

Abstract
Theiler's murine encephalomyelitis virus (TMEV) induces two distinct cell death programs, necrosis and apoptosis. The apoptotic pathway is of particular interest because TMEV persists in the central nervous system of mice, largely in infiltrating macrophages, which undergo apoptosis. Infection of murine macrophages in culture induces apoptosis that is Bax dependent through the intrinsic or mitochondrial pathway, restricting infectious-virus yields and raising the possibility that apoptosis represents a mechanism to attenuate TMEV yet promote macrophage-to-macrophage spread during persistent infection. To help define the cellular stressors and upstream signaling events leading to apoptosis during TMEV infection, we screened baby hamster kidney (BHK-21) cells transfected to express individual nonstructural genes (except 3B) of the low-neurovirulence BeAn virus strain for cell death. Only expression of the leader protein led to apoptosis, as assessed by fluorescence-activated cell sorting analysis of propidium iodide- and annexin V-stained transfected cells, immunoblot analysis of poly(ADP-ribose) polymerase and caspase cleavages, electron microscopy, and inhibition of apoptosis by the pancaspase inhibitor qVD-OPh. After transfection, Bak and not Bax expression increased, suggesting that the apical pathway leading to activation of these Bcl-2 multi-BH-domain proapoptotic proteins differs in BeAn virus infection versus L transfection. Mutation to remove the CHCC Zn finger motif from L, a motif required by L to mediate inhibition of nucleocytoplasmic trafficking, significantly reduced L-protein-induced apoptosis in both BHK-21 and M1-D macrophages.
Theiler's murine encephalomyelitis viruses (TMEV), members of the genus Cardiovirus in the family Picornaviridae, are highly cytolytic RNA viruses. Mice experimentally infected with a low-neurovirulence TMEV, such as BeAn virus, develop persistent infection in the central nervous system (CNS) and an inflammatory demyelinating disease, providing an experimental analogue for multiple sclerosis. BeAn virus persists primarily in macrophages in the CNS of infected mice. Schlitt et al. (34) found that 74% of TUNEL-positive cells in infected spinal cords (primarily in CNS lesions) were T and B lymphocytes and 8% were macrophages, although virus genomes were detected in <1% of apoptotic cells, consistent with infection of only a low percentage of macrophages and the fact that TMEV does not infect T or B lymphocytes in culture. Thus, some means other than direct infection was responsible for apoptosis of most CNS macrophages, including TMEV triggering apoptosis through tumor necrosis factor alpha or tumor necrosis factor alpha-related apoptosis-inducing ligand by binding death receptors on activated macrophages in vitro, as reported elsewhere (17).
Infection of mouse macrophages induces apoptosis (16, 26) mediated by Bax through the intrinsic or mitochondrial pathway and severely restricts the yield of progeny virus (37). Thus, apoptosis may be a mechanism to attenuate the virus yet promote macrophage-to-macrophage spread through phagocytosis of infected apoptotic blebs during persistence (37). In contrast, TMEV infection in other rodent cells tested thus far, including baby hamster kidney (BHK-21) cells, produces necrotic cell death with high virus yields. The contrasting outcomes of TMEV infection point to the existence of two distinct virus-induced cell death programs.
The genes of an increasing number of RNA viruses have been shown to encode proteins that trigger apoptosis. Among picornaviruses, coxsackievirus B3 1B (VP2) (12, 13), avian encephalomyocarditis virus 1C (VP3) (24) and 2C (25), enterovirus 71 2A (20), and poliovirus 2A (10) and 3C protease (3Cpro) (3) induce apoptosis, mostly through the intrinsic pathway. Coxsackievirus B3 VP2 has been shown to interact with the proapoptotic Siva protein in a yeast two-hybrid screen (12), but exactly how the VP2-Siva interaction or any of the other picornavirus proteins initiates the apoptotic cascade remains unknown.
To gain insight into the upstream signaling events that lead to apoptosis, we tested the ability of individual BeAn virus nonstructural genes to induce apoptosis in uninfected BHK-21 cells. Only the leader (L) protein resulted in apoptosis and mutation of the CHCC Zn finger motif in L significantly reduced L protein-induced apoptosis.
MATERIALS AND METHODS
Cells and viruses.
BHK-21 cells were grown in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (FBS), 7.5% tryptose phosphate, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in 5% CO2. Cells of the immature myelomonocytic cell line M1, derived from the SL mouse strain, were induced to differentiate into macrophages with supernatants from L929 and P388D1 cells as described previously (18). The origin and passage history of the BeAn virus stock have been described (32); generation of BeAn virus with mutation in the L-protein Zn finger domain is described below. Virus titers of clarified lysates of infected cells were determined by standard plaque assay in BHK-21 cells (32).
Virus infections.
After virus adsorption at a multiplicity of infection of 10 for 45 min at 24°C, cell monolayers were washed twice with phosphate-buffered saline (PBS) containing 1 mM CaCl2 and 0.5 mM MgCl2 and incubated in Dulbecco's modified Eagle medium containing 1% FBS at 37°C for various times. The end of the adsorption period was designated as time zero.
Reagents.
The following reagents and antibodies were purchased commercially: the pancaspase inhibitor qVD-OPh and rabbit anti-caspase 8 (R&D Systems, Minneapolis, MN); mouse anti-caspase 9, rabbit anti-caspase 3, rabbit anti-poly(ADP-ribose) polymerase (PARP), rabbit anti-Bak, rabbit anti-Flag M2, and rabbit antiactin antibodies (Cell Signaling Technology, Beverly, MA); mouse anti-Bax (Santa Cruz Biotechnology Inc., Santa Cruz, CA); goat anti-mouse immunoglobulin G (IgG)-horseradish peroxidase and goat anti-rabbit IgG-horseradish peroxidase (BD Pharmingen, San Diego, CA); anti-rabbit IgG-Alexa Fluor 568 and puromycin (Invitrogen, Carlsbad, CA); enhanced chemiluminescence solution (Amersham, Piscataway, NJ); cell-permeative proteasome inhibitor MG132 (Calbiochem, Darmstadt, Germany); and actinomycin D (Sigma, St. Louis, MO).
