


Abstract
Although Methanocaldococcus (Methanococcus) jannaschii was the first archaeon to have its genome sequenced, little is known about the promoters of its protein-coding genes. To expand our knowledge, we have experimentally identified 131 promoters for 107 protein-coding genes in this genome by mapping their transcription start sites. Compared to previously identified promoters, more than half of which are from genes for stable RNAs, the protein-coding gene promoters are qualitatively similar in overall sequence pattern, but statistically different at several positions due to greater variation among their sequences. Relative binding affinity for general transcription factors was measured for 12 of these promoters by competition electrophoretic mobility shift assays. These promoters bind the factors less tightly than do most tRNA gene promoters. When a position weight matrix (PWM) was constructed from the protein gene promoters, factor binding affinities correlated with corresponding promoter PWM scores. We show that the PWM based on our data more accurately predicts promoters in the genome and transcription start sites than could be done with the previously available data. We also introduce a PWM logo, which visually displays the implications of observing a given base at a position in a sequence.
INTRODUCTION
The transcription system of Archaea is a minimal but functionally comparable version of the RNA polymerase (RNAP) II apparatus of Eucarya (1). Initiation of basal transcription requires a promoter, a multi-subunit RNAP, and two general transcription factors—TATA box-binding protein (TBP) and transcription factor B (TFB). The archaeal RNAP is similar in architecture and subunit composition to the eukaryotic RNAP II (2–5), and archaeal TBP and TFB are homologous to eukaryotic TBP and TFIIB (1). Studies have indicated that the archaeal promoters are similar to the eukaryotic RNAP II promoters, with a TATA box and a TFB recognition element (BRE) being the core promoter elements (1,6). First, TBP binds to the TATA box, dramatically kinking the DNA in the process. TFB stabilizes this TBP/DNA complex by binding to the BRE upstream of the TATA box, and making nonsequence-specific contacts downstream. The N-terminal domain of TFB subsequently recruits RNAP to the transcription start site (TSS). In some Archaea, including methanogens and Sulfolobales, there is a third promoter element—the initiator (Inr)—located at the TSS. This element is less important; mutations at the Inr are less detrimental than those in the TATA box, and insertions or deletions between the two elements can shift the TSS relative to the original Inr (7).
Available promoter studies are scattered among various groups of Archaea, e.g. methanogens (8–10), Sulfolobales (11), Pyrobaculum (12) and haloarchaea (13,14). Because promoters from different archaeal groups have somewhat different sequence patterns (6), data from the groups cannot be combined to better resolve a universal archaeal promoter pattern. Within a single archaeal class, the largest collection of experimentally determined, naturally occurring protein gene promoters is 61 in the haloarchaea (13), but even this is a pool of data from two genera. Mutagenesis studies on some specific promoters help to define functionally important promoter elements (7,14–17), but they do not increase the sample size of natural promoters.
Recently, a genome-wide selection for naturally occurring promoters was carried out in Methanocaldococcus (Methanococcus) jannaschii (18), the first archaeon to have a fully sequenced genome (19). Genomic DNA was fragmented and promoter-containing fragments were selected by their in vitro affinity for purified transcription factors TBP and TFB using an electrophoretic mobility shift assay (EMSA). While almost all tRNA gene promoters were identified, only 23 genomic regions containing 29 presumed promoters for 27 protein-coding genes were found. A limitation of these data is that TSSs were not determined, so the locations of the promoter elements were inferred by looking within the regions for promoter-like sequences.
To elucidate the properties of protein promoters in the M. jannaschii genome, we experimentally determined the TSSs of a diverse subset of the protein-coding genes. We explored the flanking sequences of the TSSs for conserved promoter elements, and analyzed the promoters in terms of their shared sequence features and their binding affinities for general transcription factors. These promoters were compared to the in vitro selected promoters, both in their sequence features and in their utility for predicting other promoters in the genome.
MATERIALS AND METHODS
Unless otherwise stated, all enzymes and reagents were used according to the manufacturers’ instructions. Genomic sequences of M. jannaschii were retrieved from the National Center for Biotechnology Information (NCBI) Entrez system (20). The NCBI accession numbers of the sequences are NC_000909.1 (chromosome), NC_001732.1 (large extra-chromosomal element) and NC_001733.1 (small extra-chromosomal element). Primers used in this study are compiled in the Supplementary Material (Supplementary Table S1). Our perl scripts are available upon request.
Preparation of M. jannaschii total cellular RNA
Methanocaldococcus jannaschii strain JAL-1T (DSM 2661) was grown as described (21). Cells were harvested during mid-log phase by centrifugation at 5500g for 15 min at 20°C. Cell pellets were washed twice with 385 mM NaCl/38 mM MgCl2, and then rapidly frozen at –80°C. Total cellular RNA was purified from frozen cell pellets with the RNeasy Mini Kit (Qiagen). The lysozyme treatment of cells was omitted because the cell wall does not have peptidoglycan (22).
Primer extension analysis
Gene-specific primers were labeled at their 5′-ends using [γ-32P]ATP and T4 polynucleotide kinase (Invitrogen). Each labeled primer was hybridized to 10 μg M. jannaschii total cellular RNA at 75°C for 5 min and then at 50°C for 5 min. Reverse transcription was carried out by adding 200 U SuperScript II or III reverse transcriptase (Invitrogen) to the RNA/primer hybrid in 1× first strand buffer, 1 mM DTT, 0.1 mg/ml BSA, 40 U rRNasin, and 1 mM each dNTP. The mixture was incubated at 50°C for 30 min and then treated with 25 mM EDTA (pH 8.0) and 1 μg RNase A (Ambion) at 37°C for 30 min. The runoff transcripts were recovered by ethanol precipitation and then subjected to 8 M urea–6% (w/v) PAGE along with a sequencing ladder generated from the same primer. Gels were analyzed by autoradiography.
Rapid amplification of 5′ cDNA ends
The rapid amplification of 5′ cDNA ends (5′-RACE) protocol was adapted from the method of Bensing et al. (23). One aliquot of 50 μg M. jannaschii total cellular RNA was treated with 10 U tobacco acid pyrophosphatase (TAP; Epicentre Technologies) at 37°C for 3 h, while another aliquot was incubated without TAP as a control. One nmol RNA oligonucleotide (5′-CAGACUGGAUCCGUCGUC-3′; Integrated DNA Technologies) was ligated to the 5′-ends of the TAP-treated or untreated RNA by incubation at 17°C for 16 h with 50 U T4 RNA ligase (Epicentre Technologies) in the presence of 1 mM ATP and 80 U rRNasin. The oligonucleotide-ligated RNA was recovered by ethanol precipitation and then used as template for reverse transcription (RT). RT reactions were carried out with a mixture of 20–30 gene-specific primers (RACE-SP1) (Supplementary Table S1). In each batch, 10 μg oligonucleotide-ligated RNA was annealed with primers (2 pmol each) in 15 μl RT buffer at 75°C for 5 min and then at 50°C for 5 min. Full-length cDNAs were synthesized using 200 U SuperScript III reverse transcriptase (Invitrogen). The 5′ cDNA ends of individual genes were amplified by polymerase chain reaction (PCR) with a linker primer (5′-CAGACTGGATCCGTCGTC-3′; corresponding to the sequence of the RNA oligonucleotide) and a gene-specific primer, either the same as RACE-SP1 or a nested primer closer to the 5′-end (RACE-SP2) (Table S1). PCR products were resolved on a 6% (w/v) nondenaturing polyacrylamide gel, and the DNA bands present in the TAP-treated lane but absent or significantly reduced in the untreated lane were excised. DNA was eluted from the excised gel region and re-amplified by PCR, followed by direct sequencing (24,25). The 5′-terminal nucleotide of the transcript is the transition point from genomic DNA sequence to the linker primer sequence.
Sequence alignment, position weight matrices, sequence scores, information content and logos
Flanking sequences of all mapped TSSs were retrieved from the M. jannaschii genome and aligned to the TSS. Conserved motifs were identified in the upstream regions of the TSSs with MEME (26) and a perl script. The upstream sequences starting with position –16 relative to the TSS were realigned based on the identified motifs. This alignment was used to find the base usage in each column. To compensate for the fact that rare events (in this case, rare bases at a position) are missed in small samples, one extra base (a pseudocount) was added to those observed in each alignment column, distributing the extra count among the four bases in proportion to their average frequencies in the genome. Thus, the small-sample-corrected empirical frequency of base b in column i is fb, i = (nb, i + pb) / (N + 1) (27), where b is a base (A, C, G or T), nb,i is the number of occurrences of b in alignment column i, pb is the frequency of base b in the M. jannaschii genome and N is the number of aligned sequences.
