

Abstract
Cyclic nucleotide phosphodiesterase-8 (PDE8) hydrolyzes the second messenger cAMP and is involved in many biological processes such as testosterone production. Although the bacterial and mammalian expression systems have been extensively tried, production of large quantity of soluble and active PDE8 remains to be a major hurdle for pharmacological and structural studies. Reported here is a detailed protocol of refolding and purification of large quantity of the PDE8A1 catalytic domain (residues 480–820) and kinetic characterization of the refolded protein. This protocol yielded about 8 mg of the PDE8A catalytic domain from 2 liter E. coli culture, which has at least 40-fold higher activity than those reported in literature. The PDE8A1 catalytic domain has kcat of 4.0 s−1 for Mn2+ and 2.9 s−1 for Mg2+, and the KM values of 1–1.8 μM. In addition, the PDE8A1 (205–820) fragment that contains both PAS and catalytic domains was expressed in E. coli and refolded. This PDE8A1 (205–820) fragment has kcat of 1.1 s−1 and KM of 0.28 μM, but aggregated at high concentration. The KM of PDE8A1 (205–820) is 2- to 7-fold higher than the KM values of 40–150 nM for the full-length PDE8s in literature, but about 6-fold lower than that of the catalytic domain. Thus, the KM difference likely implies an allosteric regulation of the PDE8A activity by its PAS domain.
The second messengers adenosine and guanosine 3′, 5′-cyclic monophosphate (cAMP and cGMP) mediate the response of cells to a wide variety of hormones and neurotransmitters and modulate many metabolic processes [1–6]. Cyclic nucleotide phosphodiesterases (PDEs) hydrolyze cAMP and cGMP to 5′-AMP and 5′-GMP. Human genome contains 21 PDE genes that are categorized into 11 families and express over 100 isoforms of PDE proteins through alternative mRNA splicing [7–10]. PDE molecules are divided into a variable regulatory domain at the N-terminus and a conserved catalytic domain with about 300 amino acids at the C-terminus. Family selective PDE inhibitors have been widely studied as therapeutics for treatment of human diseases, including cardiotonics, vasodilators, smooth muscle relaxants, antidepressants, antithrombotics, antiasthmatics, and agents for improvement of learning and memory [11–18]. A well known example is the PDE5 inhibitor sildenafil (Viagra) that has been approved for treatment of both male erectile dysfunction and pulmonary hypertension [11, 14, 19].
Human genome expresses two PDE8 subfamilies (8A and 8B), both of which are cAMP-specific enzymes and have KM of 40–150 nM for cAMP and >100 μM for cGMP [20–23]. PDE8 is distributed in various human tissues and is abundant in testis [24–27]. Functionally, PDE8 has been reported to be involved in regulation of T-cell activation [28], chemotaxis of activated lymphocytes [29], modulation of testosterone production in Leydig cell [30], and potentiation of biphasic insulin response to glucose [31]. It has been recently reported that the H305P mutation of PDE8B1 is associated with micronodular adrenocortical hyperplasia [32] and PDE8B gene variants are involved in regulation of thyroid-stimulating hormone levels and thyroid function [33]. Molecules of PDE8 contain a Per-ARNT-Sim (PAS) domain that is a structural motif and an environmental protein sensor involved in many biological processes such as response to oxygen partial pressure and redox signaling [34, 35]. PDE8 was reported to bind IκBβ, a regulatory protein of transcription factor NF-κB [36], but the binding mode and biological function are unknown.
While crystal structures of the catalytic domains of eight PDE families have been determined [37], PDE8 remains to be one of three PDE families, whose structures of any fragments are not available. The expressions of PDE8 in the baculovirus and E. coli systems have been reported [20–23], but such procedures often produced small amounts of the PDE8 enzymes that have low catalytic activity. Lack of an effective protocol for preparation of large quantity of active PDE8 enzymes appears to be a hurdle for the structural study and inhibitor discovery of PDE8. Reported here are the details of the protein expression of the PDE8A catalytic domain in E. coli, refolding from the inclusion body, purification and kinetic characterization of the refolded PDE8A1. Our refolding protocol yielded 8 mg of the PDE8A catalytic domain from 2 liter E. coli culture, which showed at least 40-fold higher activity than those of PDE8s reported in literature [20–23]. Therefore, this study is valuable not only for basic and structural research, but also for development of PDE8 selective inhibitors.
Materials and methods
Subcloning of the PDE8A1 catalytic domain
The Expressed Sequence Tag cDNA clone of PDE8A1 (GenBank #AF332653) was purchased from American Type Culture Collection (10325182). The cDNA fragments for expression of the PDE8A1 catalytic domain (residues 480–820) and the PDE8A1 fragment (205–820) were amplified by PCR and subcloned into vector pET15b. The following oligonucleotide primers that contain the restriction sites of NdeI and XhoI were used for amplification of the desired genes: 5′-GTGCCGCGCGGCAGCCATATGGCTAGCTCCCTTGATGATGTCCCAC and 3′-GGATCCTCGAGTTACTTCATTTCGTCCAGTCCTTTC. The amplified cDNAs of PDE8A1 and the expression vector pET15b were digested by the restriction enzymes, purified with agarose gel, and ligated together by T4 DNA ligase.
The plasmid pET15b-PDE8A1 was transferred into E. coli strain BL21 (CodonPlus) for overexpression. The cell bearing the vector pET15b-PDE8A1 was grown in a modified 2xYT culture medium (16 g tryptone, 10 g yeast extract, and 5 g NaCl per liter) that was autoclaved before addition of 0.4% glucose, 100 mg ampicillin, and 20 mg chloramphenicol per liter. After the cell was grown at 37°C to A600 = 0.7, 0.1 mM IPTG (isopropyl β-D-thiogalactopyranoside) was added to induce the overexpression and the cell culture was transferred to room temperature for further growth overnight.
