

Abstract
The tumor suppressor protein PTEN plays an important role in intestinal cell proliferation and differentiation and tumor suppression by antagonizing phosphatidylinositol 3-kinase (PI3K). Despite its importance, the molecular mechanisms regulating PTEN expression are largely undefined. Here, we show that treatment of the colon cancer cell line, HT29, with the differentiating agent sodium butyrate (NaBT) increased PTEN protein and mRNA expression and induced JNK activation. Inhibition of c-Jun-NH2-terminal kinase (JNK) by chemical or genetic methods attenuated NaBT-induced PTEN expression. In addition, our findings demonstrated a cross-talk between NF-κB and JNK with respect to PTEN regulation. Overexpression of the NF-κB superrepressor increased PTEN expression and JNK activity, whereas overexpression of the p65 NF-κB subunit reduced both basal and NaBT-mediated JNK activation and PTEN expression. Moreover, we showed that overexpression of PTEN or treatment with NaBT increased expression of the cyclin dependent kinase inhibitor p27kip1 in HT29 cells; this induction was attenuated by inhibition of PTEN or JNK expression or overexpression of p65. Finally, we demonstrate a role for PTEN in NaBT-mediated cell death and differentiation. Our findings suggest that the NF-κB/JNK/PTEN pathway plays a critical role in normal intestinal homeostasis and colon carcinogenesis.
Keywords: PTEN, JNK, NF-κB, p27kip1, PI3K, intestinal cells
INTRODUCTION
The tumor suppressor protein PTEN (for Phosphatase and tensin homologue deleted on chromosome ten) antagonizes the activity of phosphatidylinositol 3-kinase (PI3K) by dephosphorylating the D3-phosphate group of lipid second messengers, thus serving as a negative regulator of the PI3K pathway (1). PTEN inhibits downstream functions mediated by the PI3K pathway, such as cell growth and survival, cell migration and invasion (2), and cell cycle progression through the regulation of the expression of the cyclin-dependent kinase inhibitor protein, p27kip1(3), which is induced by PTEN in various cells (4, 5). Previously, we demonstrated that inhibition of PI3K or overexpression of PTEN significantly enhances intestinal cell differentiation either spontaneously or induced by the short chain fatty acid sodium butyrate (NaBT) (6), a histone deacetylase (HDAC) inhibitor produced in the colon by breakdown of dietary fiber (7). PTEN expression correlates with expression of Cdx-2, a homeodomain protein required for intestinal epithelial cell differentiation, along the length of the murine colon (8). Moreover, PTEN stimulates Cdx-2 protein expression and the transcriptional activity of the Cdx-2 promoter, thus further indicating a role for PTEN in the process of intestinal differentiation. Despite the importance of PTEN in apoptosis and differentiation, little is known about the regulation of PTEN expression.
NF-κB is a heterodimer consisting of the DNA binding subunit p50 and the transactivation subunit RelA/p65. The activation pathway of NF-κB is regulated by an endogenous cytoplasmic inhibitor, IκB, which, in response to certain stimuli, is phosphorylated and degraded, leaving NF-κB to translocate into the nucleus (9). NF-κB is a central regulator of the transcriptional activation of a number of genes involved in apoptosis, differentiation, and growth; induction of these genes in intestinal epithelial cells by activated NF-κB profoundly influences mucosal inflammation, repair and inflammation-associated gastrointestinal cancers (10, 11). Recently, we have demonstrated a novel feedback regulation of PTEN through TNFα-mediated NF-κB activation (12). In agreement with our findings, Vasudevan et al (13) reported a suppressive effect of NF-κB activation on PTEN expression and the prevention of apoptosis. Given the roles of PTEN in antagonizing PI3K-mediated cell survival and tumorigenesis, these findings suggest that PTEN plays an important role in NF-κB function.
c-Jun N-terminal protein kinase (JNK) is a subfamily of the mitogen activated protein kinase (MAPK) superfamily (14). JNK has three isoforms (JNK1, 2 and 3). Among them, JNK1 and JNK2 are ubiquitously expressed while JNK3 is mainly expressed in neuronal tissues and in the heart (15). JNK was originally identified by its ability to specifically phosphorylate the transcription factor c-Jun on its N-terminal transactivation domain. The JNK pathway, together with NF-κB, play important roles in numerous physiological processes (16). For example, the balance between NF-κB and JNK activity controls dendritic cell survival (17). JNK inhibition results in NF-κB activation in multiple myeloma cell lines (18), and NF-κB activation induces MUC2 transcription whereas JNK activation inhibits this induction in human colon epithelial cells (19). Previously, we found that induction of intestinal cell differentiation is associated with increased JNK activity and c-Jun phosphorylation (20). In agreement with these findings, inhibition of JNK has been shown to attenuate intestinal cell differentiation (21).
The purpose of our present study was to determine the role of JNK in the regulation of PTEN expression. Here, we show that NaBT induces NF-κB inhibition and JNK activation, leading to PTEN expression in intestinal cells. Interestingly, our findings demonstrate a cross-talk mechanism between the NF-κB and JNK pathways on PTEN regulation. Moreover, NaBT induces p27kip1 expression through the JNK/NF-κB/ PTEN pathway. Our results identify PTEN as a downstream target of the JNK pathway. In addition, our findings suggest that the JNK/PTEN signaling pathway may regulate intestinal cell differentiation through the regulation of p27kip1 expression.
MATERIALS AND METHODS
Materials
NaBT and c-Jun protein were purchased from Sigma Chemical Company (St. Louis, MO). SP600125 was from Calbiochem (San Diego, CA). Mouse anti-human PTEN monoclonal antibody, rabbit anti-IκBα polyclonal antibody, rabbit anti-p50 polyclonal antibody, rabbit anti-p65 polyclonal antibody, rabbit anti-ERK1 polyclonal antibody, rabbit anti-JNK1 and rabbit anti-JNK2 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse anti-human JNK1/JNK2 antibody was from BD Pharmingen (San Diego, CA). Rabbit anti-β-actin antibody was from Sigma. JNK1, JNK2 and non-targeting control siRNA SMARTpool was purchased from Dharmacon, Inc. (Lafayette, CO). The SMARTpool for targeting JNK1 consisted of four pooled SMARTselection-designed siRNAs: JNK1 siRNA1 (5′-UCACAGUCCUGAAACGAUA-3′), JNK1 siRNA2 (5′-GAUUGGAGAUUCUACAUUC-3′), JNK1 siRNA3 (5′-AAGCUAAGCCGACCAUUU-3′), and JNK1 siRNA4 (5′-GAAGCAAGCGUGACAACAA-3′). The SMARTpool for targeting JNK2 consisted of four pooled SMARTselection-designed siRNAs: JNK2 siRNA1 (5′-GGAAAGAGCUAAUUUACAA-3′), JNK2 siRNA2 (5′-AAAGAGAGCUUAUCGUGAA-3′), JNK2 siRNA3 (5′-CAAGAUGUGUAUUUGGUUA-3′), and JNK2 siRNA4 (5′-GAUGAUAGGUUAGAAAUAG-3′). All siRNAs were synthesized with UU as a 3'-overhang on each strand. Adenovirus vectors encoding β-gal (AdCA-LacZ; control) and PTEN (AdCA-PTEN) were from Dr. Akira Horii (Tohoku University School of Medicine, Sendai, Japan). Adenovirus vector encoding hemagglutinin-tagged IκB-α superrepressor (Ad5IκB-AA) and its control vector (Ad5GFP) were gifts from Dr. Christian Jobin (University of North Carolina, Chapel Hill). The adenovirus vector encoding NF-κB p65 subunit (Ad5p65) and its control vector (Ad5GFP) were from Dr. Craig Logsdon (MD Anderson Cancer Center, Houston). [γ-32P] ATP (3,000 Ci/mmol) was from Amersham Pharmacia Biotech (Piscataway, NY). Total RNA was isolated using Ultraspec RNA (Biotecx Laboratories, Houston, TX). Polyvinylidene difluoride (PVDF) membranes for Western blots were from Millipore Corp. (Bedford, MA), and x-ray film was purchased from Eastman Kodak (Rochester, NY). The enhanced chemiluminescence (ECL) system for Western immunoblot analysis was from Amersham (Arlington Heights, IL). Tissue culture media and RT-PCR reagents were obtained from GIBCOBRL (Grand Island, NY). All other reagents were of molecular biology grade and purchased from Sigma.