In a position weight matrix (PWM), the score given for observing base b at position i is sb, i = log2 (fb, i / pb) (28). The total score of a sequence match to the matrix is the sum of the matrix elements corresponding to the bases observed at the respective positions. In keeping with common usage of ‘bit score’ in contexts such as an NCBI-BLAST score (20) and Workman's log-odds score (27), we refer to this total score as a PWM bit score. When cast as a Bayesian inference analysis, an increase of +1 in a PWM bit score corresponds to a 2-fold increase of the ratio P(the sequence is a promoter)/P(the sequence is random), where P(H) is the probability that hypothesis H is true.
A PWM logo displays a PWM as stacked letters (representing bases). The height of each letter at a position is proportional to that base's score at that position in the PWM. Bases with positive matrix scores are stacked as upright letters above the baseline, while bases with negative scores are stacked as reversed letters below the baseline. Bases with higher scores are stacked on top of those with lower scores, while bases with equal scores are stacked in an alphabetical order.
Following Stormo (28,29), we define the information content of column i in a set of aligned sequences as 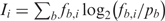 , and the total information content of the complete alignment as
, and the total information content of the complete alignment as 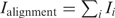 . Thus, the information content is the average of the PWM bit scores over all the aligned sequences (and a pseudocount sequence). The base 2 logarithm gives information units in bits. Each bit of information corresponds to a 2-fold increase in the probability of drawing the observed (aligned) sequences from the column-specific base frequencies relative to the probability of drawing the same sequences from the genomic base composition, averaged over all the aligned sequences. We note that some authors disagree with adjusting the calculation of the information content for the unequal frequencies of bases in the genome (30,31).
. Thus, the information content is the average of the PWM bit scores over all the aligned sequences (and a pseudocount sequence). The base 2 logarithm gives information units in bits. Each bit of information corresponds to a 2-fold increase in the probability of drawing the observed (aligned) sequences from the column-specific base frequencies relative to the probability of drawing the same sequences from the genomic base composition, averaged over all the aligned sequences. We note that some authors disagree with adjusting the calculation of the information content for the unequal frequencies of bases in the genome (30,31).
An energy-normalized sequence logo (enoLOGO) displays the information content at each position in a sequence alignment by the height of a stack of letters (representing bases) (27). The total height of the stack at position i equals Ii, and the height of each individual letter in that stack is proportional to the frequency of the corresponding base in the alignment column.
Statistical analyses
Pearson's chi-square tests were performed to compare the observed base frequencies in corresponding columns of two sequence alignments. Contingency tables were constructed from the observed counts. The expected base frequencies at a given position were based on the combined counts of the two alignments. Chi-square test P-values were calculated with Excel (Microsoft).
Correlation coefficients and regression lines were calculated with the Analysis ToolPak of Excel (Microsoft). The significance of a correlation was assessed by a Monte Carlo analysis in which the data were randomized between pairs for 106 times, and the frequency of instances in which the magnitude of the correlation coefficient equaled or exceeded that of the original data was determined.
Competition EMSA
Recombinant M. jannaschii TBP and TFBc (C-terminus of TFB) were expressed in Escherichia coli cells and purified as described (18). Competitor promoter DNAs were amplified by PCR with primers listed in the Supplementary Material (Supplementary Table S1). Competition assays were carried out as described (32). Briefly, in each assay 1 ng (∼5 fmol) labeled tRNAVal promoter DNA from Methanococcus vannielii (33) was mixed with 50 ng TBP, 20 ng TFBc, and increasing concentrations of competitor DNA in a final volume of 20 µl containing 20 mM Tris·HCl (pH 7.5), 150 mM KCl, 10 mM MgCl2, 0.05 mM EDTA, 0.5 mM DTT, 0.1 mM PMSF, 5% (w/v) glycerol and 1 µg poly(dI-dC). The reactions were incubated at 75°C for 30 min and then resolved on a 5% (w/v) nondenaturing polyacrylamide gel. Band intensities of bound and free probes were quantified by phosphorimaging. The bound/free ratios were calculated and then normalized by the ratio in the reaction without competitor DNA. Replicate experiments (n = 4 or 6) were done and the mean bound/free ratios were used. A plot of log(bound/free ratio) vs. log(concentration of competitor DNA) was generated to calculate a reference concentration (C0.1), at which the bound/free ratio was 0.1. The C0.1 of each competitor promoter was normalized by the C0.1 of unlabeled M. vannielii tRNAVal promoter, and the ratio C0.1(M. vannielii tRNAVal promoter)/C0.1(competitor promoter) was used to estimate the relative binding affinity of the competitor promoter for transcription factors TBP and TFBc.
Promoter predictions
The promoter score of any given sequence is the sum of the PWM bit scores of the promoter elements in a prediction model. The promoter prediction model was either a BRE/TATA-box PWM [covering the BRE (9 nt), the TATA box (8 nt) and an additional 4 nt on each side of them], or a combination of the BRE/TATA-box PWM and a proximal promoter element (PPE)/Inr PWM [covering the PPE (10 nt) and the Inr (2 nt)]. When the model includes the PPE/Inr PWM, a spacer score is used to penalize suboptimal spacings between the TATA box and the TSS. This spacer score is the base 2 logarithm of the frequency of the particular spacing divided by the frequency of the most common spacing observed in the mapped promoters (34,35). It has been noted that this formulation lacks a normalization that would be included in an information content or absolute probability calculation (36,37). In the present context, the correction would be the addition of a constant (–1.54 bits) to all scores. Because all scores, including the threshold, are shifted by the same amount, no results are altered.
Promoter predictions were carried out in the M. jannaschii genome and a randomized M. jannaschii genome in which the nucleotide order was shuffled. Every subsequence of appropriate length was retrieved from the genomic sequences and scored using the prediction models. When the model that includes the PPE/Inr PWM was used, TATA box to TSS spacings of 19 to 27 were tested, and the highest total score selected. A subsequence with a score greater than or equal to a threshold was counted as a predicted promoter. Throughout this work, we set the threshold to predict 50% of a testing set of known promoters.
The following conventions were used to evaluate the performance of a prediction model. The ability of a model to detect known promoters is sensitivity (= true predicted promoters/total promoters). The proportion of successful predictions of a model is precision (= true predicted promoters/total predicted promoters). The overall performance of a prediction model is accuracy [= (sensitivity + precision)/2].
RESULTS
Determining TSSs of protein-coding genes
The M. jannaschii genome is ∼1.7 Mb and includes 1738 protein-coding genes (19). To narrow our search for promoters, we focused on the genes whose immediate upstream regions are likely to contain promoters. If adjacent genes are transcribed divergently, there are likely to be divergent promoters responsible for their expression in the region between them. Also, the region between genes transcribed in the same direction might contain a promoter, particularly if there is adequate space (≥40 bp) and the flanking genes have no obvious functional connection. If the space is <40 bp and the downstream gene is obviously more highly expressed than the preceding gene, then there is likely to be a promoter. Using these criteria, we identified 1133 protein-coding genes as candidates for having a promoter immediately upstream. These candidates were compared to a list of proteins found to be expressed in mid-log phase cells in a previous proteomic study (21), so that we could emphasize genes apt to be expressed under our culture conditions. Guided by these data and a goal of diversity, we chose ∼12% of the 1133 candidate genes for experimental analyses, comprising 105 divergently transcribed genes and 30 nondivergent genes (Supplementary Table S2). These genes are distributed throughout the genome. The protein products of all the nondivergent genes and 83 of the divergent genes were observed in the proteomic study (21).
To determine TSSs, we first used conventional primer extension analysis. Of the 135 chosen genes, we performed primer extension on 77 (all of them divergent except MJ0746), and identified the TSSs of 42 (Supplementary Table S2). Examples of the primer extension data are shown in Figure 1A, and the rest are in the Supplementary Material (Supplementary Figure S1A). Of the 57 genes for which protein products were detected by the proteomic study (21), the TSSs of 39 were identified by primer extension. Of the 20 genes for which protein products were not detected, the TSSs of only three were observed (Table 1). The success rate of primer extension on the former genes was significantly (P < 0.0001) higher than that on the latter genes. The failure to identify the TSSs of 18 of the 57 genes with observed protein products suggested that our primer extension analyses were not sensitive enough for all expressed genes.
Figure 1.

