

Abstract
Francisella tularensis, the etiological agent of tularemia, is capable of infecting a wide range of animals and causes a severe, lethal disease in humans. The pathogen evades killing by cells of the innate immune system utilizing genes encoding a pathogenicity island, including iglABCD, and instead utilizes these cells as a niche for replication and dissemination to other organs within the host. Regulators of the igl genes (e.g., MglA, SspA, FevR and PmrA) have been identified, but environmental stimuli and mechanisms of regulation are as yet unknown and are likely to involve additional gene products. In this work, we more closely examine the roles that environmental iron and the ferric uptake repressor protein (Fur) play in the regulation of the iglABCD operon. We also used a genetic approach to identify and characterize a new regulator of the igl operon, designated migR (macrophage intracellular growth regulator; FTL_1542). Quantitative real-time reverse transcription-PCR in a site-directed migR mutant confirmed the reduction in the number of iglC transcripts in this strain and also demonstrated reduced expression of fevR. Comparison of the migR and fevR mutants in monocyte-derived macrophages (MDMs) and epithelial cell lines revealed a reduced ability for each mutant to grow in MDMs, yet only the fevR mutant exhibited impaired replication in epithelial cell lines. Confocal analysis of infected MDMs revealed that although neither mutant reached the MDM cytosol, the fevR mutant was trapped in lamp-1-positive phagosomes, whereas the migR mutant resided in mature phagolysosomes enriched with both lamp-1 and cathepsin D. Disruption of migR and fevR also impaired the ability of F. tularensis to prevent neutrophil oxidant production. Thus, we have identified migR, a gene that regulates expression of the iglABCD operon and is essential for bacterial growth in MDMs and also contributes to the blockade of neutrophil NADPH oxidase activity.
Francisella tularensis is a gram-negative, facultative intracellular pathogen and is the etiologic agent of tularemia. While first associated with a plague-like illness in rodents, F. tularensis is capable of causing disease in a wide range of animal hosts, including fish, birds, small mammals, and humans, and is also found in invertebrate vectors (45, 48). The most common form of human tularemia is a cutaneous, ulceroglandular disease that results from the bite of an arthropod vector carrying bacteria or contact with the blood of an infected animal through an abrasion in the skin (23). While less common, the pneumonic form of the disease is associated with the highest morbidity and mortality and can result from inhalation of as few as 10 organisms (48). These facts, coupled with the relative ease of dissemination of the bacteria, have led to F. tularensis being designated a category A select agent by the Centers for Disease Control and Prevention (CDC) (32).
Several studies have shown that F. tularensis has the ability to persist or replicate within both phagocytes and other cell types, including human monocyte-derived macrophages (MDMs), human neutrophils, bronchial airway epithelial cells, and other tissue culture cell lines, such as HEp-2 and J774A.1 cells (28, 35, 37, 40, 44, 50, 57, 60). Replication within host cells is dependent on genes located within the Francisella pathogenicity island (FPI), but intracellular survival and growth are likely to require additional genes as well. Most notably, genes comprising the iglABCD operon have been directly implicated in escape from the phagosome and/or the ability to replicate in the host cell cytosol (19, 34, 44, 53, 55). Genome-wide screens have also identified genes outside the FPI that are involved in other aspects of virulence or dissemination in animal models of tularemia (50, 61, 65).
While some genetic screens have identified genes critical to the intracellular life cycle of F. tularensis, little has been done to examine the regulation of these genes or the environmental stimuli leading to their differential expression. The first gene identified to encode a regulator of virulence gene expression was mglA (6, 10, 35). Homologous to the stringent starvation protein of Escherichia coli, MglA positively regulates several genes in the FPI, including those in the iglABCD operon (35). More recent work has shown that the regulatory activity of MglA on the igl operon also requires SspA, a second transcriptional activator capable of associating with RNA polymerase (14). Heterodimerization of these two proteins facilitates their interaction with the unique α subunits of F. tularensis RNA polymerase, which in turn stimulates the transcription of numerous genes throughout the chromosome (10, 14). A third regulatory gene, fevR, is also positively regulated by MglA and SspA and is reported to control the expression of the same set of genes as MglA and SspA (9). Finally, the disruption of an orphan response regulatory gene, pmrA, which is predicted to contain a DNA binding domain, also has been found to have a negative effect on the transcription of many genes, including the igl operon (43). The regulatory activity of pmrA is apparently not exerted by altering mglA and sspA expression; nevertheless, the pmrA regulon does overlap with the genes regulated by mglA and sspA, specifically the genes residing on the FPI. The exact mechanism of regulation by these factors is unknown, as are the environmental and/or host signals leading to this regulation.
Generally, the ability to sense and rapidly respond to environmental signals through modification of gene expression is vital to the ability of a bacterium to adapt to and survive under different conditions, including those found in various host cell environments. Studies of gene expression in F. tularensis have shown an upregulation of FPI genes when bacteria are grown intracellularly compared to when they are grown in broth (29). An increase in capsule production and surface pili has also been demonstrated when F. tularensis is grown in Chamberlain's defined medium (CDM) compared to when it is grown in rich growth medium (15, 27). While the specific signal or signals leading to these changes are unknown, iron availability has emerged as an environmental signal that influences the expression of numerous Francisella genes. Among these are the genes in the fslABCD and iglABCD operons, which are involved in iron acquisition and intracellular growth, respectively (11, 21, 62). Previous studies have shown a role for the ferric uptake regulator protein (Fur) in the iron-dependent regulation of fsl but not igl transcription (11).
We initiated the work in this study by considering iron as an environmental signal leading to regulation of the iglABCD operon and by more closely examining the role of Fur in this regulation. We conducted a genetic screen for new regulators of the igl operon and identified a new gene, migR (FTL_1542), that is involved in its regulation. Here, the effect of migR mutation on transcriptional regulation of the iglABCD operon has been partially characterized, as well as effects of this mutation on the interactions of F. tularensis with host cells. Specifically, we have examined whether this mutation alters intracellular growth in human phagocytes and epithelial cell lines. Additionally, we examined the specific effects of this mutation on phagosome maturation in macrophages and inhibition of the neutrophil oxidative burst.
MATERIALS AND METHODS
Bacterial strains, plasmid construction, growth conditions, and antibiotics.
F. tularensis LVS (ATCC 29684), Francisella novicida U112, and F. novicida fur::TnKn (26) were grown in modified Mueller-Hinton (MMH) broth (Becton Dickinson, Sparks, MD) or on Mueller-Hinton agar (Acumedia, Lansing, MI) supplemented with 1% (wt/vol) glucose, 0.025% ferric pyrophosphate, and 2% IsoVitaleX. Spectinomycin (25 μg/ml for LVS and 100 μg/ml for F. novicida), kanamycin (25 μg/ml), and hygromycin (200 μg/ml) were added to the bacterial growth media when appropriate. CDM was prepared as described previously (13) or with 28 μM, 350 nM, or no added FeSO4, as dictated by experimental parameters. Iron-replete growth conditions were achieved by overnight growth of bacterial cultures in CDM containing 28 μM FeSO4, followed by dilution into the same medium before performing Miller assays for β-galactosidase quantitation (41). Iron depletion was achieved by growing bacterial cultures in CDM containing 7 μM FeSO4, followed by a 1:1,000 dilution into CDM containing 350 nM FeSO4 (LVS), or by direct colony inoculation into CDM containing no added FeSO4 (F. novicida) before performing β-galactosidase assays. LVS strains containing chromosomal lacZ reporters in iglB and fslC were described in a previous work (11).
For a plasmid-borne study of iron-responsive DNA elements, PCR was performed on an LVS chromosomal template to amplify ∼300 nucleotide segments of DNA upstream of fslA, iglA, and iglC. Oligonucleotide primers were all designed with 5′ NcoI-BamHI and 3′ KpnI “tails” to facilitate cloning into the NcoI-KpnI restriction sites of pTrc99A (4). Successful cloning of these DNA elements was followed by the amplification of a promoterless lacZ gene from a pA23 (64) template with oligonucleotide primers containing 5′ KpnI and 3′ SalI tails. The lacZ amplicon was then cloned into the KpnI-SalI restriction sites of the pTrc99A plasmids containing the previously described DNA fragments to create reporters of transcription for each of the three genes. Finally, the entire transcriptional reporter cassettes were removed from the pTrc99A backbone by the BamHI-SalI digest and were cloned into the same sites in the Francisella-E. coli shuttle plasmid pBB103 (sequences for oligonucleotide primers available upon request). As a control, the promoterless lacZ reporter alone was cloned into the same BamHI-SalI restriction sites in pBB103. These plasmids were designated pBB119, pBB125, pBB133, and pBB134 (Table 1) and were introduced to F. novicida strains by rubidium chloride cryotransformation as described elsewhere (27).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| Strains | ||
| F. tularensis LVS | F. tularensis type B live vaccine strain | K. L. Ekins |
| F. novicida U112 | F. novicida wild-type strain | 26 |
| F. novicida fur::TnKn | F. novicida carrying an insertion mutation in fur | 26 |
| Plasmids | ||
| pBB103 | Francisella-E. coli shuttle plasmid carrying spectinomycin resistance cassette and polylinker | |
| pBB107 | Fusion of pBDJ303 and pMKM219, final Tn5 delivery plasmid for mutagenesis of F. tularensis spp. | 11 |
| pBB109 | Tn5-hygromycin delivery plasmid for mutagenesis of F. tularensis spp. derived from plasmid pBB107 | This study |
| pBB114 | Complementation plasmid carrying full-length FTL_1542 driven by the Francisella groES promoter | This study |
| pBB115 | Complementation plasmid carrying full-length FTL_0347 driven by Francisella groES promoter | This study |
| pBB119 | Francisella-E. coli shuttle plasmid containing promoterless lacZ reporter | This study |
| pBB125 | Francisella-E. coli shuttle plasmid containing the iglA-lacZ reporter | This study |
| pBB133 | Francisella-E. coli shuttle plasmid containing the fslA-lacZ reporter | This study |
| pBB134 | Francisella-E. coli shuttle plasmid containing the iglC-lacZ reporter | This study |
| pBB135 | Complementation plasmid carrying a full-length fevR gene driven by native promoter | This study |
Complementation plasmids pBB114 and pBB135 (Table 1) were created by amplifying the entire FTL_1542 or FTL_0449 gene using LVS chromosomal DNA as a template and oligonucleotide primers containing 5′ KpnI and 3′ SalI tails (sequences for oligonucleotide primers available upon request). These amplicons were cloned downstream of the F. tularensis groES promoter in pTrc99A, and the entire expression cassette was transferred to pBB103, using BamHI-SalI restriction sites present on both plasmids.