Cell culture and treatments
The human colon cancer cell lines HT29 and Caco-2 and human embryonic kidney cell line HEK293 were purchased from ATCC (Rockville, MA). Rat intestinal epithelial (RIE-1) cells were obtained from Dr. Kenneth D. Brown (Cambridge Research Station, Babraham, Cambridge, UK). HT29 cells were maintained in McCoy's 5A supplemented with 10% FCS. Caco-2 cells were incubated in MEM supplemented with 15% FBS. HEK293 and RIE-1 cells were cultured in DMEM supplemented with 10% FCS. Cells were infected with adenovirus vectors at 10 plaque-forming units (pfu)/cell as described previously (22) and incubated for 24 h prior to initiating treatment. JNK inhibitor, SP600125, was initially dissolved in dimethyl sulfoxide (Me2SO), and effects compared with cells treated with Me2SO at the same final concentration.
Transient transfection, luciferase assays
The JNK1, JNK2 and non-targeting control siRNA SMARTpool duplexes were introduced into cells by electroporation (Gene Pulser, Bio-Rad) as we have described previously (23). siRNA SMARTpool, consisting of 4 siRNA duplexes, was designed using an algorithm comprised of 33 criteria and parameters that effectively eliminate non-functional siRNA (24). For luciferase assay, HT29 cells were seeded at 1 × 105 cells into 24-well plates in triplicate 24 h prior to transfection. Cells were then transiently transfected with 0.5 μg of NF-κB reporter plasmid and 0.05 μg of Renilla reporter pRL-null to normalize for variation in transfection efficiency, using LipofectAMINE Plus transfection agent following the manufacturer's recommended protocol. Cells were harvested for measurement of firefly and Renilla luciferase activities using the dual luciferase assay system. Firefly luciferase activity was determined by subtracting background signal and normalized to the Renilla activity.
JNK activity assay
Cell lysates were incubated with 1.5 μg of anti-JNK1 or anti-JNK2 antibody overnight at 4°C. Immune complexes were recovered with protein A-Sepharose beads, washed twice with lysis buffer, and once with kinase buffer. Pellets were resuspended in 40 μl of kinase buffer (25 mM Tris, pH 7.4; 2 mM dithiothreitol; 0.1 mM Na3VO4; 10 mM MgCl2; and 5 μCi of [γ-32P]ATP) containing c-Jun protein at 30°C for 30 min as previously described (20). The kinase reaction was terminated by addition of SDS sample loading buffer (50 mM Tris, pH 6.8; 100 mM DTT; 2% SDS; 0.1% bromophenol blue and 10% gylcerol). The samples were then heated to 95°C for 5 min and resolved by SDS-PAGE. The gels were dried and the phosphorylated protein visualized by autoradiography.
RNA isolation, reverse transcription-PCR (RT-PCR) and Northern blot analysis
RNA was isolated from cells using Ultraspec RNA reagent according to the manufacturer's protocol and as we have previously described (25). Total RNA (5 μg) was reverse transcribed with Maloney Murine Leukemia Virus (M-MLV) reverse transcriptase and the PCR was performed using the following two primers: 5'-ACAGGCTCCCAGACATGACA-3′, which spans nucleotides 1018-1037 of human PTEN mRNA sequence, and 5′-TCAGACTTTTGTAATTTGTGTATG-3′, which is complementary to nucleotides 2217-2240 of the mRNA sequence (25). The PCR product was sequenced for confirmation and used as a probe for Northern blot. RNA (30 μg) was run in 1.2% agarose/formaldehyde gels and transferred to supported nitrocellulose. Membranes were hybridized to a random-primed 32P-labeled PTEN cDNA probe. After hybridization with the GAPDH probe, a control for equality of RNA loading, membranes were washed again and signals detected by autoradiography.
Protein preparation, Western immunoblot and enzymatic assay
Western immunoblot analyses were performed as described previously (26). Briefly, cells were lysed with TNN buffer at 4°C for 30 min. Lysates were clarified by centrifugation (10,000 g for 30 min at 4°C) and protein concentrations determined using the method of Bradford (27). Total protein (100 μg) was resolved on a 10% polyacrylamide gel and transferred to PVDF membranes. Filters were incubated overnight at 4°C in blotting solution (Tris-buffer saline containing 5% nonfat dried milk and 0.1% Tween 20). Expression of PTEN, JNK1, JNK2, HA-tagged IκB-AA, p50, IκBα, ERK1, p65 and p27kip1 were detected with specific antibodies following blotting with a horseradish peroxidase-conjugated secondary antibody and visualized using ECL detection. Intestinal alkaline phosphatase (IAP) activity was detected by an IAP kit from Sigma as we have described previously (6).
Preparation of nuclear extracts and electrophoretic mobility shift assays (EMSAs)
The nuclear extracts were prepared from HT29 cells according to the procedure previously described (28) with minor modifications. Nuclear extracts (10 μg) were incubated with 40,000 cpm of 32P-labeled NF-κB consensus oligonucleotide (5′-AGTTGAGGGGACTTTCCCAGG-3′) and 2 μg of poly (dA·dT) in a buffer containing 8% glycerol, 100 mM NaCl, 5 mM MgCl2, 5 mM dithiothreitol, and 0.1 μg/ml phenylmethylsulfonyl fluoride in a final volume of 20 μl, for 15 min at room temperature. The complexes were fractionated on 6% native polyacrylamide gels run in 1x TBE buffer (89 mM Tris, 89 mM boric acid, and 2.0 mM EDTA), dried, and exposed to Kodak X-AR film at −70 °C. Competition binding experiments were performed by the addition of the nonradioactive oligonucleotide, in 100-fold molar excess, at the time of addition of the labeled probe.