Mapping TSSs using primer extension and 5′-RACE. (A) Examples of primer extension. Arrowheads indicate runoff transcripts. (B) Examples of 5′-RACE. Each panel is a nondenaturing polyacrylamide gel photographed using Chemi Doc™ System (Bio-Rad). Lanes: M, 25 bp DNA ladder (Promega); +, RT–PCR products from TAP-treated RNA; –, RT–PCR products from untreated RNA (control). Solid arrowheads indicate TSSs. Open arrowheads indicate processing sites.
Table 1.
Summary of results of primer extension and 5′-RACE analyses
| Protein detecteda | Primer extension |
5′-RACE |
||
|---|---|---|---|---|
| Testedb | Successesc | Tested | Successes | |
| + | 57 | 39 | 113 | 94 |
| − | 20 | 3 | 22 | 13 |
| Total | 77 | 42 | 135 | 107 |
a+, gene product detected by proteomics; –, gene product not detected.
bNumber of genes analyzed.
cNumber of genes for which one or more transcription start sites were identified.
To aid in identifying weak promoters, we switched to 5′-RACE, a more sensitive method. In 5′-RACE, a synthetic RNA oligonucleotide is ligated to the 5′-ends of the transcripts, thereby making it possible to amplify the 5′-ends using PCR. To distinguish TSSs from the ends of RNA processing products, we adapted the modification described by Bensing et al. (23) in which total cellular RNA not treated by TAP is analyzed as a control. Primary transcripts, which have a 5′ triphosphate, can be ligated to the RNA oligonucleotide only after conversion of the 5′ triphosphates to 5′ monophosphates by TAP. Therefore, for primary transcripts, 5′-RACE yields RT–PCR products from TAP-treated RNA, but not from untreated RNA. On the other hand, RNA processing products, which already have a 5′ monophosphate, can be directly ligated to the RNA oligonucleotide and will produce RT–PCR products with or without TAP treatment. It has been found that in the decay of three E. coli RNAs, a substantial fraction of the 5′ triphosphates were converted to 5′ monophosphates by pyrophosphate removal (38). In a 5′-RACE analysis, such a mixture of 5′-end types would look like a mixture of processed and primary transcripts starting with the same 5′-terminal nucleotide. We observed such patterns in some of our data, but they do not change the inferred start site locations.
We applied 5′-RACE to the 135 genes chosen above and identified TSSs for 107 of them (Tables 1 and Supplementary Table S2). Figure 1B shows examples of the 5′-RACE results (others are in Supplementary Figure S1B). The overall success rate of 5′-RACE was higher than that of primer extension. Of the 42 genes for which TSSs were observed by primer extension, all these TSSs were also identified by 5′-RACE, with one additional TSS identified for 3 of the genes. Of the 35 genes for which TSSs were not observed by primer extension, the TSSs of 16 were identified by 5′-RACE. Of the 58 genes that were not analyzed by primer extension, the TSSs of 49 were identified by 5′-RACE. Although 5′-RACE is very sensitive, the TSSs of 28 of the 135 analyzed genes were not identified. Of these 28 genes, 3 showed only processing sites and the other 25 showed no detectable RT–PCR products. A possible explanation of the negative results is that the primary transcripts were absent or too scarce to be detected under our growth conditions. There are many alternative explanations (the TSS is far from where we predicted, sequence errors leading to bad primer design, experimental failure, etc.), but such problems would not be expected to introduce systematic biases.
In summary, we identified 131 TSSs for 107 protein-coding genes in the M. jannaschii genome. These are compiled in Supplementary Table S2, along with TSSs for three other genes (39). The distances from the TSSs to their corresponding translation start sites are summarized in Figure 2. Three quarters of the distances (101/134) are 80 nt or less. Although this is consistent with the fact that the M. jannaschii genome is very compact (∼88% coding) (19), the distances are longer than those observed in some other Archaea (see Discussion section).
Figure 2.

Histogram of distances from the TSSs to their nearest downstream translation start sites. Gene translation start locations were identified initially by the coding region identification tool CRITICA (40), and then curated manually by David E. Graham (University of Texas) using neighboring DNA features and comparative analyses of translation start codons of orthologs in related genomes (personal communication).
Sequence features of protein promoters
To identify conserved promoter elements, we retrieved the flanking sequences of the 134 experimentally determined TSSs from the M. jannaschii genome and aligned them to the TSS. Two regions were identified with base frequencies obviously different from those of the genome. One region is near the TSS itself and the other is centered ∼30 nt upstream. Since the upstream region is A + T rich, we presumed that it is the TATA box. It is known that the spacing between the TATA box and the TSS can vary by a few nucleotides (1). To refine the alignment of the upstream sequences, we searched for conserved motifs in the region from –44 to –20 relative to the TSS using MEME (26). We used a 17-nt search window to encompass both the TATA box and the adjacent BRE. MEME was set to identify exactly one motif in each sequence, and therefore 134 motifs were identified. In seven of the cases, however, with the aid of a perl script we found an alternative motif with a closer-to-optimal spacing to the TSS. These were used in the sequence alignment.
To visualize recurring features in the aligned promoter sequences, we generated logos in two different styles. Figure 3A shows an energy-normalized sequence logo (enoLOGO) of the protein promoters in this study and, for comparison, a logo of the M. jannaschii promoters previously identified by in vitro selection (18). The total height of the stack at each position is the information content, while the relative heights of the individual bases indicate their relative frequencies at that position. To more clearly show the over- and under-represented bases (relative to the genome average), we also generated a PWM logo of the protein promoters (Figure 3B). At each position, the bases above the axis (which have a positive score in the PWM) support a matching sequence being a promoter, while bases below the axis decrease support for a matching sequence being a promoter. Although many of the following observations can be seen in both logos, they are frequently more evident in the PWM logo.
Figure 3.

Logos of promoter sequences. (A) Energy-normalized sequence logos of the protein promoters in this study (top) and the promoters previously identified by the in vitro selection (bottom) (18). The horizontal axis shows nucleotide positions, and the vertical axis is information content in bits (see Materials and Methods section). Promoter elements (BRE, TATA box, PPE and Inr) are bracketed, and the TSS is indicated with a bent arrow. Of the protein promoters in this study, the total information content of BRE is 2.63 bits, TATA box 6.09 bits, PPE 1.84 bits and Inr 1.13 bits. Of the in vitro selected promoters, the total information content of BRE is 4.28 bits, and TATA box 7.05 bits. Closed circles indicate positions at which the protein promoters differ significantly from the in vitro selected promoters (P < 0.05, data from Supplementary Table S3). (B) PWM logo of the protein promoters in this study. The values on the vertical axis are bit scores (which are distinct from bits of information, see Materials and methods section). The vertical scale below the axis is reduced 6-fold relative to that above. Due to the variation in spacing between the TATA box and the TSS, the nucleotide position numbers to the left of the vertical bars in panels A and B are for the most common spacing. In both panels, the gray areas would completely cover 90% of the logos generated from random sets of nucleotides drawn from the genomic base composition, and sample size matched to the number of sequences in the particular logo (i.e. it is a measure of the random background).
The TATA box of the protein promoters shows a sequence pattern TWTATATA (W = A or T), similar to the ‘A box’ pattern TTTATATA proposed for stable RNA gene promoters in M. vannielii (9). This TATA box pattern seems to be confined to the methanogens (8), as other Archaea (e.g. haloarchaea and Sulfolobales) have TATA boxes with different patterns (6,13). Although the TATA box is the most conserved promoter element, chi-square tests show that 3 of the 8 positions in the TATA box (–28, –27 and –24) differ significantly (P < 0.05) between the protein promoters in this study and the in vitro selected promoters (18).
The BRE spans the nine positions upstream of the TATA box. Position –34 is highly conserved, consistent with the finding that position –34 is essential for specific binding of the human TFIIB to BRE (41). Besides position –34, three other positions (–37, –35 and –32) make sequence-specific contacts to the carboxy-terminal 2/3 of TFB in the crystal structure (42). Notably, base frequencies at all three of these positions differ significantly (P < 0.05) between the protein and in vitro selected promoter sets (Figure 3A). Positions –40 through –37 provide a striking illustration of the difference between the enoLOGO and the PWM logo. In the PWM logo, there are clear over-representations of A, C, C and G (respectively) at these positions, while in the enoLOGO the most abundant bases are A, T, A and A. The latter are the most abundant bases in these positions of the alignment, but A and T are exaggerated because they start out more abundant than G and C (the genome has 68.7% A + T). The preferences for A, C, C and G are more pronounced in the in vitro selected promoters (Figures 3A and S2). Positions –40 through –38 are an extension to the canonical BRE, and the crystal structure shows that TFB binds the phosphate backbone of this region (42). Although phosphate contacts are generally assumed to be non-specific, the observed base preferences suggest that the bases may contribute to a favorable spatial structure. TFB also binds to the DNA immediately downstream of the TATA box (43–45), but here there are no significant base biases in the protein promoters, consistent with these being non-specific contacts.
