

Abstract
Induced pluripotent stem cells (iPSCs) have been generated from somatic cells by transgenic expression of Oct4, Sox2, Klf4, and cMyc. A major difficulty in the application of this technology for regenerative medicine, however, is the delivery of reprogramming factors. Whereas retroviral transduction increases the risk of tumorigenicity, transient expression methods have considerably lower reprogramming efficiencies. Here we show a highly efficient piggyBac transposon-based approach to generate integration-free iPSCs. Transposons carrying 2A peptide-linked reprogramming factors induced reprogramming of mouse embryonic fibroblasts with equivalent efficiencies to retroviral transduction. Transposons were removed from these primary iPSCs by re-expressing transposase. Transgene-free iPSCs could be easily identified by HSVtk-FIAU selection. piggyBac excises without a footprint, leaving the iPSC genome without any genetic alteration. iPSCs fulfilled all criteria of pluripotency, such as expression of embryonic stem cell-specific markers, teratoma formation and contribution to chimeras. piggyBac transposon-based reprogramming may be used to generate therapeutically applicable iPSCs.
Introduction
Reprogramming of differentiated somatic cells into a pluripotent state has been carried out by cell fusion or nuclear transfer 1. The molecular basis of reprogramming has been revealed by exogenous expression of combinations of transcription factors. Four factors, namely Oct4 (also known as Pou5f1), Sox2, Klf4, and cMyc, that are important for self-renewal of embryonic stem cells (ESCs) have been shown to reprogram both mouse and human somatic cells into ESC-like pluripotent cells, named induced pluripotent stem cells (iPSCs) 2-4. Induction of reprogramming by these defined four factors is mostly carried out by co-infection with retroviral vectors 2-4. The main problems of this retrovirus-based method are oncogenicity and mutagenesis. Chimeric mice derived from iPSCs as well as their offspring developed tumors, probably because of reactivation of the proviral cMyc oncogene 5. Even though three-factor (Oct4, Sox2, and Klf4) iPSC-derived animals did not develop tumors 6, ectopic expression of any one of these genes may have deleterious consequences. For instance, ectopic expression of Oct4 in skin and intestine causes tumor development 7. Overexpression of Klf4 induces dysplasia in skin 8. Furthermore, retroviral integration itself causes insertional mutagenesis and also may alter the expression pattern of nearby genes 9. Transgene integration-free iPSCs are necessary for their future therapeutic application. To address this, two methods, namely adenoviral transduction 10 and repeated plasmid transfection 11, have been recently reported. Both methods have successfully generated integration-free iPSCs. However, their efficiencies were 100~1000-fold lower than those achieved by retroviral reprogramming 10, 11. Novel strategies to generate transgene-free iPSCs with a reasonable efficiency are required.
To achieve this, we utilized the piggyBac transposon system to deliver the reprogramming factors, which were linked by self cleaving peptides. The approximately 20-amino acid-long 2A peptides from foot-and-mouth disease virus (F2A) and Thosea asigna virus (T2A) work as self-cleaving signals and enable expression of several gene products from a single transcript 12, facilitating multi-gene delivery to target cells. The piggyBac transposon is a moth-derived DNA transposon 13, highly active in mammalian cells and mice 14, which has been used for gene delivery and mutagenesis 14. The piggyBac transposon system has a very large cargo capacity. Up to 10 kb DNA fragments can be transposed without losing transposition efficiency 14. Unlike most other DNA transposons, piggyBac does not leave “footprint” mutations upon excision 15. For instance, the TA dinucleotides used by the Sleeping Beauty transposon as integration sites are changed to TAG(T/A)CTA after excision 16. In contrast, the TTAA integration sites used by piggyBac transposons are repaired to the original sequence upon excision 15. This allows removal of transposons from the host genome without changing any nucleotide sequence. Using the piggyBac transposon system, we demonstrate the efficient generation of transgene integration- and mutation-free iPSCs, which are invaluable for therapeutic applications.
Results
Construction of transposon-based reprogramming vectors
In order to deliver the reprogramming factors to recipient cells, we constructed piggyBac transposon-based reprogramming vectors (Fig. 1a). cDNA sequences constituting the open reading frames from Oct4, Sox2, Klf4 and cMyc were combined into a single open reading frame (OSKM) using 2A peptides and placed under the constitutively active CAG promoter. We reasoned that Lin28 may further enhance reprogramming because it is an essential factor for another reprogramming cocktail consisting of Oct4, Sox2, Nanog, and Lin28 4. Therefore we also constructed a 5-factor vector by linking Lin28 to the 4 factors through 2A peptide (OSKML). A puΔtk cassette serves as a negative selection marker for later transposon removal. These expression cassettes together with puΔtk were flanked by the terminal repeats of the piggyBac transposon. To verify expression of each factor, we transfected 293 cells with the transposon vectors and analyzed protein levels by Western blots (Fig. 1b). cMyc from the 4-factor vector and Lin28 from the 5-factor vector were detected migrating at the same molecular weights as their endogenous counterparts. All other proteins were detected at slightly higher molecular weights due to the residual 2A peptides, which were still attached to their C-termini (Supplementary Fig. 1). Most antibodies also detected uncleaved proteins. The amounts, however, were far less than those of correctly processed proteins. These results show that 2A-peptide mediated cleavage of a single polypeptide chain efficiently produces individual reprogramming factors.
Figure 1. Construction of piggyBac transposon-mediated reprogramming vectors.

(a) Schematic representation of piggyBac transposons. Four or five factors were linked through 2A peptides. Expression of the factors was driven by the constitutively active CAG promoter. bpA, bovine growth hormone polyadenylation signal; puΔtk, PGK promoter-driven puΔtk expression cassette; PB5′ and PB3′, terminal repeats of the piggyBac transposon. (b) Western blot analyses of 2A peptide-mediated expression of the reprogramming factors. The left most lanes of each panel (individual) are positive control transfections using unlinked factor expression vectors. ‘−’, a negative control transfection using an eGFP expression vector.