Cell viability assay.
WST-1 reagent (Roche Applied Science, Indianapolis, IN), a tetrazolium salt, was added to the medium of monolayer cultures in 96-well microtiter plates at the indicated times, and plates were incubated for 1 to 2 h at 37°C in 5% CO2. Cell viability was determined based on the absorbance of samples at 420 nm (reference wavelength, 610 nm) measured with a Vmax kinetic microplate reader (Molecular Devices, Sunnyvale, CA), indicating cleavage of the tetrazolium salt to formazan against background and medium-alone controls. Cell death values were calculated as the ratio of BeAn virus-infected to mock-infected cultures.
Fluorescence-activated cell sorting (FACS).
At various times posttransfection, cell monolayers were washed with PBS and detached with trypsin, and 5 × 105 cells were stained with propidium iodide (PI) and annexin V-fluorescein isothiocyanate (Calbiochem, Darmstadt, Germany) according to the manufacturer's instructions. Incubation of cell monolayers with puromycin (2 μg/ml) and actinomycin D (1 μg/ml) provided positive controls for apoptosis. For each sample, 10,000 events were analyzed using a Beckman Coulter Epics Elite ESP and WinMDI 2.8 software.
Plasmid construction.
Overlap extension PCR (29) was used to assemble internal ribosome entry site (IRES) constructs expressing a BeAn virus nonstructural protein(s) by joining the 3′ end of the BeAn virus IRES to nucleotides encoding each of seven nonstructural proteins (2A, 2B, 2C, 3A, 3C, and 3D and the precursor 3CD) with an AUG codon at the N terminus and a Flag epitope at the C terminus (Fig. 1). The IRES and each virus protein were amplified from a full-length infectious BeAn virus clone (19) using the primer pairs 1 plus 3 and 2 plus 4, respectively (see Fig. S1 and Table S1 in the supplemental material). The two PCR products were gel purified, mixed in equimolar amounts, and PCR amplified with the primer pair 1 plus 2. The resulting products were digested with NheI and NotI and ligated into the pIRES vector, replacing the encephalomyocarditis (EMCV) IRES with that of BeAn virus. For assembly of the pIRES-L plasmid, the IRES and L were amplified from a full-length infectious BeAn virus clone (19) using the primer pair 1 and 2; for the pL plasmid, L was amplified with primers 1B and 2 (see Table S1 in the supplemental material). The resulting PCR products were digested with NheI and NotI, gel purified, and ligated into pIRES digested with the same restriction enzymes. All recombinant plasmids were recovered from transformed Escherichia coli, and the nucleotide sequences of the inserts were confirmed by dideoxynucleotide sequencing.
FIG. 1.

(A) TMEV final gene products arranged on the polyprotein in a standard picornavirus format of the L protein: four P1 capsid proteins and three P2 and four P3 nonstructural proteins. The 18-kDa out-of-frame L* protein, which is initiated 13 nucleotides downstream of the authentic polyprotein AUG, is shown below the polyprotein. (B) All of the BeAn virus nonstructural proteins except the 20-amino-acid 3B (VPg) were assembled using overlap extension PCR into the pIRES vector downstream of the BeAn virus IRES so that each protein is translated from a 5′ AUG codon and contains a C-terminal Flag sequence. The L protein was also assembled into the same vector without an IRES for cap-dependent translation.
Leader protein mutagenesis.
Another overlap extension PCR-based method (14) was used to introduce nucleotide changes into the sequence of the L-protein CHCC Zn finger motif in a plasmid containing the full-length wild-type BeAn virus cDNA in pGEM4 (Promega, Madison, WI). The mutagenic primers were as follows: reverse, 5′-ACGGCTGTGCGAATAGTGCGCACATCTGGG-3′, and forward, 5′-CAGATGTGCGCACTATTCCACAGCCGTTG-3′. The outside forward primer containing an NruI site was 5′-CCATCGCGACGTGGTTGGAGAT-3′ and the reverse primer containing a SalI site was 5′-AGAGTCGACCAACAGTAGATT-3′. Final PCR products were separated by electrophoresis, gel purified, and cloned into NruI and SalI restriction sites in the BeAn virus full-length plasmid.
Transfection.
Subconfluent monolayers of BHK-21 cells in 35-mm six-well plates were transfected with reaction mixtures consisting of 2 to 3 μg of DNA of an expression vector and Lipofectamine 2000 (Invitrogen) at a 2.5:1 ratio of reagent to DNA. Complexes incubated for 20 min at 24°C were added to cell monolayers for 4 h at 37°C, the growth medium was replaced, and incubation was continued for 24 to 48 h at 37°C in 5% CO2. For M1-D cell transfection, 2 × 106 cells were washed and suspended in 100 μl nucleofection solution (Amaxa, Gaithersburg, MD), 2 μg of pmaxGFP or pIRES construct DNA was added, and samples were transferred into certified cuvettes and transfected with the Amaxa Nucleofector using the program D-23. M1-D transfection medium (RPMI, 1% FBS, 0.1 mM nonessential amino acids) with 1% FBS (500 μl) warmed to 37°C was added immediately after transfection to each cuvette, and cells were collected and dispensed into wells of 12-well plates containing 1 ml prewarmed M1-D transfection medium. Plates were incubated at 37°C in 5% CO2 for the indicated times and assessed for cell viability in the presence of WST-1 or for apoptosis by flow cytometry.
Preparative in vitro RNA transcription.
