy-L-leucyl-L-leucyl-L-leucinal. Additionally, catalysis (i.e., complete T4 to T3 conversion) is not necessary for ubiquitination because under certain experimental conditions of cofactor depletion, T4 can still induce D2 ubiquitination even without T3 generation (44). Importantly, D2 activity is correlated with the amount of D2 and not Ub-D2, indicating that D2 is inactivated upon ubiquitination (115). In contrast, to this date no ubiquitinated forms of D1 or D3 have been identified under similar conditions, in agreement with the long (>12 h) half-life of these enzymes (115).
3. Identification of the D2 ubiquitinating core complex
a. Ub conjugases (E2).
Because the process of Ub-mediated proteolysis is well characterized in the yeast Saccharomyces cerevisiae, it was used to identify E2s involved in D2 ubiquitination. The use of this model system was feasible because D2 ubiquitination and degradation is intact in yeast transiently expressing D2. Additionally, D2 retained its short half-life and sensitivity to substrate-mediated degradation and to inhibitors of the proteasome in this system (116). Because yeast lacks the appropriate machinery to synthesize selenoproteins, the CysD2 mutant was used in these studies; it also undergoes ubiquitination and proteasomal degradation (110). Notably, in mutant strains lacking UBC6 or UBC7, transiently expressed D2 was stabilized and exhibited an impaired sensitivity to substrate-induced degradation relative to the wild-type strain (116). UBC6 and UBC7 are both members of the ER-associated degradation (ERAD) pathway (117,118). Thus, these findings correlated with data identifying D2 as an ER-resident protein (48,52). Because elimination of misfolded or unfolded proteins is one aspect of ERAD (119), it was important to establish that these observations with UBC6 and UBC7 were not due to D2 protein misfolding. However, the observation that the half-life of D2 under basal conditions and upon exposure to substrate were both affected by mutations in UBC6 and UBC7 suggests that this was not the case (116).
The role of UBC6 and UBC7 in D2 ubiquitination has also been assessed in human cells. Using a dominant negative strategy based on the overexpression of inactive UBC6 and/or UBC7 (120), it was found that inactivation of both UBC6 and UBC7 was required for stabilization of D2 (121). In vitro binding studies demonstrated a high-affinity, specific physical association between murine UBC7 and the cytosolic carboxyl terminal region (amino acids 169–234) of D2 (48,121). Interestingly, UBC6 did not directly bind D2, but was associated with the D2-UBC7 complex (121). These studies suggest that UBC7 is the key D2 Ub conjugase, whereas UBC6 plays a less specific, accessory role.
b. Ub ligases (E3).
A yeast two-hybrid screen of a human brain library identified WD40-repeat/SOCS box protein 1 (WSB1), a downstream target of the morphogen sonic hedgehog (Shh), as a D2 interacting protein (31,122). Structurally, this protein contains seven WD-40 repeats that can form a β-propeller-like structure and mediate protein–protein interactions (123,124). Additionally, WSB1 contains a suppressor of cytokines signaling (SOCS) box at the carboxy-terminus, making it a potential candidate E3 ligase for D2 (123). SOCS-box motifs interact with the Elongin BC complex, a multifunctional regulatory complex controlling many different pathways within the cell (125). This complex assembles with the E2-activating cullin family member Cul5 and RING-H2 finger protein Rbx1 that in turn ligates Ub to protein substrates (126,127).
Hydrophobic cluster analysis (42) was used to investigate structural features of WSB1, and these studies predicted that WSB1 contains the hallmarks of a D2-specific E3 Ub ligase component, with a propeller-like structure formed by the WD-40 repeats mediating substrate recognition whereas the SOCS box facilitates the interaction with other components of the catalytic E3 ligase complex (31). Detailed experimental studies confirmed that D2 ubiquitination requires WSB1, and that Elongin B, Elongin C, Cul5, and Rbx1 do participate in the D2 ubiquitinating catalytic core complex (ECSWSB1) (31) (Fig. 6).
Figure 6.

The D2 ubiquitination machinery: composition of the ECSWSB1 catalytic core complex. Modeling of the Ub conjugating Cul5–Rbx1—Elongin C–Elongin B–von Hippel-Lindau complex associated with the SOCS box of WSB1 while the WSB1 propeller binds D2 is shown. Rbx1 interacts with the E2-enzyme Ubc7, which in turn associates with the ER membrane via Cue1, an ER membrane-anchored protein that is required for Ubc7 function. It is currently unknown whether only the D2 subunit not bound by WSB1 is undergoing ubiquitination or whether only this subunit is catalytically active. Ub is shown docked to Ubc7. T4 substrate is shown in white at the active site of the D2 dimer. [Reprinted with permission from Dentice et al.: Nat Cell Biol 7:698–705, 2005 (31).]
The interaction between D2 and the WD-40 propeller of WSB1 requires an 18-amino acid loop in D2 that, if removed, prolongs D2 half-life and abolishes D2 sensitivity to substrate-induced degradation (31). Further analyses demonstrated that the six amino-terminal amino acids in this 18-residue stretch are specifically required for D2 recognition by WSB1. Finally, this 18-amino acid instability loop, not the subcellular localization of D2, is the key determinant of D2 susceptibility to ubiquitination and rapid turnover rate (52).
Due to its key role in the regulation of D2 protein levels, the biology of WSB1 is also relevant to D2 cell biology and physiology. With this in mind, another WSB1 substrate was recently identified, homeodomain-interacting protein kinase 2, a nuclear protein kinase involved in the induction of apoptosis (128). However, it remains to be determined whether the signaling pathways controlled by homeodomain-interacting protein kinase 2 and D2 overlap. Additionally, it is notable that increased WSB1 copy number is associated with good prognosis in patients with neuroblastoma, suggesting a role for WSB1 and by extension perhaps thyroid hormone signaling, in the favorable outcome of this disease (129).
It has recently been reported that the WSB1 gene locus found on chromosome 11 undergoes an interchromosomal interaction with the imprinted Igf2/H19 locus on chromosome 7, which is suggested to direct these distant DNA segments to a common nuclear transcription factory. This physical interaction is essential for the appropriate expression of WSB1, suggesting that the transcriptional regulation of WSB1 is closely linked to that of the imprinted Igf2/H19 locus (130,131,132). Intriguingly, the Igf2/H19 locus is part of a large coordinately regulated imprinted gene network that also includes the imprinted Dlk1-Gtl2 locus, which notably contains Dio3 (see Section V.C.2) (133,134,135). Because many of the genes in this imprinted network are relevant for the control of embryonic growth (136), it is possible that the regulation of the WSB1 and Dio3 genes share some mechanisms, given that both act to reduce thyroid hormone signaling.
Heterologous expression of D2 in yeast also led to the discovery that D2 is potentially a substrate of another Ub ligase, Doa10 (137). TEB4 (MARCH-VI) is the mammalian ortholog of the yeast Doa10 protein, and these two proteins appear to have identical topology with 14 transmembrane helices resident in the ER membrane, whereas the bulk of these proteins, including the N and C termini, are located in the cytosol (138). TEB4 has been identified as a novel ER-resident Ub ligase, containing a conserved RING-CH finger domain at the N terminus, that catalyzes Ub ligation in a reaction involving UBC7 (139). Subsequent characterization of the role of TEB4 in D2 ubiquitination concluded that TEB4 can mediate loss of D2 activity under basal and substrate-induced conditions, likely as a result of D2 ubiquitination and subsequent proteasomal degradation (140). These findings suggest that D2 ubiquitination is under dual control by WSB1 and TEB4. Of note, some D2-expressing tissues express very little WSB1 and TEB4, suggesting that alternate pathways of D2 degradation likely exist.
Interestingly, patients with defective synthesis and/or secretion of thyroglobulin with relatively high serum free T3 (FT3) concentrations and disproportionately low serum free T4 (FT4) concentrations have increased D2, but not D1, activity in the thyroid gland (141). It is unlikely that the increase in thyroid D2 activity is simply the result of the lower serum T4 concentration given that D2 mRNA levels remained unaffected. Alternatively, it is tempting to speculate that the high FT3/FT4 ratios found in these individuals are the result of impaired D2 ubiquitination and proteasomal degradation in the thyroid and that the same impairment in the UBC6- and UBC7-mediated ERAD mechanisms is also involved in the accumulation of the large amounts of defective thyroglobulin retained intracellularly in the thyroids of these patients.
4. A model for regulation of D2 activity by ubiquitination.
While monitoring the D2 homodimer and its interaction with the ECSWSB1 catalytic core complex in studies using live-cell FRET (44), it became clear that there is a conformational change in D2 upon substrate binding that exposes the two critical lysine residues K237 and K244 to ubiquitination. Given their position within the D2 structure, ubiquitination of these residues interferes with the dimerization interface between the D2 globular domains. However, ubiquitination does not interfere with the N-terminal transmembrane dimerization domain of D2. Because correct D2 dimerization is critical for enzyme activity, ubiquitination of these lysines inactivates D2 but does not cause terminal disassembly of the molecule, raising the possibility that D2 could be reactivated by deubiquitination before degradation by the proteasome.
5. Deubiquitination reactivates D2.
Deubiquitinases, also known as Ub-specific processing proteases (USPs), are cysteine proteases that specifically cleave Ub from Ub-conjugated target proteins. Deubiquitination has been recognized as an important regulatory step in ubiquitination-mediated regulation of protein function, and importantly, deubiquitinase induction has been demonstrated to regulate many different physiological processes (142,143,144,145,146). In the same yeast two-hybrid screen of a human brain library that identified WSB1, von Hippel-Lindau protein (pVHL)-interacting deubiquitinating enzyme-1 (VDUI/USP33) was also identified as a D2-interacting protein, suggesting that this USP33 and its closely related family member and pVHL-interacting deubiquitinating enzyme-2 (VDU2/USP20) might be involved in the rescue and reactivation of D2 from ubiquitination and subsequent proteasomal degradation (147).
The interaction between D2 and USP33 and USP20 has been confirmed in mammalian cells, and coexpression of USP33 or USP20 with D2 prolongs D2 half-life and increases D2 activity. It has also been shown that USP33-mediated deubiquitination of D2 is an important element of the mechanisms for cold acclimatization in brown adipocytes (147), and D2 expression is transcriptionally induced in stimulated BAT by a cAMP-dependent mechanism (25,148,149). USP33 expression is also up-regulated by cold exposure or norepinephrine, thus decreasing D2 degradation and amplifying the transcriptional induction of D2 (147).
Hundreds of deubiquitinases have been cloned, yet the substrates of only a few have been identified. Although D2 was the first identified substrate of USP33 (VDU1) and USP20 (VDU2), USP20 has also been shown to deubiquitinate and stabilize hypoxia-inducible factor (HIF)-1α (147,150). Additionally, to date, D2 is the only known deubiquitinase substrate that is an ER resident protein (151,152,153,154,155).
Because the pVHL E3 ligase complex mediates the ubiquitination of USP33 and USP20, this complex could also indirectly regulate the deubiquitination of D2 (156,157). pVHL is part of an Elongin C, Elongin B, and cullin-2 Ub ligase (E3) complex (158,159) that is very similar to the ECSWSB1 catalytic core complex. Germline mutations of the VHL gene result in a hereditary cancer syndrome, Von Hippel-Lindau disease (160). It has also been demonstrated that USP33 is overexpressed in a specific subclass of acute lymphoblastic leukemia (161). However, it is currently unknown whether changes in D2 deubiquitination contribute to either of these disease states.
6. Structure-function aspects of the D2 ubiquitination/deubiquitination complex.
The dynamics of the interactions among the specific components of the ECSWSB1 catalytic core complex during D2 ubiquitination were studied using FRET (44). Remarkably, this revealed that the D2 homodimer is continuously associated with the ECSWSB1 catalytic core complex and USP33. Exposure to D2 substrate further increased the D2:WSB1 association, while decreasing the D2:UBC7 interaction (44). These changes are in agreement with the substrate-induced ubiquitination model of D2, because WSB1 promotes D2 ubiquitination. Moreover, it is logical to assume that the UBC7:D2 interaction would be weakened after ubiquitination of D2. The continuous association of D2 with this regulatory protein complex would support rapid cycles of deiodination, followed by deactivation via conjugation to Ub and subsequent enzyme reactivation by deubiquitination, allowing for tight control of T3 production and thyroid hormone signaling. These findings provide a novel model for ubiquitination-mediated regulation of protein function in which enzymatic function is transiently inactivated through the partial conformational change of a dimeric enzyme, which then can be reversed upon deubiquitination (44).
III. Cellular Control of Thyroid Hormone Signaling
Although it is intuitive that the intensity of thyroid hormone signaling should vary according to its plasma concentration, it is well recognized that a number of cellular mechanisms can modify T3 actions, including transporters across cellular membranes, deiodinase expression level and activity, as well as other modifications of the thyroid hormone molecule.
A. Transport across membranes
Thyroid hormone is hydrophobic, and thus it has been generally assumed for decades that simple diffusion through the plasma membrane would give thyroid hormone access to the intracellular compartment. However, for years a handful of investigators have claimed that this was not the case, based on experimental evidence that thyroid hormone transporters were mandatory for crossing the plasma membrane. Although credible, it was hard to attach a physiological relevance to these data given that under no known physiological or pathophysiological settings was thyroid hormone transport across the plasma membrane limiting for thyroid hormone signaling (162).
Two discoveries changed this overall perspective almost overnight. First, the recognition that MCT8 is a very active and specific thyroid hormone transporter highly expressed in liver, kidney, brain, and heart. Initially, the rat MCT8 was cloned and shown to induce an approximately 10-fold increase in uptake of 10 nm 125I-T4, -T3, -rT3, and -T2 when expressed in Xenopus laevis oocytes, indicating a clear preference for the transport of T3. Saturation analysis provided apparent Km values of 2–5 μm for T4, T3, and rT3 (163). Notably, coexpression of a reduced nicotinamide adenine dinucleotide phosphate-dependent cytosolic T3 binding protein in the cytosol, identical to mu-crystallin, increased the observed effects of MCT8 expression (164). Second, two groups independently linked mutations in the coding region of the X-linked MCT8 gene with severe psychomotor retardation and high serum T3 concentrations in several unrelated young boys. This suggests that this novel syndrome originated from a defect in MCT8-mediated T3 entry into neurons, resulting in impaired T3 action and metabolism. In fact, fibroblasts from these patients have increased D2 activity, which reflects intracellular hypothyroidism and elevated TSH levels that are normalized with the administration of T3 but not T4, confirming a state of relative insensitivity to thyroid hormone actions (165,166). Later studies indicated that the MCT8 gene is located in Xq13 and mutations in MCT8 result in a previously recognized disease caused by an X-linked condition, Allan-Herndon-Dudley syndrome (167). This syndrome is characterized by congenital hypotonia that progresses to spasticity with severe psychomotor delays. Affected males also present with muscle hypoplasia, generalized muscle weakness, and limited speech. Importantly, these patients have elevated serum levels of FT3, low to below normal serum levels of FT4, and normal levels of TSH. Two mice strains in which the MCT8 gene has been targeted for disruption were shown to recapitulate the changes in thyroid economy observed in the affected patients (168,169). At the same time, no neurological phenotype could be identified in these animals, possibly indicating that compensatory mechanisms for the lack of MCT8 exist in mice or simply that critical steps in the development of the central nervous system in mice are differently influenced by thyroid hormone.
Today, in addition to MCTs, other families of thyroid hormone transporters have been identified using functional expression studies in X. laevis oocytes: organic anion transporters (e.g., OATPs), and L-type amino acid transporters (170). Among these, OATP1C1 has a high affinity and specificity for T4 and is expressed in capillaries throughout the brain, suggesting that it is critical for transport of T4 over the blood-brain barrier (170). In the MCT family of transporters, MCT10 has been characterized as yet another thyroid hormone transporter and transports T3 more effectively than MCT8 (164). As a result of these studies, thyroid hormone transport across the plasma membrane is recognized today as a critical mechanism for thyroid hormone action. Of note, given that thyroid hormone transporters are necessary for deiodinases to metabolize iodothyronines (55), the existence of these transporters constitutes a newly recognized mechanism to control thyroid hormone metabolism and action.
B. Deiodinase-mediated mechanisms
At the cellular level, thyroid hormone action is initiated through the binding of T3 to TR. The extent of thyroid hormone signaling in a given cell ultimately depends upon the level of TR occupancy, which is determined by the intrinsic affinity of TR for T3 and the T3 concentration in the nucleus (Fig. 7). These values are such that, at normal serum T3 concentration, the contribution from serum T3 alone results in an approximately 50% TR occupancy in most tissues (19).
Figure 7.

Role of D2 and D3 in thyroid hormone signaling. T4 and T3 are represented by blue and green circles, whereas D2 and D3 homodimers are represented by brown and yellow ovals. A, D3 catalyzes the conversion of plasma and cellular T4 and T3 to the inactive metabolites rT3 and T2, respectively, decreasing the nuclear pool of T3 available to occupy TRs. B, In D2-expressing cells, the nuclear pool of T3 available to the TRs originates from both plasma T3 and T3 generated via D2. [Modified with permission from Bianco et al.: Endocrinology 148:3077–3079, 2007 (445). ©The Endocrine Society.]
Although plasma thyroid hormones may provide a uniform signal to all tissues of the body, their biological impact is not homogeneous. Primary changes in deiodinase activity can elicit T3-dependent downstream effects in response to endogenous nonthyroidal signals (171) and xenobiotic agents (172), as well as environmental changes such as cold (148) or hypoxia (28). In this regard, D2 functions as an additional intracellular source of T3, such that the nuclear T3 concentration will be higher given the combination of T3 from the plasma and the T3 that is locally converted from T4 by D2. For example, the saturation of the TRs in the central nervous system, where D2 is expressed, is close to 95% (19). At the same time, T3 concentration in discrete areas of the brain drops by about 50% after targeted disruption of the Dio2 gene (173). Furthermore, in BAT the levels of D2 activity and TR occupancy are dynamic and change according to the metabolic activity of the tissue (discussed in Section IV. B. 1-3), increasing from a TR occupancy of approximately 65% at room temperature to approximately 100% during cold exposure (24,174). Although D1 also converts T4 to T3, its contribution to TR occupancy is negligible (19).
Although the mechanistic details underlying such discrimination between D1-generated T3 and D2-generated T3 are largely unknown, it is undisputed that D1-generated T3 equilibrates rapidly with plasma (∼30 min), whereas D2-generated T3 has a much longer intracellular residence time and will only equilibrate with plasma after several hours (19). Differences in subcellular localization of D1 and D2 (48) might contribute to this phenomenon, with D2-generated T3 having preferential access to the nucleus given its residence in the ER. At the same time, it is logical to assume that by virtue of being anchored at the plasma membrane, D1-generated T3 would rapidly equilibrate with plasma. Given the significant role played by thyroid hormone transporters in cellular T3 and T4 homeostasis, it is conceivable that these transporters also play a role as determinants of the fate of T3 within the cell.
At the same time, tissues expressing D3 have lower T3 concentrations than what would be expected from plasma contribution; thus, D3-expressing tissues have a gene expression profile typical of hypothyroid cells (28,175). This is explained by the inactivation of T3 and T4 that takes place at the plasma membrane level immediately after these hormones enter the cell. In hepatocarcinoma or neuroblastoma cells, D3 expression results in a reduced metabolic rate, which is increased upon inactivation of D3 (28) (see Section V.A.3). These findings indicate that D3 expression decreases thyroid hormone action at the cellular level, a mechanism that can be more or less stringent depending on the level of D3 activity.
Thus, deiodinases constitute a potent mechanism to control thyroid hormone signaling, allowing cells to customize their own T3 footprint in a spatial- and temporal-dependent/specific fashion. Although early conclusive evidence supporting such a mechanism was obtained in BAT of cold-exposed rodents (23,24,25,26,148,176), today other signals triggering such mechanisms have been discovered in a growing number of tissues (32,171,172,177). In addition, deiodinase-mediated control of thyroid hormone signaling has been abundantly studied during metamorphosis and embryogenesis in vertebrates (29,30), a period during which temporally and spatially controlled exposure to thyroid hormone also regulates important developmental events in the brain and other tissues (see Section IV.A). Achieving such tight tissue-specific control of thyroid hormone signaling by modulating serum T3 concentration would not be a feasible mechanism.
C. Deiodination in intact cells and cofactor availability
The vast majority of the work performed to date in which deiodination is studied includes measurement of a specific deiodinase maximum velocity in cell or tissue sonicates/homogenates under optimum conditions of substrate and cofactor concentrations, usually with a strong reducing agent such as dithiothreitol or 2-mercaptoethanol (36). Although this approach has advantages in biochemical studies of enzyme kinetics and other physicochemical properties, it does not necessarily reflect the flux of thyroid hormone through a specific deiodinase pathway. As an example, cofactor concentrations in liver might vary rapidly and become limiting for certain deiodination pathways (178); substrate availability might also be limiting given that specific thyroid hormone transporters are necessary to allow substrate access to all active centers of deiodinases.
To have access to more physiologically relevant data, investigators developed deiodinase assays in live cells, which are performed under physiological conditions (179). Deiodinase-expressing cells are incubated in the presence of physiological substrate (free) concentrations (picomolar range) for several hours, while substrate consumption as well as product formation is monitored after media samples are resolved by HPLC (180). Using this approach, in vivo scenarios of deiodinase pathways can be monitored in cell cultures, and the relative contribution of different deiodinase pathways can be quantified appropriately under physiological conditions. For example, exposure of D2-expressing cells to the flavonol kaempferol dramatically increases D2 activity as measured in cell sonicates, whereas T3 production, as measured in live cells, is only mildly elevated (172). Subsequent studies demonstrated that in addition to increasing Dio2 gene expression, exposure to this flavonol exhausted the cellular D2 cofactor, limiting the ability of the increased D2 pool to catalyze T4 to T3 conversion (172). These findings are reminiscent of the early studies of liver D1 activity in fasted or refed rats, in which changes in T4 to T3 conversion were attributed to in vivo cofactor availability (181,182,183).
Three mouse models of D2 overexpression provide strong evidence that tissues have different capacity to deiodinate thyroid hormone, even when expressing similar deiodinase levels. Two of these models are mice in which D2 is overexpressed in the myocardium, either constitutively (184) or conditionally (185) activated. In both animals, D2 activity in myocardium sonicates is extremely high, and yet only a mild cardiac phenotype was evident in both animal models. Notably, no changes in serum T4 or T3 concentrations were observed in either model. In contrast, mice in which D2 is overexpressed in liver develop spectacular changes in thyroid hormone serum concentrations, with T4 dropping to undetectable levels with a doubling of serum T3 (186). Although a number of factors could contribute to such findings, including the fact that the liver is substantially larger than the heart, it is difficult to neglect the possibility that D2 cofactor concentration in the myocardium is a limiting step in local T3 production.
D. Conjugation of iodothyronine and its effects on deiodination pathways
Thyroid hormone molecules are subject to conjugation of the outer ring hydroxyl group with glucuronic acid or sulfate, a reaction that inactivates and can also dramatically change the affinity of the deiodinases for the different iodothyronines. Thus, glucuronidated iodothyronines are excreted in the bile and then eliminated through the feces or recycled in the enterohepatic cycle, whereas sulfated iodothyronines are rapidly deiodinated. Many of the enzymes that are involved in the glucuronidation and sulfation of thyroid hormones are targets of the nuclear hormone receptor constitutive androstane receptor (187,188). This receptor mediates the induction of hepatic drug metabolism in response to xenobiotics, and treatment of mice with phenobarbital induces many of these genes and increases the rate of thyroid hormone metabolism in a constitutive androstane receptor-dependent manner (188).
Sulfotransferases are soluble cytoplasmatic enzymes expressed in various tissues, e.g., liver, kidney, intestine, and brain, and can be divided into the phenol and hydroxysteroid sulfotransferase groups (189). The phenol sulfotransferases that process iodothyronines include SULT1A1, SULT1A2, SULT1A3, and SULT1B1 (190,191). While sulfation of T4 is minimal, sulfation of 3,3′-T2 is catalyzed orders of magnitude faster than that of T3 or rT3 (191). At the same time, the estrogen sulfatase SULT1E1, expressed in liver, uterus, and mammary gland, is very efficient in sulfating 3,3′-T2 and T3, and much more efficient in sulfating rT3 and T4 (192). This pathway could contribute significantly to the high levels of iodothyronine sulfates found in human fetal plasma.
Although D2 and D3 do not process T4S and/or T3S as substrates, D1-mediated iodothyronine deiodination is markedly affected after substrate sulfation. The mechanism by which sulfation stimulates inner ring deiodination (IRD) of T4, T3, and T2 is unclear, whether increasing Vmax or decreasing the apparent Km value depending on the specific substrate (193). Although T4 is IRD or outer ring deiodinated (ORD) by D1 at equal rates, IRD of T4S by D1 is accelerated about 200-fold, and ORD becomes undetectable (193). IRD of T3 by D1 is also stimulated by about 40-fold after sulfation (193), and a similar change happens with ORD of 3,3′-T2 by D1. Because sulfated iodothyronines are processed preferentially by D1, serum levels of these molecules accumulate when the D1 pathway is impaired, e.g., in sick patients, in hypothyroid patients, or in patients receiving inhibitors of D1 such as PTU or iopanoic acid (194). Consistent with the role of D1 in the normal metabolism and elimination of sulfated iodothyronines, mice with a targeted deletion of D1 exhibit a marked shift from urinary to fecal excretion of iodothyronines (195). In the fetus, D1 activity is low and T3S accumulates in the fetal circulation and could function as a reservoir from which active T3 may be released in a tissue-specific and time-dependent manner (196); notably, a similar pathway is also catalyzed by intestinal bacteria (197).
E. Deiodinases and thyronamines
Decarboxylated iodothyronines form a new and exciting class of biologically active molecules, the thyronamines (198) (Fig. 8A). To date, two endogenous thyronamines, 3-iodothyronamine (T1AM) and thyronamine (T0AM), have been identified in rodent brain, heart, liver, and blood (198,199). Thyronamines are agonists for the G protein-coupled trace amine receptor 1 (TAR1) (198). T1AM is the most potent TAR1 activator, with an EC50 of 14 nm, whereas T0AM is about 10-fold less effective. Notably, T4 and T3 do not activate TAR1, and both T1AM and T0AM are unable to bind to the nuclear TR. In addition, thyronamines are also subject to sulfation by the sulfotransferases SULT1A2, SULT1A3, and SULT1E1 (200), but it is unclear how this affects their biological activity, metabolism by the deiodinases, and overall clearance.
Figure 8.

A, Structure and nomenclature of thyronamines. B, Schematic diagram of potential thyronamine deiodination pathways. [Reprinted with permission from Piehl et al.: Endocrinology 149:3037–3045, 2008 (203). ©The Endocrine Society.]
1. Biological effects of thyronamines.
In vivo, the biological effects of thyronamines appear to be opposite to those of thyroid hormone. Although thyroid hormone is known to increase metabolic rate, injection of either T1AM or T0AM into mice leads to a rapid drop in core temperature of up to 7.5 C, along with bradycardia and a decrease in cardiac output (198,201). Remarkably, while mice were inactive, shivering, huddling, or piloerection were not observed, and mice returned to normal 6 to 8 h after treatment. Similar studies in Siberian hamsters found that T1AM injection leads to a decrease in body temperature, metabolic rate, and respiratory quotient, along with a transient switch from carbohydrate to lipid utilization (199).
Although the effects of thyronamine injection are dramatic, it is currently unknown whether this is a reflection of an artificially high dose of thyronamine. The effects of T1AM and T0AM on hypothermia and cardiac function are more compatible with inhibition of cAMP accumulation; thus the observed response might be secondary to activation of TAR1 or mediated through another yet to be identified TAR subtype (198,201). Additionally, T1AM has been shown to inhibit dopamine and norepinephrine reuptake and transport into synaptic vesicles, indicating that modulation of other monoamines could contribute to some of the observed effects of thyronamine treatment (202).
2. Thyronamine biosynthesis.
For the production of the biologically active thyronamines T1AM and T0AM, iodothyronine precursors would need to be both decarboxylated and deiodinated. Although the order of this pathway remains to be defined, it has been postulated that the aromatic amino acid decarboxylase, which normally produces dopamine and serotonin by decarboxylation of 3,4 (OH)2-phenylalanine and 5-hydroxytrytophan, could also act on iodothyronines (198). Additionally, recent work using the novel technique of liquid chromatography and tandem mass spectrometry, in which multiple deiodination products can be identified and quantified independent of the use of radioactive substrates, demonstrated that thyronamines can be metabolized by the deiodinases (203). In this work, it was determined that sequential deiodination from thyroxine amine (T4AM) to T1AM or T0AM would be possible via multiple pathways (Fig. 8B). As would be expected, D1 deiodinated a variety of thyronamines at both the phenolic and tyrosyl rings, whereas D2 and D3 deiodinations were limited to the phenolic and tyrosyl rings of their respective thyronamine substrates. This observation indicates that deiodinases are involved in thyronamine biosynthesis, revealing yet another layer of complexity in the regulation of thyroid hormone action by these enzymes.