Generation of primary iPSCs using the transposon vector
We next investigated whether piggyBac transposons carrying the 4 or 5 factors could induce reprogramming in mouse embryonic fibroblasts (MEFs). The protocol is illustrated in Figure 2a. We used Lipofectamine2000 to transfect MEFs with 2.0 μg of piggyBac transposon and 2.0 μg of piggyBac transposase-expression vector in 6-well plates. One day post transfection, MEFs were replated onto a feeder-layer at a split ratio of 1:18 and cultured using serum-based ESC medium (F15L) with or without valproic acid (VPA) for 5 days. Culture medium was subsequently changed to serum-free ESC medium (K15L) on day 7. The serum-free culture was previously shown to enhance reprogramming of both mouse and human fibroblasts 17, 18. We found that appropriate combinations of serum-based and serum-free media could further enhance reprogramming and maximize the number of iPSC colonies compared to culture in either media alone (Supplementary Fig. 2). Under these conditions, colonies became visible around day 7 without VPA and day 10 with VPA. At day 14, colonies were sufficiently large to be picked. Although VPA was previously reported to enhance reprogramming efficiency of mouse and human fibroblasts 17, 19, we could not observe this effect (Fig. 2b, e). The striking effect of VPA was growth suppression of un-reprogrammed cells. Colonies without VPA treatment tended to be much bigger than VPA-treated colonies and their morphology was not typical for ESC colonies, although all of these colonies were positive for alkaline phosphatase (AP) staining (Fig. 2b, c, top). In contrast, VPA-treated colonies were more spherical and morphologically indistinguishable from ESC colonies (Fig. 2c, bottom). All ESC-like colonies were positive for Nanog expression as well as AP staining (Fig. 2c, d, bottom), indicating successful reprogramming of these colonies. Intriguingly, Nanog immunostaining showed that non-ESC-like colonies without VPA treatment contained many Nanog-positive patches (Fig. 2d, top). All colonies grown without VPA contained at least one Nanog-positive small sub-colony, indicating that reprogramming did occur, but that surrounding un-reprogrammed cells prevented reprogrammed cells from expanding and forming ESC-like colonies. Thus, VPA seems to suppress un-reprogrammed cell growth and facilitates expansion of reprogrammed cells to form colonies. Using the 5-factor transposon, we were able to increase the colony number approximately two-fold compared to the 4-factor transposon (Fig. 2e). We transfected 106 MEFs with a transfection efficiency of approximately 10 % and obtained approximately 1,000 colonies, therefore, the overall reprogramming efficiency is approximately 1%, virtually equivalent to the retroviral method. When the transposon amount was reduced from 2.0 μg to 0.5 μg, the colony number decreased about 10-fold (Fig. 2e).
Figure 2. Generation of primary iPSCs using piggyBac transposons.
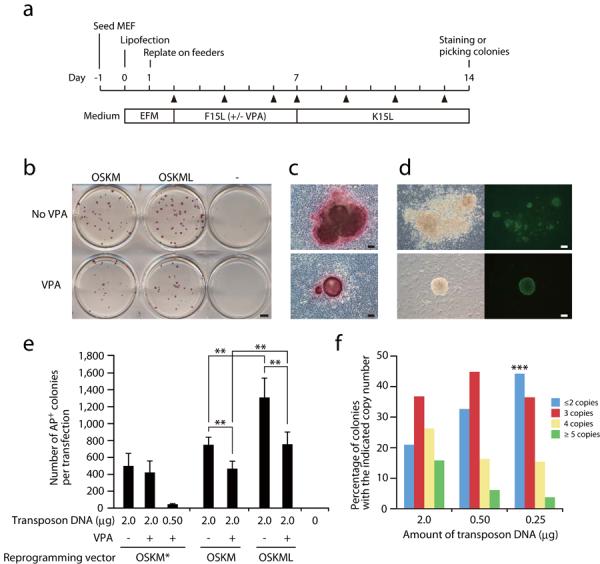
(a) Time schedule of transposon-mediated reprogramming. EFM, embryonic fibroblast medium; F15L, serum-based ESC medium; K15L, serum-free ESC medium. Triangles, medium change. (b) Alkaline phosphatase staining of day 14 colonies. ‘−’, a negative control transfection using an eGFP expression vector. (c) Enlarged view of AP-stained colonies with (bottom) and without (top) VPA treatment. (d) Immunostaining of Nanog at day 14 with (bottom) and without (top) VPA treatment. Left, phase contrast; right, anti-Nanog immunostaining. (e) The numbers of colonies obtained by transposon-based reprogramming. OSKM* is identical to OSKM except that Sox2 and Klf4 are linked by F2A instead of T2A. (f) Transposon copy number distribution depending on the transposon plasmid amount. The OSKM* vector was used for the 2.0 μg and the 0.50 μg transfection, and the OSKML vector for the 0.25 μg transfection. *** Only one single-copy transposon iPS colony was identified in the experiments. Data are shown as mean ± s.d. Experiments were performed at least 3 times per each condition. **, p<0.05 (Student's t test). Scale bars, 5 mm (b), 100 μm (c, d)
We picked colonies from the VPA-treated culture and established “primary” iPSC lines, which were morphologically indistinguishable from ESCs and positive for endogenous Nanog expression (Supplementary Fig. 3a). The efficiency of establishment of primary iPSCs was routinely more than 90%. We investigated the copy number of the integrated transposons by Southern blot analysis (Supplementary Fig. 3c). Using 2.0 μg of transposon DNA, most colonies had multiple insertions. Twenty percent of colonies had 2-copy integrations (Fig. 2f), which increased to 45% when the transposon DNA amount was reduced to 0.25 μg (Fig. 2f). We obtained only 1 colony with a single copy integration out of over 100 colonies analyzed, suggesting that at least 2 transposons are necessary for efficient reprogramming using our vectors. This may be explained by recent evidence suggesting that fibroblasts require higher levels of expression of reprogramming factors than other cell types such as hepatocytes or epithelial cells 20.
