

Abstract
In the present work, we have attempted a comprehensive analysis of cytosolic and microsomal proteomes to elucidate the signaling pathways impaired in human hepatoma (Huh7) cells upon herpes simplex virus type 1 (HSV-1; Cgal+) infection. Using a combination of differential in-gel electrophoresis and nano liquid chromatography/tandem mass spectrometry, 18 spots corresponding to 16 unique deregulated cellular proteins were unambiguously identified, which were involved in the regulation of essential processes such as apoptosis, mRNA processing, cellular structure and integrity, signal transduction, and endoplasmic-reticulum-associated degradation pathway. Based on our proteomic data and additional functional studies target proteins were identified indicating a late activation of apoptotic pathways in Huh7 cells upon HSV-1 Cgal+ infection. Additionally to changes on RuvB-like 2 and Bif-1, down-regulation of Erlin-2 suggests stimulation of Ca2+-dependent apoptosis. Moreover, activation of the mitochondrial apoptotic pathway results from a time-dependent multi-factorial impairment as inferred from the stepwise characterization of constitutive pro- and anti-apoptotic factors. Activation of serine-threonine protein phosphatase 2A (PP2A) was also found in Huh7 cells upon HSV-1 Cgal+ infection. In addition, PP2A activation paralleled dephosphorylation and inactivation of downstream mitogen-activated protein (MAP) kinase pathway (MEK½, ERK½) critical to cell survival and activation of proapoptotic Bad by dephosphorylation of Ser-112. Taken together, our results provide novel molecular information that contributes to define in detail the apoptotic mechanisms triggered by HSV-1 Cgal+ in the host cell and lead to the implication of PP2A in the transduction of cell death signals and cell survival pathway arrest.
HSV-11 is a large, double-stranded DNA virus with a genome of 153 kbp, encoding at least 89 proteins. HSV-1 replicates in the nucleus of the host cell, and its gene expression follows a temporal pattern including three stages: immediate early (IE), early (E), and late (L) genes (1). In cells productively infected with HSV-1, nucleoli, chromatin, and cellular membranes are subjected to major structural alterations (2–4), and the synthesis of most cellular proteins is progressively inhibited during the course of infection; although some specific host proteins continue to be efficiently synthesized, even during the late phase (2, 4). A remarkable effect of HSV-1 is the inhibition of cellular apoptosis mediated by cellular and viral proteins that are expressed during the apoptosis “prevention window” (5). Interestingly, removal of some antiapoptotic viral proteins, as in HSV-1 strains including rdΔ27, CgalΔ3, and vBsΔ27, results in an impaired capacity of blocking the host cell apoptotic response, preferentially in tumor cells (6–8). Although some exceptions have been reported (8), these data suggest that cancer cells are especially sensitive to apoptosis induced by modified HSV-1 strains. Moreover, HSV-1 exhibits a unique genetic flexibility. More than 40 kbp of the viral genome can be replaced by foreign DNA, yet allowing normal replication since many viral proteins are not strictly required to mediate virus multiplication in cultured cells. Furthermore, HSV-1 virulence can be modulated by modification or deletion of target genes maintaining the replicative capacity in tumor cells and consequently the cytopathic capacity during the lytic phase. In contrast to other viruses (9), the cytolytic capacity of HSV-1 in murine cells facilitates the evaluation of the toxicity and safety of newly designed vectors in murine syngenic cancer models. Anti-herpetic drugs, such as acyclovir or forscanet, are available and provide a safety mechanism to shut off viral replication in case of undesired local or systemic infection. Finally, HSV-1 does not integrate into the cellular genome and remains in an episomal state, preventing insertional mutagenesis (9). Both, genetic flexibility and oncoapoptotic capacity highlight the potential of HSV-1 in the development of therapeutic strategies for killing human cancer cells (7, 10).
There is an increasing interest in the identification of cellular intermediates orchestrating the host cell response to HSV-1 strains to promote the development of more efficient and specific vectors. Transcriptional profiling studies using cDNA microarrays have been conducted in mouse and rat embryo fibroblasts (11, 12), human foreskin fibroblasts (13), murine peritoneal cells, and inflammatory macrophages (14), human embryonic lung cells (15), and human glioma cell lines (16) to further understand the molecular alterations induced by HSV-1. However, since changes in mRNA abundance do not always correspond to changes at the protein level (17), proteomics is expected to provide a more extensive description of the cellular mechanisms de-regulated by HSV-1 infection. Recent studies have used different protein separation methods including difference gel electrophoresis (DIGE), isotope-coded affinity tag, multidimensional liquid chromatographic separations followed by liquid chromatography/tandem mass spectrometry (LC-MS/MS) or stable isotope labeling by amino acids in cell culture to study the cellular response to different viral infection (18–22). In addition, comparative proteomics based on a combination of 2-DE and immunoprecipitation with mass spectrometry has been used to describe protein profiles of HSV-1-infected cells. It has been described that HSV-1 VP19C and VP26 proteins associate to ribosomes in HeLa cells (2). Moreover, HSV-1 ICP8 and ICP27 interact directly with members of large cellular complexes involved in cellular translation, replication or damage repair, non-homologous, and homologous recombination and chromatin remodeling in human epidermoid carcinoma suggesting new insights into viral replication mechanisms (23, 24).
Hepatocellular carcinoma (HCC) is one of the most common malignancies worldwide with a global annual incidence of nearly one million cases (25). Despite new insights into the molecular pathogenesis of HCC and improvements in present-day treatments, the rising incidence of HCC along with the poor prognostic at the time of diagnosis highlight the urgent need of novel and more efficient therapeutic strategies. The development of HSV-1-based oncolytic vectors (9), the effect of which may be re-inforced by induction of oncoapoptosis (7), is a promising research avenue to target specifically and efficiently HCC cells. Our present study was aimed at identifying differentially expressed proteins in human hepatoma (Huh7) cells at different time points after HSV-1 Cgal+ infection by an organellar 2-DE proteomic study using a combination of DIGE and nanoLC-ESI-MS/MS. Based on the proteomic information and additional functional experiments, we have identified deregulation of central intermediates targeted by HSV-1 Cgal+ resulting in the impairment of apoptosis and cell survival pathways in human hepatoma cells.
EXPERIMENTAL PROCEDURES
Materials—
The following reagents and materials were used: anti-phospho-Raf (Ser-259 and Ser-338), anti Raf-1, anti-phospho-MEK½ (Ser-217/Ser-221), anti-phospho-ERK½ (Thr-202/Tyr-204), anti-phospho-Akt (Ser-473), anti-phospho-Bad (Ser-112), anti-cleaved caspase 3 and 7, anti-phospho-FAK (Tyr-576/Tyr-577), anti-ERK½, anti-Akt, and anti-PP2A subunit A were from Cell Signaling. Anti-Bcl-xL and anti-Bcl-2 were purchased from Santa Cruz Biotechnology. Anti-Bim was from Chemicon International, and anti-RuvB-like 2 was from BD Transduction Laboratories. Anti-KDEL, anti-HSV-1 ICP4, anti-erlin-2, anti-phospho-PP2A subunit C (Tyr-307), and anti-Bif-1 were from Abcam, and anti-prohibitin was from Calbiochem. Anti-Us3 was from Covalab (France). Electrophoresis reagents were purchased from GE Healthcare and trypsin from Promega.
Virus Production—
Vero cells were used for propagation and titration of HSV-1 Cgal+. HSV-1 Cgal+ is a replication-competent HSV-1 strain derived from CgalΔ3, which derives from HSV-1 17syn+ with both copies of ICP4 deleted. HSV-1 Cgal+ is obtained by repairing both copies of ICP4 and the insertion of LacZ gene from the intergenic region IGR54 (26). Cells were maintained in monolayer with Dulbecco's modified Eagle's medium containing 5–10% fetal bovine serum and penicillin/streptomycin. Huh7 cells (JCRB Genebank, Japan) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, L-glutamine and penicillin/streptomycin. Huh7 cells (1 × 106 or 5 × 106 cells/dish were used for analytical and proteomic experiments, respectively) were infected with HSV-1 Cgal+ at multiplicity of infection of 5 plaque-forming units/cell. After incubation for 1 h at 37 °C, cells were washed and incubated with fresh culture medium under the same conditions for 4, 8, and 24 h. There were no statistically significant differences between control Huh7 cells and Mock-infected Huh7 cells (incubated with a minimum quantity of non-infected Vero cells supernatant). Therefore Mock-infected Huh7 cells were used as the reference sample.