Mutagenesis, screening, and identification of regulators of iglB transcription.
The LVS strain containing a chromosomal lacZ reporter of iglB transcription was transformed with the Tn5 delivery plasmid pBB109 (Table 1). Colonies obtained after ∼3 days of growth at 30°C on MMH agar with 25 μg/ml spectinomycin were inoculated into 5 ml of MMH broth with 25 μg/ml spectinomycin and were grown at 30°C with agitation to an optical density at 600 nm of ∼0.1. Cultures were then serially diluted and plated onto MMH agar with 200 μg/ml hygromycin at 41°C to select for isolates with Tn5 insertions into the F. tularensis chromosome and a simultaneous loss of the temperature-sensitive transposon delivery plasmid. Resulting hygromycin-resistant colonies were arrayed onto 96-well cell culture plates in 100 μl of MMH broth and were incubated at 37°C until turbid. Freezer stocks were made by adding 100 μl of 2× freezing medium (1.0 M sucrose, 20% glycerol). To identify strains with reduced iglB expression, Tn5 mutants were recovered from freezer stocks and plated onto MMH or CDM agar at 37°C using a 96-prong replicator (Boekel, Feasterville, PA). After ∼24 h, reporter enzyme activity was visualized by overlaying Whatman filter paper no. 1 presoaked with 20 mg/ml X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) in dimethylformamide diluted 1:4 in water. Quantitation of β-galactosidase activity was accomplished by conducting β-galactosidase assays in triplicate on duplicate cultures of bacteria grown to mid-log phase in MMH broth. IglC was detected by Western blotting using a goat anti-IglC antibody raised against IglC purified from F. novicida generously provided by Karl Klose (University of Texas, San Antonio).
To identify the sites of Tn5 insertions affecting iglB expression, genomic DNA was isolated from individual colonies and digested with KpnI to create a DNA fragment containing the oriR6K origin, the hyg gene, and the flanking chromosomal sequence. The digested DNA was ligated, transformed into pir+ E. coli, and plated onto agar plates with 200 μg/ml hygromycin to select for transformants that carried the plasmid of interest. Plasmid DNA was isolated and sequenced using a primer with the sequence 5′-GTGACAGGGGCCTCTTTTATC-3′ that anneals to the 3′ end of the hyg gene and produces a sequence of the flanking chromosomal DNA. Sequence data were used to search the sequenced bacterial chromosome database using NCBI BLAST to identify Tn5 insertion sites within the F. tularensis chromosome.
Creation of site-directed mutants using intron-directed mutagenesis.
Site-directed insertion mutants were created using a modified TargeTron (Sigma-Aldrich, St. Louis, MO) mutagenesis system (52). In brief, the coding sequence of each gene of interest was entered into the Sigma TargeTron primer design site to determine the appropriate oligonucleotides for retargeting the intron. Importantly, an XhoI restriction site was substituted for the HindIII site when designing the intron-binding site primer. Retargeted PCR products were generated using an intron PCR template (TA0100; Sigma-Aldrich) according to the recommendations of the manufacturer. The resulting fragment was introduced into the delivery vector pKEK1140, and cloning was verified by BglII digestion. LVS transformed with the retargeted plasmid was grown at 30°C on MMH agar with 25 μg/ml kanamycin. Individual colonies were purified once by growing them at the permissive temperature, and resulting colonies were screened by PCR to identify mutants before passaging at 37°C to cure the plasmid.
Real-time reverse transcription-PCR (RT-PCR) for quantification of mglA, sspA, pmrA, fevR, and iglC transcript levels.
RNA was isolated from wild-type LVS, and the site-directed FTL_1542 mutant cultures were grown to mid-log phase in MMH broth, using a MasterPure complete DNA and RNA isolation kit (Epicentre, Madison, WI). High-quality cDNA was generated using SuperScript III reverse transcriptase and random primers (Invitrogen, Carlsbad, CA) according to the recommendations of the manufacturer. cDNA was quantified using the Power SYBR green PCR master mix (Applied Biosystems, Foster City, CA). Transcript levels for each gene were determined by comparing to a standard curve generated with each corresponding primer set, using dilutions of genomic DNA template. Relative transcript abundance was determined by normalizing the message in the mutant and wild-type strains to that of the tul4 gene.
Neutrophil and macrophage isolation.
Heparinized venous blood samples were obtained from healthy adult volunteers in accordance with a protocol approved by the Institutional Review Board for Human Subjects at the University of Iowa. Neutrophils (polymorphonuclear leukocytes [PMN]) were isolated using dextran sedimentation and density gradient separation on Ficoll-Hypaque, followed by hypotonic lysis of erythrocytes (20). PMN (∼98% purity) were resuspended in Hank's balanced salt solution (HBSS) without divalent cations, counted, and then diluted into appropriate media as indicated. Mononuclear cells were isolated by centrifugation on Ficoll-Hypaque, washed twice in RPMI 1640 (Cambrex), resuspended in RPMI plus 20% autologous serum at a concentration of 2 × 106/ml, and differentiated into MDM by incubation in Teflon jars for 5 to 7 days at 37°C (56, 57).
Intracellular growth assays.
Wild-type or mutant LVS strains were used to infect MDM (multiplicity of infection [MOI], ∼20:1), A549 cells, or HEp-2 cells (MOI, ∼100:1) in 24-well tissue culture plates. Approximately 105 MDMs were seeded into individual wells in RPMI with 10% autologous serum and allowed to adhere overnight. Wells were washed, and cells were resuspended in RPMI with 2.5% autologous serum. Bacteria grown to mid-log phase in MMH broth were quantified by absorbance at 600 nm, and quantitation was confirmed by plate counting. To optimize phagocytosis, bacteria were opsonized by incubation in 50% fresh autologous serum for 30 min at 37°C as we described previously (40, 57). The appropriate numbers of bacteria were added to each well, and infection was synchronized by centrifugation at 600 × g at 12°C for 4 min (57, 59). Initial infection efficiency was quantified after 1 h coincubation at 37oC. MDM monolayers were washed extensively with phosphate-buffered saline to remove uningested bacteria and then processed immediately or incubated for another 23 h at 37°C in fresh medium. Host cell lysis was achieved by the addition of 1% saponin to each well, and serial dilutions were plated onto MMH agar to enumerate live organisms. Similarly, 2 × 105 HEp-2 cells or A549 cells were seeded into individual wells in minimal essential medium with 10% fetal bovine serum and were allowed to adhere overnight. Bacteria were added and infection was synchronized as described above. After 4 h incubation at 37°C, gentamicin (10 μg/ml) was added for 1 h to eliminate extracellular bacteria. Host cells were lysed, and the bacteria were enumerated as described above to quantify bacterial uptake or were replenished with gentamicin-free growth medium and incubated for an additional 19 h before lysis and enumeration to quantify intracellular growth.
Confocal analysis of F. tularensis phagosomes.
Our established methods were used to assess phagosome composition in macrophages (40, 57, 59). In brief, MDMs attached to chamber slides (Lab-Tek) were infected with opsonized F. tularensis at an MOI of 20:1. Phagocytosis was synchronized as described above, and after 1 h at 37°C, monolayers were washed extensively to remove uningested bacteria. After a total of 1 to 24 h at 37°C, MDM were fixed in 10% formalin, permeabilized with cold methanol-acetone, blocked, and then double-stained to detect bacteria and lamp-1 or cathepsin D. Bacteria were detected using rabbit anti-F. tularensis antiserum (BD Biosciences) or mouse anti-F. tularensis lipopolysaccharide T-14 (Novus Biologicals). Mouse anti-human lamp-1 hybridoma supernatants (clone H4A3) were from the Developmental Studies Hybridoma Bank of the University of Iowa. Rabbit anti-cathepsin D polyclonal antibodies were from Upstate Biotechnology, Inc. Fluorescein isothiocyanate- and rhodamine-conjugated F(ab′)2 secondary antibodies were from Jackson ImmunoResearch Laboratories. Samples were viewed using an LSM 510 confocal microscope (Carl Zeiss, Inc., Thornwood, NY). For each experiment, phagosomes in 50 to 100 infected cells were scored in duplicate or triplicate samples for each marker and time point.
Neutrophil infection and measurement of respiratory burst.
Bacteria were grown on MMH agar for 48 h at 37°C in 5% CO2 and harvested into HBSS containing divalent cations. Washed bacteria were opsonized with 50% fresh autologous serum for 30 min at 37°C as described above, washed again with HBSS lacking divalent cations, and quantified by measurement of absorbance at 600 nm. Generation of reactive oxygen species (ROS) by neutrophils was assessed using luminol-enhanced chemiluminescence (CL) assays as described previously (2, 18). Briefly, neutrophils were incubated at 5 × 106/ml in RPMI 1640 (without phenol red) supplemented with 250 μM luminol and 4% human serum albumin for 10 min at room temperature. Two-hundred-microliter aliquots were dispensed in triplicate into 96-well microtiter dishes (white/opaque Perkin Elmer OptiPlate 96) and infected at 37°C with the indicated strains of bacteria at an MOI of 50:1 unless otherwise specified. CL was recorded at 30-s intervals for 1 h using a BMG Laboratories Novostar luminometer (BMG LabTech Inc., Durham, NC).