Removal of transposons from primary iPSCs
Next we attempted to eliminate transposons from the iPSC genome. Once excised, only 50% of transposons re-integrate into the genome 21. We utilized the human Herpes Virus thymidine kinase (HSVtk)-FIAU selection system to select transposon-free iPSCs after transfection of piggyBac transposase-expressing plasmid (Figs. 1a and 3a). We used three different primary iPSC clones, which had each been confirmed to carry 2 copies of the piggyBac transposon (Supplementary Fig. 3d). Splinkerette-PCR was used to identify the integration sites (Supplementary Fig. 4). After transient transposase expression and FIAU selection, resistant colonies were picked and screened by PCR using transgene-specific primer pairs. We obtained 4 transposon-free clones out of 5 FIAU-resistant colonies from the iPS25 line, 7 clones out of 7 from iPS28 and 2 clones out of 11 from iPS216. In these experiments the efficiency of transposon removal per transfected cell was approximately 10−5. However, FIAU selection allowed easy identification of integration-free iPSCs, as 50 % of FIAU resistant colonies were integration-free. Transposon-negative clones were further expanded and subjected to Southern blot analysis probing for the 5′ repeat of the piggyBac transposon. All clones that were negative by PCR were confirmed to have lost their transposons by Southern blot analysis (Fig. 3b). Transposon removal was also confirmed using a series of additional transgene probes (Supplementary Fig. 5). To further verify this, we performed genomic PCR in 2 clones from each parental primary iPSC line using a transposon-specific primer and primers specific for each integration site (Supplementary Fig. 4). In all parental iPSC lines, specific host-transposon junction amplifications were readily detected, whereas MEFs and transposon-free iPSCs did not yield any PCR products (Fig. 3c-e). This was also confirmed by using primer pairs specific for the transposon (Supplementary Fig. 6). Furthermore, exhaustive PCR analyses did not show any random integration of the transposon vector or the piggyBac transposase expression vector (Supplementary Fig. 6). Thus, we conclude that the transposons had been successfully removed in FIAU-resistant iPSC colonies.
Figure 3. Transposon removal from the established iPSC lines.
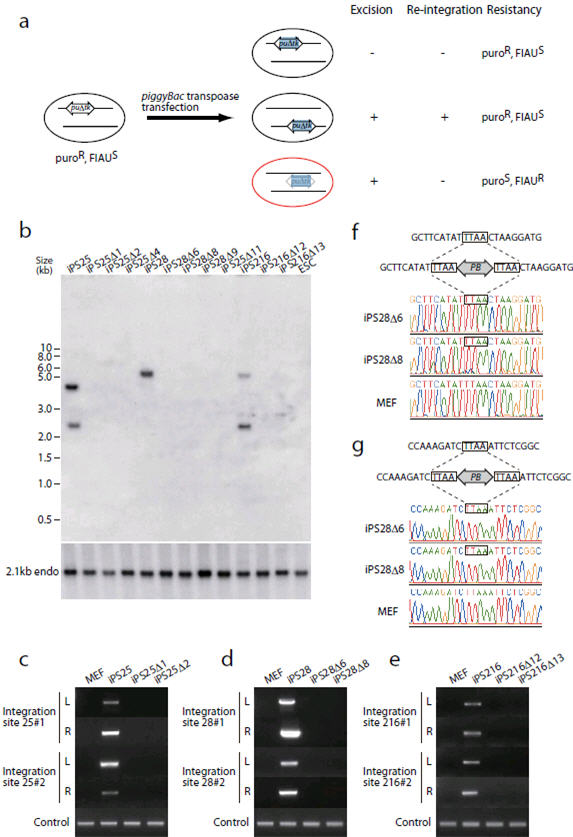
(a) Schematic representation of transposon removal. After piggyBac transposase transfection, cells in which transposons either do not transpose or mobilize into other loci are FIAU sensitive, whereas cells in which transposons do not re-integrate elsewhere become FIAU resistant. (b-e) Evidence of transposon-free iPSC lines shown by Southern blot analysis (b) and genomic PCR (c-e). (b) The piggyBac transposon 5′ repeat was used as a probe. Parental iPSC lines have 2 copies of transposon integration (iPS25, 28, and 216). Note that in the HindIII digest of iPS28, two fragments have similar sizes and migrate at the same position (see also Supplemental Fig. 3d). FIAU-resistant sub-clones (designated Δ) did not possess any transposons. The blot was re-hybridized with a control probe to show equal sample loading. (c-e) Specific primers were designed to detect individual transposon integration sites. MEFs and each primary iPSC clone serve as negative and positive controls, respectively. (f, g) Evidence of perfect restoration of the original sequences after transposon excision in iPS28-derived clones. The top panels are the original genomic sequences. The middle panels show transposon integration sites where TTAA sequences were duplicated at both ends of the transposon. The electrophoregrams in the bottom panels show that sequences in iPS28-derived clones after excision are identical to the original sequences in MEFs. Integration site 1 on chr.6 (f) and site 2 on chr.15 (g) are shown.
The piggyBac transposon does not leave a footprint at the excised site. To confirm that the transgene-free iPSC had no footprint mutations, we PCR-amplified the genomic regions encompassing the integration/excision sites and sequenced the PCR products directly. If there was a footprint mutation (for instance a nucleotide change, insertion or deletion), the sequence after TTAA, the target site of piggyBac integration, would be a mixture of sequences coming from the wildtype and the excised allele. The signals from all integration sites were identical to the original genomic sequence (Fig. 3f, g and Supplementary Fig. 7), indicating the absence of footprint mutations. Thus, we successfully “cured” transgene-harboring iPSCs by removing the transposon.