Protein refolding and purification
About 10 grams of the frozen cells from 2 liter culture were suspended in 40 mL of the extraction buffer and passed through French Press three times at 1200 psi to disrupt them. The extraction buffer was 20 mM Tris-HCl, pH 7.5, 50 mM NaCl, 1 mM β-mercaptoethanol (β-ME), and 1 mM EDTA. The homogenate was centrifuged at 15,000 rpm in a Beckman JA-20 rotor for 30 min. Unfortunately, the recombinant PDE8A1 fragments mainly existed in the pellet phase and therefore refolding was necessary to obtain soluble and active PDE8A1 protein. The pellet was dissolved in 10 mL buffer of 6 M guanidine, 0.1 M Tris-HCl pH 8.0 under slow orbital shaking at room temperature for 3 hours. The dissolved mixture was centrifuged at 15,000 rpm for 20 min to remove the insoluble debris.
The supernatant was loaded into a Ni-NTA column (φ=2.5 cm, 25 ml QIAGEN agarose beads). The column was washed with 100 mL buffer of 8 M urea, 0.1 M Tris-HCl pH 8.0, and eluted with the same buffer plus 0.5 M arginine. The fractions of the PDE8A1 catalytic domain from the elution were combined. The protein concentration was estimated by the absorption of A280 (1.097 units = 1 mg/ml), as calculated by program ProtParam [38]. Fifty milligrams of the PDE8A catalytic domain at 2 mg/ml (25 ml of protein solution) was added dropwise into 1.7 liters of the refolding buffer under mild stir. The refolding buffer was 0.5 M Tris-HCl pH 7.0, 20 mM MgCl2, 20 mM MnCl2, 20 μM ZnSO4, 0.7 M arginine, 30% glycerol, 10 mM NaCl, 1 mM KCl, and 10 mM DTT. The refolding was carried out at 30 μg/ml protein concentration without shaking at 4°C for 3 days.
To concentrate the refolded PDE8A1, the dilute refolding system was mixed with 15 grams of hydroxyapatite HTP GEL (Bio-Rad) that was presoaked in water. After stirred at room temperature for 15 min, the suspension was filtered with a filter paper. The beads were collected, re-suspended in 100 mL of 20 mM Tris-HCl, pH 8.0, 50 mM NaCl, 1 mM β-ME, and then packed into a column. The column was washed with 50 mL of the same buffer and eluted with 100 mL of 20mM Tris-HCl pH 8.0, 100 mM KH2PO4, 1 mM β-ME. The fractions were combined and dialyzed against 0.5 liter of 20 mM Tris-HCl pH 7.5, 50 mM NaCl, 1 mM β-ME twice, 1 hour and overnight.
To remove the His-tag, 2.5 mM CaCl2 and 1 μg/ml bovine thrombin (Haematologic Tech. Inc.) were added for digestion at 25°C for 1 hour. The digested PDE8A1 was loaded into a Q-Sepharose column (GE Healthcare) that was pre-equilibrated with a buffer of 50 mM NaCl, 20 mM Tris-HCl pH 7.5, 1 mM β-ME, 1 mM MgCl2. The column was washed with 200 mL of 20 mM Tris-HCl pH 7.5, 100 mM NaCl, 1 mM β-ME, 1 mM EDTA, and then PDE8A1 was eluted out with the same buffer except for 300 mM NaCl. After being concentrated to about 10 mL, the protein was loaded into a gel filtration column Sephacryl S300 (GE Healthcare) and eluted with a buffer of 20 mM Tris-HCl pH 7.5, 50 mM NaCl, 1 mM β-ME, 1 mM MgCl2. The protein was finally concentrated by Amicon cell and ultrafiltration membrane YM30 and its purity was estimated by the SDS gel.
The fragment of PDE8A1 (205–820) that contains both PAS and catalytic domains was subcloned into pET15b and expressed in E. coli under the similar procedure, and refolded in a buffer of 50 mM Tris-HCl, pH 7.0, 1.25 M urea, 1 M arginine, 40 mM MnCl2, 10 mM NaCl, 1 mM KCl, and 10 mM DTT.
Enzymatic assay
The enzymatic activities were assayed by using 3H-cAMP or 3H-cGMP as substrate, as previously reported [39]. The catalytic domain of PDE8A1 was incubated with a reaction mixture of 20 mM Tris-HCl, pH 7.5, 10 mM MgCl2 or 4 mM MnCl2, 1 mM DTT, 3H-cAMP or 3H-cGMP (20000–40000 cpm/assay) at 24°C for 15 min. The reaction was terminated by adding 0.2 M ZnSO4 and Ba(OH)2. The reaction product 3H-AMP or 3H-GMP was precipitated out while unreacted 3H-cAMP or 3H-cGMP remained in the supernatant. After centrifugation, the radioactivity in the supernatant was measured in a liquid scintillation counter. The reaction was controlled at hydrolysis of <30% substrate under suitable enzyme concentrations (0.004 to 0.085μg/ml PDE8A1). Twelve concentrations of cAMP or cGMP in a range of 0.04 to 50 μM were used to obtain the kinetic parameters. The enzymatic properties were analyzed by the steady state kinetics [40]. The non-linear regression of the Michealis-Menten equation, as well as Eadie-Hofstee plots were performed to obtain the values of KM, Vmax, and kcat.
Results
Expression and purification of PDE8A1
To express the soluble catalytic domain of PDE8A1 in E. coli, the conditions were extensively tested, including temperatures for E. coli cell growth, cell culture mediums (LB, 2xYT, etc), different expression vectors such as pET15 and pET32, various lengths of the PDE8A1 fragments, addition of various chemicals, induction at different stages of E. coli growth, etc. However, almost all the conditions mainly produced inclusion body of PDE8A1. The best expression condition yielded a small amount of the soluble protein that appeared to have reasonable activity, but was not enough for crystallization. Thus, refolding of the inclusion body was performed to produce large quantity of the soluble catalytic domain of PDE8A1, as discussed in the next section.