Two-dimensional DIGE and Imaging—
The culture medium was removed after the indicated periods of time, and the cells were washed three times with ice-cold PBS. Subcellular fractionation was performed using the Qproteome Cell Compartment kit from Qiagen. Cytosolic and microsomal fractions (including mitochondria, endoplasmic reticulum, and plasma membrane) were selectively isolated by differential centrifugation according to the manufacturer's recommendations. After acetone precipitation, protein samples were solubilized in two-dimensional DIGE sample buffer: 7 m urea, 2 m thiourea, 4% CHAPS, 30 mm Tris, buffered to pH 8. Protein concentration was determined using the Bradford assay (Bio-Rad). Then 50-μg protein was labeled with 400 pmol of CyDye DIGE Fluor minimal dyes (GE Healthcare) and incubated on ice in the dark for 30 min according to the manufacturer's instructions (Cy3, Cy5 for samples and Cy2 for internal control consisting of a mixture composed by equal amounts of protein from all samples). Paired samples were reverse-labeled in order to prevent potential dye labeling bias (see supplemental Table 1 for DIGE experimental design). The reaction was stopped by addition of 1 μl of 10 mm lysine and incubated on ice for 10 min. Samples were cup-loaded onto immobilized pH gradient strips, 24 cm, pH 3–11NL (GE Healthcare), and subjected to isoelectrofocusing in IPGphor™ isoelectro-focusing system (GE Healthcare) according to the manufacturer's recommendations. Upon IEF, strips were incubated in equilibration buffer (50 mm Tris-HCl, pH 8.8, 6 m urea, 30% glycerol, 2% SDS, a trace of bromphenol blue) containing 0.5% dithiothreitol for 15 min and thereafter in the same buffer with 4.5% iodoacetamide for 15 min. For the second dimension, strips were loaded on top of 12.5% polyacrylamide gels and run (1 watt/gel) for 12–14 h until the bromphenol blue dye reached the gel bottom-end. Subsequently, two-dimensional gels were scanned using a Typhoon™ Trio Imager (GE Healthcare) at 100 μm resolution with λex/λem of 488/520, 532/580, and 633/670 nm for Cy2, Cy3, and Cy5, respectively. The photomultiplier tube was set to ensure that the maximum pixel intensity was between 90,000 and 99,000 pixels. Image analysis was performed using DeCyder 6.5 software (GE Healthcare) as described in the user's manual. Three independent experiments were performed for each experimental setup. Briefly, the differential in-gel analysis module was used for spot detection, spot volume quantification, and volume ratio normalization of different samples in the same gel. Then the Biological Variation Analysis module was used to match protein spots among different gels and to identify protein spots that exhibited significant differences. Manual editing was performed in the Biological Variation Analysis module to ensure that spots were correctly matched between different gels and to get rid of streaks and speckles. All spots had a similar technical variation with a median coefficient variation of 5.3% and 7.2% in cytosolic and microsomal analyses, respectively. Differential expressed spots were considered for MS analysis when the fold-change was larger than 1.5 at least in one of the analyzed time points and the p value after t test was below 0.05. Preparative gels were run with 350 μg of protein following the same procedure described above. Proteins were visualized by staining with SYPRO Ruby protein gel stain (Bio-Rad), and images were acquired with a Typhoon™ Trio Imager using λex/λem of 532/560 nm. Spots differentially represented were excised manually, and gel specimens were processed with a MassPrep station (Waters) as described elsewhere (27). In-gel tryptic digestion was performed with 12.5 ng/μl trypsin in 50 mm ammonium bicarbonate for 12 h at 37 °C. The resulting peptides were extracted with 5% formic acid, 50% acetonitrile. Samples were then concentrated in a SpeedVac before MS analysis.
LC-ESI-MS/MS Analysis—
Microcapillary reversed phase LC was performed with a CapLC™ (Waters) capillary system. Reversed phase separation of tryptic digests was performed with an Atlantis, C18, 3 μm, 75 μm × 10 cm Nano Ease™-fused silica capillary column (Waters) equilibrated in 5% acetonitrile, 0.2% formic acid. After injection of 6 μl of sample, the column was washed during 5 min with the same buffer, and the peptides were eluted using a linear gradient of 5–50% acetonitrile in 30 min at a constant flow rate of 0.2 μl/min. The column was coupled online to a Q-TOF Micro (Waters) using a PicoTip nanospray ionization source (Waters). The heated capillary temperature was 80 °C, and the spray voltage was 1.8–2.2 kV. MS/MS data were collected in an automated data-dependent mode. The three most intense ions in each survey scan were sequentially fragmented by collision-induced dissociation using an isolation width of 2.5 and a relative collision energy of 35%. Data processing was performed with MassLynx 4.0. Data base searching was done with ProteinLynx Global Server 2.1 (Waters) and Phenyx 2.2 (GeneBio, Geneva, Switzerland) against UniProt Knowledgebase Release 12.3 consisting of UniProtKB/Swiss-Prot Release 54.3 and UniProtKB/TrEMBL Release 37.3 with 285.335 and 4.932.421 entries, respectively. The search was enzymatically constrained for trypsin and allowed for one missed cleavage site. Further search parameters were as follows: no restriction on molecular weight and isoelectric point; fixed modification, carbamidomethylation of cysteine; variable modification, oxidation of methionine. A summary table is available (supplemental Table 2) that contains the main submission parameters including scoring models, thresholds, and round of calculations that are specific to Phenyx.
Measurement of β-galactosidase Activity—
1 × 106 Huh7 cells grown in 6-well plates were infected with HSV-1 Cgal+ (multiplicity of infection = 5). After incubation for 1 h at 37 °C, inocula were eliminated, and cells were covered with fresh medium. At 4, 8, and 24 hpi, cells were stained in situ for β-galactosidase Culture medium was removed, and cells were washed twice with PBS. Then, cells were fixed with 0.5% glutaraldehyde in PBS for 10 min. After three washing steps with PBS, cells were incubated with a β-galactosidase activity staining solution (5 mm potassium ferricyanide, 5 mm potassium ferricyanide, 1 mm MgCl2, and 1 mg/ml X-gal) for 6 h at 37 °C. Cells were observed (original magnification ×10/0.25) in a Leica DM-IRB microscope and photographed with a Leica DC-480 camera.
Immunoblotting Analysis—
Protein extraction and Western blotting were performed as described previously (28). Equal amounts of protein (15 μg) were resolved in 12.5% SDS-polyacrylamide gels. Proteins were electrophoretically transferred to nitrocellulose membranes for 45 min at 120 V. Membranes were probed with primary antibodies at 1:1000 dilution or 1:200 for anti-Us3, in 5% bovine serum albumin or 5% non-fat milk, depending of the primary antibody used. After incubation with the appropriate horseradish peroxidase-conjugated secondary antibody (1:5000), the immunoreactivity was visualized by enhanced chemiluminescence (PerkinElmer Life Sciences). Equal loading of the gels was assessed by Ponceau staining and hybridization with a β-actin-specific antibody (Abcam).
Apoptosis Assay—
Apoptosis was estimated by the determination of soluble histone-DNA complexes using the Cell Death Detection assay (Roche). Cell death enzyme-linked immunosorbent assays were performed according to the manufacturer's instructions. Specific enrichment of mono- and oligonucleosomes released into the cytoplasm (enrichment factor) was calculated as the absorbance ratio Cgal+/Mock-infected cells. Absorbance values were normalized by the protein concentration. Five independent experiments in triplicates were performed.
RESULTS
Cytopathic Effect Induced by HSV-1 Cgal+ Infection in Huh7 Cells—
Cgal+ is a replication competent HSV-1 strain expressing β-galactosidase. The capacity of infection and replication of Cgal+ in Huh7 cells was monitored by following the expression of the viral ICP4 protein and by X-gal staining. As expected, 4 and 8 hpi HSV-1 Cgal+ increased the amount of blue-stained cells with minimal phenotypic changes with respect to Mock-infected Huh7 cells (supplemental Fig. 1). However, 24 hpi, in addition to a significant decrease in the number of cells, dramatic morphological changes including cell rounding, large cytoplasmic vacuoles and detachment were observed, which were typical manifestations of HSV-1-associated cytopathic effect (supplemental Fig. 1). Additionally, replication was assessed in Huh7 cells both in vitro and in HCC xenograft murine models (data not shown).
Cytosolic and Microsomal Differentially Expressed Proteins in HSV-1 Cgal+- infected Huh7 Cells—
Cellular pathways interfered with HSV-1 Cgal+ were investigated using a combination of DIGE and mass spectrometry. To increase the efficiency of proteomic analyses, cytosolic and microsomal subcellular fractions were isolated. Detection of ER lumen protein retaining receptor (KDEL) and prohibitin (PHB) preferentially in the microsomal fraction in addition to major glutamine synthase location in the cytosolic fraction indicated the efficiency of the enrichment procedure (supplemental Fig. 2).
Protein mixtures from cytosolic and microsomal fractions extracted from Mock and HSV-1 Cgal+-Huh7-infected cells (4 and 8 hpi) were compared by DIGE analysis, alternating Cy3 and Cy5 labeling to compensate for any differential observation resulting from the chemistry of the fluorescent dyes. DeCyder analysis allowed detection and quantification of 3090 and 3256 spots in the cytosolic and microsomal fractions, respectively (Fig. 1). Differences were accepted when t test <0.05 and the fold-change >1.5 at least in one of the analyzed time points (4 and 8 hpi). According to this criterion, 24 and 18 spots were selected from cytosolic and microsomal analysis, respectively. Differential spots were localized on preparative gels and were identified by nanoLC-ESI-MS/MS after in-gel trypsin digestion (see supplemental information for technical details). From the resulting tryptic digests 12 cytosolic and 6 microsomal proteins were unambiguously identified (Table I): Serine-threonine protein phosphatase 2A 65-KDa regulatory subunit A β isoform (PP2A), heterogeneous nuclear ribonucleoprotein K (HNRPK), septin 8 (SEPT8), serpin B6 (SPB6), macrophage capping protein (CAPG), eukaryotic translation initiation factor 3 subunit 2 (IF32), glutathione S-transferase P (GSTP1), Galectin-1 (LEG1), and an isoform of 52 KDa FK 506-binding protein (FKBP4) were up-regulated in cytosolic fraction of HSV-1 Cgal+-infected Huh7 cells. In contrast, a distinct isoform of 52 KDa FK 506-binding protein (FKBP4) and fortilin (TCTP) were down-regulated. Bif-1 (SHLB1), succinyl-CoA ligase (ADP-forming) beta chain (SUCB1), succinyl-CoA ligase (GDP-forming) beta chain (SUCB2, two isoforms) and Erlin-2 (ERLN2), were down-regulated in the microsomal fraction of Huh7-infected cells while RuvB-like 2 (RUVB2) displayed a transient increase 4–8 hpi. A linear correlation between the molecular weight and isoelectric point calculated from the sequence of the identified proteins and the experimental electrophoretic mobility of the corresponding spots, deduced from the 2-DE gels, was observed in all cases further increasing the reliability of the identifications (data not shown). As often appears on two dimensional gel-based studies, two different spots were identified as products of the same gene (FKBP4 and SUCB2). In particular FKBP4 was identified from two inversely correlated spots with different pI. These observations suggest that different isoforms or post-translational modifications of these proteins might play different roles in the course of HSV-1 infection.