Pluripotency of the integration-free iPSCs
The integration-free iPSCs had a normal karyotype (Fig. 4a). To investigate whether these transposon-free iPSCs are pluripotent, we first conducted RT-PCR analysis of 7 ESC-specific genes (Oct4, Sox2, Nanog, Ecat1, Esrrb, Dppa3, and Dnmt3l) (Fig. 4b and Supplementary Fig. 8). All genes analyzed were expressed in iPSCs at similar levels to ESCs. Immuno-fluorescence staining also showed that iPSC lines uniformly expressed Oct4 and Nanog (Fig. 4c). To further analyze reprogramming, we investigated the DNA methylation status at the Oct4 and Nanog promoter regions by bisulphite sequencing. Consistent with previous reports, these promoter regions were hypermethylated in MEFs and hypomethylated in ESCs (Fig. 4d). In the transgene-free iPSCs, these promoter regions were also demethylated (Fig. 4d), indicating that iPSCs were epigenetically reprogrammed. To further investigate whether transgene-free iPSCs are pluripotent, we performed in vivo differentiation analysis. Pluripotent cells are able to form teratomas when injected subcutaneously into immuno-deficient mice. Four weeks after injection, integration-free iPSCs gave rise to teratomas. Histological analysis of the teratomas showed that they contained endodermal (epithelial tissue), mesodermal (muscle and cartilage) and ectodermal (epidermis) tissues (Fig. 4e). To further prove the pluripotency of integration-free iPSCs, we analyzed their contribution to chimera development (Fig. 5 and Supplementary Table 1). We therefore transfected the iPS25Δ1 line with a constitutive eGFP expression vector. Chimeric embryos expressing eGFP were identified at day 10.5 post coitum (Fig. 5a). A live-born chimeric mouse expressing eGFP is shown in Figure 5b. To show that integration-free iPSCs can contribute to the germ cell lineage, we targeted a GFP cassette into the Nanog locus in iPS25Δ1 line (Supplementary Fig. 9). Chimeric embryos were dissected at day 12.5 and developing gonads were observed under the fluorescence stereomicroscope. At this stage of development, Nanog is expressed exclusively in the germ cell lineage. Out of 7 embryos analyzed, 1 embryo had GFP-positive cells in the developing gonad, indicating that integration-free iPSCs contributed into the germ cell lineage (Fig. 5c). We also obtained live-born chimeras from Nanog-knock-in iPSCs (Supplementary Table 1). Although germ line transmission in adult chimera remains to be analyzed, we concluded that these iPSCs are genuinely pluripotent. Hence, transposon-based reprogramming generates transgene integration-free iPSCs.
Figure 4. Characterization of integration-free iPSCs.
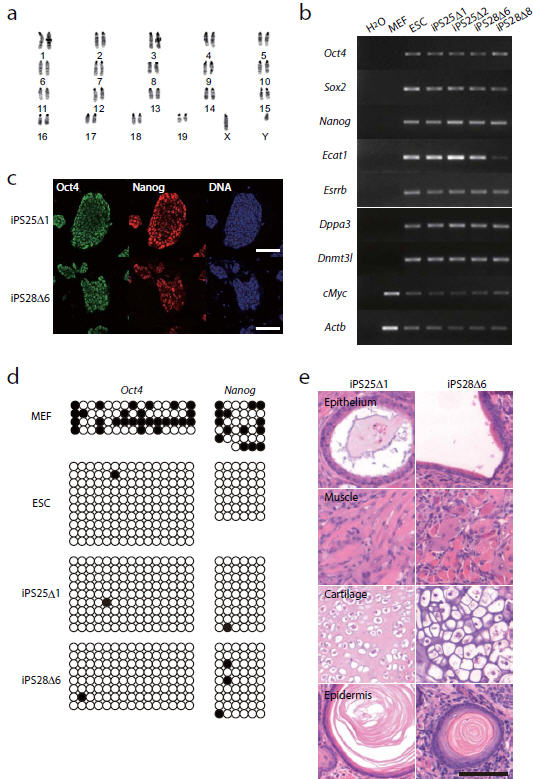
(a) Normal karyotype of the integration-free iPSC line iPS25Δ1. (b) RT-PCR analyses of integration-free iPSC lines, compared to expression in ESCs. (c) Immunofluorescent analysis of Oct4 and Nanog expression in integration-free iPSCs. (d) Bisulphite sequencing of the promoter region of Oct4 and Nanog. Open and closed circles indicate unmethylated and methylated CpG dinucleotides, respectively. (e) Teratomas generated from integration-free iPSCs, showing differentiation to all three germlayers. Scale bar, 100 μm
Figure 5. Contribution of integration-free iPSCs to somatic tissue and germ line in chimeras.

(a, b) Chimeric embryos at 10.5 d.p.c. (a) and postnatal day 5 pups (b) derived from integration-free iPSCs with constitutive eGFP expression. GFP images are shown at the bottom. (c) Developing gonad of an e12.5 embryo derived from Nanog-GFP knock-in iPSCs. GFP image is shown below. The broken line outlines the gonad. Scale bars, 1 mm (a), 5 mm (b), 200 μm (c)
Discussion
A non-mutated genome is an important requirement for iPSC-based regenerative medicine. Although retroviral delivery of the reprogramming factors is efficient and widely used, iPSCs generated using retroviral vectors have insertional mutations and are known to be tumorigenic. Transient expression methods, such as adenoviral transduction 10 and repeated plasmid transfection 11, have been developed to avoid this problem. However they also have a serious disadvantage, namely a very low reprogramming efficiency. The major problem with transient expression systems is the reduction of expression levels over a few days as the cells divide. The dox-inducible reprogramming system revealed that at least 10 days of stable expression is required to achieve reprogramming of both mouse and human fibroblasts 22-25, and that expression for more than 10 days further increased the reprogramming efficiency 22-25. These results suggest that sustained expression of the factors is beneficial to achieve reprogramming with a higher efficiency.
