

Abstract
SH2-domain-containing inositol 5′-phosphatase-1 (SHIP) deficiency significantly increases the number of hematopoietic stem cells (HSCs) present in the bone marrow (BM). However, the reconstitution capacity of these HSCs is severely impaired, suggesting that SHIP expression might be an intrinsic requirement for HSC function. To further examine this question, we developed a model in which SHIP expression is ablated in HSCs while they are resident in a SHIP-competent milieu. In this setting, we find that long-term repopulation by SHIP-deficient HSCs is not compromised. Moreover, SHIP-deficient HSCs from this model repopulate at levels comparable with wild-type HSCs upon serial transfer. However, when HSCs from mice with systemic ablation of SHIP are transplanted, they are functionally compromised for repopulation. These findings demonstrate that SHIP is not an intrinsic requirement for HSC function, but rather that SHIP is required for the BM milieu to support functionally competent HSCs. Consistent with these findings, cells that comprise the BM niche express SHIP and SHIP deficiency profoundly alters their function.
Introduction
Because of their capacity for self-renewal and multilineage potential, hematopoietic stem cells (HSCs) support blood cell production throughout life. HSCs are thought to reside in specialized endosteal and vascular niches in the bone marrow (BM) that support quiescence, self-renewal, and/or differentiation.1–4 Other HSC fates include mobilization to extramedullary sites and return to the BM.5 In addition, apoptosis, which regulates the homeostasis of differentiated tissues,6 contributes to control of the HSC compartment size, where approximately 1% to 2% of HSCs are apoptotic during steady-state hematopoiesis.7 Thus, a careful balance between these fates must be maintained to sustain a sufficient number of HSCs to maintain blood cell production throughout life. We and others have found that stem cell–restricted SH2-domain–containing inositol 5′-phosphatase-1 (s-SHIP)8,9 and SH2-domain–containing inositol 5′-phosphatase-1 (SHIP)7,10 play roles in the biology of pluripotent and adult tissue stem cells, respectively. Previously, we showed that SHIP limits the HSC compartment size in vivo. However, after transplantation, these HSCs exhibit compromised BM homing and long-term multilineage repopulation.7
SHIP or s-SHIP expression in adult physiology has been documented in HSCs,8,9 most blood cell lineages,11–14 embryonic fibroblasts,7 and endothelial cells.15 Through its enzymatic domain, SHIP can hydrolyze the PI3K products, PI (3,4,5)P3 and I (1,3,4,5)P4, and thus is capable of regulating the activity of multiple PI3K effector pathways, including the distal kinases Akt, Btk, MAP/ERK, PLC-γ, and intracellular Ca2+. The context in which SHIP mediates these activities is determined by its expression but also by its inducible recruitment to various receptor-associated signaling complexes mediated by tyrosine phosphorylation of receptor motifs or NPXY sequences present in SHIP.11,13,16,17 Because of the nearly ubiquitous expression of SHIP in differentiated hematopoietic cell types and its induced recruitment to a wide variety of receptor-associated signaling complexes, SHIP has the potential to regulate a wide variety of cellular functions in adult physiology.
SHIP is expressed throughout the hematopoietic system and in endothelial cells. From biochemical studies alone, it is difficult to determine whether SHIP might play a critical role in normal mammalian physiology. Toward this end, genetic analysis of SHIP mutant mice has revealed a pivotal role for SHIP in a variety of differentiated cell types. SHIP plays a role in the control of the NK receptor repertoire and cytolytic function,18,19 B-cell development, and antibody production,20,21 the myeloid response to bacterial mitogens,22 development of marginal zone macrophages,23 osteoclast function,24 lymph node recruitment of dendritic cells,25 mast cell degranulation,26 and the homeostasis and function of myeloid immunoregulatory cells.25,27
Previously, we found that the poor repopulating capacity of SHIP-deficient HSCs may be a result of their reduced BM homing capacity. We speculated that SHIP-deficient HSCs might have normal repopulating function if they were not required to home to their BM niche. To test this hypothesis, we developed an in situ SHIP deletion model, where SHIP expression is ablated after HSCs are resident in a SHIP-competent BM microenvironment. Analysis of the hematopoietic compartment in this model showed that SHIP deficiency does not diminish the capacity of HSCs to self-renew or mediate multilineage repopulation after serial transfer to secondary hosts. Conversely, when SHIP expression is ablated systemically in MxCreSHIPflox/flox mice, we find that SHIP deficiency compromises the repopulation potential of HSCs in a similar fashion to that in germline SHIP-deficient mice. This demonstrates an unanticipated requirement for SHIP expression in the function of the HSC BM niche. Consistent with these results, we show that SHIP is expressed in cells that comprise the BM niche, and SHIP deficiency significantly alters their production of chemokines and their ability to support HSC cycling.
Methods
Mice
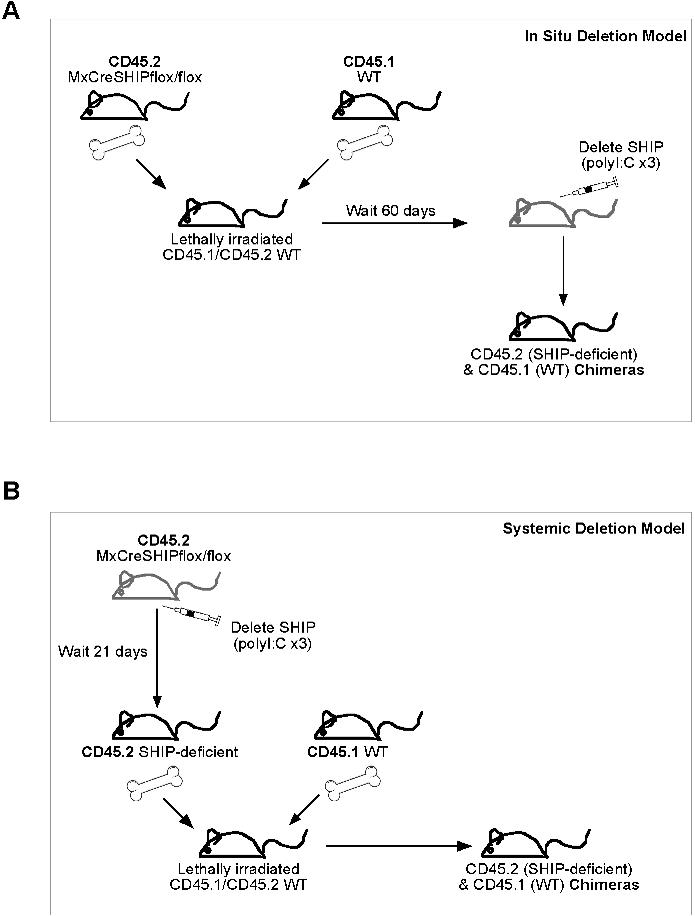
Production of SHIP−/− and MxCreSHIPflox/flox mice has been previously described.18,27 Briefly, SHIP−/− mice were generated by deletion of the promoter and first exon of SHIP via a Cre-LoxP strategy and backcrossed to a C57BL6/J background. MxCreSHIPflox/flox mice were generated using SHIPflox/flox and MxCre mice (The Jackson Laboratory, Bar Harbor, ME). MxCreSHIPflox/flox:WT-Ly5.1 and SHIPflox/flox:WT-Ly5.1 BM chimeras were created by cotransplanting 5 × 105 whole bone marrow (WBM) cells from MxCreSHIPflox/flox (CD45.2+) or SHIPflox/flox (CD45.2+) plus 5 × 105 cells from WT-Ly5.1 (CD45.1+) mice into lethally irradiated CD45.1+45.2+ recipients. All animal experiments were conducted with approval of the University of South Florida Institutional Animal Care and Use Committee.