IV. Deiodinase-Mediated Thyroid Hormone Signaling in Discrete Cell Systems
Spatial- and temporal-dependent/specific expression of activating and inactivating deiodinases has been shown to play a physiological role in a number of cell systems during development and also in adult vertebrates (29,30,33). These mechanisms provide an optimized control of thyroid hormone action on a cell-specific basis, the specificity of which could not be achieved simply by modulating serum thyroid hormone levels.
A. Development and metamorphosis
Given the importance of thyroid hormone during development and metamorphosis, there is a growing recognition of the important role played by deiodinases to modulate its signaling during these processes. This originated from observations that a wide range of growth factors and morphogens, such as the TGF-β, fibroblast growth factor, and hedgehog families of secreted proteins, regulate both deiodinase expression and activity. The importance of this local control of thyroid hormone action during embryogenesis and metamorphosis is well illustrated by studies of bone development in chickens, retina development in frogs, and skin and inner ear development in rodents.
1. Hedgehog signaling and regulation of proliferation and differentiation.
Fundamentally, the development of a specific tissue can be simplified as an initial proliferation of precursor cells followed by terminal differentiation into mature cells. In this regard, thyroid hormone is a highly potent agent. Its action involves the induction of genes promoting differentiation (e.g., nerve growth factor) as well as the reduction of the proliferative potential by decreasing cyclin D1 levels (204,205,206,207,208). Thyroid hormone signaling is also modulated locally by the deiodinases, and early studies on their role during differentiation and development have previously been reviewed (5). More recently, it was discovered that D2 and D3 activity, and thereby thyroid hormone signaling, can be modulated by Shh (31,32), a highly potent proliferation-promoting morphogen (209,210).
Shh, a member of the hedgehog (Hh) family of secreted signaling proteins, is critical for many aspects of vertebrate development. Originally identified as homolog of the morphogenic Drosophila hedgehog protein (211,212), Shh has been demonstrated to be responsible for a number of early patterning processes (213). This includes the control of left-right asymmetry, the dorso-ventral patterning of the central nervous system and somites and patterning of the limb, and regulation of development in vasculogenesis, angiogenesis, bone and cartilage formation, and lung branching morphogenesis (214,215,216,217). In addition to its functions in embryonic development, Hh signaling is also important for the maintenance and proliferation of a wide variety of progenitor cells, including (but not limited to) neural precursors in the hippocampus and subventricular zone (218,219), progenitor cells in the inner ear (220), basal epithelial cells in hair follicles (221), bone marrow-derived endothelial cells (222), hematopoietic stem cells (223), and chondrocyte progenitor cells (216).
Chondrocyte proliferation and bone development provide an excellent example of the intersection of Hh and thyroid hormone signaling. During development, chondrocytes in the developing tibial growth plate leaving the proliferative pool produce Indian hedgehog (ihh) (216). Ihh and Shh have similar biological properties and share identical target genes (224). Ihh induces the secretion of PTHrP from perichondrial cells and chondrocytes near the ends of the skeletal elements, which acts to maintain chondrocyte proliferation such that hypertrophic differentiation is limited to the cells furthest from where PTHrP is produced (216).
The regulation of PTHrP mRNA expression by the Hh pathway is at least partially mediated through the induction of WSB1, a Shh-inducible Ub ligase subunit previously identified in chicken embryonic structures (122). WSB1 is expressed in the perichondrial sheath surrounding the developing chondrocytes in the chicken tibial growth plate. Hedgehog stimulation results in D2 ubiquitination via WSB1 (see Section II.C), inactivating the protein and resulting in a local hypothyroidism that induces PTHrP, thereby regulating chondrocyte differentiation (31). This is consistent with the observations that thyroid hormone inhibits the proliferation of chondrocytes and instead stimulates their differentiation (225). Thus, Shh-mediated regulation of D2 activity is a mechanism by which the Hh pathway can modulate local thyroid hormone levels via the deiodinases and thus enhance PTHrP expression, and it thereby controls skeletogenesis (31) (Fig. 9). WSB1 has also been reported to regulate cell fate in other settings, such as promoting cell proliferation in several human cell lines of pancreatic cancer (226).
Figure 9.

D2 activity is regulated by hedgehog signaling via WSB1 in the developing chicken tibial growth plate. In the graphs, white bars indicate treatment with vehicle, whereas black bars equal treatment with Shh (left). In situ hybridizations show WSB1 expression levels in perichondrium/periosteum (PC/PO). Indian hedgehog increases WSB1 expression, in turn increasing the ubiquitination and inactivation of D2. Less D2 results in a block in T3 to T4 production and local hypothyroidism at the apical perichondrium, causing an increase in PTHrP production, resulting in chondrocyte proliferation. [Reprinted in part with permission from Dentice et al.: Nat Cell Biol 7:698–705, 2005 (31).]
Shh signaling also plays an important role in skin physiology, and Hh signaling has been found to be overactive in proliferating cells such as basal cell carcinomas and related skin tumors (210,227,228). Skin is also a known target of thyroid hormone and expresses D2 and D3 (Fig. 10) (229,230,231). In primary proliferating keratinocytes, as well as mouse and human basal cell carcinomas, Shh increases the expression of D3. In concert with the Shh-mediated selective proteolysis of D2, this results in intracellular hypothyroidism and increased cyclin D1 levels and proliferation (32).
Figure 10.

D3 expression in normal skin and basal cell carcinoma (BCC). D3 expression in normal skin during the hair follicle cycle is time- and cell type-specific and overlaps Shh targets. A, D3 staining in the mouse skin at different stages of the hair follicle cycle demonstrated that during anagen (postnatal day 5), D3 was highly expressed in the hair follicle matrix and absent from the dermal papilla. In telogen (prenatal day 21), D3 expression was almost absent from the hair follicles. B, D3 immunostaining of normal skin and a representative BCC sample. [Reprinted with permission from Dentice et al.: Proc Natl Acad Sci USA 104:14466–14471, 2007 (32).]
This attenuation of thyroid hormone signaling by the deiodinases represents a novel way for Hh signaling to regulate cellular proliferation and differentiation and suggests that such cross-talk might be present in many development systems where Hh signaling is present.
2. Other growth factors/morphogens.
Other developmental factors can also alter deiodinase activity and thyroid hormone signaling. It has long been known that phorbol esters such as 12-O-tetradecanoyl phorbol-13-acetate, which strongly promote cell proliferation, also stimulate D3 expression and activity. This is in agreement with the observation that growth factors such as epidermal growth factor (EGF) and the acid and basic fibroblast growth factors can increase D3 expression and activity in a wide variety of cell types (5). Because these growth factors are important for cell proliferation and differentiation as well as tissue development and repair, this suggests that the deiodinases and their modulation of thyroid hormone signaling have a critical role in these processes. This idea has been further strengthened by the recent discovery that both the growth factor TGF-β and the promitogenic hormone estradiol can increase D3 expression and activity in a diverse range of both untransformed and transformed human cell types, including fetal and adult fibroblasts, hemangioma cells, fetal epithelia, and skeletal muscle myoblasts, and in the case of TGF-β, they can activate D3 synergistically in the presence of acidic fibroblast growth factor or EGF (232).
Additionally, there is a growing body of evidence that D2 is also regulated by growth factors because D2 expression is positively regulated by EGF in HC11 mouse mammary epithelial cells (233), by platelet-derived growth factor and basic fibroblast growth factor in human vascular smooth muscle cells (234), and by insulin via the enhancement of norepinephrine signaling in primary brown adipocytes (235). Given the diametric roles of D2 and D3 in amplification and cessation of thyroid hormone signaling, respectively, it is surprising that both enzymes are activated by growth factor exposure. However, because no comparison of D2 and D3 induction in response to growth factors has been conducted simultaneously, it is possible that these transient increases in D2 and D3 activity occur over a different time period, resulting in a time-dependent increase or decrease in thyroid hormone signaling. Regardless, these results indicate that important inducers of differentiation and development (i.e., growth factors) can alter the expression and activity of the deiodinase family, thereby modulating thyroid hormone signaling and its transcriptional effects on cellular proliferation and differentiation.
3. Development/metamorphosis.
The critical role of thyroid hormone during amphibian metamorphosis was first identified in the beginning of the 20th century, when it was observed that extracts from thyroid glands could precociously induce metamorphosis in tadpoles and that removal of the thyroid gland inhibited metamorphosis. Subsequent research in multiple frog strains, most notably Xenopus laevis, characterized the specific metamorphic changes mediated by thyroid hormone, including such events as the reabsorption of the tadpole tail, generation of a new intestinal system, and the remodeling of skin, respiratory organs, central nervous system, and skeleton. The history and resulting conclusions have been comprehensively reviewed (236). Not surprisingly, the unique capacity of the deiodinases to amplify or decrease local thyroid hormone levels is used to regulate metamorphosis in a tissue-specific manner, thereby affecting the timing of metamorphic changes (30). This has been evidenced by experiments showing that the D2 and D3 inhibitor iopanoic acid blocks metamorphosis in Rana catesbeiana tadpoles (237) and that increases in D2 activity in thyrotrophs during the peak period of metamorphosis terminate T4 production and therefore metamorphosis (238), and by the observation that D3-expressing cells in the dorsal areas of the X. laevis visual system prevent thyroid hormone-induced proliferation and metamorphosis, thereby causing an asymmetric shift in ocular positioning (27). Recently it was revealed that deiodinase-mediated changes in thyroid hormone signaling during morphogenesis are relatively widespread because the expression of D2 in specific tissues indicates how rapidly it responds to thyroid hormone-induced metamorphosis (239).
It is possible that deiodinases are also critical early in amphibian development, well before the induction of metamorphosis. Because the deiodinases and their substrates T4 and T3 are detectable early in embryogenesis in spatially defined neurogenic regions, this suggests a developmental role in early tadpole neurogenesis (240).
In the case of mammals, thyroid hormone has been shown to be critical for early embryogenesis and development because children with congenital hypothyroidism display symptoms including stunted growth, bone and muscle dystrophy, mental retardation, respiratory difficulties, and hearing impairment. Consistent with the last phenotype, D2 was demonstrated to be necessary for proper hearing development in rodents. Initial evidence for this originated from the observation of a peak in D2 activity in the mouse cochlea at approximately postnatal d 7, which occurs immediately before the onset of hearing (241). Subsequent analysis revealed that mice with a deletion of D2 have a phenotype similar to that of hypothyroid mice, with defective auditory function and a higher threshold for auditory-evoked brain stem response. This defect stems from a retarded differentiation of the cochlear inner sulcus and sensory epithelium, which, combined with a deformity of the tectorial membrane, results in hearing impairment (242). This reveals that the peak of D2 activity and resulting thyroid hormone levels are critical for coordinating the varied developmental events in the differentiating and maturing auditory systems. It also suggests that D2, and possibly the other deiodinases, could play a role in other tissues that require precise spatial control and timing of differentiation. Such tissues include the brain or testes, where thyroid hormone is critical for control of proliferation and differentiation and deiodinase expression is observed (5,243). In fact, it has been reported that in mice with targeted inactivation of Dio2 (Dio2 null), there is half as much T3 in the brain compared with wild-type mice, with clear defects in certain agility tasks, although it is likely that compensatory mechanisms are limiting the severity of the phenotype (173).
B. Energy homeostasis
Given the generalized metabolic sensitivity to thyroid hormone documented in human subjects during hypo- and hyperthyroidism, one would anticipate a major physiological role of this hormone in energy homeostasis. However, the relative constancy of serum T3 concentration seemed to preclude a major role of T3 in the basal metabolic rate (BMR) variations observed after a meal or during sleep (244). With the realization that deiodinases, in particular D2 and D3, could alter local intracellular thyroid status without necessarily altering serum T3 concentrations, this presumption has been challenged.
1. D2 is a key molecule for cold-adaptive thermogenesis in brown adipocyte.
It is well established that D2 plays a critical role in mature brown adipocyte biology by providing an additional source of T3 during tissue activation (24,25,26,245). This tissue is the major site of adaptive thermogenesis in rodents, with heat being generated as a result of the actions of uncoupling protein 1 (UCP-1) (246,247). Cold-induced thermogenesis in BAT has been shown to depend upon the cAMP-mediated acceleration of D2-catalyzed T3 production, which in turn leads to the induction of T3-responsive thermogenic genes including UCP-1. Of note, despite the large D2-mediated increase in BAT nuclear T3 seen in cold-exposed animals, serum T3 concentrations remain fairly constant in the first several hours of cold exposure, demonstrating that D2 can have tissue-specific metabolic effects. In Dio2 null mice, survival in the cold is only possible because the mice begin shivering (149), a behavior not normally seen in small rodents (248). An additional role played by D2 and thyroid hormones in BAT is to mediate a 3- to 4-fold increase in the activity of lipogenic proteins, i.e., malic enzyme, glucose 6-phosphate dehydrogenase, and Spot14 observed in this tissue during cold exposure, a response that is blunted in hypothyroid rats (24,176,249,250,251).
2. D2 is critical in diet-induced thermogenic pathways.
Evidence for a role for D2 in the control of metabolic pathways beyond cold-induced thermogenesis was recently provided by the discovery that bile acids can confer resistance to diet-induced obesity in mice via up-regulation of D2 expression in BAT (171). In this tissue, binding of bile acids to the plasma membrane G protein-coupled receptor TGR5 triggers an increase in cAMP formation and subsequently elevates D2 expression (Fig. 11). In normal mice fed a high-fat diet supplemented with bile acids, oxygen consumption increased and the mice did not gain weight or become as insulin resistant as mice fed the high-fat diet alone. This effect is lost in Dio2 null mice. The importance of this mechanism in rodents fed a normal diet remains to be determined, and D2-independent bile acid-activated pathways may play a role. It is nonetheless noteworthy that D2 is overexpressed in two other rodent models of resistance to diet-induced obesity. UCP-1 null mice are paradoxically lean (252) and have ectopic expression of D2 in their white fat, whereas mice with a targeted deletion of both liver X receptor α and β express D2 ectopically in the liver (253). If the ectopic expression of D2 in these animals results in tissue-specific thyrotoxicosis, as is suggested by gene expression profiling in the case of the liver X receptor α and β null mice, this would support the concept that the D2 pathway increases energy expenditure.
Figure 11.

Bile acids stimulate D2 expression in brown adipocytes. Schematic representation of the bile acid-TGR5–D2 pathway in brown adipocytes. Bile acids in the general circulation derived from the enterohepatic circulation potentially may stimulate TGR5 increasing cAMP, and thus leading to an increase in D2 expression in tissues where both proteins are coexpressed, e.g., BAT and skeletal muscle. This pathway has been shown to increase energy expenditure and protect against diet-induced obesity in mice (171). [Reproduced in part with permission from Baxter and Webb: Nature 439:402–403, 2006 (446).]
3. A metabolic role for D2 in humans.
Human newborns grow less dependent on BAT thermogenesis with maturity, and adult humans, unlike small mammals, do not have substantial amounts of BAT (248). The mass of BAT in humans peaks at the time of birth, when brown adipocytes comprise almost 1% of body weight (254,255,256). It could thus be assumed that the D2 pathway is most important for thermogenesis in infants and much less important in adults except in cases where BAT mass is abnormally increased such as in patients with pheochromocytoma (257). However, the amount of BAT in adults may be greater than once thought because studies utilizing 18F-fluorodeoxyglucose positron emission tomography/computed tomography imaging have identified focal deposits in the mediastinum and in extramediastinal areas (258,259). More importantly, given the finding of D2 expression in human skeletal muscle (100,260) and cultured skeletal muscle cells (261), a larger metabolic role for D2 in humans may be anticipated because skeletal muscle is the predominant site of thermogenesis and insulin-induced glucose disposal in adult humans.
Various studies support a previously unrecognized contribution of D2 to the thyroid status and metabolic rate in humans. Earlier studies have consistently found diet-induced changes in serum thyroid hormones that could be explained by changes in D2 activity. As an example, the increase in BMR observed in subjects fed a high-carbohydrate diet is typically associated with an increase in the serum T3/T4 ratio (262), a condition that is also observed in adult subjects chronically treated with terbutaline, a β-adrenergic receptor stimulator (263). A connection with D2 can be easily imagined given that it is the only cAMP-dependent deiodinase (264). Furthermore, studies of patients receiving T4 replacement at various dosages have shown a direct correlation of the BMR with FT4 and inversely with serum TSH but not with serum T3 (265). Together, these data indicate that D2-produced T3 might be a significant physiological determinant of energy expenditure in humans.
4. D3 expression reduces energy expenditure: adaptation to hypoxia.
Hypoxia stimulates robust induction of D3 activity via a HIF-dependent pathway (28). Exposure to both normobaric hypoxia and hypoxia mimetics (which also promote HIF accumulation) markedly increases D3 mRNA and activity in cultured cells from diverse tissues including cardiomyocytes, hepatocytes, and human neurons. Of note, a number of other cell types showed no D3 response despite strong HIF induction, illustrating that the regulation of deiodinase expression by hypoxia is highly cell-type specific. Chromatin immunoprecipitation analysis of human D3-expressing neurons documents specific interaction between the HIF-1α transcription factor and sequences in the DIO3 5′ flanking region (FR), providing strong evidence that D3 is a direct HIF-1α target gene in responsive cell types. This induction of D3 by hypoxia can explain a number of clinical observations, including the previous description of D3 in the hypoxic tissues of the human fetus (266,267) and the ischemic tissues of critically ill patients (268).
Because thyroid hormone is a potent stimulator of metabolic rate and oxygen consumption, the regulation of local T3 action in hypoxic tissues is predicted to have important physiological consequences. It is notable that many previously established HIF-target genes are known to promote the survival of hypoxic cells by reducing oxygen consumption. Consistent with this function, studies of isolated cells indicate that endogenous D3 activity is a potent inhibitor of T3-dependent oxygen consumption (28). This suggests a mechanism of metabolic regulation during certain hypoxic-ischemic injuries in which HIF-induced D3 promotes the viability of hypoxic tissues by reducing T3-stimulated energy expenditure (28). The recent report of acute D3 induction in a rat model of myocardial infarction (269) suggests that cardiac ischemia may be one such scenario (see Section V.A.1), but further study of this and other injury models is needed to fully elucidate the role of both D2 and D3 in hypoxic-ischemic disease.
5. Mechanisms underlying the thermogenic effects of T3.
Thyroid hormone is one of the few truly potent stimulators of the metabolic rate, such that energy expenditure is severalfold higher in hyperthyroid compared with hypothyroid patients (270,271,272). However, little is known about how D2-generated T3, or T3 from the plasma for that matter, accelerates energy expenditure (273). Although many T3-responsive candidate genes have been identified, their exact contribution remains to be established. Increased mitochondrial uncoupling has been proposed as one mechanism, and this effect has been demonstrated in the skeletal muscle of mildly thyrotoxic human volunteers (274). Similar results were reported in hepatocytes of thyrotoxic rats (272,275), but it is unclear whether this effect is mediated by UCPs. Another general mechanism by which T3 may increase energy expenditure would be to accelerate the turnover of ATP by inducing ATP-utilizing enzymes. Several T3-responsive genes have been implicated, e.g., the Na+/K+ ATPase (276,277) and the sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA1) gene (278,279,280,281) among others (282,283). Which, if any, of these genes is most responsible for the observed increase in energy expenditure driven by T3 remains to be determined, and it is possible that yet to be identified mechanisms could play a major role.
C. Deiodinases in the hypothalamus/pituitary systems
In the brain, D2 is predominantly expressed in the different types of glial cells, whereas D3 expression is predominantly neuronal (284,285,286,287). Given that TRs are found in neurons, this spatial arrangement indicates that regulation of thyroid hormone signaling in the brain depends on these two cell types. A hypothesis is that the D2-produced T3 leaves the glial cells and interacts with nuclear TRs to trigger paracrine effects in the nearby neurons (170,288). Supporting this model is the observation that thyroid hormone transporters play a critical role in brain development (165,166). The mechanism by which enough glial-produced T3 manages to establish a T3-dependent mRNA footprint in neurons despite the D3 activity in these cells is still unknown.
The hypothalamus is the brain region in which deiodinase-mediated local control of thyroid hormone signaling is recognized. There, T3 provides important regulatory signals for the control of appetite, satiety, and reproductive function, as well as the TRH/TSH feedback mechanism (289,290,291). D2 expression is highest in the mediobasal hypothalamus (MBH) including the arcuate nucleus and median eminence, with activities three to four times higher than other areas in the hypothalamus (ventromedial nucleus, dorsomedial nucleus, lateral hypothalamus) and cerebral cortex (292). In the MBH, D2 expression is concentrated in the specialized glial cells known as tanycytes, located in the floor and infralateral walls of the third ventricles. This location suggests that the cerebrospinal fluid may be a source of T4 that could be transduced to the hypothalamus and/or pituitary gland via T3 released from the tanycyte processes into specific hypothalamic nuclei and/or the pituitary portal plexus (see Ref. 289 for review). In addition, D2 is also expressed in astrocytes within the hypothalamus, such as what is found in other brain areas (284). Notably, D2 expression is not present in the paraventricular nucleus (PVN) (292), which contains the TRH-expressing neurons, indicating that locally generated T3 diffuses to this area from other nuclei in the hypothalamus. The observation that D2 expression in tanycytes is only moderately regulated by thyroid hormone opens the possibility that in these cells D2-generated T3 could be controlled predominantly by other neuronal signals (reviewed in Ref. 289).
D3 expression is also moderately present throughout the hypothalamus, with the notable exception of the supraoptic nucleus (286). Its important role in the local regulation of thyroid hormone signaling became evident with the creation of the Dio3 null mice, which undergo complex changes in thyroid economy during the neonatal period, ultimately developing central hypothyroidism (293,294). At birth, D3 expression is concentrated in the bed nucleus of the stria terminalis and hypothalamic medial and median preoptic nuclei, whereas by postnatal d 10 its distribution is diffuse. It is conceivable that its neonatal focal expression coincides with the period during which the set point for TRH secretion is being established, explaining the disruption of the TRH/TSH axis later in life observed in the Dio3 null animals.
All deiodinase activities are present in sonicates of the rodent pituitary gland (295,296). However, given the diversity of the pituitary cell types and the low abundance of thyrotrophs, conclusive data about D2 expression only recently became available with studies that employed in situ hybridization of D2 mRNA combined with TSH immunocytochemistry (297). In these studies, thyrotrophs were distributed throughout the pars distalis of the rat pituitary gland. TSH cells were small and angulated and contained a small nucleus. D2 mRNA was widely detected in TSH cells—a feature that increases during hypothyroidism. D2 mRNA was also identified in anterior pituitary cells that were not immunoreactive for TSH, suggesting that some thyrotrophs were “empty” or that D2 expression is also present in non-TSH secreting cells. Data from human tissue are scarce and controversial. Studies of human TSH-producing tumors suggest that D3 is the predominant deiodinase in TSH-secreting tumor cells (298) or that both D1 and D2 are expressed in these cells (299).
1. Signals regulating D2 activity in the brain.
D2 mRNA in the brain is up-regulated by numerous effects including iodine deficiency, stress, trauma, light, infection, and hypothyroidism (289,290,291,300). In addition to these pretranslational events, studies demonstrated the existence of a rapid posttranslational regulatory mechanism of D2 activity in the central nervous system (96,103) through mechanisms that today we know involve D2 ubiquitination (see Section II.C). Of note, the proteins mediating D2 ubiquitination and deubiquitination are expressed in the central nervous system and exhibit some degree of tissue specificity. For example, in situ hybridization combined with immunocytochemistry of the rat brain shows that in addition to neurons, the D2-specific Ub E3-ligase WSB1 and the D2-specific deubiquitinase USP33 are differentially expressed in astrocytes and tanycytes, the two main D2-expressing cell types in this tissue (301). Tanycytes express both WSB1 and USP33, indicating the potential for D2 ubiquitination and deubiquitination in these cells. Only WSB1 is expressed in glial fibrillary acidic protein-positive astrocytes throughout the brain. Notably, changes in thyroid status do not affect the expression of WSB1 or USP33 in the central nervous system of the rat (301), indicating that changes in the expression of these genes are not part of the brain homeostatic response to hypo- or hyperthyroidism (301).
2. D2 relays serum T4 concentration to hypophysiotropic TRH neurons and thyrotrophs.
Although administration of T3 potently suppresses TSH secretion in animals and patients, there is some logic to the idea that plasma T4 must also be relevant for TSH feedback mechanism, given that T4 is the major secretory product of the thyroid gland (302). Monitoring plasma T4 levels would provide an ideal index of thyroid gland activity (303), which is the ultimate variable controlled by TSH. In fact, it has been determined experimentally that serum FT4 concentration is a key player mediating the TSH feedback mechanism (304). This is convincingly demonstrated during iodine deficiency or mild primary hypothyroidism (305), during which a declining serum T4 concentration promotes dramatic increases in serum TSH secretion while serum T3 concentrations remain unaffected.
Because T4 is only minimally active, these observations suggest that T4 to T3 conversion at the pituitary gland and/or hypothalamus must be involved. A series of subsequent studies suggested that the effects of serum T4 on TSH secretion are possible only because of the expression of D2 in the pituitary, which rapidly converts T4 to T3 locally. This is also true in the hypothalamus, given that the normalization of both circulating T3 and T4 is required to suppress TRH mRNA in the PVN of the hypothalamus. TSH suppression is not efficient with systemic administration of replacement doses of T3, indicating that T4 to T3 conversion within the brain is important (306). This model is also supported by data obtained in animals with targeted disruption of the Dio2 gene. Levels of both serum T4 and TSH were found to be significantly elevated in such mice, confirming that in the absence of D2 the thyrotrophic cell is relatively insensitive to the feedback effect of plasma T4 (307). Furthermore, whereas serum TSH levels in wild-type mice are suppressed by administration of either T4 or T3, only T3 was effective in Dio2 null mouse (307).
The corollary is that inputs from both circulating T4 and T3 are integrated by the thyrotroph and TRH-expressing neurons, with D2 expression critical for T4 input to be relayed in these cells (Fig. 12). Despite its intrinsic logic, it was only recently that this model was fully validated with the discovery of the mechanisms that control D2 activity in thyrotrophs. A major obstacle was the conciliation of the homeostatic nature of D2 activity with its role in TSH regulation, which is counterintuitive. D2 activity increases if serum T4 is low, whereas the opposite happens at high serum T4 concentrations. This is because binding of T4 to the D2 active center accelerates D2 inactivation by ubiquitination (see Section II.C.1–2). This has been understood as an adaptive mechanism in brain and other D2-expressing tissues to minimize changes in the intracellular concentration of T3 during iodine deficiency and hypothyroidism (308,309). However, such homeostatic behavior at the thyrotroph, if operational, would impair the efficient transduction of changes in serum T4. This would keep TSH secretion from changing while serum T4 concentration fluctuated.
Figure 12.

Role of D2 in TSH feedback. In the thyrotroph, the TSH gene is subject to negative feedback by T3 in the nucleus derived from two distinct sources: plasma T3, illustrated as T3(T3); and plasma T4, which is then converted to T3 intracellularly via the D2 pathway, represented as T3(T4). This schematic includes the plasma membrane, which contains thyroid hormone transporters (indicated by the pink and red circles); the cytoplasm, containing the enzymes involved in thyroid hormone metabolism; and the nucleus, containing the TRs. D2 is represented in its active form (yellow) and inactive form (red). Transition between active and inactive D2 is via ubiquitination and deubiquitination, which are catalyzed by WSB1 and VDU1/VDU2, respectively. As a result of ubiquitination, D2-mediated generation of T3(T4) occurs at variable rates, decreasing as serum T4 concentration increases. Ultimately, these processes determine nuclear TR saturation, with only a minor fraction of the TRs being unoccupied under normal conditions. [Figure modified with permission from Bianco and Kim: J Clin Invest 116:2571–2579, 2006 (33). ©American Society for Clinical Investigation.]