Our piggyBac transposon-based approach meets the criteria of high efficiency while retaining a non-mutated genome. First, the piggyBac transposon integrates into the host genome, allowing stable expression of transgenes. Second, the integrated piggyBac transposons can be excised from the host genome without leaving “footprint” mutations. These unique characteristics of the piggyBac transposon enable us not only to reprogram somatic cells with efficiencies similar to the retroviral method but also to remove the reprogramming factors from the iPSCs permanently without genetic alteration. Genomic integrity is key for the therapeutic applications of iPSCs. However, Wang et al. have shown that approximately 5 % of piggyBac excisions in mouse ESCs had microdeletions at the excision sites 21, which might be mutagenic. Also, donor cells used for the generation of iPSCs can in principle have already acquired somatic mutations. Genetic alterations could also occur during the reprogramming process. Whole genome sequencing methods could be used to identify these mutations and the most suitable iPSC line for therapeutic applications. Intriguingly, it seems that piggyBac transposition does not require host factors 26. Our method could be readily applied to other cell types such as keratinocytes, which can be reprogrammed more efficiently than fibroblasts 27 to generate integration-free iPSCs.
We observed statistically significant reduction in iPSC colony formation by VPA treatment (Fig. 2e), although VPA treatment generated more homogeneous iPSC colonies. VPA was previously reported to enhance reprogramming efficiency when retroviral methods were employed 17, 19. It is likely that VPA opens chromatin, as genes whose expression is elevated in ESCs compared to MEFs, are upregulated after 7 days treatment in MEFs 19. If this is the case, VPA treatment can be beneficial for cells, which do not receive all factors or only receive low copy numbers of individual reprogramming factors and therefore have insufficient expression levels of these factors. Since polycistronic expression by a strong promoter provides sufficient expression levels of all reprogramming factors, enhancement by VPA could not be observed. Rather, we saw a reduction of overall reprogramming efficiency. This could be due to a growth suppressive effect of VPA on a certain MEF sub-populations. We indeed observed that after VPA treatment growth of MEFs was negatively affected (data not shown).
We utilized 2A peptides for the polycistronic expression of the reprogramming factors from a single vector. This strategy was employed by recent studies and proven to be capable of generating the iPSCs 11, 28. In our study, the five known factors (Oct4, Sox2, Klf4, cMyc and Lin28) were successfully combined into a single vector and induced reprogramming of MEFs, more efficiently than the 4-factor vector. The use of additional factors in combination with the 5 factors might further enhance the reprogramming efficiency. UTF1 expression or p53 knock-down, for example, have been recently reported to substantially enhance reprogramming of human fibroblasts 29. It was also shown that either ectopic expression of C/EBPα or Pax5 knock-down was required to reprogram B lymphocytes by the 4 factors 30. The use of 2A peptide provides an easy way to link even more than 5 factors in a single expression cassette. The piggyBac transposon serves a versatile vehicle for such large and complex cargoes, as the transposition efficiency is not limited by its cargo size.
One of the most important results of our approach is the direct selection of integration-free iPSCs. By titrating the transposon DNA amount, we established a condition, in which about half of the primary iPSC colonies have two or less transposon copies. As an alternative to the clone-based approach used here (Fig. 6 Scheme A), the primary iPSC colonies can be pooled together for downstream transposon removal (Fig. 6 Scheme B). We have also succeeded in isolating transposon-free iPSCs using FIAU selection for the loss of puΔtk from a primary iPSC pool (data not shown). This simple and rapid approach will be invaluable for the generation of patient-specific integration-free iPSCs.
Figure 6. Schemes to generate transgene-free iPSCs.

Scheme A (black arrows): After transfection of transposon and transposase DNA into MEFs to generate primary iPSCs, individual iPSC colonies are picked to identify colonies with 2 copy transposon integrations, by Southern blot analysis. These clones are then expanded and the transposase is re-expressed to remove the transposons. HSVtk-FIAU negative selection is used to identify integration-free iPSCs. Scheme B (red arrows): Using an optimized protocol, analysis of individual clones can be bypassed. Primary iPSCs can be pooled and directly subjected to transposon removal and HSVtk-FIAU selection.
While this manuscript was under review, Kaji et al. and Woltjen et al. reported similar results 31, 32. These groups used 2A peptides to link the 4 factors, delivered these using the piggyBac transposon and succeeded in reprogramming of mouse and human fibroblasts. However, the recovery of transgene-free iPSCs was not facilitated by selection. The negative selection system incorporated in our vectors greatly enables efficient isolation of transgene-free iPSCs.
The piggyBac transposon-based method together with the polycistronic expression design offers an alternative strategy for the efficient generation of iPSCs with an intact genome. We envisage that this strategy will facilitate the generation of patient-specific iPSCs for the purposes of drug discovery and cell-based regenerative medicine.
Methods
Plasmid construction
All primer sequences are listed in Supplementary Table 2. All PCR-amplified fragments were verified by sequencing.
< 2A peptide-linked 4- or 5-factor cassettes>
Mouse Oct4, Sox2, Klf4 and cMyc were PCR-amplified with primer pairs Oct4-B-U1 and Oct4-F2A-S, Sox2-X and Sox2-F2A-S, Klf4-X and Klf4-F2A-S, and Myc-X and Myc-S, respectively. Oct4 PCR product was digested with BamHI and SalI, and cloned into pBluescriptIIKS+ (pBS), resulting in pPB-Oct4F2A. Sox2, Klf4 and cMyc PCR products were cloned into a TA cloning vector (Promega), resulting in pGEMT-Sox2F2A, Klf4F2A, cMyc, respectively. To combine 4 factors in a single vector, a XhoI-SalI fragment of Sox2-F2A was first inserted into the SalI site of pBS-Oct4F2A, resulting in pBS-OfSf. Then a XhoI-SalI frament of Klf4-F2A was inserted into the SalI site of pBS-OfSf, resulting in pBS-OfSfKf. Finally, a XhoI-SalI fragment of cMyc was inserted into the SalI site of pBS-OfSfKf, resulting in pBS-4F.