Cell isolation
BM cells were flushed from femurs and tibias, red blood cell lysed, centrifuged, and resuspended in staining media composed of Dulbecco phosphate-buffered saline, 3% fetal bovine serum (FBS), and 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid. For transplantation, WBM was simply flushed, washed, and resuspended in sterile phosphate-buffered saline (PBS). Spleens were manually crushed then treated like BM cells. Peripheral blood (PB) was collected in Microtainers with K2EDTA (BD Biosciences, San Jose, CA). Red blood cell lysis was performed twice to obtain PB mononuclear cells (PBMCs).
Conditional deletion of SHIP
MxCreSHIPflox/flox (CD45.2+) mice and MxCreSHIPflox/flox:WT-Ly5.1 BM chimeras were conditionally ablated for SHIP expression through intraperitoneal injection of polyinosinic-polycytidylic acid (poly I:C; Sigma-Aldrich, St Louis, MO). Mice were injected 3 times with 625 μg poly I:C on days 1, 4, and 7.
Flow cytometry
All antibodies were from BD Biosciences, eBioscience (San Diego, CA), or Invitrogen (Carlsbad, CA). PBMCs, splenocytes, or BM cells were treated with CD16/CD32 mouse Fc block (2.4G2) on ice for 20 minutes and then stained with a panel of antibodies. Global reconstitution was assessed by staining with: CD45.1-PE (A20) and CD45.2-FITC (104) or CD45.1-PE (A20), and CD45.2-PercpCy5.5 (104). Multilineage reconstitution was assessed by staining with: CD45.1-PE (A20), CD45.2-PercpCy5.5 (104), B220–Alexa Fluor 700 (RA3-6B2), CD3-PECy7 (145-2C11), Mac1-APCCy7 (M1/70), and NK1.1-APC (PK136). Myeloid, erythroid, and megakaryocytic reconstitution was assessed by staining with: CD45.1-PE (A20), CD45.2-PercpCy5.5 (104), Mac1-APCCy7 (M1/70), Gr1-FITC (RB6-8C5), or CD41-FITC (MWReg30) and Ter119-APC (TER-119). Phenotypic HSCs were assessed by staining with: CD45.1-purified (A20) conjugated to Pac Orange using zenon technology, CD45.2-PerCPCy5.5 (104), Sca1-PECy7 (D7), c-Kit-APC-AF750 (2B8), CD48-APC (HM48-1), CXCR4-PE (2B11/CXCR4), and a lineage (Lin) panel on FITC; CD2 (RM2-5), CD3ϵ (145-2C11), CD4 (GK1.5), CD5 (53-7.3), CD8α (53-6.7), B220 (RA3-6B2), Gr-1 (RB6-8C5), Mac-1 (M1/70), NK1.1 (PK136), and Ter119 (TER-119) or Sca1-FITC (E13-161.7), c-Kit-PECy7 (2B8), CD48-APC (HM48-1), CXCR4-biotin (2B11/CXCR4), and a Lin panel on PE; CD2 (RM2-5), CD3ϵ (145-2C11), CD4 (GK1.5), CD5 (53-7.3), CD8α (53-6.7), B220 (RA3-6B2), Gr-1 (RB6-8C5), Mac-1 (M1/70), NK1.1 (PK136), and Ter119 (TER-119) followed by a secondary stain of streptavidin-AF700. After 20 minutes of staining on ice, the cells were washed and resuspended in staining media containing 75 ng/mL 4′,6-diamidino-2-phenylindole dihydrochloride (Sigma-Aldrich) for dead cell exclusion. All flow cytometry acquisition was done on a fluorescence-activated cell sorter (FACS) LSRII, a FACSAria, or a FACSVantage (all from BD Biosciences). Data were analyzed using FlowJo software (TreeStar, Ashland, OR).
In situ SHIP deletion BM transplantations
A total of 5 × 105 MxCreSHIPflox/flox (CD45.2+) or SHIPflox/flox (CD45.2+) WBM cells were cotransplanted (intravenously) with 5 × 105 WBM competitor cells from WT-Ly5.1 (CD45.1+) mice into lethally irradiated CD45.1+45.2+ recipients. Recipients received a split dose of 11 Gy at least 2 hours before transplantation. After 60 days, recipient mice were treated with the poly I:C series.
Secondary BM transplantations from in situ SHIP-deleted mice
Five months after poly I:C treatment of MxCreSHIPflox/flox:WT-Ly5.1 and SHIPflox/flox:WT-Ly5.1 BM chimeras, mice were killed and 106 WBM cells were transplanted (intravenously) into lethally irradiated CD45.1+45.2+ recipients.
Systemic SHIP deletion BM transplantations
MxCreSHIPflox/flox (CD45.2+) mice and SHIPflox/flox (CD45.2+) mice were pretreated with the poly I:C series. SHIP deletion was confirmed by Western blot of PBMCs. A total of 5 × 105 WBM cells from MxCreSHIPflox/flox (CD45.2+) or SHIPflox/flox (CD45.2+) mice were cotransplanted (intravenously) with 5 × 105 WBM competitors from WT-Ly5.1 (CD45.1+) mice into lethally irradiated CD45.1+45.2+ recipients.
Western blot detection of SHIP and β-actin expression
Cell lysates were prepared from cultured or sorted cells using modified radio immunoprecipitation assay (RIPA) lysis buffer (Millipore, Billerica, MA). Lysate supernatants were resolved on a 4% to 12% Bis-Tris gel and transferred to a Hybond-ECL nitrocellulose membrane (GE Healthcare, Little Chalfont, United Kingdom). For Odyssey technology, the membrane was blocked with Odyssey blocking buffer (LICOR Biosciences, Lincoln, NE) and probed with 1 to 5 μg/mL P1C1 (Santa Cruz Biotechnology, Santa Cruz, CA) and then anti–mouse IR-700 at 1:8000 (Invitrogen). Probed blots were scanned using an Odyssey infrared imager. For chemiluminescence, the membrane was blocked with 5% nonfat milk in PBS with 0.1% Tween-20 (PBS-T) and probed with 1 μg/mL P1C1 or 1 μg/mL actin:C-11 (Santa Cruz Biotechnology) followed by anti–mouse IgG-horseradish peroxidase (HRP) or anti–goat IgG-HRP at 1:1000 (eBioscience). Protein was detected using SuperSignal West Femto Substrate (Thermo Electron, Waltham, MA).
Calculation of repopulation units
Repopulating units (RU) were calculated as per Harrison et al28:
where C = 105 competitor cells. Because we used 5 × 105 competitor cells, we used this equation:
Enzyme-linked immunosorbent assay detection of cytokines
Serum was isolated from PB and stored at −20°C or sent for analysis by a service from Charles River Laboratories (Wilmington, MA), where enzyme-linked immunosorbent assay for multiple cytokines was performed. BM plasma was assayed for cytokines by flushing cleaned femurs with 500 μL PBS. The PBS was then centrifuged at 300g for 5 minutes at 4°C. The supernatant was removed to a new tube and stored at −20°C. Cell-culture supernatants were harvested at regular intervals and stored at −20°C. Stored supernatants were then assayed for stromal cell derived factor-1 (SDF-1) using the Quantikine Immunoassay (R&D Systems, Minneapolis, MN).
Isolation and culture of stromal cells
Bone marrow was flushed from intact femurs and tibias into tissue-culture flasks containing α-modified minimum essential medium (MEM) with nucleotides (α-MEM).29 The α-MEM was supplemented with 15% FBS, 50 U penicillin-G/mL, 50 μg streptomycin/mL, 0.3 μg amphotericin-B/mL, and 50 μg ascorbate/mL. After culturing cells for 3 days undisturbed, media was changed and nonadherent cells were removed.
Isolation and culture of osteoblasts
Cleaned and flushed femurs and tibias were scraped using a scalpel to remove remaining soft tissue.30 The bones were cut into 1- to 2-mm2 pieces and incubated with shaking in a solution of 2 mg/mL collagenase-II in Dulbecco modified Eagle medium (DMEM) at 37°C for 2 hours. Complete culture medium (cCM) was added to stop the collagenase action. The cCM consisted of DMEM (pH 7.4) supplemented with 2.2 g NaHCO3/L, 10% FBS, 50 U penicillin-G/mL, 50 μg streptomycin/mL, 50 μg gentamycin/mL, 1.25 μg amphotericin-B/mL, and 100 μg ascorbate/mL. The bone pieces were washed 3 times with cCM and cultured in 25-cm2 tissue-culture flasks.