The refolded PDE8A1 was purified by three columns (Table 1). Since refolding was performed in diluted solution, it would be time-consuming if the refolded protein were concentrated by Amicon concentrator and YM30 membrane. Fortunately, the refolded PDE8A1 catalytic domain binds to hydroxyapatite HTP. Thus, mixing hydroxyapatite beads with the diluted protein solution and filtration of the suspension with filter paper quickly concentrated the folded PDE8A1. The PDE8A1 catalytic domain weakly interacted with the ionic Q-Sepharose column and was retained in the column under 100 mM NaCl, but eluted out at 300 mM NaCl. The gel filtration column S300 separated small amounts of oligomers with different molecular weights. The peak from the S300 column appeared to have molecular weight of monomeric PDE8A1. A typical batch of refolding and purification used 50 mg PDE8A1 catalytic domain from the Ni-NTA column and yielded about 8 mg active protein from the S300 column with purity >95%. The overall recovery of the catalytic activity from the purification was about 30% and the protein recovery percentage was 15.5% (Table 1). The SDS gel showed that the protein purity after each step of purification was improved slightly (Fig. 1), thus implying that partially-folded or aggregated PDE8A1 co-existed with the more completely folded enzyme and was removed during the purification.
Table 1.
PDE8A purification
| Purification Steps | Total protein (mg) | Specific activity (nmol/min/mg) | Total activity (nmol/min) | Activity recovery (%) |
|---|---|---|---|---|
| Refolding* | 50.0 | 5.6 | 280 | 100 |
| hydroxyapatite HTP | 35.0 | 6.0 | 208 | 74 |
| Q-Sepharose | 12.0 | 7.2 | 87 | 31 |
| Sephacryl S300 | 7.6 | 11.4 | 87 | 31 |
Since the PDE8A catalytic domain was expressed as pellet and the cell lysate had trace activity, the pellet from 10 grams of wet cells was dissolved in 6 M guanidine and purified by Ni-NTA affinity column. A total of 50 mg purified protein from Ni-NTA was refolded and the activity from the refolding was used as the starting unit.
Fig. 1.

Acrylamide gels of the PDE8A1 catalytic domain. Left lane, molecular weight markers; lane 1, E. coli cell before induction; lane 2, total cell after induction; lane 3, supernatant; lane 4, pellet; lane 5, proteins from the Ni-NTA column; lane 6, HTP column; lane 7, Q-Sepharose column after thrombin cleavage of the 10 residue tag; lane 8, finally concentrated PDE8A1 from Sephacryl S300 column; right, the native gel of PDE8A1.
Refolding of PDE8A1
Refolding conditions were extensively searched to obtain the most active PDE8A1 catalytic domain. Divalent metal manganese was essential for the refolding and its 10 mM or higher concentration reached a plateau of the specific activity (Fig. 2a). Surprisingly, magnesium ion up to 50 mM had little impact on the refolding (Fig. 2a), although it has been mostly used as the catalytic ion in the activity assay (Table 2). DTT was also required for the refolding and enhanced activity by about 3-fold (Fig. 2b). The pH of the buffer was an important factor for the refolding (Fig. 2c). Acidic pHs were better than the basic pHs while pH > 9 yielded inactive PDE8A1. L-arginine was another sensitive factor and at 0.7 M it resulted in 5-fold more activity than the case without L-arginine (Fig. 2d). The protein concentration and temperature were less sensitive, but could yield about 2-fold activity increase (Figs. 2e and 2f). Although the best protein concentration for the refolding was 20 μg/ml at 4°C, 30 μg/ml was more effective to prepare large quantity of PDE8A1 for crystallization. Other tested factors include slow shaking during refolding, folding time, and concentration of glycerol. The slow shaking of the refolding solution in coldroom yielded much lower activity of PDE8A1 than the case without shaking. One day of refolding produced about 90% activity of the sample from three day refolding. Glycerol did not significantly impact the refolding and thus was kept at 30% without further optimization.
Fig. 2.

Refolding of the PDE8A1 catalytic domain. The following factors were individually optimized while other variables in the final buffer conditions were held constant. (A) Dependence of the catalytic activity on concentration of Mn2+ (◆) and Mg2+ (□), (B) effect of DTT concentration, (C) pH effect, (D) impact of L-arginine concentration, (E) effect of protein concentration, and (F) effect of temperature. The final buffer for the refolding was 0.5 M Tris-HCl, pH 7.0, 30% glycerol, 0.7 M arginine, 20 mM MnCl2, 20 mM MgCl2, 20μM ZnSO4, 10 mM NaCl, 1 mM KCl, 10 mM DTT.
Table 2.
| Table 2a. Enzymatic properties of the PDE8A1 catalytic domain for cAMP as substrate | |||||
|---|---|---|---|---|---|
| PDE8A | Metal ion | KM (μM) | kcat (s−1) | Vmax (μmol/mg/min) | kcat/KM (s−1 μM−1) |
| 8ACATref | 10 mM Mg2+ | 7.0 ± 0.1 | 2.9 ± 0.1 | 4.5 ± 0.1 | 0.4 ± 0.1 |
| 8ACATref | 4 mM Mn2+ | 1.8 ± 0.1 | 4.0 ± 0.1 | 6.1 ± 0.1 | 2.2 ± 0.1 |
| 8A205ref | 4 mM Mn2+ | 0.28± 0.01 | 1.1 ± 0.1 | 0.93 ± 0.04 | 3.9 ± 0.1 |
| 8ACATnat | 10 mM Mg2+ | 1.0 ± 0.1 | |||
| 8ACATnat | 4 mM Mn2+ | 1.5 ± 0.2 | |||
| Table 2b. Enzymatic properties of the PDE8A1 catalytic domain for cGMP as substrate | |||||
|---|---|---|---|---|---|
| PDE8A | Metal ion | KM (mM) | kcat (s−1) | Vmax (μmol/mg/min) | kcat/KM (s−1 μM−1) |
| 8ACATref | 10 mM Mg2+ | 1.5 ± 0.2 | 0.4 ± 0.1 | 0.6 ± 0.1 | 0.3 ± 0.1 × 10−3 |
| 8ACATref | 4 mM Mn2+ | 1.6 ± 0.1 | 1.6 ± 0.2 | 2.5 ± 0.3 | 1.0 ± 0.1 × 10−3 |
8ACATref and 8ACATnat represent the catalytic domains of PDE8A (residues 480–820) respectively from the refolding and natural folding in E. coli.
