

Abstract
Base excision repair (BER) is an evolutionarily conserved process for maintaining genomic integrity by eliminating several dozen damaged (oxidized or alkylated) or inappropriate bases that are generated endogenously or induced by genotoxicants, predominantly, reactive oxygen species (ROS). BER involves 4–5 steps starting with base excision by a DNA glycosylase, followed by a common pathway usually involving an AP-endonuclease (APE) to generate 3′ OH terminus at the damage site, followed by repair synthesis with a DNA polymerase and nick sealing by a DNA ligase. This pathway is also responsible for repairing DNA single-strand breaks with blocked termini directly generated by ROS. Nearly all glycosylases, far fewer than their substrate lesions particularly for oxidized bases, have broad and overlapping substrate range, and could serve as back-up enzymes in vivo. In contrast, mammalian cells encode only one APE, APE1, unlike two APEs in lower organisms. In spite of overall similarity, BER with distinct subpathways in the mammals is more complex than in E. coli. The glycosylases form complexes with downstream proteins to carry out efficient repair via distinct subpathways one of which, responsible for repair of strand breaks with 3′ phosphate termini generated by the NEIL family glycosylases or by ROS, requires the phosphatase activity of polynucleotide kinase instead of APE1. Different complexes may utilize distinct DNA polymerases and ligases. Mammalian glycosylases have nonconserved extensions at one of the termini, dispensable for enzymatic activity but needed for interaction with other BER and non BER proteins for complex formation and organelle targeting. The mammalian enzymes are sometimes covalently modified that may affect activity and complex formation. The focus of this review are the early steps in mammalian BER for oxidized damage.
Keywords: DNA glycosylases, AP-endonuclease, covalent modification, BER subpathways, repair complex, BER interactome
Introduction
The genome is inherently unstable due to spontaneous chemical reactions, and its fidelity is compromised due to very low but significant replication errors. Moreover, the genomes of all organisms are continuously exposed to a wide variety of insults, e.g., endogenous reaction oxygen species (ROS), and environmental genotoxic agents including cigarette smoke, UV light, ionizing radiation. Furthermore, chemotherapeutic drugs including alkylating agents are invariably genotoxic and induce damage in both healthy and tumor cell genomes. ROS, because of their continuous generation as byproducts of respiration in the mitochondria [1] represent the predominant group of chemicals that damage genomes of aerobic organisms. ROS are also generated by ionizing radiation and during oxidative metabolism of xenobiotic agents by p450s. ROS include O2•− (superoxide radical anion), OH• (hydroxyl radical) and H2O2 (hydrogen peroxide). O2•− is also generated during inflammatory response by NADH oxidases [2]. ROS-induced cellular changes have been implicated in a multitude of diseases, ranging from cardiovascular dysfunction, arthritis, cancer, as well as in aging and age-related disorders. To counteract ROS-induced DNA damage, cells have evolved several defense mechanisms that act at different levels to prevent or repair such damage.
The base excision repair (BER) pathway is responsible for repairing most endogenous base lesions and abnormal bases in the genome as well as similar lesions generated by several groups of environmental agents, or their metabolic intermediates. The BER pathway is also involved in repair of DNA single strand breaks. Such breaks resulting from free radical reaction of deoxyribose residues invariably possess blocked termini. Contrary to the earlier concept that BER is a simple process requiring very few proteins, recent discoveries paint a more complex picture of mammalian BER with several subpathways. This review is exclusively focused on the early steps in BER, in particular, for repair of the most predominant damage namely, those induced by ROS. The review of Horton et al. in this issue is devoted to some aspects of the late steps in BER.
DNA repair: A recipe for survival
It has been generally estimated that some 104 lesions are induced in a mammalian cell genome every day by spontaneous chemical reactions and during cellular metabolism. The most predominant among these are spontaneously generated or products of ROS reaction [3]. Environmental chemicals, tobacco smoke and radiation add to the damage load. Most ROS-induced oxidized base lesions and abasic (AP) sites if left alone could be replicated by replicative or DNA translesion synthesis (TLS) polymerases [4, 5]. Misreplication of oxidized bases and noninstructional AP sites would often give rise to point and sometimes more complex mutations. Single nucleotide polymorphisms (SNPs), observed at quite high frequency (1 in 300 bp) in mammalian genomes, likely resulted from such spontaneous mutations. The origin of sporadic cancer is also very likely due to such spontaneous mutations in oncogenes and tumor suppressor genes [6]. For example, C is spontaneously deaminated to U and ROS could oxidatively deaminate C to 5′ hydroxy uracil (5-OHU). Both U and 5-OHU preferably pair with A during DNA replication by replication or TLS DNA polymerases resulting in GC→AT transition mutation. Similarly, G is oxidized to 8-oxo-7,8 dihydroguanosine (8-oxoG) by a variety of ROS [7]. Its mispairing with A would give rise to GC→TA transversion mutation [8]. Such mutations have indeed been preferentially observed in cellular genomes after oxidative stress [9]. Defense mechanisms to maintain genomic integrity in the face of continuous assault on the genome evolved in all organisms primarily in the form efficient DNA repair. Thus, repair of oxidized base lesions is essential for maintaining genome integrity and survival.
Base Excision Repair (BER): A pathway for processing small base adducts, inappropriate or oxidized bases, and DNA single-strand breaks
BER is the most versatile among excision repair pathways, and is responsible for repairing most endogenous lesions like oxidized bases and AP sites, as well as DNA single-strand breaks. The basic mechanism of BER was first elucidated in E. coli [10]. Subsequent studies showed that the process is conserved in the eukaryotes including mammals. Until recently, BER was believed to be the simplest, thoroughly characterized process among all repair pathways. Although BER is responsible for repair of a majority of cellular DNA damages [3, 11], the functional significance of BER in prevention of disease remains unclear. This is presumably because nucleotide excision repair (NER) and DNA mismatch repair (MMR) deficiencies have been linked to cancer and other diseases, whereas no disease phenotype has been linked to BER deficiency so far [12].
In addition to oxidation of DNA bases, ROS attack deoxyribose in DNA to generate strand breaks with nonligatable dirty ends (sugar fragments or 3′ phosphates). These are also repaired by BER pathway. The 3′ blocking groups include 3′ phosphate, 3′ phosphoglycolaldehyde, or 3′ phosphoglycolate [13]. The 5′ terminus normally contains phosphate but after ROS reaction the nonligatable ends include 5′OH and 5′ phosphodeoxyribose derivatives such as 2-deoxyribonolactone [14].
Basic mechanism of BER
BER requires only four or five enzymes in the basic reaction steps to carry out repair of DNA containing AP sites or base damage. These include a DNA glycosylase, an AP endonuclease, a DNA polymerase, and a DNA ligase [15]. BER is initiated with excision of a damaged base by the DNA glycosylase. An AP site is generated due to cleavage of the N-glycosidic bond of the damaged base. The APE cleaves the AP site in the second step, to generate 3′ OH and 5′ deoxyribose phosphate (dRP) terminus [16]. The third reaction in the pathway involves the DNA polymerase to fill in the single nucleotide gap generated due to lesion base removal. In mammalian cells, the repair DNA polymerase β (Polβ) has an intrinsic dRPase activity [17, 18] which cleaves the dRP residue to generate 5′ phosphate and the resulting nick after single nucleotide incorporation is sealed by the DNA ligase in the final step. These BER enzymes are conserved from E. coli to mammals.
DNA glycosylase, a key enzyme in BER
BER is unique among the excision repair processes in that the individual base lesions are recognized by distinct DNA glycosylases less than a dozen of which are expressed in E. coli or mammalian cells. However, each DNA glycosylase acts on a limited number of damaged bases. DNA glycosylases are relatively small (~30–50 kDa) monomeric proteins that do not require cofactors for their activity. These enzymes generally excise altered bases that cause only minor perturbations in the DNA helix. Table 1 provides the list of DNA glycosylases characterized in E. coli and mammals so far. The first DNA glycosylase to be discovered was uracil DNA glycosylase (UDG) in E coli after searching for an enzymatic activity that would recognize uracil whose presence in DNA as a G•U pair would be mutagenic because U is generated due to deamination of cytosine [19]. Subsequently, similar enzyme activities were identified in other bacteria, yeast, plants and mammalian cells, mitochondria-specific UDGs were also characterized [20]. In contrast to the promiscuity of most DNA glycosylases for diverse substrates, UDG is highly specific for U which tightly fits into UDG’s catalytic pocket. Elucidation of the crystal structures and mutational studies of UDG [21] have established the structural basis for substrate specificity in preference to structurally similar normal bases like C or T in DNA. DNA binds along a positively charged groove in the enzyme, but the tight-fitting uracil-binding pocket located at the base of this groove is too deep and narrow to allow binding of DNA-uracil unless it is ‘flipped out’ of the DNA helix. The structure of human UDG and bound to uracil-containing oligo has demonstrated the basis for the enzyme-assisted nucleotide flipping [21, 22]. Another DNA glycosylase SMUG1 has been characterized in human cells [23, 24], which catalyzes excision of U and also oxidized pyrimidines such as 5-hydroxycytosine (5-OHC). SMUG1 is a member of UDG family of glycosylases. Finally, a third DNA glycosylase in mammalian cells, thymine-(T•G)-DNA glycosylase (TDG) was shown to excise T and U from their base pairs with G, and may be involved in repairing T generated by deamination of 5-methyl C in methylated C•G pair [25].
Table 1.
Mammalian DNA glycosylases and their prototypes in E. coli
| Enzyme | E. coli | Human | ||||
|---|---|---|---|---|---|---|
| Type | Preferred substrate | Type | Preferred substrate | |||
| Uracil DNA glycosylase (UDGs) | Ung | Monofunctional | uracil | UNG SMUG1 TDG |
Monofunctional Monofunctional Monofunctional |
uracil uracil U and T in U(T) •G |
| Alkylbase DNA glycosylase | Tag AlkA | Monofunctional | 3-methyladenine | MPG | Monofunctional | 3-methyladenine, hypoxanthine |
| Adenine-specific mismatch DNA glycosylase | MutY | Monofunctional | A in 8-oxoG•A | MYH | Monofunctional | A in 8-oxoG•A |
| DNA glycosylases for oxidized bases | EndoIII (Nth) | Bifunctional (β lyase) | 5-OHC, 5-OHU, Tg | NTH1 OGG1 |
Bifunctional (β) Bifunctional (β) |
Tg, 5-formylU, 8-oxoG, Fapy-G |
| EndoVIII (Nei) Fpg (MutM) |
Bifunctional (βδ lyase) | Tg, 5-OHU 8-oxoG Fapy-G (−A) |
NEIL1 NEIL2 |
Bifunctional (βδ) Bifunctional (βδ) |
Fapy-A (−G), Tg, 5-OHU Hydantoin (Sp, Gu), 5-OHU |
|
SP, spiriminohydantoin; Gu, guanidinohydantoin; Tg, thymine glycol; Fapy, formamidopyrimidine
As discussed later, DNA glycosylases specific for oxidized base lesions are bifunctional with intrinsic AP lyase activity that causes cleavage of DNA strand at the AP site generated after base excision [26]. In this case, the 3′ terminus at the cleavage site is blocked with α,β-unsaturated aldehyde dehydration product of deoxyribose phosphate or only phosphate and the 5′ terminus contains a phosphate residue (Fig. 1, 2). Unlike in the case of APE cleavage product of an AP site where the 5′ terminus is blocked with deoxyribose phosphate, the 3′ terminus of ROS-induced DNA strand breaks is nonligatable, and needs to be cleaned prior to the subsequent step in BER.
Figure 1.

A schematic illustration of BER subpathways for damaged bases and DNA strand breaks. The damaged base is represented as a star(*). Divergent base excision steps converge to common steps for end processing, followed by repair DNA synthesis (represented as blue dots) and strand sealing. Polβ could be involved in LP-BER by collaborating with FEN1. Other details are discussed in the text.
Figure 2.