SHIP immunoprecipitation and Western blot for phosphotyrosine
Stromal cell cultures were lysed in modified RIPA buffer. SHIP was immunoprecipitated overnight with 0.2 μg P1C1 and protein A/G-agarose beads from precleared lysate supernatants. The immunoprecipitated protein was blotted and the membrane blocked using the protocol described in “Western blotting detection of SHIP.” It was then probed using an antiphosphotyrosine, HRP-conjugated monoclonal antibody (4G10, Santa Cruz Biotechnology) diluted 1:1000 with 3% milk in PBS-T. Phosphorylated tyrosine was detected using SuperSignal West Femto Substrate diluted 1:5 with Pierce ECL Western Blotting Substrate (Thermo Electron).
ELF97 alkaline phosphatase assay
Alkaline phosphatase (ALP) activity was measured using an ELF97 assay (Invitrogen) modified for flow cytometry.31 Osteoblasts were fixed overnight in 70% EtOH at 4°C. Cells were then washed and transferred into FACS tubes. After preincubation with developing buffer, phosphate substrate was added to the cells. The cells were incubated at room temperature for 1 to 30 minutes before adding a phosphatase inhibitor, levamisole (10 mM), to stop the reaction.
Coculture proliferation assay
Hematopoietic progenitor cells (HPCs)31 were isolated from wild-type (WT) mice by lineage depletion of BM using an antibody cocktail on PE: CD3ϵ (145-2C11), CD4 (GK1.5), CD5 (53-7.3), B220 (RA3-6B2), Gr-1 (RB6-8C5), Mac-1 (M1/70), and Ter119 (TER-119) followed by a magnetic enrichment using anti-PE microbeads (Miltenyi Biotec, Auburn, CA). HPCs were loaded with 5 μM carboxyfluorescein succinimidyl ester (CFSE). WT CFSE+ HPCs were cocultured with confluent SHIP−/− or WT stromal cells for 36 hours. Proliferation was assayed based on loss of CFSE in Lin−cKit+Flk2− cells by staining with: a lineage-PE panel (CD3, CD4, CD5, B220, Ter119, Mac1, Gr1), cKit-APC (Miltenyi Biotec), Flk2-PE (A2F10.1), and 4′,6-diamidino-2-phenylindole dihydrochloride.
Histology
Femurs were embedded in O.C.T. compound (Tissue-Tek, Torrance, CA), snap-frozen in liquid nitrogen, and stored at −80°C. Cryostat sections (10 μm) were fixed, preincubated in blocking buffer (PBS, 3% BSA, anti-CD32/CD16, 2.4G2), and stained with antibodies. Sections were embedded in fluorescent mounting medium (Dako Denmark, Glostrup, Denmark) and analyzed by confocal microscopy (Leica TCS-SL; Leica Microsystems, Deerfield, IL) using a 63× objective. Image processing was performed with Leica confocal software.
Statistical analysis
All statistical analyses were performed using Prism (GraphPad, San Diego, CA).
Results
HSC rendered SHIP-deficient in a SHIP-competent niche retain multilineage repopulation
To determine whether the repopulation defect observed for SHIP−/− HSCs is the result of an inability to home efficiently to the BM HSC niche, we developed an in situ SHIP deletion model where SHIP expression is ablated after achieving HSC engraftment, bypassing the requirement of SHIP for BM homing. In this model, we cotransplant an equal dose of SHIP-competent BM cells in which Cre-mediated deletion of SHIP is not possible. This model enables quantitation of competitive repopulating activity for HSC-rendered SHIP-deficient in situ. A similar approach was previously used to study the role of Rho family proteins in HSC function.32 In our in situ SHIP deletion model, lethally irradiated CD45.1+45.2+ recipients are transplanted with equal numbers of CD45.2+CD45.1− MxCreSHIPflox/flox and CD45.1+CD45.2− SHIP+/+ WBM cells. In these chimeras, equivalent repopulation from both the MxCreSHIPflox/flox (CD45.2+CD45.1−) and SHIP+/+ (CD45.1+CD45.2−) BM donors was observed at 60 days after transplantation, indicating comparable levels of HSC engraftment from each donor (Figure 1A,B). We then administered 3 intraperitoneal poly I:C injections to induce SHIP deletion in the MxCreSHIPflox/flox portion of these BM chimeras. Two months after deletion, CD45.2+CD45.1− and CD45.1+CD45.2− cells from the PB were FACS sorted to assess SHIP expression by Western blot analysis. SHIP deletion is highly efficient in poly I:C–treated MxCreSHIPflox/flox cells as indicated by the near-complete absence of SHIP expression in the CD45.2+CD45.1− cells derived from MxCreSHIPflox/flox HSC (data not shown). For further confirmation, we harvested spleens from all chimeras on their termination 5 months after poly I:C treatment. Western blot analysis of FACS-sorted CD45.2+CD45.1− cells (Figure 1C) confirmed nearly complete ablation of SHIP expression in the hematopoietic compartment derived from MxCreSHIPflox/flox HSC present in these BM chimeras.
Figure 1.

In situ deletion of SHIP does not compromise the capacity of HSCs to mediate long-term multilineage repopulation. (A) Contour plots for CD45.1 versus CD45.2 staining illustrating equal repopulation in PB at 60 days after transplantation before poly I:C treatment in 2 representative MxCreSHIPflox/flox (CD45.2):WT-Ly5.1 (CD45.1) BM chimeras. (B) Percentage of donor repopulation in the PB by MxCreSHIPflox/flox (CD45.2) and WT-Ly5.1 (CD45.1) HSCs in MxCreSHIPflox/flox (CD45.2):WT-Ly5.1 (CD45.1) BM chimeras at 60 days after transplantation before poly I:C treatment. (C) Representative SHIP Western blot from single-positive CD45.1+CD45.2−- or CD45.2+CD45.1−-sorted cells isolated from splenocytes from MxCreSHIPflox/flox (CD45.2):WT-Ly5.1 (CD45.1) BM chimeras 5 months after poly I:C treatment. Single-positive splenocytes were sorted and lysates were probed for SHIP expression. (D) Percentage of global repopulation in the PB by MxCreSHIPflox/flox (CD45.2) and WT-Ly5.1 (CD45.1) HSC in MxCreSHIPflox/flox (CD45.2):WT-Ly5.1 (CD45.1) BM chimeras at the indicated times after poly I:C treatment (n ≥ 5). At monthly intervals, the level of donor reconstitution was assessed in PB. (E) RU activity by MxCreSHIPflox/flox (CD45.2) and WT-Ly5.1 (CD45.1) HSCs in the MxCreSHIPflox/flox (CD45.2):WT-Ly5.1 (CD45.1) BM chimeras 5 months after poly I:C treatment (n ≥ 5). (F) Contour plots for CD45.1 versus CD45.2 staining that illustrate multilineage repopulation in PB 20 weeks after poly I:C treatment in a representative MxCreSHIPflox/flox (CD45.2):WT-Ly5.1 (CD45.1) BM chimera. (G) Percentage repopulation of the indicated lymphoid and myeloid cell lineages by MxCreSHIPflox/flox (CD45.2) and WT-Ly5.1 (CD45.1) HSC in MxCreSHIPflox/flox (CD45.2):WT-Ly5.1 (CD45.1) BM chimeras 5 months after poly I:C treatment (n ≥ 5). At death, the level of donor reconstitution was assessed in BM. Significance was established using the unpaired Student t test (* P < .05). Errors shown represent the SEM; ■, cells derived from MxCreSHIPflox/flox BM;  , cells derived from WT-Ly5.1 BM.
, cells derived from WT-Ly5.1 BM.