Fig. 1.
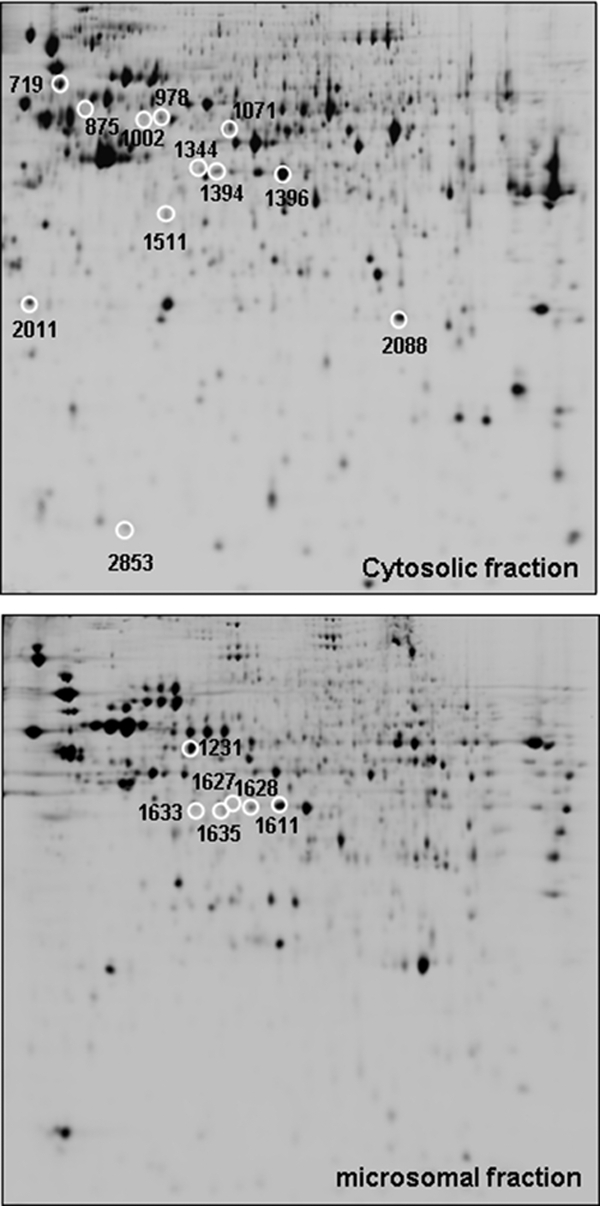
Representative two-dimensional images from cytosolic and microsomal proteomes of HSV-1 Cgal+-infected Huh7 cells (8 hpi). White circles indicate those differential spots detected in Huh7-infected cells that were subsequently identified by nanoLC-ESI-MS/MS.
Table I.
MS/MS identification of DIGE differential spots
n.s., non-significant; n.d., non-determined.
| Protein | Code | Access No. | MW/pI | Phenyx Score | 4 hpi/Mock
|
8 hpi/Mock
|
|||
|---|---|---|---|---|---|---|---|---|---|
| t test | Ratio | t test | Ratio | ||||||
| Cytosolic proteins | |||||||||
| 719 | Ser/Thr- PP2A 65 kDa regulatory subunit A | PP2A | P30154 | 66214/4.84 | 26.18 | 0.0012 | 1.71 | 0.0041 | 1.83 |
| 875 | Heterogeneous nuclear ribonucleoprotein K | HNRPK | P61978 | 50976/5.39 | 36.43 | 0.022 | 1.62 | 0.0033 | 2.97 |
| 978 | 52 kDa FK506-binding protein | FKBP4 | Q02790 | 51805/5.35 | 14.11 | 0.0035 | −2.37 | 8.80E-05 | −4.8 |
| 1002 | 52 kDa FK506-binding protein | FKBP4 | Q02790 | 51805/5.35 | 56.85 | 0.0071 | 1.72 | 0.0071 | 2.24 |
| 1071 | Septin-8. [ISOFORM 2] | SEPT8 | Q92599 | 55756/5.89 | 19.83 | 0.034 | 1.87 | 0.0031 | 2.26 |
| 1344 | UPF0160 MYG1 | MYG1 | Q9HB07 | 42450/6.2 | 21.4 | 0.028 | 1.71 | 0.0024 | 1.67 |
| 1394 | Serpin B6 | SPB6 | P35237 | 42590/5.18 | 15.28 | n.s. | n.d. | 0.013 | 1.77 |
| 1396 | Macrophage capping protein | CAPG | P40121 | 39240/6.73 | 15.33 | n.s. | n.d. | 0.0022 | 1.78 |
| 1511 | Eukaryotic translation initiation factor 3 subunit 2 | IF32 | Q13347 | 36502/5.38 | 39.46 | 0.0042 | 1.95 | 0.0011 | 1.93 |
| 2011 | Fortilin | TCTP | P63029 | 19462/4.76 | 66.69 | 0.0037 | −1.54 | 0.035 | −1.55 |
| 2088 | Glutathione S-transferase P (GST class-pi) | GSTP1 | P04906 | 23439/6.89 | 16.69 | n.s | n.d | 0.0077 | 1.56 |
| 2853 | Galectin-1 | LEG1 | P09382 | 14716/5.33 | 8.68 | n.s | n.d | 0.034 | 1.89 |
| Microsomal proteins | |||||||||
| 1231 | RuvB-like 2 | RUVB2 | Q9Y230 | 51157/5.49 | 31.84 | 0.0028 | 1.73 | n.s | n.d |
| 1611 | Bax- interacting factor 1 (Bif-1) | SHLB1 | Q9Y371 | 40796/5.78 | 15.6 | 0.045 | −1.57 | 0.034 | −1.46 |
| 1627 | Succinyl-CoA ligase [ADP-forming] beta-chain | SUCB1 | Q9P2R7 | 50317/7.05 | 12.4 | 0.031 | −1.81 | 0.026 | −1.54 |
| 1628 | Succinyl-CoA ligase [GDP-forming] beta chain | SUCB2 | Q96I99 | 46511/6.15 | 58.79 | 0.034 | −1.48 | 0.028 | −1.53 |
| 1633 | Erlin-2 | ERLN2 | O94905 | 37840/5.47 | 23.670 | 0.045 | −1.7 | 0.012 | −1.7 |
| 1635 | Succinyl-CoA ligase [GDP-forming] beta chain | SUCB2 | Q96I99 | 46511/6.15 | 21.63 | 0.0069 | −2.17 | 0.0064 | −1.75 |
Among the identified alterations, there are proteins that mediate apoptosis (TCTP, ERLN2, RUVB2, SHLB1), signal transduction (PP2A, FKBP4), mRNA processing (HNRPK, IF32), cellular structure and integrity (SEPT8, CAPG), acyclovir metabolism (SUCB1, SUCB2), and endoplasmic-reticulum-associated degradation pathway (ERLN2). Changes on apoptosis-related proteins such as Bif-1, erlin-2, and RuvB-like 2 were confirmed in total cell extracts by Western blot analysis (Fig. 2).
Fig. 2.

Changes on additional proteins participating in apoptosis were identified in HSV-1 Cgal+-infected Huh7 cells by DIGE analysis. Erlin-2, RuvB-like 2, and Bif-1 steady state levels were calculated by quantification of the corresponding spots at different post-infection time points using DeCyder software (A). The results were confirmed by Western blot analysis of total cell extracts using specific antibodies (B). Equal protein loading was ensured using an antibody against β-actin. A representative blot of three independent experiments is shown.
HSV-1 Cgal+ Induces Apoptosis in Human Hepatoma Cells—
To characterize the molecular mechanisms associated with potential HSV-1 Cgal+-induced apoptosis, the apoptotic mitochondrial pathway was investigated. Four hpi, HSV-1 Cgal+ induced Bim levels, an initiator of apoptosis in the Bcl-2-regulated pathway (Fig. 3). Moreover, HSV-1 Cgal+ promoted Bad activation at 24 hpi by decreasing phospho-Bad levels and concomitantly, down-regulation of Bcl-xL. Consequently, 24 hpi caspase 3 was activated whereas caspase 7 remained uncleaved. On the other hand, HSV-1 Cgal+ down-regulates Bcl-2, in a time-dependent manner (Fig. 3).
Fig. 3.