Two mouse thyrotroph-derived cells—TtT-97, a transplantable thyrotrophic tumor (310), and TαT1, an immortalized SV40 T-antigen-expressing pituitary cell line (311)—were instrumental in solving this apparent paradox (297). Using the TαT1 mouse tumor cell line, it was found that the absolute rate of T4-induced loss of D2 activity in these cells is offset by the combined effect of D2 reactivation and a high rate of D2 synthesis. As a result, exposure to higher T4 concentrations rapidly translates into an increase in thyrotrophic D2-mediated T3 production and suppression of TSHβ gene expression, even as absolute D2 activity is reduced, thus explaining the mechanism of TSH feedback. In other D2-expressing cell systems (GH4C1 and MSTO-211 cells) that express D2 at much lower levels, similar increases in T4 exposure suppressed D2 activity and blunted D2-generated T3 production as predicted. Only in the TαT1 thyrotrophs was T3 production sustained while T4 levels increased. Although we do not fully understand the cellular differences that allow such a mechanism to operate, a simple explanation is that the massive D2 expression in thyrotrophs surpasses the maximal rate at which T4 induces D2 ubiquitination and inactivation. Thus, because of elevated D2 expression in thyrotrophs, net local T3 production is low at lower T4 concentrations and high at high T4 concentrations, allowing appropriate TSH feedback.
3. Metabolic control by deiodinases in the hypothalamus.
The MBH is a prime target of thyroid hormone; thus, local D2 and D3 activities can affect a number of homeostatic functions (289,312). T3 administration rapidly induces hyperphagia partially due to an increase in neuropeptide Y (NPY) expression (313). In addition, localized T3 administration to the ventromedial but not to the arcuate nucleus promotes rapid and specific changes in gene expression that cause a 4-fold increase in food intake (314). Notably, hypothalamic D2 activity in rodents exhibits a circadian rhythmicity with an activity peak at night, which coincides with their peak of metabolic activity (314,315). These studies establish an important role of the deiodinases in controlling T3-dependent metabolic effects in the hypothalamus, particularly given that D2-expressing tanycytes integrate a number of hormonal and neuronal signals while altering local T3 concentrations.
Fasting induces a state of central hypothyroidism that depends on a falling serum concentration of leptin (316,317). At the same time, fasting leads to approximately 2-fold up-regulation of D2 expression and activity in the hypothalamus (318), which could contribute to the suppression in TRH/TSH secretion seen during this condition. However, the direct effect of local T3 on hypophysiotropic TRH neurons of the PVN during fasting is controversial. Importantly, both the wild-type and the TRβ2 null mice show a similar reduction in TRH expression in the PVN during fasting, although TRβ2 is the key TR isoform mediating negative feedback regulation TRH expression (319). Consequently, T3-independent feeding-related regulatory mechanisms were also proposed. These include α-MSH, agouti-related protein (AgRP), and NPY-mediated effects of leptin on TRH expression (reviewed in Ref. 291) and the direct role of leptin signaling on the TRH promoter (320).
It has also been suggested that D2 expression in the arcuate nucleus is localized in glial cells that are in direct opposition to neurons coexpressing NPY, AgRP, and UCP2. Notably, the fasting-induced increase in D2 activity and local thyroid hormone activation in the arcuate nucleus is paralleled by an increase in UCP2-dependent mitochondrial uncoupling in NPY/AgRP expressing neurons. These events were shown to be critical for increased excitability of these orexigenic neurons and consequent rebound feeding after food deprivation (321).
4. A role for D2 in the control of hypothalamic rhythmicity
a. D2 and the seasonal breeding.
Seasonal variation of the reproductive function is important to ensure that offspring are born at the appropriate time of the year, particularly for species living in temperate regions. This is accomplished by photoperiod regulation of reproduction, a mechanism that regulates gonadal function according to annual changes in day length. It was demonstrated that central thyroid hormone levels may play a significant role in the photoperiodic regulation of the reproductive axis (322). The MBH is believed to be the center for photoperiodism, and a series of studies indicates that deiodinase-mediated control of thyroid hormone signaling underlies a conserved photoperiodism mechanism in avian and mammalian species. Rapid Dio2 induction in the MBH of the Japanese quail (Coturnix japonica) is the one of the earliest recorded events in the photoperiodic signal transduction pathway (323), which seems to be induced by a preceding peak of TSH β-subunit expression in the pars tuberalis via a cAMP-dependent mechanism (324) (Fig. 13). MBH D2 expression is induced by light (long days), an effect that is mimicked by intracerebroventricular administration of T3 and blocked by the administration of the deiodinase inhibitor iopanoic acid. Similar mechanisms seem to be functional in Syrian hamster (Mesocricetus auratus) (325), and Djungarian (Siberian) hamsters (Phodopus sungorus) with the additional involvement of melatonin regulating MBH Dio2 expression (326), although in this species an independent study could not detect photoperiod-dependent change in hypothalamic D2 but observed D3 induction on short days (325). In the short-day breeder Saanen goat (Capra hircus), D2 expression was suppressed by long days (327), a situation opposite to the long-day breeder Japanese quail or Djungarian hamster. Coordinated reciprocal expression of D2 and D3 also seems to be involved in the photoperiodic mechanism in the hypothalamus of Japanese quail, where Dio3 levels are reduced after exposure to long days, with the opposite observed during short days (324). This constitutes a two-gene switch mechanism that controls local thyroid hormone signaling and consequently the photoinduction process.
Figure 13.
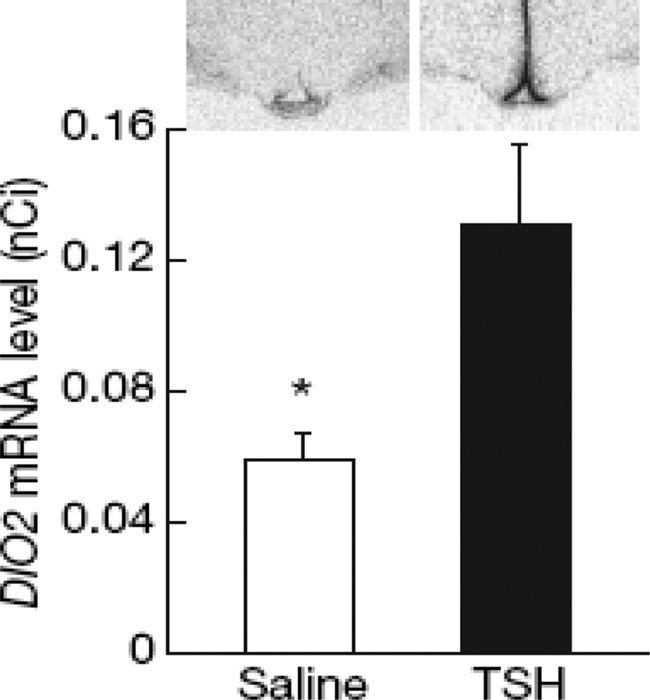
Thyrotrophin triggers photoperiodic response and long-day photoinduced seasonal breeding in the Japanese quail (Coturnix japonica). Chronic intracerebroventricular infusion of TSH increases D2 expression of the mediobasal hypothalamus. [Reprinted with permission from Nakao et al.: Nature 452:317–322, 2008 (324).]
b. D2 and the pineal gland.
The pineal gland synthesizes and secretes melatonin under the control of environmental lighting. Melatonin has the ability to entrain biological rhythms and can influence reproductive function of many animals (328). In the rat pineal gland, D2 activity increases 20–30 times during the hours of darkness (329,330), an induction that is negatively regulated by Fos-related antigen 2 (331,332) and abolished by the β-adrenergic blocker propranolol (333). As a result, there is an increase in the pineal T3/T4 ratio during the dark period that coincides with the maximal D2 activity. Dio2 induction parallels the nocturnal increase in pineal arylalkylamine N-acetyltransferase (AA-NAT), the rhythm-generating enzyme for melatonin (334), and ablation of the hypothalamic suprachiasmatic nucleus abolishes AA-NAT and D2 rhythmicities. Earlier in vivo studies in which the deiodinase inhibitor iopanoic acid was used to prevent the nocturnal increase in D2 found no modification in either the pineal AA-NAT activity or melatonin content (335). However, it is conceivable that targets outside the gland could be targeted by T3 produced in the pineal, either by diffusion or via the pineal capillary system (336). Conversely, acute administration of melatonin causes a 7-fold increase in D2 activity in BAT of ground squirrels (337), whereas it does not affect enzyme activity in the pineal gland or the frontal cortex. At the same time, melatonin injection for 7–21 d to adult Syrian hamsters substantially decreased Dio2 expression in the median eminence/arcuate nucleus region of the hypothalamus, where D2 is known to be regulated by photoperiod (338); nonphotoperiodic species do not exhibit this regulation. Thus, whereas there is a potential for pinealocyte-produced T3 to play a role in the central nervous system, melatonin affects D2 activity in discrete brain areas, as well as outside the brain, possibly playing a neuroendocrine role in photoperiod species.
D. Transgenic murine models of ectopic D2 expression in the heart
It has been known for several decades that the heart is a very sensitive target of thyroid hormone. A series of genes are regulated by T3 in the myocardium, including myosin heavy chain (MHCα, β), phospholamban, SERCA2a, and hyperpolarization-activated cyclic nucleotide-gated channel (HCN2), among others (339). In addition, thyrotoxicosis increases myocardial sensitivity or responsiveness to β-adrenergic stimuli, explaining why β-adrenergic blockade is so effective in ameliorating the cardiac symptoms of thyrotoxicosis (340). Despite this, few studies have been able to distinguish between the direct effects of thyroid hormone on myocardial tissue and the indirect cardiovascular effects that result from systemic thyrotoxicosis. These indirect, compensatory effects occur as the heart responds to a decrease in systemic vascular resistance and to changes in neurohormonal input from the central nervous system (341).
Humans express D2 in the myocardium, which could make the myocardium more sensitive to changes in circulating T4. However, unlike humans, rodents express little or no D2 in their myocardium. Thus, a murine model of constitutive cardiac-specific thyrotoxicosis was prepared to provide a mouse model that might better reflect events in the hyperthyroid human myocardium (184). This is a transgenic mouse in which human D2 is driven by the mouse MHCα promoter and is therefore expressed at extremely high levels in the myocardium. Notably, these mice are systemically euthyroid, and myocardium T3 levels are only minimally increased. However, these animals have some physiological, biochemical, and molecular changes in the heart consistent with thyrotoxicosis. Perfused hearts were tachycardic and had an increase in rate pressure product and a decrease in phosphocreatine without any changes in creatine kinase activity, whereas the expression of two T3-responsive genes, HCN2 and β-MHC, was significantly affected. Subsequent studies on adrenergic signal transduction with this mouse model determined that cAMP accumulation in response to either norepinephrine or forskolin treatment was increased in isolated ventricular cardiomyocytes and membrane-enriched fractions from these D2 transgenic hearts as compared with wild type (342). Immunoblot analysis and ADP-ribosylation studies suggest that the increase in adenylate cyclase Vmax is mediated by a decrease in the expression of inhibitory G proteins (Giα-3 and/or Goα), suggesting that cardiac thyrotoxicosis increases β-adrenergic responsiveness via alterations in G proteins. Nevertheless, the lack of a major phenotype in vivo indicates that the cardiac-specific thyrotoxicosis seen in this model can be compensated for by autonomic regulation and/or other factors.
Given the positive inotropic and chronotropic effects triggered by T3, therapy with thyroid hormone has been advocated for treating heart disease, but this remains controversial because T3 also increases myocardial oxygen consumption and could be fatal. To explore this controversy, a murine model of conditional cardiac-specific D2 expression was created using the tetracycline transactivator system (185). D2 myocardial expression increased cardiac T3 levels and enhanced myocardial contractility, accompanied by increased calcium transients and sarcoplasmic reticulum (SR) Ca++ uptake. These phenotypic changes were associated with induction of SERCA2a expression as well as decreased Na++/Ca++ exchanger, MHCβ, and sarcolipin expression. In pressure overload, targeted increases in D2 activity did not block hypertrophy but completely prevented impaired contractility and SR Ca++ cycling as well as altered expression patterns of SERCA2a, sarcolipin, and other markers of pathological hypertrophy. Thus, these data provide supportive evidence that: 1) ectopic D2 expression increases thyroid hormone signaling in a tissue-specific fashion; and 2) the locally generated T3 enhances cardiac function; perhaps most remarkably, the D2-mediated increase in thyroid hormone signaling prevented the deterioration of cardiac function and altered gene expression after pressure overload.
V. Deiodination in Disease States
In addition to modulating thyroid hormone signaling in development and in different areas of adult physiology, deiodinase pathways also modulate thyroid hormone signaling in disease states, generally decreasing the occupancy of TRs and the transcription of T3-responsive genes (Fig. 14). Although these changes can concurrently take place in different tissues, they can also exhibit a remarkable degree of tissue specificity and vary dramatically in intensity. Ultimately, disease-associated changes in deiodinase pathways can be so potent that thyroid hormone signaling is compromised both locally and systemically, as seen during nonthyroidal illness syndrome. This condition is present in about 75% of hospitalized patients (343) and is characterized by variable changes in circulating thyroid hormone levels according to the type and severity of illness, typically including a fall in serum T3 and a rise in serum rT3. It is controversial whether nonthyroidal illness syndrome is an adaptive response to catabolic stress or a pathological entity that should be treated with exogenous thyroid hormone (344,345). A number of controlled trials with thyroid hormone supplementation have shown no benefit (346) or, in one case, worsened outcome (347).
Figure 14.

Local and global changes in thyroid hormone signaling during nonthyroidal illness. A, In euthyroid individuals, thyroid hormone signaling in peripheral tissues is determined by the available nuclear T3 pool that is maintained within normal levels through the regulation of glandular secretion and the conversion of T4 into T3 by D1- and D2-containing tissues. B, During illness and the low T3 syndrome, the nuclear T3 pool is decreased due to decreased glandular secretion, decreased conversion of T4 into T3, and the inactivation of T4 and T3 by reactivated D3. The red arrows indicate decreased enzyme activity; the word “Secretion” has been hatched and lines dotted to indicate a decrease, and the question mark indicates uncertainty regarding the role played by D2 in illness. In panels A and B, some processes are shown in a simplified manner for the sake of clarity: the smaller circles on the left represent cells expressing D1 and/or D2, whereas T4 inside these cells originates from the plasma; the mRNA footprint indicates the set of genes that are regulated by thyroid hormone. [Modified with permission from Bianco et al.: Endocrinology 148:3077–3079, 2007 (445). ©The Endocrine Society.]
A. Local impact of altered deiodinase activity on thyroid hormone signaling in disease states
Several lines of evidence indicate that deiodinases contribute to local responses to tissue injury and disease (348). This concept is also supported by the observation that D2 and D3 activities are modulated by a wide variety of growth factors and morphogens, such as the fibroblast growth factors and the TGF-β ligand family, which are important mediators of tissue injury repair or response to disease (see Section IV.A). A logical extension is that perturbations in deiodinase expression and/or activity, and therefore thyroid hormone signaling, would affect the progression and outcome of disease states.
1. Cardiomyopathy.
Given the responsiveness of the heart to thyroid hormone, the involvement of thyroid hormone in the etiology of heart failure has received considerable attention in recent years. The syndrome of chronic heart failure is principally caused by hemodynamic overload of the heart as a result of hypertension, aortic stenosis, valvular dysfunction, or loss of ventricular tissue due to myocardial infarction. This triggers a complex adaptive response that includes cardiomyocyte hypertrophy and changes in contractility and metabolism, which, if not successful in normalizing wall stress, leads to progressive contractile dysfunction and heart failure. To a certain extent, the response to overload of the heart involves the recapitulation of the fetal gene program (349), and notably, many of these genes are transcriptionally controlled by thyroid hormone (350).
Thyroid hormone levels are maintained at low levels during fetal development, and therefore it is not surprising that aspects of the cardiac phenotype of heart failure resemble that of hypothyroidism. This includes the reduced expression of the Ca++-pump of the sarcoplasmic reticulum (SERCA2a), whose activity is central in beat-to-beat regulation of cytosolic Ca++, and it is thought that impaired thyroid hormone signaling may account for the approximately 50% reduction of SERCA2a expression that is a principal cause of contractile dysfunction in heart failure (351).
Recent animal studies suggest that heart disease induces local tissue hypothyroidism by altering cardiac thyroid hormone metabolism via the deiodinases. D1 and D2 activities are exceptionally low in the heart, and although D2 activity is responsive to reduced T3 levels (352), local conversion of T4 accounts for no more than 7% of cardiac T3 (353). Similarly, D3 expression is virtually undetectable in the healthy heart. However, deiodinase expression is not static because high cardiac D3 activity has been documented in several models of heart failure, including right ventricular hypertrophy from monocrotaline-induced pulmonary arterial hypertension (28,175), left ventricular pressure overload from aortic banding (185), and myocardial infarction from left coronary artery ligation (269). In the model of right ventricular failure, the increase in D3 activity was associated with anatomically specific local hypothyroidism in the right ventricle (28). Furthermore, induction of D3 mRNA and protein expression was associated with activation of the HIF-dependent pathway (28), confirming studies in this model that had shown increased nuclear HIF-1α in cardiomyocytes (354). This suggests that D3 induction in the heart is part of the HIF-orchestrated response generally seen in hypoxia and ischemia, and which is aimed at reducing oxygen consumption and energy turnover (see Section IV.B.4).
Although the effects of D3 induction on energy turnover are clearly adaptive, this need not be the case for other aspects of cardiomyocyte function. In one study (175), ventricles that developed stable compensatory hypertrophy showed significantly less induction of D3 activity compared with ventricles where hypertrophy progressed to failure. TGF-β signaling in the myocardium is particularly increased in the transition from compensatory hypertrophy to failure (355,356), and it may be speculated that this increases D3 activity to a level where reduced thyroid hormone signaling becomes maladaptive.
2. Injury to the nervous system.
Animal studies indicate a beneficial role of thyroid hormone in brain injury (357,358), suggesting that a local D2-mediated increase in thyroid hormone signaling could be neuroprotective. In this regard, several animal models demonstrate altered expression of D2 and D3 after neuronal injury. In the rat, induction of focal cerebral ischemia by transient middle cerebral artery occlusion is followed by the transient up-regulation of D2 mRNA in the ipsilateral striatum in the astrocytic cell bodies (359). In primary cultures of rat astrocytes, hypoxia had no effect on D2 mRNA accumulation; however, the half-life of D2 protein was increased (360). At the same time, it is known that in a neuronal cell line D3 activity is strongly induced by hypoxia (28). An analogous situation is found in the rat peripheral nervous system, in which basal D2 and D3 expressions are low, but mRNA levels of both D2 and D3 rise rapidly after nerve injury (361,362). Although D2 expression localizes to the peripheral nerve sheath, D3 localizes to both the peripheral connective sheath and the internal endoneural compartment. In both models of injury to the nervous system, central and peripheral, D2 and D3 expressions in response to hypoxia seem to be highly cell-specific, suggesting that the coordinated expression of these enzymes might play a role in the response to injury and/or during the healing process.
3. Tumors.
Variable deiodinase expression and activity have been reported in a large number of tumoral tissues or tumor cell lines. In some cases this is not surprising, given that similar deiodinase activity is also found in the nontumoral tissue from which the tumor originated, e.g., thyroid and brain tumors. In other cases, however, deiodinase expression is not present in the tissue from which the tumor originated, e.g., mesothelioma and hepatocarcinoma cell lines, and its significance, if any, remains largely unknown. It is conceivable that local modulation of thyroid hormone signaling in tumoral cells has a role in tumorigenesis and/or interferes with tumor development given that thyroid hormone has been shown either to inhibit or promote proliferation of cells with abnormal pathological stages depending on the particular cell type (363). D1 mRNA and/or activity have been reported in well-differentiated breast cancers (364). High D2 activity has been found in follicular thyroid carcinomas and, in certain patients with widely metastatic disease, it can cause hypertriiodothyronemia (365). D3 mRNA and/or activity has been reported in several human tumors, including astrocytomas (366), gliomas (367), TSH-secreting pituitary tumors (298), vascular anomalies (368,369), and one malignant solitary fibrous tumor (370). Expanding on these studies, it was revealed that D3 overexpression in basal cell carcinoma and the resulting cellular hypothyroidism promote proliferation in malignant cells (32).
4. Inflammation.
Turpentine injection in the hindlimb provides an animal model of local inflammation in which local D3 activity in the muscle/subcutis around the resulting abscess is increased while liver D1 and D3 activities remain unchanged (371) (Fig. 15). D3 induction could result from the increased expression of IL-1β and granulocyte-macrophage colony-stimulating factor in the abscess area, given that both activate the MAPK/ERK signaling cascade, a known pathway of D3 induction (232,372,373). Immunohistochemistry further localized the expression of D3 to the inflammatory cells surrounding the abscess, and not to the muscle itself (371). D3 induction is likely to decrease thyroid hormone signaling locally, which could favor cell proliferation and tissue repair. In addition, due to the relatively small size of this lesion, it is unlikely that this regional increase in D3 could account for the decrease in serum T3 concentrations observed.
Figure 15.

Hindlimb section showing a turpentine-induced abscess in the subcutis. A, Hematoxylin and eosin staining. Note necrosis in the middle of the abscess, surrounded by a large number of granulocytes, lymphocytes, and macrophages. B, D3 immunocytochemistry (ab 676). C, MCT8 immunohistochemistry (ab1306). D, Staining with preabsorbed D3 antiserum. E, D3 preimmune staining. F, Staining with preabsorbed MCT8 antiserum. G, MCT8 preimmune staining. Bar, 500 μm. Note D3 expression found within the inflammation in the abscess. [Reprinted with permission from Boelen et al.: Endocrinology 146:5128–5134, 2005 (371). ©The Endocrine Society.]
B. Systemic impact of abnormal deiodinase activity on thyroid hormone homeostasis
Changes in thyroid hormone signaling by the deiodinases tend to be localized to a specific cell group or tissue. However, these changes could also potentially affect systemic thyroid hormone homeostasis because the cytoplasm and plasma compartments of the cells are in equilibrium. This means that deiodinase-mediated production of T3 or consumption of T4 and/or T3—which takes place in the cytoplasm—eventually affects the plasma levels of thyroid hormones. Because changes in deiodinase expression are rarely dramatic, the changes in plasma are largely minimized by the potent homeostatic mechanisms coordinated by the hypothalamus/pituitary/thyroid gland. For example, a falling serum T3 concentration rapidly triggers an increase in TRH/TSH secretion, which prompts an increase in thyroidal secretion, compensating the fluctuation in serum T3 concentration. The relevance of such mechanisms has been elegantly illustrated by the studies in mice with single or combined targeted disruption of the deiodinase genes, all of which have normal serum T3 concentrations (35,374).
However, in some cases of massive alteration in deiodinase expression, systemic changes do occur because the deiodinase-based mechanisms overwhelm the compensatory response. This is the case in patients with large infantile hemangiomas (369) or other vascular tumors (368), which cause consumptive hypothyroidism due to the accelerated D3-mediated degradation of circulating thyroid hormone at rates exceeding the synthetic capacity of the thyroid gland (348). In other cases, the TRH/TSH compensatory mechanisms are deactivated, such as in the case of nonthyroidal illness syndrome, in which the production of ILs and other mediators of inflammation suppress TSH/TRH secretion (375), so that serum thyroid hormone concentrations are directly and unopposedly affected by altered deiodinase activity.
1. Maintenance of plasma T3 in healthy individuals
a. Thyroidal T3 production.
About 20% of the daily T3 production is derived directly from thyroid secretion, whereas the remaining is the result of deiodinase-mediated peripheral deiodination of T4. The T3 found in the thyroid secretion comes directly from the iodinated thyroglobulin (376), but it can also come from intrathyroidal deiodination of T4 to T3. It is in fact notable that in the thyrocytes, the local action of the deiodinases has systemic consequences. D1 and/or D2 can be expressed in thyrocytes (73,101,377,378), with D2 expression being species specific (379). In human but not in rat thyrocytes, D2 is abundantly expressed because the rat Dio2 gene 5′ FR lacks the two thyroid transcription factor-1 (Nkx2.1) binding sites found in the human DIO2 promoter (379). It has been difficult to ascertain how much thyrocyte deiodination contributes to the T3 content in the thyroid secretion. However, given that T4 and T3 are found in the human thyroglobulin at a molar ratio of 15:1 and that in thyroid secretion this ratio is only 11:1, it is conceivable that thyrocyte deiodination contributes significantly to the net thyroidal daily T3 production (101,380). At the same time, different disease states are known to affect the T4/T3 ratio in the thyroglobulin (381) and to increase thyrocyte deiodination of T4 (101,382). For example, in patients with a hyperactive thyroid, up to two thirds of the daily T3 production comes directly from the thyroid, with a major part of this produced by local deiodination of T4 (383). A similar situation can be found in patients with McCune-Albright syndrome, a condition caused by sporadic activating mutations in the GNAS locus encoding the α-subunit of the G stimulatory protein, Gsα, that stimulates cAMP formation (384,385). McCune-Albright syndrome patients often have increased serum T3/T4 ratio (386) in the setting of elevated thyroidal D1 and D2 activities (387). Because the Dio2 gene is responsive to cAMP, it is likely that the observed increase in D2 activity reflects an increase in cAMP-mediated D2 expression (264,388).
b. Extrathyroidal T3 production.
Initial studies performed in rodents led to the idea that D1 is the major pathway through which extrathyroidal T3 is generated. The discovery of D2 changed this, but it is still clear that the D1 pathway does play a role (389). In rodents, D1 and D2 contribute equally to plasma T3 and, in humans, D1 is likely to play an even smaller role as a source of plasma T3. The key role of D2 in humans was prompted by the observation that treatment with the D1 inhibitor PTU (1000 mg/d) only led to an approximately 30% decrease in serum T3 in patients with primary hypothyroidism receiving fixed doses of exogenous T4 (390,391,392). In addition, during hypothyroidism there is a tendency for the fractional conversion of T4 to T3 to increase, which matches the much higher D2 activity in this condition; D1 activity is in fact reduced by hypothyroidism (393,394). In vitro modeling estimates comparing T3 production in cultured cells by the D1 or D2 pathways also favor the D2 pathway as a predominant source of T3 under euthyroid conditions but also suggest that D1 becomes predominant as serum T3 levels increase, as in thyrotoxic patients (180). This study goes one step further and suggests that skeletal muscle is the main source of plasma T3 in humans, but it uses estimates of D2 activity that are at least two orders of magnitude higher than others have been able to measure in similar muscle samples (395). Thus, although the kinetic studies support a major role for D2 as a source for plasma T3 in humans, the exact location of the D2 pool remains elusive.
c. Homeostatic mechanisms defending plasma T3 levels.
The availability of genetically modified animals, i.e., D1 null, D2 null, D1/D2 null mice (35), as well as the D1-deficient C3H mouse and the C3H/D2 null mouse (374), has provided insight as to the importance of the deiodinase-based homeostatic mechanisms for the maintenance of serum T3 concentrations (Table 1). Remarkably, each of these animals has a normal serum T3 concentration and an increased serum T4 concentration. Although the elevations in serum T4 concentration may result from decreased clearance, it would seem more likely that this is the result of increased thyroidal production given that to normalize serum T3 concentration an excess of T4 will have to be secreted; recall that the T4/T3 ratio in the gland’s secretion is greater than 10. In fact, there is increased serum TSH concentration in both the D2 null and C3H/D2 null models, but not in C3H and D1 null mice, despite sizeable elevations in serum T4 (35,374). It is fascinating that the hypothalamic-pituitary-thyroid axis could be wired such that adjustments in serum T4 concentrations are tolerated to maintain serum T3 concentrations. It is conceivable that D2 ubiquitination in thyrotrophs could explain such tolerance given that higher serum T4 concentrations will reduce D2 activity, and thus relatively less T4 to T3 conversion will take place in these cells (297).
2. Abnormal deiodinase pathways in nonthyroidal illness syndrome: decreased activation vs.
increased catabolism of thyroid hormone. Over the years, it has been unclear how much the alterations in deiodinase pathways seen in patients with nonthyroidal illness syndrome result in decreased thyroid hormone activation, increased thyroid hormone inactivation, or both. Two remarkable new findings in this area brought new light into this puzzle. First, D3 activity, normally absent from most adult tissues, is reactivated in sick patients (268). Second, D2 activity, thought to be important for T3 production, is in fact increased in such patients (395). Combined, these observations indicate that increased thyroid hormone catabolism plays a significant role in the modifications observed in the serum concentrations of thyroid hormone during the nonthyroidal illness syndrome.
a. D1 in nonthyroidal illness.