8A205ref is the PDE8A fragment with residues 205–820 from the refolding.
Kinetic properties of the refolded PDE8A1
For measurement of the kinetic properties of the refolded proteins, the enzyme concentration in a range of 5.3 to 84.8 ng/ml was tested. As shown in Fig. 3, the activity had essentially a linear relationship with the tested enzyme concentrations. Thus, 10–20 ng/ml PDE8A1 was used in the later measurements of the kinetic properties.
Fig. 3.

Relationship between the PDE8A1 concentration and cAMP conversion. The assays were carried out at a protein concentration range of 5.3 to 84.8 ng/ml and in a buffer of 20 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 1 mM DTT, 3H-cAMP (20000 cpm/assay) at 24°C for 15 min.
When 4 mM Mn2+ was used as the catalytic ion, the refolded PDE8A1 catalytic domain has a KM of 1.8 μM and a kcat of 4.0 s−1 for cAMP, and a KM of 1.6 mM and a kcat of 1.6 s−1 for cGMP (Table 2, Fig. 4). The specificity constant (kcat/KM)cAMP/(kcat/KM)cGMP is 2135 fold for the manganese catalysis. When 10 mM Mg2+ was used as the catalytic ion, the refolded PDE8A1 catalytic domain has a KM of 7.0μM and a kcat of 2.9 s−1 for cAMP, and a KM of 1.5 mM and a kcat of 0.4 s−1 for cGMP (Table 2). The specificity constant (kcat/KM)cAMP/(kcat/KM)cGMP for the Mg2+ catalysis is 1680. Thus, the specificity constants measured by using manganese or magnesium consistently indicate the cAMP-specificity of PDE8A1. In addition, the kcat values suggest that manganese is more effective than magnesium as the catalytic ion to catalyze the reaction. This is more obvious at low substrate concentration. At 1 μM cAMP, the specific activity for 4 mM MnCl2 was about 4-fold higher than that of 10 mM MgCl2 (Fig. 4). However, it remains unknown if manganese serves as the physiological catalytic ion.
Fig. 4.
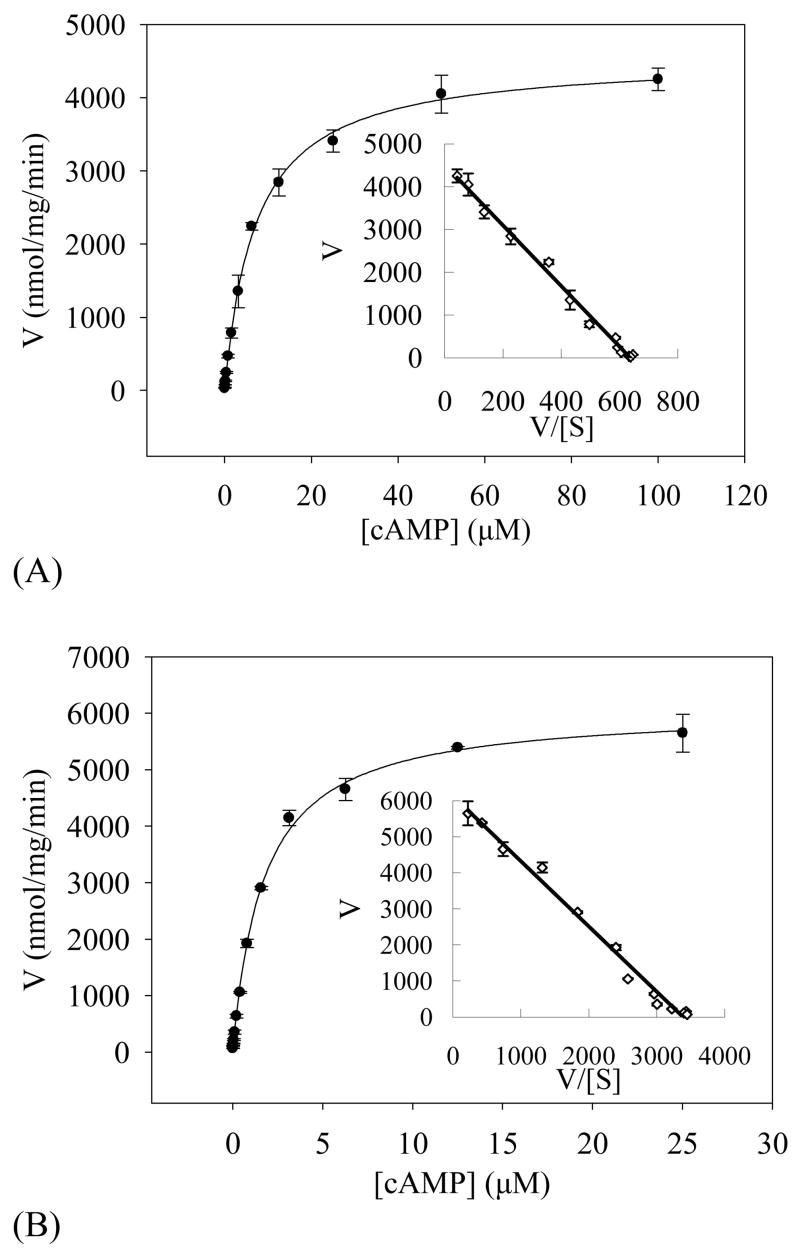
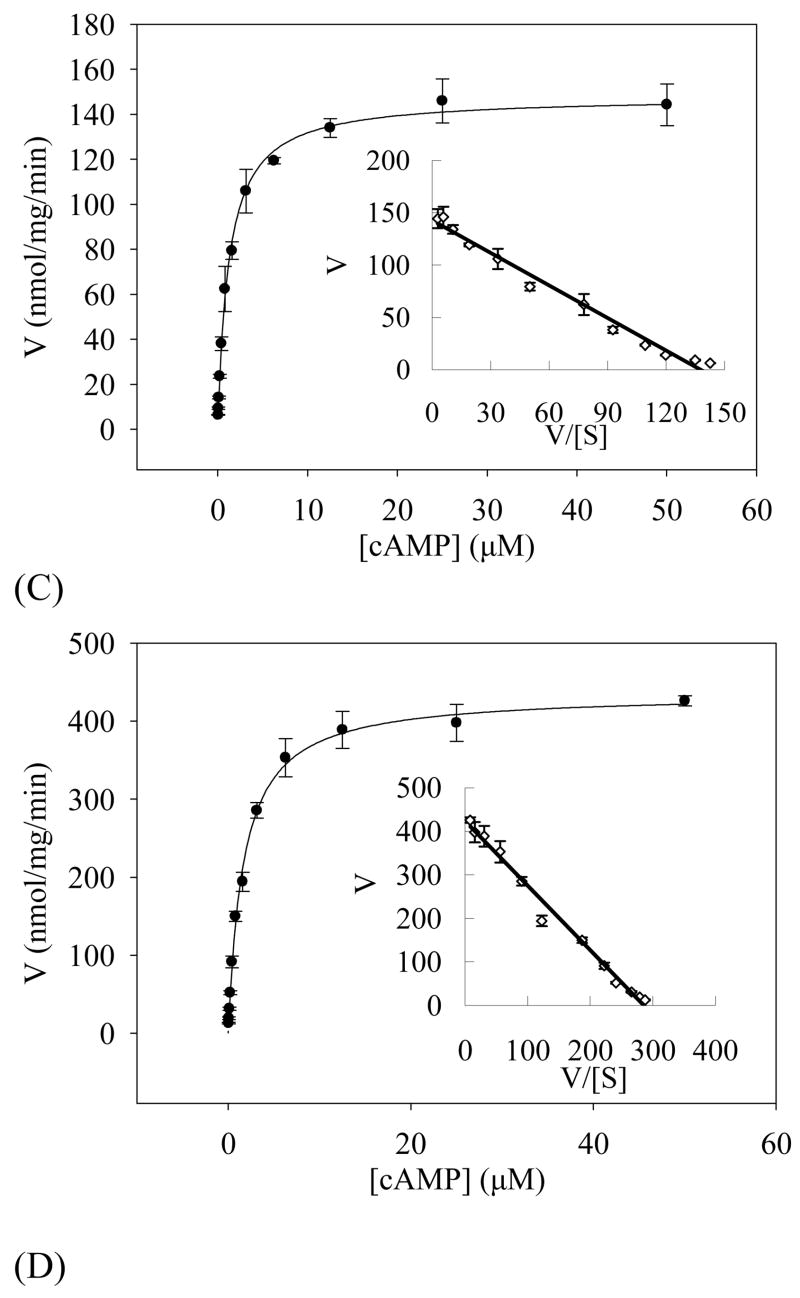