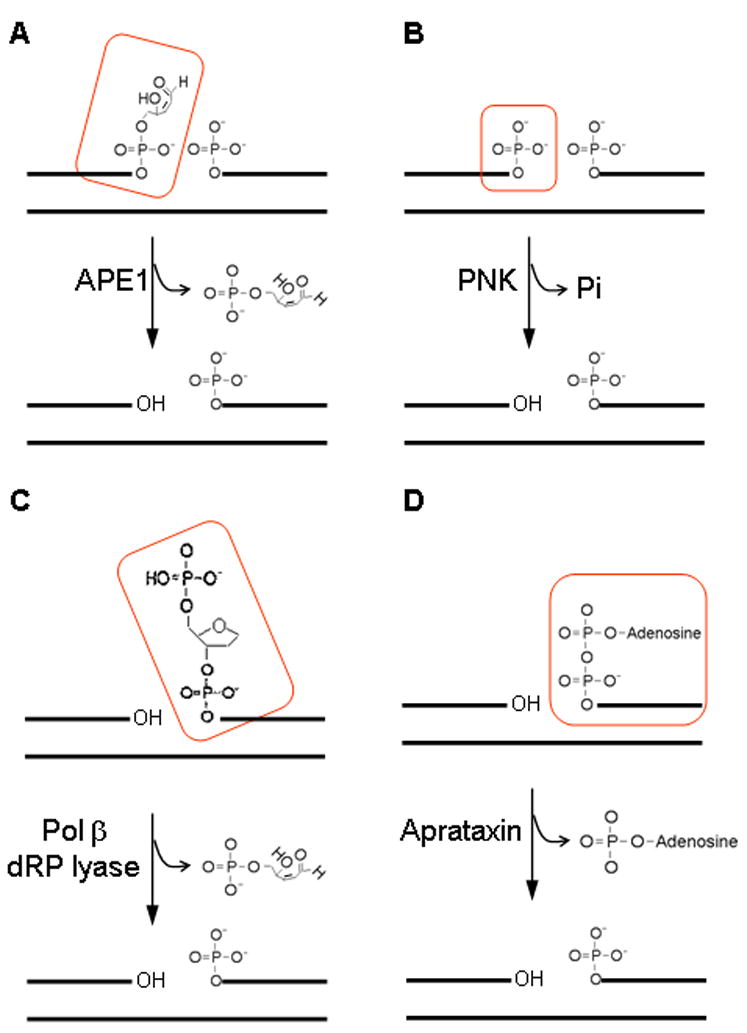
Distinct end cleaning activities of various enzymes. APE1 removes 3′ blocking PUA (boxed in red in A) while PNK removes 3′ phosphate (boxed in red in B). Polβ removes 5′ blocking dRP (boxed in red in C). Aprataxin hydrolyzes 5′-5′ linkage of AMP to the 5′ phosphate (D). The dRP lyase activity Polβ requires the aldehyde group.
Repair of alkylated bases by monofunctional DNA glycosylases and by MGMT
Endogenous base lesions also include methyl adducts which are likely to be generated by chemical reaction with S-adenosylmethionine [27]. The methylated base adducts like other base lesions could also be generated by reaction with exogenous methylating agents including many chemotherapeutic drugs and N-methylnitrosamines [28]. A multitude of methyl base adducts are produced many of which including O6-methylguanine and O4-methylthymine are highly mutagenic. The methylated adducts are repaired in all organisms via two pathways. One is a direct reversal of damage without DNA repair synthesis. This was first discovered in E. coli as an adaptive response and the protein named Ada [29] turned out to be conserved in eukaryotes and mammals and named O6-methylguanine-DNA methyltransferase (MGMT) [27]. MGMT demethylates O6-methylG in situ in DNA and also O4-methylT. A second direct reversal enzyme named AlkB in E. coli and ABH in mammals demethylates 1-methylA and 3-methylC with more complicated reaction chemistry [30]. In contrast, other methyladducts as well as hypoxanthine and xanthine, deamination products of A and G respectively, are repaired via the conventional BER pathway by N-methylpurine-DNA glycosylase (MPG) and also named MAG. This and its E. coli ortholog AlkA excise N-alkylpurines, primarily 3 methylA, 3-methylG and 7-methylA. E. coli expresses another glycosylase Tag which specifically excises 3-methyladenine. AlkA also excises O2-methyladducts of C and T [31, 32].
DNA glycosylases for normal bases
While DNA glycosylases in general recognize only abnormal bases including U in DNA as substrates, there is one exception for mismatch-specific glycosylase MutY. MutY was discovered in E. coli as a mutator gene whose deficiency induces spontaneous C•G→A•T transversion mutation. It excises primarily the normal base A (and also G) from A•G and A(G)•8-oxoG base pair mismatches [33, 34]. Thus MutY and its mammalian homolog MYH apparently serve an important function in protection against oxidative damage by removing the base A incorporated in the nascent strand opposite 8-oxoG during DNA replication [9, 35, 36].
Bifunctional DNA glycosylases
The monofunctional glycosylases excise the substrate base, leaving an intact AP site. In contrast, the bifunctional glycosylases have an additional lyase activity. This second intrinsic activity is invariably present in DNA glycosylases specific for oxidized bases. The monofunctional DNA glycosylases generate an AP site which is processed by an AP endonuclease. Bifunctional DNA glycosylases further process the AP site via β or βδ elimination reaction. A mechanistic distinction between these two types of enzymes is that monofunctional DNA glycosylases typically use an activated water molecule as a nucleophile in attacking sugar C1′ of the target nucleotide, where bifunctional glycosylases/AP lyases often use ε-NH2 of a lysine or the N-terminal proline as the active site nucleophile [37]. An intermediate step is the formation of a transient Schiff base between the amino group and the C1′ of deoxyribose for both base excision and DNA strand cleavage. Usually, the base excision and lyase reaction steps act in a concerted sequence. However, in some cases, e.g., for 8-oxoguanine-DNA glycosylase (OGG1), the lyase reaction is very weak. Thus, intact AP sites are the major product after OGG1 catalyzed cleavage of 8-oxoguanine from DNA [38, 39].
Despite differences in the specific residues used to recognize damaged bases, one unifying mechanisms for BER initiation by DNA glycosylases is the extrahelical flipping of the damaged deoxynucleotide into a lesion-specific recognition pocket. All DNA glycosylases studied so far bind to the minor groove, kink DNA at the site of damage, and flip the lesion base out of the DNA major groove. Thus each DNA glycosylase is damage specific, and only those bases after nucleotide flipping that can be accommodated in the defined binding pocket provide the necessary contacts and orientation for their excision. The initial recognition apparently exploits the deformability of the DNA at a base pair destabilized by the presence of a lesion [40]. It is likely that plasticity of the catalytic pocket allows induced fit of diverse substrates. This could also explain why DNA glycosylases (with the exception of UDG) are poor enzymes with low turnover.
Oxidized base-specific DNA glycosylases
Oxidized base-specific DNA glycosylases were also first discovered in E. coli, and along with subsequently characterized eukaryotic orthologs. These are categorized into two families based on tertiary structure, active site characteristics and AP lyase reaction, and are named after the prototype Nth [endonuclease III] and Fpg (formamidopyrimidine-DNA glycosylase [37]. The Nth family utilizes an internal Lys residue as the active site for β elimination reaction, generating a 3′ phospho α,β-unsaturated aldehyde (3′ PUA), also named 3′ phosphor 4-hydroxylpentenal, at the strand break. In contrast, the members of the E. coli Fpg family including Nei (endonuclease VIII) catalyze βδ elimination at the AP site by utilizing N-terminal Pro as the nucleophile and removes the deoxyribose residue to produce a 3′ phosphate terminus at the DNA strand break [41]. In both cases, the 5′ terminus retains the phosphate moiety. Figure 1 depicts the β and βδ elimination reactions.
DNA glycosylases could also be classified into two distinct families based on tertiary structure features, namely, the presence of either an HhH or an H2TH motif unrelated to the type of oxidized base recognized [40]. Representative structures have been determined for both families. However, some members of these families have common substrates. OGG1, MutY and Nth and their homologs have HhH structure [42–44]; while, MutM (Fpg), Nei have H2TH motif [44–47].
Mammalian DNA glycosylases for oxidized bases
Four oxidized base-specific glycosylases have been characterized in mammalian cells. NTH1 and OGG1 were characterized previously, both of which belong to the Nth family [38, 48, 49]. Thus, both NTH1, preferring oxidized pyrimidines as substrates, and OGG1, primarily responsible for the removal of 8-oxoG and ring opened quanine, i.e., formamidopyrimidine (Fapy-G), catalyze β elimination [11, 49]. Subsequently, we characterized two additional human DNA glycosylases, and independently discovered by others, which belong to the Fpg/Nei family; we named these NEIL1 and NEIL2 [50–55]. All these enzymes as already indicated are bifunctional glycosylases with broad substrate range. We also identified a third member of this family, namely NEIL3, but no glycosylase activity has been characterized for this enzyme so far (Izumi, T., unpublished observation).
Among the three NEILs, N-terminal Pro, present only in NEIL1 and NEIL2 acts as the active site nucleophile [50, 53]. NEILs are closer to Nei than Fpg, and we and others initially showed that both enzymes prefer modified pyrimidine substrates [53]. However, NEIL1 was later shown to prefer ring-opened purines, i.e., formamidopyrimidine (Fapy)-A and –G, which along with 8-oxoG are the preferred substrates for E. coli Fpg. While we initially showed 8-oxoG as a poor substrate of NEILs on duplex DNA substrate, others have shown NEIL1’s robust 8-oxoG excision activity [54, 56]. Subsequently, we showed that NEILs activity and substrate specificity depends largely on the DNA structure, and NEILs have significant 5-hydroxyuracil excision activity with single-stranded or bubble DNA [57]. Interestingly there is no evidence for NEILs in the yeast which however, has one OGG1 and two distinct NTHs (Ntg) [58].
Table 2 lists the comparative characteristics of OGG1, NTH1, NEIL1, and NEIL2 all of which have substrate preference but not absolute specificity.
Table 2.
Comparative Properties of Mammalian DNA Glycosylases for Oxidized Bases
| Properties | Nth Type | Nei Type | ||
|---|---|---|---|---|
| OGG1 | NTH1 | NEIL1 | NEIL2 | |
| Size (kD) | 38 | 36 | 43 | 36 |
| DNA structure | dsDNA | dsDNA | ssDNA; Bubble, Fork, and dsDNA | ssDNA; Bubble, Fork, and dsDNA |
| Downstream Enzyme | APE1 | APE1 | PNK | PNK |
| Conserved motif | HhH | HhH | H2TH | H2TH |
| Catalytic residue | Lys 249 | Lys 212 | Pro1 | Pro1 |
| Cell Cycle Dependence | None | None | S-phase Specific | None |
| Dispensable Sequences | C-terminal 20 & N-terminal 10 residues | N-terminal 80 residues | C-terminal 101 residues | C-terminal 10 residues |
| AP lyase | β-elimination (3′ PUA) | βδ-elimination (3′ phosphate) | ||
ds, double stranded; ss, single-stranded; HhH, helix-hairpin-helix; H2TH, helix-2-turn-helix.
Nonessentiality of individual DNA glycosylases in mammals
The genes and cDNAs of mammalian OGG1 and NTH1 were cloned in late 1990s and NEIL1 and NEIL2 genes were identified in mammalian genome databases in 2002. The genomic cloning led to the generation of mouse mutants lacking OGG1, NTH1 and NEIL1 as well as MYH [59–61]. All of the mouse mutants and their embryonic fibroblasts are viable. Furthermore, no strong phenotype was observed in the mutant mice in regards to enhanced incidence of spontaneous cancer or accelerated aging. Although a small increase in cancer incidence was observed in OGG1-null mice after exposure to bromate [62], the lack of immediate effect was unexpected, particularly because the deficiency of other BER proteins, such as APE1 and DNA Polβ, induce embryonic lethality in mutant mice [63, 64]. There is compelling evidence that the oxidized base lesions are endogenously generated at a reasonably high frequency many of which are mutagenic and thus potentially carcinogenic. Repair of such mutagenic and carcinogenic lesions is essential, which is presumably carried out by other glycosylases in the cells deficient in a single enzyme. Furthermore, enhanced accumulation of 8-oxoG, observed in DNA isolated from various tissues of OGG1 null mice, confirms the requirement of OGG1 in removing such lesions. In contrast, deficiency in NER or MMR processes significantly enhances cancer susceptibility [65–67]. One possible explanation for the lack of phenotype of the glycosylase mutants is that, unlike in NER or MMR, multiple glycosylases could provide back-up function because of their overlapping substrate range. We could argue that oxidative damage repair is too important to be left to a single enzyme for repair. At the same time, as we have already pointed out, the glycosylases do have preferred substrates and it is also likely that the individual enzyme act preferentially depending on the state of the genome, i.e., whether these are cycling or postmitotic and whether the damage is located in transcriptionally active vs. inactive sequences [68]. This could also explain why accumulation of oxidized bases in the genomes of OGG1 or NTH1 null mouse cells does not cause any obvious phenotype [69]. Although oxidized bases could possibly be repaired alternatively via the NER pathway, this is unlikely to be a major alternative repair mode for most oxidized bases [70], because such bases do not induce significant perturbation in the duplex DNA structure needed for recognition by the NER proteins. We have proposed a hypothesis that the lesions in transcriptionally active sequences are preferentially repaired by other glycosylases such as NEILs while OGG1 and NTH1 act globally in repairing both active and inactive genome sequences [71].
More than a dozen oxidatively damaged bases have been identified in the genome but only a few DNA glycosylases have been characterized. This is consistent with the observation that the glycosylases have broad substrate range. These enzymes also have very low turn-over, presumably as the price to pay for their promiscuity. This is especially true for mammalian oxidized base-specific DNA glycosylases. However, these enzymes may also have distinct in vivo functions, although the requirement is not absolute as the deficiency of a given DNA glycosylase is not lethal. Thus the distinction among different BER sub-pathways initiated with different DNA glycosylases is not absolute; the subpathways may be preferentially used for specific cellular states, damage site in inactive vs. active regions of the genome and the nature of the damage itself.