DNA clones oriented with the 5′ end of the IRES or L nucleotide sequence downstream of the T7 promoter (Fig. 1) were linearized at the NotI site within the vector downstream of the 3′ end of the BeAn virus nonstructural genes, and RNA transcripts were synthesized using the RiboMax(R) large-scale RNA production system (Promega). Reaction mixtures contained 2 mM spermidine, 80 mM HEPES-KOH (pH 7.5), 24 mM MgCl2, 7.5 mM rNTPs, 40 mM dithiothreitol, and 5 μl of enzyme mix (T7 RNA polymerase, recombinant RNasin and recombinant inorganic pyrophosphatase; Promega) in 100 μl. Incubations were for 4 h at 37°C, and RNAs were analyzed for integrity and quantity on 1% native RNA agarose gels with or without prior DNase treatment.
In vitro translation.
In vitro translation in rabbit reticulocyte lysate (RRL) (Red Nova lysate kit; Novagen, Madison, WI) was carried out in 12.5-μl reaction mixtures after salt and RNA concentrations had been optimized. Mixtures containing 20 mM HEPES-KOH (pH 7.5), 0.1 mM KCl, 0.1 mM MgSO4, 2 mM dithiothreitol, 8 mM creatine phosphate, a methionine-free amino acid mixture, and 20 μg/ml RNA in the presence of 10 μCi [35S]methionine and [35S]cysteine (Tran35S-label; ICN Biomedicals, Inc., Irvine, CA) were incubated for 1 h at 30°C. Incorporation of [35S]methionine was calculated from trichloroacetic acid precipitates, and samples were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 12% NuPAGE bis-Tris minigels (Invitrogen), which were dried and exposed to X-ray film (Denville Scientific Inc., Metuchen, NJ).
Statistical analysis.
A paired Student's t test was used to compare groups, and differences were considered significant at P < 0.05.
RESULTS
Translation of virus nonstructural proteins in RRL.
Because genes of RNA viruses may be subject to RNA splicing when expressed from a polymerase II promoter in mammalian cells, the products of BeAn virus nonstructural gene constructs were first authenticated by translation in RRL. In vitro transcription of the pIRES constructs containing individual BeAn virus nonstructural genes and the 3CD precursor gene generated RNAs of the expected sizes upon electrophoresis in agarose gels (data not shown). SDS-PAGE analysis of RNAs translated in RRL revealed [35S]methionine-labeled cognate proteins for L, 2A, 2B, 2C, and 3A (data not shown) as well as 3D polymerase (3Dpol) and 3CD but not the BeAn virus 3Cpro (Fig. 2). The latter result was not unexpected in light of the reported rapid degradation in RRL of the related cardiovirus EMCV 3Cpro protein (22). The presence of a 10-amino-acid sequence, LLVRGRTLVV, near the EMCV 3Cpro N terminus, which is recognized as a signal for ubiquitin/26S proteasomal degradation (21), and a homologous degradation sequence, LLLRAHLFVV, in BeAn virus 3Cpro would likely render 3Cpro unstable. Translation of BeAn virus 3Cpro in RRL in the presence of 100 μM lactacystin, a proteasome inhibitor that binds to a 20S subunit, revealed a faint 3Cpro protein band on SDS-PAGE (data not shown). In contrast, translation of BeAn virus 3CD in RRL revealed proteins bands for 3CD, 3Dpol, and 3Cpro on SDS-PAGE (Fig. 2). It has been reported that the presence of EMCV 3Dpol in the precursor 3CD masks the 3Cpro signal for ubiquitination, contributing to greater 3Cpro stability in RRL (22).
FIG. 2.
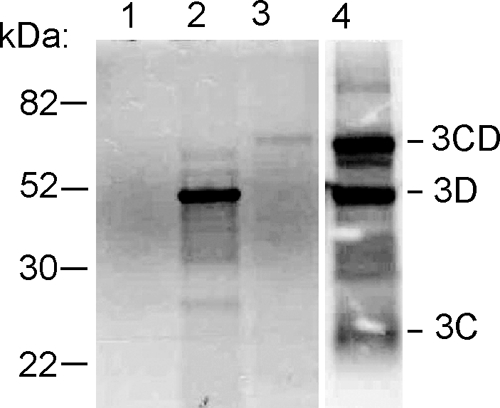
SDS-PAGE analysis of in vitro-translated RNAs transcribed from pIRES-3C, -3D, and -3CD plasmids in RRL in the presence of [35S]methionine. Translation of pIRES-3Cpro reveals no apparent labeling of 3Cpro (lane 1); pIRES-3Dpol shows labeling of the 53-kDa 3Dpro (lane 2); pIRES-3CD shows labeling of the 65-kDa 3CD (lane 3); and when pIRES 3CD is overexposed, labeling of 3CD, 3Dpro, and 23-kDa 3Cpro is shown (lane −4).
Expression of virus nonstructural proteins in BHK-21 cells.
The pIRES nonstructural protein plasmids (Fig. 1B) were transfected into BHK-21 cells with Lipofectamine, and lysates were prepared at 24 and 48 h posttransfection, electrophoresed on 12% SDS-polyacrylamide gels, transferred to membranes, and immunoblotted with rabbit polyclonal anti-Flag antibody to detect the Flag sequences. Transfection efficiency in these experiments was >80%, as determined by FACS analysis of cells cotransfected with pIRES-GFP and pIRES nonstructural protein plasmids. As shown in Fig. 3A, immunoblotting revealed expression of BeAn virus L, 2A, 2B, 2C, 3A, and 3D. BeAn virus L was expressed at 24 h, but expression had decreased at 48 h, whereas expression of 2A, 2B, 2C, and 3A at 24 h was maintained or increased at 48 h. BeAn virus 3Cpro was undetectable by immunoblotting, presumably because 3Cpro undergoes proteasomal degradation in vivo, as noted above for EMCV 3Cpro (33). Indeed, 3Cpro accumulated to detectable levels at 12 to 24 h after transfection of pIRES-3Cpro in the presence of 20 μM of the reversible proteasome inhibitor z-Leu-Leu-Leu CHO (MG132) (Fig. 3B), suggesting that 3Cpro is highly susceptible to in vivo degradation by the ubiquitin/26S proteasome. The decline in 3Cpro band intensity likely reflects progressive cell death. The BeAn virus precursor 3CD was not detected after transfection of pIRES-3CD, even in the presence of MG132, for reasons presently unresolved.