The Inr (the promoter element at the TSS) differs between the protein and stable RNA promoters. The protein promoter TSSs exhibit a strong preference for A or G, while previously characterized stable RNA promoters in the Methanococcales have a more specific preference for G (10,18). The immediately upstream nucleotide (–1) shows preference for T in both data sets. Although the stable RNA promoters display preference for C in the second nucleotide of the transcript (10,18), this is not observed in the protein promoter set.
The proximal promoter element (PPE) spans positions –11 to –2. The A + T content of the PPE is very high (86%). However, this is not simply an A + T-rich region. It shows a specific, if weak, sequence pattern AAATTWTTAT. The first two positions, –11 and –10, are the most conserved, as was observed for haloarchaeal promoters (13). For the in vitro selected promoters, no reliable data on the PPE or the Inr are available for comparison because the selection identified the transcription factor-binding regions in the genome, not the exact start sites (18).
No other elements were observed within the region from –240 to +240 except an over-representation of G in a region (+14 to +19) downstream of the TSS (data not shown). Our inspection of the corresponding sequences revealed that this is contributed by the Shine–Dalgarno sequences (ribosome binding sites) (46).
Besides the specific sequence elements, the spacing between the 3′ edge of the TATA box and the TSS is also conserved. In 94% (126/134) of the protein promoters, the spacing is 23 ± 2 nt (Figure 4).
Figure 4.

Histogram of the number of promoters versus the spacers between the 3′ edge of the TATA box and the TSS.
General transcription factors bind protein promoters less tightly than most tRNA promoters
Because relatively few protein promoters were isolated by the in vitro selection, we wanted to know whether general transcription factors would bind protein promoters under our in vitro conditions. To characterize the binding of protein promoters by transcription factors, we used competition EMSA assays, a fast method commonly used to measure binding affinities (32,47–51). We used the M. vannielii tRNAVal promoter, an extensively characterized methanococcal promoter (33), as a reference DNA in the competition assays. The transcription factors we used are M. jannaschii TBP and TFBc (the C-terminal 2/3 of TFB). TFBc is much more stable than full-length TFB, and therefore is commonly used in promoter-binding assays (18,32) and structural studies (42,52). In the presence of TBP, TFBc gel shifted DNA (at 150 mM K+) at a lower concentration (1/5 to 1/10) than did full-length TFB (at 60–90 mM K+) (18,32), though it is not known what percentage of the full-length TFB was active in those experiments. Our assays had a saturating amount of TBP and a limited amount of TFBc. Thus, the labeled tRNAVal promoter had to compete with the (unlabeled) promoter DNA being assayed to form a TBP/TFBc/promoter ternary complex (under our assay conditions, TBP binding alone was not sufficient to gel shift the labeled tRNAVal promoter). Figure 5A shows a representative competition EMSA gel. Out of the 134 identified protein promoters, we selected 12 on the basis of encompassing a wide variety of BRE/TATA-box PWM scores (Materials and Methods section) for competition EMSA (Supplementary Table S4). All of the 12 protein promoters tested measurably competed with the tRNAVal promoter for the transcription factors, while two nonspecific DNAs (pUC18 multiple cloning region and MJ0723 coding region) displayed little or no competition (data not shown).
Figure 5.

Measuring relative binding affinities of promoters for transcription factors TBP and TFBc using competition EMSA. (A) Representative EMSA gel, in which increasing concentrations of competitor DNA (0, 0.25, 0.5, 1, 2 and 4 nM unlabeled M. vannielii tRNAVal promoter) were used to compete the labeled tRNAVal promoter out of the TBP/TFBc/promoter ternary complex. Solid arrowhead, the shifted ternary complex (bound probe); open arrowhead, free probe. (B) Bound to free probe ratios on EMSA gels (as in A) were determined by phosphorimaging. A plot of log(bound/free ratio) versus log(concentration of competitor DNA) was generated, and a good correlation was observed. A reference concentration (C0.1), at which the bound/free ratio was 0.1, was calculated from the regression line.
To quantify the relative binding affinities, we calculated each promoter's reference concentration, C0.1(promoter), as explained in Figure 5B. Based on these values we summarized the relative binding affinity of each promoter by the ratio C0.1(unlabeled M. vannielii tRNAVal promoter)/C0.1(competitor promoter). Supplementary Table S4 shows the relative binding affinities of the 12 tested protein promoters, as well as those of the 19 tRNA promoters measured using the same competition EMSA assays (32). Transcription factors bind the protein promoters less tightly than the tRNA promoters recovered from the in vitro selection (18), even though 9 of the 12 protein genes were among the highly expressed genes (53). These results may provide an explanation for why relatively few protein promoters were found in in vitro selections that recovered nearly all tRNA promoters. Such a differential efficiency is consistent with the observation that the tRNA promoters isolated by in vitro selection bind TBP/TFBc more tightly than do the tRNA promoters not isolated by selection (18,32).
Correlations between promoter sequence, binding affinity and gene expression
Basal transcription in Archaea is initiated by the binding of transcription factors to the TATA box and the BRE. To check the correlation between promoter sequence and transcription factor binding, we scored the sequences of the tested promoters using the BRE/TATA-box PWM of the protein promoters (Supplementary Table S4). Figure 6 shows that there is a close relationship between the log2 (relative binding affinity) of a promoter and its BRE/TATA-box score. The correlation coefficient (r = 0.75) is significantly different from zero (P < 10−5).
Figure 6.

Correlation between promoter BRE/TATA-box score and transcription factor binding. The ‘selected’ tRNA promoters were identified by the in vitro selection (18). The ‘other’ tRNA promoters were identified computationally (32). The vertical position of a point indicates the effectiveness of a promoter, relative to the M. vannielii tRNAVal promoter, in competition for TBP/TFBc. The horizontal position is its BRE/TATA-box score. The correlation coefficient (r) is 0.75, indicating a positive correlation. The equation for the regression line is log2(relative binding affinity) = 0.40 × (BRE/TATA-box score) – 6.60.
In Archaea, little is known about the correlation between promoter sequence and gene expression. Available data show that mutations in the ‘distal promoter element’ greatly affect transcriptional activity both in vivo (14,15) and in vitro (7,17,33). In the mutational analysis of the BRE/TATA-box region of the tRNAVal promoter of M. vannielii (33), another member of the Methanococcales, the in vitro transcriptional activities were reported. We calculated the sequence scores of the tRNAVal promoter and mutants from it using the BRE/TATA-box PWM of the protein promoters of M. jannaschii (Supplementary Table S5). These data show a strong relationship between the log2(in vitro transcriptional activity) of a promoter and its BRE/TATA-box score (Supplementary Figure S3). Even though the transcription data are for the transcription system from a mesophile and the BRE/TATA-box PWM is from an extreme thermophile, the correlation coefficient (r = 0.88) is significantly different from zero (P < 10−5).
Promoter predictions
Computational prediction of promoters comes with many caveats, ranging from the simplistic nature of the promoter models used to the inability to integrate effects of regulatory features. With the exception of a small number of very strong promoters, the match to a consensus sequence or a PWM score is not a sufficiently reliable predictor of promoters. However, combined evidence approaches in which promoter profiles are combined with other data have been more successful (54–56). Thus, we pose the question: do our protein promoter data improve promoter prediction in M. jannaschii over that based on the in vitro selected promoters (18)?
We used PWMs for our prediction models (Materials and Methods section). In addition to providing the data for PWMs, the mapped protein promoters were also used to adjust the sensitivity of each model (see below). To avoid circularity we used a random quarter of the protein promoters as a testing set for setting the sensitivity, and used the rest to build a protein promoter model. The results reported are averages of 100 repetitions of this partitioning. All the in vitro selected promoters were used to construct the model because they were not used in setting the sensitivity.
To compare the prediction accuracies (see Materials and Methods section for definition) of these two models, we apply the following reasoning. In our analyses, we adjusted the threshold of each model to detect half of the promoters in the testing set, that is, the sensitivity was 50% (Materials and Methods section). With the sensitivity fixed, the accuracy of a model is solely dependent on its precision (Materials and Methods section). When two models predict the same number of true promoters, the model with the smaller total number of predicted promoters has higher precision, and therefore higher accuracy.