RESULTS
Iron-dependent, Fur-independent regulation of the iglABCD operon.
To examine the role of the iron-dependent regulator Fur in expression of the iglABCD operon, we grew strains containing either an iglB-lacZ or fslC-lacZ chromosomal reporter, which were identified using our transposon delivery system (11), in CDM containing high (28 μM) or low (350 nM) FeSO4. β-Galactosidase assays were conducted on strains grown to mid- or late-log growth phase before examining reporter expression. When grown under high-iron conditions, the expression of each reporter was unchanged from the mid-log to the late-log phase of growth. When grown under iron-limiting conditions, there was an increase in the expression of each reporter, a phenomenon that was exacerbated as the cultures progressed from mid-log to late-log growth phase. This trend is presumably observed because iron was being actively depleted in late-log phase by bacterial growth and utilization (Fig. 1). Because this growth phase-dependent increase in expression was observed in the low- but not high-iron-containing medium, we believe the induction is likely due to decreased iron availability and not the result of other growth phase effects. To assess the role of Fur in the regulation of these genes, we overexpressed Fur in trans in each strain and repeated the assay. The data in Fig. 1 clearly show repression of the fslC-lacZ reporter regardless of iron availability and growth phase, whereas the expression of iglB was unaffected.
FIG. 1.
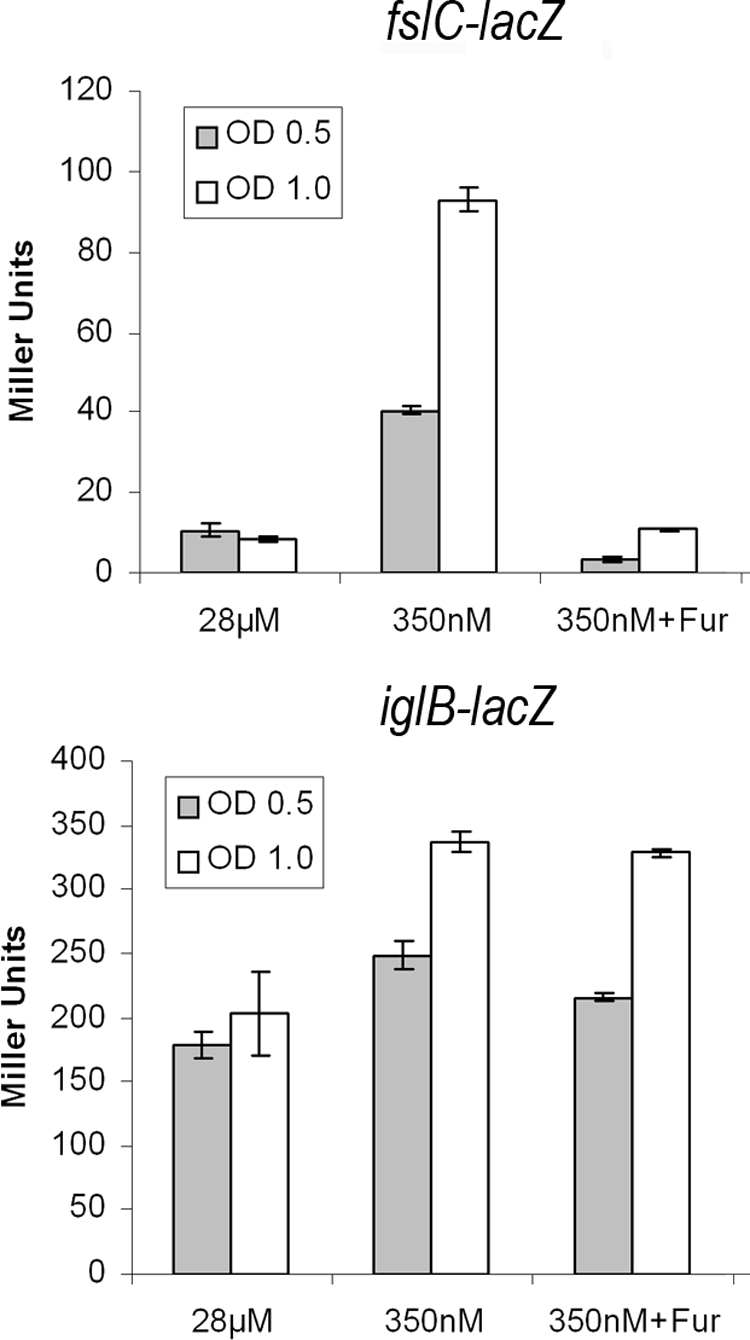
Examination of the effect of iron concentration and Fur overexpression on fslC and iglB chromosomal reporters. Duplicate cultures of each reporter strain were grown in CDM with a high (28 μM) or low (350 nM) concentration of FeSO4. β-Galactosidase assays were conducted at the mid- or late-log phases of growth. The fslC reporter is induced ∼10-fold in late-log phase at low Fe concentrations (P < 0.001). This induction was repressed by the overexpression of Fur in trans, which returned fslC-lacZ expression to the same level as that seen when grown in iron-replete broth (P = 0.103). The iglB reporter is induced ∼2-fold in late-log-phase growth at low Fe concentrations (P < 0.001). Expression of Fur in trans had no significant effect on iglB reporter expression (P = 0.112). Data are the averages of at least two independent experiments performed in triplicate ± 1 standard deviation. OD, optical density.
Identification of an iron-responsive DNA segment upstream of iglABCD.
Since the iglB reporter appeared to be responding to iron in a Fur-independent manner, we wanted to identify the region of DNA that was capable of eliciting the iron-dependent regulation. To this end, we amplified DNA upstream of iglA, iglC (containing a weak putative Fur-binding site) (62), and fslA (containing a near-consensus Fur-binding site) and cloned each fragment into a Francisella-E. coli shuttle vector in front of a promoterless lacZ reporter gene (Fig. 2B). Wild-type F. tularensis or an isogenic strain containing an insertion mutation in fur was transformed with each of three different reporter plasmids or a promoterless lacZ control and grown in high- or low-iron-containing medium. The lacZ control reporter exhibited the same low-level expression irrespective of iron availability in both genetic backgrounds. The reporter of fslA activity demonstrated an ∼33-fold induction in response to growth in iron-limiting medium and an ∼73-fold induction in the Fur deletion mutant (Fig. 2A). The iglA reporter also responded positively to growth in iron-limiting medium, albeit to a lesser extent (∼2-fold) than the fslA reporter. When assayed in the fur mutant background, the fslA reporter was further induced and lost its responsiveness to medium iron concentration. Expression of the iglA reporter in the Fur mutant background was increased approximately threefold over the level measured under iron-replete growth conditions (Fig. 2A). The reporter of iglC activity, containing a weak putative consensus Fur-binding site, was unaffected by either iron availability or genetic background.
FIG. 2.

Determination of iron-responsive DNA sequence. (A) Wild-type (WT) or fur mutant strains of F. novicida were transformed with plasmid-borne reporters of fslA, iglA, or iglC and were grown under iron-replete (+) or iron-depleted (−) conditions to measure transcriptional activity. Miller assays were carried out on at least two replicate cultures and were assayed in triplicate. The promoterless lacZ control reporter is unaffected by iron or genetic background. The fslA reporter is upregulated by growth in a low-iron medium (P < 0.001) and is further induced in the fur mutant background (P < 0.001). Expression of the iglA reporter is modestly induced both by growth in an iron-depleted medium (P < 0.001) or when in the fur mutant strain (P < 0.001). Activity of the iglC reporter remains unchanged under tested iron availabilities and in either genetic background. Data are the averages of at least two independent experiments performed in triplicate ± 1 standard deviation. (B) Schematic representation of DNA amplified and assayed for reporter activity.
Identification of iglABCD operon regulators.
Currently, four gene products that appear to control expression of the iglABCD operon have been described—MglA, SspA, FevR, and PmrA (14, 35, 43)—none of which has a known role in iron-mediated regulation. To identify additional genes encoding proteins affecting expression of this operon, we mutagenized an F. tularensis strain containing a chromosomal lacZ reporter of iglB activity (11). Using a transposon delivery system recently developed in our laboratory (11), we generated and screened a library of 2,500 mutants for changes in iglB expression. Three unique mutants were isolated, each of which exhibited significantly reduced β-galactosidase activity of the iglB-lacZ reporter when streaked onto MMH agar (data not shown).
Chromosomal transposon insertion sites were cloned and sequenced to identify the location of each insertion. One of the transposon insertions resided within the lacZ coding sequence, causing the complete loss of β-galactosidase activity. Since F. tularensis LVS has two copies of the FPI, we performed a Western blot assay for IglC to examine the effect of these transposon insertions on expression of the iglABCD operon not containing the lacZ reporter. Results from the Western blot assay indicate that, indeed, each transposon mutation affects both copies of the iglABCD operon (Fig. 3A). As expected, the transposon insertion in the lacZ reporter itself caused no obvious change in IglC levels, and this mutant was not pursued further (Fig. 3A). A second transposon insertion was mapped to nucleotide 163 of FTL_0347, an ∼0.6-kb gene that encodes a hypothetical protein with predicted transmembrane domains. This mutant displayed a seven- to eightfold reduction in iglB-lacZ expression and a concomitant reduction in cell-associated IglC accumulation observed by Western blotting (Fig. 3A and C). Unfortunately, additional work indicated that the transposon insertion was not responsible for the phenotype of this mutant, and it was not pursued further. The final transposon insertion was mapped to nucleotide 211 of FTL_1542 (Fig. 3D), a 2.1-kb gene present in all F. tularensis subspecies sequenced to date, and was predicted to encode a hypothetical protein that may contain a conserved AMP binding domain. This gene was also recently identified as a regulator of pepO, a virulence-associated gene in F. novicida (9). A transposon insertion in FTL_1542 resulted in a fivefold reduction in iglB-lacZ expression, which was similar to the reduction in cell-associated IglC accumulation observed by Western blotting (Fig. 3A and C). These findings suggest that an insertion mutation in FTL_1542 affects expression of the iglABCD operons on both pathogenicity islands.