At monthly intervals, we monitored the degree of multilineage repopulation by CD45.2+CD45.1− (SHIP-deficient) versus CD45.1+CD45.2− (SHIP-competent) cells in these chimeras. We found that SHIP-deleted HSCs retain the ability to efficiently perform long-term, multilineage repopulation at levels comparable with the competing SHIP-competent HSCs present in these chimeras. We observed no significant difference in global hematopoietic repopulation (Figure 1D) or RU activity (Figure 1E) between in situ SHIP-deleted and WT HSCs up to 5 months after ablation of SHIP expression. In addition, repopulation of both the lymphoid and myeloid arms of hematopoiesis was observed out to 5 months after induction of SHIP deficiency (Figure 1F). Thus, when HSCs are established in situ and then rendered SHIP-deficient, this does not significantly compromise their potential for long-term, multilineage repopulation. However, we did observe a small, but significant, reduction in T lymphocyte reconstitution in the CD45.2+CD45.1− SHIP-deficient compartment despite normal repopulation of the B and NK lymphoid lineages (Figure 1G). Consistent with this finding, a reduction in peripheral T-cell numbers is observed in germline SHIP−/− mice.33 Because decreased T-cell production is observed only in the SHIP-deficient portion of these chimeras and not in the other lymphoid lineages, we conclude that SHIP is required for the efficient development of T lymphocytes, and this requirement is intrinsic to the T-cell lineage.
SHIP-deficient HSCs derived from a SHIP-competent niche have normal self-renewal capacity
Analysis of the aforementioned chimeras demonstrated long-term, multilineage repopulation from the SHIP-deficient HSC compartment is intact when HSCs are resident in a SHIP-competent niche. This finding suggested that self-renewal capacity might also be intact in SHIP-deficient HSCs. To compare the self-renewal capacity of SHIP-deficient and SHIP-competent HSCs in these chimeras, we performed serial BM transfers from the initial chimeras to secondary recipients. Whole BM was harvested from the in situ deleted chimeras at 5 months after SHIP deletion and was then transplanted into lethally irradiated CD45.1+CD45.2+ secondary hosts. Before these serial transfers, we first compared the proportion of SHIP-deficient (CD45.2+CD45.1−) to SHIP-competent (CD45.1+CD45.2−) cells in the HSC compartment by multiparameter flow cytometric analysis of CD45.1 versus CD45.2 staining on Kit+Lin−Sca1+CD48− (KLSCD48) cells. This analysis revealed a statistically comparable level of contribution to the KLSCD48 HSC compartment by the SHIP-deficient and SHIP-competent HSCs within the MxCreSHIPflox/flox:WT-Ly5.1 BM chimeras (Figure 2A). SHIP-deficient HSCs in these MxCreSHIPflox/flox:WT-Ly5.1 BM chimeras were also found to represent a statistically comparable proportion of the KLSCD48 compartment compared with SHIPflox/flox HSC in the SHIPflox/flox:WT-Ly5.1 BM control chimeras analyzed in parallel (Figure 2B). We then monitored global repopulation in the secondary hosts for a period of 4 months and found that SHIP-deficient HSCs from MxCreSHIPflox/flox:WT-Ly5.1 BM chimeras competed effectively in donor blood cell repopulation compared with the WT-Ly5.1 HSC present in the same BM inoculum (Figure 2C), demonstrating that SHIP-deficient HSCs can effectively home and engraft on transplantation when they are derived from a SHIP-competent HSC niche. Thus, SHIP expression is not an intrinsic requirement for HSC homing to BM and self-renewal.
Figure 2.

In situ deletion of SHIP does not compromise homing and secondary repopulating capacity of HSCs. (A) Flow cytometric quantitation of the relative contribution to KLSCD48 HSC by MxCreSHIPflox/flox (CD45.2) or WT-Ly5.1 (CD45.1) cells in the BM of MxCreSHIPflox/flox (CD45.2):WT-Ly5.1 (CD45.1) chimeras 5 months after poly I:C treatment (n ≥ 3). BM cells were stained with markers for the Lineage−/lowc-Kit+Sca1+CD48− (KLSCD48) phenotype.4 (B) Flow cytometric quantitation of the relative contribution to KLSCD48 HSCs by MxCreSHIPflox/flox (CD45.2) cells in the BM of MxCreSHIPflox/flox (CD45.2):WT-Ly5.1 (CD45.1) chimeras versus SHIPflox/flox (CD45.2) cells in SHIPflox/flox (CD45.2):WT-Ly5.1 (CD45.1) control chimeras. Both sets of chimeras were analyzed 5 months after poly I:C treatment (n ≥ 3). (C) Percentage global repopulation of PB at the indicated times by MxCreSHIPflox/flox (CD45.2) and WT-Ly5.1 (CD45.1) HSCs after serial transplantation of 106 WBM cells from MxCreSHIPflox/flox (CD45.2):WT-Ly5.1 (CD45.1) chimeras 5 months after poly I:C treatment (n ≥ 3). Significance was established using the unpaired Student t test. Errors shown represent the SEM; ■, cells derived from MxCreSHIPflox/flox BM (Cre+); , cells derived from WT-Ly5.1 BM (WT) or SHIPflox/flox (Cre−).
Systemic induction of SHIP deficiency in adult physiology is detrimental to the repopulating capacity of BM HSCs
SHIP is expressed in embryonic, fetal, and adult tissues, including mesenchymal, endothelial, and hematopoietic lineages.8,9,11,12 Cells from all 3 of these tissues participate directly or indirectly in the HSC BM niche, and thus their development may be impaired or altered during ontogeny in mice with germline SHIP deficiency. Therefore, the HSC disruptions we observe could be the result of developmental alterations of the niche by SHIP deficiency. Alternatively, SHIP could be required in adult physiology for the normal function of the niche. To test the latter possibility, we induced systemic SHIP deficiency in adult mice and then asked whether this also impairs HSC function. Induction of SHIP deficiency in the previous chimeras showed no intrinsic requirement of SHIP for BM homing, repopulation, or self-renewal by HSC and thus, in the setting of induced, but systemic SHIP deficiency we are testing whether SHIP deficiency impairs the HSC niche in BM. For these studies, BM was harvested from MxCreSHIPflox/flox mice that had undergone systemic SHIP-ablation by 3 poly I:C injections (Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article). Induction of SHIP deficiency was confirmed by Western blot analysis of PBMCs (data not shown). Before transplantation, we observed that cells of the KLSCD48 HSC phenotype were significantly expanded in mice with induced SHIP deficiency relative to SHIPflox/flox controls that underwent an equivalent regimen of poly I:C injections (Figure 3A). Furthermore, the absolute number of KLSCD48 cells in mice with induced SHIP deficiency was significantly expanded compared with SHIPflox/flox controls (data not shown). This expansion seems to be restricted to more primitive KLSCD48 and KLS progenitor cells and was not found in cKit+Lin− cells (data not shown). Because we observed no significant expansion of the SHIP-deficient KLSCD48 cells in chimeras with a SHIP-competent niche (Figure 1), we conclude then that SHIP expression is required by the adult BM niche to limit the size of the KLSCD48 HSC compartment. To analyze the repopulating ability of BM HSCs in mice with systemic induction of SHIP deficiency, we cotransplanted equal numbers of WBM cells from SHIP-deleted (CD45.2+CD45.1−) and SHIP-competent (CD45.1+CD45.2−) donors into lethally irradiated WT (CD45.2+CD45.1+) recipients. As before, monthly PB monitoring was used to assess engraftment. Using both global repopulation (Figure 3B) and RU (Figure 3C) as measures of engraftment, the HSC from donors with induced SHIP deficiency did not compete effectively against HSC from SHIP-competent donors. Furthermore, multilineage reconstitution appears to be significantly compromised for all lymphoid lineages in the PB (Figure 3D,E) and in myeloerythroid lineages in the BM (Figure 3F). However, despite significantly decreased myelopoiesis, megakaryopoiesis, and erythropoiesis in the BM by HSCs from mice with induced SHIP deficiency (Figure 3F), we observed statistically comparable myeloid reconstitution in the PB and spleen from these HSC (Figure 3E; and data not shown). This sustained peripheral myelopoiesis by the SHIP-deficient BM graft that occurs despite significantly compromised central myelopoiesis is probably the result of robust extramedullary myelopoiesis that we and others observe in the spleens of SHIP−/− mice33 (A.L.H., W.G.K., unpublished data, December 2007).
