

Abstract
Inositol hexakisphosphate kinases (IP6Ks) phosphorylate inositol hexakisphosphate (InsP6) to yield 5-diphosphoinositol pentakisphosphate (5-[PP]-InsP5 or InsP7). In this study, we report the characterization of a selective inhibitor, N2-(m-(trifluoromethy)lbenzyl) N6-(p-nitrobenzyl)purine (TNP), for these enzymes. TNP dose-dependently and selectively inhibited the activity of IP6K in vitro and inhibited InsP7 and InsP8 synthesis in vivo without affecting levels of other inositol phosphates. TNP did not inhibit either human or yeast Vip/PPIP5K, a newly described InsP6/InsP7 1/3-kinase. Overexpression of IP6K1, -2, or -3 in cells rescued TNP inhibition of InsP7 synthesis. TNP had no effect on the activity of a large number of protein kinases, suggesting that it is selective for IP6Ks. TNP reversibly reduced InsP7/InsP8 levels. TNP in combination with genetic studies was used to implicate the involvement of two pathways for synthesis of InsP8 in yeast. TNP induced a fragmented vacuole phenotype in yeast, consistent with inhibition of Kcs1, a Saccharomyces cerevisiae IP6K. In addition, it also inhibited insulin release from Min6 cells in a dose-dependent manner further implicating InsP7 in this process. TNP thus provides a means of selectively and rapidly modulating cellular InsP7 levels, providing a new and versatile tool to study the biological function and metabolic relationships of inositol pyrophosphates.
Inositol(1,4,5)trisphosphate (Ins(1,4,5)P3)2 is the cytosolic product of inositol phospholipid-specific phospholipase Cs and serves multiple biological functions. In higher eukaryotes, it regulates Ca2+ release from intracellular stores via binding to Ins(1,4,5)P3-specific receptors located in the endoplasmic reticulum. In addition, in Saccharomyces cerevisiae and all other eukaryotes that have been studied, Ins(1,4,5)P3 also undergoes complex metabolism to generate a series of inositol polyphosphates with diverse functions (1, 2). Inositol hexakisphosphate (InsP6) can be synthesized in inositol phospholipid-specific phospholipase C/Ins(1,4,5)P3-mediated pathways in most, if not all, eukaryotes. InsP6 is further metabolized by the inositol hexakisphosphate kinases (IHPKs or IP6Ks), which add a pyrophosphate moiety at the 5-position to generate 5-[PP]-InsP5 or 5-InsP7. Dictyostelium discoideum and S. cerevisiae, each possess one IP6K gene product designated Kcs1 in yeast (3, 4). In mammals, three IP6Ks have been identified (5, 6). In addition, a second InsP6/5-InsP7 kinase, designated Vip/PPIP5K, has been identified in yeast (7) and mammalian cells (8, 9). This kinase is distinct from IP6K/Kcs1 in that it phosphorylates the 1/3-position of InsP6 and 5-[PP]-InsP5 (10).
Insights into the biological functions of inositol pyrophosphates have come from genetic studies in yeast and by manipulating expression of inositol polyphosphate kinases in mammalian cells. More recently, yeast mutants failing to synthesize inositol pyro phosphate molecules have been found to be impaired in several cellular functions, such as DNA repair, chromatin remodeling, and telomere length maintenance (11–14). Production of 1/3-InsP7 is necessary for regulating cellular phosphate starvation responses through the inhibition of the cyclin/cyclin-dependent kinase (7, 10, 15). In D. discoideum, InsP7 has been reported to be important in chemotaxis (3). In mammals, overexpression of the IP6K1 increases insulin release from pancreatic β cells, whereas overexpression of IP6K2 increases apoptosis in a variety of cell lines (16, 17).
As a means to further study the roles of inositol pyrophosphate messengers, we set out to develop pharmacological tools that permit acute inhibition of specific inositol polyphosphate kinases. Here we report the characterization of an IP6K inhibitor, which appears to be selective in vitro and in vivo and its use in furthering our understanding of the role of InsP7 synthesis in yeast and in insulin secretion in mammalian cells.
EXPERIMENTAL PROCEDURES
Materials
Ip3-3 kinase inhibitor (TNP; see “Results”) and thapsigargin (TG) were from Calbiochem. [2-3H]Inositol and [γ-32P]ATP were from GE Healthcare. HeLa cells were from ATCC. PEI-cellulose TLC sheets were from Merck. Reduced glutathione was from Sigma. Oligonucleotides were from the University of Dundee in-house DNA synthesis center.
Methods
Cloning—The human IP6K1 and the catalytic fragment of IP3-3KA were cloned from IMAGE clones using the following primers: IP6K1, 5′-GCATGAATTCATGTGTGTTTGTCAAAC-3′ and 5′-GCATGTCGACctactggttctcgtcccgcatc-3′; IP3-3KA, 5′-CatgGAATTCggcaacgtgcagctggaagcg-3′ and 5′-CATGGTCGACTCATCTCTCAGCCAGGCTGGCC-3′.
The primers include the recognition sequences for EcoRI and SalI, respectively. The genes were amplified using PCR using the Hifidelity PCR enzyme (Roche Applied Science). IP6K1 was cloned into the mammalian expression vector enhanced green fluorescence protein (Clontech), whereas the catalytic fragment of IP3-3KA was cloned into pGEX-6P-1 (for bacterial expression) as EcoRI-SalI fragments. Clones were sequenced in the in-house sequencing center.
Cell Culture—HeLa (human cervical cancer) cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with fetal bovine serum (10%, v/v) at 37 °C in a humidified 5% CO2, 95% air environment. HeLa stable cell lines were generated by transfecting cells grown on 3.5-cm dishes with appropriate amounts of vector DNA of enhanced green fluorescence protein or enhanced green fluorescence protein-IP6K1 using the transfection reagent polyethyleneimine (10 μg/well) at ∼70% confluence. We routinely achieved about 90% transfection efficiency by this method. After 48 h, the medium was replaced by similar medium as above but containing 0.8 mg/ml G418 sulfate. Stably expressing clones were selected after 3 weeks of growth in selection medium. Clones were identified by observing GFP fluorescence microscopically and by monitoring protein expression by Western blotting using anti-GFP antibody (Abcam).
Min6 cells were maintained in Dulbecco's modified Eagle's medium (high glucose) containing heat-inactivated serum (15%, v/v) and 72 μm β-mercaptoethanol at 37 °C in a humidified 5% CO2, 95% air environment. 40,000 cells were seeded into each well of a 96-well plate. They were subsequently cultured for 48 h in 200 μl of the above medium. Cells were transfected 16 h postseeding with 0.2 μg of HA-IP6K1 plasmid DNA and 0.5 μl of Lipofectamine 2000/well.