Because D1 catalyzes both T4 to T3 conversion and the clearance of rT3, the inhibition of D1 alone is theoretically sufficient to explain both decreased serum T3 and increased serum rT3 in certain patients. Consistent with this, analysis of tissues obtained from intensive care unit patients within minutes of death document that liver D1 is decreased compared with healthy controls, and this liver D1 activity correlates positively with the serum T3:rT3 ratio (268). Although D1 is normally a sensitive marker of peripheral thyroid status and is transcriptionally stimulated by T3 (396), several lines of evidence indicate that this T3-dependent D1 induction is impaired by illness-induced cytokines. In HepG2 cells, the activation of nuclear factor-κB (NF-κB) (an important mediator of immune and inflammatory responses) by TNF-α impairs T3-stimulated D1 induction, and this impairment is reversed by both a dominant-negative NF-κB and a pharmacological antagonist of NF-κB activation (397). IL-1 and IL-6 also inhibit T3-dependent induction of hepatic D1, and this IL effect is inhibited both in vitro and in vivo (398) by the forced overexpression of steroid receptor coactivator 1, indicating that defects in T3 receptor coactivators are important for the suppression of liver D1 during illness. At the same time, given that studies in healthy individuals indicate that D1 accounts for only a small fraction of circulating T3, the substantial fall in serum T3 in moderate to severe illness (even without TSH suppression) suggests that additional factors must contribute. Both the inhibition of D2-mediated T4 to T3 conversion and the acceleration of T3 clearance via D3 have been investigated in recent years.
b. D2 in nonthyroidal illness.
Given the recognition that D2 is likely to play an important role in maintenance of serum T3 levels in humans, it has also been postulated that decreases in peripheral D2 might play a role in the fall of serum T3 by reducing T4 to T3 conversion. However, available reports indicate that peripheral D2 (measured in skeletal muscle) is increased rather than decreased during critical illness (395). Thus, current data do not indicate that changes in peripheral D2 contribute significantly to the nonthyroidal illness syndrome.
c. D3 in nonthyroidal illness.
In addition to decreased T4 to T3 conversion, the acceleration of D3-mediated T3 clearance is a second mechanism that can potentially lower serum T3. This hypothesis was investigated directly by the analysis of serum and tissue specimens obtained from intensive care unit patients at the time of death. These studies documented the reactivation of D3 activity in the liver and skeletal muscle of critically ill patients and discovered that specific D3 activity in the liver correlated positively with serum rT3 (399) and negatively with the serum T3:rT3 ratio (268). D3 induction was also confirmed in skeletal muscle biopsies from still-living patients with septic shock and nonthyroidal illness (400). These data support the concept that D3 contributes to the derangement of serum thyroid function tests in critically ill patients and, more broadly, these studies indicate that D3 can be reactivated in benign human cell types in response to tissue injury.
3. TRH/TSH homeostatic mechanisms are suppressed during nonthyroidal illness.
The etiology of the changes in serum concentrations of thyroid hormone in the nonthyroidal illness syndrome is multifactorial and includes suppression of the central thyroid axis and a subsequent decrease in glandular secretion. A number of studies indicate the potential role of D2 in the suppression of the hypothalamic-pituitary-thyroid axis during nonthyroidal illness syndrome (177,401,402). This pathway could work via D2-generated localized hypothalamic thyrotoxicosis followed by the down-regulation of TRH expression in the PVN. Although there is presently no direct evidence that D2-mediated T3 production in tanycytes is responsible for the suppression of TRH secretion, circumstantial evidence supports the functionality of this pathway (39).
Systemic administration of bacterial lipopolysaccharide (LPS) to rats promotes changes in thyroid economy highly similar to nonthyroidal illness, i.e., reduced plasma T4 and T3, with minimal effect on TSH and TRH (403). In this system, D2 expression and activity are rapidly induced by LPS in tanycytes of the MBH (177) (Fig. 16), an effect also observed in hypothalami of LPS-treated mice or mice with chronic inflammation (402,404). D2 activity in the pituitary gland was slightly reduced or not affected in these animal models (402,404). Little is known about the molecular mechanism that leads to increased hypothalamic D2 expression during infection. NF-κB is a potential effector of this pathway, given that its activation potently induces transcription of the human and rat Dio2 gene, and a potent NF-κB binding site has been characterized in the human DIO2 5′ FR (177,405). At the same time, studies on the kinetics of NF-κB activation in the rat MBH suggest that the LPS-induced NF-κB activation is more involved in the maintenance of the high D2 expression rather than in the initiation process (406). Given that LPS induces the expression of a variety of cytokines in a mouse model for nonthyroidal illness (407), resolving which of these molecules is upstream to the NF-κB pathway in this system has been challenging. Currently it is known that in some settings, IL-1β levels parallel the increase in D2 expression (404), whereas circulating corticosterone levels seem not to be involved in the LPS-mediated increase of hypothalamic D2 expression (408).
Figure 16.

Infection up-regulates D2 expression in the mediobasal hypothalamus. Low-power dark-field micrographs from three different rostrocaudal levels of the median eminence (ME) showing the effect of LPS treatment on D2 mRNA expression in the mediobasal hypothalamus. A–C, Controls; D–F, lipopolysaccharide-treated animals. Note: D2 in situ hybridization signal is increased in the tanycytes lining the wall of the third ventricle (III), in the tanycyte processes in the tuberoinfundibular sulci (arrows) and in the external zone of the ME. ARC, Arcuate nucleus. [Reprinted with permission from Fekete et al.: Endocrinology 145:1649–1655, 2004 (177). ©The Endocrine Society.]
The induction of D2 expression in the hypothalamus by LPS administration is thought to increase local thyroid hormone signaling in the MBH, subsequently causing central hyperthyroidism and TSH suppression, analogous to what is observed in patients with systemic infection and nonthyroidal illness. Studies using the animal model of turpentine injection demonstrate that localized muscle infection can increase D2 mRNA levels in the arcuate nucleus/median eminence region 48 h after the turpentine injection, at the same time that D3 activity and TRH mRNA levels decreased in the PVN (404). These findings support a model where the effects of D2-induced local thyrotoxicosis on suppression of the central thyroid axis are augmented by a down-regulation of D3, resulting in a decrease in TRH expression, further contributing to low serum thyroid hormone levels.
C. Genetic basis of deiodinase-related diseases
Given all the evidence indicating that deiodinases are critical for a number of cell systems, both during development and in adult vertebrates, it is not surprising that specific diseases or conditions have been linked to modifications in the deiodinase genes or genes involved in deiodinase synthesis and function. What is surprising, however, is that only a handful of such diseases/conditions have been identified, reflecting that isolated deiodinase dysfunction is usually associated with very mild systemic phenotypes, largely compensated by strong TSH-dependent homeostatic mechanisms. This is clear from the analysis of the several mouse models in which deiodinase genes have been inactivated (35).
1. Gene polymorphisms
a. The DIO2 Thr92Ala polymorphism.
Although many single nucleotide polymorphisms (SNPs) in the deiodinase genes have been identified, their functional relevance remains to be assessed. Perhaps the best characterized deiodinase SNP is the Thr92Ala in the D2 gene (D2-G/A; Thr92Ala; GI 13654872; rs225014) (409). This SNP is common in various ethnic groups; its allele frequency is 0.35 in Caucasians and is particularly high (0.75) in Pima Indians (409). The Thr92Ala change in the coding region of D2 does not interfere with the kinetics of the transiently expressed D2 enzyme (410,411) but was found to be associated with decreased enzyme velocity in the human thyroid and skeletal muscle (411). Studies addressing functional consequences of the Thr92Ala SNP are conflicting (412). Although this SNP was initially found to be associated with insulin resistance in obese Caucasians (409) and type 2 diabetes (411), in subsequent studies it could not be linked to type 2 diabetes or insulin resistance in Amish or Danish populations or in individuals enrolled in the Framingham Heart Study (413,414,415). The available data on the association of Thr92Ala SNP with the risk for the development of hypertension are also conflicting (416,417).
It is possible that these conflicting data are the result of linkage of the Thr92Ala D2 SNP with another gene polymorphism. In this regard, it has been shown that a Pro121Ala polymorphism of the peroxisome proliferator-activated receptor γ2 is inconsistently associated with insulin resistance (418). However, a combination of the Thr92Ala D2 and Pro121Ala peroxisome proliferator-activated receptor γ2 polymorphisms modulated insulin resistance and other features (blood pressure and metabolic syndrome) in nondiabetic patients, with carriers of both polymorphisms being the most affected (418). Interestingly, this study also demonstrated a peroxisome proliferator response element in the DIO2 by which these two polymorphisms might functionally complement each other to increase the risk for insulin resistance-related abnormalities (418).
It has also been proposed that the Thr92Ala D2 SNP may either be associated or in linkage disequilibrium with a functional D2 SNP involved in the development of Graves’ disease in a Russian population (419). A study of three human DIO2 SNPs in subjects in iodine-deficient areas of China found that two of the SNPs (intronic SNPs; rs225012 and rs225010 in GenBank) were associated with mental retardation, whereas the Thr92Ala was not (420). Lastly, the Thr92Ala D2 polymorphism was also linked to susceptibility for generalized osteoarthritis, a common late-onset articular joint disease (421).
The effects of the D2 Thr92Ala polymorphism on thyroid hormone homeostasis and thyroid function have also been investigated. A correlation has been observed between the D2 Thr92Ala polymorphism and serum TSH but not serum thyroid hormone levels in healthy individuals (410). A different study on Dutch blood donors also confirmed that individuals heterozygous for the Thr92Ala D2 polymorphism have lower serum TSH compared with individuals with wild-type D2, and surprisingly, also to individuals homozygous for Thr92Ala D2 polymorphism (422). Other studies indicated that thyroidectomized patients with the Thr92Ala polymorphism required a higher dose of T4 to achieve target TSH levels (423). However, the Thr92Ala polymorphism was not correlated with differences in general condition, neurocognitive functioning, or the response to T4/T3 combination therapy in patients treated for hypothyroidism (424).
b. Other D2 polymorphisms.
The 5′ untranslated region (UTR) of the human D2 mRNA contains several short open reading frames (ORFs). The shortest and most 5′ of these, ORF-A, decreases D2 activity by interfering with the translational initiation of the D2 ORF (98). A SNP found in ORF-A, D2-ORFA-Gly3Asp (rs12885300), was linked with lower levels of plasma T4, FT4, and rT3 in a dose-dependent manner in healthy blood donors; however, no association was detected in elderly men (425). Subsequent studies also indicated that the D2-ORFA-Gly3Asp polymorphism could increase DIO2 transcription; however, the possible mechanism for this effect remains unresolved (426). In chicken, two Dio2 mRNA polymorphisms have been described. One polymorphism is found at the end of the SECIS element at position 5836 and does not affect SECIS function (99). A second silent mutation is located in the D2 coding region, in the donor site of an alternatively spliced −77 cD2 transcript (98), but no data are presently available as to whether it affects the splicing of the D2 message.
c. D1 and D3 polymorphisms.
Evidence has been presented that two SNPs identified in the 3′ UTR of the human D1 mRNA are related to altered circulating thyroid hormone serum levels (410). These SNPs include: D1a-C/T at position 785 (GI 4557521; rs11206244), and D1b-A/G at position 1814 (rs12095080), located 33 nucleotides 3′ to the SECIS element. Carriers of the T variant of the D1a-C/T polymorphism exhibited higher rT3/T4 and lower T3/rT3 ratios in serum, suggesting a negative effect of this variant on D1 expression and/or activity. In contrast, the G variant of the D1b-A/G polymorphism correlated with lower rT3/T4 and higher T3/rT3 ratios, suggesting increased D1 expression and/or activity of this variant. These alterations in thyroid hormone levels are also paralleled by serum TSH levels (410). Interestingly, the D1a-T and D1b-A haplotype allele also contributed to circulating T3 in an age-dependent manner with lower serum T3 levels associated with the haplotype in the elderly study population (mean age, 77 yr), but not in the younger one (mean age, 46 yr) (427). Additionally, a significant association of the T variant of the D1a-C/T polymorphism was observed with increased free IGF-I levels (427), and the C allele of the rs2235544 polymorphism was associated with increased D1 function and increased FT3/T4 ratio (389). A T/G polymorphism has also been identified at position 1546 of the human D3 mRNA sequence (GI 4503334), 66 nucleotides 5′ to the SECIS element, but there was no significant correlation between this polymorphism and plasma iodothyronines (410).
2. DIO3 gene imprinting may contribute to uniparental disomy.
The D3 enzyme is encoded by the Dio3 gene, located on mouse chromosome 12F1 and human 14q32 (428). This gene is composed of a single exon of approximately 1.9 kb in length (429). A second gene has also been identified, Dio3os, that is transcribed in the opposite orientation of Dio3 (430). The Dio3os is alternatively spliced in multiple ways, resulting in a complex pattern of transcripts, some of which overlap with the Dio3 promoter and transcription start site. No ORFs are conserved between any of the mouse and human Dio3os splicing products; thus it is thought that this gene is likely to be a noncoding gene (430). Although some antisense genes can act to silence gene expression, simultaneous expression of DIO3 and DIO3os has been reported in the developing fetus and in several human tissues and neuroblastoma cell lines (133,430,431,432). On the other hand, Dio3os up-regulation in the mature brown adipocyte corresponded to a loss of Dio3 transcript, indicating that concurrent expression is not mandatory (433).
Dio3 transcription is regulated by a variety of hormones and growth factors (434) but is also subject to regulation by genomic imprinting. Imprinted genes are preferentially expressed from one parental allele (136). In this case, Dio3 is mainly expressed from the paternal allele, although slight expression from the maternal allele can also be detected in the fetus (133,134,135). Dio3 is located within the imprinted Dlk1-Gtl2 gene locus (133,134,135,435). Additional paternally expressed genes found in this locus include delta-like 1 (Dlk1), an EGF family member (also called Pref1) that inhibits adipocyte differentiation, and a protein of unknown function, retrotransposon-like 1 (Rtl1) (436,437). Maternally, these genes are normally repressed, with several noncoding RNAs expressed instead (437). Transcripts expressed include those of gene trap locus 2 (Gtl2), Rtl1 antisense (Rtl1-as), RNA imprinted and accumulated in the nucleus (Rian), and microRNA containing gene (Mirg). Many of these are precursors for smaller RNAs, with Rian coding for small nucleolar RNAs, whereas Rtl1-as and Mirg are associated with microRNAs (438,439). Interestingly, Dio3os is not imprinted and instead exhibits a biallelic pattern of expression (437). This complex arrangement is consistent with the finding that many imprinted genes also contain multiply spliced, untranslated, antisense genes within the same locus (431).
The imprinting of genes is associated with differential methylation of CpG-rich DNA sequences (136). Although the Dio3 promoter contains a CpG island, there is no difference in the methylation pattern between the paternal and maternal alleles of Dio3, with this region being relatively unmethylated (133,134,135). However, an intergenic germline-derived differentially methylated region (IG-DMR) located between the Dlk1 and Gtl2 genes has been implicated in the imprinting control of this locus (440). The IG-DMR is unmethylated maternally, and maternal transmission of a deletion of this region was lethal after embryonic d 16. Additionally, this deletion resulted in the loss of expression of the normal maternal complement of noncoding transcripts and increased biallelic expression of the paternally expressed genes Dlk1, Rtl1, and Dio3. When the IG-DMR deletion was paternally inherited, no change in Dlk1, Rtl1, Dio3, or noncoding RNA expression was observed, indicating that methylation of this region appears to be functionally equivalent to its loss. Although Dio3 is paternally imprinted in these fetal studies, the Dlk1-Gtl2 locus is not imprinted in the placenta, and Dio3 is also not imprinted in adult brain (440,441). This suggests that a more complex Dio3 imprinting may exist in adult Dio3-expressing tissues.
In uniparental disomy (UPD), two copies of the same chromosome come from one parent. Models of this disease for the Dio3-containing chromosome are found in both mice (UPD12) and humans (UPD14). In embryos of mice with two copies of paternally derived chromosome 12 (pUPD12), Dio3 expression were doubled whereas Dio3 mRNA levels were reduced to approximately half of normal in models of maternal UPD12 (mUPD12) (134). Mice with pUPD12 die in late gestation and exhibit muscle hypertrophy and hypoossification of bones, whereas mice with mUPD12 die perinatally with a reciprocal skeletal muscle phenotype and additional growth, skin, and ear defects (435,442). In situ analysis did not localize Dio3 expression to tissues affected in the UPD12 fetuses such as muscle and bone (134). However, when serum total T3 levels were measured in embryonic d 15.5 UPD12 embryos, serum T3 levels in pUPD12 embryos were one third lower than controls, whereas those of the mUPD12 embryos were increased by about one third. Thus, it has been hypothesized that fetal exposure to abnormal levels of thyroid hormone may contribute to some of the abnormalities found in both mouse UPD12 and human UPD14 (133,134,440).
3. Patients with a mutation in SBP-2 have abnormal thyroid hormone metabolism.
Disruption of any of the factors that regulate selenoprotein synthesis (see Section II.B) could potentially affect deiodinase production and thyroid hormone homeostasis. Moreover, a recent finding that patients with mutations in SBP-2 have abnormal thyroid hormone metabolism strongly supports this concept (80) (Fig. 17). In these landmark studies, a family was identified in which three of seven siblings had abnormal thyroid function tests with elevated TSH and T4, whereas T3 was below the normal range. Additionally, whereas T3 administration suppressed TSH equally well in both affected and unaffected individuals, T4 was much less effective in TSH suppression of affected individuals. Using cultured skin fibroblasts, it was determined that affected individuals had much less D2 enzyme activity. However, DIO2 mRNA levels of these patients were not decreased, indicating that lower D2 protein levels were due to a posttranscriptional defect. With linkage analysis, affected individuals were found to have the missense mutation R540Q in the RNA binding domain of SBP-2. Further analysis revealed that the selenoproteins GPX and Selenoprotein P were also decreased in these individuals, suggesting a global defect in selenoprotein synthesis. These studies also reported another patient with similar thyroid function tests where the affected individual had two different alleles of SPB-2 that both result in a truncated SBP-2 protein. Taken together, this work clearly illustrates that impaired selenoprotein synthesis can result in defects in deiodinase synthesis, leading to defects in thyroid hormone metabolism.
Figure 17.

Individuals with a mutation in SBP-2 have defects in D2. A, Serum TSH (mU/liter) in affected or unaffected individuals before and during oral administration of incremental doses of l-T4. Patients with a mutation in SBP-2 required greater doses of T4 to suppress TSH. B, Serum TSH (mU/liter) and corresponding serum T3 levels (ng/dl) in affected or unaffected individuals before and during the oral administration of incremental doses of l-T3 for 5 d. TSH suppression was achieved at similar concentration of serum T3 in all four subjects. C, D2 enzymatic activity in fibroblasts from two affected children and six unaffected individuals. Baseline (open symbols) and cAMP-stimulated (filled symbols) D2 activity in unaffected (squares) and affected (circles) individuals. The dashed line indicates the limit of detection of the assay. The mean baseline activity in fibroblasts from affected individuals was reduced to near or below the limit of detection and was not inducible by cAMP. D2 mRNA levels were the same in both affected and unaffected individuals (data not shown). [Reprinted with permission from Dumitrescu et al.: Nat Genet 37:1247–1252, 2005 (80).]
Although some of the individuals in the above studies presented delayed growth, this relatively mild phenotype was unexpected because mice lacking the selenocysteine tRNA die in utero (443). This suggests that not all selenoprotein synthesis is adversely affected in these patients. Clues as to why production of some selenoproteins might be selectively more impaired in affected individuals is found in studies indicating that SBP-2 does not have the same affinity for all SECIS elements (87,89,444). In an extension of these results, recent work has shown that the R540Q SBP-2 mutant has impaired binding to some, but not all, SECIS elements (87,89). Thus, whereas the R540Q SBP-2 mutant no longer bound to the SECIS elements found in the GPx1, Dio1, and Dio2 SECIS elements, binding to those found in the phospholipid hydroperoxide GPX (PhGPx) and TRX reductase 1 were retained (87). To evaluate the functional activity of the R540Q SBP-2 mutant, in vitro translation of a luciferase reporter containing an in-frame UGA and different SECIS elements was assessed (87). Surprisingly, both the R540Q mutant and the wild type SBP-2 were able to similarly facilitate UGA read-through of reporters containing GPx1, Dio1, and PhGPX SECIS elements. In additional experiments, read-through of a Dio1 SECIS-containing UGA luciferase reporter was assessed in a competition assay in the presence of increasing amounts of other SECIS elements. Under these conditions, GPx1 and Dio2 SECIS elements competed more efficiently in wild-type SBP-2-mediated Dio1 read-through than R540Q mutant-mediated read-through. On the other hand, the PhGPx SECIS element competed both wild-type and R540Q mutant SBP-2 mediated read-through to a similar extent. These studies indicate that although the R540Q SBP-2 mutant does exhibit impaired binding to some SECIS elements, its effects on selenoprotein production will not simply mirror SECIS affinity. Instead, a more complex set of factors, including the amounts and types of other selenoprotein mRNAs, will generate the defective pattern of selenoprotein production observed in these patients.
VI. Conclusions
The body of work that has been reviewed here illustrates the importance of the deiodinases in local control of thyroid hormone action. In addition to maintaining thyroid hormone homeostasis, deiodinase expression controls thyroid hormone action relatively independent of thyroid hormone serum concentrations. From a broad perspective, this paradigm can be seen as an example of how hormones are activated or inactivated in a controlled fashion in specific extraglandular tissues, in an analogous role to 5α-reductase and P450 aromatase in sex steroid metabolism and to 11β-hydroxysteroid dehydrogenase in glucocorticoid metabolism. This has a tremendous impact on our understanding of how thyroid hormone signaling interacts with vertebrate development and metamorphosis. It also unveiled new aspects of the role played by thyroid hormone in response to injury and tissue repair, hypothalamic function, and energy homeostasis. Comparable to the situation with the steroid-metabolizing enzymes, for which multiple antagonist drugs are in widespread clinical use, drug development for the control of deiodination could provide useful therapeutic tools in these areas.
Acknowledgments
The authors are grateful for the insightful work provided by Mr. John W. Harney, all of the fellows and students that contributed to some of the studies discussed in this article, and to the different funding agencies who supported the research.
Footnotes
This work was supported by Hungarian Scientific Research Fund Grant OTKA T049081; Sixth EU Research Framework Program Grant LSHM-CT-2003-503041; János Bolyai Research Scholarship of the Hungarian Academy of Sciences; and National Institute of Diabetes, Digestive and Kidney Disorders Grants DK76117, DK58538, DK65055, DK78560, DK078476, DK076099, and DK064643.
Disclosure Statement: A.C.B. is a consultant for Exelixis; all other authors have nothing to disclose.
First Published Online September 24, 2008
The authors dedicate this article to the memory of Dr. Robert Utiger, a giant in the field of thyroid hormone metabolism and a great mentor, role model, and friend.
Abbreviations: AgRP, Agouti-related protein; BAT, brown adipose tissue; BMR, basal metabolic rate; 3D, three-dimensional; D1, type 1 iodothyronine deiodinase; D2, type 2 iodothyronine deiodinase; D3, type 3 iodothyronine deiodinase; Dio1, the gene encoding D1; Dio2, the gene encoding D2; Dio3, the gene encoding D3; Dlk1, delta-like 1; E1, Ub activating enzyme; E2, Ub conjugase; E3, Ub ligase; EFsec, elongation factor sec; EGF, epidermal growth factor; ER, endoplasmic reticulum; ERAD, ER-associated degradation; FR, flanking region; FRET, fluorescence resonance energy transfer; FT3, free T3; FT4, free T4; GPX, glutathione peroxidase; Gtl2, gene trap locus 2; HCN2, hyperpolarization-activated cyclic nucleotide-gated channel 2; Hh, hedgehog; HIF, hypoxia-inducible factor; IDUA, α-L-iduronidase; IG-DMR, intergenic germline-derived differentially methylated region; ihh, Indian hedgehog; IRD, inner ring deiodination; Km, Michaelis-Menten constant; LPS, bacterial lipopolysaccharide; MBH, mediobasal hypothalamus; MCT, monocarboxylate transporter; MHC, myosin heavy chains; NF-κB, nuclear factor-κΒ; NIS, sodium-iodine symporter; NPY, neuropeptide Y;ORD, outer ring deiodination; ORF, open reading frame; PhGPX, phospholipid hydroperoxide GPX; PTU, 6-n-propyl-2-thiouracil; pVHL, von Hippel-Lindau protein; PVN, paraventricular nucleus; Rtl1, retrotransposon-like 1; SBP-2, SECIS binding protein 2; Sec, selenocysteine; SECIS, Sec insertion sequence; SERCA1, sarco(endo)plasmic reticulum Ca2+ ATPase; Shh, sonic hedgehog; SLA, soluble liver antigen; SNP, single nucleotide polymorphism; SOCS, suppressor of cytokine signaling; SPS-1, selenophosphate synthetase-1; SPS-2, selenophosphate synthetase-2; SR, sarcoplasmic reticulum; T0AM, thyronamine; T1AM, 3-iodothyronamine; TAR, G protein-coupled trace amine receptor 1; TR, thyroid hormone receptor; TRX, thioredoxin; Ub, ubiquitin; UCP, uncoupling protein; UPD, uniparental disomy; USP, Ub-specific protease; UTR, untranslated region; WSB1, WD40-repeat/SOCS box protein 1.