Unique DNA structure preference of NEILs
There is one absolute distinction in substrate recognition specificity between two families of oxidized base-specific glycosylases. OGG1 and NTH1 excise base lesions only from duplex DNA, which is expected because the undamaged strand provides the template for repair of the damaged strand. So we were greatly surprised to observe that NEIL1 and NEIL2 preferentially excise base lesions from single-stranded DNA, including bubble or forked DNA structures [57]. Yasui’s group had made a similar observation earlier for NEIL1 [55]. Because the bubble and fork structures mimic the transcription and replication intermediate respectively, we have proposed preferential involvement of NEILs in repair of oxidized bases during transcription or DNA replication. Our initial observation of S-phase-specific activation of NEIL1 and not of other three glycosylases suggested NEIL1’s preferential involvement in repair of base lesions in template DNA prior to replication. We have named this subpathway replication-associated BER (RA-BER) [57]. A similar model was earlier proposed for repair of U in replicating DNA by UNG2 which also shows S-phase specific activation [72]. Thus SMUG1 may preferentially act in repairing U in noncycling genomes [73].
While the RA-BER model and NEIL1’s role therein are not established, our recent observations on the interaction of NEIL1 with the sliding clamp PCNA and DNA damage-inducible alternative clamp 9-1-1 and NEIL1’s activation by these proteins support our hypothesis [74, 75]
Two basic modes of BER: SN- vs. LP-BER
In the simple BER model, excision of the damaged base leaves a 1-nt gap whose repair has been named single nucleotide repair (SN-BER). This has also been referred to as short-patch repair (SP-BER) [76]. In contrast, a repair patch size of 2–8 deoxynucleotides was observed initially in reconstituted BER systems and subsequently in vivo, which was named long-patch BER (LP-BER). During in vitro repair of an AP site analog, lacking the aldehyde group, Polβ could not remove the 5′ blocking group generated by APE via its dRP lyase activity [77–80]. Matsumoto’s and Dogliotti’s groups first showed that such a 5′ blocking group could be removed by the 5′-flap endonuclease 1 (FEN-1) normally required for removing the 5′ RNA primers from Okazaki fragments during DNA replication. Subsequent steps of LP-BER are identical to that of DNA replication involving DNA polymerases δ/ε and DNA ligase I (Lig I). These enzymes including FEN-1 are recruited by the sliding clamp PCNA which enhances their activity [77–79]. The choice of LP-BER vs. SN-BER may thus depend on, among other factors, the state of the 5′-dRP terminal moiety. With unaltered aldehyde group in deoxyribose Polβ could carry out SN-BER by excising the 5′-dRP [17, 81]. On the other hand, LP-BER is necessary when the AP sites are further oxidized by ROS and after cleavage of the DNA strand by APE, the 5′ blocking groups could not be removed by Polβ’s 5′ end cleaning lyase activity. The LP-BER subpathway utilizes the DNA replication machinery where FEN-1 excises the displaced 5′-oxidized deoxyribose fragment containing segment displaced as a flap during repair [82]. More recently, FEN-1 was found to have additional 5′-exonuclease and gap-specific endonuclease activities [83]. The role of FEN-1’s exonuclease functions in LP-BER however, is yet to be clarified. PCNA, loaded on duplex DNA by replication factor-C (RFC), acts as a sliding clamp in LP-BER as it does during replication [84]. It is likely that Polδ/ε can substitute for Polβ, and carry out LP-BER because Polβ-null cells, grow normally and are not hypersensitive to oxidative stress [85]. However, Polβ also interacts with FEN-1 and Lig I and could thus carry out LP-BER [86]. These distinct pathways are schematically shown in Figure 1.
Repair of AP sites and single-strand interruption with nonligatable termini: a common step in the BER pathway
DNA glycosylases excise damaged or inappropriate bases generating repair intermediates, namely AP sites and strand breaks with blocked termini. Repair of these lesions share common steps with that of ROS-induced DNA single-strand breaks, and spontaneously generated AP sites. AP sites could be further oxidized to various products with oxidation and fragmentation of deoxyribose moiety and strand cleavage [87]. The major products include 2′-deoxyribonolactone, 2-deoxypentos-4-ulose and 3′ phosphoglycolate [14]. Unmodified AP sites are also generated by monofunctional DNA glycosylases, as already mentioned. But, all oxidized base-specific glycosylases generate 3′ phospho α,β unsaturated aldehyde or 3′ phosphate respectively, and 5′ phosphate at the strand break. In addition to the AP sites, the single-strand breaks with 3′ blocking residues have to be repaired to maintain genomic integrity.
AP-endonuclease (APE), a ubiquitous repair protein with dual nucleolytic activities
In view of continuous generation of AP sites and DNA strand breaks, all of which are more toxic than damaged bases themselves [88], it is not surprising that multiple enzymatic processes have evolved to repair these lesions among which APE is the most prominent and ubiquitous [88]. APE was discovered in E. coli as a 3′ exonuclease/DNA 3′ phosphatase and was named exonuclease III (Xth) [89] and subsequently found to be an AP site-specific endonuclease that generates 3′ OH terminus after cleaving the DNA strand 5′ to the abasic site. It is also active as a 3′ exonuclease/phosphatase and produces 3′ OH terminus at strand breaks with 3′ blocks. Another endonuclease, endonuclease IV (Nfo), was subsequently discovered in E. coli whose activities as AP site-specific endonuclease and 3′ end cleaning phosphodiesterase are very similar to that of Xth except Nfo lacks 3′ exonuclease activity and does not require Mg2+ for catalysis [90, 91]. Subsequently APEs were characterized in a variety of organisms ranging from bacteria to yeast, C. elegans and mammals. These enzymes have similar but not identical enzymatic functions and conserved structures, and could be classified as a member of the Xth or Nfo family.
Multiple APEs are also present in lower eukaryotes
The in vivo function of E. coli Nfo is not clear because its deletion in cells containing wild type Xth does not cause any phenotype. However, Xth null cells are highly sensitive to ROS confirming that the AP sites and strand breaks with 3′ blocked ends are toxic [89]. E. coli lacking both Xth and Nfo are highly sensitive to ROS and alkylating agents, as expected [92]. However, the fact that the cells are viable indicates either that AP sites and nonligatable strand breaks could be bypassed or that these are repaired via an alternative back-up mechanism. Noninstructional AP sites are highly mutagenic [93]. However, these could be bypassed by translesion synthesis (TLS) DNA polymerases in the absence of repair [94, 95].
There is no evidence so far that oxidized AP sites or strand breaks could be bypassed in vivo. However, an alternative process for repairing these in multi-allelic E. coli or post replication repair could bypass AP sites by strand switching [10, 96], or the AP sites could also be repaired via NER [97]. The observation that Xth/Nfo deficient E. coli loses viability when UvrB, a key NER protein, is also inactivated, supports this scenario [92].
Xth and Nfo type APEs, named APN2 and APN1, respectively, are expressed in yeast [98, 99]. In contrast to the situation in E. coli, APN1 is the major contributor of APE activity in these unicellular eukaryotes. However, as in E. coli, deletion of both APN genes does not cause lethality although it increases sensitivity to alkylating and oxidizing agents. However, simultaneous inactivation of the nucleotide excision repair pathway is lethal as in E. coli [100].
Two distinct types of APE possess nearly identical activity
The structures of E. coli Xth and its mammalian ortholog APE1 as well as those of Xth or Nfo of various microorganisms have been solved by X-ray crystallography [101]. It is remarkable that Xth and Nfo have nearly identical enzymatic activity and yet do not have any structural similarity. Xth is a Mg2+-dependent nuclease belonging to the superfamily of pancreatic DNase. The divalent metal bound form of the Xth and human APE1 show one metal ion bound per monomeric protein molecule [102]. However, recent molecular dynamic simulation predicts that APE1 does have two potential Mg2+-binding sites A and B, although only one Mg2+ is bound to the protein which shuttles between the two sites during catalysis [103]. Mol et al. [102] solved the structure of human APE1 bound to its cleavage product and Mg2+ was shown to be present at the A site. The structure of substrate bound active APE1 could not be solved in the presence of Mg2+ for obvious reasons. The key residues and motifs required for catalytic activity and metal binding in all Xth family members are conserved which allowed our lab to clone the S. cerevisiae Apn2 [99]. We subsequently identified cloned S. pombe APN2 based on sequence homology [104].
The Nfo type APE is extremely unusual because it is a Zn metalloenzyme with 3-Zn2+ center [105]. It does not require external Mg2+ for activity [92]. The evolutionary significance of the Nfo family is unclear.
Mammalian cells express only Xth type APE, APE1
Unlike in E. coli or yeast, there is no evidence for an Nfo ortholog in mammalian cells. Mammalian APE1 was characterized based on activity more than two decades ago [106]. Subsequent to its cloning and extensive characterization of APE1, a second Xth ortholog, named APE2 was cloned [107, 108]. However, the recombinant APE2 has negligible APE-specific activity suggests its lack of function in BER (108; Izumi T., unpublished). Nevertheless, expression of APE2 in various mammalian cells does implicate it in some other in vivo functions.
Mammalian APE1 and E. coli Xth have distinct properties
In spite of similarity in tertiary structure and conserved motifs and catalytic mechanism, APE1 has many distinctive features starting with its unique N-terminal extension of about 61 residues which are absent in Xth and dispensable for APE activity [109]. This segment contains the nuclear localization signal (NLS) [110] and is also required for the transcriptional regulation function unique to APE1 and unrelated to BER [111, 112]. We recently discovered that human APE1 (hAPE1) is acetylated in vivo at Lys 6/ Lys 7 which modulates its regulatory but not endonuclease activity [113]. The endonuclease and 3′ end cleaning phosphodiesterase activity of APE1 is weaker than of Xth, but more importantly, APE1 has barely detectable DNA 3′ phosphatase activity and has a very limited 3′ exonuclease activity [114, 115]. However, 3′ mismatch-specific exonuclease activity has been observed in APE1 although its in vivo significance has not been established [116].
APE1 is essential in mammalian cells
The attempts of several groups to generate APE1-null mice failed because of very early (4–5 days post coitus) embryonic death. This strongly suggested that unlike in lower organisms, APE1 is essential at least for early embryonic development [64, 117]. Efforts to generate cell lines from APE1 null embryos were also uniformly unsuccessful which distinguishes it from Polβ. Null mutation for Polβ in mice is also embryonic lethal (although the embryos survived longer) [118]. However, Polβ null mouse embryonic fibroblast lines could be established which have normal viability [119].
We tested whether APE1 is essential in the postembryonic stage by introducing “floxed” hAPE1 transgene in the genome of APE1 heterozygous mice. Our attempt to generate viable mouse embryo lacking endogenous APE1 but expressing ectopic hAPE1, by inbreeding the heterozygous, transgenic mice was unsuccessful. However, we were able to establish embryonic fibroblast lines from such crosses which lack the endogenous APE1 allele but have normal viability. Transgenic expression of Cre in these cells leading to deletion of the APE1 transgene triggered apoptosis. This observation and an independent study provide unequivocal evidence that APE1 is essential even for somatic cells [120, 121]. Kelley and his colleagues had earlier shown that APE1 downregulation triggers apoptosis in several human cell lines [122]. Further studies showed that both DNA repair and transcriptional regulation activity of APE1 are independently required for cell viability [120].
Accumulation of single-strand breaks in the APE1-null cell genomes
Essentiality of APE1 in mammalian cells but not in lower organisms suggests that some cytotoxic lesions normally repaired via APE1-dependent BER could be alternatively repaired by NER or other pathways in E. coli or yeast. We suggest that these include DNA strand breaks with phosphosugar fragment-containing 3′ terminus which are efficiently repaired by APE1. We had shown earlier that APE1 activity is limiting in repair of ROS-induced strand breaks but not of AP sites [123]. Using embryonic fibroblast line with conditional null mutation of APE1, we observed that APE1 inactivation led to significant increase in single-strand breaks as well as AP sites [112, 124].
PNK-dependent unique BER subpathway for mammalian cells
NTH1 and OGG1 generate 3′ PUA by β-elimination of deoxyribose phosphate which is efficiently removed by APE1 in the next step. NEILs, on the other hand, have βδ lyase activity and thus generate 3′ phosphate, a poor substrate of APE1. However, mammals, unlike E. coli, express high level of polynucleotide kinase (PNK) with dual 5′ kinase/3′phosphatase activities [125]. PNK has already been implicated in repair of ROS-induced single-strand breaks [126]. We recently showed that NEIL-initiated BER in the mammals unlike in E. coli does not involve APE, but requires PNK [51, 115]. The presence of NEIL-initiated, APE-independent BER subpathway in mammalian cells was further supported by the presence of PNK and not APE1 in the NEIL1 or NEIL2 immunocomplex isolated from cell extracts [51, 115]. We also showed that NEILs carry out SN-BER mediated by PNK, Polβ, Lig IIIα and XRCC1. Stable interaction of NEILs with the downstream repair proteins Polβ, Lig IIIα and XRCC1 led us to propose that these DNA glycosylases, as the first enzyme in the repair process, determine the specific BER sub-pathway [51, 115]. Thus for oxidized bases, DNA glycosylases/ AP lyases that carry out either β elimination or βδ elimination determine the subsequent steps. APE1 is responsible for processing the β elimination product while PNK is required for generating 3′-OH termini from 3′-phosphate, a product of βδ elimination, as in the case of NEIL1 and NEIL2. Furthermore, AP sites and 3′ PUA generated by other DNA glycosylases can also be processed through a NEIL-PNK dependent pathway [127]. This alternative route or repair thus may provide important redundancy in mammalian BER, a critical safeguard against oxidative and spontaneous DNA damage [71].