Figure 3.

Systemic induction of SHIP deficiency compromises the capacity of HSCs to mediate long-term multilineage repopulation. (A) Flow cytometric quantitation of KLSCD48 HSCs in the BM of MxCreSHIPflox/flox (CD45.2) mice and SHIPflox/flox (CD45.2) controls 21 days after poly I:C treatment (n ≥ 3). (B) Percentage of global repopulation in PB at the indicated times after transplantation by WBM cells from SHIP-ablated MxCreSHIPflox/flox (CD45.2) donors and WT-Ly5.1 (CD45.1) control donors (n ≥ 9). (C) RU activity from SHIP-ablated MxCreSHIPflox/flox (CD45.2) donors and WT-Ly5.1 (CD45.1) control donors (n ≥ 9). (D) Representative dual-color contour plots that illustrate the level of PB reconstitution at 24 weeks after transplantation from SHIP-ablated MxCreSHIPflox/flox (CD45.2) BM donors and WT-Ly5.1 (CD45.1) BM competitor donors. (E) Percentage PB repopulation for the indicated lymphoid or myeloid lineage by WBM cells in a competitive transplantation of SHIP-ablated MxCreSHIPflox/flox (CD45.2) and WT-Ly5.1 (CD45.1) BM competitor cells (n ≥ 9). Before death, the level of donor reconstitution was assessed in PB. (F) Percentage BM repopulation for the indicated myeloid (Mac1), granulocytic (Gr1), megakaryocytic (CD41), or erythroid (Ter119) lineage by WBM cells in a competitive transplantation of SHIP-ablated MxCreSHIPflox/flox (CD45.2) and WT-Ly5.1 (CD45.1) BM competitor cells (n ≥ 9). At death, the level of donor reconstitution was assessed in BM. Significance was established using the unpaired Student t test (*P < .05; **P < .005; ***P < .001). Errors shown represent the SEM; ■, cells derived from MxCreSHIPflox/flox BM; , cells derived from SHIPflox/flox (A) or WT-Ly5.1 BM (B,C,E,F).
A SHIP-deficient BM microenvironment expands phenotypic HSCs and reduces surface expression of CXCR4
Expansion of the phenotypic HSC compartment and functional impairment of HSCs in mice after the induction of systemic SHIP deficiency described herein was reminiscent of our previous findings for HSCs in germline SHIP−/− mice. To examine whether CXCR4 surface expression on HSCs might also be altered by the SHIP status of the BM niche, we assessed CXCR4 surface expression on in situ SHIP-deleted HSCs where the niche remains SHIP-competent and in HSCs from mice after induction of systemic SHIP deficiency where the BM microenvironment is also rendered SHIP-deficient. In the in situ SHIP-deleted model described in Figure 1, we found that the SHIP-deficient KLSCD48 cells do not show significant reductions in CXCR4 surface expression compared with SHIP-competent KLSCD48 cells present in the same mice (Figure 4A,B). However, when we analyzed the surface expression of CXCR4 on KLSCD48 cells in mice with induced systemic SHIP deficiency, we found that the CXCR4 surface expression is significantly reduced relative to SHIPflox/flox controls that received an equivalent regimen of poly I:C (Figure 4C,D). As shown in Figure 3, KLSCD48 cells are also significantly expanded in mice when systemic SHIP deficiency is induced. Thus, expansion of the phenotypic HSC compartment and down-modulation of CXCR4 are triggered only by induced, systemic SHIP deficiency, demonstrating regulation of these HSC properties by the BM niche in adult physiology.
Figure 4.

Systemic, but not in situ, deletion of SHIP impairs CXCR4 surface expression on KLSCD48 HSCs. (A) A representative histogram of CXCR4 surface expression on KLSCD48 HSC cells derived from MxCreSHIPflox/flox (CD45.2) or WT-Ly5.1 (CD45.1) donors in the BM of MxCreSHIPflox/flox (CD45.2):WT-Ly5.1 (CD45.1) chimeras 5 months after poly I:C treatment. (B) Median fluorescence intensity (MFI) of CXCR4 staining on KLSCD48 HSCs derived from MxCreSHIPflox/flox (CD45.2) or WT-Ly5.1 (CD45.1) donors in the BM of MxCreSHIPflox/flox (CD45.2):WT-Ly5.1 (CD45.1) chimeras 5 months after poly I:C treatment. (C) Representative histograms of CXCR4 surface expression on KLSCD48 HSC in the BM of MxCreSHIPflox/flox (CD45.2) mice or SHIPflox/flox (CD45.2) controls 21 days after poly I:C treatment. (D) MFI of CXCR4 staining on KLSCD48 HSC in the BM of MxCreSHIPflox/flox (CD45.2) or SHIPflox/flox (CD45.2) controls 21 days after poly I:C treatment. Significance was established using the unpaired Student t test (*P < .05). ■ represent cells derived from MxCreSHIPflox/flox BM (Cre+); , cells derived from SHIPflox/flox (Cre−) or WT-Ly5.1 BM (WT).
Production of soluble factors that influence HSC behavior are altered in SHIP-deficient mice
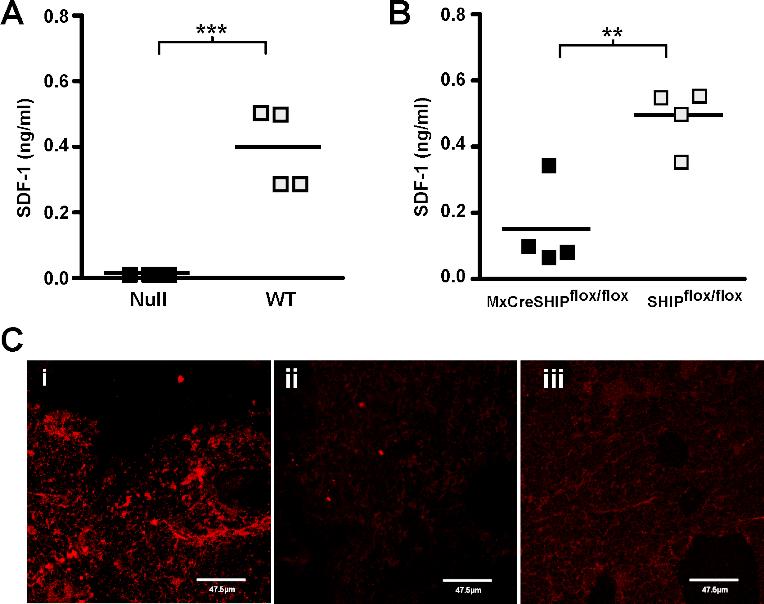
Although the BM niche can influence the behavior of HSCs by elaborating cell bound ligands for HSC receptors, it also mediates effects through the production of soluble factors that influence HSC proliferation, survival, mobilization, and BM homing. Consistent with our proposed disruption of niche function by SHIP deficiency, SHIP−/− mice exhibit significantly increased plasma levels of several growth and survival factors for HSCs (thrombopoietin [TPO], granulocyte colony-stimulating factor [G-CSF], and interleukin-6 [IL-6]) (Table 1). The increase in these factors and others may contribute to the increased number of HSCs present in both the medullary and extramedullary compartments of SHIP−/− mice. We also observed increased concentrations of soluble factors that promote HSC mobilization, including G-CSF, IL-5, matrix metalloproteinase-9, and soluble vascular cell adhesion molecule-1 (Table 1). In addition, homing and retention of HSC by the SHIP-deficient BM niche may be compromised by reduced production of SDF-1/CXCL12. We find significantly reduced SDF-1/CXCL12 in both the blood and BM plasma of SHIP-deficient mice (Figure 5). SDF-1/CXCL12 is produced by both the osteoblastic and vascular niche in BM.3,34 Indeed, decreased SDF-1 production in the SHIP-deficient BM niche could be triggered by increased G-CSF production, as G-CSF is known to reduce SDF-1 expression in BM niche cells.35 Taken together, these findings provide evidence for several significant perturbations in the SHIP-deficient BM niche.
