

Abstract
To eliminate unavoidable contamination of purified recombinant proteins by DnaK, we present a unique approach employing a BL21(DE3) ΔdnaK strain of Escherichia coli. Selected representative purified proteins remained soluble, correctly assembled, and active. This finding establishes DnaK dispensability for protein production in BL21(DE3), which is void of Lon protease, key to eliminating unfolded proteins.
Obtaining substantial amounts of pure protein is essential in innumerable biological studies and indispensable to the biochemical characterization of proteins. The ease of growth, well-characterized genetics, and the large number of tools for gene expression have long made Escherichia coli the organism of choice for protein overproduction. The BL21(DE3) strain is widely used for recombinant protein production because of its engineered capacity to produce T7 polymerase and its deficiency in Lon and OmpT proteases.
DnaK is an abundant protein (about 1% of the total protein of E. coli) (17) that interacts with a wide range of newly synthesized polypeptides (28) and assists their proper folding and assembly into oligomers by preventing protein aggregation. DnaK, together with ClpB ATPase, is also required to disaggregate preformed protein aggregates (12, 20), and it participates in the degradation of damaged proteins by Lon and ClpP (26, 27).
The inactivation of dnaK has been shown previously to increase the insoluble fractions of certain aggregation-prone recombinant proteins (7), and DnaK alleviates the aggregation of certain heterologous proteins when coproduced with the protein of interest (8). However, fruitful coproduction of recombinant proteins with chaperones has been challenged by recent findings demonstrating that chaperones increase the solubility but not necessarily the quality of proteins (13, 15).
The DnaK binding site, a five-residue hydrophobic core flanked by two basic residue-enriched regions, occurs on average every 36 residues in protein sequences (25). One consequence is unwanted DnaK contamination of recombinant proteins during purification in E. coli, even after several chromatographic steps (1, 3, 11, 14, 16, 21, 23).
One challenge in protein purification is to obtain the highest level of purity in the fewest steps. Biologically active impurities can jeopardize research or therapeutic applications even if present in trace amounts. One approach developed to circumvent DnaK contamination is extensive washing of columns with ATP since DnaK in its ATP-bound state has low affinity for protein (3). However, this strategy lengthens the purification procedure, is expensive, and is of inconsistent effectiveness (1, 14).
In an attempt to eliminate DnaK contamination, we have investigated whether recombinant proteins could be produced in the absence of DnaK. Toward that end, we constructed a ΔdnaK derivative of the extensively employed E. coli B host strain BL21(DE3). The consequences of the absence of DnaK for the production, solubilities, correct assembly, and activities of several recombinant proteins in BL21(DE3) have been studied. Obtaining a BL21(DE3) ΔdnaK strain has allowed us to elucidate to what extent such a major E. coli chaperone is indispensable to protein overproduction in the particular genetic context of an E. coli strain that lacks Lon, an ATP-dependent protease responsible for degrading unfolded proteins (10).
dnaK in BL21(DE3) was inactivated by the introduction of a null allele, ΔdnaK::Kan, from the E. coli PopC4617 strain by P1 transduction (see Table S1 in the supplemental material). Transductants were selected at 30°C in Luria-Bertani medium complemented with kanamycin. The absence of dnaK was verified by colony PCR using the specific primers dnaK-Nter (5′-GGTAAAATAATTGGTATCGACCTGG-3′) and dnaK-Cter (5′-GTCTTTGACTTCTTCAAATTCAGCG-3′) (see Fig. S1 in the supplemental material). Immunoblotting using an anti-DnaK antibody showed that the obtained transductant (EN2) did not produce DnaK (see Fig. S1 in the supplemental material). The EN2 strain has been deposited at the Collection Nationale de Culture de Microorganismes at the Institut Pasteur (with identification number CNCM I-3863).
E. coli K-12 dnaK mutants usually have a narrow range of permissive temperatures for growth (around 30°C) and exhibit multiple cellular defects, such as impaired cell division and the inhibition of DNA and RNA synthesis (4, 6, 18, 22). Inactivating dnaK in the genetic background of BL21(DE3), an E. coli B strain which is already deficient in OmpT and Lon proteases, did not lead to a dramatic difference in the exponential growth rate at 30°C, but at stationary phase, EN2 cells exhibited slightly reduced ability to form colonies on plates (data not shown). As expected for dnaK mutants, EN2 cells demonstrated impaired growth at 42°C (data not shown). Inactivating dnaK in BL21(DE3) did not induce major morphological defects, and EN2 cells were never found to form long filaments, as dnaK mutants with other genetic backgrounds have previously been reported to do (5) (data not shown). Therefore, the EN2 strain can easily be cultivated at 30°C.