Expression and Assay of IP6K1 Activity—HA-tagged IP6K1 plasmid (10 μg) was transfected into 0.75 × 106 HeLa cells plated on 10-cm2 dishes. 48 h after transfection, cells were lysed in lysis buffer (20 mm Tris-Cl, pH 7.5, 100 mm NaCl, 1% (v/v) Triton X-100, 1 mm Na3VO4, 1 mm NaF, 1 mm sodium pyrophosphate, 5 mm sodium β-glycerophosphate, 1 mm β-mercaptoethanol, and 20% (v/v) glycerol). Lysates were clarified by centrifugation at 10,000 × g at 4 °C for 30 min. 5 μg of mouse monoclonal anti-HA antibody was incubated with 0.5 mg of the above lysate with 50 μl of protein G beads on a shaking nutator for 3 h at 4 °C. The lysate was aspirated off, and the beads were washed twice with 1 ml of lysis buffer and twice with 1 ml of assay buffer (20 mm Tris-Cl, pH 7.5, 100 mm NaCl, 5 mm MgCl2, 1 mm Na2OVO4, 1 mm sodium pyrophosphate, 1 mm NaF, 5 mm sodium β-glycerophosphate, and 1 mm β-mercaptoethanol) before electrophoresis for Western blot or activity assays. For activity assays, the beads were finally suspended in 200 μl of lysis buffer containing 5 μl of [2-3H]InsP6 (specific activity = 21.4Ci/mmol, 5 μCi/0.5 ml) and appropriate concentrations of TNP or DMSO. Final concentration of DMSO was 1% (v/v). The reaction was started by the addition of ATP at a final concentration of 1 mm. Control reactions contained no ATP. Tubes were incubated at 37 °C on a shaking nutator for 1 h. Reactions were stopped by the addition of 300 μl of 1 mm EDTA. Reactions were centrifuged briefly, and the supernatant was transferred to a fresh tube. [2-3H]InsP7 produced was separated from the [2-3H]InsP6, using HPLC (see below). [2-3H]InsP7 produced was estimated using the ratio of the DPM in the corresponding peak to the total DPM in both peaks. [2-3H]InsP7 was plotted against the corresponding TNP concentration for curve fitting (Sigma Plot software) and calculation of IC50 values.
Expression and Purification of the Catalytic Fragment of IP3-3KA—The catalytic fragment of IP3-3KA was expressed as a GST fusion protein in the BL21 Escherichia coli strain. 1 liter of E. coli was grown to an OD of 1.0 at 37 °C. Protein expression was induced by the addition of 0.5 mm isopropyl 1-thio-β-d-galactopyranoside and reduction of the bacterial growth temperature to 26 °C. The bacteria were harvested 18 h after induction with isopropyl 1-thio-β-d-galactopyranoside by centrifugation at 4000 rpm for 40 min at 4 °C. The pellet was resuspended in lysis buffer (50 mm Tris-HCl, pH 7.5, containing 100 mm NaCl, 1% (v/v) Triton X-100, 1 mm benzamidine, 1 mm phenylmethylsulfonyl fluoride, 1 mm β-mercaptoethanol, and complete protease inhibitor tablet from Roche Applied Science). Cells were ruptured by sonication (six 15-s bursts with 45 s of cooling between two bursts) on an Ultrasonic processor (VCX-750W) sonicator at 65% power. Subsequently, the sonicate was centrifuged at 18,590 × g for 40 min at 4 °C on an Avanti J-25 centrifuge. The supernatant was incubated with 1 ml of washed (with lysis buffer) glutathione-Sepharose 6B beads (Amersham Biosciences) for 2 h at 4 °C. Following incubation, the supernatant was aspirated off, and beads were washed with 50 ml of wash buffer (lysis buffer with no Triton X-100). Finally, the protein was eluted from beads with elution buffer (wash buffer plus 2 mm reduced glutathione). Protein was electrophoresed on a Novex 4–12% BisTris gel in 1× MOPS running buffer (Invitrogen), and the gel was Coomassie-stained for verifying the purity of the protein. The protein was concentrated and stored at a final concentration of 2 mg/ml in wash buffer plus 50% glycerol (v/v) at -20 °C.
IP3-3K Assays—30 nm IP3-3KA was incubated with 0.1 μCi of [γ-32P]ATP and 5 μm Ins(1,4,5)P3 in assay buffer (20 mm Tris-HCl, pH 7.5, plus 100 mm NaCl, 1 mm dithiothreitol, 5 mm MgCl2). 1 μl of the assay was spotted onto PEI-cellulose TLC sheets. Samples were chromatographed in 2.5 m ammonium formate plus 1.0 m formic acid to separate out [32P]Ins(1,3,4,5)P4, [γ-32P]ATP, and any inorganic phosphate ([32P]Pi). The sheets were dried and then scanned using a PhosphorImager. The [32P]Ins(1,3,4,5)P4 formed was measured by calculating density corresponding to [32P]Ins(1,3,4,5)P4 as a percentage of the total density of [γ-32P]ATP plus [32P]Ins(1,3,4,5)P4. Percentage of [32P]Ins(1,3,4,5)P4 produced was plotted against TNP concentration. Curve fitting to the data was done using the Sigma Plot software. Km/IC50 values were calculated from the equation provided by the software.
Calculation of Ki Values—Ki values were determined using the equation,
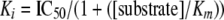 |
(Eq. 1) |
where IC50 represents the value determined from curve fit analysis, Km is the value for ATP, and [substrate] is the concentration of ATP used in the IP3-3K or IP6K1 assay, respectively.
Vip/PPIP5K Assays—100 ng of purified Homo sapiens Vip2 fragment (amino acids 1–365) or S. cerevisiae Vip1 was preincubated with vehicle (DMSO) or 10 μm TNP for 30 min on ice. Following the pretreatment, [γ-32P]ATP and 100 μm InsP6 was added to the enzyme, and the assay was carried out at 37 °C for 60 min as per the protocol reported earlier by Fridy et al. (28). [32P]Pi, [32P]ATP, [32P]InsP7, and [32P]InsP8 formed during the assay were visualized after separation by PEI-cellulose thin layer chromatography in a tank containing 1.09 m KH2PO4, 0.72 m K2HPO4, and 2.13 m HCl and subsequent phosphorimaging.
[2-3H]Inositol Labeling of Cells—HeLa cells were seeded at 1 × 106 cells on 60-mm plates in 3 ml of inositol-free medium supplemented with dialyzed fetal calf serum (10% (v/v), molecular weight cut-off = 10,000; Invitrogen) and 50 μCi of [2-3H]-inositol for 4 days. At the time of TNP treatment or sorbitol stimulation, the plates were 90–95% confluent. TNP treatment of cells was carried out as follows: 3 μl from a 1000× stock of TNP (in DMSO) was added to cells labeled with [2-3H]inositol as mentioned above and gently swirled. For controls, 3 μl of DMSO was added to cells. The DMSO concentration was 0.1% of the total volume of medium in all cases. Cell treatment with TG was also carried out using a similar procedure. For stimulation with sorbitol, appropriate aliquots from a 2 m stock solution of sorbitol was added to cells with or without TNP at various concentrations for 2 h at 37 °C. To check for reversibility of TNP inhibition, after the labeled cells were treated with TNP, medium was aspirated off, and cells were washed three times with 2 ml of inositol-free medium supplemented with dialyzed fetal calf serum. Cells were then allowed to recover for 2 h before the inositol phosphates were extracted.
Extraction and Analysis of Inositol Polyphosphates by HPLC—Stimulated or transfected cells were washed with ice-cold PBS and then lysed with 0.5 m trichloroacetic acid. Subsequently, cells were scraped, and the supernatant was separated from the debris by centrifugation in a cold microcentrifuge for 10 min at full speed. The supernatant was neutralized by the addition of 1 m K2CO3. The neutralized solution was once again centrifuged, as mentioned above. Finally, the supernatant was loaded onto a Whatman Partisphere SAX column. The inositol phosphates were eluted from the column using 1 mm EDTA (buffer A) and 1 mm EDTA plus 1.3 m (NH4)2HPO4, pH 3.85 (buffer B). The gradient was as follows: initial loading at 0% buffer B for 10 min, 0–35% buffer B in the next 15 min, followed by 35–100% buffer B in the next 65 min, followed by a high salt wash of 100% buffer B for the next 5 min and a wash with 100% buffer A for the final 10 min. The flow rate was 1 ml/min throughout the gradient, and 1-ml fractions were collected. This gradient was standardized for optimal separation of InsP6, which typically elutes at fraction 61, InsP7 ([PP]-InsP5) at fraction 71, and InsP8 ([PP]2-InsP4) at fraction 81.