FIG. 3.

Identification of genes affecting iglB transcription. (A) Western blot of cell lysates isolated from each indicated strain, using anti-IglC antibody. (B) Coomassie blue staining of a sodium dodecyl sulfate-polyacrylamide gel run on a cell lysate from each indicated strain as the loading control. (C) β-Galactosidase assay results and ORF number of each identified putative iglB regulator mutant. Miller assay results demonstrate an ∼5 to 7-fold reduction in iglB transcription in FTL_1542 and FTL_0347 mutant strains (P < 0.001). The IglC Western blot confirms that the mutations affect the expression of iglC as well as that of iglB and suggests that the mutations affect both chromosomal copies of the FPI. (D) Schematic representation of the transposon insertion (black arrowhead) at nucleotide 211 and the site-directed insertion (white arrowhead) at nucleotide 1458 of FTL_1542 and the surrounding genes. FTL_1542 is upstream of genes encoding MraW, a hypothetical protein, and FtsI. The intragenic regions separating these genes are 6, −3, and −7 nucleotides long, respectively. A gene encoding a 30S ribosomal protein is 106 nucleotides downstream of ftsI.
In vitro growth and complementation and creation of site-directed mutants.
The FTL_1542 (migR) mutant was transformed with a complementation plasmid (pBB114) containing its full-length gene, whose expression was driven by the F. tularensis groES promoter. The complementing clone was initially identified by the restoration of blue color when exposed to X-Gal. β-Galactosidase assays were conducted to quantify the extent to which complementation restored iglB transcription (data not shown). Consistent with the X-Gal plate screen, expression of the iglB-lacZ reporter in the FTL_1542 mutant was restored by a functional copy of the FTL_1542 gene provided in trans; however, the level of iglB-lacZ activity reached only 50% of that of the parent strain (data not shown). The maximal doubling time of the FTL_1542 mutant, under our growth conditions, was ∼72% of that of the parent iglB-lacZ strain. The effect of this mutation on maximal in vitro growth rate is similar to that of a fevR mutant (data not shown) and those reported for mglA and sspA mutants (14). Complementation of the FTL_1542 mutant restored the growth rate to 93% of that of the parent (data not shown). The apparent lack of complete complementation in the FTL_1542 strain in each of these assays may be the result of inappropriate protein stoichiometry or some other unforeseen effect of the overexpression of this protein from the complementation plasmid.
To further demonstrate that the transposon insertion in FTL_1542 was responsible for the reduced expression of iglB and iglC, we utilized a modified intron-directed mutagenesis system to create a site-directed mutant of F. tularensis (52). Creation of this mutant allowed us to assess the mutant phenotype in the absence of the compounding iglB mutation present in the original mutant strain. Wild-type and mutant strains created using the intron-directed mutagenesis system were transformed with pBB125, which carries a plasmid-borne iglA-lacZ reporter, and β-galactosidase assays were conducted. The iglA-lacZ reporter exhibited a fivefold reduction in activity in the site-directed FTL_1542 (migR) mutant compared to that in the wild-type strain. This finding is in agreement with our chromosomal reporter data for a mutation in this gene.
Effect of migR mutation on the expression of known regulators of virulence genes.
Gene products encoded by mglA, sspA, fevR, and pmrA have all been shown to affect the expression of genes within the FPI, including the iglABCD operon (6, 9, 10, 35, 43). The precise mechanism of action of these regulators is unknown, but the available data suggest that additional gene products may play a significant role in activating the expression of genes within these overlapping regulons (14, 43). Therefore, we examined the effect of the migR mutation on the transcript levels of each of these four known virulence gene regulators, using real-time quantitative RT-PCR (qRT-PCR). We also examined the expression of iglC by using qRT-PCR to confirm our results from lacZ reporter and Western blot assays. The qRT-PCR analysis confirmed the reduction in expression of iglC, which is consistent with our previous observations shown in Fig. 3B and C. Specifically, the data indicate that the FTL_1542 mutant strain contains 8.5-fold less iglC transcripts than the wild-type strain (P < 0.001; two-tailed Student's t test) (Fig. 4). The relative numbers of transcripts of mglA and pmrA were not significantly different in the mutant versus the wild-type strain (P > 0.05; two-tailed Student's t test). There was a slight but significant (P = 0.034; two-tailed Student's t test) reductions in the number of sspA transcripts in the mutant, which contained 0.74-fold as many sspA transcripts as the wild-type strain. Most interesting was the 15-fold reduction in the number of fevR transcripts in the migR mutant strain (P < 0.001; two-tailed Student's t test) (Fig. 4). Since FevR is essential for expression of the iglABCD operon (9), it is likely that the reduction in igl expression observed in the FTL_1542 (migR) mutant is due to a reduction in fevR expression in this strain.
FIG. 4.

Effect of migR mutation on the expression of virulence regulators. Real-time qRT-PCR was conducted on an mRNA template obtained from wild-type (WT) LVS (closed diamonds) or the site-directed migR mutant (open squares). The transcript of each gene was normalized to the transcript of tul4, and the wild-type transcript for each gene was set to 1.0. The numbers of mglA and pmrA transcripts were unaffected by the mutation in migR (P > 0.1). The migR mutation resulted in a modest 1.4-fold decrease in the number of sspA transcripts (P = 0.034). The iglC and fevR transcript levels were reduced in the migR mutant strain by 8.5- and 15-fold, respectively (P < 0.001).
Intracellular survival and growth in HEp-2 cells, A549 cells, and MDMs.
As mutations in migR reduced expression of the iglABCD operon, we examined the effect of these mutations on intracellular survival and growth in both epithelial cells and macrophages. Additionally, since the regulatory effect of the migR mutation on iglABCD seemed to be through fevR, we created a fevR mutant by using the intron-directed mutagenesis system as a means for comparison. First, MDMs were infected with opsonized wild-type LVS, the original migR transposon mutant, or the site-directed migR and fevR mutant strains and their trans-complemented counterparts. Infection efficiencies were similar for all six strains after 1 h of coincubation with MDMs. To quantify intracellular replication, host cells were lysed 24 h postinfection, and viable bacteria were enumerated. Wild-type LVS multiplied approximately 50- to 70-fold over the 24-h time course of the experiment, similar to the rate, reported by us and others, in primary macrophages or macrophage cell lines (29, 39, 47, 58). The migR and fevR mutants were greatly impaired for growth in MDMs, reaching a maximum of fivefold or twofold replication over the course of the experiment (P < 0.001; two-tailed Student's t test), respectively (Fig. 5A). This was an expected result since igl expression is greatly reduced in the migR strain (Fig. 3B). There was no significant difference between the original FTL_1542 Tn5 insertion mutant and the site-directed migR mutant (P = 0.951). Complementation of the migR mutant significantly restored the intracellular growth (P < 0.001; two-tailed Student's t test), albeit to a level lower than that of the parent strain (Fig. 5A). These data are consistent with the incomplete complementation observed in iglB-lacZ expression and in vitro growth experiments. Comparable results were also obtained using the original transposon insertion mutant strain and its isogenic trans-complemented strain (data not shown).
FIG. 5.
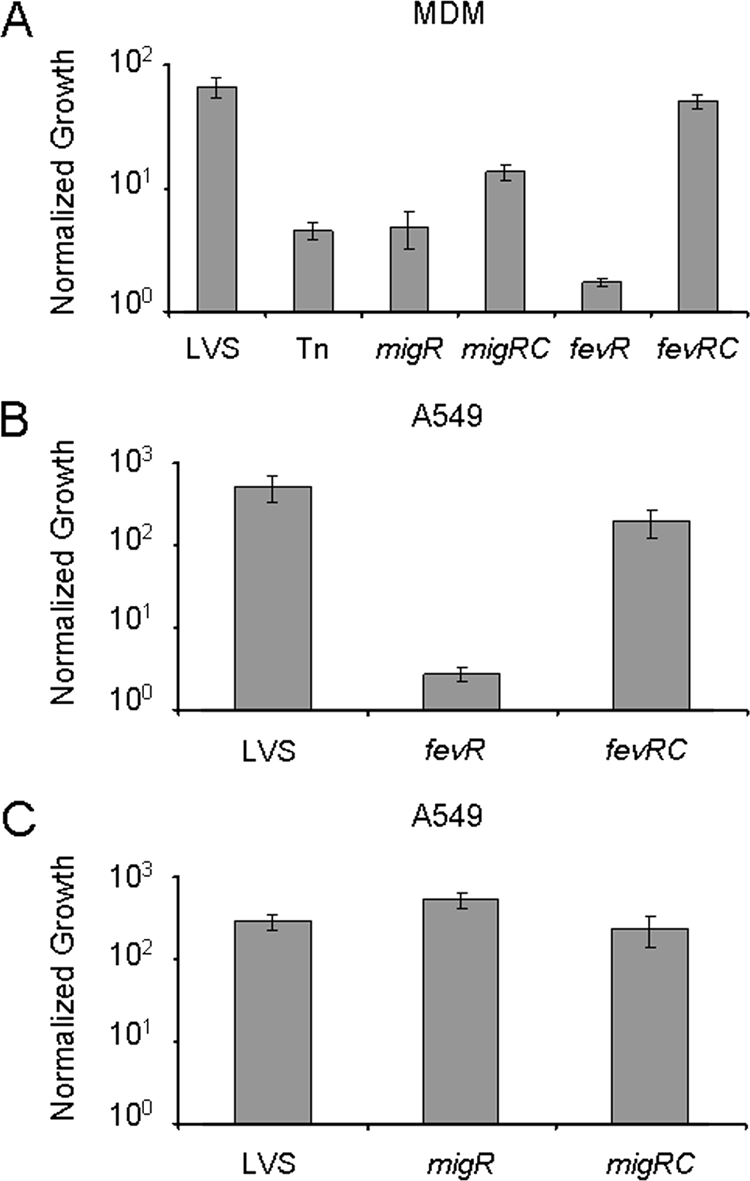
Intracellular growth of migR and fevR mutant strains. (A) Wild-type LVS, the original Tn5 migR mutant (Tn), site-directed migR (migR) and fevR (fevR) mutants, and their corresponding complemented migRc and fevRc strains were used to infect MDM cells (MOI, 20:1) in vitro, and intracellular growth was quantified as described in Materials and Methods. There was no significant difference in growth between the original Tn5 migR mutant and the site-directed migR mutant (P = 0.951). Complementation of the migR strain with the full-length FTL_1542 gene in trans restored growth in MDMs (P < 0.001). (B and C) Wild-type LVS, site-directed migR and fevR mutants, and their corresponding complemented strains migRc and fevRc were used to infect A549 (MOI, 100:1) cells in vitro. Uptake of each strain was quantified after 1 h for MDMs or 4 h for A549 cells. Intracellular growth for each cell type was determined 24 h after infection. Data were normalized by dividing the results at the 24 h time point by those at the 1 h or 4 h time point. Both migR and fevR mutant strains were impaired for growth in MDMs, while only the fevR mutant was defective for growth defect in A549 cells. Representative data from one of three experiments performed in triplicate are presented.