To change F2A between Oct4 and Sox2 to T2A, the NsiI-AatII fragment of pBS-4F was replaced with a PCR fragment generated with a primer pair of OctT2ASox and OtS-seqL, resulting in pBS-4FT.
To further change F2A between Sox2 and Klf4 to T2A, the AatII-MscI fragment of pBS-4FT was replaced with a PCR fragment generated by fusion-PCR. Briefly, two PCRs were performed using primer pairs StK-U1 and StK-L2, and StK-U2 and StK-L1 in separate tubes. These PCR products were then column-purified and used as templates for a 2nd PCR with a primer pair StK-U1 and StK-L1. The resulting vector was called pBS-4FTT.
To add Lin28 to the 2A peptide-liked 4-factor vector, Lin28 cDNA was PCR-amplified by using Lin28-U1 and Lin28-L1 and cloned into the BamHI-EcoRI site of pBS. A T2A-Lin28 fragment flanked by BstBI and XhoI sites was amplified by PCR (primer pair MtL-U1/MtL-L1) and inserted into the BstBI-XhoI site of pBS-4FTT, resulting in pBS-4FTTL.
OSKM, OSKM*, and OSKML in Figure 2 correspond to 4FTT, 4FT, and 4FTTL, respectively.
<piggyBac transposon-based expression vector>
First, a CAG promoter-driven expression vector, pCAG.EBNXN, was generated by replacing the EcoRI-NsiI fragment of pCAG-IP 33 with a linker containing multicloning sites (EcoRI, BglII, NotI, XhoI, NsiI). Second, a piggyBac vector carrying a CAG promoter-driven expression cassette, pPB-CAG.EBNXN, was generated by inserting the SalI-BamHI fragment of pCAG.EBNXN into the XhoI-BamHI site of pPB-LR.
<4-factor or 5-factor expression vectors>
Four- or five-factor expression vectors were constructed by inserting the BamHI-SalI fragment of the 4-factor construct or the BamHI-XhoI fragment of the 5-factor construct into the BglII-XhoI site of pPB-CAG.EBNXN, resulting in pPB-CAG.4F, pPB-CAG.4FT, pPB-CAG.4FTT, and pPB-CAG.4FTTL, respectively. Finally, a negative selection marker, PGK-puΔtk cassette, was excised from pFlexible with XhoI digestion and inserted into the SalI site of pPB-CAG-based factor expression vectors.
< piggyBac transposon vector with an eGFP cassette>
First, a piggyBac transposon vector carrying the human ubiquitin C (UbC) promoter-driven expression cassette was constructed. UbC promoter and bovine growth hormone polyadenylation signal sequence (bpA) were PCR amplified with a primer pair of UbC-U and -L and bpA-U and -L, respectively. An NheI-EcoRI fragment of UbC promoter and an EcoRI-SalI fragment of bpA were ligated into the NheI-SalI site of pPB-LR5, resulting in pPB-UbC. Second, an eGFP fragment was excised from pCX-GFP and inserted into the EcoRI site of pPB-UbC, resulting in pPB-UbC.eGFP. Finally, PGKneo cassette was excised from PL452 34 by XhoI digestion and inserted into the SalI site of pPB-UbC.eGFP, resulting in pPB-UbC.eGFP-neo
Cell culture
MEFs were prepared from e14.5 wildtype embryos (inbred C57Bl/6-Tyr c-Brd/c-Brd for the piggyBac study, B6129S6F1 and ICRB6F1 for the retroviral study) and cultured in DMEM containing 10 % FBS (HyClone), 2 mM L-glutamine, 1 × non-essential amino acid, and 0.1 mM 2-mercaptoethanol. A germline-competent mouse ESC line (KY1.1, B6129S6F1 background; unpublished) and mouse iPSC lines were cultured on mitomycin C-treated MEFs in serum-based ESC medium (F15L), which is DMEM containing 15 % FBS (HyClone), 2 mM L-glutamine, 1 × non-essential amino acid, 0.1 mM 2-mercaptoethanol and 1000 U ml−1 LIF (Chemicon).
Reprogramming of MEFs using transposon vectors
MEFs were plated onto 6-well plates (5 × 105 cells/well) one day before transfection. The next day (day 0), 2.0 μg of pCMV-mPBase 35 and various amounts of plasmids harboring the piggyBac transposon were transfected using Lipofectamine2000 (Invitrogen) according to the manufacturer's instructions. On day 1, transfected MEFs were trypsinised and replated onto feeder layers at a split ratio of 1:18 in MEF medium. On day 2, F15L medium was applied. VPA was added to culture medium at 2 mM 19 from day 2 to day 7. The medium was refreshed every other day. On day 7, medium was changed to serum-free ESC medium (K15L), which contains 15% Knockout serum replacement (Invitrogen) instead of FBS. Medium was refreshed every other day. On day 14, colonies were either stained using the Alkaline phosphatase detection kit (Chemicon) and counted, or picked and further expanded.
Reprogramming of MEFs by retroviral vectors
Retroviral vectors (pMXs-Oct3/4, pMXs-Sox2, pMXs-Klf4, pMXs-c-Myc) 2 were obtained from Addgene. Preparation of retroviruses and infection of MEFs was described in ref. 2. One day post infection cells were replated onto 6-well plates containing a feeder layer at 3,000 cells/well. Subsequent culture conditions are described in Supplementary Figure 2.
Transposon removal from primary iPSCs
The piggyBac transposase-expression vector was electroporated into 2 × 106 iPSCs. Cells were maintained for 3 days to allow turnover of the puΔtk protein. 5 × 105 cells were then seeded onto 10 cm dishes containing feeder cells. The next day FIAU was added to the culture medium (0.2 μM final concentration) and selection was continued for 5 days. After an additional 5 days of culture without FIAU, the resulting colonies were picked and expanded. Transposon removal was examined by PCR with primers listed in Supplementary Table 2 and confirmed by Southern blot analysis using the 5′ terminal repeat of the piggyBac transposon as a probe. The probe for the 5′ terminal repeat of piggyBac transposon specifically detects the 5′ repeat and does not detect the 3′ terminal repeat in the Southern blot analysis.