Our first comparison of the two models was based on the predictions in a randomized M. jannaschii genome. To the extent that the randomized genome contains some sequences that might act as promoters, the number of instances (true predicted promoters) does not depend on the promoter prediction model (each was set to detect 50% of the testing set promoters). Our prediction results are summarized in the first two data rows of Table 2. Following the reasoning above, we compare the values in column 4, and a smaller value (fewer total predicted promoters) indicates better performance of the corresponding model. In this case, the model based on promoters of protein-coding genes is more accurate than the model derived from the in vitro selected promoters. This was observed for 97 of the 100 partitionings of the mapped promoters between training and testing sets.
Table 2.
Promoter predictions of different models
| Training promoters used in model | PWM regions used in model | Predicted promotersa |
Differenceb | |
|---|---|---|---|---|
| M. jannaschii genome | Randomized genome | |||
| In vitro selectionc | BRE/TATAd | 5067 ± 2286 | 4042 ± 1940 | 1026 ± 348 |
| Protein-coding genese | BRE/TATA | 2204 ± 849 | 1436 ± 665 | 768 ± 191 |
| Protein-coding genes | Extendedf | 1386 ± 561 | 551 ± 323 | 835 ± 243 |
aThe mean number ± SD of total predicted promoters for 100 random assortments of the 134 mapped protein promoters between a training set (100 or 101) and a testing set (34 or 33). For each assortment, the threshold for each model was set to predict 50% of the 34 or 33 testing set promoters.
bDifference in number of predicted promoters between the M. jannaschii and randomized genomes.
cSixty promoters from Li et al. (14). Over the 100 replicates (see footnote a), the PWM of the model did not change, but the threshold was adjusted to detect 50% of the testing set promoters.
dThe BRE (9 nt) and the TATA box (8 nt) plus 4 nt on each side.
eThe 100 or 101 promoters in the training set for each of the 100 replicates (see footnote a).
fBRE/TATA-box, PPE/Inr, and spacer score (see Materials and Methods section).
We performed the same analysis on the unshuffled M. jannaschii genome. Because we do not know all the actual promoters in the genome, we cannot distinguish true predicted promoters from false predicted promoters on a case-by-case basis. Therefore we have made a reasonable, but untested, assumption that the true predicted promoter detection rates of the two models are similar (each was adjusted to detect 50% of the promoters in the testing set). The protein-promoter-based model had fewer total predicted promoters (first two data rows in column 3), so it is more accurate. This was observed for 94 of the 100 partitionings of the mapped promoters between training and testing sets.
As a check on the reasonableness of our approach to building and evaluating these models, we used them to estimate the number of authentic promoters in the M. jannaschii genome. Each model predicted approximately 800–1000 more promoters, on average, in the actual genome than in the randomized genome. Because the threshold was set to predict 50% of the testing set promoters, the approximately 800–1000 difference in the numbers of promoters predicted at 50% sensitivity would suggest a total of approximately 1600–2000 authentic promoters in the M. jannaschii genome, a seemingly reasonable number. If the randomized genome contains a large number of true promoter sequences, we would be subtracting too large of a background value, and this suggested number of authentic promoters would be an underestimate.
Because we do not have experimentally determined PPE or Inr data for the in vitro selected promoters, these elements were not part of the above models. However, since these elements are known for the transcriptionally mapped protein promoters, we added them to the prediction model to see if this improves prediction accuracy. When we added a PWM for the PPE and Inr, and a TATA box to TSS spacer score to the protein-promoter-based model, the total number of predicted promoters was decreased for both the M. jannaschii genome and the randomized genome (Table 2), and therefore the resulting model is more accurate. Given that at 50% sensitivity the actual genome has approximately 800 more predicted promoters than its randomization, this again suggests approximately 1600 promoters in the genome.
Due to the variation in spacing between the TATA box and the TSS (Figure 4), the BRE and the TATA box alone do not unambiguously predict the TSS; only 34% of the TSSs are at the most common spacing. However, the introduction of the PPE/Inr PWM and the spacer score resulted in a model with a start site prediction precision of 73% (true TSSs/predicted TSSs) (data not shown).
DISCUSSION
Methanocaldococcus jannaschii protein gene promoters
To expand our knowledge of protein-coding gene promoters in M. jannaschii, we have identified the TSSs and promoters of over 100 of its genes, the largest collection for any archaeon.
The promoters of protein-coding genes in M. jannaschii look like a more variable version of the promoters previously identified by the in vitro selection (18). Most protein-coding gene promoters differ at multiple positions from the TATA-box consensus sequence (TWTATATA). The variation is even greater in the BRE. In spite of their variations in sequence, these DNA regions compete for TBP and TFBc in the absence of other protein factors, such as TFE (57,58), single-stranded DNA binding protein (59), or an activator (39).
The PPE and the Inr are often neglected, but are important promoter elements, both biologically and computationally. The PPE largely overlaps with the open complex region that spans at least positions –11 to –1 (60). Broadly speaking, the open complex region is in interaction with many transcription-related proteins, such as RNAP (43,45), TFB (43,45), TFE (61), TFIIE (62,63) and single-stranded DNA binding protein (59). Although the high A + T content of the PPE might facilitate the formation of an open complex, the sequence of the PPE is also important. Mutations in this region can dramatically change transcription efficiency (7,17), and also affect start site selection within limits (64). Our analyses show that the inclusion of the PPE, the Inr and the spacer score improves the accuracy of promoter prediction and increases the precision of predicting start sites.
Promoter sequence and intrinsic promoter strength
Available experimental data are too limited to fully resolve the relationships between promoter sequence, transcription factor binding and transcriptional activity. Our results show that the transcription factor binding affinity of a promoter (as measured by competition EMSA) correlates with the promoter's PWM score. This systematic relationship between DNA sequence and protein-binding affinity is consistent with both theoretical predictions (30) and experimental data in other systems (47). However, because transcriptional activity depends on much more than transcription factor binding, this leaves unanswered the relationship between a DNA sequence and its transcriptional activity in vivo.
In M. jannaschii, many proteins appear to have little or no gene-specific regulation. Relative abundances of most observed proteins remain unchanged in spite of variations in growth media, growth conditions and growth phases (53,65 and Giometti, C. S. et al., unpublished observations), supporting a picture in which expression levels of many or most genes are set by intrinsic promoter strength. To the extent that this is true, it is relevant to ask whether promoter sequence scores are correlated with their corresponding gene transcription levels. Although we do not have quantitative data for in vivo promoter activity or for transcript abundances in M. jannaschii, we do have an indirect datum regarding some RNA levels. Of the TSSs mapped by 5′-RACE, 73 were also analyzed by the primer extension method. Primer extension successfully mapped 47 of these TSSs, and failed to map 26 of them. Although many factors can influence the success rates of these methods, the most obvious source for a systematic difference between primer extension and 5′-RACE results is transcript abundance; primer extension is expected to be less successful with lower abundance transcripts. The average promoter bit score for the 47 promoters for which primer extension succeeded is 11.62 bits, while that for the 26 promoters for which primer extension failed (9.43 bits) is significantly lower (P < 0.01). This systematic trend would not be expected unless transcription level was positively correlated with the PWM score of the corresponding promoter.
Another observation that points in the same direction is our analysis showing that the in vitro transcriptional activities of the M. vannielii tRNAVal promoter and variants of it (33) correlate with their PWM scores (P < 10−5) (Figure S3). Although these in vitro activities were measured in M. vannielii and the PWM was derived from M. jannaschii promoters, it would be difficult to argue that the observed relationship is coincidental.
Protein promoters are not evolved for maximal binding to transcription factors
The in vitro selected promoters have higher binding affinities for the transcription initiation factors than do the protein promoters, but our data show that the in vitro selected promoters are less effective (accurate) as a pattern for identifying promoters in the genome. This suggests that naturally occurring promoters are not evolved for maximal binding to transcription factors. It is not uncommon for researchers to experimentally seek the optimal binding site for a protein, for example by using a SELEX approach (66,67), and then use this as a profile for attempting to identify naturally occurring binding sites in genomes (68). Although the approach is very effective at ‘evolving’ a high-affinity binding site, the subsequent prediction of natural sites is often less successful (39,69,70). Biological functions are not necessarily evolved for maximal activity.
The 5′-untranslated regions
In M. jannaschii, 130 of the 134 mapped protein-coding gene transcripts have 5′-untranslated regions (5′-UTR) of ≥10 nt (and the remaining 4 are all 9 nt). This differs dramatically from observations in haloarchaea (13) and Pyrobaculum (12), where 67% and 100% (respectively) of the experimentally mapped transcripts have 5′-UTRs of <10 nt. The 5′-UTR can play an important role in determining the translational efficiency of an mRNA via mechanisms that include the ribosome binding site (RBS), RNA folding, and upstream open reading frames (uORF).