Velocity of the PDE8A1 catalysis versus substrate concentrations. The KM and kcat values were obtained by non-linear regression and Eadie-Hofstie plot (insert). (A) The catalysis of the refolded PDE8A1 by using 10 mM Mg2+ as the catalytic ion. (B) The catalysis of the refolded PDE8A1 by using 4 mM Mn2+ as the catalytic ion. (C) The catalysis of the supernatant fraction of PDE8A1 from the E. coli expression by using 10 mM Mg2+ as the catalytic ion. (D) The catalysis of the supernatant fraction of PDE8A1 from the E. coli expression by using 4 mM Mn2+ as the catalytic ion.
An unusual observation is that magnesium had KM of 7 μM, about 4 times bigger than KM of 1.8 μM for manganese. To study whether the apparent affinity constant KM is dependent on nature of the divalent ions, the PDE8A1 catalytic domain that was expressed and naturally folded in E. coli without addition of divalent ions was partially purified by the similar procedure as that of the refolded and the kinetic parameters were measured. This naturally folded PDE8A1 had the KM values of 1.0μM for Mg2+ and 1.5μM for Mn2+ when cAMP was used as the substrate (Table 2). Thus, the KM difference between the magnesium and manganese catalyses of the refolded PDE8A catalytic domain apparently resulted from the refolding process, but is not biologically relevant.
The kinetic properties of the refolded PDE8A1 catalytic domain are comparable with those of other cAMP-specific PDE families. For example, the full length PDE4D2 and the catalytic domain of PDE7A1 have the KM values of 1.5 and 0.2 μM, the kcat values of 3.9 and 1.6 s−1 for cAMP, and the specificity constants (kcat/KM)cAMP/(kcat/KM)cGMP of 500 and 4000, respectively [39]. A notable difference among these three cAMP-specific PDEs is that the kcat values of PDE8A1 for cGMP is 2.5- to 7.9-fold lower than that for cAMP (Table 2), in comparison to comparable or even better kcat for cGMP than that for cAMP in the PDE4D2 and PDE7A1 catalyses [39]. Therefore, the substrate specificity of the three cAMP-specific PDE families is dominantly determined by KM, while kcat might have some minor contribution in the case of PDE8A1.