Table 1.
Disrupted cytokine distribution in peripheral blood of SHIP-deficient mice
| Cytokine | SHIP−/− | WT | P |
|---|---|---|---|
| TPO (ng/mL) | 13.36 ± 0.3636 (n = 11) | 11.79 ± 0.2143 (n = 14) | < .001 |
| G-CSF (pg/mL) | 749.1 ± 39.13 (n = 4) | 161.9 ± 24.62 (n = 4) | < .001 |
| IL-6 (pg/mL) | 39.82 ± 6.201 (n = 11) | 13.67 ± 0.5205 (n = 13) | < .001 |
| IL-5 (ng/mL) | 0.2147 ± 0.0235 (n = 11) | 0.1511 ± 0.01634 (n = 14) | .031 |
| MMP-9 (ng/mL) | 476.3 ± 60.59 (n = 11) | 175.6 ± 23.3 (n = 14) | < .001 |
| VCAM-1 (ng/mL) | 2771 ± 82.06 (n = 11) | 1862 ± 70.14 (n = 14) | < .001 |
Values are mean plus or minus SEM. Various factors were measured in serum obtained from PB by ELISA. The data are pooled from multiple assays derived from at least 4 different plasma samples for each genotype. Significance was established using the unpaired Student t test.
Figure 5.

SHIP deficiency impairs in vivo production of SDF-1/CXCL12. (A) Scatter plots indicating the mean and range for the concentration of SDF-1/CXCL12 in the blood plasma of SHIP−/− and WT littermates. (B) Scatter plots indicating the mean and range for the concentration of SDF-1/CXCL12 in the BM plasma of SHIP−/− and WT littermates. The data are pooled from multiple assays derived from a minimum of 7 different samples for each genotype. Significance was established using the unpaired Student t test (**P < .005; ***P < .001). ■ represent SHIP−/− samples; , WT samples.
A role for SHIP in BM microenvironment signaling and function
Our transplantation studies indicated that SHIP deficiency disrupts the BM niche that supports normal HSC function and BM homing. SHIP is expressed primarily in hematopoietic tissues in both humans and mice; however, direct analysis of SHIP expression in cells that constitute BM niche cell types was not directly tested.11–14,36 We sought more direct evidence of a role for SHIP in the BM microenvironment by assessing whether it is expressed in cells that constitute key nonhematopoietic cell types in the BM niche. Thus, we prepared osteoblast (OB) and stromal cell cultures from SHIP−/− and WT BM. There were no substantive differences in morphology of stromal cells between the 2 genotypes. However, in OB cultures, we consistently observe readily apparent differences, with SHIP−/− OBs exhibiting elongated and nonrandomly oriented protrusions (Figure S2: day 11) and organized growth along axes (Figure S2: days 13 and 23). We readily detect SHIP protein expression in WT stromal (Figure 6A) and OB cultures (Figure 6B). In addition, we detect tyrosine phosphorylation of SHIP in WT BM stromal cells, indicating that SHIP participates in signaling pathways active in cells of the BM niche (Figure 6C). Consistent with our detection of SHIP expression in BM niche cell cultures, we confirmed that SHIP protein expression is also readily detected in stromal cultures derived from the BM of SHIPflox/flox mice but not MxCreSHIPflox/flox mice treated with poly I:C (Figure 6D). These results indicate that SHIP is expressed and participates in signaling in BM niche cells and thus is capable of influencing their function.
Figure 6.

SHIP is expressed and phosphorylated in BM niche cells. (A) Representative SHIP Western blot from stromal cells cultured from the BM of SHIP−/− and WT littermates. (B) SHIP Western blot from osteoblast cells cultured from the BM of SHIP−/− and WT littermates. (C) pTyr Western blot on a SHIP immunoprecipitation of stromal cells cultured from the BM of SHIP−/− and WT littermates. (D) SHIP Western blot from stromal cells cultured from the BM of MxCreSHIPflox/flox and SHIPflox/flox littermates. Each lane on the Western blots represents a culture lysate derived from an independent mouse.
To further assess a functional role for SHIP in the BM microenvironment, we assayed SHIP−/− and WT OBs for ALP activity. SHIP-deficient OBs exhibit significantly less ALP activity compared with WT OBs (Figure 7A,B), indicating that they are immature as ALP activity is closely correlated with maturation in the OB lineage.37–40 As a further test of SHIP-deficient BM niche function, we examined the cell cycle kinetics of WT hematopoietic stem/progenitor cells (HS/PCs) placed on BM stromal layers prepared from either SHIP−/− or WT mice. When we place1 million HS/PCs in coculture with confluent SHIP−/− stromal layers and measure proliferation by loss of CFSE, we find a clear defect in the ability of SHIP−/− BM stroma to support normal proliferation of WT HS/PCs (Figure 7C,D). These results demonstrate a significant and qualitative change in BM stromal cells that lack SHIP expression such that they are unable to support normal proliferation by SHIP-competent HS/PCs. In addition, we analyzed the supernatants of multiple independent SHIP-deficient and WT stromal cultures for SDF-1 production and found profoundly compromised SDF-1 production in all SHIP-deficient cultures analyzed compared with WT cultures analyzed in parallel (Figure S3). Accordingly, immunohistology on femur sections from SHIP−/− mice showed little to no SDF-1 production in the BM (Figure S3). When taken together, these results demonstrate that SHIP is required for multiple functions of BM niche cells, including production of factors that mediate BM recruitment, HSC retention, or HSC proliferation.
Figure 7.

SHIP deficiency alters the function of BM niche cells. (A) ALP-positive cells from cultures of SHIP−/− and WT osteoblasts showing altered phosphatase kinetics at 1, 15, and 30 minutes. (B) Representative FACS plots illustrating gating strategy for ALP-positive cells based on ALP-negative BM controls and showing ALP kinetics at increasing time points. (C) Frequency of Lin−cKit+Flk2− (KFL) stem/progenitor cells among Lin-depleted BM cells (HPCs) cultured alone (□), or with SHIP−/− (■) or WT () stromal cells in each cell division after 36 hours. (D) Representative histograms showing decreased proliferation of CFSE+ KFL cells cultured with SHIP−/− stromal cells (top) compared with those cultured with WT stromal cells (bottom). Significance was established using the unpaired Student t test (*P < .05; **P < .005; ***P < .001). ■ represent SHIP−/− osteoblasts (A); , WT osteoblasts (A); □, WT HPCs alone (C); ■, HPCs cultured with SHIP−/− stroma (C); , HPCs cultured with WT stroma (C).
Discussion
In this study, we show that induction of SHIP deficiency in the adult compromises HSC function only when SHIP deficiency is systemic, indicating a requirement for SHIP expression by the HSC niche. Indeed, when adult HSCs residing in a SHIP-competent niche are rendered SHIP-deficient, they completely retain their capacity for multilineage repopulation and self-renewal. Thus, SHIP expression is not an intrinsic requirement for control of HSC homeostasis or function in the BM but is required for normal function by the BM niche. Consistent with this, we find that SHIP is expressed and tyrosine phosphorylated in niche cells and plays an intrinsic role in their production of chemokines and their function. Furthermore, we find that, after induction of systemic SHIP deficiency in the adult, the SHIP-deficient BM microenvironment promotes expansion of cells of the KLSCD48 HSC phenotype. Despite this phenotypic expansion, BM HSCs from these donors are fundamentally altered such that they are no longer able to mediate efficient multilineage repopulation after intravenous transplantation.