We observed an RBS in 86 of the 134 M. jannaschii 5′-UTRs (71 of the 110 genes) (Supplementary Table S6). Even two of the four 9-nt long leaders (classified as ‘leaderless’ in 13) include an RBS. In haloarchaea, fewer than 10% of the transcripts have an RBS, yet these RBS-lacking transcripts are efficiently translated (13). In Sulfolobus, the first protein-coding gene of an operon usually lacks an RBS, while later genes in the operon have one (71). These variations in RBS utilization reinforce the fact that the Archaea comprise a diverse domain.
Because M. jannaschii has a very low G + C content (31.3%) and grows optimally at 85°C (19,72), stably folded RNAs tend to be quite obvious due to a local increase in G + C (32,73,74). We examined the 134 5′-UTRs for potentially stable structures using the Vienna RNA secondary structure prediction package (75). Only the 5′-UTR of the gene MJ1260 (SSU ribosomal protein S6E) is predicted to have a stable secondary structure at 85°C (data not shown). This region corresponds to an experimentally identified noncoding RNA (32). These results suggest that RNA secondary structures do not commonly play a role in the regulation of M. jannaschii gene expression.
The translation (versus nontranslation) of a uORF can influence expression of downstream coding sequences. Eukaryotes initiate translation with a ribosome scanning mechanism, and the translation of a uORF tends to alter that of a downstream coding region (76,77). Thus, in human mRNAs, the occurrence of uORFs is significantly suppressed relative to random expectation (78). We found 232 uORFs in the 5′-UTRs of the 134 mapped M. jannaschii transcripts. When we replaced all the 5′-UTRs with random sequences computationally (10 000 repetitions), we found an average of 226 uORFs with a length distribution similar to those in the actual leaders (data not shown). Thus, the M. jannaschii 5′-UTRs are neither enriched nor depleted of uORFs relative to random sequences. Additional evidence that few, if any, of these M. jannaschii uORFs are translated is that only one 2-amino acid uORF (in the 5′-UTR of MJ1260) has a potential RBS, in striking contrast to the >60% frequency of an RBS upstream of the annotated coding sequence. These observations do not exclude an important role of uORFs in M. jannaschii, but they suggest that such regulation is not common among the genes sampled here.
Logos and the representation of shared sequence elements
The sequence logo, as introduced by Schneider and Stephens (79), provides a vivid method to portray the recurring sequence features of a set of aligned sequences. At each position in the alignment, the sequence logo displays the information content at that position by the height of the stacked letters, and the relative frequency of each base type by the fraction of the stack height devoted to the corresponding letter. The information content displayed in a sequence logo (as defined in 79) differs from the one that we have used here (Materials and Methods section), unless the genomic G + C content is 50% (all bases are equally abundant). However, an alignment of random sequences drawn from the M. jannaschii genome will have a 31.3% G + C content, the genomic composition (19). Yet, even with an unlimited number of sequences (no sampling error), the corresponding sequence logo would have a height of 0.10 bits (out of a possible 2) at every sequence position, a value that is three times the height of the gray area in top part of Figure 3A. Only when drawn from a pool of equal-frequency bases (a good approximation for E. coli, but not for M. jannaschii) is the height of a sequence logo (79) of random sequences expected to go to zero.
To avoid this behavior, we have used the enoLOGO introduced by Workman et al. (27). The enoLOGO is very similar to the traditional sequence logo, but analyzes how the aligned bases differ from the composition of the genome being analyzed (a more precise statement can be found in Materials and Methods section). A perhaps surprising aspect of the corresponding measure of information content is that the maximum value attainable depends on the identity of the base. In the case of M. jannaschii, an overabundance of G or C in an alignment column distinguishes the position more from the rest of the genome than does an overabundance of A or T, and this is reflected in a potential for higher information content. To this extent, we find this measure to be a more meaningful representation of how a collection of sequences differ from the genome in which they were found.
However, there remains one very unintuitive aspect of the presentation. If we were to observe an alignment column of sequences from the M. jannaschii genome such that A, C, G and T were equally frequent, this would be non-random and would have an information content sensu Stormo (28,29) of 0.11 bits. Since this column systematically departs from the genome average, this makes sense. What is confusing is that the four bases will appear as equal height letters, and thus it will not be obvious in an enoLOGO how this is not random. For this reason we have introduced the PWM logo. It is a graphical display of the scores assigned to each of the bases in evaluating whether a candidate sequence belongs with those in the alignment or it is drawn randomly from the genome. Thus, the height of each letter reflects how observing that base would affect the decision as to whether the new sequence should be categorized with those in the alignment or not. In our hypothetical example of a column of equally abundant bases in an alignment of M. jannaschii sequences, G and C would be given positive bit scores (0.68) because they are over-represented relative to the genome average, and would be displayed above the axis. Conversely, A and T would be given negative scores (–0.46) because they are under-represented, and would be displayed below the axis. It is important to realize that the bits of information in an enoLOGO are not the same as the bits in a ‘bit score’ displayed by a PWM logo. Also, (i) information content of an enoLOGO can never be negative, whereas every position in a PWM logo will possibly have one or more bases with negative values, and (ii) information content as displayed in an enoLOGO asymptotically approaches a maximum value with increased sampling, whereas the scores in a PWM and hence in a PWM logo have no such limit.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Aeronautics and Space Administration (NAG 5-12334 to G.J.O., partial); the Department of Energy (DE-FG02-01ER63201 to G.J.O., partial). Funding for open access charge: Gary J. Olsen.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Claudia I. Reich for suggestions, assistance with the experiments, unpublished results, and critical review of the article. We thank Ying Jiang, James J. Davis, and other members of the laboratory for helpful discussions. We thank David E. Graham (University of Texas) for providing his curated translation start locations for M. jannaschii proteins. We also thank Carol S. Giometti, Sandra L. Tollaksen, Gyorgy Babnigg (all at Argonne National Laboratory), Hanjo Lim, Wenhong Zhu and John R. Yates, 3rd (all at Scripps Research Institute) for sharing their unpublished observations.