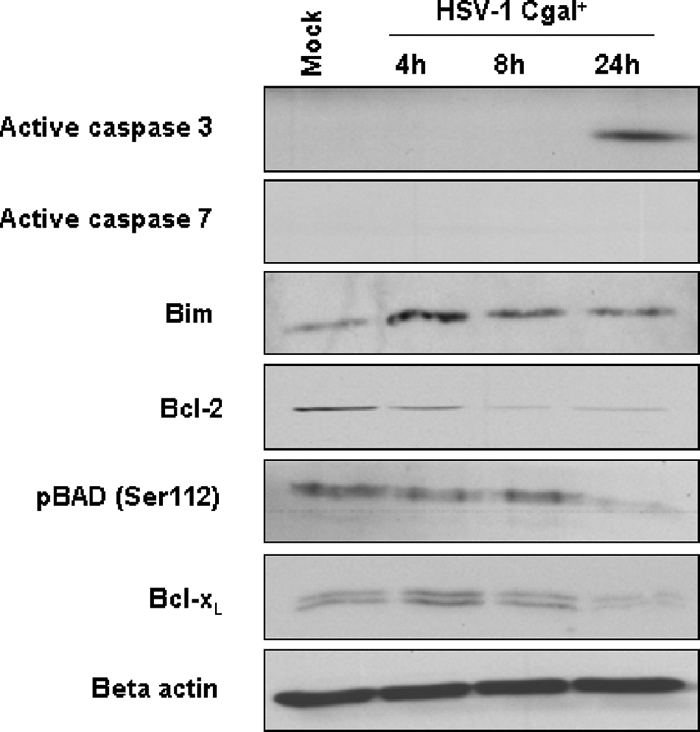
Impairment of apoptosis intermediates in Huh7 cells upon HSV-1 Cgal+ infection. Steady state levels of active caspase 3 and 7, Bim, Bcl-2, phosphorylated Bad (Ser-112), Bcl-xL in Mock, and HSV-Cgal+-infected Huh7 cells at 4, 8, and 24 hpi. Equal protein loading was demonstrated using an antibody against β-actin. Representative blots from three independent experiments are shown.
Since other studies have reported mechanisms by which wild-type HSV-1 blocks cellular apoptosis (7), and considering that HSV-1 Cgal+ vector derives from CgalΔ3 after repair of both ICP4 copies and insertion of LacZ in the IGR54 region, we decided to investigate whether the activation of apoptosis intermediates correlated with an increase in programmed cell death. The analysis of HSV-Cgal+- infected Huh7 cells at 16 and 24 hpi showed values 3- and 8-fold higher, respectively, than those found in Mock and HSV-Cgal+-infected Huh7 cells at 4 and 8 hpi demonstrating that HSV-1 Cgal+ induced apoptosis in Huh7 cells (Fig. 4). Since some apoptotic intermediates were only patent at 24 hpi, their activation was also measured at 16 hpi (see supplemental Fig. 3). All these data point out that HSV-1 Cgal+ -induced apoptosis within the first 24 h occurs without cleavage of caspase 7 and involves the stimulation of the apoptotic mitochondrial pathway through the activation of pro-apoptotic Bad and Bim, down-regulation of anti-apoptotic Bcl-xL and Bcl-2, and activation of caspase 3. To determine if the proapoptotic behavior of Cgal+ resulted from the impairment of the well known anti-apoptotic viral capacity, Us3 expression levels were investigated. The sequence of Us3 gene of Cgal+ did not show differences when compared with the reported WT sequence (data not shown). However, the steady state levels of Us3 were significantly lower in Huh7 cells after infection with Cgal+ than with 17syn+ (Fig. 5). Impairment of Us3 expression might indicate that while Cgal+ is replication positive, it does differ from 17syn+.
Fig. 4.
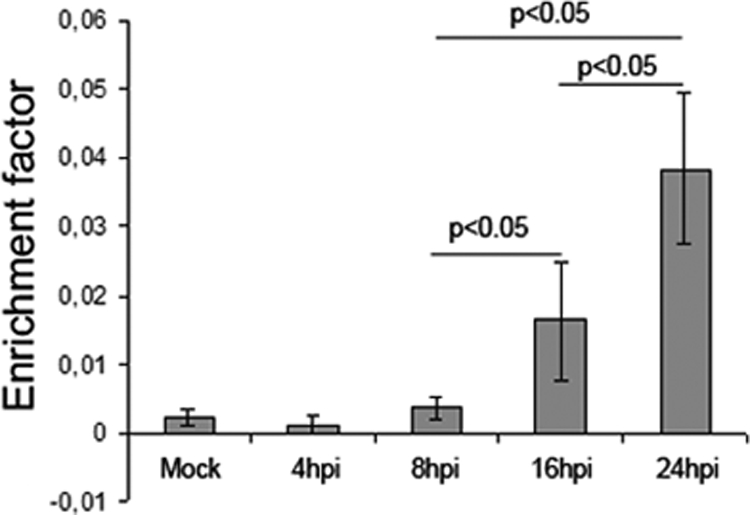
Quantitative apoptosis measurement in HSV-1 Cgal+-infected Huh7 cells. Specific enrichment of mono- and oligonucleosomes released into the cytoplasm (enrichment factor) was calculated as the ratio between the absorbance values of the samples obtained from infected and Mock cells corrected by protein concentration. Data are means ± S. D. of five independent experiments performed in triplicate.
Fig. 5.

Cgal+ induces lower levels of Us3 than the parental WT strain 17syn+. Us3 levels were determined in Cgal+ or 17syn+-infected Huh7 cells 4 and 24 hpi. Additionally, levels of ICP4 protein were determined as references. Equal protein loading was assessed with β-actin-specific antibody. A representative Western blot from five independent experiments is shown.
HSV-1 Cgal+ Activates Protein Phosphatase 2A in Huh7-infected Cells: Implications in Survival Pathways—
DIGE analysis revealed the increment of the serine/threonine PP2A subunit A in HSV-1 Cgal+-infected Huh7 cells. Up-regulation of PP2A subunit A was validated in total cell extracts by Western blot (Fig. 6A). To investigate the potential activation of PP2A, the phosphorylation of the catalytic subunit in Tyr-307 was measured using specific antibodies. A time-dependent decrease of Tyr-307 phosphorylation was observed, indicating the activation of PP2A in Huh7 cells upon HSV-1 Cgal+ infection (Fig. 6A). Since it has been demonstrated that transient activation of PP2A slows down the transmission of growth and survival signals through dephosphorylation of MAP kinases (29, 30), experiments were performed to analyze the interaction of our vector with MAP kinase pathway. HSV-1 Cgal+ induced dephosphorylation of MEK½ 24 hpi and ERK½ in a time-dependent manner (Fig. 6B). However, the virus induced upstream Raf-1 activation by promoting, 24 hpi, the phosphorylation of Ser-338, the activating site, and reducing the phosphorylation level of the inhibitory site Ser-259, which were essential events for Raf-1 switch on (Fig. 6B). Despite Raf-1 activation, these observations suggest that HSV-1 Cgal+ critically down-regulates cell proliferation through a PP2A-mediated arrest of Raf/MEK/ERK pathway. Activation of focal adhesion kinase (FAK)/phosphatidylinositol 3-kinase (PI3K)/Akt survival pathway has been reported to be common to many viral species, including herpes viruses (31–33). In agreement with these observations, HSV-1 Cgal+ induced Akt activation in Huh7 cells through phosphorylation of Ser-473 with maximum effect, 24 hpi, in parallel with FAK activation (Fig. 6C). Interestingly, FAK, Akt, and Raf-1 are also impaired 16 hpi (supplemental Fig. 3).
Fig. 6.
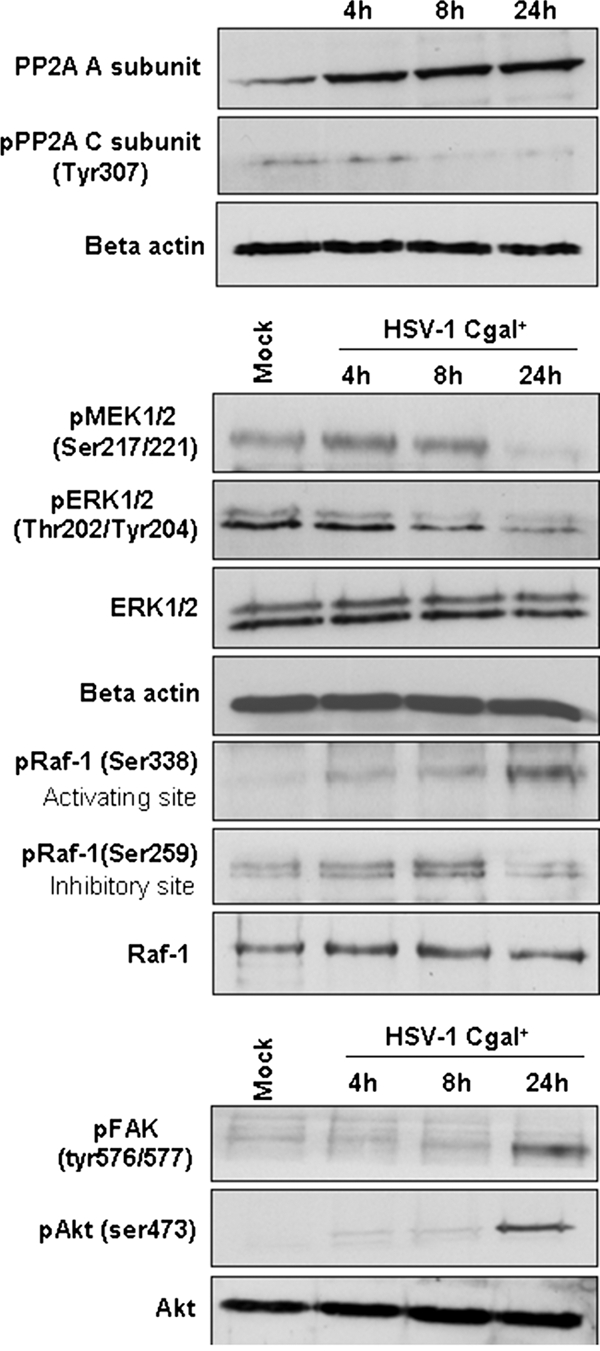
Impairment of signaling intermediates in HSV-infected Huh7 human hepatoma cells. Levels and residue-specific phosphorylation of PP2A scaffold and catalytic subunits (A), Raf/MAP kinase pathway (B), and FAK/PI3K/Akt pathway (C) intermediates were determined by Western blot in HSV-Cgal+-infected Huh7 cells. Differential phosphorylation of intermediates was confirmed by normalization with the total amount of the corresponding protein. Equal protein loading was demonstrated using an antibody against β-actin. Experiments were performed in triplicate. Representative blots are shown.