DNA fragmentation assay
Cells were plated in 96-well plates 24 h before treatment. After treatment, DNA fragmentation was evaluated by examination of cytoplasmic histone-associated DNA fragments (mononucleosomes and oligonucleosomes) using a Cell Death Detection ELISAPlus kit (Roche Molecular Biochemicals, Indianapolis, IN) according to the manufacturer's instructions and as we have described previously (29).
Statistical analysis
Due to heterogeneous variability among treatment groups, the data in figure 4C was transformed using logarithm to the base 10 and analyzed using the two-way classification analysis of variance. The Fisher's least significant difference procedure was used for multiple comparisons with Bonferroni adjustment for the number of comparisons. Data was assessed at the 0.05 level of significance.
Fig. 4. Crosstalk between JNK and NF-κB activation.

A, B. HT29 cells were infected with control adenovirus encoding GFP or an adenovirus encoding the NF-κB superrepressor IκB-AA (A) or the p65 NF-κB subunit (B). After incubation for 24 h, cells were treated with or without NaBT for an additional 24 h. Whole cell protein was extracted for immunoprecipitation using anti-JNK1 or anti-JNK2 antibody. JNK1 or JNK2 activity was assessed by an in vitro kinase assay using GST-c-Jun protein as substrate. JNK1 or JNK2 expression levels were assessed by Western blotting. C. HT29 cells were transfected with a NF-κB-luciferase reporter plasmid. Twenty-four h after transfection, cells were pre-treated with the JNK inhibitor, SP600125, for 30 min followed by treatment with NaBT for an additional 24 h. Cells were harvested and luciferase activity assayed. D. HT29 cells were transfected with siRNA directed to JNK1 plus siRNA directed to JNK2 or control siRNA. Twenty-four h after siRNA transfection, cells were transfected with a NF-κB-luciferase reporter plasmid. Twenty-four h later, cells were harvested and luciferase activity was assayed. (Data represent mean ± SD; * = p < 0.05 vs. control; † = p < 0.05 vs. NaBT alone.). Results are representative of three independent experiments.
RESULTS
NaBT increases PTEN expression in HT29 cells
Treatment with NaBT induces intestinal cell differentiation, whereas overexpression of PTEN enhances this process (6). To better delineate upstream signaling pathways responsible for PTEN induction, we used the human colon cancer cell line HT29, which has wild type (ie, nonmutated) PTEN (30). HT29 is used extensively as a model of intestinal epithelial cell proliferation and differentiation (31). Cells were treated with NaBT (5 mM) for various times and protein was extracted. Western blotting demonstrated a NaBT-mediated increase in PTEN expression which was apparent by 8 h after treatment and continued for the 48 h time course (Fig. 1A). To examine whether the induction of PTEN protein was associated with an increase in mRNA expression, HT29 cells were treated with NaBT (5 mM) over a time course, and total cellular RNA was extracted for analysis of PTEN expression by Northern blot. As shown in Fig. 1B, NaBT increased PTEN mRNA levels compared with vehicle control, which was apparent by 4 h after treatment and continued for the 48 h time course. Together, these results identify induction of PTEN mRNA and protein levels with NaBT treatment. In spite of the increased mRNA and protein levels in NaBT-treated cells, a time-dependent decrease of PTEN protein levels was noted in control cells, which we speculate may be due to decreased serum concentration over the incubation period (32-34).
Fig. 1. NaBT increases PTEN expression.

A. HT29 cells were treated with NaBT for various times. Total protein was extracted and Western blot was performed for analysis of PTEN protein expression. B. HT29 cells were treated with NaBT for various times. RNA was isolated and analyzed by Northern blotting. C. RIE-1, Caco-2 and HEK-293 cells were treated with NaBT for 24 h. Cell lysates were fractionated by SDS-PAGE and blotted with anti-PTEN and anti-β-actin antibodies. PTEN signals from three separate experiments were quantitated densitometrically and expressed as fold-change with respect to β-actin for protein or GAPDH for mRNA.
To determine whether this induction of PTEN is limited to HT29 cells or occurs in other cells, the human colon cancer cell line, Caco-2, and normal rat intestinal epithelial cell line RIE-1 and human embryonic kidney cell line HEK293 were incubated in the presence or absence of NaBT for 24 h (Fig. 1C). Induction of PTEN protein expression was noted in all cells. These results confirm our findings in HT29 cells and, moreover, suggest a general regulation of PTEN expression by NaBT.
Inhibition of JNK attenuates PTEN induction by NaBT
Previously, we have shown that treatment with NaBT increased JNK activity and c-Jun phosphorylation which was associated with enterocyte-like differentiation in Caco-2 intestinal cells (6, 20). In agreement with our results, Orchel et al (21) have shown that inhibition of JNK attenuated NaBT-induced differentiation in HT29 cells. To investigate the possible regulatory effect of JNK on PTEN expression, HT29 cells were pretreated with a selective JNK inhibitor, SP600125 (35), followed by treatment with NaBT for 4, 8, or 24 h as early as 4 h after treatment with maximal induction at 24 h. As expected, NaBT increased PTEN protein expression (Fig. 2A) and mRNA expression (Fig. 2B); this induction was dramatically attenuated by pretreatment with SP600125. These results suggest that NaBT-induced PTEN expression requires JNK activation. Previously, we showed that NaBT increased JNK1 activity at 8 h and sustained for 72 h after treatment in Caco-2 cells. To determine if NaBT increases JNK1 and JNK2 activity, HT29 cells were treated with NaBT and then harvested for in vitro kinase assays using GST-c-Jun as substrate (Fig. 2C). Increased JNK activity was noted for both JNK1 and JNK2 over the time course (4 to 48 h).
Fig. 2. NaBT-induced PTEN expression requires JNK activation.

A. HT29 cells were pretreated with a specific JNK inhibitor, SP600125, for 30 min followed by treatment with NaBT for 4, 8 or 24 h. Total protein was extracted and Western blot was performed for analysis of PTEN protein expression. B. HT29 cells were pretreated with a specific JNK inhibitor, SP600125, for 30 min followed by treatment with NaBT for 4, 8 or 24 h. Total RNA was extracted and Northern blot was performed for analysis of PTEN mRNA expression. C. HT29 cells were treated with NaBT from 4 to 48 h. Whole cell protein was extracted for immunoprecipitation using anti-JNK1 or JNK2 antibody. JNK1 and JNK2 activity was assessed by an in vitro kinase assay using GST-c-Jun protein as substrate. D. HT29 cells were transfected with siRNA directed to JNK1 or JNK2 or control siRNA. Cells were harvested 12 or 24 h after transfection (left) or 24 h after transfection, cells were treated with NaBT for an additional 24 h (middle & right). Whole cell protein was extracted and Western blot was performed for analysis of PTEN protein expression. Knockdown of either JNK1 or JNK2 attenuated PTEN induction by NaBT. PTEN signals from three separate experiments were quantitated densitometrically and expressed as fold-change with respect to β-actin.