Radioactive Labeling of Yeast and Inositol Phosphate Extraction—2 × 103 cells/ml WT, kcs1Δ, vip1Δ, kcs1vip1ΔΔ, ddp1Δ, and ddp1kcs1ΔΔ were grown in synthetic minimal medium supplemented with 50 μCi/ml [3H]inositol. Inositol phosphates were extracted, as explained above. TNP at a final concentration of 10 μm was added to growth medium, and cells were allowed to grow overnight at 30 °C. For controls, equal amounts of DMSO were added to the culture, and the final concentration of DMSO was 0.001% (v/v). The cells went through ∼8–9 divisions in labeling medium. Cells were pelleted by centrifugation at 5000 rpm for 10 min in a refrigerated microcentrifuge. They were washed with YPD once and lysed by incubation with 0.5 m trichloroacetic acid on ice for 30 min. The debris was pelleted by centrifugation at full speed in a refrigerated microcentrifuge. The supernatant was neutralized by the addition of 1 m K2CO3, and then the inositol phosphates were separated by strong anion exchange chromatography, as explained above.
Yeast Culture and Fluorescence Labeling of Vacuoles—Wild type yeast, Ipk2Δ, and Kcs1Δ (all isogenic to BY4741) were grown in YPD (YP + 2% glucose (w/v)) medium at 30 °C. TNP at a final concentration of 10 μm was added to growth medium, and cells were allowed to grow for 2 h. For controls, equal amounts of DMSO were added to the culture, and the final concentration of DMSO was 0.001% (v/v). For labeling vacuoles, the yeast vacuole marker sampler kit from Molecular Probes was used. Briefly, 5 ml of yeast in exponential growth phase (A600 = 0.4–0.6) were pelleted by centrifugation and resuspended in 10 mm Hepes-NaOH buffer, pH 7.4, containing 5% glucose (w/v). Vacuoles were stained with Cell Tracker Blue CMAC and yeast vacuole membrane marker MDY64 according to the manufacturer's instructions. 1 μl was mounted onto slides, and cells were visualized using a Delta Vision microscope with appropriate filter sets.
Insulin Release Assay from Min6 Cells—Medium was removed, and cells were washed 10 times with glucose-free KREBS (10 mm Hepes-NaOH, pH 7.4, 119 mm NaCl, 4.74 mm KCl, 2.54 mm CaCl2, 1.19 mm MgCl2, 1.19 mm KH2PO4, 25 mm NaHCO3, and 1 mm EGTA) to remove traces of culture medium and serum. Cells were treated with various concentrations of TNP for 2 h in 200 μl of glucose-free KREBS under the same conditions of temperature and CO2 as under culturing conditions. Following treatment with TNP, the glucose-free KREBS was replaced with KREBS containing 2.5 mm glucose and TNP at the same concentrations. 5 min later, aliquots of cell supernatants were taken out and used for measurement of insulin release. Insulin released was measured using the rat/mouse insulin enzyme-linked immunosorbent assay kit (Linco Laboratories), as per the manufacturer's instructions. For each assay, a standard curve was generated using purified insulin provided in the kit. Aliquots of cell supernatants withdrawn during the assay were diluted 1:10 with glucose-free KREBS to give a final absorbance value within the linear range of the standard curve. The amount of insulin released was calculated from the standard curve generated. Blanks were stimulated with glucose-free KREBS.
RESULTS
IP6Ks belong to the IPK superfamily of inositol polyphosphate kinases, also comprising IP3-3Ks and IPMKs by virtue of significant homology in their sequences. A common structural fold has been reported for IP3-3KA from mammals and Ipk2p from S. cerevisiae, where the N and C termini of the proteins coordinate ATP binding, and the middle of the protein forms the inositol phosphate binding domain (18, 19). Since IP6Ks share sequence homology in the ATP and inositol phosphate binding regions with the other members of the IPK family, it is possible that they share a similar structural fold. Alignment of the ATP and inositol phosphate (IP) binding regions from human IP3-3KA, IP6K1-3, Kcs1p, and Ipk2p are shown in Fig. 1. Although both IP3-3KA and Kcs1p contain N-terminal sequences (not shown) that do not exist in the IP6Ks or Ipk2p, all of the members of the IPK family show high levels of conservation in their ATP binding regions and IP binding regions but little sequence conservation elsewhere (18, 20). For example, each contains the conserved “GH” (residues 34 and 35 in IP6K1) and the “SLL” motifs (residues 334–336) important for ATP binding and the fully conserved “PXXXDXK” motif (residues 221–226) required for IP binding.
FIGURE 1.

ClustalW alignment of the IPK family members. The following proteins are aligned: hIP3-3KA, hIP6K1, hIP6K2, hIP6K3, Sckcs1, and Scipk2, where the prefix “h” represents H. sapiens and “Sc” represents S. cerevisiae. Accession numbers are as follows: IP3-3KA, NP_002211; IP6K1, NP_695005; IP6K2, NP_057375; IP6K3, NP_473452; Sckcs1, NP_010300; Scipk2p, NP_010458. Amino acids highlighted in black boxes indicate identical residues. Amino acids highlighted in gray boxes indicate the conserved nature of the amino acids (such as hydrophobicity, charge, etc.). The numbers indicate the amino acids from the corresponding protein shown in that line.
A family of molecules having a purine base with either a nitrobenzyl (TNP; Fig. S1A), aminobenzyl (TAP; Fig. S1B), or benzyl (TBP; Fig. S1C) side chain attached at the N-6 position and an (m-(trifluoromethyl)benzyl) attached at the N-2 group were characterized as inhibitors for IP3-3K from a panel of purine-based molecules. Of these, TNP was found to have the lowest Ki value (4.3 μm) for IP3-3K (21). We have now characterized this compound as a potential inhibitor for IP6Ks.
TNP Is a Selective Inhibitor of the IP6Ks in Vitro and in Vivo—To determine if IP6Ks and IP3-3Ks are inhibited by TNP, we performed in vitro activity assays with both enzymes in the presence of increasing concentrations of TNP. For this purpose, HA-tagged IP6K1 was expressed and immunoprecipitated from HeLa cells. The Western blot of the immunoprecipitated enzyme is shown in Fig. 2A. The immunoprecipitated enzyme was incubated with 1 mm ATP and [3H]InsP6, and production of [3H]InsP7 was monitored using HPLC separation (Fig. 2B). The formation of InsP7 was inhibited by the presence of TNP with an IC50 value of 0.47 μm determined from curve fitting (Fig. 2, B and C). Taking account of the Km for ATP and the ATP concentration in the assay, this translates to a Ki of 0.24 μm (Table 1).
FIGURE 2.