DISCUSSION
The use of HSV-1-based therapies is arising as a promising strategy to kill cancer cells (7, 10), but the intermediates underlying the tumoral cell response must be elucidated to promote the development of more efficient and selective vectors. In this report, we have used an organellar proteomic approach to analyze global alterations in protein expression that arise in human hepatoma cells infected with HSV-1 Cgal+ virus. Novel HSV-1 Cgal+ targets involved in the regulation of human hepatoma cell death have been identified. Early up-regulation of RuvB-like 2 and a time-dependent decrease of Bif-1 were induced on Huh7 cells upon HSV-1 Cgal+ infection. RuvB-like 2 is an ATPase and putative DNA helicase, the down-regulation of which has been shown to reduce cell growth in Huh7 cells and increases the expression of proapoptotic genes leading to an enhanced conformational activation of Bak-1 and Bax (34). Reduction of endogenous Bif-1 that might result from the HSV-1 virion host shutoff prevents activation of Bax and Bak, cytochrome c release, and caspase 3 activation through various intrinsic death signals (35). Down-regulation of the pro-apoptotic Bif-1 and the transient increase of RuvB-like 2 might provide mechanisms to evade or retard the cellular apoptotic response to HSV-1 Cgal+ infection, until progeny virions are produced. In contrast, down-regulation of anti-apoptotic intermediates such as Bcl-2 family proteins, Bcl-xL and Bcl-2 (36), up-regulation of pro-apoptotic proteins including the Bcl-2-interacting mediator Bim (37) and Galectin-1, as well as activation of caspase 3 might explain the apoptotic response of Huh7 cells to HSV-1 Cgal+ infection. Interestingly, up-regulation of Galectin-1 (38) and activation of caspase 3 (5, 39) have been previously associated to HSV-1-induced apoptosis. However, data have been also reported indicating that there is a disconnection in HSV-1-infected cells between caspase 3 cleavage and other apoptotic events (5). Although it has been claimed that both KOS 1.1 (WT) and vBSΔ27 HSV-1 strains induce reduction of procaspase 7 levels in the cervical carcinoma cell line Hep-2 (39), the apoptotic events observed in Huh7 cells upon Cgal+ infection within the first 24 h do not require caspase 7 processing. However, we cannot exclude activation of caspase 7 at longer hpi. Different HSV-1 backbones might induce specific cellular responses as suggested by studies showing that while Bcl-xL and Bcl-2 proteins remain unchanged in human epithelial cells infected with a highly apoptotic HSV-1 mutant (vBSΔ27) (40), other apoptotic HSV-1 mutants such as tsk and 27LacZ induce a decrease of Bcl-2 RNA and protein levels, and a concomitant caspase 3 cleavage, in infected hamster kidney and HeLa cells (41). These observations suggest that the cellular responses depend strongly on the HSV-1 strain used and are orchestrated by cell type specific mechanisms involving pathways with common and dissimilar intermediates (Fig. 7).
Fig. 7.

Schematic representation of signaling pathways altered in human hepatoma cells upon HSV-1 Cgal+ infection. The arrows indicate the alteration of specific targets.
The impairment of apoptotic mediators observed in Huh7-infected cells is in agreement with the late (16–24 hpi) apoptotic cell death as determined by the specific enrichment of mono- and oligonucleosomes released into the cytoplasm. To date, seven viral gene products (ICP4, ICP27, Us3, ICP22, gD, gJ, and LAT) and two cellular proteins (Bcl-2, NFκB) have been proposed to participate in the HSV-1 strategy to control host cell apoptosis (7, 42). In particular, the role of ICP4 in apoptosis prevention is demonstrated using the wild-type HSV-1 17syn+-derived, ICP4-null CgalΔ3 strain (7). HSV-1 Cgal+ results from the reinsertion of both ICP4 copies in CgalΔ3 backbone, and therefore it might be able to block host cell apoptosis upon infection. The observed proapoptotic behavior of HSV-1 Cgal+ in human liver cancer Huh7 cells might be explained assuming that liver cells are specially sensitive to HSV-induced apoptosis, as previously proposed (43–45). In addition, proapoptotic HSV-1 variants lacking viral antiapoptotic proteins induce programmed cell death preferentially in human cancer cells (7). However, reduced Us3 kinase expression mediate, at least in part, Cgal+-induced apoptosis in Huh7 cells. Alterations in Us3 have been previously reported as related with modifications in genetically engineered HSV-1 mutants (46). Additionally to the reinsertion of ICP4, LacZ gene was inserted in the intergenic region IGR54 to follow Cgal+ infectivity and replication in HCC xenograft murine models, resulting in a replication-competent, non-attenuated, HSV strain. IGR54 is close to Us3 locus, and hence we suggest that Cgal+-impaired Us3 expression may result from alterations in the Us3 promoter region that, to our knowledge, has not been completely characterized. Dephosphorylation of Bad may be also explained by reduction of Us3 activity, although it is still controversial if Bad can be considered as a substrate of Us3 kinase (47–49).
Apoptosis mediated by caspase 3 in HSV-1 Cgal+-infected Huh7 cells was further confirmed by the activation of the upstream proapoptotic Bad as assessed by its reduced phosphorylation upon infection. Recent evidences suggest that PP2A activates Bad by dephosphorylation of Ser-112 (50). Accordingly, we found early up-regulation of PP2A scaffold subunit A and subsequent dephosphorylation of Tyr-307 in the catalytic subunit, suggesting PP2A activation in Huh7 infected cells (29, 51). Different viruses target PP2A in order to deregulate selected cellular pathways in the host and promote viral progeny (52) including human cytomegalovirus (53), parainfluenza virions (54), adenovirus (55–57), human immunodeficiency virus (58), hepatitis C virus (59, 60), and simian virus 40 and polyomavirus (61). Activation of PP2A is induced by hepatitis viruses through mechanisms dependent on endoplasmic reticulum calcium release (59). Interestingly, HSV-1 activates calcium-signaling pathways (62, 63), although little is known about the mechanism involved. Our results suggest that decrease in fortilin and erlin-2 levels in Huh7-infected cells may explain in part the uncontrolled Ca2+ efflux produced in HSV-1 Cgal+ infection leading to Huh7 cell death by apoptosis. Fortilin is an anti-apoptotic protein that binds calcium and blocks Ca2+-dependent apoptosis (64, 65). Down-regulation of erlin-2 expression markedly inhibited IP3R receptor polyubiquitination and degradation leading to pathological changes in Ca2+ signaling causing abnormal growth and apoptosis (66, 67). On the whole, activation of caspase 3, up-regulation of Bim and Galectin-1, the transient increase of RuvB-like 2 and down-regulation of fortilin, erlin-2, Bif-1, phospho-Bad, Bcl-2, and Bcl-xL provide new insights into the delicate balance between pro- and antiapoptotic factors in human hepatoma cells upon HSV-1 Cgal+ infection.
A well characterized PP2A function is the regulation of Raf/MAP kinase pathways, that have central functions in cell proliferation and cell differentiation processes (68). Paralleling the HSV-1 Cgal+ induced activation of PP2A, we found inactivation of MAP kinase downstream cascade as evidenced by MEK½ and ERK½ dephosphorylation, compromising the cell survival potential. However, apparent activation of upstream Raf-1 was suggested by both, phosphorylation of the activating site (Ser-338) and dephosphorylation of the inhibitory site (Ser-259), one of the substrates of PP2A (69–72). Sustained phosphorylation of Raf-1 has been also reported when MEK signaling is blocked by pharmacological inhibition (73) and in cells overexpressing impedes mitogenic signal propagation, an inhibitor of KSR that disrupts the Raf-MEK interaction (74). These observations might be explained by the lack of feedback inhibition provided by the phosphorylation of the inhibitory site by active ERK½ (75). Nevertheless, it has been recently proposed that regulation of Raf-1 involves a complex phosphorylation pattern of thirteen specific sites (75, 76), and therefore the analysis restricted to Ser-338 and Ser-259 must be extended to extract a reliable functional conclusion.