Impact of the PAS domain on the catalysis of PDE8A
An unusual observation of this study is that the KM of our PDE8A1 catalytic domain is significantly larger than those for the full length PDE8. Our catalytic domain of PDE8A1 from the refolding has KM of 1.0–1.8μM when Mg2+ or Mn2+ was used as the catalytic ion, which are 7- to 45-fold larger than the KM values of 40–150 nM for full length PDE8A and PDE8B, as reported by four groups [20–23]. This KM incomparability of PDE8 is in contrast to our early observations that other PDE families have comparable KM values between their catalytic domains and the full length proteins, as shown in the cases of PDE4 [39, 41], PDE7 [39, 42], PDE9 [43], and PDE10 [44, 45]. Although the proteins of full-length PDE8s were partially purified and had lower activity, the different KM values do not appear to be the experimental artifacts. Rather, the different KM values may reflect the allosteric behavior of PDE8 and the regulation of the catalytic activity by its PAS domain. This argument is supported by the observation that the PDE8A catalytic domains prepared by either refolding or natural folding in E. coli have the similar KM values. Since the E. coli systems have been widely used to express proteins for biochemical and structural studies, the similar KM values imply that the refolding procedure likely produced biologically relevant conformation of the PDE8A catalytic domain. Indeed, the crystal of the refolded PDE8A1 revealed a topological folding comparable with the structures of other PDE families [46]. The argument of the allosteric catalysis is consistent with the early report that the full-length PDE8A had about 5-fold better activity than its catalytic domain and the binding of the partner protein IκBβ further stimulated catalytic activity about 2-fold [36]. To verify our assumption of the allosteric behavior, a PDE8A1 (205–820) fragment that contains both PAS and catalytic domains was expressed in E. coli and refolded using similar protocols. This fragment has KM of 280 nM and kcat of 1.1 s−1 when cAMP is used as the substrate and 4 mM MnCl2 is the catalytic ion. Thus, the KM of 280 nM for the PDE8A (205–820) fragment is 2- to 7-fold to the KM values of 40 – 150 nM for the full-length PDE8 [20–23], in contrast to 7- to 45-fold difference between the catalytic domain and full-length PDE8. This comparison implies the impact of the PAS domain on the substrate affinity and the catalysis.
Discussion
PDE8 remains one of three PDE families whose crystal structures of any fragments are not available [35], apparently due to the difficulty in preparation of large quantity of the active enzyme. Several research groups have reported the expression of PDE8A and 8B [20–23], but these protocols yielded proteins with low catalytic activities. For example, the expression of a 545-residue fragment of PDE8A in the baculovirus system showed Vmax of 0.15 μmol/min/mg [20], which is about 40-fold less active than Vmax of 6.1μmol/min/mg for our PDE8A catalytic domain. The fragment of C-terminal 584 amino acids of PDE8B had Vmax of only 0.14 nmol/min/mg [22], which is 40000-fold worse than that of our PDE8A. Thus, the refolding protocol in this paper produces large quantity of highly active PDE8A catalytic domain and is valuable for basic studies of PDE8 and also for discovery of PDE8 inhibitors.
All PDE molecules contain two divalent metals that are separated by about 4 Å [37]. The first metal was identified as zinc ion by the anomalous diffraction experiment [47]. The zinc ion forms an octahedron with four PDE residues and two water molecules and has thus been assumed to play both structural and catalytic roles [48]. The second metal ion remains to be identified, in spite of that almost all structure refinements have used magnesium as the catalytic ion [37]. The second metal ion also forms six coordinations, one with an aspartic acid and five with bound water molecules [37]. Although magnesium has effectively catalyzed most PDE families [7–10], our study showed that manganese has kcat about 1.5-fold better than that of magnesium for the catalysis of the PDE8A1 catalytic domain (Table 2), and the specific activity of manganese is about 4-fold to that of magnesium at concentration of 1 μM cAMP. Thus, our observation, together with the early report that manganese activated PDE9A twice as much as magnesium [49–51], suggest that PDE families may have different preferences for the divalent metal ions, yet to be further studied.
Acknowledgments
This work was supported by NIH grant GM59791 to HK and by the 985 project of Science Foundation of Sun Yat-sen University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Antoni F. Molecular diversity of cyclic AMP signaling. Front Neuroendocrin. 2000;21:103–132. doi: 10.1006/frne.1999.0193. [DOI] [PubMed] [Google Scholar]
- 2.O’Neill JS, Maywood ES, Chesham JE, Takahashi JS, Hastings MH. cAMP-dependent signaling as a core component of the mammalian circadian pacemaker. Science. 2008;320:949–953. doi: 10.1126/science.1152506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piper M, van Horck F, Holt C. The role of cyclic nucleotides in axon guidance. Adv Exp Med Biol. 2007;621:134–143. doi: 10.1007/978-0-387-76715-4_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zaccolo M, Movsesian MA. cAMP and cGMP signaling cross-talk: role of phosphodiesterases and implications for cardiac pathophysiology. Circ Res. 2007;100:1569–1578. doi: 10.1161/CIRCRESAHA.106.144501. [DOI] [PubMed] [Google Scholar]