FIG. 3.

SDS-PAGE of the expression of pIRES-BeAn virus nonstructural gene constructs in transiently transfected BHK-12 cells. (A) Immunoblot analysis at 24 h (even lanes) and 48 h (odd lanes) posttransfection. Lanes: 1, empty vector; 2 and 3, L; 4 and 5, 2A; 6 and 7, 2B; 8 and 9, 2C; 10 and 11, 3A; 12 and 13, 3D polymerase. (B) Time course of the expression of 3Cpro in the presence of 20 μM of the reversible 26S proteasome inhibitor MG132. MW, molecular weight (in thousands).
Expression of L but not other nonstructural proteins in BHK-21 cells is cytotoxic.
We used BHK-21 cells transfected with individual BeAn virus nonstructural genes (except 3B) to screen for cytotoxicity with the intent of testing constructs positive for apoptosis in M1-D cells. Transfection of BeAn virus pIRES-L but not pIRES constructs for 2A, 2B, 2C, 3A, and 3Dpol produced significant cell death at 24 h (P < 0.5) and 48 h (P < 0.01) compared to transfection of pIRES-empty vector (Fig. 4A). Efforts to assess cell death by 3Cpro in the presence of MG132 were complicated by MG132-dependent cytotoxicity (39). Cytopathic effect was observed by light microscopy as early as 12 to 16 h of incubation with MG132 alone, and cell death beyond that induced by MG132 alone was detected in the WST-1 cell survival assay from 24 to 48 h after transfection of 3Cpro in the presence of MG132 (P < 0.05), although the amount of cell death exceeding that of MG132 was not impressive. Thus, BeAn virus 3Cpro in the absence of 26S proteasomal degradation appears to be cytotoxic, as reported for poliovirus 3Cpro (3).
FIG. 4.

Cytotoxicity determined by the WST-1 assay at 24 and 48 h after transfection of plasmid BeAn virus nonstructural gene constructs in BHK-21 cells. *, P < 0.05; **, P < 0.01. (A) Cell death after IRES-dependent expression of virus nonstructural genes (mean ± standard error; n = 3 to 6). (B) Cell death after cap-dependent expression of L (mean ± standard error; n = 3). (C) Comparison of cytotoxicity after cap-dependent expression of L and the L CHCC mutant (mean ± standard error; n = 3). (D) Time course of cap-dependent expression of L up to 20 h posttransfection.
Transfection of BHK-21 cells with the pL plasmid construct (Fig. 1B), in which L expression is cap dependent, also led to progressive cytotoxicity at 24 h (P < 0.05) and 48 h (P < 0.01) compared to transfection with pEV (Fig. 4B). Temporal analysis of the expression of L by immunoblotting first revealed L expression at 12 h, with increased expression at 16 to 20 h posttransfection as cell death commenced (Fig. 4D).
A BeAn virus L Zn finger mutant is not cytotoxic.
Inhibition of active nucleocytoplasmic trafficking by cardiovirus L was initially recognized as inhibition of innate immunity by blocking the type I interferon response (11, 38, 43). Mutation of the TMEV L CHCC Zn finger motif compromises the antinucleocytoplasmic trafficking activity of L (6, 23). To determine the role of the L Zn finger in cell death, we mutated the domain to RHRC without changing the amino acid sequence of the L* protein encoded by an alternative overlapping open reading frame (Fig. 1A), as initially reported for the TMEV DA strain (38). The DA L* protein has been reported to be antiapoptotic (9, 15). As shown in Fig. 5, transfection of a plasmid with a green fluorescent protein nuclear localization signal (pGFPNLS) followed 24 h later by transfection of pL resulted in efflux of GFP from the nucleus (Fig. 5A), whereas GFP accumulated and remained in the nucleus after transfection of only pGFPNLS (Fig. 5B) or after transfection of the pL mutant (Fig. 5C), indicating that the mutated L protein no longer disrupted the nuclear pore complex. Expression of L was also detected throughout the cytoplasm of transfected BHK-21 cells immunostained with anti-Flag antibody (Fig. 5D and E), but in some cells L was perinuclear. The BeAn virus pL mutant construct in transfected cells was not cytotoxic (Fig. 4C).
FIG. 5.

Digital confocal microscopic images of nuclei of cells loaded with the marker protein GFP (GFPNLS) (A to C) and Flag-tagged BeAn virus L (D and E) in BHK-21 cells 24 h posttransfection. Cells transfected with pL showed efflux of GFP from the nucleus to the cytoplasm (A), whereas cells transfected with pEV (B) and the pL mutant plasmid DNA (C) retained GFP in the nucleus. Indirect immunofluorescent antibody staining of L in the cytoplasm of BHK-21 cells 24 h after pL transfection showed DAPI-stained nuclei (D) and L stained with rabbit anti-Flag M2 and anti-rabbit IgG-Alexa Fluor 568 and nuclei stained with DAPI (E). Bars = 10 μm.
A portion of BeAn virus L cytotoxicity is due to apoptosis.