To assess the effect of these mutations on bacterial growth in epithelial cell lines, a similar set of experiments was carried out using human epithelial (HEp-2) and human airway epithelial (A549) tissue culture cells. The uptakes of the five strains by HEp-2 and A549 cells were comparable after a 4-h incubation period. Thereafter, wild-type LVS underwent rapid replication, multiplying about 500-fold in A549 cells (Fig. 5B and C) and nearly 1,000-fold in HEp-2 cells (data not shown) by 24 h postinfection. Interestingly, while the fevR mutant achieved only minimal growth in these cell lines (Fig. 5B), the kinetics of growth for the migR mutant were indistinguishable from those of the parent strain over the course of the experiment (Fig. 5C). This was an unexpected result, given the dramatic reduction in igl expression in this strain and the reduced ability of the mutant to grow in primary human macrophages. Thus, we report the identification of a mutant defective for growth in MDMs that replicates normally in HEp-2 and A549 epithelial cells.
Intramacrophage trafficking of migR and fevR mutants.
After uptake by MDMs, F. tularensis prevents phagosome-lysosome fusion and resides in a compartment that accumulates late endosome membrane glycoproteins prior to phagosome egress and replication in the cytosol (16, 17, 54, 55). In contrast, mutant bacteria that lack functional mglA or an intact FPI are defective for phagosome escape and reside in mature phagolysosomes (8, 54, 55). Since the data in Fig. 5A indicated that intracellular growth of the migR and fevR mutants are impaired, we used confocal microscopy to assess the intracellular fate of these mutants and to begin to determine whether either strain exhibited an aberrant trafficking phenotype. Infected MDMs were analyzed 1 h or 18 to 22 h after initiation of infection, and in each case, cells containing wild-type LVS, mutant organisms, or their trans-complemented counterparts were directly compared. Representative images are shown in Fig. 6A to C, and pooled data from three independent experiments are summarized in Fig. 6D and E.
FIG. 6.


Composition of migR and fevR mutant phagosomes in MDMs. (A to C) Representative confocal sections of MDMs infected for 1 h or 19 to 22 h (overnight) at 37°C with LVS (A), the migR mutant or its trans-complemented migRc strain (B), or the fevR mutant and its trans-complemented fevRc strain (C). In each case, samples were stained to detect bacteria and lamp-1 (lamp) or cathepsin D (catD), as indicated. Arrows indicate positive phagosomes. (D to E) Percentage of bacteria inside MDMs that were infected for 1 h (D) or overnight (E) that were inside lamp-1- or cathepsin D-positive phagosomes. Data are the averages ± the standard errors of the means of the results from three independent experiments performed in triplicate.
Our findings indicate that early in infection (1 h after uptake), the vast majority of the migR and fevR mutants, as well as the trans-complemented migRc and fevRc strains, resided in phagosomes that accumulated the late endosome membrane glycoprotein lamp-1 but not the lysosomal marker cathepsin D and, in this manner, resembled wild-type LVS (Fig. 6A to D). No significant differences in initial infection efficiency were detected, and ∼75% of the macrophages in each monolayer contained intracellular bacteria (data not illustrated). However, as infection progressed, it became apparent that the migR and fevR mutants had distinct fates in MDMs that differed both from one another and from wild-type LVS. Thus, by 5 h after uptake, ∼50% of wild-type LVS had breached the phagosome membrane (data not shown); after 18 to 22 h at 37°C, the robust growth of wild-type bacteria in the MDM cytosol was readily apparent (Fig. 6A), and these organisms did not colocalize with lamp-1 or cathepsin D (Fig. 6A and E). Analysis of MDMs infected for 19 to 22 h with the migR or fevR mutants demonstrated that the vast majority of both strains remained trapped inside phagosomes and did not reach the cytosol (Fig. 6E). Compartments containing the migR mutant resembled mature phagolysosomes as indicated by their accumulation of lamp-1 and cathepsin D (Fig. 6B and E). Similar data were obtained for the original Tn5 insertion mutation in FTL_1542 (data not shown). In contrast, the fevR mutant appeared trapped in a more immature compartment that lacked cathepsin D despite a sustained accumulation of lamp-1 (Fig. 6C and E). Robust replication of both complemented strains (migRc and fevRc strains) in the MDM cytosol 18 to 22 h after uptake (Fig. 6B and C) confirms a role for both migR and fevR in the manipulation of macrophage membrane trafficking by F. tularensis strain LVS.
Mutations in migR and fevR affect the ability of F. tularensis to block NADPH oxidase activity in neutrophils.
An important aspect of F. tularensis virulence is its ability to prevent neutrophil activation (1, 40). To assess whether the migR or fevR mutants were compromised in their ability to block NADPH oxidase activity, we performed luminol-enhanced CL assays to measure the production of oxidants during the infection of PMN. Concordant with our published data (3, 40, 58), infection of PMN with wild-type LVS did not trigger a respiratory burst (Fig. 7). In marked contrast, both the strain carrying the Tn5 insertion in FTL_1542 and the site-directed migR mutant stimulated similar levels of NADPH oxidase activation in PMN, as judged by the luminol CL assay. Not surprisingly, the fevR mutant was also unable to prevent oxidant production (Fig. 7). For both migR and fevR mutant strains, PMN NADPH oxidase inhibition was restored by complementation with the wild-type gene in trans. Collectively, these data demonstrate that migR and fevR play a role in the disruption of neutrophil function, likely via effects on the expression of genes in overlapping regulons.
FIG. 7.

migR and fevR mutants activate human neutrophils. Neutrophils were left untreated (UN) or were infected with LVS, the original Tn5 FTL_1542 insertion mutant (Tn), the migR mutant (migR), its trans-complemented strain migRc, the fevR mutant (fevR), or its trans-complemented strain fevRc at 37°C, and ROS production was measured at 30-s intervals for 1 h using the luminol assay. Data indicate luminol CL in counts per second (cps) and are the averages ± the standard errors of the means (gray bars) of triplicate samples from the results of one representative experiment.
DISCUSSION
The facultative intracellular pathogen F. tularensis is capable of subverting the early innate immune response and replicating within macrophages and epithelial cells to cause significant morbidity and mortality in humans. Previous studies have demonstrated that genes within the iglABCD operon are induced in response to growth under iron-limiting conditions (11, 21, 36, 42, 62); however, the underlying mechanism remained obscure. One research group has identified sequences upstream of the iglC gene with similarity to the consensus Fur binding site (21), although a Fur binding site in the middle of an operon would be an unusual regulatory arrangement. We have examined the effect of iron concentration and overexpression of the Fur repressor in strains containing chromosomal reporters of either iglB or fslC transcription. The fslC gene is part of an operon that is regulated by iron and carries a strong consensus Fur binding site upstream of fslA, the first gene of the operon (21, 62). Chromosomal reporters of iglB and fslC were induced when grown under iron-limiting conditions, although induction of the iglB reporter was mild. As expected, the chromosomal reporter of fslC activity was repressed by the overexpression of Fur in both iron-replete and iron-depleted growth media. In contrast, overexpression of Fur had no significant effect on the expression of iglB regardless of the iron concentration in the media.
To identify the region of DNA containing iron-responsive regulatory sequences, we cloned DNA fragments upstream of iglA, iglC, and fslA into a Francisella-E. coli shuttle plasmid containing a lacZ reporter gene. Wild-type or fur mutant Francisella strains were transformed with each reporter plasmid. The fslA DNA fragment contains a well-conserved Fur box DNA sequence. As expected, this reporter plasmid produced much more lacZ activity (∼73-fold) in the fur mutant than in the wild-type background, regardless of the iron concentration in the growth medium. Furthermore, reporter activity is increased ∼33-fold in the wild-type strain when grown in iron-limiting medium. These data provide compelling support for the notion that the fslA operon is regulated by Fur in response to iron in a conventional manner. A reporter containing DNA upstream of the iglA gene, likely to include promoter and regulatory sequences, underwent a mild ∼2- to 3-fold induction in the wild-type background, but was also induced slightly in the fur mutant genetic background when grown in iron-limiting medium. These data are consistent with the results from our fur overexpression studies and suggest, at best, a modest role for Fur in the regulation of the iglABCD operon. The lacZ reporter plasmid containing DNA sequences upstream of the iglC gene with a weak putative Fur box showed no responsiveness to changes in iron concentration, or to the presence or absence of Fur. We conclude that Fur binding sequences are present upstream of iglA, but not iglC. Together, these data suggest a minor role for iron and the Fur protein in the regulation of iglABCD and that the regulatory effects are exerted through the DNA sequence upstream of the operon.