- 5.De Felice FG, Wasilewska-Sampaio AP, Barbosa AC, Gomes FC, Klein WL, Ferreira ST. Cyclic AMP enhancers and Abeta oligomerization blockers as potential therapeutic agents in Alzheimer’s disease. Curr Alzheimer Res. 2007;4:263–271. doi: 10.2174/156720507781077287. [DOI] [PubMed] [Google Scholar]
- 6.Horvath A, Stratakis CA. Unraveling the molecular basis of micronodular adrenal hyperplasia. Curr Opin Endocrinol Diabetes Obes. 2008;15:227–233. doi: 10.1097/MED.0b013e3282fe7416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 8.Lugnier C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents. Pharmacol Ther. 2006;109:366–398. doi: 10.1016/j.pharmthera.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res. 2007;100:309–327. doi: 10.1161/01.RES.0000256354.95791.f1. [DOI] [PubMed] [Google Scholar]
- 10.Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Ann Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- 11.Rotella DP. Phosphodiesterase 5 inhibitors: current status and potential applications. Nature Rev Drug Discovery. 2002;1:674–682. doi: 10.1038/nrd893. [DOI] [PubMed] [Google Scholar]
- 12.Schrör K. The pharmacology of cilostazol. Diabetes Obes Metab. 2002;4:S14–19. doi: 10.1046/j.1463-1326.2002.0040s2s14.x. [DOI] [PubMed] [Google Scholar]
- 13.Lipworth BJ. Phosphodiesterase-4 inhibitors for asthma and chronic obstructive pulmonary disease. Lancet. 2005;365:167–175. doi: 10.1016/S0140-6736(05)17708-3. [DOI] [PubMed] [Google Scholar]
- 14.Gales BJ, Gales MA. Phosphodiesterase-5 inhibitors for lower urinary tract symptoms in men. Ann Pharmacother. 2008;42:111–115. doi: 10.1345/aph.1K422. [DOI] [PubMed] [Google Scholar]
- 15.Houslay MD, Schafer P, Zhang KY. Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov Today. 2005;10:1503–1519. doi: 10.1016/S1359-6446(05)03622-6. [DOI] [PubMed] [Google Scholar]
- 16.Blokland A, Schreiber R, Prickaerts J. Improving memory: a role for phosphodiesterases. Curr Pharm Des. 2006;12:2511–2523. doi: 10.2174/138161206777698855. [DOI] [PubMed] [Google Scholar]
- 17.Menniti FS, Faraci WS, Schmidt CJ. Phosphodiesterases in the CNS: targets for drug development. Nat Rev Drug Discov. 2006;5:660–670. doi: 10.1038/nrd2058. [DOI] [PubMed] [Google Scholar]
- 18.Sharma RK, Das SB, Lakshmikuttyamma A, Selvakumar P, Shrivastav A. Regulation of calmodulin-stimulated cyclic nucleotide phosphodiesterase (PDE1): review. Int J Mol Med. 2006;18:95–105. [PubMed] [Google Scholar]
- 19.Galie N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, Fleming T, Parpia T, Burgess G, Branzi A, Grimminger F, Kurzyna M, Simonneau G. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148–2157. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 20.Fisher DA, Smith JF, Pillar JS, St Denis SH, Cheng JB. Isolation and characterization of PDE8A, a novel human cAMP-specific phosphodiesterase. Biochem Biophys Res Commun. 1998;246:570–577. doi: 10.1006/bbrc.1998.8684. [DOI] [PubMed] [Google Scholar]
- 21.Soderling SH, Bayuga SJ, Beavo JA. Cloning and characterization of a cAMP-specific cyclic nucleotide phosphodiesterase. Proc Natl Acad Sci USA. 1998;95:8991–8996. doi: 10.1073/pnas.95.15.8991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hayashi M, Matsushima K, Ohashi H, Tsunoda H, Murase S, Kawarada Y, Tanaka T. Molecular cloning and characterization of human PDE8B, a novel thyroid-specific isozyme of 3′,5′-cyclic nucleotide phosphodiesterase. Biochem Biophys Res Commun. 1998;250:751–756. doi: 10.1006/bbrc.1998.9379. [DOI] [PubMed] [Google Scholar]
- 23.Gamanuma M, Yuasa K, Sasaki T, Sakurai N, Kotera J, Omori K. Comparison of enzymatic characterization and gene organization of cyclic nucleotide phosphodiesterase 8 family in humans. Cell Signal. 2003;15:565–574. doi: 10.1016/s0898-6568(02)00146-8. [DOI] [PubMed] [Google Scholar]
- 24.Wang P, Wu P, Egan RW, Billah MM. Human phosphodiesterase 8A splice variants: cloning, gene organization, and tissue distribution. Gene. 2001;280:183–194. doi: 10.1016/s0378-1119(01)00783-1. [DOI] [PubMed] [Google Scholar]
- 25.Hayashi M, Shimada Y, Nishimura Y, Hama T, Tanaka T. Genomic organization, chromosomal localization, and alternative splicing of the human phosphodiesterase 8B gene. Biochem Biophys Res Commun. 2002;297:1253–1258. doi: 10.1016/s0006-291x(02)02371-9. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi T, Gamanuma M, Sasaki T, Yamashita Y, Yuasa K, Kotera J, Omori K. Molecular comparison of rat cyclic nucleotide phosphodiesterase 8 family: unique expression of PDE8B in rat brain. Gene. 2003;319:21–31. doi: 10.1016/s0378-1119(03)00809-6. [DOI] [PubMed] [Google Scholar]
- 27.Pérez-Torres S, Cortés R, Tolnay M, Probst A, Palacios JM, Mengod G. Alterations on phosphodiesterase type 7 and 8 isozyme mRNA expression in Alzheimer’s disease brains examined by in situ hybridization. Exp Neurol. 2003;182:322–334. doi: 10.1016/s0014-4886(03)00042-6. [DOI] [PubMed] [Google Scholar]
- 28.Glavas NA, Ostenson C, Schaefer JB, Vasta V, Beavo JA. T cell activation up-regulates cyclic nucleotide phosphodiesterases 8A1 and 7A3. Proc Natl Acad Sci USA. 2001;98:6319–6324. doi: 10.1073/pnas.101131098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong H, Osmanova V, Epstein PM, Brocke S. Phosphodiesterase 8 (PDE8) regulates chemotaxis of activated lymphocytes. Biochem Biophys Res Commun. 2006;345:713–719. doi: 10.1016/j.bbrc.2006.04.143. [DOI] [PubMed] [Google Scholar]