References
- Gross J, Pitt-Rivers R 1952 The identification of 3,5, 3′-L-triiodothyronine in human plasma. Lancet 1:439 [DOI] [PubMed] [Google Scholar]
- Braverman LE, Ingbar SH, Sterling K 1970 Conversion of thyroxine (T4) to triiodothyronine (T3) in athyreotic subjects. J Clin Invest 49:855–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Koenig RJ 2000 Gene regulation by thyroid hormone. Trends Endocrinol Metab 11:207–211 [DOI] [PubMed] [Google Scholar]
- Yen PM, Ando S, Feng X, Liu Y, Maruvada P, Xia X 2006 Thyroid hormone action at the cellular, genomic and target gene levels. Mol Cell Endocrinol 246:121–127 [DOI] [PubMed] [Google Scholar]
- Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR 2002 Biochemistry, cellular and molecular biology and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev 23:38–89 [DOI] [PubMed] [Google Scholar]
- Visser TJ, van der Does-Tobe I, Docter R, Hennemann G 1975 Conversion of thyroxine into tri-iodothyronine by rat liver homogenate. Biochem J 150:489–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra IJ 1977 A study of extrathyroidal conversion of thyroxine (T4) to 3,3′,5-triiodothyronine (T3) in vitro. Endocrinology 101:453–463 [DOI] [PubMed] [Google Scholar]
- Kaplan MM, Utiger RD 1978 Iodothyronine metabolism in rat liver homogenates. J Clin Invest 61:459–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmel M, Utiger RD 1977 Thyroidal and peripheral production of thyroid hormones. Review of recent findings and their clinical implications. Ann Intern Med 87:760–768 [DOI] [PubMed] [Google Scholar]
- Chopra IJ, Solomon DH, Chopra U, Wu SY, Fisher DA, Nakamira Y 1978 Pathways of metabolism of thyroid hormones. Recent Prog Horm Res 34:521–657 [DOI] [PubMed] [Google Scholar]
- Chopra IJ, Hershman JM, Pardridge WM, Nicoloff JT 1983 Thyroid function in nonthyroidal illnesses. Ann Intern Med 98:946–957 [DOI] [PubMed] [Google Scholar]
- Leonard JL, Visser TJ 1986 Biochemistry of deiodination. In: Hennemann G, ed. Thyroid hormone metabolism. New York: Marcel Dekker; 189–229 [Google Scholar]
- Hennemann G 1986 Thyroid hormone deiodination in healthy man. In: Hennemann G, ed. Thyroid hormone metabolism. New York: Marcel Dekker; 277–295 [Google Scholar]
- Sugawara M, Lau R, Wasser HL, Nelson AM, Kuma K, Hershman JM 1984 Thyroid T4 5′-deiodinase activity in normal and abnormal human thyroid glands. Metabolism 33:332–336 [DOI] [PubMed] [Google Scholar]
- Takamatsu J, Sugawara M, Kuma K, Kobayashi A, Matsuzuka F, Mozai T, Hershman JM 1984 Ratio of serum triiodothyronine to thyroxine and the prognosis of triiodothyronine-predominant Graves’ disease. Ann Intern Med 100:372–375 [DOI] [PubMed] [Google Scholar]
- Nishikawa M, Toyoda N, Yonemoto T, Ogawa Y, Tabata S, Sakaguchi N, Tokoro T, Gondou A, Yoshimura M, Yoshikawa N, Inada M 1998 Quantitative measurements for type 1 deiodinase messenger ribonucleic acid in human peripheral blood mononuclear cells: mechanism of the preferential increase of T3 in hyperthyroid Graves’ disease. Biochem Biophys Res Commun 250:642–646 [DOI] [PubMed] [Google Scholar]
- Oppenheimer JH, Schwartz HL, Surks MI 1972 Propylthiouracil inhibits the conversion of L-thyroxine to L-triiodothyronine: an explanation of the antithyroxine effect of propylthiouracil and evidence supporting the concept that triiodothyronine is the active thyroid hormone. J Clin Invest 51:2493–2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiersinga WM, Trip MD 1986 Amiodarone and thyroid hormone metabolism. Postgrad Med J 62:909–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen PR, Silva JE, Kaplan MM 1981 Relationships between circulating and intracellular thyroid hormones: physiological and clinical implications. Endocr Rev 2:87–102 [DOI] [PubMed] [Google Scholar]
- Kaplan MM, Yaskoski KA 1981 Maturational patterns of iodothyronine phenolic and tyrosyl ring deiodinase activities in rat cerebrum, cerebellum, and hypothalamus. J Clin Invest 67:1208–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker KB, Schneider MJ, Davey JC, Galton VA 1995 The type III 5-deiodinase in Rana catesbeiana tadpoles is encoded by a thyroid hormone-responsive gene. Endocrinology 136:4424–4431 [DOI] [PubMed] [Google Scholar]
- St. Germain DL, Galton VA 1997 The deiodinase family of selenoproteins. Thyroid 7:655–668 [DOI] [PubMed] [Google Scholar]
- Silva JE, Larsen PR 1985 Potential of brown adipose tissue type II thyroxine 5′-deiodinase as a local and systemic source of triiodothyronine in rats. J Clin Invest 76:2296–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco AC, Silva JE 1987 Nuclear 3,5,3′-triiiodothyronine (T3) in brown adipose tissue: receptor occupancy and sources of T3 as determined by in vivo techniques. Endocrinology 120:55–62 [DOI] [PubMed] [Google Scholar]
- Bianco AC, Sheng X, Silva JE 1988 Triiodothyronine amplifies norepinephrine stimulation of uncoupling protein gene transcription by a mechanism not requiring protein synthesis. J Biol Chem 263:18168–18175 [PubMed] [Google Scholar]
- Carvalho SD, Kimura ET, Bianco AC, Silva JE 1991 Central role of brown adipose tissue thyroxine 5′-deiodinase on thyroid hormone-dependent thermogenic response to cold. Endocrinology 128:2149–2159 [DOI] [PubMed] [Google Scholar]
- Marsh-Armstrong N, Huang H, Remo BF, Liu TT, Brown DD 1999 Asymmetric growth and development of the Xenopus laevis retina during metamorphosis is controlled by type III deiodinase. Neuron 24:871–878 [DOI] [PubMed] [Google Scholar]
- Simonides WS, Mulcahey MA, Redout EM, Muller A, Zuidwijk MJ, Visser TJ, Wassen FW, Crescenzi A, da-Silva WS, Harney J, Engel FB, Obregon MJ, Larsen PR, Bianco AC, Huang SA 2008 Hypoxia-inducible factor induces local thyroid hormone inactivation during hypoxic-ischemic disease in rats. J Clin Invest 118:975–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galton VA 2005 The roles of the iodothyronine deiodinases in mammalian development. Thyroid 15:823–834 [DOI] [PubMed] [Google Scholar]
- Brown DD 2005 The role of deiodinases in amphibian metamorphosis. Thyroid 15:815–821 [DOI] [PubMed] [Google Scholar]
- Dentice M, Bandyopadhyay A, Gereben B, Callebaut I, Christoffolete MA, Kim BW, Nissim S, Mornon JP, Zavacki AM, Zeold A, Capelo LP, Curcio-Morelli C, Ribeiro R, Harney JW, Tabin CJ, Bianco AC 2005 The hedgehog-inducible ubiquitin ligase subunit WSB-1 modulates thyroid hormone activation and PTHrP secretion in the developing growth plate. Nat Cell Biol 7:698–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dentice M, Luongo C, Huang S, Ambrosio R, Elefante A, Mirebeau-Prunier D, Zavacki AM, Fenzi G, Grachtchouk M, Hutchin M, Dlugosz AA, Bianco AC, Missero C, Larsen PR, Salvatore D 2007 Sonic hedgehog-induced type 3 deiodinase blocks thyroid hormone action enhancing proliferation of normal and malignant keratinocytes. Proc Natl Acad Sci USA 104:14466–14471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco AC, Kim BW 2006 Deiodinases: implications of the local control of thyroid hormone action. J Clin Invest 116:2571–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez A, St Germain DL 2003 Thyroid hormone deiodinases: physiology and clinical disorders. Curr Opin Pediatr 15:416–420 [DOI] [PubMed] [Google Scholar]
- St Germain DL, Hernandez A, Schneider MJ, Galton VA 2005 Insights into the role of deiodinases from studies of genetically modified animals. Thyroid 15:905–916 [DOI] [PubMed] [Google Scholar]
- Kuiper GG, Kester MH, Peeters RP, Visser TJ 2005 Biochemical mechanisms of thyroid hormone deiodination. Thyroid 15:787–798 [DOI] [PubMed] [Google Scholar]
- Kohrle J 2007 Thyroid hormone transporters in health and disease: advances in thyroid hormone deiodination. Best Pract Res Clin Endocrinol Metab 21:173–191 [DOI] [PubMed] [Google Scholar]
- Bianco AC, Larsen PR 2005 Cellular and structural biology of the deiodinases. Thyroid 15:777–786 [DOI] [PubMed] [Google Scholar]
- Gereben B, Zeold A, Dentice M, Salvatore D, Bianco AC 2008 Activation and inactivation of thyroid hormone by deiodinases: local action with general consequences. Cell Mol Life Sci 65:570–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnidehou S, Caillou B, Talbot M, Ohayon R, Kaniewski J, Noel-Hudson MS, Morand S, Agnangji D, Sezan A, Courtin F, Virion A, Dupuy C 2004 Iodotyrosine dehalogenase 1 (DEHAL1) is a transmembrane protein involved in the recycling of iodide close to the thyroglobulin iodination site. FASEB J 18:1574–1576 [DOI] [PubMed] [Google Scholar]
- Gaboriaud C, Bissery V, Benchetrit T, Mornon JP 1987 Hydrophobic cluster analysis: an efficient new way to compare and analyse amino acid sequences. FEBS Lett 224:149–155 [DOI] [PubMed] [Google Scholar]
- Callebaut I, Labesse G, Durand P, Poupon A, Canard L, Chomilier J, Henrissat B, Mornon JP 1997 Deciphering protein sequence information through hydrophobic cluster analysis (HCA): current status and perspectives. Cell Mol Life Sci 53:621–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callebaut I, Curcio-Morelli C, Mornon JP, Gereben B, Buettner C, Huang S, Castro B, Fonseca TL, Harney JW, Larsen PR, Bianco AC 2003 The iodothyronine selenodeiodinases are thioredoxin-fold family proteins containing a glycoside hydrolase clan GH-A-like structure. J Biol Chem 278:36887–36896 [DOI] [PubMed] [Google Scholar]
- Sagar GD, Gereben B, Callebaut I, Mornon JP, Zeold A, da Silva WS, Luongo C, Dentice M, Tente SM, Freitas BC, Harney JW, Zavacki AM, Bianco AC 2007 Ubiquitination-induced conformational change within the deiodinase dimer is a switch regulating enzyme activity. Mol Cell Biol 27:4774–4783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard JL, Rosenberg IN 1978 Subcellular distribution of thyroxine 5′-deiodinase in the rat kidney: a plasma membrane location. Endocrinology 103:274–280 [DOI] [PubMed] [Google Scholar]
- Maciel RM, Ozawa Y, Chopra IJ 1979 Subcellular localization of thyroxine and reverse triiodothyronine outer ring monodeiodinating activities. Endocrinology 104:365–371 [DOI] [PubMed] [Google Scholar]
- Prabakaran D, Ahima RS, Harney JW, Berry MJ, Larsen PR, Arvan P 1999 Polarized targeting of epithelial cell proteins in thyrocytes and MDCK cells. J Cell Sci 112:1247–1256 [DOI] [PubMed] [Google Scholar]
- Baqui MM, Gereben B, Harney JW, Larsen PR, Bianco AC 2000 Distinct subcellular localization of transiently expressed types 1 and 2 iodothyronine deiodinases as determined by immunofluorescence confocal microscopy. Endocrinology 141:4309–4312 [DOI] [PubMed] [Google Scholar]
- Curcio C, Baqui MM, Salvatore D, Rihn BH, Mohr S, Harney JW, Larsen PR, Bianco AC 2001 The human type 2 iodothyronine deiodinase is a selenoprotein highly expressed in a mesothelioma cell line. J Biol Chem 276:30183–30187 [DOI] [PubMed] [Google Scholar]
- Baqui M, Botero D, Gereben B, Curcio C, Harney JW, Salvatore D, Sorimachi K, Larsen PR, Bianco AC 2003 Human type 3 iodothyronine selenodeiodinase is located in the plasma membrane and undergoes rapid internalization to endosomes. J Biol Chem 278:1206–1211 [DOI] [PubMed] [Google Scholar]
- Silva JE, Larsen PR 1978 Contributions of plasma triiodothyronine and local thyroxine monodeiodination to triiodothyronine to nuclear triiodothyronine receptor saturation in pituitary, liver, and kidney of hypothyroid rats. Further evidence relating saturation of pituitary nuclear triiodothyronine receptors and the acute inhibition of thyroid-stimulating hormone release. J Clin Invest 61:1247–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeold A, Pormuller L, Dentice M, Harney JW, Curcio-Morelli C, Tente SM, Bianco AC, Gereben B 2006 Metabolic instability of type 2 deiodinase is transferable to stable proteins independently of subcellular localization. J Biol Chem 281:31538–31543 [DOI] [PubMed] [Google Scholar]
- Silva JE, Dick TE, Larsen PR 1978 Contribution of local tissue thyroxine monodeiodination to the nuclear 3,5,3′-triiodothyronine in pituitary, liver, and kidney of euthyroid rats. Endocrinology 103:1196–1207 [DOI] [PubMed] [Google Scholar]
- Oppenheimer JH, Schwartz HL 1985 Stereospecific transport of triiodothyronine from plasma to cytosol and from cytosol to nucleus in rat liver, kidney, brain, and heart. J Clin Invest 75:147–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesema EC, Kuiper GG, Jansen J, Visser TJ, Kester MH 2006 Thyroid hormone transport by the human monocarboxylate transporter 8 and its rate-limiting role in intracellular metabolism. Mol Endocrinol 20:2761–2772 [DOI] [PubMed] [Google Scholar]
- Martin JL 1995 Thioredoxin—a fold for all reasons. Structure 3:245–250 [DOI] [PubMed] [Google Scholar]
- Coutinho PM, Henrissat B 1999 Carbohydrate-active enzymes (http://afmb.cnrs-mrs.fr/∼cazy/CAZY/index.html) [Google Scholar]
- Leonard JL, Rosenberg IN 1981 Solubilization of a phospholipid-requiring enzyme, iodothyronine 5′-deiodinase, from rat kidney membranes. Biochim Biophys Acta 659:205–218 [DOI] [PubMed] [Google Scholar]
- Safran M, Leonard JL 1991 Comparison of the physicochemical properties of type I and type II iodothyronine 5′-deiodinase. J Biol Chem 266:3233–3238 [PubMed] [Google Scholar]
- Farwell AP, Safran M, Dubord S, Leonard JL 1996 Degradation and recycling of the substrate-binding subunit of type II iodothyronine 5′-deiodinase in astrocytes. J Biol Chem 271:16369–16374 [PubMed] [Google Scholar]
- Leonard DM, Stachelek SJ, Safran M, Farwell AP, Kowalik TF, Leonard JL 2000 Cloning, expression, and functional characterization of the substrate binding subunit of rat type II iodothyronine 5′-deiodinase. J Biol Chem 275:25194–25201 [DOI] [PubMed] [Google Scholar]
- Leonard JL, Leonard DM, Safran M, Wu R, Zapp ML, Farwell AP 1999 The mammalian homolog of the frog type II selenodeiodinase does not encode a functional enzyme in the rat. Endocrinology 140:2206–2215 [DOI] [PubMed] [Google Scholar]
- Safran M, Farwell AP, Leonard JL 1996 Catalytic activity of type II iodothyronine 5′-deiodinase polypeptide is dependent upon a cyclic AMP activation factor. J Biol Chem 271:16363–16368 [PubMed] [Google Scholar]
- Leonard JL, Visser TJ, Leonard DM 2001 Characterization of the subunit structure of the catalytically active type I iodothyronine deiodinase. J Biol Chem 276:2600–2607 [DOI] [PubMed] [Google Scholar]
- Schoenmakers CH, Pigmans IG, Poland A, Visser TJ 1993 Impairment of the selenoenzyme type I iodothyronine deiodinase in C3H/He mice. Endocrinology 132:357–361 [DOI] [PubMed] [Google Scholar]
- Salvatore D, Low SC, Berry M, Maia AL, Harney JW, Croteau W, St. Germain DL, Larsen PR 1995 Type 3 iodothyronine deiodinase: cloning, in vitro expression, and functional analysis of the placental selenoenzyme. J Clin Invest 96:2421–2430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenmakers CH, Pigmans IG, Visser TJ 1995 Investigation of type I and type III iodothyronine deiodinases in rat tissues using N-bromoacetyl-iodothyronine affinity labels. Mol Cell Endocrinol 107:173–180 [DOI] [PubMed] [Google Scholar]
- Curcio-Morelli C, Gereben B, Zavacki AM, Kim BW, Huang S, Harney JW, Larsen PR, Bianco AC 2003 In vivo dimerization of types 1, 2, and 3 iodothyronine selenodeiodinases. Endocrinology 144:937–946 [DOI] [PubMed] [Google Scholar]
- Leonard JL, Simpson G, Leonard DM 2005 Characterization of the protein dimerization domain responsible for assembly of functional selenodeiodinases. J Biol Chem 280:11093–11100 [DOI] [PubMed] [Google Scholar]
- Simpson GI, Leonard DM, Leonard JL 2006 Identification of the key residues responsible for the assembly of selenodeiodinases. J Biol Chem 281:14615–14621 [DOI] [PubMed] [Google Scholar]
- Weichsel A, Gasdaska JR, Powis G, Montfort WR 1996 Crystal structures of reduced, oxidized, and mutated human thioredoxins: evidence for a regulatory homodimer. Structure 4:735–751 [DOI] [PubMed] [Google Scholar]
- Sagar GD, Gereben B, Callebaut I, Mornon JP, Zeold A, Curcio-Morelli C, Harney JW, Luongo C, Mulcahey MA, Larsen PR, Huang SA, Bianco AC 2008 The thyroid hormone-inactivating deiodinase functions as a homodimer. Mol Endocrinol 22:1382–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry MJ, Banu L, Larsen PR 1991 Type I iodothyronine deiodinase is a selenocysteine-containing enzyme. Nature 349:438–440 [DOI] [PubMed] [Google Scholar]
- Behne D, Kyriakopoulos A, Meinhold H, Kohrle J 1990 Identification of type I iodothyronine 5′-deiodinase as a selenoenzyme. Biochem Biophys Res Commun 173:1143–1149 [DOI] [PubMed] [Google Scholar]
- Berry MJ, Larsen PR 1992 The role of selenium in thyroid hormone action. Endocr Rev 13:207–219 [DOI] [PubMed] [Google Scholar]
- Berry MJ, Banu L, Chen YY, Mandel SJ, Kieffer JD, Harney JW, Larsen PR 1991 Recognition of UGA as a selenocysteine codon in type I deiodinase requires sequences in the 3′ untranslated region. Nature 353:273–276 [DOI] [PubMed] [Google Scholar]
- Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN 2003 Characterization of mammalian selenoproteomes. Science 300:1439–1443 [DOI] [PubMed] [Google Scholar]
- Hoffmann PR, Berry MJ 2005 Selenoprotein synthesis: a unique translational mechanism used by a diverse family of proteins. Thyroid 15:769–775 [DOI] [PubMed] [Google Scholar]
- Berry MJ, Maia AL, Kieffer JD, Harney JW, Larsen PR 1992 Substitution of cysteine for selenocysteine in type I iodothyronine deiodinase reduces the catalytic efficiency of the protein but enhances its translation. Endocrinology 131:1848–1852 [DOI] [PubMed] [Google Scholar]
- Dumitrescu AM, Liao XH, Abdullah MS, Lado-Abeal J, Majed FA, Moeller LC, Boran G, Schomburg L, Weiss RE, Refetoff S 2005 Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat Genet 37:1247–1252 [DOI] [PubMed] [Google Scholar]
- Forchhammer K, Bock A 1991 Selenocysteine synthase from Escherichia coli. Analysis of the reaction sequence. J Biol Chem 266:6324–6328 [PubMed] [Google Scholar]
- Guimaraes MJ, Peterson D, Vicari A, Cocks BG, Copeland NG, Gilbert DJ, Jenkins NA, Ferrick DA, Kastelein RA, Bazan JF, Zlotnik A 1996 Identification of a novel selD homolog from eukaryotes, bacteria, and archaea: is there an autoregulatory mechanism in selenocysteine metabolism? Proc Natl Acad Sci USA 93:15086–15091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low SC, Harney JW, Berry MJ 1995 Cloning and functional characterization of human selenophosphate synthetase, an essential component of selenoprotein synthesis. J Biol Chem 270:21659–21664 [DOI] [PubMed] [Google Scholar]
- Carlson BA, Xu XM, Kryukov GV, Rao M, Berry MJ, Gladyshev VN, Hatfield DL 2004 Identification and characterization of phosphoseryl-tRNA[Ser]Sec kinase. Proc Natl Acad Sci USA 101:12848–12853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XM, Carlson BA, Mix H, Zhang Y, Saira K, Glass RS, Berry MJ, Gladyshev VN, Hatfield DL 2007 Biosynthesis of selenocysteine on its tRNA in eukaryotes. PLoS Biol 5:e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Palioura S, Salazar JC, Su D, O'Donoghue P, Hohn MJ, Cardoso AM, Whitman WB, Soll D 2006 RNA-dependent conversion of phosphoserine forms selenocysteine in eukaryotes and archaea. Proc Natl Acad Sci USA 103:18923–18927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubenik JL, Driscoll DM 2007 Altered RNA binding activity underlies abnormal thyroid hormone metabolism linked to a mutation in selenocysteine insertion sequence-binding protein 2. J Biol Chem 282:34653–34662 [DOI] [PubMed] [Google Scholar]
- Gupta M, Copeland PR 2007 Functional analysis of the interplay between translation termination, selenocysteine codon context, and selenocysteine insertion sequence-binding protein 2. J Biol Chem 282:36797–36807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squires JE, Stoytchev I, Forry EP, Berry MJ 2007 SBP2 binding affinity is a major determinant in differential selenoprotein mRNA translation and sensitivity to nonsense-mediated decay. Mol Cell Biol 27:7848–7855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavatte L, Brown BA, Driscoll DM 2005 Ribosomal protein L30 is a component of the UGA-selenocysteine recoding machinery in eukaryotes. Nat Struct Mol Biol 12:408–416 [DOI] [PubMed] [Google Scholar]
- de Jesus LA, Hoffmann PR, Michaud T, Forry EP, Small-Howard A, Stillwell RJ, Morozova N, Harney JW, Berry MJ 2006 Nuclear assembly of UGA decoding complexes on selenoprotein mRNAs: a mechanism for eluding nonsense-mediated decay? Mol Cell Biol 26:1795–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp LV, Lu J, Striebel F, Kennedy D, Holmgren A, Khanna KK 2006 The redox state of SECIS binding protein 2 controls its localization and selenocysteine incorporation function. Mol Cell Biol 26:4895–4910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzy SA, Caban K, Copeland PR 2005 Characterization of the SECIS binding protein 2 complex required for the co-translational insertion of selenocysteine in mammals. Nucleic Acids Res 33:5172–5180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small-Howard A, Morozova N, Stoytcheva Z, Forry EP, Mansell JB, Harney JW, Carlson BA, Xu XM, Hatfield DL, Berry MJ 2006 Supramolecular complexes mediate selenocysteine incorporation in vivo. Mol Cell Biol 26:2337–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XM, Mix H, Carlson BA, Grabowski PJ, Gladyshev VN, Berry MJ, Hatfield DL 2005 Evidence for direct roles of two additional factors, SECp43 and soluble liver antigen, in the selenoprotein synthesis machinery. J Biol Chem 280:41568–41575 [DOI] [PubMed] [Google Scholar]
- Burmeister LA, Pachucki J, St Germain DL 1997 Thyroid hormones inhibit type 2 iodothyronine deiodinase in the rat cerebral cortex by both pre- and posttranslational mechanisms. Endocrinology 138:5231–5237 [DOI] [PubMed] [Google Scholar]
- Kim SW, Harney JW, Larsen PR 1998 Studies of the hormonal regulation of type 2 5′-iodothyronine deiodinase messenger ribonucleic acid in pituitary tumor cells using semiquantitative reverse transcription-polymerase chain reaction. Endocrinology 139:4895–4905 [DOI] [PubMed] [Google Scholar]
- Gereben B, Kollar A, Harney JW, Larsen PR 2002 The mRNA structure has potent regulatory effects on type 2 iodothyronine deiodinase expression. Mol Endocrinol 16:1667–1679 [DOI] [PubMed] [Google Scholar]
- Gereben B, Bartha T, Tu HM, Harney JW, Rudas P, Larsen PR 1999 Cloning and expression of the chicken type 2 iodothyronine 5′- deiodinase. J Biol Chem 274:13768–13776 [DOI] [PubMed] [Google Scholar]
- Salvatore D, Bartha T, Harney JW, Larsen PR 1996 Molecular biological and biochemical characterization of the human type 2 selenodeiodinase. Endocrinology 137:3308–3315 [DOI] [PubMed] [Google Scholar]
- Salvatore D, Tu H, Harney JW, Larsen PR 1996 Type 2 iodothyronine deiodinase is highly expressed in human thyroid. J Clin Invest 98:962–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Germain DL 1988 The effects and interactions of substrates, inhibitors, and the cellular thiol-disulfide balance on the regulation of type II iodothyronine 5′-deiodinase. Endocrinology 122:1860–1868 [DOI] [PubMed] [Google Scholar]
- Leonard JL, Kaplan MM, Visser TJ, Silva JE, Larsen PR 1981 Cerebral cortex responds rapidly to thyroid hormones. Science 214:571–573 [DOI] [PubMed] [Google Scholar]
- Koenig RJ, Leonard JL, Senator D, Rappaport N, Watson AY, Larsen PR 1984 Regulation of thyroxine 5′-deiodinase activity by 3,5,3′- triiodothyronine in cultured rat anterior pituitary cells. Endocrinology 115:324–329 [DOI] [PubMed] [Google Scholar]
- Silva JE, Leonard JL 1985 Regulation of rat cerebrocortical and adenohypophyseal type II 5′- deiodinase by thyroxine, triiodothyronine, and reverse triiodothyronine. Endocrinology 116:1627–1635 [DOI] [PubMed] [Google Scholar]
- Obregon MJ, Larsen PR, Silva JE 1986 The role of 3,3′,5′-triiodothyronine in the regulation of type II iodothyronine 5′-deiodinase in the rat cerebral cortex. Endocrinology 119:2186–2192 [DOI] [PubMed] [Google Scholar]
- Halperin Y, Shapiro LE, Surks MI 1994 Down-regulation of type II L-thyroxine, 5′-monodeiodinase in cultured GC cells: different pathways of regulation by L-triiodothyronine and 3,3′,5′-triiodo-L-thyronine. Endocrinology 135:1464–1469 [DOI] [PubMed] [Google Scholar]
- Leonard JL, Silva JE, Kaplan MM, Mellen SA, Visser TJ, Larsen PR 1984 Acute posttranscriptional regulation of cerebrocortical and pituitary iodothyronine 5′-deiodinases by thyroid hormone. Endocrinology 114:998–1004 [DOI] [PubMed] [Google Scholar]
- Steinsapir J, Harney J, Larsen PR 1998 Type 2 iodothyronine deiodinase in rat pituitary tumor cells is inactivated in proteasomes. J Clin Invest 102:1895–1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinsapir J, Bianco AC, Buettner C, Harney J, Larsen PR 2000 Substrate-induced down-regulation of human type 2 deiodinase (hD2) is mediated through proteasomal degradation and requires interaction with the enzyme’s active center. Endocrinology 141:1127–1135 [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A 1998 The ubiquitin system. Annu Rev Biochem 67:425–479 [DOI] [PubMed] [Google Scholar]
- Coux O, Tanaka K, Goldberg AL 1996 Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem 65:801–847 [DOI] [PubMed] [Google Scholar]
- Pickart CM 2001 Mechanisms underlying ubiquitination. Annu Rev Biochem 70:503–533 [DOI] [PubMed] [Google Scholar]
- Pines J, Lindon C 2005 Proteolysis: anytime, any place, anywhere? Nat Cell Biol 7:731–735 [DOI] [PubMed] [Google Scholar]
- Gereben B, Goncalves C, Harney JW, Larsen PR, Bianco AC 2000 Selective proteolysis of human type 2 deiodinase: a novel ubiquitin- proteasomal mediated mechanism for regulation of hormone activation. Mol Endocrinol 14:1697–1708 [DOI] [PubMed] [Google Scholar]