Since our initial data demonstrated that Fur did not have a dominant regulatory role on the igl operon, we mutagenized an F. tularensis LVS strain carrying a chromosomal reporter of iglB transcription, in an attempt to identify new regulators of this operon. A mutant library screen identified one mutant of interest with a transposon insertion in the F. tularensis LVS gene migR (FTL_1542) that resulted in a reduced expression of genes in the igl operon as indicated by use of transcriptional reporters as well as Western blotting. migR was also identified in an F. novicida screen for regulators of pepO (9), and it is noteworthy that the pepO gene is thought to be nonfunctional in F. tularensis subspecies tularensis and holarctica (31). Based on limited homology, migR was annotated as caiC in the Schu S4 chromosome in GenBank, a designation that has since been removed. Because of its regulatory effects on fevR and iglABCD, and its intracellular growth phenotype in macrophages, we have given the FTL_1542 open reading frame (ORF) the designation of migR (macrophage intracellular growth regulator).
Transposon and site-directed mutagenesis of migR resulted in decreased expression of igl genes and a reduced ability of mutant bacteria to grow in primary MDMs. Gene orientation and the short intragenic regions between predicted genes suggest that migR could be the first gene in an operon that includes FTL_1541 (mraW, S-adenosyl-methyltransferase), FTL_1539 (hypothetical protein), and FTL_1538 (ftsI, penicillin binding protein) (Fig. 3D). This gene arrangement is shared among F. tularensis subspecies. MigR is annotated as a hypothetical protein that shares some similarity to acyl coenzyme A ligases and contains a conserved AMP-binding domain. Proteins sharing this functional domain are commonly involved in fatty acid modifications, such as the activation of fatty acids by the addition of coenzyme A as they enter the cytoplasm to both sequester the fatty acid as well as initiate metabolism of the energy-rich substrate (33). Although it is possible that migR is operonic with one or more of the downstream genes, the ability to complement the various phenotypes of this mutant with just the FTT_1542 ORF alone in several assays is strong genetic evidence that the mutation is not polar or that downstream genes are not strongly associated with the regulatory function of migR. Furthermore, the regulatory effect exerted by migR is specific and not due to a general downregulation of gene expression in F. tularensis, since other genes examined by qRT-PCR are unaffected by this mutation. While it is not immediately clear how this gene product is exerting a regulatory effect on fevR and, thus, on iglABCD, proteins sharing domain homology regulate gene expression in E. coli through the modification of fatty acids, enabling them to interact with DNA binding regulatory proteins (7, 22).
To determine if the regulatory effect of the migR mutant was exerted through any of the previously identified regulators of the iglABCD operon, we used real-time qRT-PCR to compare the levels of mglA, sspA, pmrA, or fevR transcription in the migR mutant with the parent strain. As expected, based on the results of our reporter and protein blot assays, we found that the number of iglC transcripts was reduced in the migR mutant. Interestingly, the level of fevR transcripts was significantly reduced in this strain, and to a much lesser extent, the level of sspA transcripts was also reduced. The reduction in fevR was dramatic in the mutant strain (15-fold; P < 0.001), while the reduction in the level of sspA transcripts was 1.4-fold (P = 0.043) and may not be biologically significant. This led us to conclude that the regulatory effect of migR on the igl operon is indirect, likely via the downregulation of FevR (Fig. 8). Previous studies have also shown that fevR expression requires MglA, SspA, and PmrA (9, 43). Expression of fevR in an mglA mutant is reduced 5- to 10-fold (9, 10), while expression of fevR in a pmrA mutant is reduced only 2.4-fold compared to that in the wild-type strain (43). These observations suggest that FevR is a central regulator of FPI gene expression with several other gene products, in turn, modulating the expression of fevR. It is attractive to speculate that different stimuli encountered by F. tularensis alter fevR expression through different signaling cascades, although no specific data are available to support this hypothesis at this time.
FIG. 8.
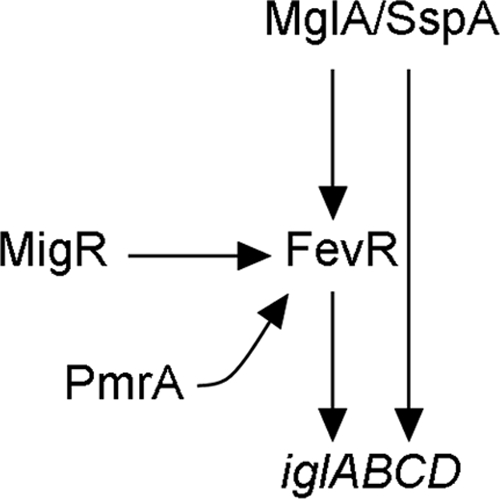
Model for the role of MigR in the regulation of iglABCD. MglA and SspA form a heterodimer that is required for fevR and iglABCD expression. MigR does not affect the expression of mglA, sspA, or pmrA. However, fevR expression is reduced 15-fold in a migR mutant. This reduction in fevR expression results in an eightfold reduction in the expression of the iglABCD operon.
Wild-type F. tularensis is capable of rapid, robust growth in both epithelial cells and macrophages (17, 37). Mutations in genes in the iglABCD operon nearly ablate replication in macrophages in vitro and markedly attenuate virulence in a mouse model of infection (30, 65). Given the finding that the migR mutation described here resulted in the net reduction of igl expression, we expected that the mutant strain would be impaired for intracellular replication within host cells. Upon infection of primary human macrophages, we found the migR mutant to be capable of only modest replication beyond the initial infection numbers. This was in sharp contrast to wild-type LVS, which multiplied up to 70-fold over the same 24-h time period. As indicated by measurement of CFU, the defect in intra-MDM growth was partially ameliorated by complementation with the full-length wild-type migR (FTL_1542) (Fig. 5A); confocal analysis revealed that 18 h after uptake, MDMs infected with wild-type LVS contained ∼50% more bacteria than did cells infected with the migRc strain (Fig. 6A and B) (L. Allen, unpublished data). Nevertheless, by 30 to 48 h postinfection, both the migRc strain and wild-type LVS led to destruction of the MDM monolayer (Allen, unpublished). Thus, although the migRc strain grew more slowly than did wild-type bacteria in MDM, our data strongly suggest that the mutation of migR accounts in large part for the reduced igl gene expression and the intracellular growth defects we describe.
Similar infection and intracellular growth experiments were also carried out with HEp-2 and A549 epithelial cell lines. These cell lines were more permissive for the intracellular replication of the wild type, which increased 500- to 1,000-fold over the 24-h experiment. Surprisingly, the migR mutant grew normally in both of these cell types, while the fevR mutant did not. These data indicate that migR is required for intracellular survival and replication in MDMs but not for growth in cultured epithelial cells. In contrast, FevR is required for growth in all cell types tested. FevR is not only a regulator of the iglABCD operon but also requires MglA and SspA for activity (9). Because of this regulatory mechanism, the fevR mutation may produce a more pronounced phenotype in macrophages than does the migR mutation, which only downregulates fevR. We know of no studies to date that examine the Francisella-containing compartment in epithelial cells or the effect of mutations in igl genes on the composition of this compartment. The contrasting phenotypes of fevR and migR mutants, along with those of igl mutants, will enable future studies to examine the role of the igl operon in the fate of F. tularensis in epithelial cells.
The distinct phenotypes of the mutant strains in epithelial cell lines could be due to at least one of the following mechanisms. First, reduced iglABCD expression in the migR mutant, as opposed to a complete loss of iglABCD expression in the fevR mutant, could account for the different intracellular growth phenotypes of the strains in MDMs and A549 cells. This explanation would require that a low level of iglABCD expression in the migR mutant would be sufficient to allow endosomal escape and replication in the less hostile environment of epithelial cells, while not being sufficient to avert the phagosomal maturation process of macrophages. Alternatively, the different growth phenotypes of migR and fevR in epithelial cells could be the result of MigR responding to different signals that are present in one cell type but not another.
Neutrophils provide an essential first line of defense against invading microbes, and a key component of their killing arsenal is the NADPH oxidase. This enzyme catalyzes the conversion of molecular oxygen into superoxide anions, which are then converted into other ROS, including H2O2 and HOCl (5). We have shown previously that F. tularensis strain LVS evades killing by neutrophils via its ability to inhibit NADPH oxidase assembly and activation at the phagosome membrane (3, 40). Virulence factors that prevent neutrophil activation during tularemia are not well defined. We now show that while wild-type bacteria prevent ROS production, phagocytosis of the migR or fevR mutant strains triggers a moderate respiratory burst that is abrogated by expression of a full-length copy of FTL_1542 (migR) or FTL_0449 (fevR) in trans. These data suggest a role for migR in the disruption of neutrophil NADPH oxidase activity. Indeed, we also identified migR by direct screening of an LVS Tn5 mutant library (11, 58) for mutants that no longer prevent neutrophil activation (Allen, unpublished). Thus, we favor a model in which migR regulates genes in addition to those in the iglABCD operon, perhaps including acpA, which encodes an acid phosphatase that can inhibit porcine NADPH oxidase activity in vitro (51).