We next investigated whether inactivating dnaK in BL21(DE3) would impair the production and solubilities of different recombinant proteins (whose features are summarized in Table S2 in the supplemental material). These proteins belong to organisms of different kingdoms, and their molecular masses range from 19 to 51 kDa; therefore, they potentially correspond to DnaK substrates since the masses of polypeptides interacting with DnaK range from 14 to 90 kDa (28). Many of them exist as oligomers and may require the assistance of DnaK for proper assembly. These proteins were also chosen for their different levels of production and solubility in E. coli. Four of them (CpxP, ClpP1, PA28α, and proteasome-activating nucleotidase [PAN]) are totally soluble, and two of them (ClpP2 and green fluorescent protein [GFP]) are aggregation prone and may require the presence of DnaK to prevent their aggregation. Importantly, all these proteins were contaminated by DnaK when purified from E. coli (see Fig. 3).
FIG. 3.
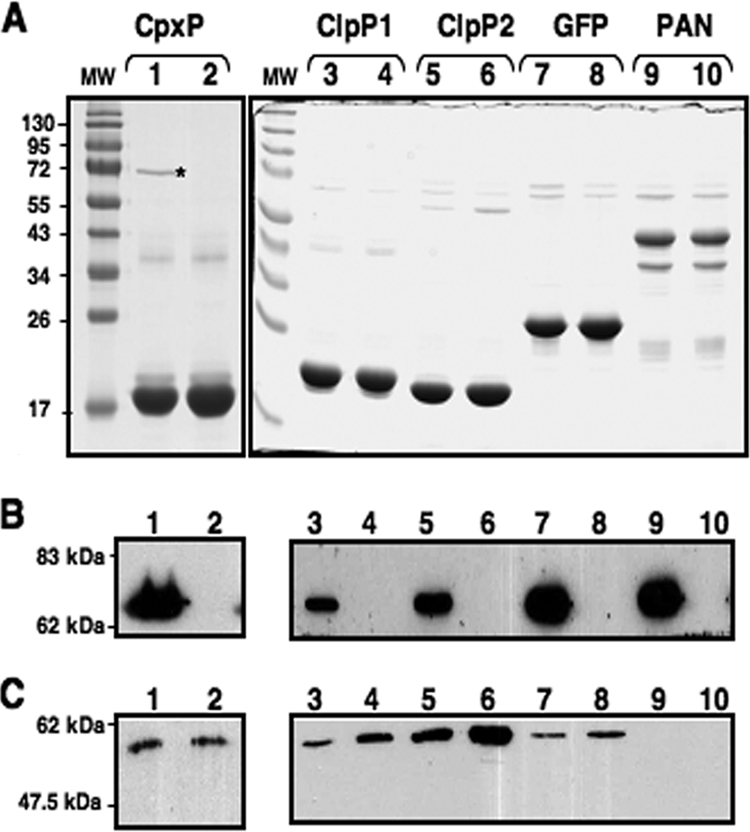
Purification of recombinant proteins in the absence of DnaK. Aliquots of 10 μg of CpxP, ClpP1, ClpP2, GFP, and PAN purified from BL21(DE3) cells (lanes 1, 3, 5, 7, and 9) or EN2 cells (lanes 2, 4, 6, 8, and 10) were loaded onto an SDS-12% PAGE gel. (A) Proteins were revealed by Coomassie blue staining. (B and C) DnaK (B) and GroEL (C) were detected by Western blotting. Sizes of molecular mass markers (lanes MW) are given in kilodaltons and indicated to the left of the gel. The asterisk indicates the position of DnaK on the gel. The two major bands in the purified PAN sample correspond to full-length 50-kDa His-PAN and the 40-kDa PAN fragment resulting from the internal initiation of translation, which copurify as oligomeric complexes (30). The gels shown are representative of results from at least three independent experiments.