BHK-21 cells transfected with the pL and pL Zn finger mutant plasmids were assessed for apoptosis by FACS analysis of PI- and annexin V-stained cells at 20 h. In a representative experiment whose results are shown in Fig. 6A, the percent PI-negative, annexin V-positive cells (lower right quadrant) was elevated after pL compared to pEV transfection. PI-positive, annexin V-positive cells (upper right quadrant) are considered necrotic because of the potential exposure of phosphoserine on the inner leaflet of membrane fragments during necrosis. Use of only cells remaining on the monolayer for FACS (Fig. 6A) explains the marked difference in the percent necrotic cells compared to the WST-1 assay (Fig. 4A and 5B). In cells transfected with the pL mutant, the percent apoptotic cells was the same as that after pEV transfection, indicating that a functional Zn finger is necessary for L-induced apoptosis. The percent apoptotic cells for pEV, pL, and the pL mutant was 2.6 ± 0.62, 13.9 ± 2.74, and 5.9 ± 2.15 (mean ± standard deviation for five experiments; P < 0.001 for differences between pEV and pL and between pL and the pL mutant). Immunoblot analysis revealed cleavage of PARP and caspases 3 and 9 to respective 17- and 37-kDa activated forms at 16 to 24 h after pL transfection (Fig. 6B). The less intense bands of the activated forms of caspases 3 and 9 after transfection of pEV and the pL mutant were probably due to the stress of transfection. The band reflecting cleavage of caspase 8 to its active 18-kDa form was inconsistently seen (Fig. 6B). Incubation of pL-transfected cells with the pan-caspase inhibitor qVD-OPh protected against apoptosis (Fig. 6C) and prevented cleavage of PARP and caspase 3 (Fig. 6D). Electron microscopic examination of cells at 24 h after transfection of pL revealed necrotic debris, probably as the result of transfection and normal-appearing cells and some cells undergoing apoptosis (Fig. 6E, top), whereas only normal-appearing cells were observed after transfection with the pL mutant (Fig. 6E, bottom). Together, these results indicate that the transfected L gene induced apoptosis through the intrinsic apoptotic pathway and that removal of the Zn finger significantly reduced L-induced apoptosis.
FIG. 6.

Effect of pL and the pL Zn finger mutant in transfected BHK-21 cells. (A) FACS analysis of PI- and annexin V-stained cells at 24 h reveals increased numbers of apoptotic cells (right lower quadrant) after pL transfection but not after transfection with the pL mutant. (B) Immunoblots revealing PARP cleavage and caspase 3 and caspase 9 cleavage to their active forms, p17 and p37, respectively, but no caspase 8 cleavage to the active p18 form. (C) Effect of treatment of pL-transfected cells with the pancaspase inhibitor qVD-OPh, showing a significant reduction (P < 0.01) in cell death compared to treatment with PBS at 24 and 48 h. (D) Immunoblot of pL-transfected cells treated with qVD-OPh, revealing a lack of PARP and caspase 3 cleavages at 20 to 24 h. (E) Electron photomicrographs of pL (top)- and pEV (bottom)-transfected cells at 24 h showing an apoptotic cell with condensed nuclear chromatin, two cells in the early stages of apoptosis, one with loss of marginated nuclear chromatin, and necrotic cell fragments. Cells in the lower frame appear normal except for ruffling of the cell membrane. The photomicrographs are at the same magnification. Bar = 5 μm.
In BeAn virus infection of M1-D cells, intrinsic apoptosis is mediated by Bcl-2 multidomain proapoptotic Bax and not Bak (37). However, expression of Bak was increased at 16 to 24 h, suggesting that Bak is activated, whereas levels of Bax decreased at 20 to 24 h during L-induced apoptosis in BHK-21 cells (Fig. 7). Although the apical pathway leading to activation of these Bcl-1 multi-BH-domain proapoptotic proteins, which permeabilize the outer mitochondrial membrane, appears to differ in BeAn virus infection versus L transfection, this could merely reflect a difference in cell type.
FIG. 7.
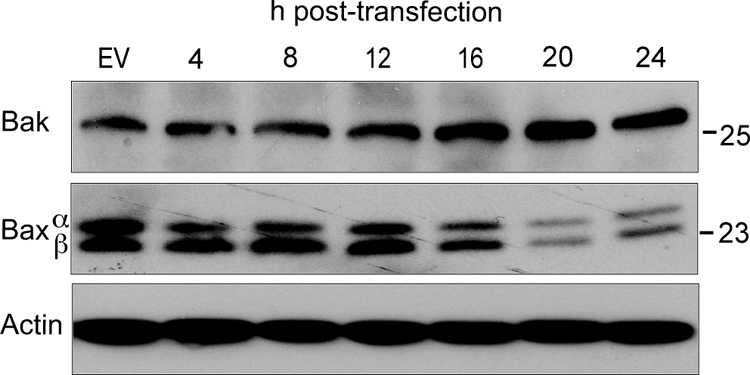
Bak and Bax expression in BHK-21 cells transfected with pL. The immunoblot reveals increased Bak expression at 12 to 24 h but decreased Bax over the same period.
BeAn virus L induces apoptosis in macrophages.
TMEV persists in the CNS of mice, primarily in macrophages, and infected macrophages provide an in vitro model for BeAn virus-induced apoptosis (37). FACS analysis of PI- and annexin V-stained M1-D macrophages electroporated with pL or the pL mutant revealed an increased percent apoptotic cells only in the pL-transfected cells 20 h (Fig. 8A). The percent apoptotic cells for pEV, pL, and the pL mutant was 12.06 ± 5.29, 34.7 ± 13.41, and 13.38 ± 3.28 (mean ± SD for three experiments; P < 0.01 for differences between pEV and pL and between pL and the pL mutant). At this time, PARP cleavage was observed on immunoblots (Fig. 8B), whereas cleavage of caspases 9 and 3 to their active forms was not observed, possibly due to a lower transient-transfection efficiency (∼55%) of this macrophage cell line. Nonetheless, these results suggest that L induces apoptosis in M1-D cells through a pathway that also requires an intact Zn finger.
FIG. 8.

Effect of pL and the pL Zn finger mutant in transfected M1-D macrophages. (A) FACS analysis of PI- and annexin V-stained cells at 24 h revealed increased numbers of apoptotic cells (right lower quadrant) after pL transfection but not after transfection of the pL mutant. (B) Immunoblot revealing PARP cleavage at 24 h.