GFP marking of an integration-free iPSC line
A cured iPSC line iPS25Δ1 was electroporated with 25 μg of a linearized Nanog-GFP targeting vector (unpublished, K.Y. and J.T.) in Hepes-buffered saline (20 mM Hepes, pH 7.05, 137 mM NaCl, 5 mM KCl, 0.7 mM Na2HPO4, 6mM dextrose) at 230 V and 500 μF using Gene Pulser II (Bio-Rad). Cells were selected with puromycin (1 μg ml−1) and resulting colonies were picked and further expanded. Homologous recombination was verified by PCR as well as Southern blot analysis. For ubiquitous expression of GFP, iPS25Δ1 was electroporated with 1 μg of pPB-UbC.eGFP-neo and 5 μg of pCMV-mPBase using the same conditions as above and selected with G418 (180 μg ml−1). Resulting colonies were picked and further expanded.
Preparation of splinkerettes
Splinkerettes were prepared by annealing Spl-top and Spl-sau (for enzymes generating 5′-GATC protruding ends), Spl-blunt (for enzymes generating blunt ends), or Spl-CG (for enzymes generating 5′-CG protruding ends). Sequences of these oligonucleotides are listed in Supplementary Table 2. Following heat denaturation at 95 °C for 10 min, annealing was performed by cooling down a mixture of 11 pmol of each strand in 10 mM Tris-HCl (pH7.4) and 5 mM MgCl2 in total volume of 100 μl.
Transposon integration site analysis
Transposon integration sites were determined by Splinkerette PCR 36. Genomic DNAs of primary iPSCs (0.5 μg) were digested by one of 4-base pair cutters, MboI, HaeIII, AluI, RsaI, MspI TaqI, and HinP1I for 2 hrs in 20 μl. After heat inactivation, 2 μl of the digestion mixture were used for ligation with the corresponding splinkerette adaptors (11pmol) in 20 μl reactions. One μl of the ligation mixture was then subjected to nested-PCR. Primer pairs Spl-P1/PB3-P1 or Spl-P1/PB5-P1 were used for the first PCR. In the second PCR, pairs of Spl-P2/PB3-P2 or Spl-P2/PB5-P2 were used. Finally, PCR products were directly sequenced to determine genomic sequences flanking the piggyBac terminal repeats. Sequences were analyzed by Blat search on the UCSC genome browser.
RT-PCR analysis
Total RNA was extracted by using TriZol reagent (Invitrogen). One μg of total RNA was reverse-transcribed using an oligo(dT) primer by SuperScriptII (Invitrogen), and subjected to PCR using primers listed in Supplementary Table 2. Quantitative RT-PCR was performed using Platinum SYBR Green qPCR superMix (Invitrogen) on the ABI7900HT sequence detector (Applied Biosystems). Serial dilutions of each RT-PCR product were used to generate a standard curve. Expression levels of individual transcripts were normalized to Gapdh expression.
Bisulphite sequencing
Bisulphite sequencing was performed by using EpiTect Bisulfite Sequencing kit (Qiagen). Primers used were listed in Supplementary Table 2.
Western blot analysis
The 4-factor or 5-factor expression vectors were introduced into 293T cells by using Lipofectamine2000 according to the manufacturer's instructions. Forty-eight hours post transfection, cells were harvested and suspended in LDS sample buffer (Invitrogen) and proteins were separated on 4-15% gradient gels. Protein blots were analyzed using anti-Oct4 (mouse monoclonal, C-10, Santa Cruz), anti-Sox2 (rabbit polyclonal, H-65, Santa Cruz), anti-Klf4 (rabbit polyclonal, H-180, Santa Cruz), anti-cMyc (rabbit polyclonal, ab11917, Abcam), anti-Lin28 (rabbit polyclonal, ab46020, Abcam), or anti-ß-actin antibodies (mouse monoclonal, AC-15, Sigma).
Immunostaining
Cells were fixed by 4% paraformaldehyde/PBS for 15 min at room temperature and permeabilized by 0.05% Triton-X100/PBS for 10 min at room temperature, then blocked by 1% BSA/PBS for 1 hr at room temperature. Cells were washed with PBS and incubated with anti-Nanog antibody (1:300, rabbit polyclonal, ab21603, Abcam) and/or anti-Oct4 antibody (1:50, mouse monoclonal, C-10, Santa Cruz) over night at 4°C. After washing with PBS (6 × 10 min), cells were labeled with Alexa488 or 555-conjugated secondary antibodies (Invitrogen) for 1 hr at room temperature. Cells were then washed with PBS (3 × 10 min) and nuclei were counterstained with Toto-3 at 0.5 μM in PBS for 1 hr at room temperature. After final washing with PBS (3 × 10 min), specimens were analyzed using a fluorescence microscope or Radiance2000 confocal microscope (Bio-Rad). The Vectastain ABC kit (Vector laboratory) was used for chromogenic detection according to the manufacturer's instructions.
Teratoma formation and blastocyst injection
Approximately 1 × 106 integration-free iPSCs were injected subcutaneously into dorsal flanks of recipient SCID mice. Tumors were isolated 4 weeks later and subjected to histological analysis. GFP-marked integration-free iPSCs were microinjected into C57Bl/6-Tyr c-Brd/c-Brd blastocysts and embryos were transplanted into CBA×B6F1 pseudopregnant females. In some experiments, embryos were dissected at the indicated time points and observed under the fluorescent stereomicroscope. Chimerism of newborn mice was analyzed by PCR using tail DNAs with GFP primers. All animal studies were carried out at the Wellcome Trust Sanger Institute under the UK Home Office license 80/2020.