The production of recombinant proteins in exponentially growing BL21(DE3) and EN2 cells in Luria-Bertani medium at 30°C was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 2 h. The same biomasses of BL21(DE3) and EN2 cells were sonicated in 1 ml of lysis buffer (50 mM Tris, pH 7.5, 100 mM KCl, 1 mM dithiothreitol). Soluble proteins were separated from aggregated proteins and cellular debris by 30 min of centrifugation at 14,000 × g and 4°C. Pellets containing protein aggregates were resuspended in 1 ml of Tris, pH 7.5, containing 1% sodium dodecyl sulfate (SDS). Total extracts and soluble and insoluble fractions were analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) (Fig. 1).
FIG. 1.

Levels of production and solubility of recombinant proteins in the absence of DnaK. Aliquots of 10 μg of total extracts (T-un and T) and soluble (S) and insoluble (P) fractions from uninduced (T-un) and IPTG-induced (T, S, and P) BL21(DE3) and EN2 cells overexpressing CpxP (A), ClpP1 (B), PA28α (C), ClpP2 (D), GFP (E), or PAN (F) were analyzed by SDS-12% PAGE on gels stained by Coomassie blue. Sizes of molecular mass markers (lanes MW) are given in kilodaltons and indicated to the left of each gel. Arrowheads indicate the positions of recombinant proteins. The gels shown are representative of results from at least three independent experiments.
The levels of production of all tested proteins in EN2 and BL21(DE3) cells were similar, as demonstrated by the protein amounts in total extracts (Fig. 1, lanes T). Moreover, dnaK inactivation did not affect the solubilities of recombinant proteins, even those such as CpxP (Fig. 1A), ClpP1 (Fig. 1B), and PA28α (Fig. 1C) produced in high amounts or those such as ClpP2 (Fig. 1D) and GFP (Fig. 1E) prone to aggregation. These findings were surprising since the function of the DnaK chaperone is to prevent protein aggregation during synthesis and to cooperate with DnaJ, GrpE, and ClpB in the disaggregation of aggregates. It seems that, even for aggregation-prone recombinant proteins, solubility may not necessarily be dependent on endogenous DnaK. This finding may reflect the different folding requirements of specific proteins. Another explanation may be the presence of another chaperone with an overlapping conjoint function. In fact, a consequence of the absence of DnaK in cells is higher levels of production of heat shock proteins such as GroEL/GroES (29). Consistent with these data, EN2 cells produced higher amounts of GroEL than BL21(DE3) cells (data not shown), and these higher amounts may compensate for the absence of DnaK in preventing protein aggregation, as was shown previously for endogenous E. coli proteins and other recombinant proteins (8, 28). An abundance of different chaperones playing nonspecialized roles in recombinant protein folding in E. coli cells may permit toleration of the loss of DnaK, without impairing cell capacity as a protein production factory.
Since the examined proteins could fold and assemble independently of DnaK, we next tested whether a protein known to interact with DnaK could be produced in the absence of this chaperone. Nemo, the IκB kinase complex regulatory component of the NF-κB signaling pathway in eukaryotes, was shown previously to tightly bind and be contaminated by DnaK when produced in E. coli (1). When recombinant His-tagged Nemo was produced in BL21(DE3) under our conditions, it was barely detectable on electrophoresis gel (Fig. 2A and B). However, immunodetection using an anti-His6 antibody (Roche) at a 1:2,000 dilution showed that the absence of DnaK resulted in an increase in Nemo production (Fig. 2D). When Nemo was produced in higher amounts, most of the protein was found in the soluble fraction, indicating that it could be produced as a soluble species in the absence of DnaK (Fig. 2D, lane 5). Increased production of Nemo in the absence of DnaK could be explained by a role of this chaperone in Nemo degradation. Producing Nemo in a BL21(DE3) strain that is deficient in the protease ClpP did not increase its cellular amount (data not shown), indicating that if Nemo was degraded in a DnaK-dependent manner in BL21(DE3) (which already lacks Lon protease), ClpP was not responsible for this proteolysis or the absence of ClpP was compensated for by another protease.
FIG. 2.