DISCUSSION
Our analysis of the ability of the TMEV nonstructural genes except that encoding 3B to induce apoptosis in transiently transfected BHK-21 cells showed that only L, which was first detected on immunoblots at 12 h posttransfection, induced progressive cell death between 24 and 48 h. Apoptosis occurred through the intrinsic apoptotic pathway, as demonstrated by FACS analysis of PI- and annexin V-stained transfected cells, immunoblot analysis of PARP and caspase cleavages, and the protection against apoptosis by the pancaspase inhibitor qVD-OPh.
The cardioviruses EMCV and TMEV regulate nuclear trafficking of cellular proteins and RNA through the L protein, which contains a Zn finger motif near the N terminus, an acidic domain with potential Thr and Tyr phosphorylation sites, and, only in TMEV, a C-terminal Ser/Thr domain (7, 43). Unlike the L of aphthoviruses, the cardiovirus L does not have a proteolytic function. Recently, studies have shown that cardiovirus L binds Ran-GTPase and phosphorylates nucleoporins, leading to disruption of the nuclear pore complex that spans the nuclear envelope and the Ran gradient (27, 28, 30). The inhibition of nuclear import allows measurement of the efflux of nuclear proteins into the cytoplasm after loading the nucleus with a marker protein (pGFPNLS). We detected efflux of GFP from the nucleus to the cytoplasm after transfection of BeAn virus pL, confirming the functionality of its Zn finger, but not after transfection of the pL Zn finger mutant.
L-induced apoptosis was also dependent on a functional Zn finger, since mutation to remove the Zn finger abrogated apoptosis. Apoptosis following expression of L was associated with activation of caspases 9 and 3 but not caspase 8, indicating engagement of the intrinsic apoptotic pathway; however, a role for the extrinsic pathway has not been totally excluded. The increased expression of proapoptotic Bak but not Bax after transfection suggests differences between L-induced apoptosis in BHK-21 cells and BeAn virus infection in M1-D cells, where apoptosis is Bax dependent (37), or inherent differences in the apoptotic machinery in the two cell types. In L-induced apoptosis, the exact signaling events upstream of Bak, which is held in check by Bcl-2 antiapoptotic family proteins, remain to be determined.
Although inducible expression of poliovirus 2A and 3Cpro leads to apoptosis in mammalian cells (3, 10), we found that their TMEV counterparts did not induce progressive cytotoxicity. For cardiovirus 2A, this finding might stem from its lack of protease activity, in contrast to that of poliovirus 2A. Regarding TMEV 3Cpro, the mature form has a 26S proteasomal degradation signal near the N terminus which is not present in poliovirus 3Cpro, so that any 3Cpro diffusing away from replication complexes would likely be rapidly degraded and unavailable for induction of apoptosis. Finally, while the avian encephalomyocarditis virus 2C has been shown to be apoptotic (25), BeAn virus 2C did not induce progressive cytotoxicity (Fig. 4A).
Preliminary studies in our laboratory indicate that BeAn virus infection in M1-D cells leads to phosphorylation of p53 at Ser15. In turn, Noxa, a Bcl-2 family BH3-only proapoptotic protein that binds to Mcl-1, a Bcl-2 antiapoptotic family member, is transcriptionally activated, releasing Bax, which then permeabilizes the outer mitochondrial membrane to activate caspase 9 (unpublished data). There is evidence that mitogen-activated protein kinases (MAPK) are involved in EMCV L-induced phosphorylation of nucleoporins (A. C. Palmenberg, personal communication); therefore, it is conceivable that p53, a downstream target of MAPK, might be activated by L. In addition, since poliovirus 3Cpro cleaves transcription factors (TFIIIC, TFIID, Oct-1, and CREB) to inhibit cellular RNA synthesis (5, 36, 40-42), it is reasonable to consider whether TMEV 3Cpro may also activate p53. 3D proteins cardioviruses as well as other picornaviruses contain a basic NLS near the N terminus, enabling 3Cpro to enter the nucleus in the form of its precursors 3BCD and 3CD, which then generates 3Cpro by autoproteolysis (1, 2, 35). Moreover, Lawson et al. (22) showed in in vitro translation reactions that the EMCV 3Cpro proteasomal ubiquitination signal is masked in 3CD rendering 3Cpro stable. Our RRL experiments indicated the analogous finding. However, we did not observe 3CD expression in BHK-21 cells transfected with our pIRES-3CD construct, possibly because of RNA splicing, although 3Cpro and 3D individually were not subject to RNA splicing. Expression of BeAn virus 3CD in the cytoplasm of BHK-21 cells stably expressing T7 RNA polymerase might help enable 3CD expression and elucidate a potential role of 3CD in inducing apoptosis (4). Expression of 3Cpro in the presence of MG132 as shown here indicates that BeAn virus 3Cpro is cytotoxic. Therefore, 3CD in addition to L are potential inducers of apoptosis during TMEV infection.
Finally, in contrast to L-induced apoptosis, BeAn virus infection of BHK-21 cells leads primarily to necrosis, with approximately only ∼20% of cell undergoing apoptosis (8; also unpublished data), the apparent default cell death pathway from infection of this mammalian cell line by TMEV. Romanova et al. (31) have shown that the cell death pathway in poliovirus infection varies in different cell lines and that a rhabdomyosarcoma cell line did not have the “necessary machinery” to undergo apoptosis. BHK-21 cells are able to undergo apoptosis based on both nonviral (puromycin and actinomycin D) and viral stimuli in this study. Clearly, a balance between pro- and antiapoptotic virus genes determines the outcome of apoptosis. Since the TMEV out-of-frame L* protein has been reported to be antiapoptotic (9, 15), it is possible that apoptosis is held in check by L* during infection in BHK-21 cells. Insight into the potential antiapoptotic role of other BeAn virus nonstructural proteins in addition to L* awaits analysis in transfected M1-D cells.
Supplementary Material
Acknowledgments
We thank Patricia Kallio for technical assistance and Robert P. Becker for electron microscopy experiments.