Activation of host FAK/PI3K/Akt signaling pathway has been associated to many viral types including herpes viruses (31–33). In agreement with these observations, mainly considering the deficient Us3 expression, we have found hyperphosphorylation of Akt at 24 hpi (31) in HSV-1 Cgal+-infected Huh7 cells. Ser-259 of Raf-1 (77) as well as Ser-112 of Bad (78) are canonical targets of active Akt but they are dephosphorylated after HSV-1 Cgal+ infection. Although additional studies are needed to reconcile these contradictory observations, it might be speculated that activation of Akt may require additional modifications, as has been reported for other kinases (79–84), or alternatively, the apparent inactivity of Akt might result from association with viral proteins (85–88) preventing the interaction with its natural targets.
Taken together, our results provide novel molecular information that contributes to define in detail the apoptotic mechanisms triggered by HSV-1 Cgal+ in human hepatoma cells and lead to the identification of PP2A as a pivotal target that mediates the transduction of cell death signals and promotes cell survival pathway arrest. This information may prove to be useful in designing novel HSV-1-based anticancer strategies to improve the specificity and efficiency of HCC treatment.
Supplementary Material
Acknowledgments
We thank Carmen Miqueo and Manuela Molina for their technical assistance.
Footnotes
Published, MCP Papers in Press, December 19, 2008, DOI 10.1074/mcp.M800202-MCP200
The abbreviations used are: Huh, human hepatoma; HSV-1, herpes simplex virus type-1; DIGE, differential in-gel electrophoresis; PP2A, protein phosphatase 2A; MAP, mitogen-activated protein; LC-MS/MS, liquid chromatography/tandem mass spectrometry; PBS, phosphate-buffered saline; hepatocellular carcinoma; X-gal, 5-bromo-4-chloro-3-indolyl-β- d-galactopyranoside; WT, wild-type; FAK, focal adhesion kinase; PI3K, phosphatidylinositol 3-kinase; hpi, hours post-infection; MEK, mitogen-activated ERK kinase; ERK, extracellular signal-related kinase.
This work was supported in part by the agreement between Foundation for Applied Medical Research and the “UTE project Centre for Applied Medical Research”, Grants Plan Nacional I+D+I 2004-01855 from Ministerio de Educación y Ciencia (to F. J. C.), Grant PROFIT FIT-340000-2005-353 from Ministerio de Industria, Turismo y Comercio (to F. J. C.), LSHB-CT-2005-018649 (THOVLEN) from the 6th framework program of the UE (to F. J. C. and A. L. E.), ISCIII-RETIC RD06/00200061 (to M. A. A.) and (F. J. C.). This laboratory is member of the National Institute of Proteomics Facilities, ProteoRed.
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
REFERENCES
- 1.Taylor, T. J., Brockman, M. A., McNamee, E. E., and Knipe, D. M. ( 2002) Herpes simplex virus. Front Biosci. 7, 752–764 [DOI] [PubMed] [Google Scholar]
- 2.Greco, A., Bienvenut, W., Sanchez, J. C., Kindbeiter, K., Hochstrasser, D., Madjar, J. J., and Diaz, J. J. ( 2001) Identification of ribosome-associated viral and cellular basic proteins during the course of infection with herpes simplex virus type 1. Proteomics 1, 545–549 [DOI] [PubMed] [Google Scholar]
- 3.Simpson-Holley, M., Colgrove, R. C., Nalepa, G., Harper, J. W., and Knipe, D. M. ( 2005) Identification and functional evaluation of cellular and viral factors involved in the alteration of nuclear architecture during herpes simplex virus 1 infection. J. Virol. 79, 12840–12851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calle, A., Ugrinova, I., Epstein, A. L., Bouvet, P., Diaz, J. J., and Greco, A. ( 2008) Nucleolin is required for an efficient herpes simplex virus type 1 infection. J. Virol. 82, 4762–4773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aubert, M., O'Toole, J., and Blaho, J. A. ( 1999) Induction and prevention of apoptosis in human HEp-2 cells by herpes simplex virus type 1. J. Virol. 73, 10359–10370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aubert, M., and Blaho, J. A. ( 2003) Viral oncoapoptosis of human tumor cells. Gene Ther. 10, 1437–1445 [DOI] [PubMed] [Google Scholar]
- 7.Nguyen, M. L., and Blaho, J. A. ( 2007) Apoptosis during herpes simplex virus infection. Adv. Virus Res. 69, 67–97 [DOI] [PubMed] [Google Scholar]
- 8.Nguyen, M. L., Kraft, R. M., and Blaho, J. A. ( 2007) Susceptibility of cancer cells to herpes simplex virus-dependent apoptosis. J. Gen. Virol. 88, 1866–1875 [DOI] [PubMed] [Google Scholar]
- 9.Mullen, J. T., and Tanabe, K. K. ( 2003) Viral oncolysis for malignant liver tumors. Ann. Surg. Oncol. 10, 596–605 [DOI] [PubMed] [Google Scholar]
- 10.Shen, Y., and Nemunaitis, J. ( 2006) Herpes simplex virus 1 (HSV-1) for cancer treatment. Cancer Gene Ther. 13, 975–992 [DOI] [PubMed] [Google Scholar]
- 11.Pasieka, T. J., Baas, T., Carter, V. S., Proll, S. C., Katze, M. G., and Leib, D. A. ( 2006) Functional genomic analysis of herpes simplex virus type 1 counteraction of the host innate response. J. Virol. 80, 7600–7612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ray, N., and Enquist, L. W. ( 2004) Transcriptional response of a common permissive cell type to infection by two diverse alphaherpesviruses. J. Virol. 78, 3489–3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taddeo, B., Esclatine, A., and Roizman, B. ( 2002) The patterns of accumulation of cellular RNAs in cells infected with a wild-type and a mutant herpes simplex virus 1 lacking the virion host shutoff gene. Proc. Natl. Acad. Sci. U. S. A. 99, 17031–17036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paludan, S. R., Melchjorsen, J., Malmgaard, L., and Mogensen, S. C. ( 2002) Expression of genes for cytokines and cytokine-related functions in leukocytes infected with Herpes simplex virus: comparison between resistant and susceptible mouse strains. Eur. Cytokine Netw. 13, 306–316 [PubMed] [Google Scholar]
- 15.Mossman, K. L., Macgregor, P. F., Rozmus, J. J., Goryachev, A. B., Edwards, A. M., and Smiley, J. R. ( 2001) Herpes simplex virus triggers and then disarms a host antiviral response. J. Virol. 75, 750–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khodarev, N. N., Advani, S. J., Gupta, N., Roizman, B., and Weichselbaum, R. R. ( 1999) Accumulation of specific RNAs encoding transcriptional factors and stress response proteins against a background of severe depletion of cellular RNAs in cells infected with herpes simplex virus 1. Proc. Natl. Acad. Sci. U. S. A. 96, 12062–12067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tian, Q., Stepaniants, S. B., Mao, M., Weng, L., Feetham, M. C., Doyle, M. J., Yi, E. C., Dai, H., Thorsson, V., Eng, J., Goodlett, D., Berger, J. P., Gunter, B., Linseley, P. S., Stoughton, R. B., Aebersold, R., Collins, S. J., Hanlon, W. A., and Hood, L. E. ( 2004) Integrated genomic and proteomic analyses of gene expression in mammalian cells. Mol. Cell. Proteomics 3, 960–969 [DOI] [PubMed] [Google Scholar]
- 18.Jiang, X. S., Tang, L. Y., Dai, J., Zhou, H., Li, S. J., Xia, Q. C., Wu, J. R., and Zeng, R. ( 2005) Quantitative analysis of severe acute respiratory syndrome (SARS)-associated coronavirus-infected cells using proteomic approaches: implications for cellular responses to virus infection. Mol. Cell. Proteomics 4, 902–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baas, T., Baskin, C. R., Diamond, D. L., Garcia-Sastre, A., Bielefeldt-Ohmann, H., Tumpey, T. M., Thomas, M. J., Carter, V. S., Teal, T. H., Van Hoeven, N., Proll, S., Jacobs, J. M., Caldwell, Z. R., Gritsenko, M. A., Hukkanen, R. R., Camp, D. G., 2nd, Smith, R. D., and Katze, M. G. ( 2006) Integrated molecular signature of disease: analysis of influenza virus-infected macaques through functional genomics and proteomics. J. Virol. 80, 10813–10828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Go, E. P., Wikoff, W. R., Shen, Z., O'Maille, G., Morita, H., Conrads, T. P., Nordstrom, A., Trauger, S. A., Uritboonthai, W., Lucas, D. A., Chan, K. C., Veenstra, T. D., Lewicki, H., Oldstone, M. B., Schneemann, A., and Siuzdak, G. ( 2006) Mass spectrometry reveals specific and global molecular transformations during viral infection. J. Proteome Res. 5, 2405–2416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacobs, J. M., Diamond, D. L., Chan, E. Y., Gritsenko, M. A., Qian, W., Stastna, M., Baas, T., Camp, D. G., 2nd, Carithers, R. L., Jr., Smith, R. D., and Katze, M. G. ( 2005) Proteome analysis of liver cells expressing a full-length hepatitis C virus (HCV) replicon and biopsy specimens of posttransplantation liver from HCV-infected patients. J. Virol. 79, 7558–7569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mannova, P., Fang, R., Wang, H., Deng, B., McIntosh, M. W., Hanash, S. M., and Beretta, L. ( 2006) Modification of host lipid raft proteome upon hepatitis C virus replication. Mol. Cell. Proteomics 5, 2319–2325 [DOI] [PubMed] [Google Scholar]
