

Abstract
Quantitative, chemically specific images of biological systems would be invaluable in unraveling the bioinorganic chemistry of biological tissues. Here we report the spatial distribution and chemical forms of selenium in Astragalus bisulcatus (two-grooved poison or milk vetch), a plant capable of accumulating up to 0.65% of its shoot dry biomass as Se in its natural habitat. By selectively tuning incident x-ray energies close to the Se K-absorption edge, we have collected quantitative, 100-μm-resolution images of the spatial distribution, concentration, and chemical form of Se in intact root and shoot tissues. To our knowledge, this is the first report of quantitative concentration-imaging of specific chemical forms. Plants exposed to 5 μM selenate for 28 days contained predominantly selenate in the mature leaf tissue at a concentration of 0.3–0.6 mM, whereas the young leaves and the roots contained organoselenium almost exclusively, indicating that the ability to biotransform selenate is either inducible or developmentally specific. While the concentration of organoselenium in the majority of the root tissue was much lower than that of the youngest leaves (0.2–0.3 compared with 3–4 mM), isolated areas on the extremities of the roots contained concentrations of organoselenium an order of magnitude greater than the rest of the root. These imaging results were corroborated by spatially resolved x-ray absorption near-edge spectra collected from selected 100 × 100 μm2 regions of the same tissues.
Selenium is now known to play essential metabolic roles in most or perhaps all eukaryotes (1), but its toxic properties were recognized earlier in the poisoning of range animals eating selenium-hyperaccumulating plants (2). Hyperaccumulating plants actively and specifically accumulate high levels of metal or metalloid compounds into their tissues (3), and there is considerable interest in the biochemical mechanisms of hyperaccumulation, particularly with regard to potential phytoremediation strategies for contaminated environments (4) and for use as dietary supplements or “nutraceuticals” (5). The two-grooved milk- or poison-vetch, Astragalus bisulcatus, also known by cattle ranchers as “locoweed,” hyperaccumulates up to 0.65% of its shoot dry biomass as Se (6) from inorganic selenium naturally present in soil in parts of its range. The major Se-containing compound in the leaves is Se-methylselenocysteine, with smaller amounts of selenocystathione and γ-glutamyl-Se-methylselenocysteine localized in the seed pods (7). Other organoselenium compounds are also produced but in smaller amounts (dimethyldiselenide in particular is clearly indicated by its characteristic odor). The purification, cloning, and characterization of a specific selenocysteine methyltransferase involved in synthesizing Se-methylselenocysteine from selenocysteine and S-methylmethionine has recently been reported (8). Despite this progress, much remains to be learned about the details of Se uptake, transport, and storage in hyperaccumulating plants. X-ray absorption spectroscopy provides a unique probe for examining the chemical species of an element present in whole tissues of plants (e.g., refs. 9–11). Here we present images of the concentration and chemical biotransformation of selenate in A. bisulcatus.
Materials and Methods
Plant Growth.
A. bisulcatus was cultivated by using a modification of the method previously described (5). Seedlings were grown initially in culture solution supplemented with 0.1 μM K2SeO4, then at weekly intervals the concentration of K2SeO4 was increased through 1.0, 2.0, 2.5, and 5.0 μM. All plants were illuminated with a mixture of tungsten and cool white fluorescent lamps at a light intensity of 150 μE (1 E = 1 mol of photons) and a day/night cycle of 16/8 h at 25/20°C. After 28 days exposure to 5 μM K2SeO4, plants were transported live to Stanford Synchrotron Radiation Laboratory (SSRL).
Data Collection.
Chemically specific imaging and spatially resolved x-ray absorption spectra were collected on the Structural Molecular Biology beamline 9-3 of SSRL, with a double crystal Si(220) monochromator, an upstream vertically collimating, harmonic-rejecting Rh-coated mirror and a downstream refocusing Rh-coated mirror. The beam was collimated to 100 × 100 μm2 by using two sets of motorized x–y tantalum slits, calibrated by using the Fraunhöfer interference fringes from a He–Ne laser. N2-filled ion chambers were located downstream of the double slits and downstream of the sample to measure incident and transmitted intensities, respectively. Plant tissues were mounted onto Mylar tape, covered with 6.3-μm polypropylene to minimize sample dehydration, and mounted at 90° to the incident beam on a stepper-motor-driven x–y positioning stage to achieve raster scanning. Se Kα fluorescence and scatter from the sample were monitored by using a 30-element germanium detector (Canberra Industries, Meriden, CT) at 125° to the transmitted beam to collect the forward-fluorescing/scattering signals. Beam exposure for the imaging experiments was less than 5 sec per pixel at each energy, whereas for the spatially resolved spectroscopy it varied from two 12-min scans for the most concentrated spots to three 20-min scans for the more dilute areas. Energy was calibrated by using the spectrum of hexagonal Se(0), assuming the first inflection to be 12658 eV (1 eV = 1.60219 × 10−19 J). X-ray absorption spectroscopy of bulk tissues was carried out on SSRL beamline 7-3 at 10K essentially as described before (5, 12).
Chemically Specific Image Data Reduction.
Briefly, the method for obtaining chemically specific concentration images of two chemical species requires spatially scanning the sample at two fixed energies that are sensitive to the different species present. This is similar to the methods used by others (13, 14) to obtain relative concentrations. In our case, 12661.2 and 12667.5 eV maximize the differences between selenomethionine and selenate (Fig. 1). At each pixel we recorded the Se Kα fluorescence- and scatter-windowed counts, summed over all detector channels and divided by I0, together with the x-ray absorbance. The windowed fluorescence images were corrected for scatter contributions, and the absorbance was background-corrected and converted to an effective thickness. The corrected fluorescence intensities at the two energies, together with a concentration calibration and the thickness, were then combined to yield an estimate of absolute concentrations of the two species at each pixel. A customized suite of computer programs was written to accomplish these steps, which are detailed below and shown pictorially in Fig. 2.
Figure 1.