We find that 3 poly I:C injections results in highly efficient SHIP deletion in MxCreSHIPflox/flox cells. If a small percentage of cells in the in situ deletion chimeras (Figure 1) remained SHIP-competent and repopulated as well as their WT counterparts in the serial transplantations shown in Figure 2, then it also logically follows that we should observe comparable repopulation with BM from systemically deleted MxCreSHIPflox/flox mice (Figure 3C). On the contrary, we observe significantly compromised repopulation by this SHIP-deficient graft despite the potential presence of residual SHIP-competent cells. These data combined with robust SHIP deletion in splenocytes (Figure 1A) in the in situ deletion model strongly suggest that normal repopulation by in situ SHIP-deleted HSCs is not the result of residual, undeleted HSC clones.
Our findings demonstrate that the HSC BM niche is profoundly altered by SHIP deficiency. SHIP expression has previously been documented in primary fibroblasts and endothelial cells, indicating the potential for SHIP to regulate the activity of these cell types.9,15 Here we document that SHIP is expressed in OBs and stromal cells derived from the BM niche. SHIP has previously been implicated in the regulation of osteoclast function and their resorption of osteoblasts.24 However, SHIP expression in primary osteoclasts has not been demonstrated, suggesting the possibility that accelerated bone resorption exhibited by SHIP−/− osteoclasts could be caused by some of the cytokine alterations that we observe in SHIP−/− mice.
The immature state of SHIP−/− OBs is consistent with the deregulated cell cycle kinetics and repopulation defect we previously reported for SHIP−/− HSCs as mature OBs are required to maintain HSCs in a quiescent state31,41–43 in which their reconstituting potential is maximal.42–45 Consistent with this functional OB impairment, we see a significant functional defect in the ability of SHIP−/− stromal cells to support normal HSC cycling, indicating an inability to support normal HSC function in a SHIP-deficient BM niche.
A previous report found that VEGF-A can induce SHIP expression in endothelial cells,15 and thus SHIP expression, or function, could be induced in niche cells to limit effector functions of BM niche cells. This could include SHIP regulating the production of key factors produced by the niche, including G-CSF, TPO, IL-5, and SDF-1. Consistent with this, we observe increased levels of G-CSF, TPO, IL-6, and matrix metalloproteinase-9, and decreased SDF-1/CXCL12 in the plasma of SHIP-deficient mice. Furthermore, we show direct evidence of compromised SDF-1 production by SHIP-deficient BM niche cells. This probably contributes to the profound mobilization and peripheralization of phenotypic HSCs that we previously observed in SHIP-deficient mice. It has not escaped our attention that increased G-CSF production by SHIP-deficient microenvironment cells could in turn down-regulate SDF-1 production by other microenvironment cells. This would cause HSC mobilization while simultaneously reducing HSC recruitment to niche cells, such as OBs, that maintain HSC quiescence.
The cellular and molecular pathways in the BM microenvironment altered by SHIP deficiency remain to be defined. Nonetheless, these findings are the first to implicate inositol phospholipid-signaling pathways as being critical to the function of the BM microenvironment that supports HSC function. It remains to be determined whether other members of phosphoinositide signaling pathways also impact niche function or whether they have an impact solely through intrinsic effects in HSCs. The analysis of phosphatase and tensin homolog (PTEN)–deficient mice suggests that PTEN plays an intrinsic role in the control of HSC homeostasis and function.46,47 Thus, PTEN is probably the dominant inositol phosphatase that opposes effector pathways distal to PI3K in HSC, whereas SHIP appears to be a dominant inositol phosphatase in cells of the BM niche that support HSCs. It will be intriguing to determine whether this segregation of PTEN and SHIP function between “seed” and “soil” is also conserved in other stem cell populations and their supporting milieu. Functional segregation of these signaling components suggests that selective targeting of these 2 phosphoinositide signaling enzymes might be of value in clinical settings where specific modulation of HSC or niche function is warranted.
Supplementary Material
Acknowledgments
The authors thank Daniela Wood and Kim Paraiso for technical assistance, discussion, and support, Michelle Collazo for assistance in editing the manuscript, and Nathan Watts for genotyping.
This work was supported in part by grants from the National Institutes of Health (RO1 HL72523, R01HL085580, and R21 DK071872) and academic development funds from Moffitt Cancer Center and the University of South Florida. During much of this study, W.G.K. was the Newman Scholar of the Leukemia & Lymphoma Society of America (White Plains, NY).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.L.H. performed experiments, analyzed results, and made the figures; M.J.S. aided in experiments shown in Figures 6 and S2; C.D. aided in experiments for Figure 3 and contributed ideas; O.W. and K.M. performed staining and microscopy for Figure S3; and A.L.H. and W.G.K. designed the research and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: William G. Kerr, PhD, Microbiology and Immunology Dept, SUNY Upstate Medical University, 2204 Weiskotten Hall, 750 E Adams St, Syracuse, NY 13210; e-mail: KerrW@upstate.edu.
References
- 1.Calvi LM, Adams GB, Weibrecht KW, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 2.Zhang J, Niu C, Ye L, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–841. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
- 3.Ara T, Tokoyoda K, Sugiyama T, Egawa T, Kawabata K, Nagasawa T. Long-term hematopoietic stem cells require stromal cell-derived factor-1 for colonizing bone marrow during ontogeny. Immunity. 2003;19:257–267. doi: 10.1016/s1074-7613(03)00201-2. [DOI] [PubMed] [Google Scholar]
- 4.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 5.Wright DE, Wagers AJ, Gulati AP, Johnson FL, Weissman IL. Physiological migration of hematopoietic stem and progenitor cells. Science. 2001;294:1933–1936. doi: 10.1126/science.1064081. [DOI] [PubMed] [Google Scholar]
- 6.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Desponts C, Hazen AL, Paraiso KH, Kerr WG. SHIP deficiency enhances HSC proliferation and survival but compromises homing and repopulation. Blood. 2006;107:4338–4345. doi: 10.1182/blood-2005-12-5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tu Z, Ninos JM, Ma Z, et al. Embryonic and hematopoietic stem cells express a novel SH2-containing inositol 5′-phosphatase isoform that partners with the Grb2 adapter protein. Blood. 2001;98:2028–2038. doi: 10.1182/blood.v98.7.2028. [DOI] [PubMed] [Google Scholar]
- 9.Desponts C, Ninos JM, Kerr WG. s-SHIP associates with receptor complexes essential for pluripotent stem cell growth and survival. Stem Cells Dev. 2006;15:641–646. doi: 10.1089/scd.2006.15.641. [DOI] [PubMed] [Google Scholar]
- 10.Helgason CD, Antonchuk J, Bodner C, Humphries RK. Homeostasis and regeneration of the hematopoietic stem cell pool are altered in SHIP-deficient mice. Blood. 2003;102:3541–3547. doi: 10.1182/blood-2002-12-3939. [DOI] [PubMed] [Google Scholar]
- 11.Damen JE, Liu L, Rosten P, et al. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-triphosphate 5-phosphatase. Proc Natl Acad Sci U S A. 1996;93:1689–1693. doi: 10.1073/pnas.93.4.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geier SJ, Algate PA, Carlberg K, et al. The human SHIP gene is differentially expressed in cell lineages of the bone marrow and blood. Blood. 1997;89:1876–1885. [PubMed] [Google Scholar]