Levels of production and solubility of recombinant Nemo in the absence of DnaK. Aliquots of 10 μg (A and C) or 20 μg (B and D) of total extracts (T) and soluble (S) and insoluble (P) fractions from uninduced and IPTG-induced BL21(DE3) and EN2 cells overexpressing Nemo were loaded onto an SDS-10% PAGE gel. Proteins were detected by Coomassie blue staining (A and C), and His-tagged Nemo was detected by Western blotting (B and D). Sizes of molecular mass markers (lanes MW) are given in kilodaltons and indicated to the left of each gel. The gels shown are representative of results from at least three independent experiments.
We next tested whether recombinant proteins produced in the absence of DnaK would remain soluble and active during their purification. Samples of 200 ml of cells overproducing CpxP, ClpP1, ClpP2, or GFP or 500 ml of PAN-overproducing cells were sonicated in 2 ml of lysis buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 10 mM imidazole). The soluble fraction obtained after 30 min of centrifugation at 38,000 × g and 4°C was loaded onto 400 μl of nickel-nitrilotriacetic acid resin, and His-tagged proteins were purified according to the recommendations of the resin manufacturer (Qiagen). After elution, His-tagged proteins were dialyzed against 50 mM Tris, pH 7.5, concentrated, and analyzed by electrophoresis.
By this procedure, recombinant proteins were purified to the levels of homogeneity indicated in Fig. 3A. Samples of 10 μg of purified proteins were used for the immunodetection of contamination by DnaK (using an anti-DnaK antibody from Stressgen at a 1:2,000 dilution). We found that DnaK in BL21(DE3) cells contaminated all preparations of purified recombinant proteins, albeit to different extents (Fig. 3B, lanes 1, 3, 5, 7, and 9). As expected, dnaK inactivation prevented such contamination (Fig. 3B, lanes 2, 4, 6, 8, and 10). It is noteworthy that most of the proteins purified from BL21(DE3) were also contaminated by GroEL, although this contamination was minor. In EN2 cells, where GroEL expression is increased, we did not systematically observe greater contamination by GroEL (Fig. 3C). Moreover, CpxP and GFP, the proteins that exhibited the greatest DnaK contamination, were not the most contaminated by GroEL, and GroEL did not copurify with PAN in the absence of DnaK. Thus, the absence of DnaK did not necessarily lead to a higher level of contamination by GroEL.
Despite the absence of DnaK, all purified recombinant proteins remained soluble even after being concentrated. Since some aggregates are soluble and solubility does not always guarrantee a native active conformation (15, 19), the activities (when readily measurable) or native conformations of some of the purified proteins were examined. One microgram of purified PAN was used to measure ATP hydrolysis at 55°C as described earlier (2). PAN proteins purified from BL21(DE3) and EN2 cells had comparable ATPase activities, with means ± standard errors of 762.33 ± 145.51 and 968.32 ± 198.85 nmol mg−1 h−1 (n = 3), respectively. The fluorescence emission spectrum (at an excitation wavelength of 400 nm) of GFP purified from EN2 cells was indistinguishable from that of GFP purified from BL21(DE3) cells (Fig. 4A), indicating that GFP remained correctly folded when produced in the absence of DnaK. CpxP, a component of the Cpx signal transduction pathway, was the protein that exhibited the greatest DnaK contamination (11). It self-associates into dimers (M. Miot and J.-M. Betton, unpublished data), and to test its correct assembly, 100 μl of purified CpxP at 1 mg/ml in a buffer of 25 mM Tris, pH 7.5, and 150 mM NaCl was loaded onto a size exclusion chromatography column (Superdex 200 HR10/30; GE Healthcare) and eluted with the same buffer at a flow rate of 0.5 ml/min. Recombinant CpxP purified from BL21(DE3) eluted at a volume of 14.98 ml (Fig. 4B), corresponding to a species with an apparent molecular mass of 39.85 kDa (a dimer of Cpx). Recombinant CpxP purified from EN2 eluted at a nearly identical volume of 14.97 ml (Fig. 4B). Thus, the absence of DnaK did not alter CpxP dimeric assembly and did not produce any soluble higher-molecular-mass aggregate species.
FIG. 4.