This work was supported by NIH grant NS23349, the Modestus Bauer Foundation, and the Wershkoff Multiple Sclerosis Research Fund.
The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
Footnotes
Published ahead of print on 29 April 2009.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Aminev, A. G., S. P. Amineva, and A. C. Palmenberg. 2003. Encephalomyocarditis virus (EMCV) proteins 2A and 2BCD localize to nuclei and inhibit cellular mRNA transcription but not rRNA transcription. Virus Res. 9559-73. [DOI] [PubMed] [Google Scholar]
- 2.Amineva, S. P., A. G. Aminev, A. C. Palmenberg, and J. E. Gern. 2004. Rhinovirus 3C protease precursors 3CD and 3CD' localize to the nuclei of infected cells. J. Gen. Virol. 852969-2979. [DOI] [PubMed] [Google Scholar]
- 3.Barco, A., E. Feduchi, and L. Carrasco. 2000. Poliovirus protease 3Cpro kills cells by apoptosis. Virology 266352-360. [DOI] [PubMed] [Google Scholar]
- 4.Buchholz, U. J., S. Finke, and K.-K. Conzelman. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73251-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das, S., and A. Dasgupta. 1993. Identification of the cleavage site and determinants required for poliovirus 3Cpro-catalyzed cleavage of human TBP. J. Virol. 673326-3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delhaye, S., V. van Pesch, and T. Michiels. 2004. The leader protein of Theiler's virus interferes with nucleocytoplasmic trafficking of cellular proteins. J. Virol. 784357-4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dvorak, C. M., D. J. Hall, M. Riddle, A. Pranter, J. Dillman, M. Deibel, and A. C. Palmenberg. 2001. Leader protein of encephalomyocarditis virus binds zinc, is phosphorylated during viral infection, and affects the efficiency of genome translation. Virology 290261-271. [DOI] [PubMed] [Google Scholar]
- 8.Friedmann, A., and H. L. Lipton. 1980. Replication of GDVII and DA strains of Theiler's murine encephalomyelitis virus in BHK 21 cells: An electron microscopic study. Virology 101389-398. [DOI] [PubMed] [Google Scholar]
- 9.Ghadge, G. D., L. Ma, S. Sato, J. Kim, and R. P. Roos. 1998. A protein critical for a Theiler's virus-induced immune system-mediated demyelinating disease has a cell type-specific antiapoptotic effect and a key role in virus persistence. J. Virol. 728605-8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldstaub, D., A. Gradi, Z. Bercovitch, Z. Grosmann, Y. Nophar, S. Luria, N. Sonenberg, and C. Kahana. 2000. Poliovirus 2A protease induces cell death. Mol. Cell. Biol. 201271-1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hato, S. V., C. Ricour, B. M. Schulte, K. H. Lanke, M. deBruijni, J. Zoll, W. J. G. Melchers, T. Michiels, and F. J. M. van Kuppeveld. 2007. The mengovirus leader protein blocks interferon a/b gene transcription and inhibits activation of interferon regulatory factor 3. Cell. Microbiol. 92921-2930. [DOI] [PubMed] [Google Scholar]
- 12.Henke, A., H. Launhardt, K. Klement, A. Stelzner, R. Zell, and T. Munder. 2000. Apoptosis in Coxsackie B3-caused diseases: interaction between the capsid protein VP2 and the proapoptotic protein Siva. J. Virol. 744284-4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henke, A., M. Nestler, S. Strunze, H. P. Saluz, P. Hortschansky, B. Menzel, U. Martin, R. Zell, A. Stelzner, and T. Munder. 2001. The apoptotic capability of Coxsackievirus B3 is influenced by the efficient interaction between the capsid protein VP2 and the proapoptotic host protein Siva. Virology 28915-22. [DOI] [PubMed] [Google Scholar]
- 14.Higuchi, R., B. Krummel, and R. K. Saiki. 1988. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 167351-7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Himeda, T., Y. Ohara, K. Asahura, Y. Kontani, and M. Sawada. 2005. A lentiviral expression system demonstrates that L* protein of Theiler's murine encephalomyelitis virus (TMEV) has an anti-apoptotic effect in a macrophage cell line. Virus Res. 38201-207. [DOI] [PubMed] [Google Scholar]
- 16.Jelachich, M. L., P. Bandyopadhyay, K. Blum, and H. L. Lipton. 1995. Theiler's virus growth in murine macrophage cell lines depends on the state of differentiation. Virology 209437-444. [DOI] [PubMed] [Google Scholar]
- 17.Jelachich, M. L., and H. L. Lipton. 2001. Theiler's murine encephalomyelitis virus induces apoptosis in gamma interferon activated M1 differentiated myelomonocytic cells through a mechanism involving tumor necrosis factor alpha (TNF-α) and TNF-α-related apoptosis-inducing ligand. J. Virol. 755930-5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jelachich, M. L., and H. L. Lipton. 1996. Theiler's murine encephalomyelitis virus kills restrictive but not permissive cells by apoptosis. J. Virol. 706856-6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar, A. S., P. Kallio, M. Luo, and H. L. Lipton. 2003. Amino acid substitutions of low neurovirulence VP2 residues contacting sialic acid dramatically reduce viral binding and spread. J. Virol. 772709-2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuo, R.-L., S.-H. Kung, Y.-Y. Hsu, and W.-T. Liu. 2002. Infection of enterovirus 71 or expression of its 2A protease induces apoptotic cell death. J. Gen. Virol. 831367-1376. [DOI] [PubMed] [Google Scholar]
- 21.Lawson, T. G., D. L. Gronros, P. E. Evans, M. C. Bastien, K. M. Michalewich, J. K. Clark, J. H. Edmonds, K. H. Graber, J. A. Werner, B. A. Lurvey, and J. M. Cate. 1999. Identification and characterization of a protein destruction signal in the Encephalomyocarditis virus 3C protease. J. Biol. Chem. 2749871-9880. [PubMed] [Google Scholar]