Inhibition of IP6K1 and the IP3-3KA activity by TNP in vitro. A, Western blot (WB) analyses of HA-tagged IP6K1 immunoprecipitated (IP) from HeLa cell lysates transiently transfected with HA-tagged IP6K1, which was used for the activity assays shown in Fig. 3B. The protein was immunoprecipitated using anti-HA antibody (5 μg/mg lysate) and analyzed using the anti-Ip6 kinase panantibody. The molecular mass standards (Prescision Plus Protein dual colored standards from Bio-Rad) are indicated. B, HPLC separation of input [2-3H]InsP6 and [2-3H]InsP7 formed during the IP6K1 assay in the presence of DMSO (filled squares) or TNP at 100 nm (open squares), 1 μm (filled circles), or 10 μm (open circles). During the assay, the immunoprecipitated enzyme was incubated with [2-3H]InsP6 and 1 mm ATP in 200 μl of assay buffer for 1 h at 37 °C on a shaking nutator in the presence of DMSO or TNP (various concentrations). C, determination of IC50 of TNP for IP6K. The percentage InsP7 formed is plotted against TNP concentration. InsP7 formed is calculated as the fraction of the radioactivity in the InsP7 peak to the total radioactivity in InsP6 + InsP7 peaks and expressed as a percentage. The error bars show the range of data obtained from duplicates shown in the chromatogram. Curve fitting to the data was done using the Sigma Plot software. D, purity of the IP3-3KA. 50 ng of purified recombinant fragment of IP3-3KA containing the catalytic domain (∼61 kDa) was electrophoresed on a 4–12% Novex BisTris gel in MOPS running buffer. The gel was stained with Coomassie Blue. The molecular mass standards (same as in A) are indicated. E, dose-dependent inhibition of IP3-3KA activity by TNP. Purified IP3-3KA was incubated with [γ-32P] ATP and 5 μm Ins(1,4,5)P3 in assay buffer at 37 °C for 30 min in the presence of the indicated concentrations of TNP. [32P]Pi,[32P]ATP, and [32P]Ins(1,3,4,5)P4 formed during the IP3-3KA activity assay were visualized after separation by PEI-cellulose thin layer chromatography and phosphorimaging. Positions of reactants and origin are marked on the right. The two leftmost lanes marked C are γ-[32P]ATP controls in assay buffer lacking enzyme. F, determination of IC50 of TNP for IP3-3KA. Ins(1,3,4,5)P4 formed was plotted against the corresponding TNP concentration, by quantifying chromatograms in E using the AIDA software. Curve fitting to the data was done using the Sigma Plot software. The error bars show the range of data obtained from duplicates assayed separately.
TABLE 1.
Inhibition of different IPK family members by TNP
For in vitro studies with IP3-3KA, a fragment of the kinase containing the catalytic domain was purified according to a protocol published earlier (19). The Coomassie Blue-stained gel shows that the major band consists of the recombinant fragment of IP3-3KA (Fig. 2D). The purified fragment was incubated with [γ-32P]ATP and Ins(1,4,5)P3, and production of [32P]Ins(1,3,4,5)P4 was monitored in the presence of increasing doses of TNP by PEI-TLC separation of ATP and Ins(1,3,4,5)P4 and subsequent radioimaging (Fig. 2E). The formation of Ins(1,3,4,5)P4 decreased with increasing TNP, and the IC50 value calculated from curve fitting was around 18 μm from two different batches of protein purified (Fig. 2, E and F). In this case, the IC50 and Ki value are equivalent due to the low ATP concentrations in the assay. The Ki of 18 μm (Table 1) is somewhat higher than the values reported by other authors (21, 22).
The in vitro data translate to an ∼30-fold difference in the Ki values for TNP between IP6K1 and IP3-3KA (Table 1), whereas the Km values for ATP were ∼100-fold apart. From these data, the following conclusions were drawn. 1) When the ATP concentration is in the millimolar range (such as that within cells), the Km values indicate that the IP3-3KA would be fully saturated at the ATP binding site, whereas the IP6K1 would only be partially saturated at the same site. 2) Since TNP is a purine analog (Fig. S1A) and is an ATP competitor, these observations lead to the prediction that the in vivo selectivity of TNP should be ∼1000-fold in favor of the IP6K1 over IP3-3KA. The above prediction was tested by treating [3H]inositol-labeled HeLa cells with increasing doses of TNP and quantifying changes in inositol phosphate levels. Fig. 3A shows the HPLC separation of the various inositol phosphates from equilibrium-radiolabeled cells. Since InsP6 levels remained fairly constant over the range of TNP concentrations used in this experiment, we used the radioactivity in the InsP6 peak to normalize the levels of the other inositol phosphates. The levels of InsP3 and InsP4 (containing a mixture of isomers) were fairly constant over the tested range of TNP concentrations (Fig. 3B). InsP5 levels were also constant, whereas InsP7 levels decreased in response to the same range of TNP concentrations. Curve fitting yielded an IC50 value of 0.55 μm for the InsP7 response (Fig. 3C). Hence, the predictions from the in vitro biochemical data appear to be ratified by the in vivo data. The unchanging InsP5 levels provide further support for in vivo selectivity of TNP for IP6Ks, because InsP5 is the product of the multikinase, another member of the IPK family that has a relatively low Km (micromolar order) for ATP (23–25).
FIGURE 3.

Inhibition of InsP7 formation in TNP-treated HeLa cells. A, HPLC separation of inositol phosphates extracted from HeLa cells stimulated with DMSO (filled circles) or TNP at 100 nm (open circles), 1 μm (filled squares), or 10 μm (open squares) for 2 h at 37 °C. The cells were grown in inositol-free medium supplemented with 10% (v/v) dialyzed fetal calf serum and 50 μCi of [2-3H]inositol for 3 days, and inositol phosphates were extracted (see “Experimental Procedures”). Since the InsP6 peak did not change by more than 10% between all of the conditions (average in control cells = 564,651 dpm and average in cells treated to 10 μm TNP = 599,011 dpm), the dpm in the InsP6 peak was used to normalize levels of other inositol phosphates. In control HeLa cells, the percentages of InsP3, InsP4, InsP7, and InsP8 with respect to InsP6 are 12, 4, 6, and 0.7%, respectively. Similar values for InsP7 and InsP8 have been reported in DDT1-MF2 cells and HEK cells (30). B, change in InsP5 (filled triangles), InsP3 (filled squares), and InsP4 (open squares) normalized to InsP6 in the presence of increasing TNP. In each case, the total radioactivity under the corresponding peak (base line-subtracted) was divided by the total radioactivity under the InsP6 peak (base line-subtracted) and expressed as a percentage. Data are the mean of two different experiments, each an average of triplicate assays, and error bars represent the S.D. of the data. C, TNP-induced dose-dependent decrease in InsP7. For each concentration of TNP, radioactivity in the [3H]InsP7 peak was normalized to that in the [3H]InsP6 peak. Data are the average of triplicate experiments, and error bars represent the S.D. of the data. Sigma Plot software was used to carry out curve fitting to the InsP7 data, and the IC50 calculation was carried out from the equation used to fit the curve.
We next tested if the decreases in cellular InsP7 caused by TNP treatment could be rescued by overexpression of IP6Ks. HeLa cells transformed with IP6K1 were labeled with [3H]inositol to equilibrium, and InsP7 levels were evaluated as a function of TNP concentration. Fig. 4A shows the HPLC separation of InsP6 and InsP7 isolated from cells overexpressing IP6K1 and control cells, treated with different concentrations of TNP. IP6K1-overexpressing cells had 2-fold more InsP7 than control cells (Fig. 4B). Following TNP treatment, InsP7 levels decreased in parallel in the two cell systems. As expected, IC50 values were not significantly different (2.0 and 1.2 μm), but at the IC50 value, IP6K1-overexpressing cells had InsP7 levels comparable with noninhibited control cells, and at 10 μm TNP, the residual InsP7 was ∼50% of that in untreated controls. Hence, IP6K1 overexpression rescues InsP7 synthesis in the face of treatment with TNP. TNP had a similar effect on InsP7 levels in cells overexpressing IP6K2 and -3 as those overexpressing IP6K1 (Fig. S2). Since all three mammalian IP6Ks share significant homology with each other and all of them have similar Km values for ATP, the 90% reduction in InsP7 levels in cells overexpressing IP6K1 (Fig. 4B) or IP6K2 or -3 (Fig. S2) strongly suggests that the effect of TNP on InsP7 occurs via direct inhibition of one or all three kinases.