- Botero D, Gereben B, Goncalves C, De Jesus LA, Harney JW, Bianco AC 2002 Ubc6p and ubc7p are required for normal and substrate-induced endoplasmic reticulum-associated degradation of the human selenoprotein type 2 iodothyronine monodeiodinase. Mol Endocrinol 16:1999–2007 [DOI] [PubMed] [Google Scholar]
- Chen P, Johnson P, Sommer T, Jentsch S, Hochstrasser M 1993 Multiple ubiquitin-conjugating enzymes participate in the in vivo degradation of the yeast MAT α 2 repressor. Cell 74:357–369 [DOI] [PubMed] [Google Scholar]
- Biederer T, Volkwein C, Sommer T 1997 Role of Cue1p in ubiquitination and degradation at the ER surface. Science 278:1806–1809 [DOI] [PubMed] [Google Scholar]
- Meusser B, Hirsch C, Jarosch E, Sommer T 2005 ERAD: the long road to destruction. Nat Cell Biol 7:766–772 [DOI] [PubMed] [Google Scholar]
- Tiwari S, Weissman AM 2001 Endoplasmic reticulum (ER)-associated degradation of T cell receptor subunits. Involvement of ER-associated ubiquitin-conjugating enzymes (E2s). J Biol Chem 276:16193–16200 [DOI] [PubMed] [Google Scholar]
- Kim BW, Zavacki AM, Curcio-Morelli C, Dentice M, Harney JW, Larsen PR, Bianco AC 2003 ER-associated degradation of the human type 2 iodothyronine deiodinase (D2) is mediated via an association between mammalian UBC7 and the carboxyl region of D2. Mol Endocrinol 17:2603–2612 [DOI] [PubMed] [Google Scholar]
- Vasiliauskas D, Hancock S, Stern CD 1999 SWiP-1: novel SOCS box containing WD-protein regulated by signalling centres and by Shh during development. Mech Dev 82:79–94 [DOI] [PubMed] [Google Scholar]
- Hilton DJ, Richardson RT, Alexander WS, Viney EM, Willson TA, Sprigg NS, Starr R, Nicholson SE, Metcalf D, Nicola NA 1998 Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci USA 95:114–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neer EJ, Schmidt CJ, Nambudripad R, Smith TF 1994 The ancient regulatory-protein family of WD-repeat proteins. Nature 371:297–300 [DOI] [PubMed] [Google Scholar]
- Kamura T, Sato S, Haque D, Liu L, Kaelin Jr WG, Conaway RC, Conaway JW 1998 The Elongin BC complex interacts with the conserved SOCS-box motif present in members of the SOCS, ras, WD-40 repeat, and ankyrin repeat families. Genes Dev 12:3872–3881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamura T, Burian D, Yan Q, Schmidt SL, Lane WS, Querido E, Branton PE, Shilatifard A, Conaway RC, Conaway JW 2001 Muf1, a novel Elongin BC-interacting leucine-rich repeat protein that can assemble with Cul5 and Rbx1 to reconstitute a ubiquitin ligase. J Biol Chem 276:29748–29753 [DOI] [PubMed] [Google Scholar]
- Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, Conaway RC, Conaway JW, Harper JW, Pavletich NP 2002 Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 416:703–709 [DOI] [PubMed] [Google Scholar]
- Choi DW, Seo YM, Kim EA, Sung KS, Ahn JW, Park SJ, Lee SR, Choi CY 2007 Ubiquitination and degradation of homeodomain-interacting protein kinase 2 (HIPK2) by WD40-repeat/SOCS box protein WSB-1. J Biol Chem 283:4682–4689 [DOI] [PubMed] [Google Scholar]
- Chen QR, Bilke S, Wei JS, Greer BT, Steinberg SM, Westermann F, Schwab M, Khan J 2006 Increased WSB1 copy number correlates with its over-expression which associates with increased survival in neuroblastoma. Genes Chromosomes Cancer 45:856–862 [DOI] [PubMed] [Google Scholar]
- Krueger C, Osborne CS 2006 Raising the curtains on interchromosomal interactions. Trends Genet 22:637–639 [DOI] [PubMed] [Google Scholar]
- Ling JQ, Li T, Hu JF, Vu TH, Chen HL, Qiu XW, Cherry AM, Hoffman AR 2006 CTCF mediates interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1. Science 312:269–272 [DOI] [PubMed] [Google Scholar]
- Spilianakis CG, Flavell RA 2006 Molecular biology. Managing associations between different chromosomes. Science 312:207–208 [DOI] [PubMed] [Google Scholar]
- Hernandez A, Fiering S, Martinez E, Galton VA, St Germain D 2002 The gene locus encoding iodothyronine deiodinase type 3 (Dio3) is imprinted in the fetus and expresses antisense transcripts. Endocrinology 143:4483–4486 [DOI] [PubMed] [Google Scholar]
- Tsai CE, Lin SP, Ito M, Takagi N, Takada S, Ferguson-Smith AC 2002 Genomic imprinting contributes to thyroid hormone metabolism in the mouse embryo. Curr Biol 12:1221–1226 [DOI] [PubMed] [Google Scholar]
- Yevtodiyenko A, Carr MS, Patel N, Schmidt JV 2002 Analysis of candidate imprinted genes linked to Dlk1-Gtl2 using a congenic mouse line. Mamm Genome 13:633–638 [DOI] [PubMed] [Google Scholar]
- da Rocha ST, Ferguson-Smith AC 2004 Genomic imprinting. Curr Biol 14:R646–R649 [DOI] [PubMed] [Google Scholar]
- Ravid T, Kreft SG, Hochstrasser M 2006 Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J 25:533–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreft SG, Wang L, Hochstrasser M 2006 Membrane topology of the yeast endoplasmic reticulum-localized ubiquitin ligase Doa10 and comparison with its human ortholog TEB4 (MARCH-VI). J Biol Chem 281:4646–4653 [DOI] [PubMed] [Google Scholar]
- Hassink G, Kikkert M, van Voorden S, Lee SJ, Spaapen R, van Laar T, Coleman CS, Bartee E, Fruh K, Chau V, Wiertz E 2005 TEB4 is a C4HC3 RING finger-containing ubiquitin ligase of the endoplasmic reticulum. Biochem J 388:647–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavacki AM, Harney JW, Li J, Gereben B, Fekete C, Bianco AC, TEB4 is an endoplasmic reticulum ubiquitin ligase that mediates type 2 iodothyronine deiodinase (D2) degradation. Program of the 89th Annual Meeting of the Endocrine Society, Toronto, Canada, 2007, OR 7-1 [Google Scholar]
- Kanou Y, Hishinuma A, Tsunekawa K, Seki K, Mizuno Y, Fujisawa H, Imai T, Miura Y, Nagasaka T, Yamada C, Ieiri T, Murakami M, Murata Y 2007 Thyroglobulin gene mutations producing defective intracellular transport of thyroglobulin are associated with increased thyroidal type 2 iodothyronine deiodinase activity. J Clin Endocrinol Metab 92:1451–1457 [DOI] [PubMed] [Google Scholar]
- Huang Y, Baker RT, Fischer-Vize JA 1995 Control of cell fate by a deubiquitinating enzyme encoded by the fat facets gene. Science 270:1828–1831 [DOI] [PubMed] [Google Scholar]
- Moazed D, Johnson D 1996 A deubiquitinating enzyme interacts with SIR4 and regulates silencing in S. cerevisiae. Cell 86:667–677 [DOI] [PubMed] [Google Scholar]
- Naviglio S, Mattecucci C, Matoskova B, Nagase T, Nomura N, Di Fiore PP, Draetta GF 1998 UBPY: a growth-regulated human ubiquitin isopeptidase. EMBO J 17:3241–3250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KC, Kim JH, Choi EJ, Min SW, Rhee S, Baek SH, Chung SS, Bang O, Park D, Chiba T, Tanaka K, Chung CH 2002 Antagonistic regulation of myogenesis by two deubiquitinating enzymes, UBP45 and UBP69. Proc Natl Acad Sci USA 99:9733–9738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Carroll M, Papa FR, Hochstrasser M, D'Andrea AD 1996 DUB-1, a deubiquitinating enzyme with growth-suppressing activity. Proc Natl Acad Sci USA 93:3275–3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcio-Morelli C, Zavacki AM, Christofollete M, Gereben B, de Freitas BC, Harney JW, Li Z, Wu G, Bianco AC 2003 Deubiquitination of type 2 iodothyronine deiodinase by von Hippel-Lindau protein-interacting deubiquitinating enzymes regulates thyroid hormone activation. J Clin Invest 112:189–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco AC, Silva JE 1987 Intracellular conversion of thyroxine to triiodothyronine is required for the optimal thermogenic function of brown adipose tissue. J Clin Invest 79:295–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jesus LA, Carvalho SD, Ribeiro MO, Schneider M, Kim SW, Harney JW, Larsen PR, Bianco AC 2001 The type 2 iodothyronine deiodinase is essential for adaptive thermogenesis in brown adipose tissue. J Clin Invest 108:1379–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Wang D, Messing EM, Wu G 2005 VHL protein-interacting deubiquitinating enzyme 2 deubiquitinates and stabilizes HIF-1α. EMBO Rep 6:373–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zhang B, Fischer JA 2002 A specific protein substrate for a deubiquitinating enzyme: liquid facets is the substrate of fat facets. Genes Dev 16:289–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnesutta N, Ceriani M, Innocenti M, Mauri I, Zippel R, Sturani E, Borgonovo B, Berruti G, Martegani E 2001 Cloning and characterization of mouse UBPy, a deubiquitinating enzyme that interacts with the ras guanine nucleotide exchange factor CDC25(Mm)/Ras-GRF1. J Biol Chem 276:39448–39454 [DOI] [PubMed] [Google Scholar]
- Ideguchi H, Ueda A, Tanaka M, Yang J, Tsuji T, Ohno S, Hagiwara E, Aoki A, Ishigatsubo Y 2002 Structural and functional characterization of the USP11 deubiquitinating enzyme, which interacts with the RanGTP-associated protein RanBPM. Biochem J 367:87–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taya S, Yamamoto T, Kanai-Azuma M, Wood SA, Kaibuchi K 1999 The deubiquitinating enzyme Fam interacts with and stabilizes β-catenin. Genes Cells 4:757–767 [DOI] [PubMed] [Google Scholar]
- Taya S, Yamamoto T, Kano K, Kawano Y, Iwamatsu A, Tsuchiya T, Tanaka K, Kanai-Azuma M, Wood SA, Mattick JS, Kaibuchi K 1998 The Ras target AF-6 is a substrate of the fam deubiquitinating enzyme. J Cell Biol 142:1053–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Wang D, Na X, Schoen SR, Messing EM, Wu G 2002 Identification of a deubiquitinating enzyme subfamily as substrates of the von Hippel-Lindau tumor suppressor. Biochem Biophys Res Commun 294:700–709 [DOI] [PubMed] [Google Scholar]
- Li Z, Na X, Wang D, Schoen SR, Messing EM, Wu G 2002 Ubiquitination of a novel deubiquitinating enzyme requires direct binding to von Hippel-Lindau tumor suppressor protein. J Biol Chem 277:4656–4662 [DOI] [PubMed] [Google Scholar]
- Pause A, Lee S, Worrell RA, Chen DY, Burgess WH, Linehan WM, Klausner RD 1997 The von Hippel-Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, a member of the Cdc53 family of proteins. Proc Natl Acad Sci USA 94:2156–2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamura T, Koepp DM, Conrad MN, Skowyra D, Moreland RJ, Iliopoulos O, Lane WS, Kaelin Jr WG, Elledge SJ, Conaway RC, Harper JW, Conaway JW 1999 Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science 284:657–661 [DOI] [PubMed] [Google Scholar]
- Maher ER, Kaelin Jr WG 1997 von Hippel-Lindau disease. Medicine (Baltimore) 76:381–391 [DOI] [PubMed] [Google Scholar]
- De Pitta C, Tombolan L, Campo Dell'Orto M, Accordi B, te Kronnie G, Romualdi C, Vitulo N, Basso G, Lanfranchi G 2005 A leukemia-enriched cDNA microarray platform identifies new transcripts with relevance to the biology of pediatric acute lymphoblastic leukemia. Haematologica 90:890–898 [PubMed] [Google Scholar]
- Hennemann G, Docter R, Friesema EC, de Jong M, Krenning EP, Visser TJ 2001 Plasma membrane transport of thyroid hormones and its role in thyroid hormone metabolism and bioavailability. Endocr Rev 22:451–476 [DOI] [PubMed] [Google Scholar]
- Friesema EC, Ganguly S, Abdalla A, Manning Fox JE, Halestrap AP, Visser TJ 2003 Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem 278:40128–40135 [DOI] [PubMed] [Google Scholar]
- Friesema EC, Jansen J, Jachtenberg JW, Visser WE, Kester MH, Visser TJ 2008 Effective cellular uptake and efflux of thyroid hormone by human monocarboxylate transporter 10. Mol Endocrinol 22:1357–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S 2004 A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet 74:168–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, Reeser M, Barrett TG, Mancilla EE, Svensson J, Kester MH, Kuiper GG, Balkassmi S, Uitterlinden AG, Koehrle J, Rodien P, Halestrap AP, Visser TJ 2004 Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 364:1435–1437 [DOI] [PubMed] [Google Scholar]
- Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, Bialer MG, Ward J, Sanabria J, Marsa S, Lewis JA, Echeverri R, Lubs HA, Voeller K, Simensen RJ, Stevenson RE 2005 Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet 77:41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrescu AM, Liao XH, Weiss RE, Millen K, Refetoff S 2006 Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology 147:4036–4043 [DOI] [PubMed] [Google Scholar]
- Trajkovic M, Visser TJ, Mittag J, Horn S, Lukas J, Darras VM, Raivich G, Bauer K, Heuer H 2007 Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest 117:627–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen J, Friesema EC, Milici C, Visser TJ 2005 Thyroid hormone transporters in health and disease. Thyroid 15:757–768 [DOI] [PubMed] [Google Scholar]
- Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, Schoonjans K, Bianco AC, Auwerx J 2006 Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439:484–489 [DOI] [PubMed] [Google Scholar]
- da-Silva WS, Harney JW, Kim BW, Li J, Bianco SD, Crescenzi A, Christoffolete MA, Huang SA, Bianco AC 2007 The small polyphenolic molecule kaempferol increases cellular energy expenditure and thyroid hormone activation. Diabetes 56:767–776 [DOI] [PubMed] [Google Scholar]
- Galton VA, Wood ET, St Germain EA, Withrow CA, Aldrich G, St Germain GM, Clark AS, St Germain DL 2007 Thyroid hormone homeostasis and action in the type 2 deiodinase-deficient rodent brain during development. Endocrinology 148:3080–3088 [DOI] [PubMed] [Google Scholar]
- Bianco AC, Silva JE 1988 Cold exposure rapidly induces virtual saturation of brown adipose tissue nuclear T3 receptors. Am J Physiol 255:E496–E503 [DOI] [PubMed] [Google Scholar]
- Wassen FW, Schiel AE, Kuiper GG, Kaptein E, Bakker O, Visser TJ, Simonides WS 2002 Induction of thyroid hormone-degrading deiodinase in cardiac hypertrophy and failure. Endocrinology 143:2812–2815 [DOI] [PubMed] [Google Scholar]
- Bianco AC, Silva JE 1987 Optimal response of key enzymes and uncoupling protein to cold in BAT depends on local T3 generation. Am J Physiol 253:E255–E263 [DOI] [PubMed] [Google Scholar]
- Fekete C, Gereben B, Doleschall M, Harney JW, Dora JM, Bianco AC, Sarkar S, Liposits Z, Rand W, Emerson C, Kacskovics I, Larsen PR, Lechan RM 2004 Lipopolysaccharide induces type 2 iodothyronine deiodinase in the mediobasal hypothalamus: implications for the nonthyroidal illness syndrome. Endocrinology 145:1649–1655 [DOI] [PubMed] [Google Scholar]
- Iwase K, Hummel BC, Walfish PG 1989 Rat hepatic and renal 5′-deiodination of rT3 during fasting: supportive role of intermediate Mr cytosolic non-glutathione thiol cofactor and NADPH. Metabolism 38:230–237 [DOI] [PubMed] [Google Scholar]
- Croteau W, Bodwell JE, Richardson JM, St Germain DL 1998 Conserved cysteines in the type 1 deiodinase selenoprotein are not essential for catalytic activity. J Biol Chem 273:25230–25236 [DOI] [PubMed] [Google Scholar]
- Maia AL, Kim BW, Huang SA, Harney JW, Larsen PR 2005 Type 2 iodothyronine deiodinase is the major source of plasma T3 in euthyroid humans. J Clin Invest 115:2524–2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsam A, Ingbar SH 1979 Observations on the factors that control the generation of triiodothyronine from thyroxine in rat liver and the nature of the effect induced by fasting. J Clin Invest 63:1145–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris ARC, Fang SL, Hinerfeld L, Braverman LE, Vagenakis AG 1979 The role of sulfhydryl groups on the impaired hepatic 3′,3,5-triiodothyronine generation from thyroxine in the hypothyroid, starved, fetal and neonatal rodent. J Clin Invest 63:516–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra IJ 1980 Alterations in monodeiodination of iodothyronines in the fasting rat: effects of reduced nonprotein sulfhydryl groups and hypothyroidism. Metabolism 29:161–167 [DOI] [PubMed] [Google Scholar]
- Pachucki J, Hopkins J, Peeters R, Tu H, Carvalho SD, Kaulbach H, Abel ED, Wondisford FE, Ingwall JS, Larsen PR 2001 Type 2 iodothyronine deiodinase transgene expression in the mouse heart causes cardiac-specific thyrotoxicosis. Endocrinology 142:13–20 [DOI] [PubMed] [Google Scholar]
- Trivieri MG, Oudit GY, Sah R, Kerfant BG, Sun H, Gramolini AO, Pan Y, Wickenden AD, Croteau W, Morreale de Escobar G, Pekhletski R, St Germain D, Maclennan DH, Backx PH 2006 Cardiac-specific elevations in thyroid hormone enhance contractility and prevent pressure overload-induced cardiac dysfunction. Proc Natl Acad Sci USA 103:6043–6048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallud S, Kelly R, DiStefano J, Parlow A, Galton VA, St. Germain D, Metabolic effects of targeted expression of type 2 iodothyronine deiodinase (D2) to rodent liver. Proc 74th Annual Meeting of the American Thyroid Association, Los Angeles, CA, 2002, Abstract P202 [Google Scholar]
- Maglich JM, Watson J, McMillen PJ, Goodwin B, Willson TM, Moore JT 2004 The nuclear receptor CAR is a regulator of thyroid hormone metabolism during caloric restriction. J Biol Chem 279:19832–19838 [DOI] [PubMed] [Google Scholar]
- Qatanani M, Zhang J, Moore DD 2005 Role of the constitutive androstane receptor in xenobiotic-induced thyroid hormone metabolism. Endocrinology 146:995–1002 [DOI] [PubMed] [Google Scholar]
- Visser TJ, Kaptein E, Glatt H, Bartsch I, Hagen M, Coughtrie MW 1998 Characterization of thyroid hormone sulfotransferases. Chem Biol Interact 109:279–291 [DOI] [PubMed] [Google Scholar]
- Wang J, Falany JL, Falany CN 1998 Expression and characterization of a novel thyroid hormone-sulfating form of cytosolic sulfotransferase from human liver. Mol Pharmacol 53:274–282 [DOI] [PubMed] [Google Scholar]
- Kester MH, Kaptein E, Roest TJ, van Dijk CH, Tibboel D, Meinl W, Glatt H, Coughtrie MW, Visser TJ 1999 Characterization of human iodothyronine sulfotransferases. J Clin Endocrinol Metab 84:1357–1364 [DOI] [PubMed] [Google Scholar]
- Kester MH, van Dijk CH, Tibboel D, Hood AM, Rose NJ, Meinl W, Pabel U, Glatt H, Falany CN, Coughtrie MW, Visser TJ 1999 Sulfation of thyroid hormone by estrogen sulfotransferase. J Clin Endocrinol Metab 84:2577–2580 [DOI] [PubMed] [Google Scholar]
- Visser TJ 1994 Role of sulfation in thyroid hormone metabolism. Chem Biol Interact 92:293–303 [DOI] [PubMed] [Google Scholar]
- Wu SY, Green WL, Huang WS, Hays MT, Chopra IJ 2005 Alternate pathways of thyroid hormone metabolism. Thyroid 15:943–958 [DOI] [PubMed] [Google Scholar]
- Schneider MJ, Fiering SN, Thai B, Wu SY, St Germain E, Parlow AF, St Germain DL, Galton VA 2006 Targeted disruption of the type 1 selenodeiodinase gene (Dio1) results in marked changes in thyroid hormone economy in mice. Endocrinology 147:580–589 [DOI] [PubMed] [Google Scholar]
- Santini F, Chopra IJ, Wu SY, Solomon DH, Chua Teco GN 1992 Metabolism of 3,5,3′-triiodothyronine sulfate by tissues of the fetal rat: a consideration of the role of desulfation of 3,5,3′-triiodothyronine sulfate as a source of T3. Pediatr Res 31:541–544 [DOI] [PubMed] [Google Scholar]
- Hazenberg MP, de Herder WW, Visser TJ 1988 Hydrolysis of iodothyronine conjugates by intestinal bacteria. FEMS Microbiol Rev 4:9–16 [DOI] [PubMed] [Google Scholar]
- Scanlan TS, Suchland KL, Hart ME, Chiellini G, Huang Y, Kruzich PJ, Frascarelli S, Crossley DA, Bunzow JR, Ronca-Testoni S, Lin ET, Hatton D, Zucchi R, Grandy DK 2004 3-Iodothyronamine is an endogenous and rapid-acting derivative of thyroid hormone. Nat Med 10:638–642 [DOI] [PubMed] [Google Scholar]
- Braulke LJ, Klingenspor M, Debarber A, Tobias SC, Grandy DK, Scanlan TS, Heldmaier G 2008 3-Iodothyronamine: a novel hormone controlling the balance between glucose and lipid utilisation. J Comp Physiol [B] 178:167–177 [DOI] [PubMed] [Google Scholar]
- Pietsch CA, Scanlan TS, Anderson RJ 2007 Thyronamines are substrates for human liver sulfotransferases. Endocrinology 148:1921–1927 [DOI] [PubMed] [Google Scholar]
- Chiellini G, Frascarelli S, Ghelardoni S, Carnicelli V, Tobias SC, DeBarber A, Brogioni S, Ronca-Testoni S, Cerbai E, Grandy DK, Scanlan TS, Zucchi R 2007 Cardiac effects of 3-iodothyronamine: a new aminergic system modulating cardiac function. FASEB J 21:1597–1608 [DOI] [PubMed] [Google Scholar]
- Snead AN, Santos MS, Seal RP, Miyakawa M, Edwards RH, Scanlan TS 2007 Thyronamines inhibit plasma membrane and vesicular monoamine transport. ACS Chem Biol 2:390–398 [DOI] [PubMed] [Google Scholar]
- Piehl S, Heberer T, Balizs G, Scanlan TS, Smits R, Koksch B, Kohrle J 2008 Thyronamines are isozyme-specific substrates of deiodinases. Endocrinology 149:3037–3045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheimer JH, Schwartz HL 1997 Molecular basis of thyroid hormone-dependent brain development. Endocr Rev 18:462–475 [DOI] [PubMed] [Google Scholar]
- Furumoto H, Ying H, Chandramouli GV, Zhao L, Walker RL, Meltzer PS, Willingham MC, Cheng SY 2005 An unliganded thyroid hormone β receptor activates the cyclin D1/cyclin-dependent kinase/retinoblastoma/E2F pathway and induces pituitary tumorigenesis. Mol Cell Biol 25:124–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porlan E, Vega S, Iglesias T, Rodriguez-Pena A 2004 Unliganded thyroid hormone receptor β1 inhibits proliferation of murine fibroblasts by delaying the onset of the G1 cell-cycle signals. Oncogene 23:8756–8765 [DOI] [PubMed] [Google Scholar]
- Bernal J, Guadano-Ferraz A, Morte B 2003 Perspectives in the study of thyroid hormone action on brain development and function. Thyroid 13:1005–1012 [DOI] [PubMed] [Google Scholar]
- Fernandez M, Pirondi S, Manservigi M, Giardino L, Calza L 2004 Thyroid hormone participates in the regulation of neural stem cells and oligodendrocyte precursor cells in the central nervous system of adult rat. Eur J Neurosci 20:2059–2070 [DOI] [PubMed] [Google Scholar]
- Marti E, Bovolenta P 2002 Sonic hedgehog in CNS development: one signal, multiple outputs. Trends Neurosci 25:89–96 [DOI] [PubMed] [Google Scholar]
- Athar M, Tang X, Lee JL, Kopelovich L, Kim AL 2006 Hedgehog signalling in skin development and cancer. Exp Dermatol 15:667–677 [DOI] [PubMed] [Google Scholar]
- Fietz MJ, Concordet JP, Barbosa R, Johnson R, Krauss S, McMahon AP, Tabin C, Ingham PW 1994 The hedgehog gene family in Drosophila and vertebrate development. Dev Suppl 1994:43–51 [PubMed] [Google Scholar]
- Nusslein-Volhard C, Wieschaus E 1980 Mutations affecting segment number and polarity in Drosophila. Nature 287:795–801 [DOI] [PubMed] [Google Scholar]
- Ericson J, Rashbass P, Schedl A, Brenner-Morton S, Kawakami A, van Heyningen V, Jessell TM, Briscoe J 1997 Pax6 controls progenitor cell identity and neuronal fate in response to graded Shh signaling. Cell 90:169–180 [DOI] [PubMed] [Google Scholar]
- Mariani FV, Martin GR 2003 Deciphering skeletal patterning: clues from the limb. Nature 423:319–325 [DOI] [PubMed] [Google Scholar]
- Hammerschmidt M, Brook A, McMahon AP 1997 The world according to hedgehog. Trends Genet 13:14–21 [DOI] [PubMed] [Google Scholar]
- Kronenberg HM 2003 Developmental regulation of the growth plate. Nature 423:332–336 [DOI] [PubMed] [Google Scholar]
- Riobo NA, Manning DR 2007 Pathways of signal transduction employed by vertebrate Hedgehogs. Biochem J 403:369–379 [DOI] [PubMed] [Google Scholar]
- Lai K, Kaspar BK, Gage FH, Schaffer DV 2003 Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat Neurosci 6:21–27 [DOI] [PubMed] [Google Scholar]
- Palma V, Lim DA, Dahmane N, Sanchez P, Brionne TC, Herzberg CD, Gitton Y, Carleton A, Alvarez-Buylla A, Ruiz i Altaba A 2005 Sonic hedgehog controls stem cell behavior in the postnatal and adult brain. Development 132:335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wang Y, Wang Z, Liu H, Shen Y, Li W, Heller S, Li H 2006 Sonic hedgehog promotes mouse inner ear progenitor cell proliferation and hair cell generation in vitro. Neuroreport 17:121–124 [DOI] [PubMed] [Google Scholar]
- Chiang C, Swan RZ, Grachtchouk M, Bolinger M, Litingtung Y, Robertson EK, Cooper MK, Gaffield W, Westphal H, Beachy PA, Dlugosz AA 1999 Essential role for Sonic hedgehog during hair follicle morphogenesis. Dev Biol 205:1–9 [DOI] [PubMed] [Google Scholar]
- Fu JR, Liu WL, Zhou JF, Sun HY, Xu HZ, Luo L, Zhang H, Zhou YF 2006 Sonic hedgehog protein promotes bone marrow-derived endothelial progenitor cell proliferation, migration and VEGF production via PI 3-kinase/Akt signaling pathways. Acta Pharmacol Sin 27:685–693 [DOI] [PubMed] [Google Scholar]
- Gering M, Patient R 2005 Hedgehog signaling is required for adult blood stem cell formation in zebrafish embryos. Dev Cell 8:389–400 [DOI] [PubMed] [Google Scholar]
- Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ 1996 Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 273:613–622 [DOI] [PubMed] [Google Scholar]
- Robson H, Siebler T, Stevens DA, Shalet SM, Williams GR 2000 Thyroid hormone acts directly on growth plate chondrocytes to promote hypertrophic differentiation and inhibit clonal expansion and cell proliferation. Endocrinology 141:3887–3897 [DOI] [PubMed] [Google Scholar]
- Archange C, Nowak J, Garcia S, Moutardier V, Calvo EL, Dagorn JC, Iovanna JL 2008 The WSB1 gene is involved in pancreatic cancer progression. PLoS ONE 3:e2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khavari PA 2006 Modelling cancer in human skin tissue. Nat Rev Cancer 6:270–280 [DOI] [PubMed] [Google Scholar]
- St-Jacques B, Dassule HR, Karavanova I, Botchkarev VA, Li J, Danielian PS, McMahon JA, Lewis PM, Paus R, McMahon AP 1998 Sonic hedgehog signaling is essential for hair development. Curr Biol 8:1058–1068 [DOI] [PubMed] [Google Scholar]
- Huang TS, Chopra IJ, Beredo A, Solomon DH, Chua Teco GN 1985 Skin is an active site for the inner ring monodeiodination of thyroxine to 3,3′,5′-triiodothyronine. Endocrinology 117:2106–2113 [DOI] [PubMed] [Google Scholar]