- 22.Lawson, T. G., D. L. Gronros, J. A. Werner, A. C. Wey, A. M. DiGeorge, J. L. Lockhart, J. W. Wilson, and P. L. Wintrode. 1994. The Encephalomyocarditis virus 3C protease is a substrate for the ubiquitin-mediated proteolytic system. J. Biol. Chem. 26928429-28435. [PubMed] [Google Scholar]
- 23.Lidsky, P. V., S. Hato, M. V. Bardina, A. G. Aminev, A. C. Palmenberg, E. V. Sheval, V. Y. Polyakov, F. J. M. van Kuppeveld, and V. I. Agol. 2006. Nucleocytoplasmic traffic disorder induced by cardioviruses. J. Virol. 802705-2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu, J., T. Wei, and J. Kwang. 2002. Avian encephalomyelitis virus induces apoptosis via major structural VP3. Virology 30039-49. [DOI] [PubMed] [Google Scholar]
- 25.Liu, J., T. Wei, and J. Kwang. 2004. Avian encephalomyelitis virus nonstructural protein 2C induces apoptosis by activating cytochrome c/caspase-9 pathway. Virology 318169-182. [DOI] [PubMed] [Google Scholar]
- 26.Obuchi, M., Y. Ohara, T. Takegami, T. Murayama, H. Takada, and H. Iizuka. 1997. Theiler's murine encephalomyelitis virus subgroup strain-specific infection in a murine macrophage-like cell line. J. Virol. 71729-733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Porter, F. W., Y. A. Bochkov, A. J. Albee, C. Wiese, and A. C. Palmenberg. 2006. A picornavirus protein interacts with Ran-GTPase and disrupts nucleocytoplasmic transport. Proc. Natl. Acad. Sci. USA 103. [DOI] [PMC free article] [PubMed]
- 28.Porter, F. W., and A. C. Palmenberg. 2009. Leader-induced phosphorylation of nucleoporins correlated with nuclear trafficking by cardioviruses. J. Virol. 831941-1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qi, D., and K. B. Scholthof. 2008. A one-step PCR-based method for rapid and efficient site-directed fragment deletion, insertion and substitution mutagenesis. J. Virol. Methods 14985-90. [DOI] [PubMed] [Google Scholar]
- 30.Ricour, C., S. Delhaye, S. V. Hato, T. Olenyik, B. Michel, F. J. M. van Kuppeveld, K. E. Gustin, and T. Michiels. 2009. Inhibition of mRNA export and dimerization of interferon regulatory factor 3 by Theiler's virus leader protein. J. Gen. Virol. 90177-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romanova, L. I., G. A. Belov, P. V. Lidsky, E. A. Tokskaya, M. S. Kolesnikova, A. G. Evstafieva, A. B. Vartapetian, D. Egger, K. Bienz, and V. I. Agol. 2005. Variability in apoptotic response to poliovirus infection. Virology 331292-306. [DOI] [PubMed] [Google Scholar]
- 32.Rozhon, E. J., J. D. Kratochvil, and H. L. Lipton. 1983. Analysis of genetic variation in Theiler's virus during persistent infection in the mouse central nervous system. Virology 12816-32. [DOI] [PubMed] [Google Scholar]
- 33.Schlax, P. E., J. Zhang, E. Lewis, A. Planchart, and T. G. Lawson. 2007. Degradation of encephalomyocarditis virus and hepatitis A virus 3C protease by the ubquitin/26S proteosomal system in vivo. Virology 360350-363. [DOI] [PubMed] [Google Scholar]
- 34.Schlitt, B. P., M. Felrice, M. L. Jelachich, and H. L. Lipton. 2003. Apoptotic cells, including macrophages, are prominent in Theiler's virus-induced inflammatory, demyelinating lesions. J. Virol. 774383-4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma, R., S. Raychaudhuri, and A. Dasgupta. 2004. Nuclear import of poliovirus protease-polymerase precursor 3CD: implications for host cell transcription shut-off. Virology 320195-205. [DOI] [PubMed] [Google Scholar]
- 36.Shen, Y., M. Igo, P. Yalamanchili, A. J. Berk, and A. Dasgupta. 1996. DNA binding domain and subunit interactions of transcription factor IIIC revealed by dissection with poliovirus 3C protease. Mol. Cell. Biol. 164163-4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Son, K.-N., R. P. Becker, P. Kallio, and H. L. Lipton. 2008. Theiler's virus-induced apoptosis in M1-D macrophages is Bax-mediated through the mitochondrial pathway, resulting in loss of infectious virus: A model for persistence in the mouse central nervous system. J. Virol. 824502-4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Pesch, V., O. van Eyll, and T. Michiels. 2001. The leader protein of Theiler's virus inhibits immediate-early alpha/beta interferon. J. Virol. 757811-7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wojcik, C. 2002. Regulation of apoptosis by the ubiquitin and proteasome pathway. J. Cell. Mol. Med. 625-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yalamanchili, P., U. Datta, and A. Dasgupta. 1997. Inhibition of host transcription by poliovirus cleavage of transcription factor CREB by poliovirus-encoded protease 3Cpro. J. Virol. 711220-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yalamanchili, P., K. S. Harris, E. Wimmer, and A. Dasgupta. 1996. Inhibition of basal transcription by poliovirus: a virus-encoded protease (3Cpro) inhibits formation of TBP-TATA box complex in vitro. J. Virol. 702922-2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yalamanchili, P., K. Weidman, and A. Dasgupta. 1997. Cleavage of transcriptional activator Oct-1 by poliovirus encoded protease 3Cpro. Virology 239176-185. [DOI] [PubMed] [Google Scholar]
- 43.Zoll, J., W. J. G. Melchers, J. M. D. Galama, and F. J. M. van Kuppevold. 2002. The Mengovirus leader protein suppresses alpha/beta interferon production by inhibition of the iron/ferritin-mediated activation of NFk B. J. Virol. 769664-9672. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.