To further demonstrate the role of JNK in NaBT-mediated PTEN induction, we transfected cells with siRNA directed to either JNK1 or JNK2 to suppress JNK1 and JNK2 expression, respectively. First, the efficacy of JNK siRNA inhibition was analyzed. JNK1 and JNK2 expression was decreased ∼30% at 12 h after transfection with JNK1 or JNK2 siRNA, respectively, and a 65% decrease was noted 24 h after transfection (Fig. 2D, left). Transfection with siRNA directed to JNK1 or JNK2 did not affect expression of JNK2 or JNK1, respectively. Based on these results, we then treated cells with NaBT 24 h after transfection with siRNA. Suppression of JNK attenuated NaBT-mediated PTEN induction in HT29 cells as compared to cells transfected with non-targeting control siRNA (Fig. 2D, middle & right panels). Immunoblotting analysis confirmed that expression of JNK1 or JNK2 was significantly inhibited by JNK1 or JNK2 siRNA for 48 h after transfection. Increased JNK2 expression was noted with NaBT treatment. Transfection of cells with non-targeting control siRNA had no effect on PTEN expression (data not shown). Together, our data indicate that JNK1 and JNK2 are important regulators of PTEN expression.
NF-κB regulation of NaBT-mediated PTEN expression in HT29 cells
We have previously demonstrated an important role for NF-κB in PTEN regulation (12). We next determined the effect of NaBT on NF-κB activity. Cells were treated with a single dose of NaBT (5 mM) and analyzed over a time course; nuclear protein was extracted and analyzed by EMSA. Treatment with NaBT increased PTEN mRNA expression from 2 to 48 h after treatment; however, results from EMSA showed no change in NF-κB binding activity from 2-8 h after NaBT treatment (Fig. 3A, left panel) and inhibition of NF-κB binding activity after 24 h (Fig. 3A, right panel). The specificity of DNA binding was confirmed by competition assays using unlabeled probe in molar excess. To further assess the mechanisms involved in NaBT-mediated inhibition of NF-κB binding activity, HT29 cells were treated with NaBT for 24 or 48 h; cytosolic and nuclear protein were extracted and p65, p50 and IκBα expression was detected by Western blotting. As shown in Fig. 3B, the decreased level of p50 expression was noted in the cytosolic and nuclear fractions with no change in either of p65 or IκBα expression. These data indicate that NaBT inhibition of NF-κB binding activity may be due to decreased levels of nuclear p50.
Fig. 3. NaBT regulates NF-κB binding activity and overexpression of the p65 NF-κB subunit blocks NaBT-induced PTEN expression.

A. HT29 cell were treated with NaBT over a time course. Nuclear protein was extracted and EMSA was performed to assess NF-κB binding activity. B. HT29 cell were treated with NaBT for 24 or 48 h. Cytosolic and nuclear protein were extracted and p65, p50 and IκBα levels were detected by Western blotting. β-actin and ERK1 was reprobed as loading control for the cytosol and nuclear fractions, respectively. C. HT29 cells were infected with an adenovirus encoding the NF-κB superrepressor, IκB-AA, or control adenovirus encoding GFP. After incubation for 24 h, cells were treated with NaBT for an additional 24 h. Total protein was isolated and Western blot was performed for analysis of PTEN protein expression and HA-tagged IκB-AA. D. HT29 cells were infected with an adenovirus encoding the p65 NF-κB subunit or control adenovirus encoding GFP. After incubation for 24 h, cells were treated with NaBT for an additional 24 h. Total protein was isolated and Western blot was performed for analysis of PTEN and p65 NF-κB subunit protein expression. E. HT29 cells were infected with adenovirus constructs and treated with NaBT as described above. Nuclear protein was extracted and EMSA was performed; increased NF-κB binding activity by overexpression of the p65 NF-κB subunit was confirmed. PTEN signals from three separate experiments were quantitated densitometrically and expressed as fold-change with respect to β-actin.
To assess the involvement of NF-κB in NaBT-mediated PTEN regulation, an adenovirus expressing the superrepressor of IκB-α (IκB-AA) was used (12). HT29 cells were infected with either the adenovirus encoding HA-tagged IκB-AA or the adenoviral control vector encoding GFP. Infection was carried out for 1 h followed by the replacement of fresh medium. After incubation for 24 h, cells were treated with NaBT for an additional 24 h. As expected, IκB-AA overexpression alone increased PTEN expression and enhanced NaBT-mediated PTEN induction (Fig. 3C, top panel); IκB-AA expression was confirmed by Western blot (Fig. 3C, bottom panel). The inhibition of NF-κB activation was confirmed by EMSA (data not shown). Conversely, we analyzed the effect of the expression of the p65 subunit of NF-κB after infection with the recombinant adenovirus Ad5p65. We found that overexpressing p65 protein decreased PTEN expression and attenuated NaBT-induced PTEN expression (Fig. 3D, top panel); p65 overexpression was confirmed and β-actin expression was constant indicating equal protein loading (Fig. 3C, middle and bottom panels, respectively). Consistently, NaBT-mediated NF-κB inhibition was reversed by p65 overexpression as shown in Fig. 3E. These results provide evidence for the involvement of NF-κB in the regulation of NaBT-mediated PTEN induction.
Cross-talk between JNK and NF-κB in HT29 cells
Next, we assessed whether NF-κB affects JNK activity in HT29 cells. HT29 cells were infected with adenovirus encoding IκB-AA (Fig. 4A), adenovirus encoding p65 (Fig. 4B) or the adenoviral control vector encoding GFP. Activity of JNK1 or JNK2 was examined using GST-c-Jun protein as the substrate. NF-κB inhibition by overexpression of IκB-AA increased JNK1 and JNK2 activity in HT29 cells as shown in Fig. 4A. Consistent with these results, NF-κB activation by overexpression of p65 in HT29 cells decreased basal JNK1 activity and attenuated NaBT-mediated JNK activation (Fig. 4B). JNK1 and JNK2 expression levels were not altered with the transfections as noted in the Western blot (Fig. 4B, bottom panel).
As JNK inhibition results in NF-κB activation in multiple myeloma cell lines (18), we next determined the effect of JNK inhibition on NF-κB activation by transfection of the NF-κB-luciferase plasmid, which contains four tandem copies of the NF-κB consensus sequence. Twenty-four h after transfection, HT29 cells were pretreated with or without SP600125 (10 μM) for 30 min followed by combination treatment with NaBT (5 mM) for an additional 24 h. Treatment with NaBT decreased NF-κB transactivation compared with control (Fig. 4C). Moreover, treatment with the JNK inhibitor, SP600125, significantly increased NF-κB reporter activity but only partially attenuated the NaBT-mediated NF-κB repression. NF-κB activation by JNK inhibition was further confirmed by the increased NF-κB reporter activity in cells transfected with JNK1 and JNK2 siRNA as compared with control siRNA transfection (Fig. 4D). Together, these results identify a negative regulation between JNK and NF-κB signaling which plays an important role in the regulation of basal PTEN expression in HT29 cells.