Common mechanism of BER and single-strand break repair: End cleaning at strand breaks
The importance of end cleaning at a strand break as a vital step in BER was recently highlighted [115]. Irrespective of the type, the intermediate step in BER is a ‘strand break with either 3′ or 5′ or both ends blocked’ by residues that are not compatible for subsequent gap filling and ligation. The normal substrate for a DNA polymerase is a gap at a strand break with 3′ OH and 5′ phosphate residues. The common pathway for providing such a repair substrate are as follows: (i) Removal of a base lesion by a monofunctional glycosylase, followed by the action of APE on the resulting AP site would generate a strand break with 3′-OH and 5′ dRP group. Though unmodified 5′ dRP moiety in mammalian cells is removed by Polβ’s intrinsic dRP lyase activity, nuclear replicative DNA polymerases Polδ/ε lack this activity. Repair synthesis by these enzymes follows the LP-BER subpathway, where the 5′-dRP containing moiety and 4–6 additional deoxynucleotides are displaced as a single stranded DNA flap, which is then cleaved by FEN-1. Furthermore, the AP site could be oxidized by ROS and its cleavage by APE would generate 5′ blocking groups which have to be processed by FEN-1 via LP-BER pathway. (ii) The action of a bifunctional glycosylase with intrinsic β lyase activity would result in a strand break with 3′ PUA, a blocking residue and 5′ phosphate. The 3′ PUA is removed by the 3′ phosphodiesterase activity of APE1 to generate 3′ OH. (iii) The bifunctional glycosylase with an intrinsic βδ elimination property would generate 3′ phosphate, and 5′ phosphate at the strand break. The 3′ end cleaning enzyme here is PNK instead of APE1 as already discussed (Figs. 1, 2).
In addition, the 3′- or 5′-termini of most if not all DNA single-strand breaks directly induced by ROS or radiation are ‘blocked’ or ‘damaged’ and must be restored to the conventional 3′-OH and 5′-phosphate moieties in order for gap filling and subsequent ligation to occur [128]. The common blocks at ROS induced strand break are 3′ phosphoglycolate and 3′ phosphoglycolaldehyde, which are processed by APE1 or 3′ phosphate which is removed by PNK. However, the ‘end cleaning’ is perhaps the most diverse enzymatic step in strand break repair, due to the variety of termini generated. As mentioned earlier, Polβ (5′ dRP lyase) and FEN-1 are used for cleaning the 5′ blocked termini.
Eukaryotic cells express other DNA end cleaning enzymes
Tyrosyl phosphodiesterase 1 (TDP1), has been implicated in end processing at DNA double-strand breaks in budding yeast [129] but the yeast and human TDP1 can also process single strand breaks in vitro [130, 131]. The primary substrate for TDP1 is the product of abortive topoisomerase 1 (top1) reaction, namely, top1-linked 3′-termini [132, 133]. 3′ end processing by TDP1 generates 3′ phosphate, which is a substrate for PNK. Finally, under some circumstances some types of blocked 3′ termini could be removed by ERCC1/XPF nuclease, a functional homologue of the yeast Rad1/Rad10 complex proposed to conduct this role in the budding yeast [134].
Aprataxin, a novel DNA end cleaning enzyme, prevents neurodegenerative disease
Aprataxin is a family of nucleotide hydrolases and transferases [135, 136]. Mutations in the aprataxin gene causes ataxia oculomotor apraxia, a neurological disorder. Recent evidences have linked aprataxin to DNA repair [137]. Aprataxin has been shown to associate with the DNA repair proteins XRCC1 and XRCC4, which are partners of DNA Lig III and Lig IV, respectively [138]. Recent studies from Stephen West’s laboratory documented a direct role of aprataxin in DNA repair in resolving abortive DNA ligation intermediates [137]. Specifically, aprataxin catalyzes the nucleophilic release of adenylate groups covalently linked to 5′ phosphate termini at single strand nicks and gaps, resulting in the production of 5′ phosphate termini that can be efficiently rejoined. This is supported by the interaction of aprataxin with DNA ligase complexes, and the sensitivity of aprataxin-defective cells to agents that cause DNA single strand breaks, such as ROS.
PALF, a distinct mammalian APE with potentially broad repair function
Akira Yasui’s group has recently identified a new protein with APE activity which they have named PALF because of some similarity with aprataxin and PNK [139]. PALF accumulate at DNA strand breaks which is dependent on poly ADP-ribose polymerase (PARP) and directly interacts with the latter. Because PALF interacts with Ku86, XRCC4 and DNA Lig IV, all involved in nonhomologous end joining of DNA double-strand breaks, PALF may a broad function in repair of both DNA single- and double-strand breaks.
Thus the ‘end cleaning’ is an important intermediate step in both BER and strand break repair, to prepare the substrate for subsequent steps polymerase and provides a link between DNA glycosylase and DNA polymerase in BER. The four major DNA end cleaning proteins and their activity are represented in Figure 2.
TREX1 and TREX2, mammalian 3′ exonucleases
TREX1 and TREX2 were identified as DNA 3′ exonucleases ubiquitously expressed in mammalian tissues [140, 141]. Their primary function may be in editing during replication by Polβ (and α) which lack constitutive 3′ exonuclease activity. Recently, TREX1 was indicated to be the product of AGS1 gene whose deficiency causes severe brain disease [142].
XRCC1 and PARP, single-strand break sensor proteins
XRCC1, a scaffold for recruiting BER proteins
XRCC1 and PARP1 serve as single-strand break sensor proteins. However, XRCC1 functions very differently by acting as a scaffold for recruiting. BER proteins for single-strand break repair. XRCC1 physically interacts with several BER proteins and activates many of them. Our results showing interaction of XRCC1 with NEILs implicates XRCC1 in the base excision step of BER as well [51, 115]. At the same time, XRCC1 interacts with the end-cleaning enzymes including APE1, PNK and possibly others [126, 143]. Moreover, the processing of 5′ OH and/ or 3′ phosphate termini is rate limiting for single strand break repair in XRCC1-mutant cells and cell extracts [126, 135, 144]. Its interaction with Polβ and Lig IIIα suggests that XRCC1 is involved primarily in SN-BER [76, 145].
XRCC1 was shown to interact with and to stimulate APE1, and thus may stimulate this activity during end processing [143]. However, others did not observe this interaction, and APE1 activity appears to be normal in XRCC1-mutant cells [146, 147].
PARP-1, -2 activated by SSBs
PARP present in mammals but not in E. coli nor yeast transfers ADP-ribose moiety from NAD to a variety of proteins including itself and predominantly histones, nonhistone HMGs and a number of other nuclear proteins including those involved in DNA metabolism to form linear or branch polymer chains using ribose-α (1′-2′) ribose linkages. The branched structure involves ribose-α (1′-1′) ribose linkage. The first ADP-ribose is usually added to the γ-carboxyl residue of Glu in acceptor proteins [148]. Some 15 other members of the PARP superfamily including distinct proteins such as tankyrases with functions unrelated to DNA repair have been identified. Genotoxic agents that generate SSB in the genome directly or indirectly activate PARP-1 and PARP-2 leading to depletion of NAD and indirectly of ATP and induce necrosis. The overlapping function of DNA damage-dependent PARPs, PARP-1 or PARP-2 is essential in mouse cells. PARP1 is recruited very early to the site of DNA strand-break and the activated enzyme may help recruit other repair proteins involved in BER. Poly ADP-ribose (PAR) is removed by the single enzyme, poly(ADP-ribose) glycol hydrolase (PARG) which has both endo- and exoglycosidase activities.
BER in Mitochondria
Mitochondrial DNA (mtDNA) contains higher steady-state level of oxidative damage and mutates at rates significantly greater than the nuclear DNA because mitochondria are the predominant site for production of endogenous ROS and tend to accumulate toxic xenobiotics. In addition to their proximity to the sites of ROS generation, it is likely that the mitochondrial genomes are more prone to oxidative damage because histones and other chromatin-associated proteins, present in nuclear genomes and acting as scavengers of oxygen radicals, are absent in the mitochondria.
BER is the only excision repair process active for mitochondrial genomes. All mtDNA repair proteins are nuclear encoded and imported. Most mtDNA repair proteins so far discovered are either identical to nuclear DNA repair proteins or isoforms of nuclear proteins arising from differential splicing [149, 150]. The existence of repair of oxidative damage to mtDNA was established several years ago [151–153]. It was also reported that mitochondrial DNA polymerase gamma (Polγ) and mtDNA ligase (Lig IIIβ), which are involved in mtDNA replication are also functional in mitochondrial BER [154, 155]. Lig IIIβ is a splice variant of the nuclear Lig IIIα, where as Polγ is unique for the mitochondria [156]. Similarly, splice variants of nuclear OGG1 and NTH1 have been characterized in mitochondria [69, 157]. Mitochondrial transport of proteins generally requires an N-terminal mitochondrial targeting sequence (MTS) which is cleaved off in the mitochondrial matrix by specific peptidases [158] although, some mitochondrial proteins lacking MTS, utilize an internal targeting sequence which is not removed during mitochondrial import [159]. Further, mitochondrial isozymes are generated primarily to eliminate the nuclear localization signal of such proteins, required for their nuclear targeting [158].
Although most mitochondrial BER enzymes were characterized and significant BER activity reported in mitochondrial extracts several years ago [152–154], the identity of APE, a key BER enzyme was not established. Recently, our lab showed that the mtAPE lacks the N-terminal 33 amino acid residues including the NLS [150]. However, the full-length APE1 could also be detected in mitochondria of some cells. This indicates that deletion of NLS is not absolutely required for mitochondrial import of APE1. We have further shown that the extract of mitochondria (containing processing peptidases) but not of nuclei or cytosol, cleaves recombinant hAPE1 to generate a mtAPE-sized product which imply that the removal of NLS is a prerequisite for mitochondrial import of APE. At the same time, fine structure deletion mapping of hAPE1 helped us identify a bipartite NLS as well as a nuclear export signal in the N-terminal segment of the APE1 polypeptide [110].
Repair interactome - A new paradigm in BER
The broad view of BER in the last decade was that of a sequential process in which individual repair enzymes carried out reactions independently of one another [112]. Based on the emerging evidence for the existence of the ‘protein interactome’, we propose a new paradigm in BER that postulates collaboration of multiple proteins in coordinated fashion involving specific protein-protein interactions to enhance the efficiency of repair. It is now evident that cellular processes involving repair of both endogenous and induced genomic damage are essential for maintaining genomic integrity and homeostasis, which involve dynamic and complex interactions among a multitude of proteins in the cellular ‘proteome’ in response to both endogenous cellular activities and environmental stress [160]. Such protein complexes are not unique to BER, and similar situations appear to exist for other DNA excision repair pathways. There is increasing evidence that the mammalian BER proteins are organized in temporally controlled complexes for cross-talks among themselves and with DNA, presumably for optimum efficiency of damaged base recognition and repair.
Formation of the multiprotein complexes for oxidative damage repair is enhanced by ROS [51, 161]. It was previously shown that NEILs carry out SN-BER mediated by PNK, Polβ, DNA Lig IIIα and XRCC1 by stably interacting with Polβ, DNA Lig IIIα and XRCC1 [51, 115]. Other interactions between OGG1 and XRCC1 was reported earlier [162]. It now appears that additional proteins including those of the DNA replication machinery are also involved in BER [26, 163]. Several DNA glycosylases interact with replication related-proteins, namely, PCNA and replication protein A (RPA). MYH and UNG interact with both PCNA and RPA [164–166]. We observed stable and functional association between NEIL1 and WRN protein [167]. Although these associations may be merely for carrying out LP-BER, several observations, e.g., (i) colocalization of UNG-PCNA-RPA complex at replication foci and (ii) S-phase-specific increase in the levels of UNG2, MYH and NEIL1 [50, 165, 168], supports our hypothesis that certain glycosylases are involved in RA-BER.
A key unanswered question is whether there is coordinated hand-over among interacting partners in smaller sub-complexes or a larger complex is preformed to carry out efficient repair. The coordination in protein complex formation was originally compared to the passing of the baton, where the repair product of each enzyme in the BER pathway is “handed” over to the next enzyme in the pathway [169]. It would be important to unravel how NEIL1 or any DNA glycosylase can simultaneously and binarily interact with several downstream proteins in the repair pathway initiated by NEIL1. All of NEIL1’s interacting partners identified so far bind to the C-terminus of NEIL1 [51, 57,115]. Though NEIL1’s interaction with its multiprotein partners using a common interaction interface (C-terminus) may be explained based the unfolded conformation/ flexibility of its C-terminus, further structural characterization of the C-terminus of NEIL1 and structure-function studies are required to elucidate the mechanism in detail.
Unique Structural features of mammalian DNA glycosylases
Mammalian DNA glycosylases possess unique structural features compared their counterparts in lower organisms and bacteria. The major characteristic of mammalian glycosylases (which may also be true for other mammalian BER enzymes) is an unfolded extension or tail which participates in subcellular translocation, and more importantly in protein-protein interactions. The sequence alignment of hNTH1 with NTHs from E. coli, archea and other lower organisms shows that the human and other mammalian NTH1 has an N-terminal tail, absent in E. coli and archea. This N-terminal tail (residues 1–95) contains putative nuclear and mitochondrial localization signals [170]. hNTH1 was found to have low activity compared to E. coli Nth [49, 171]. This is because of the inhibitory role of N-terminal tail of hNTH1 on its turnover. Thus, the N-terminal tail of hNTH1 could regulate the overall catalytic turnover by reducing the rate of product release without affecting glycosylase or AP lyase activities [172].