- 30.Vasta V, Shimizu-Albergine M, Beavo JA. Modulation of Leydig cell function by cyclic nucleotide phosphodiesterase 8A. Proc Natl Acad Sci USA. 2006;103:19925–19930. doi: 10.1073/pnas.0609483103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dov A, Abramovitch E, Warwar N, Nesher R. Diminished phosphodiesterase-8B potentiates biphasic insulin response to glucose. Endocrinology. 2008;149:741–748. doi: 10.1210/en.2007-0968. [DOI] [PubMed] [Google Scholar]
- 32.Horvath A, Mericq V, Stratakis CA. Mutation in PDE8B, a cyclic AMP-specific phosphodiesterase in adrenal hyperplasia. N Engl J Med. 2008;358:750–752. doi: 10.1056/NEJMc0706182. [DOI] [PubMed] [Google Scholar]
- 33.Arnaud-Lopez L, Usala G, Ceresini G, Mitchell BD, Pilia MG, Piras MG, Sestu N, Maschio A, Busonero F, Albai G, Dei M, Lai S, Mulas A, Crisponi L, Tanaka T, Bandinelli S, Guralnik JM, Loi A, Balaci L, Sole G, Prinzis A, Mariotti S, Shuldiner AR, Cao A, Schlessinger D, da MU, Abecasis GR, Nagaraja R, Sanna S, Naitza S. Phosphodiesterase 8B gene variants are associated with serum TSH levels and thyroid function. Am J Hum Genet. 2008;82:1270–1280. doi: 10.1016/j.ajhg.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crews ST, Fan CM. Remembrance of things PAS: regulation of development by bHLH-PAS proteins. Curr Opin Genet Dev. 1999;9:580–587. doi: 10.1016/s0959-437x(99)00003-9. [DOI] [PubMed] [Google Scholar]
- 35.Wenger RH, Katschinski DM. The hypoxic testis and post-meiotic expression of PAS domain proteins. Semin Cell Dev Biol. 2005;16:547–553. doi: 10.1016/j.semcdb.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 36.Wu P, Wang P. Per-Arnt-Sim domain-dependent association of cAMP-phosphodiesterase 8A1 with IkappaB proteins. Proc Natl Acad Sci USA. 2004;101:17634–17639. doi: 10.1073/pnas.0407649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ke H, Wang H. Crystal structures of phosphodiesterases and implications on substrate specificity and inhibitor selectivity. Curr Top Med Chem. 2007;7:391–403. doi: 10.2174/156802607779941242. [DOI] [PubMed] [Google Scholar]
- 38.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker John M., editor. The Proteomics Protocols Handbook. Humana Press; 2005. pp. 571–607. [Google Scholar]
- 39.Wang H, Liu Y, Chen Y, Robinson H, Ke H. Multiple elements jointly determine inhibitor selectivity of cyclic nucleotide phosphodiesterases 4 and 7. J Biol Chem. 2005;280:30949–30955. doi: 10.1074/jbc.M504398200. [DOI] [PubMed] [Google Scholar]
- 40.Fersht A. The basic equations of enzyme kinetics, in Structure and mechanism in protein science. Freeman and Company; New York: 1999. pp. 103–132. [Google Scholar]
- 41.Bolger GB, Erdogan S, Jones RE, Loughney K, Scotland G, Hoffmann R, Wilkinson I, Farrell C, Houslay MD. Characterization of five different proteins produced by alternatively spliced mRNAs from the human cAMP-specific phosphodiesterase PDE4D gene. Biochem J. 1997;328:539–548. doi: 10.1042/bj3280539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michaeli T, Bloom TJ, Martins T, Loughney K, Ferguson K, Riggs M, Rodgers L, Beavo JA, Wigler M. Isolation and characterization of a previously undetected human cAMP phosphodiesterase by complementation of cAMP phosphodiesterase-deficient Saccharomyces cerevisiae. J Biol Chem. 1993;268:12925–12932. [PubMed] [Google Scholar]
- 43.Huai Q, Wang H, Zhang W, Colman R, Robinson H, Ke H. Crystal structure of phosphodiesterase 9 shows orientation variation of inhibitor 3-isobutyl-1-methylxanthine binding. Proc Natl Acad Sci USA. 2004;101:9624–9629. doi: 10.1073/pnas.0401120101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang H, Liu Y, Hou J, Zheng M, Robinson H, Ke H. Structural insight into substrate specificity of phospodiesterase 10. Proc Natl Acad Sci, USA. 2007;104:5782–5787. doi: 10.1073/pnas.0700279104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soderling SH, Bayuga SJ, Beavo JA. Isolation and characterization of a dual-substrate phosphodiesterase gene family: PDE10A. Proc Natl Acad Sci USA. 1999;96:7071–7076. doi: 10.1073/pnas.96.12.7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang H, Yan Z, Yang S, Cai J, Robinson H, Ke H. Kinetic and structural studies of phosphodiesterase-8A and implication on the inhibitor selectivity. Biochemictry. 2008 doi: 10.1021/bi801487x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu RX, Hassell AM, Vanderwall D, Lambert MH, Holmes WD, Luther MA, Rocque WJ, Milburn MV, Zhao Y, Ke H, Nolte RT. Atomic structure of PDE4: Insight into phosphodiesterase mechanism and specificity. Science. 2000;288:1822–1825. doi: 10.1126/science.288.5472.1822. [DOI] [PubMed] [Google Scholar]
- 48.Huai Q, Colicelli J, Ke H. The crystal structure of AMP-bound PDE4 suggests a mechanism for phosphodiesterase catalysis. Biochemistry. 2003;42:13220–13226. doi: 10.1021/bi034653e. [DOI] [PubMed] [Google Scholar]
- 49.Fisher DA, Smith JF, Pillar JS, St Denis SH, Cheng JB. Isolation and characterization of PDE9A, a novel human cGMP-specific phosphodiesterase. J Biol Chem. 1998;273:15559–15564. doi: 10.1074/jbc.273.25.15559. [DOI] [PubMed] [Google Scholar]
- 50.Wang P, Wu P, Egan RW, Billah MM. Identification and characterization of a new human type 9 cGMP-specific phosphodiesterase splice variant (PDE9A5). Differential tissue distribution and subcellular localization of PDE9A variants. Gene. 2003;314:15–27. doi: 10.1016/s0378-1119(03)00733-9. [DOI] [PubMed] [Google Scholar]
- 51.Huai Q, Wang H, Zhang W, Colman RW, Robinson H, Ke H. Crystal structure of phosphodiesterase 9 shows orientation variation of inhibitor 3-isobutyl-1-methylxanthine binding. Proc Natl Acad Sci USA. 2004;101:9624–9629. doi: 10.1073/pnas.0401120101. [DOI] [PMC free article] [PubMed] [Google Scholar]