- 13.Lioubin MN, Algate PA, Tsai S, Carlberg K, Aebersold A, Rohrschneider LR. p150Ship, a signal transduction molecule with inositol polyphosphate-5-phosphatase activity. Genes Dev. 1996;10:1084–1095. doi: 10.1101/gad.10.9.1084. [DOI] [PubMed] [Google Scholar]
- 14.Kerr WG, Heller M, Herzenberg LA. Analysis of lipopolysaccharide-response genes in B-lineage cells demonstrates that they can have differentiation stage-restricted expression and contain SH2 domains. Proc Natl Acad Sci U S A. 1996;93:3947–3952. doi: 10.1073/pnas.93.9.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zippo A, De Robertis A, Bardelli M, Galvagni F, Oliviero S. Identification of Flk-1 target genes in vasculogenesis: Pim-1 is required for endothelial and mural cell differentiation in vitro. Blood. 2004;103:4536–4544. doi: 10.1182/blood-2003-11-3827. [DOI] [PubMed] [Google Scholar]
- 16.Sattler M, Verma S, Pride YB, Salgia R, Rohrschneider LR, Griffin JD. SHIP1, an SH2 domain containing polyinositol-5-phosphatase, regulates migration through two critical tyrosine residues and forms a novel signaling complex with DOK1 and CRKL. J Biol Chem. 2001;276:2451–2458. doi: 10.1074/jbc.M006250200. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Jenkins B, Shin JL, Rohrschneider LR. Scaffolding protein Gab2 mediates differentiation signaling downstream of Fms receptor tyrosine kinase. Mol Cell Biol. 2001;21:3047–3056. doi: 10.1128/MCB.21.9.3047-3056.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang JW, Howson JM, Ghansah T, et al. Influence of SHIP on the NK repertoire and allogeneic bone marrow transplantation. Science. 2002;295:2094–2097. doi: 10.1126/science.1068438. [DOI] [PubMed] [Google Scholar]
- 19.Wahle JA, Paraiso KH, Costello AL, Goll EL, Sentman CL, Kerr WG. Cutting edge: dominance by an MHC-independent inhibitory receptor compromises NK killing of complex targets. J Immunol. 2006;176:7165–7169. doi: 10.4049/jimmunol.176.12.7165. [DOI] [PubMed] [Google Scholar]
- 20.Brauweiler A, Tamir I, Dal Porto J, et al. Differential regulation of B cell development, activation, and death by the src homology 2 domain-containing 5′ inositol phosphatase (SHIP). J Exp Med. 2000;191:1545–1554. doi: 10.1084/jem.191.9.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Helgason CD, Kalberer CP, Damen JE, et al. A dual role for Src homology 2 domain-containing inositol-5-phosphatase (SHIP) in immunity: aberrant development and enhanced function of B lymphocytes in ship −/− mice. J Exp Med. 2000;191:781–794. doi: 10.1084/jem.191.5.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sly LM, Rauh MJ, Kalesnikoff J, Song CH, Krystal G. LPS-induced upregulation of SHIP is essential for endotoxin tolerance. Immunity. 2004;21:227–239. doi: 10.1016/j.immuni.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 23.Karlsson MC, Guinamard R, Bolland S, Sankala M, Steinman RM, Ravetch JV. Macrophages control the retention and trafficking of B lymphocytes in the splenic marginal zone. J Exp Med. 2003;198:333–340. doi: 10.1084/jem.20030684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takeshita S, Namba N, Zhao JJ, et al. SHIP-deficient mice are severely osteoporotic due to increased numbers of hyper-resorptive osteoclasts. Nat Med. 2002;8:943–949. doi: 10.1038/nm752. [DOI] [PubMed] [Google Scholar]
- 25.Ghansah T, Paraiso KH, Highfill S, et al. Expansion of myeloid suppressor cells in SHIP-deficient mice represses allogeneic T cell responses. J Immunol. 2004;173:7324–7330. doi: 10.4049/jimmunol.173.12.7324. [DOI] [PubMed] [Google Scholar]
- 26.Huber M, Helgason CD, Scheid MP, Duronio V, Humphries RK, Krystal G. Targeted disruption of SHIP leads to Steel factor-induced degranulation of mast cells. EMBO J. 1998;17:7311–7319. doi: 10.1093/emboj/17.24.7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paraiso KH, Ghansah T, Costello A, Engelman RW, Kerr WG. Induced SHIP deficiency expands myeloid regulatory cells and abrogates graft-versus-host disease. J Immunol. 2007;178:2893–2900. doi: 10.4049/jimmunol.178.5.2893. [DOI] [PubMed] [Google Scholar]
- 28.Harrison DE, Jordan CT, Zhong RK, Astle CM. Primitive hemopoietic stem cells: direct assay of most productive populations by competitive repopulation with simple binomial, correlation and covariance calculations. Exp Hematol. 1993;21:206–219. [PubMed] [Google Scholar]
- 29.Hughes FJ, Aubin JE. Methods in Bone Biology. London, United Kingdom: Chapman and Hall; 1998. [Google Scholar]
- 30.Bakker A, Klein-Nulend J. Osteoblast isolation from murine calvariae and long bones. Methods Mol Med. 2003;80:19–28. doi: 10.1385/1-59259-366-6:19. [DOI] [PubMed] [Google Scholar]
- 31.Mayack SR, Wagers AJ. Osteolineage niche cells initiate hematopoietic stem cell mobilization. Blood. 2008;112:519–531. doi: 10.1182/blood-2008-01-133710. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Cancelas JA, Lee AW, Prabhakar R, Stringer KF, Zheng Y, Williams DA. Rac GTPases differentially integrate signals regulating hematopoietic stem cell localization. Nat Med. 2005;11:886–891. doi: 10.1038/nm1274. [DOI] [PubMed] [Google Scholar]
- 33.Helgason CD, Damen JE, Rosten P, et al. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998;12:1610–1620. doi: 10.1101/gad.12.11.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petit I, Szyper-Kravitz M, Nagler A, et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. 2002;3:687–694. doi: 10.1038/ni813. [DOI] [PubMed] [Google Scholar]
- 35.Semerad CL, Christopher MJ, Liu F, et al. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood. 2005;106:3020–3027. doi: 10.1182/blood-2004-01-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ware MD, Rosten P, Damen JE, Liu L, Humphries RK, Krystal G. Cloning and characterization of human SHIP, the 145-kD inositol 5-phosphatase that associates with SHC after cytokine stimulation. Blood. 1996;88:2833–2840. [PubMed] [Google Scholar]
- 37.Quarles LD, Yohay DA, Lever LW, Caton R, Wenstrup RJ. Distinct proliferative and differentiated stages of murine MC3T3-E1 cells in culture: an in vitro model of osteoblast development. J Bone Miner Res. 1992;7:683–692. doi: 10.1002/jbmr.5650070613. [DOI] [PubMed] [Google Scholar]
- 38.Raouf A, Seth A. Ets transcription factors and targets in osteogenesis. Oncogene. 2000;19:6455–6463. doi: 10.1038/sj.onc.1204037. [DOI] [PubMed] [Google Scholar]
- 39.Vary CP, Li V, Raouf A, et al. Involvement of Ets transcription factors and targets in osteoblast differentiation and matrix mineralization. Exp Cell Res. 2000;257:213–222. doi: 10.1006/excr.2000.4879. [DOI] [PubMed] [Google Scholar]
- 40.Stewart K, Walsh S, Screen J, et al. Further characterization of cells expressing STRO-1 in cultures of adult human bone marrow stromal cells. J Bone Miner Res. 1999;14:1345–1356. doi: 10.1359/jbmr.1999.14.8.1345. [DOI] [PubMed] [Google Scholar]
- 41.Bradford GB, Williams B, Rossi R, Bertoncello I. Quiescence, cycling, and turnover in the primitive hematopoietic stem cell compartment. Exp Hematol. 1997;25:445–453. [PubMed] [Google Scholar]
- 42.Fleming WH, Alpern EJ, Uchida N, Ikuta K, Spangrude GJ, Weissman IL. Functional heterogeneity is associated with the cell cycle status of murine hematopoietic stem cells. J Cell Biol. 1993;122:897–902. doi: 10.1083/jcb.122.4.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Glimm H, Oh IH, Eaves CJ. Human hematopoietic stem cells stimulated to proliferate in vitro lose engraftment potential during their S/G (2)/M transit and do not reenter G (0). Blood. 2000;96:4185–4193. [PubMed] [Google Scholar]
- 44.Passegue E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202:1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carlesso N, Aster JC, Sklar J, Scadden DT. Notch1-induced delay of human hematopoietic progenitor cell differentiation is associated with altered cell cycle kinetics. Blood. 1999;93:838–848. [PubMed] [Google Scholar]
- 46.Yilmaz OH, Valdez R, Theisen BK, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 47.Zhang J, Grindley JC, Yin T, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}