- 23.Fontaine-Rodriguez, E. C., Taylor, T. J., Olesky, M., and Knipe, D. M. ( 2004) Proteomics of herpes simplex virus infected cell protein 27: association with translation initiation factors. Virology 330, 487–492 [DOI] [PubMed] [Google Scholar]
- 24.Taylor, T. J., and Knipe, D. M. ( 2004) Proteomics of herpes simplex virus replication compartments: association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J. Virol. 78, 5856–5866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El-Serag, H. B., and Rudolph, K. L. ( 2007) Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 132, 2557–2576 [DOI] [PubMed] [Google Scholar]
- 26.Johnson, P. A., Best, M. G., Friedmann, T., and Parris, D. S. ( 1991) Isolation of a herpes simplex virus type 1 mutant deleted for the essential UL42 gene and characterization of its null phenotype. J. Virol. 65, 700–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santamaria, E., Avila, M. A., Latasa, M. U., Rubio, A., Martin-Duce, A., Lu, S. C., Mato, J. M., and Corrales, F. J. ( 2003) Functional proteomics of nonalcoholic steatohepatitis: mitochondrial proteins as targets of S-adenosylmethionine. Proc. Natl. Acad. Sci. U. S. A. 100, 3065–3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruiz, F., Corrales, F. J., Miqueo, C., and Mato, J. M. ( 1998) Nitric oxide inactivates rat hepatic methionine adenosyltransferase in vivo by S-nitrosylation. Hepatology 28, 1051–1057 [DOI] [PubMed] [Google Scholar]
- 29.Chen, J., Martin, B. L., and Brautigan, D. L. ( 1992) Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science 257, 1261–1264 [DOI] [PubMed] [Google Scholar]
- 30.Letourneux, C., Rocher, G., and Porteu, F. ( 2006) B56-containing PP2A dephosphorylate ERK and their activity is controlled by the early gene IEX-1 and ERK. EMBO J. 25, 727–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benetti, L., and Roizman, B. ( 2006) Protein kinase B/Akt is present in activated form throughout the entire replicative cycle of δU(S)3 mutant virus but only at early times after infection with wild-type herpes simplex virus 1. J. Virol. 80, 3341–3348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheshenko, N., Liu, W., Satlin, L. M., and Herold, B. C. ( 2005) Focal adhesion kinase plays a pivotal role in herpes simplex virus entry. J. Biol. Chem. 280, 31116–31125 [DOI] [PubMed] [Google Scholar]
- 33.Liu, T. C., Wakimoto, H., Martuza, R. L., and Rabkin, S. D. ( 2007) Herpes simplex virus Us3(−) mutant as oncolytic strategy and synergizes with phosphatidylinositol 3-kinase-Akt-targeting molecular therapeutics. Clin. Cancer Res. 13, 5897–5902 [DOI] [PubMed] [Google Scholar]
- 34.Rousseau, B., Menard, L., Haurie, V., Taras, D., Blanc, J. F., Moreau-Gaudry, F., Metzler, P., Hugues, M., Boyault, S., Lemiere, S., Canron, X., Costet, P., Cole, M., Balabaud, C., Bioulac-Sage, P., Zucman-Rossi, J., and Rosenbaum, J. ( 2007) Overexpression and role of the ATPase and putative DNA helicase RuvB-like 2 in human hepatocellular carcinoma. Hepatology 46, 1108–1118 [DOI] [PubMed] [Google Scholar]
- 35.Takahashi, Y., Karbowski, M., Yamaguchi, H., Kazi, A., Wu, J., Sebti, S. M., Youle, R. J., and Wang, H. G. ( 2005) Loss of Bif-1 suppresses Bax/Bak conformational change and mitochondrial apoptosis. Mol. Cell. Biol. 25, 9369–9382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim, R. ( 2005) Unknotting the roles of Bcl-2 and Bcl-xL in cell death. Biochem. Biophys. Res. Commun. 333, 336–343 [DOI] [PubMed] [Google Scholar]
- 37.Willis, S. N., and Adams, J. M. ( 2005) Life in the balance: how BH3-only proteins induce apoptosis. Curr. Opin. Cell Biol. 17, 617–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gonzalez, M. I., Rubinstein, N., Ilarregui, J. M., Toscano, M. A., Sanjuan, N. A., and Rabinovich, G. A. ( 2005) Regulated expression of galectin-1 after in vitro productive infection with herpes simplex virus type 1: implications for T cell apoptosis. Int. J. Immunopathol. Pharmacol. 18, 615–623 [DOI] [PubMed] [Google Scholar]
- 39.Kraft, R. M., Nguyen, M. L., Yang, X. H., Thor, A. D., and Blaho, J. A. ( 2006) Caspase 3 activation during herpes simplex virus 1 infection. Virus Res. 120, 163–175 [DOI] [PubMed] [Google Scholar]
- 40.Aubert, M., Pomeranz, L. E., and Blaho, J. A. ( 2007) Herpes simplex virus blocks apoptosis by precluding mitochondrial cytochrome c release independent of caspase activation in infected human epithelial cells. Apoptosis 12, 19–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zachos, G., Koffa, M., Preston, C. M., Clements, J. B., and Conner, J. ( 2001) Herpes simplex virus type 1 blocks the apoptotic host cell defense mechanisms that target Bcl-2 and manipulates activation of p38 mitogen-activated protein kinase to improve viral replication. J. Virol. 75, 2710–2728 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Goodkin, M. L., Morton, E. R., and Blaho, J. A. ( 2004) Herpes simplex virus infection and apoptosis. Int. Rev. Immunol. 23, 141–172 [DOI] [PubMed] [Google Scholar]
- 43.Irie, H., Kiyoshi, A., and Koyama, A. H. ( 2004) A role for apoptosis induced by acute herpes simplex virus infection in mice. Int. Rev. Immunol. 23, 173–185 [DOI] [PubMed] [Google Scholar]
- 44.Irie, H., Koyama, H., Kubo, H., Fukuda, A., Aita, K., Koike, T., Yoshimura, A., Yoshida, T., Shiga, J., and Hill, T. ( 1998). Herpes simplex virus hepatitis in macrophage-depleted mice: the role of massive, apoptotic cell death in pathogenesis. J. Gen. Virol. 79, 1225–1231 [DOI] [PubMed] [Google Scholar]
- 45.Pretet, J. L., Pelletier, L., Bernard, B., Coumes-Marquet, S., Kantelip, B., and Mougin, C. ( 2003) Apoptosis participates to liver damage in HSV-induced fulminant hepatitis. Apoptosis 8, 655–663 [DOI] [PubMed] [Google Scholar]
- 46.Leopardi, R., Van Sant, C., and Roizman, B. ( 1997) The herpes simplex virus 1 protein kinase US3 is required for protection from apoptosis induced by the virus. Proc. Natl. Acad. Sci. U. S. A. 94, 7891–7896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benetti, L., Munger, J., and Roizman, B. ( 2003) The herpes simplex virus 1 US3 protein kinase blocks caspase-dependent double cleavage and activation of the proapoptotic protein BAD. J. Virol. 77, 6567–6573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cartier, A., Komai, T., and Masucci, M. G. ( 2003) The Us3 protein kinase of herpes simplex virus 1 blocks apoptosis and induces phosporylation of the Bcl-2 family member Bad. Exp. Cell Res. 291, 242–250 [DOI] [PubMed] [Google Scholar]
- 49.Munger, J., and Roizman, B. ( 2001) The US3 protein kinase of herpes simplex virus 1 mediates the posttranslational modification of BAD and prevents BAD-induced programmed cell death in the absence of other viral proteins. Proc. Natl. Acad. Sci. U. S. A. 98, 10410–10415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chiang, C. W., Kanies, C., Kim, K. W., Fang, W. B., Parkhurst, C., Xie, M., Henry, T., and Yang, E. ( 2003) Protein phosphatase 2A dephosphorylation of phosphoserine 112 plays the gatekeeper role for BAD-mediated apoptosis. Mol. Cell. Biol. 23, 6350–6362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen, J., Parsons, S., and Brautigan, D. L. ( 1994) Tyrosine phosphorylation of protein phosphatase 2A in response to growth stimulation and v-src transformation of fibroblasts. J. Biol. Chem. 269, 7957–7962 [PubMed] [Google Scholar]
- 52.Sontag, E. ( 2001) Protein phosphatase 2A: the Trojan Horse of cellular signaling. Cell. Signal. 13, 7–16 [DOI] [PubMed] [Google Scholar]
- 53.Michelson, S., Turowski, P., Picard, L., Goris, J., Landini, M. P., Topilko, A., Hemmings, B., Bessia, C., Garcia, A., and Virelizier, J. L. ( 1996) Human cytomegalovirus carries serine/threonine protein phosphatases PP1 and a host-cell-derived PP2A. J. Virol. 70, 1415–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De, B. P., Gupta, S., Gupta, S., and Banerjee, A. K. ( 1995) Cellular protein kinase C isoform zeta regulates human parainfluenza virus type 3 replication. Proc. Natl. Acad. Sci. U. S. A. 92, 5204–5208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roopchand, D. E., Lee, J. M., Shahinian, S., Paquette, D., Bussey, H., and Branton, P. E. ( 2001) Toxicity of human adenovirus E4orf4 protein in Saccharomyces cerevisiae results from interactions with the Cdc55 regulatory B subunit of PP2A. Oncogene 20, 5279–5290 [DOI] [PubMed] [Google Scholar]