REFERENCES
- 1.Soppa J. Basal and regulated transcription in Archaea. Adv. Appl. Microbiol. 2001;50:171–217. doi: 10.1016/s0065-2164(01)50006-4. [DOI] [PubMed] [Google Scholar]
- 2.Zillig W, Palm P, Langer D, Klenk HP, Lanzendorfer M, Hudepohl U, Hain J. RNA polymerases and transcription in archaebacteria. Biochem. Soc. Symp. 1992;58:79–88. [PubMed] [Google Scholar]
- 3.Langer D, Hain J, Thuriaux P, Zillig W. Transcription in Archaea: similarity to that in Eucarya. Proc. Natl Acad. Sci. USA. 1995;92:5768–5772. doi: 10.1073/pnas.92.13.5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Darcy TJ, Hausner W, Awery DE, Edwards AM, Thomm M, Reeve JN. Methanobacterium thermoautotrophicum RNA polymerase and transcription in vitro. J. Bacteriol. 1999;181:4424–4429. doi: 10.1128/jb.181.14.4424-4429.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Best AA, Olsen GJ. Similar subunit architecture of archaeal and eukaryal RNA polymerases. FEMS Microbiol. Lett. 2001;195:85–90. doi: 10.1111/j.1574-6968.2001.tb10502.x. [DOI] [PubMed] [Google Scholar]
- 6.Soppa J. Normalized nucleotide frequencies allow the definition of archaeal promoter elements for different archaeal groups and reveal base-specific TFB contacts upstream of the TATA box. Mol. Microbiol. 1999;31:1589–1592. doi: 10.1046/j.1365-2958.1999.01274.x. [DOI] [PubMed] [Google Scholar]
- 7.Reiter WD, Hudepohl U, Zillig W. Mutational analysis of an archaebacterial promoter: essential role of a TATA box for transcription efficiency and start-site selection in vitro. Proc. Natl Acad. Sci. USA. 1990;87:9509–9513. doi: 10.1073/pnas.87.24.9509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown JW, Daniels CJ, Reeve JN. Gene structure, organization, and expression in archaebacteria. Crit. Rev. Microbiol. 1989;16:287–338. doi: 10.3109/10408418909105479. [DOI] [PubMed] [Google Scholar]
- 9.Thomm M, Wich G. An archaebacterial promoter element for stable RNA genes with homology to the TATA box of higher eukaryotes. Nucleic Acids Res. 1988;16:151–163. doi: 10.1093/nar/16.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wich G, Hummel H, Jarsch M, Bar U, Böck A. Transcription signals for stable RNA genes in Methanococcus. Nucleic Acids Res. 1986;14:2459–2479. doi: 10.1093/nar/14.6.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reiter WD, Palm P, Zillig W. Analysis of transcription in the archaebacterium Sulfolobus indicates that archaebacterial promoters are homologous to eukaryotic pol II promoters. Nucleic Acids Res. 1988;16:1–19. doi: 10.1093/nar/16.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slupska MM, King AG, Fitz-Gibbon S, Besemer J, Borodovsky M, Miller JH. Leaderless transcripts of the crenarchaeal hyperthermophile Pyrobaculum aerophilum. J. Mol. Biol. 2001;309:347–360. doi: 10.1006/jmbi.2001.4669. [DOI] [PubMed] [Google Scholar]
- 13.Brenneis M, Hering O, Lange C, Soppa J. Experimental characterization of cis-acting elements important for translation and transcription in halophilic Archaea. PLoS Genet. 2007;3:e229. doi: 10.1371/journal.pgen.0030229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Danner S, Soppa J. Characterization of the distal promoter element of halobacteria in vivo using saturation mutagenesis and selection. Mol. Microbiol. 1996;19:1265–1276. doi: 10.1111/j.1365-2958.1996.tb02471.x. [DOI] [PubMed] [Google Scholar]
- 15.Palmer JR, Daniels CJ. In vivo definition of an archaeal promoter. J. Bacteriol. 1995;177:1844–1849. doi: 10.1128/jb.177.7.1844-1849.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baliga NS, DasSarma S. Saturation mutagenesis of the TATA box and upstream activator sequence in the haloarchaeal bop gene promoter. J. Bacteriol. 1999;181:2513–2518. doi: 10.1128/jb.181.8.2513-2518.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hain J, Reiter WD, Hudepohl U, Zillig W. Elements of an archaeal promoter defined by mutational analysis. Nucleic Acids Res. 1992;20:5423–5428. doi: 10.1093/nar/20.20.5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li E, Reich CI, Olsen GJ. A whole-genome approach to identifying protein binding sites: promoters in Methanocaldococcus (Methanococcus) jannaschii. Nucleic Acids Res. 2008;36:6948–6958. doi: 10.1093/nar/gkm499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bult CJ, White O, Olsen GJ, Zhou L, Fleischmann RD, Sutton GG, Blake JA, FitzGerald LM, Clayton RA, Gocayne JD, et al. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 20.Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2007;35:D5–D12. doi: 10.1093/nar/gkl1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu W, Reich CI, Olsen GJ, Giometti CS, Yates JR., III Shotgun proteomics of Methanococcus jannaschii and insights into methanogenesis. J. Proteome Res. 2004;3:538–548. doi: 10.1021/pr034109s. [DOI] [PubMed] [Google Scholar]
- 22.Howland JL. The Surprising Archaea: Discovering Another Domain of Life. New York, NY: Oxford University Press; 2000. [Google Scholar]
- 23.Bensing BA, Meyer BJ, Dunny GM. Sensitive detection of bacterial transcription initiation sites and differentiation from RNA processing sites in the pheromone-induced plasmid transfer system of Enterococcus faecalis. Proc. Natl Acad. Sci. USA. 1996;93:7794–7799. doi: 10.1073/pnas.93.15.7794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rhodius VA, Suh WC, Nonaka G, West J, Gross CA. Conserved and variable functions of the σE stress response in related genomes. PLoS Biol. 2006;4:e2. doi: 10.1371/journal.pbio.0040002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tabansky I, Nurminsky DI. Mapping of transcription start sites by direct sequencing of SMART RACE products. Biotechniques. 2003;34:482. doi: 10.2144/03343bm06. 485–486. [DOI] [PubMed] [Google Scholar]
- 26.Bailey TL, Williams N, Misleh C, Li WW. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006;34:W369–W373. doi: 10.1093/nar/gkl198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Workman CT, Yin Y, Corcoran DL, Ideker T, Stormo GD, Benos PV. enoLOGOS: a versatile web tool for energy normalized sequence logos. Nucleic Acids Res. 2005;33:W389–W392. doi: 10.1093/nar/gki439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stormo GD, Fields DS. Specificity, free energy and information content in protein-DNA interactions. Trends Biochem. Sci. 1998;23:109–113. doi: 10.1016/s0968-0004(98)01187-6. [DOI] [PubMed] [Google Scholar]
- 29.Stormo GD. DNA binding sites: representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- 30.Schneider TD. Theory of molecular machines. II. Energy dissipation from molecular machines. J. Theor. Biol. 1991;148:125–137. doi: 10.1016/s0022-5193(05)80467-9. [DOI] [PubMed] [Google Scholar]
- 31.Erill I, O'Neill MC. A reexamination of information theory-based methods for DNA-binding site identification. BMC Bioinformatics. 2009;10:57. doi: 10.1186/1471-2105-10-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li E. PhD Dissertation. Urbana, IL: University of Illinois; 2007. Non-coding genomics of Methanocaldococcus jannaschii: a survey of promoters, non-coding RNA genes, and repetitive DNA elements. [Google Scholar]
- 33.Hausner W, Frey G, Thomm M. Control regions of an archaeal gene. A TATA box and an initiator element promote cell-free transcription of the tRNAVal gene of Methanococcus vannielii. J. Mol. Biol. 1991;222:495–508. doi: 10.1016/0022-2836(91)90492-o. [DOI] [PubMed] [Google Scholar]
- 34.Staden R. Computer methods to locate signals in nucleic acid sequences. Nucleic Acids Res. 1984;12:505–519. doi: 10.1093/nar/12.1part2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hertz GZ, Stormo GD. Escherichia coli promoter sequences: analysis and prediction. Methods Enzymol. 1996;273:30–42. doi: 10.1016/s0076-6879(96)73004-5. [DOI] [PubMed] [Google Scholar]
- 36.Shultzaberger RK, Bucheimer RE, Rudd KE, Schneider TD. Anatomy of Escherichia coli ribosome binding sites. J. Mol. Biol. 2001;313:215–228. doi: 10.1006/jmbi.2001.5040. [DOI] [PubMed] [Google Scholar]
- 37.Shultzaberger RK, Chen Z, Lewis KA, Schneider TD. Anatomy of Escherichia coli σ70 promoters. Nucleic Acids Res. 2007;35:771–788. doi: 10.1093/nar/gkl956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Celesnik H, Deana A, Belasco JG. Initiation of RNA decay in Escherichia coli by 5′ pyrophosphate removal. Mol. Cell. 2007;27:79–90. doi: 10.1016/j.molcel.2007.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ouhammouch M, Dewhurst RE, Hausner W, Thomm M, Geiduschek EP. Activation of archaeal transcription by recruitment of the TATA-binding protein. Proc. Natl Acad. Sci. USA. 2003;100:5097–5102. doi: 10.1073/pnas.0837150100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Badger JH, Olsen GJ. CRITICA: coding region identification tool invoking comparative analysis. Mol. Biol. Evol. 1999;16:512–524. doi: 10.1093/oxfordjournals.molbev.a026133. [DOI] [PubMed] [Google Scholar]