The current model for lethal infection by F. tularensis involves the inhalation of as few as 1 to 10 viable bacteria, which are engulfed by lung alveolar macrophages (49, 66). F. tularensis rapidly replicates in this niche, and infected macrophages mediate dissemination to the liver and spleen (25). Accordingly, research efforts and infection models have focused on understanding the interactions between F. tularensis and MDMs, elicited murine peritoneal macrophages and murine bone marrow-derived macrophages, as well as human and murine macrophage-like cell lines. Since the endotoxin of F. tularensis is nonstimulatory, it is generally believed that pathogenesis is mediated by the ability of the bacteria to replicate intracellularly, the principal target of growth being macrophages (46, 63). In general, mutants defective for growth in macrophages are also attenuated for in vivo virulence. However, a few mutants that grow normally inside macrophages yet exhibit reduced virulence in mouse infection models have been reported (12, 24, 38, 65). In this study, we report the identification of mutations that differentially affect PMN activation and intramacrophage growth and are dispensable for replication in A549 and HEp-2 cells. We show for the first time that mutants lacking a functional migR (FTL_1542) reside in compartments with features of mature phagolysosomes in MDMs and, in this manner, resemble mutants that lack functional mglA or iglC (8, 54, 55). At the same time, the fact that this mutant grew normally in A549 and HEp-2 cells suggests that replication of F. tularensis in epithelial cells may represent an important, yet understudied, aspect of tularemia. In this same vein, we recently demonstrated that uracil auxotrophic mutants of LVS are killed by MDMs and neutrophils, yet replicate normally in macrophage- and epithelial-like cell lines (58). What accounts for these cell type-specific virulence defects is unknown and merits further investigation. In this regard it is also noteworthy that the fevR and migR mutants, while both defective in phagosome escape and intracellular growth, appeared to reside in distinct compartments in MDMs since phagosomes containing the migR mutant accumulated the lysosomal marker cathepsin D whereas phagosomes containing the fevR mutant did not. Although these data suggest that the migR and fevR mutants differentially affect macrophage membrane trafficking, additional studies are needed to ascertain whether fevR mutants exhibit specific defects in phagosome escape while retaining at least partial capacity to induce phagosome maturation arrest.
In summary, we conclude that reduced iron availability and Fur play modest roles in the regulation of the iglABCD operon. Perhaps this represents a “fine tuning” of the expression of these genes necessary in a specific survival niche. Importantly, we have identified a gene in F. tularensis LVS that regulates the expression of iglABCD, likely through the reduced expression of fevR, and may also influence expression of other genes throughout the F. tularensis chromosome. Mutations in migR result in the reduced expression of fevR and genes in the igl operon and also negatively affect growth in primary human macrophages but not that in epithelial cells. Moreover, the effects of the fevR and migR mutants on neutrophil NADPH oxidase activity and macrophage membrane trafficking are novel and provide avenues for further study.
Acknowledgments
We thank Martin Pavelka (University of Rochester Medical Center) for providing pMP529 containing the hyg gene and Karl Klose (University of Texas at San Antonio) for the anti-IglC antibody.
This work was supported, in part, by funds from the NIH to L.-A.H.A. (R01AI073835). B.W.B. was supported by the NIH Training in Mechanisms of Parasitism grant (5T32AI007511). S.R.L. was supported by the University of Iowa Presidential Fellowship. B.D.J. was supported by NIH funds from the Midwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases (2U54 AI057160-06) and by a University of Iowa Carver Collaborative Pilot Grant. This work was also supported by U.S. Public Health Service grant PO1 (AI044642).
Editor: A. J. Bäumler
Footnotes
Published ahead of print on 6 April 2009.
REFERENCES
- 1.Allen, L. A. 2006. Interview with Lee-Ann Allen regarding pivotal advance: Francisella tularensis LVS evades killing by human neutrophils via inhibition of the respiratory burst and phagosome escape. Interview by Helene F. Rosenberg. J. Leukoc. Biol. 801222-1223. [DOI] [PubMed] [Google Scholar]
- 2.Allen, L. A., B. R. Beecher, J. T. Lynch, O. V. Rohner, and L. M. Wittine. 2005. Helicobacter pylori disrupts NADPH oxidase targeting in human neutrophils to induce extracellular superoxide release. J. Immunol. 1743658-3667. [DOI] [PubMed] [Google Scholar]
- 3.Allen, L. A., and R. L. McCaffrey. 2007. To activate or not to activate: distinct strategies used by Helicobacter pylori and Francisella tularensis to modulate the NADPH oxidase and survive in human neutrophils. Immunol. Rev. 219103-117. [DOI] [PubMed] [Google Scholar]
- 4.Amann, E., B. Ochs, and K. J. Abel. 1988. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene 69301-315. [DOI] [PubMed] [Google Scholar]
- 5.Babior, B. M. 1999. NADPH oxidase: an update. Blood 931464-1476. [PubMed] [Google Scholar]
- 6.Baron, G. S., and F. E. Nano. 1998. MglA and MglB are required for the intramacrophage growth of Francisella novicida. Mol. Microbiol. 29247-259. [DOI] [PubMed] [Google Scholar]
- 7.Black, P. N., N. J. Faergeman, and C. C. DiRusso. 2000. Long-chain acyl-CoA-dependent regulation of gene expression in bacteria, yeast and mammals. J. Nutr. 130305S-309S. [DOI] [PubMed] [Google Scholar]
- 8.Bönquist, L., H. Lindgren, I. Golovliov, T. Guina, and A. Sjostedt. 2008. MglA and Igl proteins contribute to the modulation of Francisella tularensis live vaccine strain-containing phagosomes in murine macrophages. Infect. Immun. 763502-3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brotcke, A., and D. M. Monack. 2008. Identification of fevR, a novel regulator of virulence gene expression in Francisella novicida. Infect. Immun. 763473-3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brotcke, A., D. S. Weiss, C. C. Kim, P. Chain, S. Malfatti, E. Garcia, and D. M. Monack. 2006. Identification of MglA-regulated genes reveals novel virulence factors in Francisella tularensis. Infect. Immun. 746642-6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buchan, B. W., M. K. McLendon, and B. D. Jones. 2008. Identification of differentially regulated Francisella tularensis genes by use of a newly developed Tn5-based transposon delivery system. Appl. Environ. Microbiol. 742637-2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakraborty, S., M. Monfett, T. M. Maier, J. L. Benach, D. W. Frank, and D. G. Thanassi. 2008. Type IV pili in Francisella tularensis: roles of pilF and pilT in fiber assembly, host cell adherence, and virulence. Infect. Immun. 762852-2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chamberlain, R. E. 1965. Evaluation of live tularemia vaccine prepared in a chemically defined medium. Appl. Microbiol. 13232-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Charity, J. C., M. M. Costante-Hamm, E. L. Balon, D. H. Boyd, E. J. Rubin, and S. L. Dove. 2007. Twin RNA polymerase-associated proteins control virulence gene expression in Francisella tularensis. PLoS Pathog. 3e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cherwonogrodzky, J. W., M. H. Knodel, and M. R. Spence. 1994. Increased encapsulation and virulence of Francisella tularensis live vaccine strain (LVS) by subculturing on synthetic medium. Vaccine 12773-775. [DOI] [PubMed] [Google Scholar]
- 16.Clemens, D. L., and M. A. Horwitz. 2007. Uptake and intracellular fate of Francisella tularensis in human macrophages. Ann. N. Y. Acad. Sci. 1105160-186. [DOI] [PubMed] [Google Scholar]
- 17.Clemens, D. L., B. Y. Lee, and M. A. Horwitz. 2004. Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect. Immun. 723204-3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dahlgren, C., and A. Karlsson. 1999. Respiratory burst in human neutrophils. J. Immunol. Methods 2323-14. [DOI] [PubMed] [Google Scholar]
- 19.de Bruin, O. M., J. S. Ludu, and F. E. Nano. 2007. The Francisella pathogenicity island protein IglA localizes to the bacterial cytoplasm and is needed for intracellular growth. BMC Microbiol. 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeLeo, F. R., L. A. Allen, M. Apicella, and W. M. Nauseef. 1999. NADPH oxidase activation and assembly during phagocytosis. J. Immunol. 1636732-6740. [PubMed] [Google Scholar]
- 21.Deng, K., R. J. Blick, W. Liu, and E. J. Hansen. 2006. Identification of Francisella tularensis genes affected by iron limitation. Infect. Immun. 744224-4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DiRusso, C. C., and P. N. Black. 2004. Bacterial long chain fatty acid transport: gateway to a fatty acid-responsive signaling system. J. Biol. Chem. 27949563-49566. [DOI] [PubMed] [Google Scholar]
- 23.Ellis, J., P. C. Oyston, M. Green, and R. W. Titball. 2002. Tularemia. Clin. Microbiol. Rev. 15631-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forslund, A. L., K. Kuoppa, K. Svensson, E. Salomonsson, A. Johansson, M. Bystrom, P. C. Oyston, S. L. Michell, R. W. Titball, L. Noppa, E. Frithz-Lindsten, M. Forsman, and A. Forsberg. 2006. Direct repeat-mediated deletion of a type IV pilin gene results in major virulence attenuation of Francisella tularensis. Mol. Microbiol. 591818-1830. [DOI] [PubMed] [Google Scholar]