- 56.Shtrichman, R., Sharf, R., Barr, H., Dobner, T., and Kleinberger, T. ( 1999) Induction of apoptosis by adenovirus E4orf4 protein is specific to transformed cells and requires an interaction with protein phosphatase 2A. Proc. Natl. Acad. Sci. U. S. A. 96, 10080–10085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shtrichman, R., Sharf, R., and Kleinberger, T. ( 2000) Adenovirus E4orf4 protein interacts with both Balpha and B` subunits of protein phosphatase 2A, but E4orf4-induced apoptosis is mediated only by the interaction with Balpha. Oncogene 19, 3757–3765 [DOI] [PubMed] [Google Scholar]
- 58.Tung, H. Y., De Rocquigny, H., Zhao, L. J., Cayla, X., Roques, B. P., and Ozon, R. ( 1997) Direct activation of protein phosphatase-2A0 by HIV-1 encoded protein complex NCp7:vpr. FEBS Lett. 401, 197–201 [DOI] [PubMed] [Google Scholar]
- 59.Christen, V., Treves, S., Duong, F. H., and Heim, M. H. ( 2007) Activation of endoplasmic reticulum stress response by hepatitis viruses up-regulates protein phosphatase 2A. Hepatology 46, 558–565 [DOI] [PubMed] [Google Scholar]
- 60.Georgopoulou, U., Tsitoura, P., Kalamvoki, M., and Mavromara, P. ( 2006) The protein phosphatase 2A represents a novel cellular target for hepatitis C virus NS5A protein. Biochimie 88, 651–662 [DOI] [PubMed] [Google Scholar]
- 61.Pallas, D. C., Shahrik, L. K., Martin, B. L., Jaspers, S., Miller, T. B., Brautigan, D. L., and Roberts, T. M. ( 1990) Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell 60, 167–176 [DOI] [PubMed] [Google Scholar]
- 62.Cheshenko, N., Del Rosario, B., Woda, C., Marcellino, D., Satlin, L. M., and Herold, B. C. ( 2003) Herpes simplex virus triggers activation of calcium-signaling pathways. J. Cell Biol. 163, 283–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cheshenko, N., Liu, W., Satlin, L. M., and Herold, B. C. ( 2007) Multiple receptor interactions trigger release of membrane and intracellular calcium stores critical for herpes simplex virus entry. Mol. Biol. Cell 18, 3119–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Graidist, P., Yazawa, M., Tonganunt, M., Nakatomi, A., Lin, C. C., Chang, J. Y., Phongdara, A., and Fujise, K. ( 2007) Fortilin binds Ca2+ and blocks Ca2+-dependent apoptosis in vivo. Biochem. J. 408, 181–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li, F., Zhang, D., and Fujise, K. ( 2001) Characterization of fortilin, a novel antiapoptotic protein. J. Biol. Chem. 276, 47542–47549 [DOI] [PubMed] [Google Scholar]
- 66.Choe, C. U., and Ehrlich, B. E. ( 2006). The inositol 1,4,5-trisphosphate receptor (IP3R) and its regulators: sometimes good and sometimes bad teamwork. Sci. STKE 2006, re15 [DOI] [PubMed]
- 67.Pearce, M. M., Wang, Y., Kelley, G. G., and Wojcikiewicz, R. J. ( 2007) SPFH2 mediates the endoplasmic reticulum-associated degradation of inositol 1,4,5-trisphosphate receptors and other substrates in mammalian cells. J. Biol. Chem. 282, 20104–20115 [DOI] [PubMed] [Google Scholar]
- 68.Garcia, A., Cayla, X., and Sontag, E. ( 2000) Protein phosphatase 2A: a definite player in viral and parasitic regulation. Microbes Infect. 2, 401–407 [DOI] [PubMed] [Google Scholar]
- 69.Abraham, D., Podar, K., Pacher, M., Kubicek, M., Welzel, N., Hemmings, B. A., Dilworth, S. M., Mischak, H., Kolch, W., and Baccarini, M. ( 2000) Raf-1-associated protein phosphatase 2A as a positive regulator of kinase activation. J. Biol. Chem. 275, 22300–22304 [DOI] [PubMed] [Google Scholar]
- 70.Adams, D. G., Coffee, R. L., Jr., Zhang, H., Pelech, S., Strack, S., and Wadzinski, B. E. ( 2005) Positive regulation of Raf1-MEK1/2-ERK1/2 signaling by protein serine/threonine phosphatase 2A holoenzymes. J. Biol. Chem. 280, 42644–42654 [DOI] [PubMed] [Google Scholar]
- 71.Jaumot, M., and Hancock, J. F. ( 2001) Protein phosphatases 1 and 2A promote Raf-1 activation by regulating 14–3-3 interactions. Oncogene 20, 3949–3958 [DOI] [PubMed] [Google Scholar]
- 72.Ory, S., Zhou, M., Conrads, T. P., Veenstra, T. D., and Morrison, D. K. ( 2003) Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14–3-3 binding sites. Curr. Biol. 13, 1356–1364 [DOI] [PubMed] [Google Scholar]
- 73.Alessi, D. R., Cuenda, A., Cohen, P., Dudley, D. T., and Saltiel, A. R. ( 1995) PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 270, 27489–27494 [DOI] [PubMed] [Google Scholar]
- 74.Matheny, S. A., Chen, C., Kortum, R. L., Razidlo, G. L., Lewis, R. E., and White, M. A. ( 2004) Ras regulates assembly of mitogenic signalling complexes through the effector protein IMP. Nature 427, 256–260 [DOI] [PubMed] [Google Scholar]
- 75.Dougherty, M. K., Muller, J., Ritt, D. A., Zhou, M., Zhou, X. Z., Copeland, T. D., Conrads, T. P., Veenstra, T. D., Lu, K. P., and Morrison, D. K. ( 2005) Regulation of Raf-1 by direct feedback phosphorylation. Mol. Cell 17, 215–224 [DOI] [PubMed] [Google Scholar]
- 76.Chang, F., Steelman, L. S., Shelton, J. G., Lee, J. T., Navolanic, P. M., Blalock, W. L., Franklin, R., and McCubrey, J. A. ( 2003) Regulation of cell cycle progression and apoptosis by the Ras/Raf/MEK/ERK pathway (Review). Int. J. Oncol. 22, 469–480 [PubMed] [Google Scholar]
- 77.Zimmermann, S., and Moelling, K. ( 1999) Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 286, 1741–1744 [DOI] [PubMed] [Google Scholar]
- 78.del Peso, L., Gonzalez-Garcia, M., Page, C., Herrera, R., and Nunez, G. ( 1997) Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 278, 687–689 [DOI] [PubMed] [Google Scholar]
- 79.Chen, R., Kim, O., Yang, J., Sato, K., Eisenmann, K. M., McCarthy, J., Chen, H., and Qiu, Y. ( 2001) Regulation of Akt/PKB activation by tyrosine phosphorylation. J. Biol. Chem. 276, 31858–31862 [DOI] [PubMed] [Google Scholar]
- 80.Conus, N. M., Hannan, K. M., Cristiano, B. E., Hemmings, B. A., and Pearson, R. B. ( 2002) Direct identification of tyrosine 474 as a regulatory phosphorylation site for the Akt protein kinase. J. Biol. Chem. 277, 38021–38028 [DOI] [PubMed] [Google Scholar]
- 81.Olsen, J. V., Blagoev, B., Gnad, F., Macek, B., Kumar, C., Mortensen, P., and Mann, M. ( 2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 82.Scheid, M. P., Marignani, P. A., and Woodgett, J. R. ( 2002) Multiple phosphoinositide 3-kinase-dependent steps in activation of protein kinase B. Mol. Cell. Biol. 22, 6247–6260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shao, Z., Bhattacharya, K., Hsich, E., Park, L., Walters, B., Germann, U., Wang, Y. M., Kyriakis, J., Mohanlal, R., Kuida, K., Namchuk, M., Salituro, F., Yao, Y. M., Hou, W. M., Chen, X., Aronovitz, M., Tsichlis, P. N., Bhattacharya, S., Force, T., and Kilter, H. ( 2006) c-Jun N-terminal kinases mediate reactivation of Akt and cardiomyocyte survival after hypoxic injury in vitro and in vivo. Circ. Res. 98, 111–118 [DOI] [PubMed] [Google Scholar]
- 84.Yang, X. J. ( 2005) Multisite protein modification and intramolecular signaling. Oncogene 24, 1653–1662 [DOI] [PubMed] [Google Scholar]
- 85.Hale, B. G., Jackson, D., Chen, Y. H., Lamb, R. A., and Randall, R. E. ( 2006) Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc. Natl. Acad. Sci. U. S. A. 103, 14194–14199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Street, A., Macdonald, A., Crowder, K., and Harris, M. ( 2004) The Hepatitis C virus NS5A protein activates a phosphoinositide 3-kinase-dependent survival signaling cascade. J. Biol. Chem. 279, 12232–12241 [DOI] [PubMed] [Google Scholar]
- 87.Wang, G., Barrett, J. W., Stanford, M., Werden, S. J., Johnston, J. B., Gao, X., Sun, M., Cheng, J. Q., and McFadden, G. ( 2006) Infection of human cancer cells with myxoma virus requires Akt activation via interaction with a viral ankyrin-repeat host range factor. Proc. Natl. Acad. Sci. U. S. A. 103, 4640–4645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Werden, S. J., Barrett, J. W., Wang, G., Stanford, M. M., and McFadden, G. ( 2007) M-T5, the ankyrin repeat, host range protein of myxoma virus, activates Akt and can be functionally replaced by cellular PIKE-A. J. Virol. 81, 2340–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.