Regulation of p27kip1 expression through the JNK/PTEN pathway in NaBT-treated HT29 cells
PTEN increases p27kip1 expression in various cells (4, 5, 36). We have shown that NaBT increases p27kip1 expression in intestinal cells (37). We next determined whether elevated PTEN expression contributed to the increase of p27kip1 expression. First, we assessed the expression of p27kip1 in HT29 cells overexpressing PTEN. Cells were infected with adenovirus vectors encoding β-gal (AdCA-LacZ; control) or PTEN (AdCA-PTEN). Twenty-four h after infection, cells were lysed and p27kip1 expression was analyzed by Western blotting. As shown in Fig. 5A, overexpression of PTEN increased p27kip1 expression. To further confirm the role of PTEN in p27kip1 regulation, HT29 cells were transfected with non-targeting control siRNA or siRNA directed to PTEN. Twenty-four h after transfection, cells were treated with NaBT for an additional 24 h, and PTEN and p27kip1 expression determined by Western blotting (Fig. 5B). Treatment with NaBT increased PTEN and p27kip1 expression. In agreement with the induction of p27kip1 by overexpression of PTEN, transfection with PTEN siRNA completely blocked PTEN expression and attenuated p27kip1 induction by NaBT, suggesting a role for PTEN in p27kip1 regulation in intestinal cells. Knockdown of PTEN did not completely block NaBT-induced p27kip1 expression suggesting that a PTEN-independent pathway(s) is involved in NaBT-induced p27kip1 expression. In preliminary findings, we have shown that NaBT treatment increases expression of the forkhead transcription factor, FOXO1; knockdown of FOXO1 attenuated NaBT-induced p27kip1 expression suggesting that NaBT regulation of p27kip1 is through PTEN as well as FOXO1 (Bose, et al, unpublished data).
Fig. 5. Regulation of p27kip1 expression by the NF-κB/JNK/PTEN pathway.

A. HT29 cells were infected with control adenovirus encoding β-gal or an adenovirus encoding PTEN. After incubation for 24 h, whole cell protein was extracted and Western blot was performed for analysis of PTEN and p27kip1 protein expression. B. HT29 cells were transfected with PTEN siRNA or control siRNA duplexes. Twenty-four h after transfection, cells were treated with NaBT for an additional 24 h. Whole cell protein was extracted and Western blot was performed for analysis of PTEN and p27kip1 protein expression. C. HT29 cells were infected with an adenovirus encoding the p65 NF-κB subunit or control adenovirus encoding GFP. After incubation for 24 h, cells were treated with NaBT for an additional 24 h. Total protein was isolated and Western blot was performed for analysis of PTEN protein expression. D. HT29 cells were transfected with JNK1 or JNK2 siRNA. Results are representative of three independent experiments.
We have shown that NF-κB activation decreased PTEN expression (12). To determine the effect of NF-κB activation on p27kip1 expression, HT29 cells were infected with adenovirus vectors encoding GFP (Ad5GFP; control) or p65 (Ad5p65). Twenty-four h after infection, cells were treated with NaBT for an additional 24 h, and p27kip1 expression was analyzed by Western blot. In agreement with the inhibition of PTEN expression, overexpression of p65 decreased basal expression of p27kip1 and attenuated NaBT-induced p27kip1 expression (Fig. 5C).
Given that inhibition of JNK attenuated PTEN expression, we next determined the effect of JNK knockdown on p27kip1 expression. HT29 cells were transfected with non-targeting control siRNA or siRNA specifically directed to JNK1 or JNK2. Twenty-four h after transfection, cells were treated with NaBT for an additional 24 h, and JNK1, JNK2 and p27kip1 expression was assayed by Western blot (Fig. 5D). Treatment with NaBT increased p27kip1 expression. Transfection with either JNK1 or JNK2 siRNA attenuated NaBT-mediated p27kip1 induction; the knockdown of JNK1 or JNK2 was confirmed after treatment with JNK1 or JNK2 siRNA, respectively. Taken together, our findings demonstrate that NaBT induced PTEN through JNK activation and NF-κB inhibition which was associated with the induction of p27kip1.
PTEN function in NaBT induced differentiation and apoptosis
Since PTEN is a potential tumor suppressor for colorectal cancer (1) and inhibition of PI3K enhances NaBT-mediated intestinal cell differentiation (6), we analyzed the role of PTEN in NaBT-induced differentiation in HT29 cells. Treatment with NaBT increased IAP activity (Fig. 6A), a differentiation marker for intestinal cells, (6) and this increase was attenuated by transfection of siRNA directed to PTEN, suggesting a role for PTEN in NaBT-mediated differentiation.
Fig. 6. Inhibition of PTEN attenuates NaBT-mediated intestinal cell differentiation and cell death.
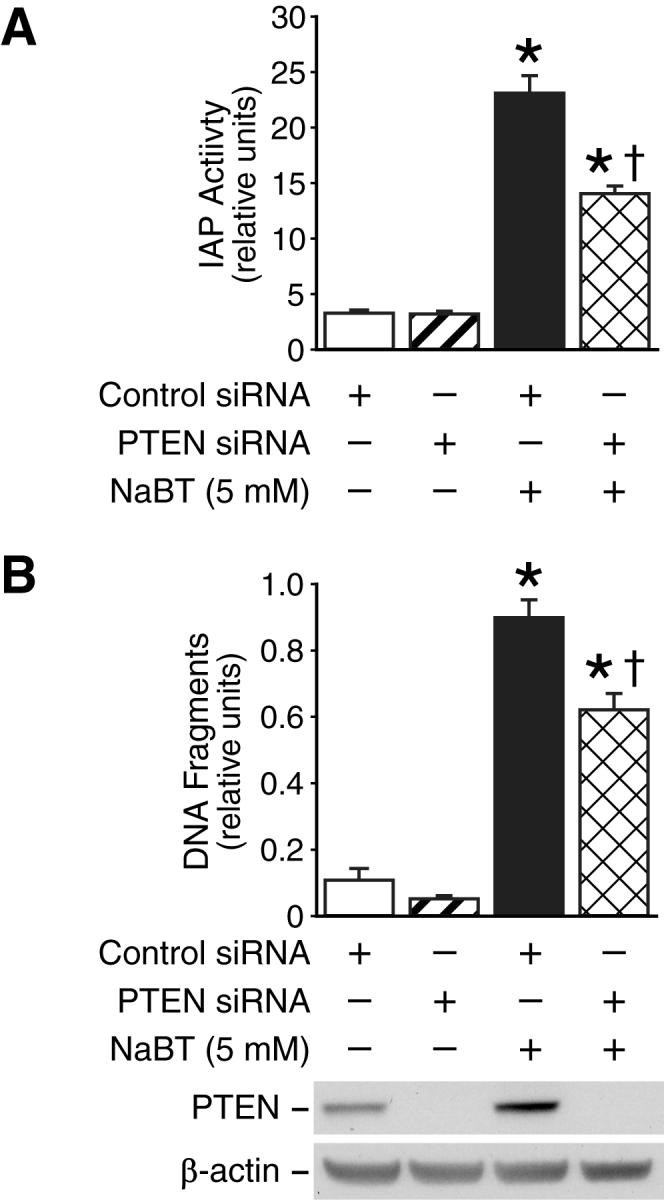
A. HT29 cells were transfected with PTEN siRNA or control siRNA duplexes. Twenty-four h after transfection, cells were treated with NaBT for an additional 40 h. Whole cell proteins were extracted and IAP enzymatic activities in the HT29 cells with or without doxycycline were analyzed. B. After transfection with PTEN siRNA or control siRNA, HT29 cells were treated with NaBT (5 mM) for 24 h. Apoptosis was analyzed by determination of DNA fragmentation by an ELISA method as described in “Materials and Methods.” (Data represent mean ± SD; * = p < 0.05 vs. control siRNA; † = p < 0.05 vs. NaBT plus control siRNA.). Results are representative of three independent experiments.