FIGURE 4.

IP6K1 over-expression rescues TNP inhibition of InsP7 synthesis. A, HPLC of InsP6 and InsP7 from HeLa cells overexpressing GFP-IP6K1 labeled with [3H]inositol. Cells were stimulated with DMSO (filled circles) or TNP at 100 nm (open circles); 1 μm (open triangles), and 10 μm (closed triangles) for 2 h at 37 °C. The soluble inositol phosphates were extracted and separated as explained under “Experimental Procedures.” B, decrease in InsP7 from HeLa cells overexpressing GFP (filled circles) or GFP-IP6K1 (open circles) as a function of TNP concentration. Data are the average of triplicate experiments, and error bars represent the S.D. of the data. Curve fitting to the InsP7 data was done using the Sigma Plot software, and IC50 calculations were carried out from the equation used to fit the curve.
Previous studies reported an increase in basal calcium levels and a subsequent decrease in the amounts of ATP-stimulated calcium release in cells treated with TNP (21). Such an increase in intracellular calcium could conceivably lead to the observed decrease in InsP7 through indirect mechanisms. To investigate this possibility, InsP7 levels in equilibrium-labeled cells treated with TNP were compared with those in cells treated with TG, which increases intracellular basal levels of calcium by inhibiting the Ca2+ pumps on the ER (26). HPLC separation of the various inositol phosphates isolated from cells treated with TNP and TG are shown in Fig. 5A. Quantification of InsP3, InsP4, and InsP7 as a percentage of InsP6 shows that there are significant differences in InsP7 levels between TG- and TNP-treated cells. TNP-treated cells showed a dramatic 90% reduction in InsP7, whereas TG-treated cells showed only minor changes in InsP7 (20–30%) when compared with InsP7 levels in controls. There was no significant change in InsP3 levels in TG-treated cells (94% of control), and this observation serves as a good negative control, since it has been comprehensively established that exposure to TG increases intracellular calcium without any change in Ins(1,4,5)P3 levels. These data further support the hypothesis that decreases in InsP7 levels following TNP treatment occur due to a specific inhibition of IP6Ks.
FIGURE 5.

Effect of TNP and thapsigargin on InsP7 isolated from equilibrium-labeled cells. A, HPLC profiles of inositol phosphates isolated from HeLa cells equilibrium-labeled with [2-3H]inositol and stimulated with DMSO (filled circles) or TNP (10 μm; filled triangles) or TG(open circles) for 2 h at 37 °C. B, bar chart showing InsP3 (white bars), InsP4 (hatched bars), InsP7 (checkered bars), and InsP8 (black bars) as a percentage of InsP6 in cells treated with DMSO, TNP, or TG. Data are the average of duplicate experiments, and error bars show the range of data.
Additionally, we sought to determine the effects of TNP on the Vip/PPIP5K class of kinases (7–9). In vitro assays were conducted with an active fragment of H. sapiens Vip2 or S. cerevisiae Vip1, in the presence and absence of TNP. Vip2 produced either [PP]-InsP5 (InsP7) or [PP]2-InsP4 (InsP8) when InsP6 was used as the substrate for the reaction, and its activity was unaffected by the presence of TNP (Fig. 6). The activity of S. cerevisiae Vip1 was also unaffected when 5-[PP]-InsP5 was used as a substrate (data not shown). These data demonstrate that Vip/PPIP5K enzymes are not targets of TNP.
FIGURE 6.

Vip/PPIP5Ks are not inhibited by TNP. 100 ng of purified Vip2 fragment (amino acids 1–365 from the human Vip2) was preincubated with vehicle (DMSO) or 10 μm TNP for 30 min on ice. Following the pretreatment, [γ-32P]ATP and 100 μm InsP6 were added to the enzyme, and the assay was carried out at 37 °C for 60 min as per the protocol reported earlier by Fridy et al. (28). [32P]Pi [32P]ATP, [32P]InsP7, and [32P]InsP8 formed during the Vip/PPIP5K activity assay were visualized after separation by PEI-cellulose thin layer chromatography and phosphorimaging. Positions of ATP and products are marked on the right. The leftmost lane marked C is the [γ-32P]ATP control in assay buffer lacking enzyme.
Furthermore, we tested the effects of TNP on a panel of protein kinases. In vitro assays were performed in the presence of 1, 10, and 100 μm TNP, using protocols described earlier (27–29). None of the kinases in the panel were inhibited by TNP even at 100 μm. Since all experiments in this study were carried out at 10 μm concentrations, this observation underlines the fact that TNP is specific for the IPK family.
Since TNP is a purine analog and inhibits IP6Ks via binding to the ATP binding site, the effect of TNP should be reversible. The reversibility of TNP inhibition was monitored by treating HeLa cells with TNP and allowing them to recover. Fig. 7 shows the results of this experiment. Following treatment with TNP, InsP7 levels fell, and when cells were allowed to recover for 2 h after the removal of TNP, InsP7 levels returned close to control levels, showing that TNP inhibition of IP6Ks is reversible.
FIGURE 7.

TNP inhibition of InsP7 synthesis is reversible. A bar chart shows InsP7 as a percentage of InsP6 in HeLa cells treated with DMSO (Control), treated with TNP, or treated with TNP and allowed to recover for 2 h after TNP treatment (Washed). Data are the average of duplicate experiments, and error bars show the range of data.
TNP Is a Useful Pharmacological Tool for Examining the Metabolism and Biological Functions of Inositol Pyrophosphates—Genetic and biochemical analyses demonstrate that InsP8 is produced by the combined action of IP6K and Vip/PPIP5K activities. Two pathways have been proposed. In pathway I, InsP6 is first pyrophosphorylated at the 5-position by IP6K and then at the 1/3-position by Vip/PPIP5K, whereas pathway II is the reciprocal order (Fig. 8A). Given the selective effects of TNP on the IP6K proteins, we postulated that the drug would be a useful probe for acutely monitoring the relative contributions of each InsP8 biosynthetic pathway. TNP inhibition of pathway I would deplete both InsP7 and InsP8, whereas only InsP8 levels would change if TNP were to act through pathway II. Since basal levels of InsP8 in many cell types are near or below the detection limits, we first enhanced its production using hyperosmotic stress, as previously reported (30). To this end, HeLa cells labeled to equilibrium with [3H]inositol were stressed by treatment with 0.2 m sorbitol in the presence or absence of TNP, and inositol polyphosphates were separated and analyzed by HPLC. Sorbitol treatment caused a 5-fold increase in the peak corresponding to InsP8 (Figs. 8, B and C). TNP reduced the levels of both InsP7 and InsP8 in a dose-dependent manner with IC50 values of ∼3.0 and 1.0 μm. The extent of inhibition at the highest dose of TNP used was ∼90%, consistent with the notion that pathway I accounts for the majority of InsP8 synthesis under the equilibrium labeling conditions tested.
FIGURE 8.