- 41.Lagrange T, Kapanidis AN, Tang H, Reinberg D, Ebright RH. New core promoter element in RNA polymerase II-dependent transcription: sequence-specific DNA binding by transcription factor IIB. Genes Dev. 1998;12:34–44. doi: 10.1101/gad.12.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Littlefield O, Korkhin Y, Sigler PB. The structural basis for the oriented assembly of a TBP/TFB/promoter complex. Proc. Natl Acad. Sci. USA. 1999;96:13668–13673. doi: 10.1073/pnas.96.24.13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Renfrow MB, Naryshkin N, Lewis LM, Chen HT, Ebright RH, Scott RA. Transcription factor B contacts promoter DNA near the transcription start site of the archaeal transcription initiation complex. J. Biol. Chem. 2004;279:2825–2831. doi: 10.1074/jbc.M311433200. [DOI] [PubMed] [Google Scholar]
- 44.Tsai FT, Sigler PB. Structural basis of preinitiation complex assembly on human pol II promoters. EMBO J. 2000;19:25–36. doi: 10.1093/emboj/19.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bartlett MS, Thomm M, Geiduschek EP. Topography of the euryarchaeal transcription initiation complex. J. Biol. Chem. 2004;279:5894–5903. doi: 10.1074/jbc.M311429200. [DOI] [PubMed] [Google Scholar]
- 46.Shine J, Dalgarno L. Terminal-sequence analysis of bacterial ribosomal RNA. Correlation between the 3′-terminal-polypyrimidine sequence of 16-S RNA and translational specificity of the ribosome. Eur. J. Biochem. 1975;57:221–230. doi: 10.1111/j.1432-1033.1975.tb02294.x. [DOI] [PubMed] [Google Scholar]
- 47.Shultzaberger RK, Roberts LR, Lyakhov IG, Sidorov IA, Stephen AG, Fisher RJ, Schneider TD. Correlation between binding rate constants and individual information of E. coli Fis binding sites. Nucleic Acids Res. 2007;35:5275–5283. doi: 10.1093/nar/gkm471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roulet E, Bucher P, Schneider R, Wingender E, Dusserre Y, Werner T, Mermod N. Experimental analysis and computer prediction of CTF/NFI transcription factor DNA binding sites. J. Mol. Biol. 2000;297:833–848. doi: 10.1006/jmbi.2000.3614. [DOI] [PubMed] [Google Scholar]
- 49.Wiley SR, Kraus RJ, Mertz JE. Functional binding of the “TATA” box binding component of transcription factor TFIID to the –30 region of TATA-less promoters. Proc. Natl Acad. Sci. USA. 1992;89:5814–5818. doi: 10.1073/pnas.89.13.5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wen JD, Gray DM. Selection of genomic sequences that bind tightly to Ff gene 5 protein: primer-free genomic SELEX. Nucleic Acids Res. 2004;32:e182. doi: 10.1093/nar/gnh179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vierke G, Engelmann A, Hebbeln C, Thomm M. A novel archaeal transcriptional regulator of heat shock response. J. Biol. Chem. 2003;278:18–26. doi: 10.1074/jbc.M209250200. [DOI] [PubMed] [Google Scholar]
- 52.Kosa PF, Ghosh G, DeDecker BS, Sigler PB. The 2.1-Å crystal structure of an archaeal preinitiation complex: TATA-box-binding protein/transcription factor (II)B core/TATA-box. Proc. Natl Acad. Sci. USA. 1997;94:6042–6047. doi: 10.1073/pnas.94.12.6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Giometti CS, Reich C, Tollaksen S, Babnigg G, Lim H, Zhu W, Yates J, Olsen G. Global analysis of a “simple” proteome: Methanococcus jannaschii. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2002;782:227–243. doi: 10.1016/s1570-0232(02)00568-8. [DOI] [PubMed] [Google Scholar]
- 54.Huerta AM, Collado-Vides J. Sigma70 promoters in Escherichia coli: specific transcription in dense regions of overlapping promoter-like signals. J. Mol. Biol. 2003;333:261–278. doi: 10.1016/j.jmb.2003.07.017. [DOI] [PubMed] [Google Scholar]
- 55.Hannenhalli S, Levy S. Promoter prediction in the human genome. Bioinformatics. 2001;17(Suppl 1):S90–S96. doi: 10.1093/bioinformatics/17.suppl_1.s90. [DOI] [PubMed] [Google Scholar]
- 56.Burden S, Lin YX, Zhang R. Improving promoter prediction for the NNPP2.2 algorithm: a case study using Escherichia coli DNA sequences. Bioinformatics. 2005;21:601–607. doi: 10.1093/bioinformatics/bti047. [DOI] [PubMed] [Google Scholar]
- 57.Hanzelka BL, Darcy TJ, Reeve JN. TFE, an archaeal transcription factor in Methanobacterium thermoautotrophicum related to eucaryal transcription factor TFIIEα. J. Bacteriol. 2001;183:1813–1818. doi: 10.1128/JB.183.5.1813-1818.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bell SD, Brinkman AB, van der Oost J, Jackson SP. The archaeal TFIIEα homologue facilitates transcription initiation by enhancing TATA-box recognition. EMBO Rep. 2001;2:133–138. doi: 10.1093/embo-reports/kve021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Richard DJ, Bell SD, White MF. Physical and functional interaction of the archaeal single-stranded DNA-binding protein SSB with RNA polymerase. Nucleic Acids Res. 2004;32:1065–1074. doi: 10.1093/nar/gkh259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hausner W, Thomm M. Events during initiation of archaeal transcription: open complex formation and DNA-protein interactions. J. Bacteriol. 2001;183:3025–3031. doi: 10.1128/JB.183.10.3025-3031.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grunberg S, Bartlett MS, Naji S, Thomm M. Transcription factor E is a part of transcription elongation complexes. J. Biol. Chem. 2007;282:35482–35490. doi: 10.1074/jbc.M707371200. [DOI] [PubMed] [Google Scholar]
- 62.Robert F, Forget D, Li J, Greenblatt J, Coulombe B. Localization of subunits of transcription factors IIE and IIF immediately upstream of the transcriptional initiation site of the adenovirus major late promoter. J. Biol. Chem. 1996;271:8517–8520. doi: 10.1074/jbc.271.15.8517. [DOI] [PubMed] [Google Scholar]
- 63.Okuda M, Watanabe Y, Okamura H, Hanaoka F, Ohkuma Y, Nishimura Y. Structure of the central core domain of TFIIEβ with a novel double-stranded DNA-binding surface. EMBO J. 2000;19:1346–1356. doi: 10.1093/emboj/19.6.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bell SD, Jackson SP. The role of transcription factor B in transcription initiation and promoter clearance in the archaeon Sulfolobus acidocaldarius. J. Biol. Chem. 2000;275:12934–12940. doi: 10.1074/jbc.275.17.12934. [DOI] [PubMed] [Google Scholar]
- 65.Giometti CS, Reich CI, Tollaksen SL, Babnigg G, Lim H, Yates JR, 3rd, Olsen GJ. Structural modifications of Methanococcus jannaschii flagellin proteins revealed by proteome analysis. Proteomics. 2001;1:1033–1042. doi: 10.1002/1615-9861(200108)1:8<1033::AID-PROT1033>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 66.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 67.Cui Y, Wang Q, Stormo GD, Calvo JM. A consensus sequence for binding of Lrp to DNA. J. Bacteriol. 1995;177:4872–4880. doi: 10.1128/jb.177.17.4872-4880.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ouhammouch M, Geiduschek EP. A thermostable platform for transcriptional regulation: the DNA-binding properties of two Lrp homologs from the hyperthermophilic archaeon Methanococcus jannaschii. EMBO J. 2001;20:146–156. doi: 10.1093/emboj/20.1.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shimada T, Fujita N, Maeda M, Ishihama A. Systematic search for the Cra-binding promoters using genomic SELEX system. Genes Cells. 2005;10:907–918. doi: 10.1111/j.1365-2443.2005.00888.x. [DOI] [PubMed] [Google Scholar]
- 70.Shultzaberger RK, Schneider TD. Using sequence logos and information analysis of Lrp DNA binding sites to investigate discrepancies between natural selection and SELEX. Nucleic Acids Res. 1999;27:882–887. doi: 10.1093/nar/27.3.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tolstrup N, Sensen CW, Garrett RA, Clausen IG. Two different and highly organized mechanisms of translation initiation in the archaeon Sulfolobus solfataricus. Extremophiles. 2000;4:175–179. doi: 10.1007/s007920070032. [DOI] [PubMed] [Google Scholar]
- 72.Jones WJ, Leigh JA, Mayer F, Woese CR, Wolfe RS. Methanococcus jannaschii sp. nov., an extremely thermophilic methanogen from a submarine hydrothermal vent. Arch. Microbiol. 1983;136:254–261. [Google Scholar]
- 73.Klein RJ, Misulovin Z, Eddy SR. Noncoding RNA genes identified in AT-rich hyperthermophiles. Proc. Natl Acad. Sci. USA. 2002;99:7542–7547. doi: 10.1073/pnas.112063799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schattner P. Searching for RNA genes using base-composition statistics. Nucleic Acids Res. 2002;30:2076–2082. doi: 10.1093/nar/30.9.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Morris DR, Geballe AP. Upstream open reading frames as regulators of mRNA translation. Mol. Cell Biol. 2000;20:8635–8642. doi: 10.1128/mcb.20.23.8635-8642.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vilela C, McCarthy JE. Regulation of fungal gene expression via short open reading frames in the mRNA 5′ untranslated region. Mol. Microbiol. 2003;49:859–867. doi: 10.1046/j.1365-2958.2003.03622.x. [DOI] [PubMed] [Google Scholar]
- 78.Iacono M, Mignone F, Pesole G. uAUG and uORFs in human and rodent 5′ untranslated mRNAs. Gene. 2005;349:97–105. doi: 10.1016/j.gene.2004.11.041. [DOI] [PubMed] [Google Scholar]
- 79.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.