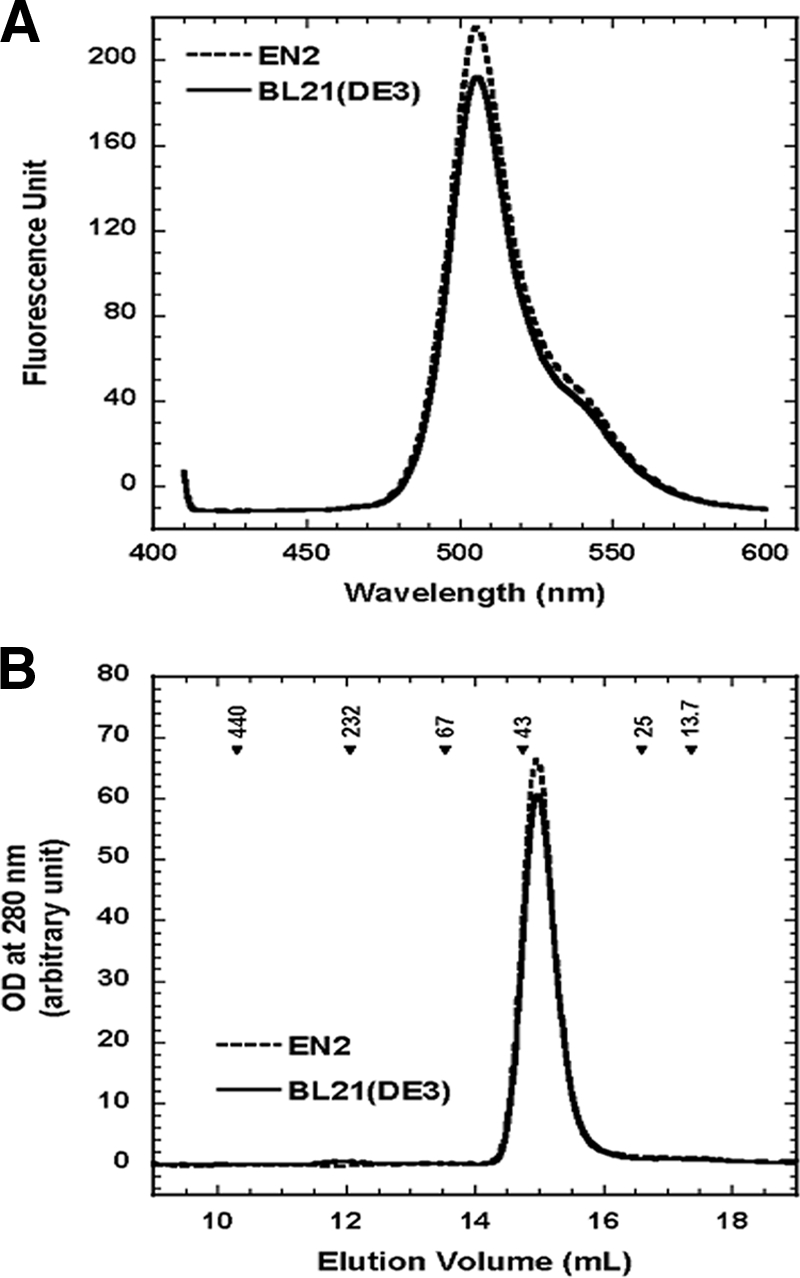
Folding and assembly of proteins in the absence of DnaK. (A) Fluorescence emission spectra of 8-μg/ml GFP preparations purified from BL21(DE3) and EN2 cells, recorded with an FP-6200 spectrofluorimeter (Jasco) at a scan rate of 250 nm min−1 using a bandwidth of 5 nm for both excitation and emission beams. The spectra shown are representative of results from at least two independent experiments. (B) Size exclusion chromatograms for CpxP proteins purified from BL21(DE3) and EN2 cells. Arrowheads indicate the elution volumes of the standards, and their masses are given in kilodaltons. The chromatograms shown are representative of results from at least three independent experiments.
Altogether, these findings indicate that high levels of correctly folded, assembled, and active soluble recombinant proteins can be produced in the absence of endogenous DnaK chaperone in BL21(DE3). Surprisingly, our study showed that the inactivation of dnaK in BL21(DE3), which does not contain Lon, did not result in an increase in the aggregation of recombinant proteins, as was seen previously in E. coli K-12 (24). It seems that in BL21(DE3) cells, and in E. coli B cells in general, factors other than DnaK and Lon may be fundamental in managing the accumulation of aggregated proteins. Through the detailed characterization of a BL21(DE3) ΔdnaK strain and testing of the production of proteins of different natures, origins, and sizes, including aggregation-prone proteins, our study demonstrates that this EN2 strain offers a strategy that can be generally and extensively used to avoid unwanted contamination by DnaK. In addition, since DnaK has ATPase activity, the EN2 strain is particularly well suited for the production and purification of recombinant ATPases, eliminating the undifferentiable ATPase contamination. Given that GroEL, another major chaperone in E. coli, has also been found to contaminate purified recombinant proteins (9), it would be of additional interest to find conditions under which both dnaK and groEL could be eliminated in the BL21(DE3) strain without impairing its survival and its remarkable protein factory capacities.