Determination of the predominant pathway to InsP8 synthesis in mammalian cells using TNP. A, metabolic pathways that lead to the synthesis of InsP8 or [PP]2-InsP4 in mammalian cells. Pathway I consists of InsP6 being phosphorylated by IP6K to [PP]-InsP5 or InsP7 and further phosphorylation of InsP7 to InsP8 by Vip/PPIP5Ks. Pathway II consists of the Vip/PPIP5Ks phosphorylating InsP6 to InsP7 and the IP6Ks subsequently phosphorylating the InsP7 to InsP8 (7–9). Ddp represents the inositol pyrophosphate phosphatase (34, 35). B, HPLC profiles of higher inositol polyphosphates isolated from [3H]inositol-labeled HeLa cells treated with DMSO (“no sorbitol” controls; closed circles), sorbitol (0.2 m plus DMSO; open circles), and sorbitol (0.2 m) plus 10 μm TNP (filled squares) for 2 h at 37°C. C, the InsP8 peak from A plotted on a smaller scale. The different curves are the same as that described in A. D and E, decrease in InsP7 (filled circles) and InsP8 (open circles) normalized to InsP6 as a function of TNP concentration. Data are the average of triplicate experiments, and error bars represent the S.D. of the data. Curve fitting and calculation of IC50 values for the InsP7 and InsP8 data were done as before.
However, because previous studies have shown that loss of the inositol pyrophosphatase (Ddp1) unmasks or exposes pathway II (12, 31), which is otherwise undetectable by equilibrium radiolabeling methods, we tested TNP inhibition of InsP7 metabolism in a variety of budding yeast strains. In WT cells, the basal level of InsP7 is about 3% of that of InsP6. Upon treatment of wild-type cells with TNP, we observed a loss of InsP7 (Fig. 9A). Similarly, when Kcs1 is deleted, this peak of InsP7 is lost, consistent with inhibition by TNP of pathway I. In further support of pathway I, we observed an ∼3-fold increase in the level of InsP7 in cells deficient for Vip1 (vip1Δ) and found that treatment with TNP or concomitantly deleting Kcs1 (vip1kcs1ΔΔ) ablated InsP7 accumulation (Fig. 9B).
FIGURE 9.

Inhibition of InsP7 formation in WT yeast cells mimics the defectin vacuole morphology reported for ipk2ΔIPK2 and kcs1Δ strains. WT, kcs1Δ, vip1Δ, and kcs1vip1ΔΔ cells were grown in yeast minimal medium supplemented with 50 μCi/ml [3H]inositol overnight at 30 °C. Inositol phosphates were extracted using a protocol similar to that with mammalian cells. HPLC traces are shown in Fig. S3. For vacuole labeling experiments, WT, ipk2Δ, or kcs1Δ strains of haploid yeast cells were grown in YPD. WT cells were grown in the presence of vehicle (DMSO) or TNP. They were then stained with cell tracker CMAC (blue), which stains the vacuolar lumen and membrane marker MDY-64 (green) according to the manufacturer's instructions. Shown is a comparison of InsP7 levels in WT, WT plus TNP, and kcs1Δ cells (A); vip1Δ, vip1Δ+ TNP, and kcs1vip1ΔΔ cells (B); and ddp1Δ, ddp1Δ+TNP and ddp1kcs1ΔΔ cells (C). D (i), WT cells grown in the presence of vehicle (DMSO). D (ii), WT cells grown in the presence of TNP (10 μm). D (iii), ipk2Δ grown in the presence of vehicle (DMSO). D (iv), kcs1Δ grown in presence of vehicle (DMSO).
We confirmed that pathway II could be unmasked by analyzing strains deficient for Ddp1 (ddp1Δ) and observed a 3-fold increase in InsP7 levels upon loss of the phosphatase (Fig. 9C). Treatment with TNP or co-deletion of Kcs1 (ddp1kcsΔΔ) further increased InsP7 (presumably 3-InsP7) levels 5–7-fold (15–21-fold as compared with wild-type cells) (Fig. 9C), consistent with a major flux of metabolism through pathway II, which could only be visualized by equilibrium labeling when Ddp1 was inactivated. Together, our experiments offer support for the presence of both pathway I and II in yeast.
In order to study the phenotypic effects of TNP on cellular functions proposed for [PP]-InsP5, we utilized two biological systems. In S. cerevisiae, loss of the IP6K, Kcs1, results in aberrant vacuole fragmentation and morphology (4, 32). Wild-type yeast strains were therefore treated with TNP for 2 h, and vacuoles were analyzed using the fluorescent probe, Cell Tracker CMAC (blue), which selectively stains the vacuolar lumen. Treatment with TNP rapidly caused the development of multifragmented vacuoles in wild type yeast (Fig. 9D (i)) compared with DMSO-treated controls (Fig. 9D (ii)), a result that is very similar to that which is observed in ipk2Δ and kcs1Δ strains (Figs. 9D, iii and iv). The fact that TNP mimics the vacuolar phenotype observed in these KO strains confirms the utility of TNP as a probe for the biological functions of inositol pyrophosphates and shows for the first time that a relatively acute loss of InsP7 rapidly affects vacuolar dynamics.
We next used a second biological system developed from observations that alterations in InsP7 influence the sensitivity of pancreatic β cells and insulinoma cell lines to glucose-stimulated insulin secretion (16). We therefore followed insulin release from Min6 pancreatoma cells simulated with glucose. These cells showed a robust response to glucose that increased insulin release by about 5-fold compared with basal level, a response that was dose-dependently inhibited by TNP. Overexpression of IP6K1 significantly increased insulin secretion in this model and substantially protected these cells from the inhibitory effects of TNP, suggesting that TNP acts to inhibit secretion via its inhibition of one or more IP6Ks (Figs. 10, A and B). These results strongly support the conclusions of previous work implicating InsP7 in insulin secretion and show that these effects are acute and not due to adaptive responses to sustained alterations in the cellular level of this compound.
FIGURE 10.

Inhibition of insulin released from Min6 cells. A, bar chart showing insulin released by Min6 cells. Cells were pretreated with different concentrations of TNP in glucose-free KREBS for 2 h prior to stimulation of insulin release by 2.5 mm glucose. 5 min after glucose stimulation, insulin contained in the buffer was measured and quantitated using the rat/mouse insulin enzyme-linked immunosorbent assay kit as per the manufacturer's instructions. Data shown are the average of duplicate readings in one experiment. Three such experiments were carried out with similar results. B, bar chart showing insulin released by Min6 cells transfected with IP6K1 and treated with vehicle (DMSO) or TNP (10 μm). Insulin released was measured under similar conditions and at the same time as in A.
DISCUSSION
In the present study, we show that a purine analogue, TNP, originally characterized as an IP3-3K inhibitor (21), is a relatively selective and reversible inhibitor of IP6Ks. The selectivity of TNP for Ip6Ks arises from two factors. Ki values given in Table 1 show that TNP has an absolute potency for IP6K1 ∼70-fold greater than that for IP3-3KA. Km values, however, show that the affinities of these two enzymes for substrate, ATP, also differ markedly but in the reverse direction, with IP3-3Ks having a Km that is 100-fold lower than that of IP6Ks. Because TNP is an ATP site competitive inhibitor of these enzymes and, because cellular ATP levels are in the mm range, well above the Km for IP3-3Ks, these data predict in vivo selectivity of ∼1000-fold. This prediction appears to be borne out by experiments analyzing inositol phosphate levels in [3H]-inositol-labeled HeLa cells where TNP dose-dependently reduced InsP7 and InsP8 levels by at least 90% without significantly affecting any other inositol phosphate analyzed.