Supplementary Material
Acknowledgments
We thank Ruby Banerjee for karyotype analysis and Meng Li, Chikara Kokubu, and Kyoji Horie for help, suggestion, and comments. We also thank everybody in Team 82 and the RSF of the Wellcome Trust Sanger Institute for their support. K.Y. is funded by postdoctoral fellowship of Japan Society for the Promotion of Science. This work is supported by the Wellcome Trust (WT077187).
Footnotes
Competing interests statement
The authors declare no conflict of interest.
Editorial Summaries
AOP: piggyBac transposons carrying reprogramming factors are used to reprogram mouse embryonic fibroblasts, with efficiencies equivalent to retroviral transduction, and then removed from the iPS cell genome without a trace.
Issue: piggyBac transposons carrying reprogramming factors are used to reprogram mouse embryonic fibroblasts, with efficiencies equivalent to retroviral transduction, and then removed from the iPS cell genome without a trace.
References
- 1.Yamanaka S. Strategies and new developments in the generation of patient-specific pluripotent stem cells. Cell Stem Cell. 2007;1:39–49. doi: 10.1016/j.stem.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 4.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 5.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 6.Nakagawa M, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- 7.Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell. 2005;121:465–477. doi: 10.1016/j.cell.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 8.Foster KW, et al. Induction of KLF4 in basal keratinocytes blocks the proliferation-differentiation switch and initiates squamous epithelial dysplasia. Oncogene. 2005;24:1491–1500. doi: 10.1038/sj.onc.1208307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nair V. Retrovirus-induced oncogenesis and safety of retroviral vectors. Curr Opin Mol Ther. 2008;10:431–438. [PubMed] [Google Scholar]
- 10.Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008;322:945–949. doi: 10.1126/science.1162494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okita K, Nakagawa M, Hyenjong H, Ichisaka T, Yamanaka S. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322:949–953. doi: 10.1126/science.1164270. [DOI] [PubMed] [Google Scholar]
- 12.Szymczak AL, et al. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol. 2004;22:589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- 13.Cary LC, et al. Transposon mutagenesis of baculoviruses: analysis of Trichoplusia ni transposon IFP2 insertions within the FP-locus of nuclear polyhedrosis viruses. Virology. 1989;172:156–169. doi: 10.1016/0042-6822(89)90117-7. [DOI] [PubMed] [Google Scholar]
- 14.Ding S, et al. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell. 2005;122:473–483. doi: 10.1016/j.cell.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 15.Fraser MJ, Ciszczon T, Elick T, Bauser C. Precise excision of TTAA-specific lepidopteran transposons piggyBac (IFP2) and tagalong (TFP3) from the baculovirus genome in cell lines from two species of Lepidoptera. Insect Mol Biol. 1996;5:141–151. doi: 10.1111/j.1365-2583.1996.tb00048.x. [DOI] [PubMed] [Google Scholar]
- 16.Ivics Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–510. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- 17.Huangfu D, et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol. 2008;26:1269–1275. doi: 10.1038/nbt.1502. [DOI] [PubMed] [Google Scholar]
- 18.Blelloch R, Venere M, Yen J, Ramalho-Santos M. Generation of induced pluripotent stem cells in the absence of drug selection. Cell Stem Cell. 2007;1:245–247. doi: 10.1016/j.stem.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huangfu D, et al. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Biotechnol. 2008;26:795–797. doi: 10.1038/nbt1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aoi T, et al. Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science. 2008;321:699–702. doi: 10.1126/science.1154884. [DOI] [PubMed] [Google Scholar]
- 21.Wang W, et al. Chromosomal transposition of PiggyBac in mouse embryonic stem cells. Proc Natl Acad Sci U S A. 2008;105:9290–9295. doi: 10.1073/pnas.0801017105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brambrink T, et al. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell. 2008;2:151–159. doi: 10.1016/j.stem.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hockemeyer D, et al. A drug-inducible system for direct reprogramming of human somatic cells to pluripotency. Cell Stem Cell. 2008;3:346–353. doi: 10.1016/j.stem.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stadtfeld M, Maherali N, Breault DT, Hochedlinger K. Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell. 2008;2:230–240. doi: 10.1016/j.stem.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wernig M, et al. A drug-inducible transgenic system for direct reprogramming of multiple somatic cell types. Nat Biotechnol. 2008;26:916–924. doi: 10.1038/nbt1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitra R, Fain-Thornton J, Craig NL. piggyBac can bypass DNA synthesis during cut and paste transposition. EMBO J. 2008;27:1097–1109. doi: 10.1038/emboj.2008.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aasen T, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26:1276–1284. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- 28.Carey BW, et al. Reprogramming of murine and human somatic cells using a single polycistronic vector. Proc Natl Acad Sci U S A. 2009;106:157–162. doi: 10.1073/pnas.0811426106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Y, et al. Two supporting factors greatly improve the efficiency of human iPSC generation. Cell Stem Cell. 2008;3:475–479. doi: 10.1016/j.stem.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 30.Hanna J, et al. Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell. 2008;133:250–264. doi: 10.1016/j.cell.2008.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaji K, et al. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009 doi: 10.1038/nature07864. doi:10.1038/nature07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woltjen K, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009 doi: 10.1038/nature07863. doi:10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niwa H, Masui S, Chambers I, Smith AG, Miyazaki J. Phenotypic complementation establishes requirements for specific POU domain and generic transactivation function of Oct-3/4 in embryonic stem cells. Mol Cell Biol. 2002;22:1526–1536. doi: 10.1128/mcb.22.5.1526-1536.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cadinanos J, Bradley A. Generation of an inducible and optimized piggyBac transposon system. Nucleic Acids Res. 2007;35:e87. doi: 10.1093/nar/gkm446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Devon RS, Porteous DJ, Brookes AJ. Splinkerettes--improved vectorettes for greater efficiency in PCR walking. Nucleic Acids Res. 1995;23:1644–1645. doi: 10.1093/nar/23.9.1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.