Supplementary Material
Acknowledgments
We thank F. Agou and P. Delepelaire for plasmids and strains. We are indebted to A. Ullmann, C. Wandersman, and G. Karimova for critically reading the manuscript. We are also grateful to J.-H. Alix and T. Msadek for fruitful discussions.
Footnotes
Published ahead of print on 3 April 2009.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Agou, F., F. Ye, S. Goffinont, G. Courtois, S. Yamaoka, A. Israel, and M. Veron. 2002. NEMO trimerizes through its coiled-coil C-terminal domain. J. Biol. Chem. 277:17464-17475. [DOI] [PubMed] [Google Scholar]
- 2.Benaroudj, N., P. Zwickl, E. Seemuller, W. Baumeister, and A. L. Goldberg. 2003. ATP hydrolysis by the proteasome regulatory complex PAN serves multiple functions in protein degradation. Mol. Cell 11:69-78. [DOI] [PubMed] [Google Scholar]
- 3.Birringer, M. S., R. Perozzo, E. Kut, C. Stillhart, W. Surber, L. Scapozza, and G. Folkers. 2006. High-level expression and purification of human thymidine kinase 1: quaternary structure, stability, and kinetics. Protein Expr. Purif. 47:506-515. [DOI] [PubMed] [Google Scholar]
- 4.Bukau, B., and G. C. Walker. 1989. Cellular defects caused by deletion of the Escherichia coli dnaK gene indicate roles for heat shock protein in normal metabolism. J. Bacteriol. 171:2337-2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bukau, B., and G. C. Walker. 1989. ΔdnaK52 mutants of Escherichia coli have defects in chromosome segregation and plasmid maintenance at normal growth temperatures. J. Bacteriol. 171:6030-6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bukau, B., and G. C. Walker. 1990. Mutations altering heat shock specific subunit of RNA polymerase suppress major cellular defects of E. coli mutants lacking the DnaK chaperone. EMBO J. 9:4027-4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carrio, M. M., and A. Villaverde. 2003. Role of molecular chaperones in inclusion body formation. FEBS Lett. 537:215-221. [DOI] [PubMed] [Google Scholar]
- 8.de Marco, A., E. Deuerling, A. Mogk, T. Tomoyasu, and B. Bukau. 2007. Chaperone-based procedure to increase yields of soluble recombinant proteins produced in E. coli. BMC Biotechnol. 7:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Marco, A., S. Volrath, T. Bruyere, M. Law, and R. Fonne-Pfister. 2000. Recombinant maize protoporphyrinogen IX oxidase expressed in Escherichia coli forms complexes with GroEL and DnaK chaperones. Protein Expr. Purif. 20:81-86. [DOI] [PubMed] [Google Scholar]
- 10.Donch, J., Y. S. Chung, and J. Greenberg. 1969. Locus for radiation resistance in Escherichia coli strain B-r. Genetics 61:363-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fleischer, R., R. Heermann, K. Jung, and S. Hunke. 2007. Purification, reconstitution, and characterization of the CpxRAP envelope stress system of Escherichia coli. J. Biol. Chem. 282:8583-8593. [DOI] [PubMed] [Google Scholar]
- 12.Goloubinoff, P., A. Mogk, A. P. Zvi, T. Tomoyasu, and B. Bukau. 1999. Sequential mechanism of solubilization and refolding of stable protein aggregates by a bichaperone network. Proc. Natl. Acad. Sci. USA 96:13732-13737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez-Montalban, N., E. Garcia-Fruitos, and A. Villaverde. 2007. Recombinant protein solubility—does more mean better? Nat. Biotechnol. 25:718-720. [DOI] [PubMed] [Google Scholar]
- 14.Guo, L. W., F. M. Assadi-Porter, J. E. Grant, H. Wu, J. L. Markley, and A. E. Ruoho. 2007. One-step purification of bacterially expressed recombinant transducin α-subunit and isotopically labeled PDE6 γ-subunit for NMR analysis. Protein Expr. Purif. 51:187-197. [DOI] [PubMed] [Google Scholar]