- Kaplan MM, Pan CY, Gordon PR, Lee JK, Gilchrest BA 1988 Human epidermal keratinocytes in culture convert thyroxine to 3,5,3′-triiodothyronine by type II iodothyronine deiodination: a novel endocrine function of the skin. J Clin Endocrinol Metab 66:815–822 [DOI] [PubMed] [Google Scholar]
- Slominski A, Wortsman J, Kohn L, Ain KB, Venkataraman GM, Pisarchik A, Chung JH, Giuliani C, Thornton M, Slugocki G, Tobin DJ 2002 Expression of hypothalamic-pituitary-thyroid axis related genes in the human skin. J Invest Dermatol 119:1449–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SA, Mulcahey MA, Crescenzi A, Chung M, Kim BW, Barnes C, Kuijt W, Turano H, Harney J, Larsen PR 2005 Transforming growth factor-β promotes inactivation of extracellular thyroid hormones via transcriptional stimulation of type 3 iodothyronine deiodinase. Mol Endocrinol 19:3126–3136 [DOI] [PubMed] [Google Scholar]
- Song S, Oka T 2003 Regulation of type II deiodinase expression by EGF and glucocorticoid in HC11 mouse mammary epithelium. Am J Physiol Endocrinol Metab 284:E1119–E1124 [DOI] [PubMed] [Google Scholar]
- Maeda A, Toyoda N, Yasuzawa-Amano S, Iwasaka T, Nishikawa M 2003 Type 2 deiodinase expression is stimulated by growth factors in human vascular smooth muscle cells. Mol Cell Endocrinol 200:111–117 [DOI] [PubMed] [Google Scholar]
- Martinez-de Mena R, Obregon MJ 2005 Insulin increases the adrenergic stimulation of 5′ deiodinase activity and mRNA expression in rat brown adipocytes; role of MAPK and PI3K. J Mol Endocrinol 34:139–151 [DOI] [PubMed] [Google Scholar]
- Brown DD, Cai L 2007 Amphibian metamorphosis. Dev Biol 306:20–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker KB, Stephens KC, Davey JC, Schneider MJ, Galton VA 1997 The type 2 and type 3 iodothyronine deiodinases play important roles in coordinating development in Rana catesbeiana tadpoles. Endocrinology 138:2989–2997 [DOI] [PubMed] [Google Scholar]
- Huang H, Cai L, Remo BF, Brown DD 2001 Timing of metamorphosis and the onset of the negative feedback loop between the thyroid gland and the pituitary is controlled by type II iodothyronine deiodinase in Xenopus laevis. Proc Natl Acad Sci USA 98:7348–7353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Brown DD 2004 Expression of type II iodothyronine deiodinase marks the time that a tissue responds to thyroid hormone-induced metamorphosis in Xenopus laevis. Dev Biol 266:87–95 [DOI] [PubMed] [Google Scholar]
- Morvan Dubois G, Sebillot A, Kuiper GG, Verhoelst CH, Darras VM, Visser TJ, Demeneix BA 2006 Deiodinase activity is present in Xenopus laevis during early embryogenesis. Endocrinology 147:4941–4949 [DOI] [PubMed] [Google Scholar]
- Campos-Barros A, Amma LL, Faris JS, Shailam R, Kelley MW, Forrest D 2000 Type 2 iodothyronine deiodinase expression in the cochlea before the onset of hearing. Proc Natl Acad Sci USA 97:1287–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng L, Goodyear RJ, Woods CA, Schneider MJ, Diamond E, Richardson GP, Kelley MW, Germain DL, Galton VA, Forrest D 2004 Hearing loss and retarded cochlear development in mice lacking type 2 iodothyronine deiodinase. Proc Natl Acad Sci USA 101:3474–3479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajner SM, dos Santos Wagner M, Melo RC, Parreira GG, Chiarini-Garcia H, Bianco AC, Fekete C, Sanchez E, Lechan RM, Maia AL 2007 Type 2 iodothyronine deiodinase is highly expressed in germ cells of adult rat testis. J Endocrinol 194:47–54 [DOI] [PubMed] [Google Scholar]
- Silva JE 2006 Thermogenic mechanisms and their hormonal regulation. Physiol Rev 86:435–464 [DOI] [PubMed] [Google Scholar]
- Bianco AC, Kieffer JD, Silva JE 1992 Adenosine 3′,5′-monophosphate and thyroid hormone control of uncoupling protein messenger ribonucleic acid in freshly dispersed brown adipocytes. Endocrinology 130:2625–2633 [DOI] [PubMed] [Google Scholar]
- Lowell BB, Spiegelman BM 2000 Towards a molecular understanding of adaptive thermogenesis. Nature 404:652–660 [DOI] [PubMed] [Google Scholar]
- Obregon MJ 2008 Thyroid hormone and adipocyte differentiation. Thyroid 18:185–195 [DOI] [PubMed] [Google Scholar]
- Bruck K 1998 Neonatal thermal regulation. In: Polin RA, Fox WW, eds. Fetal and neonatal physiology. Philadelphia: W. B. Saunders Co.; 676–702 [Google Scholar]
- Bianco AC, Carvalho SD, Carvalho CR, Rabelo R, Moriscot AS 1998 Thyroxine 5′-deiodination mediates norepinephrine-induced lipogenesis in dispersed brown adipocytes. Endocrinology 139:571–578 [DOI] [PubMed] [Google Scholar]
- Carvalho SD, Negrao N, Bianco AC 1993 Hormonal regulation of malic enzyme and glucose-6-phosphate dehydrogenase in brown adipose tissue. The American Journal of Physiology 264:E874–881 [DOI] [PubMed] [Google Scholar]
- Christoffolete MA, Linardi CC, de Jesus L, Ebina KN, Carvalho SD, Ribeiro MO, Rabelo R, Curcio C, Martins L, Kimura ET, Bianco AC 2004 Mice with targeted disruption of the Dio2 gene have cold-induced overexpression of the uncoupling protein 1 gene but fail to increase brown adipose tissue lipogenesis and adaptive thermogenesis. Diabetes 53:577–584 [DOI] [PubMed] [Google Scholar]
- Liu X, Rossmeisl M, McClaine J, Riachi M, Harper ME, Kozak LP 2003 Paradoxical resistance to diet-induced obesity in UCP1-deficient mice. J Clin Invest 111:399–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaany NY, Gauthier KC, Zavacki AM, Mammen PP, Kitazume T, Peterson JA, Horton JD, Garry DJ, Bianco AC, Mangelsdorf DJ 2005 LXRs regulate the balance between fat storage and oxidation. Cell Metab 1:231–244 [DOI] [PubMed] [Google Scholar]
- Hull D 1977 Brown adipose tissue and the newborn infant’s response to cold. In: Philipp EE, Barnes J, Newton M, eds. Scientific foundation of obstetrics and gynaecology. London: William Heinemann; 545–550 [Google Scholar]
- Houstek J, Vizek K, Pavelka S, Kopecky J, Krejcova E, Hermanska J, Cermakova M 1993 Type II iodothyronine 5′-deiodinase and uncoupling protein in brown adipose tissue of human newborns. J Clin Endocrinol Metab 77:382–387 [DOI] [PubMed] [Google Scholar]
- Heaton JM 1972 The distribution of brown adipose tissue in the human. J Anat 112:35–39 [PMC free article] [PubMed] [Google Scholar]
- Ricquier D, Nechad M, Mory G 1982 Ultrastructural and biochemical characterization of human brown adipose tissue in pheochromocytoma. J Clin Endocrinol Metab 54:803–807 [DOI] [PubMed] [Google Scholar]
- Cohade C, Mourtzikos KA, Wahl RL 2003 “USA-Fat”: prevalence is related to ambient outdoor temperature-evaluation with 18F-FDG PET/CT. J Nucl Med 44:1267–1270 [PubMed] [Google Scholar]
- Hany TF, Gharehpapagh E, Kamel EM, Buck A, Himms-Hagen J, von Schulthess GK 2002 Brown adipose tissue: a factor to consider in symmetrical tracer uptake in the neck and upper chest region. Eur J Nucl Med Mol Imaging 29:1393–1398 [DOI] [PubMed] [Google Scholar]
- Croteau W, Davey JC, Galton VA, St Germain DL 1996 Cloning of the mammalian type II iodothyronine deiodinase. A selenoprotein differentially expressed and regulated in human and rat brain and other tissues. J Clin Invest 98:405–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi Y, Murakami M, Mizuma H, Ogiwara T, Imamura M, Mori M 1999 Expression and regulation of type II iodothyronine deiodinase in cultured human skeletal muscle cells. J Clin Endocrinol Metab 84:3293–3300 [DOI] [PubMed] [Google Scholar]
- Danforth Jr E, Horton ES, O'Connell M, Sims EA, Burger AG, Ingbar SH, Braverman L, Vagenakis AG 1979 Dietary-induced alterations in thyroid hormone metabolism during overnutrition. J Clin Invest 64:1336–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidegger K, O'Connell M, Robbins DC, Danforth Jr E 1984 Effects of chronic β-receptor stimulation on sympathetic nervous system activity, energy expenditure, and thyroid hormones. J Clin Endocrinol Metab 58:895–903 [DOI] [PubMed] [Google Scholar]
- Bartha T, Kim SW, Salvatore D, Gereben B, Tu HM, Harney JW, Rudas P, Larsen PR 2000 Characterization of the 5′-flanking and 5′-untranslated regions of the cyclic adenosine 3′,5′-monophosphate-responsive human type 2 iodothyronine deiodinase gene. Endocrinology 141:229–237 [DOI] [PubMed] [Google Scholar]
- al-Adsani H, Hoffer LJ, Silva JE 1997 Resting energy expenditure is sensitive to small dose changes in patients on chronic thyroid hormone replacement. J Clin Endocrinol Metab 82:1118–1125 [DOI] [PubMed] [Google Scholar]
- Richard K, Hume R, Kaptein E, Sanders JP, van Toor H, De Herder WW, den Hollander JC, Krenning EP, Visser TJ 1998 Ontogeny of iodothyronine deiodinases in human liver. J Clin Endocrinol Metab 83:2868–2874 [DOI] [PubMed] [Google Scholar]
- Huang SA, Dorfman DM, Genest DR, Salvatore D, Larsen PR 2003 Type 3 iodothyronine deiodinase is highly expressed in the human uteroplacental unit and in fetal epithelium. J Clin Endocrinol Metab 88:1384–1388 [DOI] [PubMed] [Google Scholar]
- Peeters RP, Wouters PJ, Kaptein E, van Toor H, Visser TJ, Van den Berghe G 2003 Reduced activation and increased inactivation of thyroid hormone in tissues of critically ill patients. J Clin Endocrinol Metab 88:3202–3211 [DOI] [PubMed] [Google Scholar]
- Olivares EL, Marassi MP, Fortunato RS, da Silva AC, Costa-e-Sousa RH, Araújo IG, Mattos EC, Masuda MO, Mulcahey MA, Huang SA, Bianco AC, Carvalho DP 2007 Thyroid function disturbance and type 3 iodothyronine deiodinase induction after myocardial infarction in rats—a time course study. Endocrinology 148:4786–4792 [DOI] [PubMed] [Google Scholar]
- Silva JE 2005 Thyroid hormone and the energetic cost of keeping body temperature. Biosci Rep 25:129–148 [DOI] [PubMed] [Google Scholar]
- Crunkhorn S, Patti ME 2008 Links between thyroid hormone action, oxidative metabolism, and diabetes risk? Thyroid 18:227–237 [DOI] [PubMed] [Google Scholar]
- Harper ME, Seifert EL 2008 Thyroid hormone effects on mitochondrial energetics. Thyroid 18:145–156 [DOI] [PubMed] [Google Scholar]
- Kim B 2008 Thyroid hormone as a determinant of energy expenditure and the basal metabolic rate. Thyroid 18:141–144 [DOI] [PubMed] [Google Scholar]
- Lebon V, Dufour S, Petersen KF, Ren J, Jucker BM, Slezak LA, Cline GW, Rothman DL, Shulman GI 2001 Effect of triiodothyronine on mitochondrial energy coupling in human skeletal muscle. J Clin Invest 108:733–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper ME, Brand MD 1993 The quantitative contributions of mitochondrial proton leak and ATP turnover reactions to the changed respiration rates of hepatocytes from rats of different thyroid status. J Biol Chem 268:14850–14860 [PubMed] [Google Scholar]
- Desai-Yajnik V, Zeng J, Omori K, Sherman J, Morimoto T 1995 The effect of thyroid hormone treatment on the gene expression and enzyme activity of rat liver sodium-potassium dependent adenosine triphosphatase. Endocrinology 136:629–639 [DOI] [PubMed] [Google Scholar]
- Folke M, Sestoft L 1977 Thyroid calorigenesis in isolated, perfused rat liver: minor role of active sodium-potassium transport. J Physiol 269:407–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonides WS, Thelen MH, van der Linden CG, Muller A, van Hardeveld C 2001 Mechanism of thyroid-hormone regulated expression of the SERCA genes in skeletal muscle: implications for thermogenesis. Biosci Rep 21:139–154 [DOI] [PubMed] [Google Scholar]
- de Meis L 2001 Role of the sarcoplasmic reticulum Ca2+-ATPase on heat production and thermogenesis. Biosci Rep 21:113–137 [DOI] [PubMed] [Google Scholar]
- Reis M, Farage M, de Meis L 2002 Thermogenesis and energy expenditure: control of heat production by the Ca(2+)-ATPase of fast and slow muscle. Mol Membr Biol 19:301–310 [DOI] [PubMed] [Google Scholar]
- Simonides WS, van Hardeveld C 2008 Thyroid hormone as a determinant of metabolic and contractile phenotype of skeletal muscle. Thyroid 18:205–216 [DOI] [PubMed] [Google Scholar]
- Izumo S, Nadal-Ginard B, Mahdavi V 1986 All members of the MHC multigene family respond to thyroid hormone in a highly tissue-specific manner. Science 231:597–600 [DOI] [PubMed] [Google Scholar]
- Oppenheimer JH, Schwartz HL, Lane JT, Thompson MP 1991 Functional relationship of thyroid hormone-induced lipogenesis, lipolysis, and thermogenesis in the rat. J Clin Invest 87:125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guadano-Ferraz A, Obregon MJ, St Germain DL, Bernal J 1997 The type 2 iodothyronine deiodinase is expressed primarily in glial cells in the neonatal rat brain. Proc Natl Acad Sci USA 94:10391–10396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu HM, Kim SW, Salvatore D, Bartha T, Legradi G, Larsen PR, Lechan RM 1997 Regional distribution of type 2 thyroxine deiodinase messenger ribonucleic acid in rat hypothalamus and pituitary and its regulation by thyroid hormone. Endocrinology 138:3359–3368 [DOI] [PubMed] [Google Scholar]
- Tu HM, Legradi G, Bartha T, Salvatore D, Lechan RM, Larsen PR 1999 Regional expression of the type 3 iodothyronine deiodinase messenger ribonucleic acid in the rat central nervous system and its regulation by thyroid hormone. Endocrinology 140:784–790 [DOI] [PubMed] [Google Scholar]
- Gereben B, Pachucki J, Kollar A, Liposits Z, Fekete C 2004 Ontogenic redistribution of type 2 deiodinase messenger ribonucleic acid in the brain of chicken. Endocrinology 145:3619–3625 [DOI] [PubMed] [Google Scholar]
- Bernal J 2005 The significance of thyroid hormone transporters in the brain. Endocrinology 146:1698–1700 [DOI] [PubMed] [Google Scholar]
- Lechan RM, Fekete C 2005 Role of thyroid hormone deiodination in the hypothalamus. Thyroid 15:883–897 [DOI] [PubMed] [Google Scholar]
- Fliers E, Alkemade A, Wiersinga WM, Swaab DF 2006 Hypothalamic thyroid hormone feedback in health and disease. Prog Brain Res 153:189–207 [DOI] [PubMed] [Google Scholar]
- Fekete C, Lechan RM 2007 Negative feedback regulation of hypophysiotropic thyrotropin-releasing hormone (TRH) synthesizing neurons: role of neuronal afferents and type 2 deiodinase. Front Neuroendocrinol 28:97–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riskind PN, Kolodny JM, Larsen PR 1987 The regional hypothalamic distribution of type II 5′-monodeiodinase in euthyroid and hypothyroid rats. Brain Res 420:194–198 [DOI] [PubMed] [Google Scholar]
- Hernandez A, Martinez ME, Fiering S, Galton VA, St Germain D 2006 Type 3 deiodinase is critical for the maturation and function of the thyroid axis. J Clin Invest 116:476–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez A, Martinez ME, Liao XH, Van Sande J, Refetoff S, Galton VA, St Germain DL 2007 Type 3 deiodinase deficiency results in functional abnormalities at multiple levels of the thyroid axis. Endocrinology 148:5680–5687 [DOI] [PubMed] [Google Scholar]
- Kaplan MM 1984 The role of thyroid hormone deiodination in the regulation of hypothalamo-pituitary function. Neuroendocrinology 38:254–260 [DOI] [PubMed] [Google Scholar]
- St Germain DL, Adler RA, Galton VA 1985 Thyroxine 5′-deiodinase activity in anterior pituitary glands transplanted under the renal capsule in the rat. Endocrinology 117:55–63 [DOI] [PubMed] [Google Scholar]
- Christoffolete MA, Ribeiro R, Singru P, Fekete C, da Silva WS, Gordon DF, Huang SA, Crescenzi A, Harney JW, Ridgway EC, Larsen PR, Lechan RM, Bianco AC 2006 Atypical expression of type 2 iodothyronine deiodinase in thyrotrophs explains the thyroxine-mediated pituitary thyrotropin feedback mechanism. Endocrinology 147:1735–1743 [DOI] [PubMed] [Google Scholar]
- Tannahill LA, Visser TJ, McCabe CJ, Kachilele S, Boelaert K, Sheppard MC, Franklyn JA, Gittoes NJ 2002 Dysregulation of iodothyronine deiodinase enzyme expression and function in human pituitary tumours. Clin Endocrinol (Oxf) 56:735–743 [DOI] [PubMed] [Google Scholar]
- Baur A, Buchfelder M, Kohrle J 2002 Expression of 5′-deiodinase enzymes in normal pituitaries and in various human pituitary adenomas. Eur J Endocrinol 147:263–268 [DOI] [PubMed] [Google Scholar]
- Gereben B, Salvatore D 2005 Pretranslational regulation of type 2 deiodinase. Thyroid 15:855–864 [DOI] [PubMed] [Google Scholar]
- Fekete C, Freitas BC, Zeold A, Wittmann G, Kadar A, Liposits Z, Christoffolete MA, Singru P, Lechan RM, Bianco AC, Gereben B 2007 Expression patterns of WSB-1 and USP-33 underlie cell-specific posttranslational control of type 2 deiodinase in the rat brain. Endocrinology 148:4865–4874 [DOI] [PubMed] [Google Scholar]
- Shupnik MA 2000 Thyroid hormone suppression of pituitary hormone gene expression. Rev Endocr Metab Disord 1:35–42 [DOI] [PubMed] [Google Scholar]
- Ridgway EC 1996 Modern concepts of primary thyroid gland failure. Clin Chem 42:179–182 [PubMed] [Google Scholar]
- Larsen PR 1982 Thyroid-pituitary interaction: feedback regulation of thyrotropin secretion by thyroid hormones. N Engl J Med 306:23–32 [DOI] [PubMed] [Google Scholar]
- Riesco G, Taurog A, Larsen R, Krulich L 1977 Acute and chronic responses to iodine deficiency in rats. Endocrinology 100:303–313 [DOI] [PubMed] [Google Scholar]
- Kakucska I, Rand W, Lechan RM 1992 Thyrotropin-releasing hormone gene expression in the hypothalamic paraventricular nucleus is dependent upon feedback regulation by both triiodothyronine and thyroxine. Endocrinology 130:2845–2850 [DOI] [PubMed] [Google Scholar]
- Schneider MJ, Fiering SN, Pallud SE, Parlow AF, St Germain DL, Galton VA 2001 Targeted disruption of the type 2 selenodeiodinase gene (DIO2) results in a phenotype of pituitary resistance to T4. Mol Endocrinol 15:2137–2148 [DOI] [PubMed] [Google Scholar]
- Silva JE, Larsen PR 1982 Comparison of iodothyronine 5′-deiodinase and other thyroid-hormone-dependent enzyme activities in the cerebral cortex of hypothyroid neonatal rat. Evidence for adaptation to hypothyroidism. J Clin Invest 70:1110–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obregon MJ, Escobar del Rey F, Morreale de Escobar G 2005 The effects of iodine deficiency on thyroid hormone deiodination. Thyroid 15:917–929 [DOI] [PubMed] [Google Scholar]
- Ridgway EC, Kieffer JD, Ross DS, Downing M, Mover H, Chin WW 1983 Mouse pituitary tumor line secreting only the α-subunit of the glycoprotein hormones: development from a thyrotropic tumor. Endocrinology 113:1587–1591 [DOI] [PubMed] [Google Scholar]
- Yusta B, Alarid ET, Gordon DF, Ridgway EC, Mellon PL 1998 The thyrotropin β-subunit gene is repressed by thyroid hormone in a novel thyrotrope cell line, mouse T αT1 cells. Endocrinology 139:4476–4482 [DOI] [PubMed] [Google Scholar]
- Hollenberg AN 2008 The role of the thyrotropin-releasing hormone (TRH) neuron as a metabolic sensor. Thyroid 18:131–139 [DOI] [PubMed] [Google Scholar]
- Ishii S, Kamegai J, Tamura H, Shimizu T, Sugihara H, Oikawa S 2003 Hypothalamic neuropeptide Y/Y1 receptor pathway activated by a reduction in circulating leptin, but not by an increase in circulating ghrelin, contributes to hyperphagia associated with triiodothyronine-induced thyrotoxicosis. Neuroendocrinology 78:321–330 [DOI] [PubMed] [Google Scholar]
- Kong WM, Martin NM, Smith KL, Gardiner JV, Connoley IP, Stephens DA, Dhillo WS, Ghatei MA, Small CJ, Bloom SR 2004 Triiodothyronine stimulates food intake via the hypothalamic ventromedial nucleus independent of changes in energy expenditure. Endocrinology 145:5252–5258 [DOI] [PubMed] [Google Scholar]
- Campos-Barros A, Musa A, Flechner A, Hessenius C, Gaio U, Meinhold H, Baumgartner A 1997 Evidence for circadian variations of thyroid hormone concentrations and type II 5′-iodothyronine deiodinase activity in the rat central nervous system. J Neurochem 68:795–803 [DOI] [PubMed] [Google Scholar]
- Legradi G, Emerson CH, Ahima RS, Flier JS, Lechan RM 1997 Leptin prevents fasting-induced suppression of prothyrotropin-releasing hormone messenger ribonucleic acid in neurons of the hypothalamic paraventricular nucleus. Endocrinology 138:2569–2576 [DOI] [PubMed] [Google Scholar]
- Mantzoros CS, Ozata M, Negrao AB, Suchard MA, Ziotopoulou M, Caglayan S, Elashoff RM, Cogswell RJ, Negro P, Liberty V, Wong ML, Veldhuis J, Ozdemir IC, Gold PW, Flier JS, Licinio J 2001 Synchronicity of frequently sampled thyrotropin (TSH) and leptin concentrations in healthy adults and leptin-deficient subjects: evidence for possible partial TSH regulation by leptin in humans. J Clin Endocrinol Metab 86:3284–3291 [DOI] [PubMed] [Google Scholar]
- Diano S, Naftolin F, Goglia F, Horvath TL 1998 Fasting-induced increase in type II iodothyronine deiodinase activity and messenger ribonucleic acid levels is not reversed by thyroxine in the rat hypothalamus. Endocrinology 139:2879–2884 [DOI] [PubMed] [Google Scholar]
- Abel ED, Ahima RS, Boers ME, Elmquist JK, Wondisford FE 2001 Critical role for thyroid hormone receptor β2 in the regulation of paraventricular thyrotropin-releasing hormone neurons. J Clin Invest 107:1017–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Bakal K, Minokoshi Y, Hollenberg AN 2004 Leptin signaling targets the thyrotropin-releasing hormone gene promoter in vivo. Endocrinology 145:2221–2227 [DOI] [PubMed] [Google Scholar]
- Coppola A, Liu ZW, Andrews ZB, Paradis E, Roy MC, Friedman JM, Ricquier D, Richard D, Horvath TL, Gao XB, Diano S 2007 A central thermogenic-like mechanism in feeding regulation: an interplay between arcuate nucleus T3 and UCP2. Cell Metab 5:21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson A 1993 Thyroidectomy progressively renders the reproductive system of starlings (Sturnus vulgaris) unresponsive to changes in daylength. J Endocrinol 139:51–55 [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Yasuo S, Watanabe M, Iigo M, Yamamura T, Hirunagi K, Ebihara S 2003 Light-induced hormone conversion of T4 to T3 regulates photoperiodic response of gonads in birds. Nature 426:178–181 [DOI] [PubMed] [Google Scholar]
- Nakao N, Ono H, Yamamura T, Anraku T, Takagi T, Higashi K, Yasuo S, Katou Y, Kageyama S, Uno Y, Kasukawa T, Iigo M, Sharp PJ, Iwasawa A, Suzuki Y, Sugano S, Niimi T, Mizutani M, Namikawa T, Ebihara S, Ueda HR, Yoshimura T 2008 Thyrotrophin in the pars tuberalis triggers photoperiodic response. Nature 452:317–322 [DOI] [PubMed] [Google Scholar]
- Barrett P, Ebling FJ, Schuhler S, Wilson D, Ross AW, Warner A, Jethwa P, Boelen A, Visser TJ, Ozanne DM, Archer ZA, Mercer JG, Morgan PJ 2007 Hypothalamic thyroid hormone catabolism acts as a gatekeeper for the seasonal control of body weight and reproduction. Endocrinology 148:3608–3617 [DOI] [PubMed] [Google Scholar]
- Watanabe M, Yasuo S, Watanabe T, Yamamura T, Nakao N, Ebihara S, Yoshimura T 2004 Photoperiodic regulation of type 2 deiodinase gene in Djungarian hamster: possible homologies between avian and mammalian photoperiodic regulation of reproduction. Endocrinology 145:1546–1549 [DOI] [PubMed] [Google Scholar]
- Yasuo S, Nakao N, Ohkura S, Iigo M, Hagiwara S, Goto A, Ando H, Yamamura T, Watanabe M, Watanabe T, Oda S, Maeda K, Lincoln GA, Okamura H, Ebihara S, Yoshimura T 2006 Long-day suppressed expression of type 2 deiodinase gene in the mediobasal hypothalamus of the Saanen goat, a short-day breeder: implication for seasonal window of thyroid hormone action on reproductive neuroendocrine axis. Endocrinology 147:432–440 [DOI] [PubMed] [Google Scholar]
- Maronde E, Stehle JH 2007 The mammalian pineal gland: known facts, unknown facets. Trends Endocrinol Metab 18:142–149 [DOI] [PubMed] [Google Scholar]
- Tanaka K, Murakami M, Greer MA 1986 Type-II thyroxine 5′-deiodinase is present in the rat pineal gland. Biochem Biophys Res Commun 137:863–868 [DOI] [PubMed] [Google Scholar]
- Murakami M, Hosoi Y, Negishi T, Kamiya Y, Ogiwara T, Mizuma H, Yamada M, Iriuchijima T, Mori M 1997 Expression and nocturnal increase of type II iodothyronine deiodinase mRNA in rat pineal gland. Neurosci Lett 227:65–67 [DOI] [PubMed] [Google Scholar]
- Chik CL, Wloka MT, Price DM, Ho AK 2007 The role of repressor proteins in the adrenergic induction of type II iodothyronine deiodinase in rat pinealocytes. Endocrinology 148:3523–3531 [DOI] [PubMed] [Google Scholar]
- Smith M, Burke Z, Humphries A, Wells T, Klein D, Carter D, Baler R 2001 Tissue-specific transgenic knockdown of fos-related antigen 2 (fra-2) expression mediated by dominant negative fra-2. Mol Cell Biol 21:3704–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya Y, Murakami M, Araki O, Hosoi Y, Ogiwara T, Mizuma H, Mori M 1999 Pretranslational regulation of rhythmic type II iodothyronine deiodinase expression by β-adrenergic mechanism in the rat pineal gland. Endocrinology 140:1272–1278 [DOI] [PubMed] [Google Scholar]
- Klein DC 2007 Arylalkylamine N-acetyltransferase: “the Timezyme.” J Biol Chem 282:4233–4237 [DOI] [PubMed] [Google Scholar]
- Guerrero JM, Reiter RJ 1992 Iodothyronine 5′-deiodinating activity in the pineal gland. Int J Biochem 24:1513–1523 [DOI] [PubMed] [Google Scholar]
- Duvernoy HM, Parratte B, Tatu L, Vuillier F 2000 The human pineal gland: relationships with surrounding structures and blood supply. Neurol Res 22:747–790 [DOI] [PubMed] [Google Scholar]
- Puig-Domingo M, Guerrero JM, Reiter RJ, Tannenbaum MJ, Hurlbut EC, Gonzalez-Brito A, Santana C 1988 Thyroxine 5′-deiodination in brown adipose tissue and pineal gland: implications for thermogenic regulation and role of melatonin. Endocrinology 123:677–680 [DOI] [PubMed] [Google Scholar]
- Revel FG, Saboureau M, Pevet P, Mikkelsen JD, Simonneaux V 2006 Melatonin regulates type 2 deiodinase gene expression in the Syrian hamster. Endocrinology 147:4680–4687 [DOI] [PubMed] [Google Scholar]
- Pachucki J, Burmeister LA, Larsen PR 1999 Thyroid hormone regulates hyperpolarization-activated cyclic nucleotide-gated channel (HCN2) mRNA in the rat heart. Circ Res 85:498–503 [DOI] [PubMed] [Google Scholar]