Previously, we have found that inhibition of PI3K enhances NaBT-mediated apoptosis in human colon cancer cells (29). Next, we accessed the effect of PTEN knockdown on NaBT-induced HT29 cell death. NaBT induced obvious cell death as shown by increased DNA fragmentation (Fig. 6B) and this increase was attenuated by knockdown of PTEN using PTEN siRNA transfection. Collectively, our results suggest that PTEN plays a role in NaBT-mediated intestinal cell death and differentiation.
DISCUSSION
In previous studies, we demonstrated enhanced intestinal cell differentiation by PTEN (6) and defined a novel feedback regulation of PTEN by NF-κB activation (12). In the present study, we further delineated the signaling pathways involved in this regulation. We show that NaBT, which induces intestinal cell differentiation, increased PTEN expression. This NaBT-mediated PTEN induction was a consequence of NF-κB inhibition and JNK activation. Treatment of the HT29 colon cancer cell line with NaBT activates JNK (20) and inhibits NF-κB activation. Consistent with these findings, inhibition of JNK activity and activation of NF-κB attenuated NaBT induction of PTEN, whereas inhibition of NF-κB enhanced this induction. In addition, negative regulation between NF-κB and JNK signaling on PTEN expression was demonstrated. Inhibition of NF-κB increased JNK activity, while activation of NF-κB inhibited JNK activity. In contrast, inhibition of JNK increased NF-κB activation. Finally, we show that NaBT increased p27kip1 expression, a downstream target of PTEN by regulation of NF-κB and JNK. Importantly, our findings identify regulation of PTEN by JNK and NF-κB in intestinal cells.
Recently, we have shown that treatment with NaBT induced intestinal cell differentiation which was associated with the activation of JNK (20). Conversely, inhibition of JNK attenuated NaBT-induced intestinal cell differentiation (21). JNK activity has been associated with intestinal cell death. For instance, TNFα-induced apoptosis in IEC-6 intestinal cells was accompanied by the activation of JNK, and inhibition of JNK protected against TNFα-induced apoptosis (38). Inhibition of JNK attenuated hydrogen peroxide-induced cell death in HT29 cells (39). Consistently, our previous results showed that overexpression of PTEN enhanced intestinal cell differentiation (6). Overexpression of PTEN in colorectal cancer cells resulted in cell-cycle arrest and enhanced cell death through inhibition of PI3K (40). Our observation that the blockade of the JNK pathway by pharmacological (ie, SP600125) or genetic mechanisms (ie, transfection with JNK siRNA) attenuated NaBT induction of PTEN argues strongly for a functional role for JNK activation in PTEN induction in intestinal cells.
Although in some cells (eg, leukemia cells), HDAC inhibitor exposure has been associated with NF-κB activation (41), several reports have shown inactivation of NF-κB by NaBT in intestinal epithelial cells (42, 43). In agreement with these reports, we also showed an inhibitory role of NaBT in NF-κB activity in HT29 cells. Although NaBT has been shown to decrease p50 in nuclear but not in cytosolic fractions of HT29 cells 12 h after NaBT treatment (44), our results showed that NaBT reduced p50 in both nuclear and cytosolic fractions with NaBT treatment for 48 h. NaBT inhibits TNFα-induced NF-κB activation in part by preventing the complete degradation of IκBα by reducing proteasome activity (42); however, our results showed that NaBT alone did not affect IκBα expression in HT29 cells. As NF-κB inhibition resulted in the induction of PTEN expression, our results strongly suggest the involvement of NF-κB inhibition in NaBT-mediated PTEN induction.
Several studies have reported a crosstalk between the NF-κB and JNK pathways (45, 46). The NF-κB-mediated inhibition of JNK signaling is crucial for numerous physiological processes, such as the response of the liver to injury and the survival of cells during an inflammatory reaction, as well as for chronic inflammatory diseases and cancers (16). In contrast, it has been reported that inhibition of NF-κB leads to JNK activation and potentiates the lethality of certain apoptotic stimuli (eg, TNF-α) (47). The importance of this antagonistic crosstalk between NF-κB and JNK has also been documented recently in animal models (44). In agreement with these findings, we showed that inhibition of NF-κB or treatment with NaBT resulted in JNK activation. Activation of NF-κB decreased JNK activity, whereas inhibition of JNK increased NF-κB activity. Therefore, our results reveal a possible crosstalk mechanism by NF-κB and JNK on PTEN regulation in intestinal cells. Nevertheless, this mechanism may not contribute completely to NaBT-induced PTEN expression since our results show that JNK inhibition does not reverse NaBT-mediated NF-κB inhibition. Furthermore, NaBT increased JNK activity and, at the same time, did not affect NF-κB binding activity from 2 to 8 h. These results suggest that NF-κB and JNK are differentially involved in the regulation of NaBT-mediated PTEN expression. Our results demonstrate that JNK is involved at early time points and together with NF-κB at later stages in the regulation of NaBT-mediated expression.
NaBT induces intestinal cell differentiation associated with the induction of p27kip1 expression (37). The function of p27kip1 is required for intestinal cell differentiation (48). Overexpression of PTEN or inhibition of PI3K increased p27kip1 expression in various cells (4, 5). Considering that intestinal cell differentiation was enhanced by PTEN overexpression, we determined whether JNK activation increased p27kip1 and, if so, whether this effect was through JNK-dependent PTEN expression. We found that overexpression of PTEN or treatment with NaBT increased p27kip1 expression in HT29 cells; this induction was attenuated by transfection with PTEN siRNA, activation of NF-κB, or inhibition of JNK by transfection with JNK1 or JNK2 siRNA. Our findings demonstrate that NaBT increased p27kip1 expression through PTEN induction which was mediated by JNK activation and NF-κB inhibition. Previously, we showed that inhibition of PI3K or overexpression of PTEN increased TNF-related apoptosis inducing ligand (TRAIL) expression in intestinal cells (22); TRAIL treatment results in increased intestinal cell differentiation and p27kip1 expression (49). Recently, Yanase et al (50) showed that activation of JNK leads to TRAIL induction in Daudi B lymphoma cells. Our future studies will determine the regulation of TRAIL expression by the JNK/PTEN pathway and the possible relationship to intestinal cell differentiation.