- 15.Haacke, A., G. Fendrich, P. Ramage, and M. Geiser. 2008. Chaperone over-expression in Escherichia coli: apparent increased yields of soluble recombinant protein kinases are due mainly to soluble aggregates. Protein Expr. Purif. 64:185-193. [DOI] [PubMed] [Google Scholar]
- 16.Hellebust, H., M. Uhlen, and S. O. Enfors. 1990. Interaction between heat shock protein DnaK and recombinant staphylococcal protein A. J. Bacteriol. 172:5030-5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hesterkamp, T., and B. Bukau. 1998. Role of the DnaK and HscA homologs of Hsp70 chaperones in protein folding in E. coli. EMBO J. 17:4818-4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Itikawa, H., and J. Ryu. 1979. Isolation and characterization of a temperature-sensitive dnaK mutant of Escherichia coli B. J. Bacteriol. 138:339-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinez-Alonso, M., E. Garcia-Fruitos, and A. Villaverde. 2008. Yield, solubility and conformational quality of soluble proteins are not simultaneously favored in recombinant Escherichia coli. Biotechnol. Bioeng. 101:1353-1358. [DOI] [PubMed] [Google Scholar]
- 20.Mogk, A., T. Tomoyasu, P. Goloubinoff, S. Rudiger, D. Roder, H. Langen, and B. Bukau. 1999. Identification of thermolabile Escherichia coli proteins: prevention and reversion of aggregation by DnaK and ClpB. EMBO J. 18:6934-6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murby, M., L. Cedergren, J. Nilsson, P. A. Nygren, B. Hammarberg, B. Nilsson, S. O. Enfors, and M. Uhlen. 1991. Stabilization of recombinant proteins from proteolytic degradation in Escherichia coli using a dual affinity fusion strategy. Biotechnol. Appl. Biochem. 14:336-346. [PubMed] [Google Scholar]
- 22.Paek, K. H., and G. C. Walker. 1987. Escherichia coli dnaK null mutants are inviable at high temperature. J. Bacteriol. 169:283-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rial, D. V., and E. A. Ceccarelli. 2002. Removal of DnaK contamination during fusion protein purifications. Protein Expr. Purif. 25:503-507. [DOI] [PubMed] [Google Scholar]
- 24.Rosen, R., D. Biran, E. Gur, D. Becher, M. Hecker, and E. Z. Ron. 2002. Protein aggregation in Escherichia coli: role of proteases. FEMS Microbiol. Lett. 207:9-12. [DOI] [PubMed] [Google Scholar]
- 25.Rudiger, S., L. Germeroth, J. Schneider-Mergener, and B. Bukau. 1997. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 16:1501-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sherman, M., and A. L. Goldberg. 1992. Involvement of the chaperonin dnaK in the rapid degradation of a mutant protein in Escherichia coli. EMBO J. 11:71-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Straus, D. B., W. A. Walter, and C. A. Gross. 1988. Escherichia coli heat shock gene mutants are defective in proteolysis. Genes Dev. 2:1851-1858. [DOI] [PubMed] [Google Scholar]
- 28.Teter, S. A., W. A. Houry, D. Ang, T. Tradler, D. Rockabrand, G. Fischer, P. Blum, C. Georgopoulos, and F. U. Hartl. 1999. Polypeptide flux through bacterial Hsp70: DnaK cooperates with trigger factor in chaperoning nascent chains. Cell 97:755-765. [DOI] [PubMed] [Google Scholar]
- 29.Tilly, K., N. McKittrick, M. Zylicz, and C. Georgopoulos. 1983. The dnaK protein modulates the heat-shock response of Escherichia coli. Cell 34:641-646. [DOI] [PubMed] [Google Scholar]
- 30.Zwickl, P., D. Ng, K. M. Woo, H. P. Klenk, and A. L. Goldberg. 1999. An archaebacterial ATPase, homologous to ATPases in the eukaryotic 26S proteasome, activates protein breakdown by 20S proteasomes. J. Biol. Chem. 274:26008-26014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.