Of the several purine-based molecules that were tested for IP3-3K inhibition, in addition to TNP, two other structurally related molecules had IC50 values of around 13.5 μm (Fig. 2, B and C) (21). It seems quite likely that these compounds would be similarly effective as inhibitors of IP6Ks. Apart from TNP and the analogues shown in Fig. 2, adriamycin, a topoisomerase inhibitor, was reported to inhibit production of Ins(1,3,4,5)P4 (33). In our hands, adriamycin did not show any inhibition of IP3-3KA in vitro (data not shown), which may be compatible with speculation that its mode of inhibition was nonspecific. Several plant polyphenols have also been reported to be inhibitors of both IP3-3Ks and IPMKs (24). Although this inhibition might also extend to IP6Ks, most plant polyphenols have other cellular targets that would render them unsuitable for intracellular studies.
The mechanisms of InsP7 generation in S. cerevisiae can help us elucidate similar pathways of InsP7 generation in mammals. Yeast Kcs1p was initially presumed to be the only IP6K phosphorylating the monoester phosphate on the 5-position of the inositol ring to generate 5-[PP]-InsP5. However, kcs1Δ cells retained low but detectable levels of an InsP7. This led to the identification and cloning of Vip/PPIP5K, a novel InsP6 kinase contributing to a novel pool of cellular InsP7 comprising two isomers that is masked in WT strains during metabolic labeling experiments by the Ddp1 phosphatase (7). A clue that a similar mechanism of InsP7/InsP8 synthesis existed in mammalian cells was provided by Pesesse et al. (30) and is seen in our data (Fig. 7), where sorbitol-induced increase in the InsP8 peak was at the cost of the InsP7 peak. This suggested that the enzymes involved in InsP7 and InsP8 synthesis were different. Two enzymes, Vip1/PPIP5K1 and Vip2/PPIP5K2 (8, 9), which are the human homologues of the yeast enzyme, have now been identified, which synthesize 1/3-[PP]-InsP5. Hence, as shown in Fig. 8A, both IP6Ks and Vips are required to complete the bifurcating pathways from InsP6 to InsP8.
TNP treatment of radiolabeled HeLa cells brings the dual pathways into focus. 10 μm TNP is 40-fold greater than the Ki for IP6Ks and should be similar to the expected cellular IC50 because of its high Km for ATP. Although this concentration of TNP should be sufficient to wipe out all cellular InsP7, 10% residual InsP7 could be observed. This can be either due to incomplete inhibition of the cellular IP6K or due to the production of other InsP7 isomers by the Vip/PPIP5K enzymes. Adding credence to the latter alternative are the following observations. 1) Vip/PPIP5K proteins contain the ATP-“GRASP” domains, which differ from the ATP binding regions of IPK family members in both sequence and in structure; it is very unlikely that they will prove to be efficient targets of TNP, and this was confirmed by a direct in vitro assay (Fig. 6). 2) The InsP7 isomers, which are the products of IP6Ks and Vip/PPIP5Ks (viz. 5-[PP]-InsP5 and 1/3-[PP]InsP5 (10) respectively), would be likely to be indistinguishable by the gradients used in our HPLC separation.
Under the sorbitol-stimulated conditions that were required to visualize and accurately measure InsP8 levels, pathway I appears to be the dominant pathway (Fig. 8A). However, pathway II could be revealed in yeast strains deficient for Ddp1 in which InsP7 levels were greatly elevated. Under these circumstances, TNP actually further increased InsP7 levels, establishing the occurrence of pathway II, albeit as a quantitatively minor component in wild type cells (Fig. 9). In mammalian cells, it is most likely that if all of the Dipps (four genes of which have been identified (34, 35)) are completely inhibited, the pool of InsP7 generated by Vip/PPIP5K will be unmasked, in a manner analogous to that seen in ddp1Δ or kcs1ddp1ΔΔ cells in S. cerevisiae.
The concept of “pharmalogs,” which are enzymes having similar affinities for inhibitors because they share structural similarities in substrate binding domains irrespective of whether they share sequence similarity, was introduced by Knight et al. (36), who characterized numerous inhibitors of the PIKK and PI 3-kinase family of enzymes. Our data suggest that this concept can be extended to the IPK family of inositol polyphosphate kinases. Although these concepts are valuable when considering the development of potent kinase inhibitors, for such compounds to prove useful as probes of biological function and ultimately, in some cases, as drugs, they must have a limited tendency toward off-target effects. Regarding kinases, a key problem is the existence of large families of genes encoding related enzymes. Protein kinases make up by far the largest group of kinases with ∼500 representatives in the human genome. We therefore tested the selectivity of TNP against a panel of 70 protein serine/threonine and tyrosine kinases (Table S1). None were significantly inhibited at 10 μm, the largest dose used in the cell-based studies described here. This strongly supports the value of TNP as a highly selective pharmacological tool in future studies of the functions of inositol polyphosphates. The vacuolar defect observed when wild type yeast were treated with TNP closely phenocopies yeast strains lacking either Kcs1p or Ipk2p, confirming the utility of this compound in studying the biological functions of inositol pyrophosphates in a range of cell types and organisms. The acute effects of an inhibitor of IP6K clearly implicate a direct rather than an adaptive effect of loss of InsP7 and should allow further studies of the dynamics of vacuole formation and turnover that would not be possible using genetic approaches. Similarly, inhibition of insulin release from Min6 cells confirms the broad utility of TNP, suggests that the function of InsP7 in this process is likely to be direct, and offers the possibility to study the dynamics of the insulin secretory machinery in more detail.
The last few years have seen major advances in our understanding of the cellular and organismal functions of inositol polyphosphates fueled primarily by studies in genetically tractable organisms, especially yeast and most recently in mice. Overexpression and/or small interfering/short hairpin RNA-based knockdown of key enzymes has also been used to infer new functions, although the identification of the major molecular targets that account for these proposed roles, especially of the pyrophosphates, has not yet been realized. The availability of a selective, cell-penetrant inhibitor of IP6Ks greatly augments the available approaches to studying the significance of inositol pyrophosphate species and will complement genetic studies. TNP rapidly reduces InsP7 levels in mammalian cells, allowing quantitative and dynamic manipulation of the pathway, and will contribute to pinpointing the molecular mechanisms it controls.
Supplementary Material
Acknowledgments
We thank Prof. Junichi Miyazaki (Osaka University) for providing Min6 cells. We thank Dr. Adele Ibrahim and Dr. Jennifer Bain of the DSTT consortium for the HA-tagged IP6K1 construct and for the protein kinase panel data. We also thank Prof. Mike Ashford (Neurosciences Institute, Division of Pathology and Neuroscience, Ninewells Hospital and Medical School, Dundee) and Dr. Ian Batty and Dr. Mike Paterson (Division of Molecular Physiology, University of Dundee) for helpful suggestions and critical discussions.
This work was supported by the Division of Signal Transduction Therapy consortium (University of Dundee).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1–S3.
Footnotes
The abbreviations used are: Ins(1,4,5)P3, inositol 1,4,5-trisphosphate; Ins(1,3,4,5)P4, inositol 1,3,4,5-tetraphosphate; InsP6, inositol hexakisphosphate; IP6K, inositol hexakisphosphate kinase; 5-[PP]-InsP5 or InsP7, bisdiphosphoinositol pentakisphosphate; TNP, N2-(m-(trifluoromethy)lbenzyl) N6-(p-nitrobenzyl)purine; TG, thapsigargin; PEI, polyethyleneimine; [PP]2-InsP4 or InsP8, bisdiphosphoinositol tetrakisphosphate; GFP, green fluorescent protein; BisTris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol; MOPS, 4-morpholinepropanesulfonic acid; PPIP5K, diphosphoinositol pentakisphosphate kinase; HPLC, high pressure liquid chromatography; IP, inositol phosphate; IP3-3K, Ip3-3 kinase, inositol(1,4,5)trisphosphate-3kinase; Ip3-3KA, isoform A of Ip3-3K; IPMK, inositol polyphosphate multikinase; InsP3, inositol trisphosphate; InsP4, inositol tetraphosphate; InsP5, inositol pentaphosphate.