The crystal structure of an enzymatically active deletion construct of hNEIL1 (lacking 56 C-terminal residues) and other modeling studies indicated that hNEIL1 has an unfolded C-terminal extension (~100 residues) which is absent in bacteria Nei [173]. The crystal structure of hNEIL1 also indicates the presence of internal Zn-less finger, unlike the C-terminal Zn finger present in other Nei orthologs. Our data show that NEIL1 interacts with downstream SN-BER proteins, via its unfolded C-terminal extension (residues 289-349) [115].
We used the software of Molecular Kinetics, Inc. [174] for prediction of prediction of naturally disordered regions (PONDR) to compare the secondary structures of hNEIL1 and hNTH1 with their E. coli prototypes Nei and Nth respectively (Fig. 3). Extended disordered regions are present only in the human proteins and their location at the N-terminus of hNTH1 and at C-terminus of hNEIL1 that are consistent with the experimental evidence about the absence of defined structure using X-ray crystallography. The prediction about the disordered structure in the C-terminal region of NEIL1 supports the hypothesis that the region individually interacting with multiple proteins is intrinsically disordered.
Figure 3.
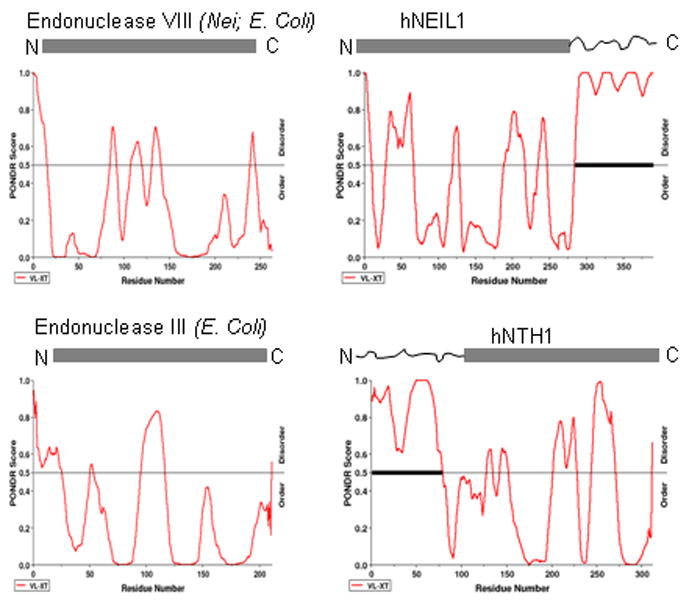
PONDR Plot of the predicted secondary structures of hNEIL1, hNTH1 and their E. coli prototypes, endonuclease VIII (Nei) and endonuclease III (Nth) respectively. The protein sequences were obtained from NCBI database. PONDR. score of 0.5 and higher indicates disordered structure [174]. The disordered C-terminal segment of hNEIL1 and N-terminal segment of hNTH1 are depicted by wiggly lines.
Figure 3 shows the PONDR©plot of the predicted secondary structure in hNEIL1 and hNTH1. The disordered structures of hNEIL1 at the C-terminus and of hNTH1 at the N-terminus are absent in their E. coli prototypes.
DNA glycosylases interact with non-BER proteins
Several other non-BER proteins have also been shown to be involved in BER, however their precise in vivo role in repair is yet to be understood. NEIL2 interacts with YB-1, a Y-box binding protein and it was suggested that YB-1 may be required for the fine-tuning of repair [161]. NTH1 was also shown to interact with and stimulated by YB-1 [175]. We have recently shown a stable interaction between NEIL1 and 9-1-1 complex and NEIL1’s stimulation as a result of this interaction [75]. 9-1-1 is a heterotrimeric protein complex and it acts as a sliding clamp similar to PCNA, but is activated by DNA damage and cell cycle checkpoints. NEIL1’s interaction with 9-1-1 highlights the linkage between NEIL1-initiated BER and damage signaling pathways. Our recent studies also showed that NEIL1’s stable association with PCNA [74]. It is highly likely that NEIL1’s interactome contains a large number of non BER proteins. Whether and how these proteins participate in BER poses an important topic for future investigation.
Covalent modification of DNA glycosylases and modulation of BER
Regulation of BER may be further fine tuned through posttranslational modifications of the BER proteins. Such modifications include phosphorylation, acetylation, mono- or poly-ADP-ribosylation, mono- and poly-ubiquitylation sumoylation and methylation among others [176]. Covalent modifications could have multiple physiological effects on proteins, including stability, interaction, organelle targeting, enzymatic activity etc. [177]. Such studies are still in an early stage. Some initial observations on modification of mammalian DNA glycosylases are summarized as follows.
Sumoylation of TDG
Sumoylation and ubiquitination alter activities of target proteins [178]. SUMO-1 and -3 sumoylate modify TDG, a glycosylase that removes uracil (or thymine) from its substrate U(T)•G pair [179]. TDG remains bound to the AP site after the excision of uracil and sumoylation reduces its affinity, thus releasing the product. The modified TDG is recycled by deconjugation, restoring its activity.
Acetylation of DNA glycosylases
Presence of multiple species of OGG1 in 2-D gel analysis of HeLa extracts suggested its covalent modification, which we showed to be acetylation [180]. We also showed that about 20% of OGG1 is acetylated in HeLa cells, and identified two acetylation acceptor lysine residues (Lys 338 and Lys 341). We further showed that acetylation enhances OGG1 turnover by decreasing its affinity for the product. We earlier observed that APE1 enhances OGG1 turnover which is higher when OGG1 is acetylated [39]. ROS induced an increase in the AcOGG1 level, indirectly by activation of p300. We proposed that acetylation of OGG1 provides a mechanism for rapid cellular response to handle enhanced oxidized base load induced by ROS [180].
Studies from our lab showed that NEIL2 is also acetylated, predominantly at Lys 49 and 153 [181]. Lys 49 is conserved among Fpg family members and its mutation inactivates the enzyme. Lys 49 acetylation strongly inhibits NEIL2 activity. We have proposed that acetylation at Lys 49 acts as a regulatory switch for NEIL2’s activity, Lys 153 acetylation could regulate its interactions.
It has been shown that TDG is acetylated both in vivo and in vitro by CBP/p300 which forms a functional complex with TDG [182]. Strong acetylation sites were identified at Lys residues 70, 94, 95 and 98. TDG acetylation does not affect binding and cleavage of G•T, G•U mispaired DNA but leads to release of CBP/p300 from the DNA bound complex and abrogates interaction with APE suggesting a role in suppression of APE-dependent repair by blocking the next reaction that follows the removal of the mispaired base. Thus acetylation appears to deregulate TDG-coupled DNA repair and contribute to the genomic instability commonly associated with cancer.
Summary and Future Perspectives
BER is distinct from the other excision repair pathways because the first step of lesion recognition by a DNA glycosylase is unique. This review on the early steps in base excision and strand break repair aims at highlighting the key roles of DNA glycosylases and APE1 (and other end-cleaning activities) in maintenance of genomic integrity in mammalian genomes. Furthermore, BER represents the predominant process for repairing endogenous DNA lesions mostly induced by ROS. While BER was earlier believed to be the simplest repair process, its complexities are now becoming increasingly evident. In this short review, we have touched upon a few of these complexities including interaction of BER enzymes with nonBER proteins, their covalent modifications, formation of pair-wise and multiprotein repair complexes and its regulation. Future research will undoubtedly be focused on these and other aspects of BER. For example, we have shown ROS-mediated activation of NEIL1 and APE1 [183, 184]. We have shown that ROS enhances NEIL2 complex formation [161]. ROS also enhances modification of OGG1 [180] and possibly other BER proteins. How such modifications affect repair efficiency in vivo should be addressed. Preliminary studies showing preferential repair of oxidized bases in replicating genomes need to be extended. The linkage of BER to susceptibility to various diseases also warrants extensive exploration. Finally, the mechanism recognition of BER substrates, which cause subtle deformation in DNA structure in chromatin, followed by their repair requiring access of the repair complex to the lesion needs to be explored. These challenges will keep the investigators busy for many years.
Acknowledgments
This article with a limited focus was not meant to provide comprehensive review of BER. Because of this and also space limitation, many appropriate references could not be included, for which the authors apologize. The authors’ research is supported by U.S. Public Health Services Research grants R01 CA81063, R01 CA53791, R01 CA102271 (TKH), P01 CA92584 and P30 ES06676. The authors thank the Mitra Lab Members for scientific discussion and Ms. Wanda Smith for expert secretarial assistance.
List of Abbreviations
- AP
abasic
- APE
AP-endonuclease
- BER
base excision repair
- dRP
deoxyribose phosphate
- 8-oxoG
8-oxo-7,8 dihydroguanine
- 5-OHU
5′ hydroxy uracil
- Fpg
formamidopyrimidine-DNA glycosylase
- Lig I
DNA ligase I
- Lig IIIα
DNA ligase IIIα
- LP-BER
long-patch BER
- MGMT
O6-methylguanine-DNA methyltransferase
- MPG
N-methylpurine-DNA glycosylase
- mtDNA
mitochondrial DNA
- MTS
mitochondrial targeting sequence
- Nfo
endonuclease IV
- Nei
endonuclease VIII
- NLS
localization signal
- Nth
endonuclease III
- O2•−
superoxide radical anion
- OGG1
8 -oxoguanine-DNA glycosylase
- OH•
hydroxy radical
- PARG
poly(ADP-ribose) glycol hydrolase
- PARP
poly(ADP-ribose) polymerase
- PNK
polynucleotide kinase
- Polγ
mitochondrial DNA polymerase gamma
- Polβ
DNA polymerase β
- PONDR
prediction of naturally disordered regions
- RA-BER
replication-associated BER
- RFC
replication factor-C
- ROS
reactive oxygen species
- RPA
replication protein A
- SN-BER
single nucleotide BER
- SNP
single nucleotide polymorphisms
- TDG
thymine-(T•G)-DNA glycosylase
- TLS
translesion synthesis
- Top1
topoisomerase 1
- 3′ PUA
3′ phosphoα,β-unsaturated aldehyde (4-hydroxylpentenal)
- TDP1
Tyrosyl phosphodiesterase 1
- UDG
uracil DNA glycosylase
- Xth
exonuclease III
References
- 1.Dawson TL, Gores GJ, Nieminen AL, Herman B, Lemasters JJ. Mitochondria as a source of reactive oxygen species during reductive stress in rat hepatocytes. Am J Physiol. 1993;264:C961–967. doi: 10.1152/ajpcell.1993.264.4.C961. [DOI] [PubMed] [Google Scholar]
- 2.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 4.Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 5.Wallace DJ. A reason for the rarity of male lupus. Lupus. 1998;7:60. doi: 10.1191/096120398678919697. [DOI] [PubMed] [Google Scholar]
- 6.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 7.Helbock HJ, Beckman KB, Ames BN. 8-Hydroxydeoxyguanosine and 8-hydroxyguanine as biomarkers of oxidative DNA damage. Methods Enzymol. 1999;300:156–166. doi: 10.1016/s0076-6879(99)00123-8. [DOI] [PubMed] [Google Scholar]
- 8.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 9.Michaels ML, Miller JH. The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine) J Bacteriol. 1992;174:6321–6325. doi: 10.1128/jb.174.20.6321-6325.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. 2. ASM Press; Washington, D.C: 2006. [Google Scholar]
- 11.Krokan HE, Nilsen H, Skorpen F, Otterlei M, Slupphaug G. Base excision repair of DNA in mammalian cells. FEBS Lett. 2000;476:73–77. doi: 10.1016/s0014-5793(00)01674-4. [DOI] [PubMed] [Google Scholar]
- 12.Papadopoulos N, Lindblom A. Molecular basis of HNPCC: mutations of MMR genes. Hum Mutat. 1997;10:89–99. doi: 10.1002/(SICI)1098-1004(1997)10:2<89::AID-HUMU1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 13.Breen AP, Murphy JA. Reactions of oxyl radicals with DNA. Free Rad Biol Med. 1995;18:1033–1077. doi: 10.1016/0891-5849(94)00209-3. [DOI] [PubMed] [Google Scholar]
- 14.Demple B, DeMott MS. Dynamics and diversions in base excision DNA repair of oxidized abasic lesions. Oncogene. 2002;21:8926–8934. doi: 10.1038/sj.onc.1206178. [DOI] [PubMed] [Google Scholar]
- 15.Mitra S, Hazra TK, Roy R, et al. Complexities of DNA base excision repair in mammalian cells. Mol cells. 1997;7:305–312. [PubMed] [Google Scholar]
- 16.Doetsch PW, Cunningham RP. The enzymology of apurinic/apyrimidinic endonucleases. Mutat Res. 1990;236:173–201. doi: 10.1016/0921-8777(90)90004-o. [DOI] [PubMed] [Google Scholar]
- 17.Matsumoto Y, Kim K. Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science. 1995;269:699–702. doi: 10.1126/science.7624801. [DOI] [PubMed] [Google Scholar]
- 18.Prasad R, Beard WA, Strauss PR, Wilson SH. Human DNA polymerase beta deoxyribose phosphate lyase. Substrate specificity and catalytic mechanism. J Biol Chem. 1998;273:15263–15270. doi: 10.1074/jbc.273.24.15263. [DOI] [PubMed] [Google Scholar]
- 19.Lindahl T. An N-glycosidase from Escherichia coli that releases free uracil from DNA containing deaminated cytosine residues. Proc Natl Acad Sci USA. 1974;71:3649–3653. doi: 10.1073/pnas.71.9.3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krokan HE, Drablos F, Slupphaug G. Uracil in DNA-occurrence, consequences and repair. Oncogene. 2002;21:8935–8948. doi: 10.1038/sj.onc.1205996. [DOI] [PubMed] [Google Scholar]