In summary, our current study provides important insights regarding the signaling mechanisms regulating PTEN expression and function in intestinal cells. Our results identify PTEN as a downstream target of the JNK and NF-κB pathway. Given the importance of p27kip1 in intestinal cell differentiation, the JNK/NF-κB/PTEN signaling pathway may alter intestinal cell differentiation through the regulation of p27kip1 expression. Collectively, our results have important implications for normal intestinal homeostasis and colon carcinogenesis.
ACKNOWLEDGMENTS
The authors thank Christian Jobin (University of North Carolina, Chapel Hill) for the adenovirus Ad5GFP and Ad5IκB-AA, Craig Logsdon (MD Anderson Cancer Center, Houston, TX) for adenovirus Ad5p65, Akira Horii (Tohoku University School of Medicine, Sendai, Japan) for adenoviruses encoding β-gal (AdCA-LacZ) and PTEN (AdCA-PTEN). We also thank Karen Martin for manuscript preparation, Tatsuo Uchida for statistical analysis and Devin Leake (Dharmacon) for helpful advice with the siRNA experiments. This work was supported by grants RO1 DK48498, R01 CA104748, R37 AG10885 and PO1 DK35608 from the National Institutes of Health.
REFERENCES
- 1.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96:4240–5. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280:1614–7. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- 3.Sun H, Lesche R, Li DM, et al. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci U S A. 1999;96:6199–204. doi: 10.1073/pnas.96.11.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Motti ML, Califano D, Troncone G, et al. Complex regulation of the cyclin-dependent kinase inhibitor p27kip1 in thyroid cancer cells by the PI3K/AKT pathway: regulation of p27kip1 expression and localization. Am J Pathol. 2005;166:737–49. doi: 10.1016/S0002-9440(10)62295-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li DM, Sun H. PTEN/MMAC1/TEP1 suppresses the tumorigenicity and induces G1 cell cycle arrest in human glioblastoma cells. Proc Natl Acad Sci U S A. 1998;95:15406–11. doi: 10.1073/pnas.95.26.15406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Q, Wang X, Hernandez A, Kim S, Evers BM. Inhibition of the phosphatidylinositol 3-kinase pathway contributes to HT29 and Caco-2 intestinal cell differentiation. Gastroenterology. 2001;120:1381–92. doi: 10.1053/gast.2001.24044. [DOI] [PubMed] [Google Scholar]
- 7.Hague A, Manning AM, Hanlon KA, Huschtscha LI, Hart D, Paraskeva C. Sodium butyrate induces apoptosis in human colonic tumour cell lines in a p53-independent pathway: implications for the possible role of dietary fibre in the prevention of large-bowel cancer. Int J Cancer. 1993;55:498–505. doi: 10.1002/ijc.2910550329. [DOI] [PubMed] [Google Scholar]
- 8.Kim S, Domon-Dell C, Wang Q, et al. PTEN and TNF-alpha regulation of the intestinal-specific Cdx-2 homeobox gene through a PI3K, PKB/Akt, and NF-kappaB-dependent pathway. Gastroenterology. 2002;123:1163–78. doi: 10.1053/gast.2002.36043. [DOI] [PubMed] [Google Scholar]
- 9.Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 10.Jobin C, Sartor RB. The I kappa B/NF-kappa B system: a key determinant of mucosalinflammation and protection. Am J Physiol Cell Physiol. 2000;278:C451–62. doi: 10.1152/ajpcell.2000.278.3.C451. [DOI] [PubMed] [Google Scholar]
- 11.Greten FR, Eckmann L, Greten TF, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–96. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 12.Kim S, Domon-Dell C, Kang J, Chung DH, Freund JN, Evers BM. Down-regulation of the tumor suppressor PTEN by the tumor necrosis factor-alpha/nuclear factor-kappaB (NF-kappaB)-inducing kinase/NF-kappaB pathway is linked to a default IkappaB-alpha autoregulatory loop. J Biol Chem. 2004;279:4285–91. doi: 10.1074/jbc.M308383200. [DOI] [PubMed] [Google Scholar]
- 13.Vasudevan KM, Gurumurthy S, Rangnekar VM. Suppression of PTEN expression by NF-kappaB prevents apoptosis. Mol Cell Biol. 2004;24:1007–21. doi: 10.1128/MCB.24.3.1007-1021.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–48. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 15.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 16.Nakano H. Signaling crosstalk between NF-kappaB and JNK. Trends Immunol. 2004;25:402–5. doi: 10.1016/j.it.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 17.Kriehuber E, Bauer W, Charbonnier AS, et al. Balance between NF-kappaB and JNK/AP-1 activity controls dendritic cell life and death. Blood. 2005;106:175–83. doi: 10.1182/blood-2004-08-3072. [DOI] [PubMed] [Google Scholar]
- 18.Hideshima T, Hayashi T, Chauhan D, Akiyama M, Richardson P, Anderson K. Biologic sequelae of c-Jun NH(2)-terminal kinase (JNK) activation in multiple myeloma cell lines. Oncogene. 2003;22:8797–801. doi: 10.1038/sj.onc.1206919. [DOI] [PubMed] [Google Scholar]
- 19.Ahn DH, Crawley SC, Hokari R, et al. TNF-alpha activates MUC2 transcription via NF-kappaB but inhibits via JNK activation. Cell Physiol Biochem. 2005;15:29–40. doi: 10.1159/000083636. [DOI] [PubMed] [Google Scholar]
- 20.Ding Q, Wang Q, Evers BM. Alterations of MAPK activities associated with intestinal cell differentiation. Biochem Biophys Res Commun. 2001;284:282–8. doi: 10.1006/bbrc.2001.4969. [DOI] [PubMed] [Google Scholar]
- 21.Orchel A, Dzierzewicz Z, Parfiniewicz B, Weglarz L, Wilczok T. Butyrate-induced differentiation of colon cancer cells is PKC and JNK dependent. Dig Dis Sci. 2005;50:490–8. doi: 10.1007/s10620-005-2463-6. [DOI] [PubMed] [Google Scholar]
- 22.Wang Q, Wang X, Hernandez A, Hellmich MR, Gatalica Z, Evers BM. Regulation of TRAIL expression by the phosphatidylinositol 3-kinase/Akt/GSK-3 pathway in human colon cancer cells. J Biol Chem. 2002;277:36602–10. doi: 10.1074/jbc.M206306200. [DOI] [PubMed] [Google Scholar]