References
- 1.Irvine, R. F. (2005) J. Physiol. (Lond.) 566 295-300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shears, S. B. (2004) Biochem. J. 377 265-280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo, H. R., Huang, Y. E., Chen, J. C., Saiardi, A., Iijima, M., Ye, K., Huang, Y., Nagata, E., Devreotes, P., and Snyder, S. H. (2003) Cell 114 559-572 [DOI] [PubMed] [Google Scholar]
- 4.Saiardi, A., Caffrey, J. J., Snyder, S. H., and Shears, S. B. (2000) J. Biol. Chem. 275 24686-24692 [DOI] [PubMed] [Google Scholar]
- 5.Saiardi, A., Erdjument-Bromage, H., Snowman, A. M., Tempst, P., and Snyder, S. H. (1999) Curr. Biol. 9 1323-1326 [DOI] [PubMed] [Google Scholar]
- 6.Saiardi, A., Nagata, E., Luo, H. R., Snowman, A. M., and Snyder, S. H. (2001) J. Biol. Chem. 276 39179-39185 [DOI] [PubMed] [Google Scholar]
- 7.Mulugu, S., Bai, W., Fridy, P. C., Bastidas, R. J., Otto, J. C., Dollins, D. E., Haystead, T. A., Ribeiro, A. A., and York, J. D. (2007) Science 316 106-109 [DOI] [PubMed] [Google Scholar]
- 8.Fridy, P. C., Otto, J. C., Dollins, D. E., and York, J. D. (2007) J. Biol. Chem. 282 30754-30762 [DOI] [PubMed] [Google Scholar]
- 9.Choi, J. H., Williams, J., Cho, J., Falck, J. R., and Shears, S. B. (2007) J. Biol. Chem. 282 30763-30765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin, H., Fridy, P. C., Ribeiro, A. A., Choi, J. H., Barma, D. K., Vogel, G., Falck, J. R., Shears, S. B., York, J. D., and Mayr, G. W. (2009) J. Biol. Chem. 284 1863-1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang, K. N., and Symington, L. S. (1995) Genetics 141 1275-1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.York, S. J., Armbruster, B. N., Greenwell, P., Petes, T. D., and York, J. D. (2005) J. Biol. Chem. 280 4264-4269 [DOI] [PubMed] [Google Scholar]
- 13.Steger, D. J., Haswell, E. S., Miller, A. L., Wente, S. R., and O'Shea, E. K. (2003) Science 299 114-116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen, X., Xiao, H., Ranallo, R., Wu, W. H., and Wu, C. (2003) Science 299 112-114 [DOI] [PubMed] [Google Scholar]
- 15.Lee, Y. S., Mulugu, S., York, J. D., and O'Shea, E. K. (2007) Science 316 109-112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Illies, C., Gromada, J., Fiume, R., Leibiger, B., Yu, J., Juhl, K., Yang, S. N., Barma, D. K., Falck, J. R., Saiardi, A., Barker, C. J., and Berggren, P. O. (2007) Science 318 1299-1302 [DOI] [PubMed] [Google Scholar]
- 17.Nagata, E., Luo, H. R., Saiardi, A., Bae, B. I., Suzuki, N., and Snyder, S. H. (2005) J. Biol. Chem. 280 1634-1640 [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez, B., Schell, M. J., Letcher, A. J., Veprintsev, D. B., Irvine, R. F., and Williams, R. L. (2004) Mol. Cell 15 689-701 [DOI] [PubMed] [Google Scholar]
- 19.Miller, G. J., and Hurley, J. H. (2004) Mol. Cell 15 703-711 [DOI] [PubMed] [Google Scholar]
- 20.Holmes, W., and Jogl, G. (2006) J. Biol. Chem. 281 38109-38116 [DOI] [PubMed] [Google Scholar]
- 21.Chang, Y. T., Choi, G., Bae, Y. S., Burdett, M., Moon, H. S., Lee, J. W., Gray, N. S., Schultz, P. G., Meijer, L., Chung, S. K., Choi, K. Y., Suh, P. G., and Ryu, S. H. (2002) ChemBioChem 3 897-901 [DOI] [PubMed] [Google Scholar]
- 22.Poinas, A., Backers, K., Riley, A. M., Mills, S. J., Moreau, C., Potter, B. V., and Erneux, C. (2005) ChemBioChem 6 1449-1457 [DOI] [PubMed] [Google Scholar]
- 23.Saiardi, A., Nagata, E., Luo, H. R., Sawa, A., Luo, X., Snowman, A. M., and Snyder, S. H. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 2306-2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayr, G. W., Windhorst, S., and Hillemeier, K. (2005) J. Biol. Chem. 280 13229-13240 [DOI] [PubMed] [Google Scholar]
- 25.Chang, S. C., Miller, A. L., Feng, Y., Wente, S. R., and Majerus, P. W. (2002) J. Biol. Chem. 277 43836-43843 [DOI] [PubMed] [Google Scholar]
- 26.Thastrup, O., Dawson, A. P., Scharff, O., Foder, B., Cullen, P. J., Drobak, B. K., Bjerrum, P. J., Christensen, S. B., and Hanley, M. R. (1994) Agents Actions 43 187-193 [DOI] [PubMed] [Google Scholar]
- 27.Hastie, C. J., McLauchlan, H. J., and Cohen, P. (2006) Nat. Protoc. 1 968-971 [DOI] [PubMed] [Google Scholar]
- 28.Bain, J., McLauchlan, H., Elliott, M., and Cohen, P. (2003) Biochem. J. 371 199-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davies, S. P., Reddy, H., Caivano, M., and Cohen, P. (2000) Biochem. J. 351 95-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pesesse, X., Choi, K., Zhang, T., and Shears, S. B. (2004) J. Biol. Chem. 279 43378-43381 [DOI] [PubMed] [Google Scholar]
- 31.Seeds, A. M., Bastidas, R. J., and York, J. D. (2005) J. Biol. Chem. 280 27654-27661 [DOI] [PubMed] [Google Scholar]
- 32.Dubois, E., Scherens, B., Vierendeels, F., Ho, M. M., Messenguy, F., and Shears, S. B. (2002) J. Biol. Chem. 277 23755-23763 [DOI] [PubMed] [Google Scholar]
- 33.da Silva, C. P., Emmrich, F., and Guse, A. H. (1994) J. Biol. Chem. 269 12521-12526 [PubMed] [Google Scholar]
- 34.Safrany, S. T., Caffrey, J. J., Yang, X., Bembenek, M. E., Moyer, M. B., Burkhart, W. A., and Shears, S. B. (1998) EMBO J. 17 6599-6607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Safrany, S. T., Ingram, S. W., Cartwright, J. L., Falck, J. R., McLennan, A. G., Barnes, L. D., and Shears, S. B. (1999) J. Biol. Chem. 274 21735-21740 [DOI] [PubMed] [Google Scholar]
- 36.Knight, Z. A., Gonzalez, B., Feldman, M. E., Zunder, E. R., Goldenberg, D. D., Williams, O., Loewith, R., Stokoe, D., Balla, A., Toth, B., Balla, T., Weiss, W. A., Williams, R. L., and Shokat, K. M. (2006) Cell 125 733-747 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.