- 25.Fortier, A. H., M. V. Slayter, R. Ziemba, M. S. Meltzer, and C. A. Nacy. 1991. Live vaccine strain of Francisella tularensis: infection and immunity in mice. Infect. Immun. 592922-2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallagher, L. A., E. Ramage, M. A. Jacobs, R. Kaul, M. Brittnacher, and C. Manoil. 2007. A comprehensive transposon mutant library of Francisella novicida, a bioweapon surrogate. Proc. Natl. Acad. Sci. USA 1041009-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gil, H., J. L. Benach, and D. G. Thanassi. 2004. Presence of pili on the surface of Francisella tularensis. Infect. Immun. 723042-3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Golovliov, I., V. Baranov, Z. Krocova, H. Kovarova, and A. Sjostedt. 2003. An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect. Immun. 715940-5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Golovliov, I., M. Ericsson, G. Sandstrom, A. Tarnvik, and A. Sjostedt. 1997. Identification of proteins of Francisella tularensis induced during growth in macrophages and cloning of the gene encoding a prominently induced 23-kilodalton protein. Infect. Immun. 652183-2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Golovliov, I., A. Sjostedt, A. Mokrievich, and V. Pavlov. 2003. A method for allelic replacement in Francisella tularensis. FEMS Microbiol. Lett. 222273-280. [DOI] [PubMed] [Google Scholar]
- 31.Hager, A. J., D. L. Bolton, M. R. Pelletier, M. J. Brittnacher, L. A. Gallagher, R. Kaul, S. J. Skerrett, S. I. Miller, and T. Guina. 2006. Type IV pili-mediated secretion modulates Francisella virulence. Mol. Microbiol. 62227-237. [DOI] [PubMed] [Google Scholar]
- 32.Khan, A. S., S. Morse, and S. Lillibridge. 2000. Public-health preparedness for biological terrorism in the USA. Lancet 3561179-1182. [DOI] [PubMed] [Google Scholar]
- 33.Klein, K., R. Steinberg, B. Fiethen, and P. Overath. 1971. Fatty acid degradation in Escherichia coli. An inducible system for the uptake of fatty acids and further characterization of old mutants. Eur. J. Biochem. 19442-450. [DOI] [PubMed] [Google Scholar]
- 34.Lai, X. H., I. Golovliov, and A. Sjostedt. 2004. Expression of IglC is necessary for intracellular growth and induction of apoptosis in murine macrophages by Francisella tularensis. Microb. Pathog. 37225-230. [DOI] [PubMed] [Google Scholar]
- 35.Lauriano, C. M., J. R. Barker, S. S. Yoon, F. E. Nano, B. P. Arulanandam, D. J. Hassett, and K. E. Klose. 2004. MglA regulates transcription of virulence factors necessary for Francisella tularensis intraamoebae and intramacrophage survival. Proc. Natl. Acad. Sci. USA 1014246-4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lenco, J., M. Hubalek, P. Larsson, A. Fucikova, M. Brychta, A. Macela, and J. Stulik. 2007. Proteomics analysis of the Francisella tularensis LVS response to iron restriction: induction of the F. tularensis pathogenicity island proteins IglABC. FEMS Microbiol. Lett. 26911-21. [DOI] [PubMed] [Google Scholar]
- 37.Lindemann, S. R., M. K. McLendon, M. A. Apicella, and B. D. Jones. 2007. An in vitro model system used to study adherence and invasion of Francisella tularensis live vaccine strain in nonphagocytic cells. Infect. Immun. 753178-3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ludu, J. S., O. M. de Bruin, B. N. Duplantis, C. L. Schmerk, A. Y. Chou, K. L. Elkins, and F. E. Nano. 2008. The Francisella pathogenicity island protein PdpD is required for full virulence and associates with homologues of the type VI secretion system. J. Bacteriol. 1904584-4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maier, T. M., M. S. Casey, R. H. Becker, C. W. Dorsey, E. M. Glass, N. Maltsev, T. C. Zahrt, and D. W. Frank. 2007. Identification of Francisella tularensis Himar1-based transposon mutants defective for replication in macrophages. Infect. Immun. 755376-5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCaffrey, R. L., and L. A. Allen. 2006. Francisella tularensis LVS evades killing by human neutrophils via inhibition of the respiratory burst and phagosome escape. J. Leukoc. Biol. 801224-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 42.Milne, T. S., S. L. Michell, H. Diaper, P. Wikstrom, K. Svensson, P. C. Oyston, and R. W. Titball. 2007. A 55 kDa hypothetical membrane protein is an iron-regulated virulence factor of Francisella tularensis subsp. novicida U112. J. Med. Microbiol. 561268-1276. [DOI] [PubMed] [Google Scholar]
- 43.Mohapatra, N. P., S. Soni, B. L. Bell, R. Warren, R. K. Ernst, A. Muszynski, R. W. Carlson, and J. S. Gunn. 2007. Identification of an orphan response regulator required for the virulence of Francisella spp. and transcription of pathogenicity island genes. Infect. Immun. 753305-3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nano, F. E., N. Zhang, S. C. Cowley, K. E. Klose, K. K. Cheung, M. J. Roberts, J. S. Ludu, G. W. Letendre, A. I. Meierovics, G. Stephens, and K. L. Elkins. 2004. A Francisella tularensis pathogenicity island required for intramacrophage growth. J. Bacteriol. 1866430-6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nylund, A., K. F. Ottem, K. Watanabe, E. Karlsbakk, and B. Krossoy. 2006. Francisella sp. (family Francisellaceae) causing mortality in Norwegian cod (Gadus morhua) farming. Arch. Microbiol. 185383-392. [DOI] [PubMed] [Google Scholar]
- 46.Oyston, P. C., A. Sjostedt, and R. W. Titball. 2004. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat. Rev. Microbiol. 2967-978. [DOI] [PubMed] [Google Scholar]
- 47.Pechous, R., J. Celli, R. Penoske, S. F. Hayes, D. W. Frank, and T. C. Zahrt. 2006. Construction and characterization of an attenuated purine auxotroph in a Francisella tularensis live vaccine strain. Infect. Immun. 744452-4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petersen, J. M., and M. E. Schriefer. 2005. Tularemia: emergence/re-emergence. Vet. Res. 36455-467. [DOI] [PubMed] [Google Scholar]
- 49.Polsinelli, T., M. S. Meltzer, and A. H. Fortier. 1994. Nitric oxide-independent killing of Francisella tularensis by IFN-gamma-stimulated murine alveolar macrophages. J. Immunol. 1531238-1245. [PubMed] [Google Scholar]
- 50.Qin, A., and B. J. Mann. 2006. Identification of transposon insertion mutants of Francisella tularensis tularensis strain Schu S4 deficient in intracellular replication in the hepatic cell line HepG2. BMC Microbiol. 669-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reilly, T. J., G. S. Baron, F. E. Nano, and M. S. Kuhlenschmidt. 1996. Characterization and sequencing of a respiratory burst-inhibiting acid phosphatase from Francisella tularensis. J. Biol. Chem. 27110973-10983. [DOI] [PubMed] [Google Scholar]
- 52.Rodriguez, S. A., J. J. Yu, G. Davis, B. P. Arulanandam, and K. E. Klose. 2008. Targeted inactivation of Francisella tularensis genes by group II introns. Appl. Environ. Microbiol. 742619-2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santic, M., R. Asare, I. Skrobonja, S. Jones, and Y. Abu Kwaik. 2008. Acquisition of the vacuolar ATPase proton pump and phagosome acidification are essential for escape of Francisella tularensis into the macrophage cytosol. Infect. Immun. 762671-2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Santic, M., M. Molmeret, K. E. Klose, and Y. Abu Kwaik. 2006. Francisella tularensis travels a novel, twisted road within macrophages. Trends Microbiol. 1437-44. [DOI] [PubMed] [Google Scholar]
- 55.Santic, M., M. Molmeret, K. E. Klose, S. Jones, and Y. A. Kwaik. 2005. The Francisella tularensis pathogenicity island protein IglC and its regulator MglA are essential for modulating phagosome biogenesis and subsequent bacterial escape into the cytoplasm. Cell Microbiol. 7969-979. [DOI] [PubMed] [Google Scholar]
- 56.Schlesinger, L. S. 1993. Macrophage phagocytosis of virulent but not attenuated strains of Mycobacterium tuberculosis is mediated by mannose receptors in addition to complement receptors. J. Immunol. 1502920-2930. [PubMed] [Google Scholar]
- 57.Schulert, G. S., and L. A. Allen. 2006. Differential infection of mononuclear phagocytes by Francisella tularensis: role of the macrophage mannose receptor. J. Leukoc. Biol. 80563-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schulert, G. S., R. L. McCaffrey, B. W. Buchan, S. R. Lindemann, C. Hollenback, B. D. Jones, and L. A. Allen. 2009. Francisella tularensis genes required for inhibition of the neutrophil respiratory burst and intramacrophage growth identified by random transposon mutagenesis of LVS. Infect. Immun. 771324-1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schwartz, J. T., and L. A. Allen. 2006. Role of urease in megasome formation and Helicobacter pylori survival in macrophages. J. Leukoc. Biol. 791214-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sjöstedt, A. 2003. Virulence determinants and protective antigens of Francisella tularensis. Curr. Opin. Microbiol. 666-71. [DOI] [PubMed] [Google Scholar]
- 61.Su, J., J. Yang, D. Zhao, T. H. Kawula, J. A. Banas, and J. R. Zhang. 2007. Genome-wide identification of Francisella tularensis virulence determinants. Infect. Immun. 753089-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sullivan, J. T., E. F. Jeffery, J. D. Shannon, and G. Ramakrishnan. 2006. Characterization of the siderophore of Francisella tularensis and role of fslA in siderophore production. J. Bacteriol. 1883785-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Titball, R. W., A. Johansson, and M. Forsman. 2003. Will the enigma of Francisella tularensis virulence soon be solved? Trends Microbiol. 11118-123. [DOI] [PubMed] [Google Scholar]
- 64.Urbanowski, M. L., L. T. Stauffer, and G. V. Stauffer. 2000. The gcvB gene encodes a small untranslated RNA involved in expression of the dipeptide and oligopeptide transport systems in Escherichia coli. Mol. Microbiol. 37856-868. [DOI] [PubMed] [Google Scholar]
- 65.Weiss, D. S., A. Brotcke, T. Henry, J. J. Margolis, K. Chan, and D. M. Monack. 2007. In vivo negative selection screen identifies genes required for Francisella virulence. Proc. Natl. Acad. Sci. USA 1046037-6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.White, J. D., J. R. Rooney, P. A. Prickett, E. B. Derrenbacher, C. W. Beard, and W. R. Griffith. 1964. Pathogenesis of experimental respiratory tularemia in monkeys. J. Infect. Dis. 114277-283. [DOI] [PubMed] [Google Scholar]