- Silva JE, Bianco SD 2008 Thyroid-adrenergic interactions: physiological and clinical implications. Thyroid 18:157–165 [DOI] [PubMed] [Google Scholar]
- Kim B, Carvalho-Bianco SD, Larsen PR 2004 Thyroid hormone and adrenergic signaling in the heart. Arq Bras Endocrinol Metabol 48:171–175 [DOI] [PubMed] [Google Scholar]
- Carvalho-Bianco SD, Kim BW, Zhang JX, Harney JW, Ribeiro RS, Gereben B, Bianco AC, Mende U, Larsen PR 2004 Chronic cardiac-specific thyrotoxicosis increases myocardial β-adrenergic responsiveness. Mol Endocrinol 18:1840–1849 [DOI] [PubMed] [Google Scholar]
- Adler SM, Wartofsky L 2007 The nonthyroidal illness syndrome. Endocrinol Metab Clin North Am 36:657–672, vi [DOI] [PubMed] [Google Scholar]
- Utiger RD 1980 Decreased extrathyroidal triiodothyronine production in nonthyroidal illness: benefit or harm? Am J Med 69:807–810 [DOI] [PubMed] [Google Scholar]
- De Groot LJ 1999 Dangerous dogmas in medicine: the nonthyroidal illness syndrome. J Clin Endocrinol Metab 84:151–164 [DOI] [PubMed] [Google Scholar]
- Brent GA, Hershman JM 1986 Thyroxine therapy in patients with severe nonthyroidal illnesses and low serum thyroxine concentration. J Clin Endocrinol Metab 63:1–8 [DOI] [PubMed] [Google Scholar]
- Acker CG, Singh AR, Flick RP, Bernardini J, Greenberg A, Johnson JP 2000 A trial of thyroxine in acute renal failure. Kidney Int 57:293–298 [DOI] [PubMed] [Google Scholar]
- Huang SA, Bianco AC 2008 Reawakened interest in type III iodothyronine deiodinase in critical illness and injury. Nat Clin Pract Endocrinol Metab 4:148–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H 2007 Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail Rev 12:331–343 [DOI] [PubMed] [Google Scholar]
- Klein I, Ojamaa K 2001 Thyroid hormone and the cardiovascular system. N Engl J Med 344:501–509 [DOI] [PubMed] [Google Scholar]
- Muller A, Simonides WS 2005 Regulation of myocardial SERCA2a expression in ventricular hypertrophy and heart failure. Future Cardiol 1:543–553 [DOI] [PubMed] [Google Scholar]
- Wagner MS, Morimoto R, Dora JM, Benneman A, Pavan R, Maia AL 2003 Hypothyroidism induces type 2 iodothyronine deiodinase expression in mouse heart and testis. J Mol Endocrinol 31:541–550 [DOI] [PubMed] [Google Scholar]
- Escobar-Morreale HF, Obregon MJ, Escobar del Rey F, Morreale de Escobar G 1999 Tissue-specific patterns of changes in 3,5,3′-triiodo-L-thyronine concentrations in thyroidectomized rats infused with increasing doses of the hormone. Which are the regulatory mechanisms? Biochimie 81:453–462 [DOI] [PubMed] [Google Scholar]
- Des Tombe AL, Van Beek-Harmsen BJ, Lee-De Groot MB, Van Der Laarse WJ 2002 Calibrated histochemistry applied to oxygen supply and demand in hypertrophied rat myocardium. Microsc Res Tech 58:412–420 [DOI] [PubMed] [Google Scholar]
- Boluyt MO, O'Neill L, Meredith AL, Bing OH, Brooks WW, Conrad CH, Crow MT, Lakatta EG 1994 Alterations in cardiac gene expression during the transition from stable hypertrophy to heart failure. Marked upregulation of genes encoding extracellular matrix components. Circ Res 75:23–32 [DOI] [PubMed] [Google Scholar]
- Lim H, Zhu YZ 2006 Role of transforming growth factor-β in the progression of heart failure. Cell Mol Life Sci 63:2584–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rami A, Krieglstein J 1992 Thyroxine attenuates hippocampal neuronal damage caused by ischemia in the rat. Life Sci 50:645–650 [DOI] [PubMed] [Google Scholar]
- D'Alecy LG 1997 Thyroid hormone in neural rescue. Thyroid 7:115–124 [DOI] [PubMed] [Google Scholar]
- Margaill I, Royer J, Lerouet D, Ramauge M, Le Goascogne C, Li WW, Plotkine M, Pierre M, Courtin F 2005 Induction of type 2 iodothyronine deiodinase in astrocytes after transient focal cerebral ischemia in the rat. J Cereb Blood Flow Metab 25:468–476 [DOI] [PubMed] [Google Scholar]
- Lamirand A, Mercier G, Ramauge M, Pierre M, Courtin F 2007 Hypoxia stabilizes type 2 deiodinase activity in rat astrocytes. Endocrinology 148:4745–4753 [DOI] [PubMed] [Google Scholar]
- Li WW, Le Goascogne C, Ramauge M, Schumacher M, Pierre M, Courtin F 2001 Induction of type 3 iodothyronine deiodinase by nerve injury in the rat peripheral nervous system. Endocrinology 142:5190–5197 [DOI] [PubMed] [Google Scholar]
- Li WW, Le Goascogne C, Schumacher M, Pierre M, Courtin F 2001 Type 2 deiodinase in the peripheral nervous system: induction in the sciatic nerve after injury. Neuroscience 107:507–518 [DOI] [PubMed] [Google Scholar]
- Puzianowska-Kuznicka M, Pietrzak M, Turowska O, Nauman A 2006 Thyroid hormones and their receptors in the regulation of cell proliferation. Acta Biochim Pol 53:641–650 [PubMed] [Google Scholar]
- Debski MG, Pachucki J, Ambroziak M, Olszewski W, Bar-Andziak E 2007 Human breast cancer tissue expresses high level of type 1 5′-deiodinase. Thyroid 17:3–10 [DOI] [PubMed] [Google Scholar]
- Kim BW, Daniels GH, Harrison BJ, Price A, Harney JW, Larsen PR, Weetman AP 2003 Overexpression of type 2 iodothyronine deiodinase in follicular carcinoma as a cause of low circulating free thyroxine levels. J Clin Endocrinol Metab 88:594–598 [DOI] [PubMed] [Google Scholar]
- Mori K, Yoshida K, Kayama T, Kaise N, Fukazawa H, Kiso Y, Kikuchi K, Aizawa Y, Abe K 1993 Thyroxine 5-deiodinase in human brain tumors. J Clin Endocrinol Metab 77:1198–1202 [DOI] [PubMed] [Google Scholar]
- Nauman P, Bonicki W, Michalik R, Warzecha A, Czernicki Z 2004 The concentration of thyroid hormones and activities of iodothyronine deiodinases are altered in human brain gliomas. Folia Neuropathol 42:67–73 [PubMed] [Google Scholar]
- Huang SA, Fish SA, Dorfman DM, Salvatore D, Kozakewich HP, Mandel SJ, Larsen PR 2002 A 21-year-old woman with consumptive hypothyroidism due to a vascular tumor expressing type 3 iodothyronine deiodinase. J Clin Endocrinol Metab 87:4457–4461 [DOI] [PubMed] [Google Scholar]
- Huang SA, Tu HM, Harney JW, Venihaki M, Butte AJ, Kozakewich HP, Fishman SJ, Larsen PR 2000 Severe hypothyroidism caused by type 3 iodothyronine deiodinase in infantile hemangiomas. N Engl J Med 343:185–189 [DOI] [PubMed] [Google Scholar]
- Ruppe MD, Huang SA, Jan de Beur SM 2005 Consumptive hypothyroidism caused by paraneoplastic production of type 3 iodothyronine deiodinase. Thyroid 15:1369–1372 [DOI] [PubMed] [Google Scholar]
- Boelen A, Kwakkel J, Alkemade A, Renckens R, Kaptein E, Kuiper G, Wiersinga WM, Visser TJ 2005 Induction of type 3 deiodinase activity in inflammatory cells of mice with chronic local inflammation. Endocrinology 146:5128–5134 [DOI] [PubMed] [Google Scholar]
- Suzuki K, Hino M, Kutsuna H, Hato F, Sakamoto C, Takahashi T, Tatsumi N, Kitagawa S 2001 Selective activation of p38 mitogen-activated protein kinase cascade in human neutrophils stimulated by IL-1β. J Immunol 167:5940–5947 [DOI] [PubMed] [Google Scholar]
- Pallud S, Ramauge M, Gavaret JM, Lennon AM, Munsch N, St Germain DL, Pierre M, Courtin F 1999 Regulation of type 3 iodothyronine deiodinase expression in cultured rat astrocytes: role of the Erk cascade. Endocrinology 140:2917–2923 [DOI] [PubMed] [Google Scholar]
- Christoffolete MA, Arrojo e Drigo R, Gazoni F, Tente SM, Goncalves V, Amorim BS, Larsen PR, Bianco AC, Zavacki AM 2007 Mice with impaired extrathyroidal thyroxine to 3,5,3′-triiodothyronine conversion maintain normal serum 3,5,3′-triiodothyronine concentrations. Endocrinology 148:954–960 [DOI] [PubMed] [Google Scholar]
- Mebis L, Debaveye Y, Visser TJ, Van den Berghe G 2006 Changes within the thyroid axis during the course of critical illness. Endocrinol Metab Clin North Am 35:807–821, x [DOI] [PubMed] [Google Scholar]
- Izumi M, Larsen PR 1977 Triiodothyronine, thyroxine, and iodine in purified thyroglobulin from patients with Graves’ disease. J Clin Invest 59:1105–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boye N, Laurberg P 1984 Deiodination of T4 to T3 and rT3 by microsomes from normal human thyroid tissue. Mol Cell Endocrinol 37:295–299 [DOI] [PubMed] [Google Scholar]
- Murakami M, Araki O, Hosoi Y, Kamiya Y, Morimura T, Ogiwara T, Mizuma H, Mori M 2001 Expression and regulation of type II iodothyronine deiodinase in human thyroid gland. Endocrinology 142:2961–2967 [DOI] [PubMed] [Google Scholar]
- Gereben B, Salvatore D, Harney JW, Tu HM, Larsen PR 2001 The human, but not rat, dio2 gene is stimulated by thyroid transcription factor-1 (TTF-1). Mol Endocrinol 15:112–124 [DOI] [PubMed] [Google Scholar]
- Laurberg P 1984 Mechanisms governing the relative proportions of thyroxine and 3,5,3′-triiodothyronine in thyroid secretion. Metabolism 33:379–392 [DOI] [PubMed] [Google Scholar]
- Laurberg P 1987 Thyroxine and 3,5,3′-triiodothyronine content of thyroglobulin in thyroid needle aspirates in hyperthyroidism and hypothyroidism. J Clin Endocrinol Metab 64:969–974 [DOI] [PubMed] [Google Scholar]
- Ishii H, Inada M, Tanaka K, Mashio Y, Naito K, Nishikawa M, Matsuzuka F, Kuma K, Imura H 1982 Sequential deiodination of thyroxine in human thyroid gland. J Clin Endocrinol Metab 55:890–896 [DOI] [PubMed] [Google Scholar]
- Laurberg P, Vestergaard H, Nielsen S, Christensen SE, Seefeldt T, Helleberg K, Pedersen KM 2007 Sources of circulating 3,5,3′-triiodothyronine in hyperthyroidism estimated after blocking of type 1 and type 2 iodothyronine deiodinases. J Clin Endocrinol Metab 92:2149–2156 [DOI] [PubMed] [Google Scholar]
- Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM 1991 Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med 325:1688–1695 [DOI] [PubMed] [Google Scholar]
- Schwindinger WF, Francomano CA, Levine MA 1992 Identification of a mutation in the gene encoding the α subunit of the stimulatory G protein of adenylyl cyclase in McCune-Albright syndrome. Proc Natl Acad Sci USA 89:5152–5156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congedo V, Celi FS 2007 Thyroid disease in patients with McCune-Albright syndrome. Pediatr Endocrinol Rev 4(Suppl 4):429–433 [PubMed] [Google Scholar]
- Celi FS, Coppotelli G, Chidakel A, Kelly M, Brillante BA, Shawker T, Cherman N, Feuillan PP, Collins MT 2008 The role of type-1 and type-2 5′deiodinase in the pathophysiology of the T3 toxicosis of McCune-Albright syndrome. J Clin Endocrinol Metab 93:2383–2389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canettieri G, Celi FS, Baccheschi G, Salvatori L, Andreoli M, Centanni M 2000 Isolation of human type 2 deiodinase gene promoter and characterization of a functional cyclic adenosine monophosphate response element. Endocrinology 141:1804–1813 [DOI] [PubMed] [Google Scholar]
- Panicker V, Cluett C, Shields B, Murray A, Parnell KS, Perry JR, Weedon MN, Singleton A, Hernandez D, Evans J, Durant C, Ferrucci L, Melzer D, Saravanan P, Visser TJ, Ceresini G, Hattersley AT, Vaidya B, Dayan CM, Frayling TM 2008 A common variation in deiodinase 1 gene DIO1 is associated with the relative levels of free thyroxine and triidothyronine. J Clin Endocrinol Metab 93:3075–3081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geffner DL, Azukizawa M, Hershman JM 1975 Propylthiouracil blocks extrathyroidal conversion of thyroxine to triiodothyronine and augments thyrotropin secretion in man. J Clin Invest 55:224–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saberi M, Sterling FH, Utiger RD 1975 Reduction in extrathyroidal triiodothyronine production by propylthiouracil in man. J Clin Invest 55:218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPresti JS, Eigen A, Kaptein E, Anderson KP, Spencer CA, Nicoloff JT 1989 Alterations in 3,3′5′-triiodothyronine metabolism in response to propylthiouracil, dexamethasone, and thyroxine administration in man. J Clin Invest 84:1650–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum SM, Nicoloff JT, Spencer CA, Kaptein EM 1984 Peripheral tissue mechanism for maintenance of serum triiodothyronine values in a thyroxine-deficient state in man. J Clin Invest 73:570–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoloff JT, Lum SM, Spencer CA, Morris R 1984 Peripheral autoregulation of thyroxine to triiodothyronine conversion in man. Horm Metab Res Suppl 14:74–79 [PubMed] [Google Scholar]
- Mebis L, Langouche L, Visser TJ, Van den Berghe G 2007 The type II iodothyronine deiodinase is up-regulated in skeletal muscle during prolonged critical illness. J Clin Endocrinol Metab 92:3330–3333 [DOI] [PubMed] [Google Scholar]
- Zavacki AM, Ying H, Christoffolete MA, Aerts G, So E, Harney JW, Cheng SY, Larsen PR, Bianco AC 2005 Type 1 iodothyronine deiodinase is a sensitive marker of peripheral thyroid status in the mouse. Endocrinology 146:1568–1575 [DOI] [PubMed] [Google Scholar]
- Nagaya T, Fujieda M, Otsuka G, Yang JP, Okamoto T, Seo H 2000 A potential role of activated NF-κB in the pathogenesis of euthyroid sick syndrome. J Clin Invest 106:393–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Koenig RJ 2000 Regulation of hepatocyte thyroxine 5′-deiodinase by T3 and nuclear receptor coactivators as a model of the sick euthyroid syndrome. J Biol Chem 275:38296–38301 [DOI] [PubMed] [Google Scholar]
- Peeters RP, Wouters PJ, van Toor H, Kaptein E, Visser TJ, Van den Berghe G 2005 Serum 3,3′,5′-triiodothyronine (rT3) and 3,5,3′-triiodothyronine/rT3 are prognostic markers in critically ill patients and are associated with postmortem tissue deiodinase activities. J Clin Endocrinol Metab 90:4559–4565 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Perez A, Palos-Paz F, Kaptein E, Visser TJ, Dominguez-Gerpe L, Alvarez-Escudero J, Lado-Abeal J 2008 Identification of molecular mechanisms related to nonthyroidal illness syndrome in skeletal muscle and adipose tissue from patients with septic shock. Clin Endocrinol (Oxf) 68:821–827 [DOI] [PubMed] [Google Scholar]
- Fekete C, Sarkar S, Christoffolete MA, Emerson CH, Bianco AC, Lechan RM 2005 Bacterial lipopolysaccharide (LPS)-induced type 2 iodothyronine deiodinase (D2) activation in the mediobasal hypothalamus (MBH) is independent of the LPS-induced fall in serum thyroid hormone levels. Brain Res 1056:97–99 [DOI] [PubMed] [Google Scholar]
- Boelen A, Kwakkel J, Thijssen-Timmer DC, Alkemade A, Fliers E, Wiersinga WM 2004 Simultaneous changes in central and peripheral components of the hypothalamus-pituitary-thyroid axis in lipopolysaccharide-induced acute illness in mice. J Endocrinol 182:315–323 [DOI] [PubMed] [Google Scholar]
- Kakucska I, Romero LI, Clark BD, Rondeel JM, Qi Y, Alex S, Emerson CH, Lechan RM 1994 Suppression of thyrotropin-releasing hormone gene expression by interleukin-1-β in the rat: implications for nonthyroidal illness. Neuroendocrinology 59:129–137 [DOI] [PubMed] [Google Scholar]
- Boelen A, Kwakkel J, Wiersinga WM, Fliers E 2006 Chronic local inflammation in mice results in decreased TRH and type 3 deiodinase mRNA expression in the hypothalamic paraventricular nucleus independently of diminished food intake. J Endocrinol 191:707–714 [DOI] [PubMed] [Google Scholar]
- Zeold A, Doleschall M, Haffner MC, Capelo LP, Menyhert J, Liposits Z, da Silva WS, Bianco AC, Kacskovics I, Fekete C, Gereben B 2006 Characterization of the nuclear factor-κB responsiveness of the human dio2 gene. Endocrinology 147:4419–4429 [DOI] [PubMed] [Google Scholar]
- Sanchez E, Singru PS, Fekete C, Lechan RM 2007 Importance of NF-kB signaling in activation of type 2 Iodothyronine deiodinase (D2) in the mediobasal hypothalamus after lipopolysaccharide (LPS) administration. Program of the 89th Annual Meeting of The Endocrine Society, Toronto, Canada, 2007, P2-657 (Abstract) [Google Scholar]
- Boelen A, Platvoet-ter Schiphorst MC, Bakker O, Wiersinga WM 1995 The role of cytokines in the lipopolysaccharide-induced sick euthyroid syndrome in mice. J Endocrinol 146:475–483 [DOI] [PubMed] [Google Scholar]
- Sanchez E, Singru PS, Fekete C, Lechan RM 2008 Induction of type 2 iodothyronine deiodinase in the mediobasal hypothalamus by bacterial lipopolysaccharide: role of corticosterone. Endocrinology 149:2484–2493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentuccia D, Proietti-Pannunzi L, Tanner K, Bacci V, Pollin TI, Poehlman ET, Shuldiner AR, Celi FS 2002 Association between a novel variant of the human type 2 deiodinase gene Thr92Ala and insulin resistance: evidence of interaction with the Trp64Arg variant of the β-3-adrenergic receptor. Diabetes 51:880–883 [DOI] [PubMed] [Google Scholar]
- Peeters RP, van Toor H, Klootwijk W, de Rijke YB, Kuiper GG, Uitterlinden AG, Visser TJ 2003 Polymorphisms in thyroid hormone pathway genes are associated with plasma TSH and iodothyronine levels in healthy subjects. J Clin Endocrinol Metab 88:2880–2888 [DOI] [PubMed] [Google Scholar]
- Canani LH, Capp C, Dora JM, Meyer EL, Wagner MS, Harney JW, Larsen PR, Gross JL, Bianco AC, Maia AL 2005 The type 2 deiodinase A/G (Thr92Ala) polymorphism is associated with decreased enzyme velocity and increased insulin resistance in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab 90:3472–3478 [DOI] [PubMed] [Google Scholar]
- Chidakel A, Mentuccia D, Celi FS 2005 Peripheral metabolism of thyroid hormone and glucose homeostasis. Thyroid 15:899–903 [DOI] [PubMed] [Google Scholar]
- Mentuccia D, Thomas MJ, Coppotelli G, Reinhart LJ, Mitchell BD, Shuldiner AR, Celi FS 2005 The Thr92Ala deiodinase type 2 (DIO2) variant is not associated with type 2 diabetes or indices of insulin resistance in the old order of Amish. Thyroid 15:1223–1227 [DOI] [PubMed] [Google Scholar]
- Grarup N, Andersen MK, Andreasen CH, Albrechtsen A, Borch-Johnsen K, Jorgensen T, Auwerx J, Schmitz O, Hansen T, Pedersen O 2007 Studies of the common DIO2 Thr92Ala polymorphism and metabolic phenotypes in 7342 Danish white subjects. J Clin Endocrinol Metab 92:363–366 [DOI] [PubMed] [Google Scholar]
- Maia AL, Dupuis J, Manning A, Liu C, Meigs JB, Cupples LA, Larsen PR, Fox CS 2007 The type 2 deiodinase (DIO2) A/G polymorphism is not associated with glycemic traits: the Framingham Heart Study. Thyroid 17:199–202 [DOI] [PubMed] [Google Scholar]
- Gumieniak O, Perlstein TS, Williams JS, Hopkins PN, Brown NJ, Raby BA, Williams GH 2007 Ala92 type 2 deiodinase allele increases risk for the development of hypertension. Hypertension 49:461–466 [DOI] [PubMed] [Google Scholar]
- Maia AL, Hwang SJ, Levy D, Larson MG, Larsen PR, Fox CS 2008 Lack of association between the type 2 deiodinase A/G polymorphism and hypertensive traits: The Framingham Heart Study. Hypertension 51:e22–e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorito M, Torrente I, De Cosmo S, Guida V, Colosimo A, Prudente S, Flex E, Menghini R, Miccoli R, Penno G, Pellegrini F, Tassi V, Federici M, Trischitta V, Dallapiccola B 2007 Interaction of DIO2 T92A and PPARγ2 P12A polymorphisms in the modulation of metabolic syndrome. Obesity (Silver Spring) 15:2889–2895 [DOI] [PubMed] [Google Scholar]
- Chistiakov DA, Savost'anov KV, Turakulov RI 2004 Screening of SNPs at 18 positional candidate genes, located within the GD-1 locus on chromosome 14q23-q32, for susceptibility to Graves’ disease: a TDT study. Mol Genet Metab 83:264–270 [DOI] [PubMed] [Google Scholar]
- Guo TW, Zhang FC, Yang MS, Gao XC, Bian L, Duan SW, Zheng ZJ, Gao JJ, Wang H, Li RL, Feng GY, St Clair D, He L 2004 Positive association of the DIO2 (deiodinase type 2) gene with mental retardation in the iodine-deficient areas of China. J Med Genet 41:585–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meulenbelt I, Min JL, Bos S, Riyazi N, Houwing-Duistermaat JJ, van der Wijk HJ, Kroon HM, Nakajima M, Ikegawa S, Uitterlinden AG, van Meurs JB, van der Deure WM, Visser TJ, Seymour AB, Lakenberg N, van der Breggen R, Kremer D, van Duijn CM, Kloppenburg M, Loughlin J, Slagboom PE 2008 Identification of DIO2 as new susceptibility locus for symptomatic osteoarthritis. Hum Mol Genet 17:1867–1875 [DOI] [PubMed] [Google Scholar]
- Brouwer JP, Appelhof BC, Peeters RP, Hoogendijk WJ, Huyser J, Schene AH, Tijssen JG, Van Dyck R, Visser TJ, Wiersinga WM, Fliers E 2006 Thyrotropin, but not a polymorphism in type II deiodinase, predicts response to paroxetine in major depression. Eur J Endocrinol 154:819–825 [DOI] [PubMed] [Google Scholar]
- Torlontano M, Durante C, Torrente I, Crocetti U, Augello G, Ronga G, Montesano T, Travascio L, Verrienti A, Bruno R, Santini S, D'Arcangelo P, Dallapiccola B, Filetti S, Trischitta V 2007 Type 2 deiodinase polymorphism (Thr92ala) predicts L-thyroxine dose to achieve target Tsh levels in thyroidectomized patients. J Clin Endocrinol Metab 93:910–913 [DOI] [PubMed] [Google Scholar]
- Appelhof BC, Peeters RP, Wiersinga WM, Visser TJ, Wekking EM, Huyser J, Schene AH, Tijssen JG, Hoogendijk WJ, Fliers E 2005 Polymorphisms in type 2 deiodinase are not associated with well-being, neurocognitive functioning, and preference for combined thyroxine/3,5,3′-triiodothyronine therapy. J Clin Endocrinol Metab 90:6296–6299 [DOI] [PubMed] [Google Scholar]
- Peeters RP, van den Beld AW, Attalki H, Toor H, de Rijke YB, Kuiper GG, Lamberts SW, Janssen JA, Uitterlinden AG, Visser TJ 2005 A new polymorphism in the type II deiodinase gene is associated with circulating thyroid hormone parameters. Am J Physiol Endocrinol Metab 289:E75–E81 [DOI] [PubMed] [Google Scholar]
- Coppotelli G, Summers A, Chidakel A, Ross JM, Celi FS 2006 Functional characterization of the 258 A/G (D2-ORFa-Gly3Asp) human type-2 deiodinase polymorphism: a naturally occurring variant increases the enzymatic activity by removing a putative repressor site in the 5′ UTR of the gene. Thyroid 16:625–632 [DOI] [PubMed] [Google Scholar]
- Peeters RP, van den Beld AW, van Toor H, Uitterlinden AG, Janssen JA, Lamberts SW, Visser TJ 2005 A polymorphism in type I deiodinase is associated with circulating free insulin-like growth factor I levels and body composition in humans. J Clin Endocrinol Metab 90:256–263 [DOI] [PubMed] [Google Scholar]
- Hernandez A, Park JP, Lyon GJ, Mohandas TK, St Germain DL 1998 Localization of the type 3 iodothyronine deiodinase (DIO3) gene to human chromosome 14q32 and mouse chromosome 12F1. Genomics 53:119–121 [DOI] [PubMed] [Google Scholar]
- Hernandez A, Lyon GJ, Schneider MJ, St Germain DL 1999 Isolation and characterization of the mouse gene for the type 3 iodothyronine deiodinase. Endocrinology 140:124–130 [DOI] [PubMed] [Google Scholar]
- Hernandez A, Martinez ME, Croteau W, St Germain DL 2004 Complex organization and structure of sense and antisense transcripts expressed from the DIO3 gene imprinted locus. Genomics 83:413–424 [DOI] [PubMed] [Google Scholar]
- Edwards CA, Ferguson-Smith AC 2007 Mechanisms regulating imprinted genes in clusters. Curr Opin Cell Biol 19:281–289 [DOI] [PubMed] [Google Scholar]
- Kester MH, Kuiper GG, Versteeg R, Visser TJ 2006 Regulation of type III iodothyronine deiodinase expression in human cell lines. Endocrinology 147:5845–5854 [DOI] [PubMed] [Google Scholar]
- Hernandez A, Garcia B, Obregon MJ 2007 Gene expression from the imprinted Dio3 locus is associated with cell proliferation of cultured brown adipocytes. Endocrinology 148:3968–3976 [DOI] [PubMed] [Google Scholar]
- Hernandez A 2005 Structure and function of the type 3 deiodinase gene. Thyroid 15:865–874 [DOI] [PubMed] [Google Scholar]
- da Rocha ST, Edwards CA, Ito M, Ogata T, Ferguson-Smith AC 2008 Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet 24:306–316 [DOI] [PubMed] [Google Scholar]
- Smas CM, Sul HS 1993 Pref-1, a protein containing EGF-like repeats, inhibits adipocyte differentiation. Cell 73:725–734 [DOI] [PubMed] [Google Scholar]
- Tierling S, Dalbert S, Schoppenhorst S, Tsai CE, Oliger S, Ferguson-Smith AC, Paulsen M, Walter J 2006 High-resolution map and imprinting analysis of the Gtl2-Dnchc1 domain on mouse chromosome 12. Genomics 87:225–235 [DOI] [PubMed] [Google Scholar]
- Cavaille J, Seitz H, Paulsen M, Ferguson-Smith AC, Bachellerie JP 2002 Identification of tandemly-repeated C/D snoRNA genes at the imprinted human 14q32 domain reminiscent of those at the Prader-Willi/Angelman syndrome region. Hum Mol Genet 11:1527–1538 [DOI] [PubMed] [Google Scholar]
- Seitz H, Youngson N, Lin SP, Dalbert S, Paulsen M, Bachellerie JP, Ferguson-Smith AC, Cavaille J 2003 Imprinted microRNA genes transcribed antisense to a reciprocally imprinted retrotransposon-like gene. Nat Genet 34:261–262 [DOI] [PubMed] [Google Scholar]
- Lin SP, Youngson N, Takada S, Seitz H, Reik W, Paulsen M, Cavaille J, Ferguson-Smith AC 2003 Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the Dlk1-Gtl2 imprinted cluster on mouse chromosome 12. Nat Genet 35:97–102 [DOI] [PubMed] [Google Scholar]
- Labialle S, Yang L, Ruan X, Villemain A, Schmidt JV, Hernandez A, Wiltshire T, Cermakian N, Naumova AK 2008 Coordinated diurnal regulation of genes from the Dlk1-Dio3 imprinted domain: implications for regulation of clusters of non-paralogous genes. Hum Mol Genet 17:15–26 [DOI] [PubMed] [Google Scholar]
- Georgiades P, Watkins M, Surani MA, Ferguson-Smith AC 2000 Parental origin-specific developmental defects in mice with uniparental disomy for chromosome 12. Development 127:4719–4728 [DOI] [PubMed] [Google Scholar]
- Bosl MR, Takaku K, Oshima M, Nishimura S, Taketo MM 1997 Early embryonic lethality caused by targeted disruption of the mouse selenocysteine tRNA gene (Trsp). Proc Natl Acad Sci USA 94:5531–5534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low SC, Grundner-Culemann E, Harney JW, Berry MJ 2000 SECIS-SBP2 interactions dictate selenocysteine incorporation efficiency and selenoprotein hierarchy. EMBO J 19:6882–6890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco AC, Ribich S, Kim BW 2007 An inside job. Endocrinology 148:3077–3079 [DOI] [PubMed] [Google Scholar]
- Baxter JD, Webb P 2006 Metabolism: bile acids heat things up. Nature 439:402–403 [DOI] [PubMed] [Google Scholar]