- 21.Slupphaug G, Mol CD, Kavli B, et al. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature. 1996;384:87–92. doi: 10.1038/384087a0. [DOI] [PubMed] [Google Scholar]
- 22.Parker JB, Bianchet MA, Krosky DJ, et al. Enzymatic capture of an extrahelical thymine in the search for uracil in DNA. Nature. 2007;449:433–437. doi: 10.1038/nature06131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masaoka A, Matsubara M, Hasegawa R, et al. Mammalian 5-formyluracil-DNA glycosylase. 2. Role of SMUG1 uracil-DNA glycosylase in repair of 5-formyluracil and other oxidized and deaminated base lesions. Biochemistry. 2003;42:5003–5012. doi: 10.1021/bi0273213. [DOI] [PubMed] [Google Scholar]
- 24.Wibley JE, Waters TR, Haushalter K, Verdine GL, Pearl LH. Structure and specificity of the vertebrate anti-mutator uracil-DNA glycosylase SMUG1. Mol Cell. 2003;11:1647–1659. doi: 10.1016/s1097-2765(03)00235-1. [DOI] [PubMed] [Google Scholar]
- 25.Hardeland U, Bentle M, Jiricny J, Schar P. The versatile thymine DNA-glycosylase: a comparative characterization of the human, Drosophila and fission yeast orthologs. Nucleic Acids Res. 2003;31:2261–2271. doi: 10.1093/nar/gkg344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitra S, Boldogh I, Izumi T, Hazra TK. Complexities of the DNA base excision repair pathway for repair of oxidative DNA damage. Environ Mol Mutagen. 2001;38:180–190. doi: 10.1002/em.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitra S. MGMT: a personal perspective. DNA Repair. 2007;6:1064–1070. doi: 10.1016/j.dnarep.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mitra S, Kaina B, Margison GP. MGMT: Reversal of Fortune - From Basic Biochemistry to Clinical Exploitation of the DNA Damage Reversal Protein, O6-Methylguanine-DNA Methyltransferase. DNA Repair. 2007;6:1063–1228. [Google Scholar]
- 29.Samson L, Cairns J. A new pathway for DNA repair in Escherichia coli. Nature. 1977;267:281–283. doi: 10.1038/267281a0. [DOI] [PubMed] [Google Scholar]
- 30.Sedgwick B, Bates PA, Paik J, Jacobs SC, Lindahl T. Repair of alkylated DNA: recent advances. DNA Repair. 2007;6:429–442. doi: 10.1016/j.dnarep.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 31.Mitra S, Kaina B. Regulation of repair of alkylation damage in mammalian genomes. Prog Nucleic Acids Res Mol Biol. 1993;44:109–142. doi: 10.1016/s0079-6603(08)60218-4. [DOI] [PubMed] [Google Scholar]
- 32.Roy R, Kennel SJ, Mitra S. Distinct substrate preference of human and mouse N-methylpurine-DNA glycosylases. Carcinogenesis. 1996;17:2177–2182. doi: 10.1093/carcin/17.10.2177. [DOI] [PubMed] [Google Scholar]
- 33.Lu AL, Chang DY. Repair of single base-pair transversion mismatches of Escherichia coli in vitro: correction of certain A/G mismatches is independent of dam methylation and host mutHLS gene functions. Genetics. 1988;118:593–600. doi: 10.1093/genetics/118.4.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nghiem Y, Cabrera M, Cupples CG, Miller JH. The mutY gene: a mutator locus in Escherichia coli that generates G.C-T. A transversions. Proc Natl Acad Sci USA. 1988;85:2709–2713. doi: 10.1073/pnas.85.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McGoldrick JP, Yeh YC, Solomon M, Essignmann JM, Lu AL. Characterization of a mammalian homolog of the E. coli MutY mismatch repair protein. Mol Cell Biol. 1995;15:989–996. doi: 10.1128/mcb.15.2.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohtsubo T, Nishioka K, Imaiso Y, et al. Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res. 2000;15:1355–1364. doi: 10.1093/nar/28.6.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCullough AK, Dodson ML, Lloyd RS. Initiation of base excision repair: glycosylase mechanisms and structures. Annu Rev Biochem. 1999;68:255–285. doi: 10.1146/annurev.biochem.68.1.255. [DOI] [PubMed] [Google Scholar]
- 38.Radicella JP, Dherin C, Desmaze C, Fox MS, Boiteux S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1997;94:8010–8015. doi: 10.1073/pnas.94.15.8010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hill JW, Hazra TK, Izumi T, Mitra S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001;29:430–438. doi: 10.1093/nar/29.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huffman JL, Sundheim O, Tainer JA. DNA base damage recognition and removal: new twists and grooves. Mutat Res. 2005;577:55–76. doi: 10.1016/j.mrfmmm.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 41.Zharkov DO, Shoham G, Grollman AP. Structural characterization of the Fpg family of DNA glycosylases. DNA Repair. 2003;2:839–862. doi: 10.1016/s1568-7864(03)00084-3. [DOI] [PubMed] [Google Scholar]
- 42.Guan Y, Manuel RC, Arvai AS, et al. MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nat Struct Biol. 1998;5:1058–1064. doi: 10.1038/4168. [DOI] [PubMed] [Google Scholar]
- 43.Bruner SD, Norman DP, Verdine GL. Structural basis recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature. 2000;403:859–866. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- 44.Hitomi K, Iwai S, Tainer JA. The intricate structural chemistry of base excision repair machinery: implications for DNA damage recognition, removal, and repair. DNA Repair. 2007;6:410–428. doi: 10.1016/j.dnarep.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 45.Sugahara M, Mikawa T, Kumasaka T, et al. Crystal structure of a repair enzyme of oxidatively damaged DNA, MutM (Fpg), from an extreme thermophile, Thermus thermophilus HB8. EMBO J. 2000;19:3857–3869. doi: 10.1093/emboj/19.15.3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gilboa R, Zharkov DO, Golan G, et al. Structure of formamidopyrimidine-DNA glycosylase covalently complexed to DNA. J Biol Chem. 2002;277:19811–19816. doi: 10.1074/jbc.M202058200. [DOI] [PubMed] [Google Scholar]
- 47.Zharkov DO, Golan G, Gilboa R, et al. Structural analysis of an Escherichia coli endonuclease VIII covalent reaction intermediate. EMBO J. 2002;21:789–800. doi: 10.1093/emboj/21.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aburatani H, Hippo Y, Ishida T, et al. Cloning and characterization of mammalian 8-hydroxyguanine-specific DNA glycosylase/apurinic, apyrimidinic lyase, a functional mutM homologue. Cancer Res. 1997;57:2151–2156. [PubMed] [Google Scholar]
- 49.Ikeda S, Biswas T, Roy R, et al. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. J Biol Chem. 1998;273:21585–21593. doi: 10.1074/jbc.273.34.21585. [DOI] [PubMed] [Google Scholar]
- 50.Hazra TK, Izumi T, Boldogh I, et al. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc Natl Acad Sci USA. 2002;99:3523–3528. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Das A, Wiederhold L, Leppard JB, et al. NEIL2-initiated, APE-independent repair of oxidized bases in DNA: Evidence for a repair complex in human cells. DNA Repair. 2006;5:1439–1448. doi: 10.1016/j.dnarep.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bandaru V, Sunkara S, Wallace SS, Bond JP. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair. 2002;1:517–529. doi: 10.1016/s1568-7864(02)00036-8. [DOI] [PubMed] [Google Scholar]
- 53.Hazra TK, Kow YW, Hatahet Z, et al. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J Biol Chem. 2002;277:30417–30420. doi: 10.1074/jbc.C200355200. [DOI] [PubMed] [Google Scholar]
- 54.Morland I, Rolseth V, Luna L, et al. Human DNA glycosylases of the bacterial Fpg/MutM superfamily: an alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res. 2002;30:4926–4936. doi: 10.1093/nar/gkf618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takao M, Kanno S, Kobayashi K, et al. A back-up glycosylase in Nth1 knock-out mice is a functional Nei (endonuclease VIII) homologue. J Biol Chem. 2002;277:42205–42213. doi: 10.1074/jbc.M206884200. [DOI] [PubMed] [Google Scholar]
- 56.Parsons JL, Zharkov DO, Dianov GL. NEIL1 excises 3′ end proximal oxidative DNA lesions resistant to cleavage by NTH1 and OGG1. Nucleic Acids Res. 2005;33:4849–4856. doi: 10.1093/nar/gki816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dou H, Mitra S, Hazra TK. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J Biol Chem. 2003;278:49679–49684. doi: 10.1074/jbc.M308658200. [DOI] [PubMed] [Google Scholar]
- 58.Alseth I, Eide L, Pirovano M, et al. The Saccharomyces cerevisiae homologues of endonuclease III from Escherichia coli, Ntg1 and Ntg2, are both required for efficient repair of spontaneous and induced oxidative DNA damage in yeast. Mol Cell Biol. 1999;19:3779–3787. doi: 10.1128/mcb.19.5.3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klungland A, Rosewell I, Hollenbach S, et al. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci USA. 1999;96:13300–13305. doi: 10.1073/pnas.96.23.13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Minowa O, Arai T, Hirano M, et al. Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc Natl Acad Sci USA. 2000;97:4156–4161. doi: 10.1073/pnas.050404497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Osterod M, Hollenbach S, Hengstler JG, et al. Age-related and tissue-specific accumulation of oxidative DNA base damage in 7,8-dihydro-8-oxoguanine-DNA glycosylase (Ogg1) deficient mice. Carcinogenesis. 2001;22:1459–1463. doi: 10.1093/carcin/22.9.1459. [DOI] [PubMed] [Google Scholar]
- 62.Arai T, Kelly VP, Minowa O, Noda T, Nishimura S. High accumulation of oxidative DNA damage, 8-hydroxyguanine, in Mmh/Ogg1 deficient mice by chronic oxidative stress. Carcinogenesis. 2002;23:2005–2010. doi: 10.1093/carcin/23.12.2005. [DOI] [PubMed] [Google Scholar]
- 63.Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science. 1994;265:103–106. doi: 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- 64.Xanthoudakis S, Smeyne RJ, Wallace JD, Curran T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc Natl Acad Sci USA. 1996;93:8919–8923. doi: 10.1073/pnas.93.17.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 66.Friedberg EC, Aguilera A, Gellert M, et al. DNA repair: from molecular mechanism to human disease. DNA Repair. 2006;5:986–996. doi: 10.1016/j.dnarep.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 67.Crew KD, Gammon MD, Terry MB, et al. Polymorphisms in nucleotide excision repair genes, polycyclic aromatic hydrocarbon-DNA adducts, and breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2007;16:2033–2041. doi: 10.1158/1055-9965.EPI-07-0096. [DOI] [PubMed] [Google Scholar]
- 68.Mitra S, Izumi T, Boldogh I, et al. Choreography of oxidative damage repair in mammalian genomes. Free Radic Biol Med. 2002;33:15–28. doi: 10.1016/s0891-5849(02)00819-5. [DOI] [PubMed] [Google Scholar]
- 69.Karahalil B, de Souza-Pinto NC, Parsons JL, Elder RH, Bohr VA. Compromised incision of oxidized pyrimidines in liver mitochondria of mice deficient in NTH1 and OGG1 glycosylases. J Biol Chem. 2003;278:33701–33707. doi: 10.1074/jbc.M301617200. [DOI] [PubMed] [Google Scholar]
- 70.Reardon JT, Bessho T, Kung HC, Bolton PH, Sancar A. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proc Natl Acad Sci USA. 1997;94:9463–9468. doi: 10.1073/pnas.94.17.9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hazra TK, Das A, Das S, et al. Oxidative DNA damage repair in mammalian cells: a new perspective. DNA Repair. 2007;6:470–480. doi: 10.1016/j.dnarep.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haug T, Skorpen F, Aas PA, et al. Regulation of expression of nuclear and mitochondrial forms of human uracil-DNA glycosylase. Nucleic Acids Res. 1998;26:1449–1457. doi: 10.1093/nar/26.6.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kavli B, Sundheim O, Akbari M, et al. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J Biol Chem. 2002;277:39926–39936. doi: 10.1074/jbc.M207107200. [DOI] [PubMed] [Google Scholar]
- 74.Dou H, Theriot CA, Das A, et al. Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen: the potential for replication-associated repair of oxidized bases in mammalian genomes. J Biol chem. 2007 doi: 10.1074/jbc.M709186200. In press. [DOI] [PubMed] [Google Scholar]