- 23.Wang Q, Zhou Y, Wang X, Evers BM. Glycogen synthase kinase-3 is a negative regulator of extracellular signal-regulated kinase. Oncogene. 2006;25:43–50. doi: 10.1038/sj.onc.1209004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational siRNA design for RNA interference. Nat Biotechnol. 2004;22:326–30. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- 25.Wu X, Senechal K, Neshat MS, Whang YE, Sawyers CL. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc Natl Acad Sci U S A. 1998;95:15587–91. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Q, Ding Q, Dong Z, Ehlers RA, Evers BM. Downregulation of mitogen-activated protein kinases in human colon cancers. Anticancer Res. 2000;20:75–83. [PubMed] [Google Scholar]
- 27.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 28.Wang Q, Wang X, Evers BM. Induction of cIAP-2 in human colon cancer cells through PKC delta/NF-kappa B. J Biol Chem. 2003;278:51091–9. doi: 10.1074/jbc.M306541200. [DOI] [PubMed] [Google Scholar]
- 29.Wang Q, Li N, Wang X, Kim MM, Evers BM. Augmentation of sodium butyrate-induced apoptosis by phosphatidylinositol 3′-kinase inhibition in the KM20 human colon cancer cell line. Clin Cancer Res. 2002;8:1940–7. [PubMed] [Google Scholar]
- 30.Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 31.Wilson CA, Browning JL. Death of HT29 adenocarcinoma cells induced by TNF family receptor activation is caspase-independent and displays features of both apoptosis and necrosis. Cell Death Differ. 2002;9:1321–33. doi: 10.1038/sj.cdd.4401107. [DOI] [PubMed] [Google Scholar]
- 32.Lorenz P, Ackermann K, Simoes-Wuest P, Pyerin W. Serum-stimulated cell cycle entry of fibroblasts requires undisturbed phosphorylation and non-phosphorylation interactions of the catalytic subunits of protein kinase CK2. FEBS Lett. 1999;448:283–8. doi: 10.1016/s0014-5793(99)00388-9. [DOI] [PubMed] [Google Scholar]
- 33.Yu M, Yeh J, Van Waes C. Protein kinase casein kinase 2 mediates inhibitor-kappaB kinase and aberrant nuclear factor-kappaB activation by serum factor(s) in head and neck squamous carcinoma cells. Cancer Res. 2006;66:6722–31. doi: 10.1158/0008-5472.CAN-05-3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. 2001;276:993–8. doi: 10.1074/jbc.M009134200. [DOI] [PubMed] [Google Scholar]
- 35.Bennett BL, Sasaki DT, Murray BW, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–6. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gottschalk AR, Basila D, Wong M, et al. p27Kip1 is required for PTEN-induced G1 growth arrest. Cancer Res. 2001;61:2105–11. [PubMed] [Google Scholar]
- 37.Litvak DA, Evers BM, Hwang KO, Hellmich MR, Ko TC, Townsend CM., Jr. Butyrate-induced differentiation of Caco-2 cells is associated with apoptosis and early induction of p21Waf1/Cip1 and p27Kip1. Surgery. 1998;124:161–9. discussion 9-70. [PubMed] [Google Scholar]
- 38.Bhattacharya S, Ray RM, Viar MJ, Johnson LR. Polyamines are required for activation of c-Jun NH2-terminal kinase and apoptosis in response to TNF-alpha in IEC-6 cells. Am J Physiol Gastrointest Liver Physiol. 2003;285:G980–91. doi: 10.1152/ajpgi.00206.2003. [DOI] [PubMed] [Google Scholar]
- 39.Salh BS, Martens J, Hundal RS, et al. PD98059 attenuates hydrogen peroxide-induced cell death through inhibition of Jun N-Terminal Kinase in HT29 cells. Mol Cell Biol Res Commun. 2000;4:158–65. doi: 10.1006/mcbr.2001.0271. [DOI] [PubMed] [Google Scholar]
- 40.Saito Y, Gopalan B, Mhashilkar AM, et al. Adenovirus-mediated PTEN treatment combined with caffeine produces a synergistic therapeutic effect in colorectal cancer cells. Cancer Gene Ther. 2003;10:803–13. doi: 10.1038/sj.cgt.7700644. [DOI] [PubMed] [Google Scholar]
- 41.Dai Y, Rahmani M, Dent P, Grant S. Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-kappaB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol Cell Biol. 2005;25:5429–44. doi: 10.1128/MCB.25.13.5429-5444.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yin L, Laevsky G, Giardina C. Butyrate suppression of colonocyte NF-kappa B activation and cellular proteasome activity. J Biol Chem. 2001;276:44641–6. doi: 10.1074/jbc.M105170200. [DOI] [PubMed] [Google Scholar]
- 43.Inan MS, Rasoulpour RJ, Yin L, Hubbard AK, Rosenberg DW, Giardina C. The luminal short-chain fatty acid butyrate modulates NF-kappaB activity in a human colonic epithelial cell line. Gastroenterology. 2000;118:724–34. doi: 10.1016/s0016-5085(00)70142-9. [DOI] [PubMed] [Google Scholar]
- 44.Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. IKKbeta is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFalpha. Immunity. 2003;19:725–37. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- 45.Tang G, Minemoto Y, Dibling B, et al. Inhibition of JNK activation through NF-kappaB target genes. Nature. 2001;414:313–7. doi: 10.1038/35104568. [DOI] [PubMed] [Google Scholar]
- 46.Javelaud D, Besancon F. NF-kappa B activation results in rapid inactivation of JNK in TNF alpha-treated Ewing sarcoma cells: a mechanism for the anti-apoptotic effect of NF-kappa B. Oncogene. 2001;20:4365–72. doi: 10.1038/sj.onc.1204570. [DOI] [PubMed] [Google Scholar]
- 47.Reuther-Madrid JY, Kashatus D, Chen S, et al. The p65/RelA subunit of NF-kappaB suppresses the sustained, antiapoptotic activity of Jun kinase induced by tumor necrosis factor. Mol Cell Biol. 2002;22:8175–83. doi: 10.1128/MCB.22.23.8175-8183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deschenes C, Vezina A, Beaulieu JF, Rivard N. Role of p27(Kip1) in human intestinal cell differentiation. Gastroenterology. 2001;120:423–38. doi: 10.1053/gast.2001.21199. [DOI] [PubMed] [Google Scholar]
- 49.Rimondi E, Secchiero P, Quaroni A, Zerbinati C, Capitani S, Zauli G. Involvement of TRAIL/TRAIL-receptors in human intestinal cell differentiation. J Cell Physiol. 2005;206:647–54. doi: 10.1002/jcp.20512. [DOI] [PubMed] [Google Scholar]
- 50.Yanase N, Hata K, Shimo K, Hayashida M, Evers BM, Mizuguchi J. Requirement of c-Jun NH2-terminal kinase activation in interferon-alpha-induced apoptosis through upregulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) in Daudi B lymphoma cells. Exp Cell Res. 2005;310:10–21. doi: 10.1016/j.yexcr.2005.06.021. [DOI] [PubMed] [Google Scholar]