- 75.Guan X, Bai H, Shi G, et al. The human checkpoint sensor Rad9-Rad1-Hus1 interacts with and stimulates NEIL1 glycosylase. Nucleic Acids Res. 2007;35:2463–2472. doi: 10.1093/nar/gkm075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kubota Y, Nash RA, Klungland A, et al. Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase beta and the XRCC1 protein. EMBO J. 1996;15:6662–6670. [PMC free article] [PubMed] [Google Scholar]
- 77.Frosina G, Fortini P, Rossi O, et al. Two pathways for base excision repair in mammalian cells. J Biol Chem. 1996;271:9573–9578. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- 78.Matsumoto T, Yokote K, Tamura K, et al. Platelet-derived growth factor activates p38 mitogen-activated protein kinase through a Ras-dependent pathway that is important for actin reorganization and cell migration. J Biol Chem. 1999;274:13954–13960. doi: 10.1074/jbc.274.20.13954. [DOI] [PubMed] [Google Scholar]
- 79.Matsumoto Y. Molecular mechanism of PCNA-dependent base excision repair. Prog Prog Nucleic Acid Res Mol Biol. 2001;68:129–138. doi: 10.1016/s0079-6603(01)68095-4. [DOI] [PubMed] [Google Scholar]
- 80.Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN-1) EMBO J. 1997;16:3341–3348. doi: 10.1093/emboj/16.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Piersen CE, Prasad R, Wilson SH, Lloyd RS. Evidence for an imino intermediate in the DNA polymerase beta deoxyribose phosphate excision reaction. J Biol Chem. 1996;271:17811–17815. doi: 10.1074/jbc.271.30.17811. [DOI] [PubMed] [Google Scholar]
- 82.Levin DS, Vijayakumar S, Liu X, et al. A conserved interaction between the replicative clamp loader and DNA ligase in eukaryotes: implications for Okazaki fragment joining. J Biol Chem. 2004;279:55196–55201. doi: 10.1074/jbc.M409250200. [DOI] [PubMed] [Google Scholar]
- 83.Singh P, Zheng L, Chavez V, Qiu J, Shen B. Concerted action of exonuclease and Gap-dependent endonuclease activities of FEN-1 contributes to the resolution of triplet repeat sequences (CTG)n- and (GAA)n-derived secondary structures formed during maturation of Okazaki fragments. J Biol Chem. 2007;282:3465–3477. doi: 10.1074/jbc.M606582200. [DOI] [PubMed] [Google Scholar]
- 84.Levin DS, McKenna AE, Motycka TA, Matsumoto Y, Tomkinson AE. Interaction between PCNA and DNA ligase I is critical for joining of Okazaki fragments and long-patch base-excision repair. Curr Biol. 2000;10:919–922. doi: 10.1016/s0960-9822(00)00619-9. [DOI] [PubMed] [Google Scholar]
- 85.Ochs K, Sobol RW, Wilson SH, Kaina B. Cells deficient in DNA polymerase beta are hypersensitive to alkylating agent-induced apoptosis and chromosomal breakage. Cancer Res. 1999;59:1544–1551. [PubMed] [Google Scholar]
- 86.Liu Y, Kao HI, Bambara RA. Flap endonuclease 1: a central component of DNA metabolism. Annu Rev Biochem. 2004;73:589–615. doi: 10.1146/annurev.biochem.73.012803.092453. [DOI] [PubMed] [Google Scholar]
- 87.Haring M, Rudiger H, Demple B, Boiteux S, Epe B. Recognition of oxidized abasic sites by repair endonucleases. Nucleic Acids Res. 1994;22:2010–2015. doi: 10.1093/nar/22.11.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hanna M, Chow BL, Morey NJ, Jinks-Robertson S, Doetsch PW, Xiao W. Involvement of two endonuclease III homologs in the base excision repair pathway for the processing of DNA alkylation damage in Saccharomyces cerevisiae. DNA Repair. 2004;3:51–59. doi: 10.1016/j.dnarep.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 89.Demple B, Harrison L. Repair of oxidative damage to DNA: enzymology and biology. Annu Rev Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 90.Demple B, Johnson A, Fung D. Exonuclease III and endonuclease IV remove 3′ blocks from DNA synthesis primers in H2O2-damaged Escherichia coli. Proc Natl Acad Sci USA. 1986;83:7731–7735. doi: 10.1073/pnas.83.20.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ljungquist S. A new endonuclease from Escherichia coli acting at apurinic sites in DNA. J Biol Chem. 1977;252:2808–2814. [PubMed] [Google Scholar]
- 92.Cunningham RP, Saporito SM, Spitzer SG, Weiss B. Endonuclease IV (nfo) mutant of Escherichia coli. J Bacteriol. 1986;168:1120–1127. doi: 10.1128/jb.168.3.1120-1127.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Auerbach P, Bennett RA, Bailey EA, Krokan HE, Demple B. Mutagenic specificity of endogenously generated abasic sites in Saccharomyces cerevisiae chromosomal DNA. Proc Natl Acad Sci USA. 2005;102:17711–17716. doi: 10.1073/pnas.0504643102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang Y, Yuan F, Wu X, et al. Error-free and error-prone lesion bypass by human DNA polymerase kappa in vitro. Nucleic Acids Res. 2000;28:4138–4146. doi: 10.1093/nar/28.21.4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kokoska RJ, McCulloch SD, Kunkel TA. The efficiency and specificity of apurinic/apyrimidinic site bypass by human DNA polymerase eta and Sulfolobus solfataricus Dpo4. J Biol Chem. 2003;278:50537–50545. doi: 10.1074/jbc.M308515200. [DOI] [PubMed] [Google Scholar]
- 96.Smith CE, Llorente B, Symington LS. Template switching during break-induced replication. Nature. 2007;447:102–105. doi: 10.1038/nature05723. [DOI] [PubMed] [Google Scholar]
- 97.Selby CP, Sancar A. Structure and function of the (A)BC excinuclease of Escherichia coli. Mutat Res. 1990;236:203–211. doi: 10.1016/0921-8777(90)90005-p. [DOI] [PubMed] [Google Scholar]
- 98.Popoff SC, Spira AI, Johnson AW, Demple B. Yeast structural gene (APN1) for the major apurinic endonuclease: homology to Escherichia coli endonuclease IV. Proc Natl Acad Sci USA. 1990;87:4193–4197. doi: 10.1073/pnas.87.11.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Johnson RE, Torres-Ramos CA, Izumi T, et al. Identification of APN2, the Saccharomyces cerevisiae homolog of the major human AP endonuclease HAP1, and its role in the repair of abasic sites. Genes Dev. 1998;12:3137–3143. doi: 10.1101/gad.12.19.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guillet M, Boiteux S. Endogenous DNA abasic sites cause cell death in the absence of Apn1, Apn2 and Rad1/Rad10 in Saccharomyces cerevisiae. EMBO J. 2002;21:2833–2841. doi: 10.1093/emboj/21.11.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gorman MA, Morera S, Rothwell DG, et al. The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J. 1997;16:6548–6558. doi: 10.1093/emboj/16.21.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 103.Oezguen N, Schein CH, Peddi SR, et al. A “moving metal mechanism” for substrate cleavage by the DNA repair endonuclease APE-1. Proteins. 2007;68:313–323. doi: 10.1002/prot.21397. [DOI] [PubMed] [Google Scholar]
- 104.Ribar B, Izumi T, Mitra S. The major role of human AP-endonuclease homolog Apn2 in repair of abasic sites in Schizosaccharomyces pombe. Nucleic Acids Res. 2004;32:115–126. doi: 10.1093/nar/gkh151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ivanov I, Tainer JA, McCammon JA. Unraveling the three-metal-ion catalytic mechanism of the DNA repair enzyme endonuclease IV. Proc Natl Acad Sci USA. 2007;104:1465–1470. doi: 10.1073/pnas.0603468104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Demple B, Herman T, Chen DS. Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: definition of a family of DNA repair enzymes. Proc Natl Acad Sci USA. 1991;88:11450–11454. doi: 10.1073/pnas.88.24.11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hadi MZ, Wilson DM., 3rd Second human protein with homology to the Escherichia coli abasic endonuclease exonuclease III. Environ Mol Mutagen. 2000;36:312–324. [PubMed] [Google Scholar]
- 108.Tsuchimoto D, Sakai Y, Sakumi K, et al. Human APE2 protein is mostly localized in the nuclei and to some extent in the mitochondria, while nuclear APE2 is partly associated with proliferating cell nuclear antigen. Nucleic Acids Res. 2001;29:2349–2360. doi: 10.1093/nar/29.11.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Izumi T, Mitra S. Deletion analysis of human AP-endonuclease: minimum sequence required for the endonuclease activity. Carcinogenesis. 1998;19:525–527. doi: 10.1093/carcin/19.3.525. [DOI] [PubMed] [Google Scholar]
- 110.Jackson EB, Theriot CA, Chattopadhyay R, Mitra S, Izumi T. Analysis of nuclear transport signals in the human apurinic/apyrimidinic endonuclease (APE1/Ref1) Nucleic Acids Res. 2005;33:3303–3312. doi: 10.1093/nar/gki641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kelley MR, Parsons SH. Redox regulation of the DNA repair function of the human AP endonuclease Ape1/ref-1. Antioxid Redox Signal. 2001;3:671–683. doi: 10.1089/15230860152543014. [DOI] [PubMed] [Google Scholar]
- 112.Izumi T, Wiederhold LR, Roy G, et al. Mammalian DNA base excision repair proteins: their interactions and role in repair of oxidative DNA damage. Toxicology. 2003;193:43–65. doi: 10.1016/s0300-483x(03)00289-0. [DOI] [PubMed] [Google Scholar]
- 113.Bhakat KK, Izumi T, Yang SH, Hazra TK, Mitra S. Role of acetylated human AP-endonuclease (APE1/Ref-1) in regulation of the parathyroid hormone gene. EMBO J. 2003;22:6299–6309. doi: 10.1093/emboj/cdg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Marenstein DR, Wilson DM, 3rd, Teebor GW. Human AP endonuclease (APE1) demonstrates endonucleolytic activity against AP sites in single-stranded DNA. DNA Repair. 2004;3:527–533. doi: 10.1016/j.dnarep.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 115.Wiederhold L, Leppard JB, Kedar P, et al. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 116.Wong D, DeMott MS, Demple B. Modulation of the 3′ → 5′-exonuclease activity of human apurinic endonuclease (Ape1) by its 5′-incised Abasic DNA product. J Biol Chem. 2003;278:36242–36249. doi: 10.1074/jbc.M306065200. [DOI] [PubMed] [Google Scholar]
- 117.Meira LB, Devaraj S, Kisby GE, et al. Heterozygosity for the mouse Apex gene results in phenotypes associated with oxidative stress. Cancer Res. 2001;61:5552–5557. [PubMed] [Google Scholar]
- 118.Sugo N, Aratani Y, Nagashima Y, Kubota Y, Koyama H. Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase beta. EMBO J. 2000;19:1397–1404. doi: 10.1093/emboj/19.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sobol RW, Horton JK, Kuhn R, et al. Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- 120.Izumi T, Brown DB, Naidu CV, et al. Two essential but distinct functions of the mammalian abasic endonuclease. Proc Natl Acad Sci USA. 2005;102:5739–5743. doi: 10.1073/pnas.0500986102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fung H, Demple B. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol Cell. 2005;17:463–470. doi: 10.1016/j.molcel.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 122.Evans AR, Limp-Foster M, Kelley MR. Going APE over ref-1. Mutat Res. 2000;461:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 123.Izumi T, Hazra TK, Boldogh I, et al. Requirement for human AP endonuclease 1 for repair of 3′-blocking damage at DNA single-strand breaks induced by reactive oxygen species. Carcinogenesis. 2000;21:1329–1334. [PubMed] [Google Scholar]
- 124.Mitra S, Izumi T, Boldogh I, et al. Intracellular trafficking and regulation of mammalian AP-endonuclease 1 (APE1), an essential DNA repair protein. DNA Repair. 2007;6:461–469. doi: 10.1016/j.dnarep.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 125.Meijer M, Karimi-Busheri F, Huang TY, Weinfeld M, Young D. Pnk1, a DNA kinase/phosphatase required for normal response to DNA damage by gamma-radiation or camptothecin in Schizosaccharomyces pombe. J Biol Chem. 2002;277:4050–4055. doi: 10.1074/jbc.M109383200. [DOI] [PubMed] [Google Scholar]
- 126.Whitehouse CJ, Taylor RM, Thistlethwaite A, et al. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001;104:107–117. doi: 10.1016/s0092-8674(01)00195-7. [DOI] [PubMed] [Google Scholar]
- 127.Mokkapati SK, Wiederhold L, Hazra TK, Mitra S. Stimulation of DNA glycosylase activity of OGG1 by NEIL1: functional collaboration between two human DNA glycosylases. Biochemistry. 2004;43:11596–11604. doi: 10.1021/bi049097i. [DOI] [PubMed] [Google Scholar]
- 128.Caldecott KW. Mammalian single-strand break repair: mechanisms and links with chromatin. DNA Repair. 2007;6:443–453. doi: 10.1016/j.dnarep.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 129.Vance JR, Wilson TE. Yeast Tdp1 and Rad1-Rad10 function as redundant pathways for repairing Top1 replicative damage. Proc Natl Acad Sci USA. 2002;99:13669–13674. doi: 10.1073/pnas.202242599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pouliot JJ, Robertson CA, Nash HA. Pathways for repair of topoisomerase I covalent complexes in Saccharomyces cerevisiae. Genes Cells. 2001;6:677–687. doi: 10.1046/j.1365-2443.2001.00452.x. [DOI] [PubMed] [Google Scholar]
- 131.El-Khamisy SF, Saifi GM, Weinfeld M, et al. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature. 2005;434:108–113. doi: 10.1038/nature03314. [DOI] [PubMed] [Google Scholar]
- 132.Yang SW, Burgin AB, Jr, Huizenga BN, et al. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc Natl Acad Sci USA. 1996;93:11534–11539. doi: 10.1073/pnas.93.21.11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pouliot JJ, Yao KC, Robertson CA, Nash HA. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science. 1999;286:552–555. doi: 10.1126/science.286.5439.552. [DOI] [PubMed] [Google Scholar]
- 134.Guzder SN, Torres-Ramos C, Johnson RE, et al. Requirement of yeast Rad1-Rad10 nuclease for the removal of 3′-blocked termini from DNA strand breaks induced by reactive oxygen species. Genes Dev. 2004;18:2283–2291. doi: 10.1101/gad.1232804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ahel I, Rass U, El-Khamisy SF, et al. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006;443:713–716. doi: 10.1038/nature05164. [DOI] [PubMed] [Google Scholar]
- 136.Brenner C. Hint, Fhit, and GalT: function, structure, evolution, and mechanism of three branches of the histidine triad superfamily of nucleotide hydrolases and transferases. Biochemistry. 2002;41:9003–9014. doi: 10.1021/bi025942q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rass U, Ahel I, West SC. Actions of aprataxin in multiple DNA repair pathways. J Biol Chem. 2007;282:9469–9474. doi: 10.1074/jbc.M611489200. [DOI] [PubMed] [Google Scholar]
- 138.Sano Y, Date H, Igarashi S, et al. Aprataxin, the causative protein for EAOH is a nuclear protein with a potential role as a DNA repair protein. Ann Neurol. 2004;55:241–249. doi: 10.1002/ana.10808. [DOI] [PubMed] [Google Scholar]
- 139.Kanno S, Kuzuoka H, Sasao S, et al. A novel human AP endonuclease with conserved zinc-finger-like motifs involved in DNA strand break responses. EMBO J. 2007;26:2094–2103. doi: 10.1038/sj.emboj.7601663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mazur DJ, Perrino FW. Structure and expression of the TREX1 and TREX2 3′ → 5′ exonuclease genes. J Biol Chem. 2001;276:14718–14727. doi: 10.1074/jbc.M010051200. [DOI] [PubMed] [Google Scholar]
- 141.Hoss M, Robins P, Naven TJ, et al. A human DNA editing enzyme homologous to the Escherichia coli DnaQ/MutD protein. EMBO J. 1999;18:3868–3875. doi: 10.1093/emboj/18.13.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Crow YJ, Hayward BE, Parmar R, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 2006;38:917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 143.Vidal AE, Boiteux S, Hickson ID, Radicella JP. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. EMBO J. 2001;20:6530–6539. doi: 10.1093/emboj/20.22.6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lan L, Nakajima S, Oohata Y, et al. In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc Natl Acad Sci USA. 2004;101:13738–13743. doi: 10.1073/pnas.0406048101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Caldecott KW, McKeown CK, Tucker JD, Ljungquist S, Thompson LH. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol Cell Biol. 1994;14:68–76. doi: 10.1128/mcb.14.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fan J, Otterlei M, Wong HK, Tomkinson AE, Wilson DM., 3rd XRCC1 co-localizes and physically interacts with PCNA. Nucleic Acids Res. 2004;32:2193–2201. doi: 10.1093/nar/gkh556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wong HK, Kim D, Hogue BA, McNeill DR, Wilson DM., 3rd DNA damage levels and biochemical repair capacities associated with XRCC1 deficiency. Biochemistry. 2005;44:14335–14343. doi: 10.1021/bi051161o. [DOI] [PubMed] [Google Scholar]
- 148.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 149.Stuart JA, Hashiguchi K, Wilson DM, et al. DNA base excision repair activities and pathway function in mitochondrial and cellular lysates from cells lacking mitochondrial DNA. Nucleic Acids Res. 2004;32:2181–2192. doi: 10.1093/nar/gkh533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chattopadhyay R, Wiederhold L, Szczesny B, et al. Identification and characterization of mitochondrial abasic (AP)-endonuclease in mammalian cells. Nucleic Acids Res. 2006;34:2067–2076. doi: 10.1093/nar/gkl177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Driggers WJ, LeDoux SP, Wilson GL. Repair of oxidative damage within the mitochondrial DNA of RINr 38 cells. J Biol Chem. 1993;268:22042–22045. [PubMed] [Google Scholar]
- 152.Dianov GL, Souza-Pinto N, Nyaga SG, et al. Base excision repair in nuclear and mitochondrial DNA. Prog Nucleic Acids Res Mol Biol. 2001;68:285–297. doi: 10.1016/s0079-6603(01)68107-8. [DOI] [PubMed] [Google Scholar]
- 153.LeDoux SP, Wilson GL. Base excision repair of mitochondrial DNA damage in mammalian cells. Prog Nucleic Acid Res Mol Biol. 2001;68:273–284. doi: 10.1016/s0079-6603(01)68106-6. [DOI] [PubMed] [Google Scholar]
- 154.Pinz KG, Bogenhagen DF. Efficient repair of abasic sites in DNA by mitochondrial enzymes. Mol Cell Biol. 1998;18:1257–1265. doi: 10.1128/mcb.18.3.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kaguni LS. DNA polymerase gamma, the mitochondrial replicase. Annu Rev Biochem. 2004;73:293–320. doi: 10.1146/annurev.biochem.72.121801.161455. [DOI] [PubMed] [Google Scholar]
- 156.Tomkinson AE, Mackey ZB. Structure and function of mammalian DNA ligases. Mutat Res. 1998;407:1–9. doi: 10.1016/s0921-8777(97)00050-5. [DOI] [PubMed] [Google Scholar]
- 157.Nishioka K, Ohtsubo T, Oda H, et al. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol Biol Cell. 1999;10:1637–1652. doi: 10.1091/mbc.10.5.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pfanner N. Protein sorting: recognizing mitochondrial presequences. Curr Biol. 2000;10:R412–415. doi: 10.1016/s0960-9822(00)00507-8. [DOI] [PubMed] [Google Scholar]
- 159.Diekert K, Kispal G, Guiard B, Lill R. An internal targeting signal directing proteins into the mitochondrial intermembrane space. Proc Natl Acad Sci USA. 1999;96:11752–11757. doi: 10.1073/pnas.96.21.11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Andersen JS, Lam YW, Leung AK, et al. Nucleolar proteome dynamics. Nature. 2005;433:77–83. doi: 10.1038/nature03207. [DOI] [PubMed] [Google Scholar]
- 161.Das S, Chattopadhyay R, Bhakat KK, et al. Stimulation of NEIL2-mediated oxidized base excision repair via YB-1 interaction during oxidative stress. J Biol Chem. 2007;282:28474–28484. doi: 10.1074/jbc.M704672200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Marsin S, Vidal AE, Sossou M, et al. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. J Biol Chem. 2003;278:44068–44074. doi: 10.1074/jbc.M306160200. [DOI] [PubMed] [Google Scholar]
- 163.Parlanti E, Locatelli G, Maga G, Dogliotti E. Human base excision repair complex is physically associated to DNA replication and cell cycle regulatory proteins. Nucleic Acids Res. 2007;35:1569–1577. doi: 10.1093/nar/gkl1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Nagelhus TA, Haug T, Singh KK, et al. A sequence in the N-terminal region of human uracil-DNA glycosylase with homology to XPA interacts with the C-terminal part of the 34-kDa subunit of replication protein A. J Biol Chem. 1997;272:6561–6566. doi: 10.1074/jbc.272.10.6561. [DOI] [PubMed] [Google Scholar]
- 165.Otterlei M, Warbrick E, Nagelhus TA, et al. Post-replicative base excision repair in replication foci. EMBO J. 1999;18:3834–3844. doi: 10.1093/emboj/18.13.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Parker A, Gu Y, Mahoney W, et al. Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long patch DNA base excision repair. J Biol Chem. 2001;276:5547–5555. doi: 10.1074/jbc.M008463200. [DOI] [PubMed] [Google Scholar]
- 167.Das A, Boldogh I, Lee JW, et al. The human Werner syndrome protein stimulates repair of oxidative DNA base damage by the DNA glycosylase NEIL1. J Biol Chem. 2007;282:26591–26602. doi: 10.1074/jbc.M703343200. [DOI] [PubMed] [Google Scholar]
- 168.Boldogh I, Milligan D, Lee MS, et al. hMYH cell cycle-dependent expression, subcellular localization and association with replication foci: evidence suggesting replication-coupled repair of adenine:8-oxoguanine mispairs. Nucleic Acids Res. 2001;29:2802–2809. doi: 10.1093/nar/29.13.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wilson SH, Kunkel TA. Passing the baton in base excision repair. Nat Struct Biol. 2000;7:176–178. doi: 10.1038/73260. [DOI] [PubMed] [Google Scholar]
- 170.Sarker AH, Ikeda S, Nakano H, et al. Cloning and characterization of a mouse homologue (mNthl1) of Escherichia coli endonuclease III. J Mol Biol. 1998;282:761–774. doi: 10.1006/jmbi.1998.2042. [DOI] [PubMed] [Google Scholar]
- 171.Thayer MM, Ahern H, Xing D, Cunningham RP, Tainer JA. Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. EMBO J. 1995;14:4108–4120. doi: 10.1002/j.1460-2075.1995.tb00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Liu X, Roy R. Truncation of amino-terminal tail stimulates activity of human endonuclease III (hNTH1) J Mol Biol. 2002;321:265–276. doi: 10.1016/s0022-2836(02)00623-x. [DOI] [PubMed] [Google Scholar]
- 173.Doublie S, Bandaru V, Bond JP, Wallace SS. The crystal structure of human endonuclease VIII-like 1 (NEIL1) reveals a zincless finger motif required for glycosylase activity. Proc Natl Acad Sci USA. 2004;101:10284–10289. doi: 10.1073/pnas.0402051101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Li X, Romero P, Rani M, Dunker AK, Obradovic Z. Predicting Protein Disorder for N-, C-, and Internal Regions. Genome Inform Ser Workshop Genome Inform. 1999;10:30–40. [PubMed] [Google Scholar]
- 175.Marenstein DR, Ocampo MT, Chan MK, et al. Stimulation of human endonuclease III by Y box-binding protein 1 (DNA-binding protein B). Interaction between a base excision repair enzyme and a transcription factor. J Biol Chem. 2001;276:21242–21249. doi: 10.1074/jbc.M101594200. [DOI] [PubMed] [Google Scholar]
- 176.Krueger KE, Srivastava S. Posttranslational protein modifications: current implications for cancer detection, prevention, and therapeutics. Mol Cell Proteomics. 2006;5:1799–1810. doi: 10.1074/mcp.R600009-MCP200. [DOI] [PubMed] [Google Scholar]
- 177.Seet BT, Dikic I, Zhou MM, Pawson T. Reading protein modifications with interaction domains. Nat Rev. 2006;7:473–483. doi: 10.1038/nrm1960. [DOI] [PubMed] [Google Scholar]
- 178.Muller S, Hoege C, Pyrowolakis G, Jentsch S. SUMO, ubiquitin’s mysterious cousin. Nat Rev. 2001;2:202–210. doi: 10.1038/35056591. [DOI] [PubMed] [Google Scholar]
- 179.Hardeland U, Steinacher R, Jiricny J, Schar P. Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J. 2002;21:1456–1464. doi: 10.1093/emboj/21.6.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bhakat KK, Mokkapati SK, Boldogh I, Hazra TK, Mitra S. Acetylation of human 8-oxoguanine-DNA glycosylase by p300 and its role in 8-oxoguanine repair in vivo. Mol Cell Biol. 2006;26:1654–1665. doi: 10.1128/MCB.26.5.1654-1665.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bhakat KK, Hazra TK, Mitra S. Acetylation of the human DNA glycosylase NEIL2 and inhibition of its activity. Nucleic Acids Res. 2004;32:3033–3039. doi: 10.1093/nar/gkh632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Tini M, Benecke A, Um SJ, Torchia J, Evans RM, Chambon P. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol Cell. 2002;9:265–277. doi: 10.1016/s1097-2765(02)00453-7. [DOI] [PubMed] [Google Scholar]
- 183.Ramana CV, Boldogh I, Izumi T, Mitra S. Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proc Natl Acad Sci USA. 1998;95:5061–5066. doi: 10.1073/pnas.95.9.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Das A, Hazra TK, Boldogh I, Mitra S, Bhakat KK. Induction of the human oxidized base-specific DNA glycosylase NEIL1 by reactive oxygen species. J Biol Chem. 2005;280:35272–35280. doi: 10.1074/jbc.M505526200. [DOI] [PubMed] [Google Scholar]