Se K x-ray absorption near-edge spectrum of A. bisulcatus root tissue treated with 25 μM for 4 days (points) fit to the spectra of models. The solid line shows the best fit, which corresponds to 53% selenate, 47% selenomethionine (dashed and dot-dash lines, respectively, both scaled according to their contributions) and the dotted line shows the difference spectrum. The vertical lines show the energies of 12661.2 and 12667.5 eV, which were used for the chemically specific imaging. (Inset) Emission spectrum at 12667.5 eV for a single element of the 30-element detector for 0.2 μl of 10 mM selenate on filter paper. The fluorescence window (FW) and scatter window (SW) are indicated, together with the Compton and Rayleigh scattering and the Se fluorescence lines.
Figure 2.

Data reduction scheme for the method of chemically specific imaging. See text for details. E1 and E2 indicate energies of 12661.2 and 12667.5 eV, respectively, for the collection of images. The four fluorescence images have the same scale, the unit of which is proportional to counts divided by I0. The same units are used for the scatter image. Obs., observed; Corr., corrected; Conc., concentration.
Fluorescent Window Calibration.
Fig. 1 Inset shows the emission spectrum of 10 mM selenate on filter paper. Despite good energy resolution, some scatter counts (the tail of the Compton scattering) fall within the fluorescence window (FW) and conversely some fluorescence counts (mainly from the Se Kβ line) fall within the scatter window (SW). In general, Fo = F + Fs and So = S + Sf, where Fo and So are the observed intensities (normalized by the incident intensity, I0) in the FW and SW, respectively. F and S are the corrected intensities of fluorescence in the FW and scatter in the SW, respectively, and Fs and Sf are the intensities of scatter in the FW and fluorescence in the SW, respectively. It is assumed that, for modest and comparable count rates, Sf = τfF and Fs = τsS, where τs and τf are energy-dependent constants. Solving:
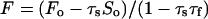 |
1 |
Raster scans similar to those of the plant specimens were used to determine τs and τf, by using a 1 × 1 mm2 filter paper wetted with 0.2 μl of 10 mM selenate, and a Se-free control. τs was determined as the mean of Fo/So = Fs/S for the Se-free scan. τf was determined as (So − Sm)/(Fo − Fm) = Sf/F for the Se-doped sample, where Fm and Sm are the means of Fo and So for the Se-free sample. Values of 0.038 and 0.049 were obtained for τs and τf, respectively, at 12667.5 eV. The observed fluorescence intensities for all imaging data sets were corrected by using Eq. 1. Even though τs is small, the effect of the scatter in Eq. 1 is significant because of the magnitude of So. Fig. 2 compares the observed fluorescence images at the two energies with the corresponding corrected images. The observed scatter is also shown.
Derivation of Effective Thickness.
When an absorbance signal is measured by using ion chambers (I0 upstream and I1 downstream), the measured absorbance, Ao = log(I0/I1), for each pixel of a specimen can be written as Ao = A + Ar + ki, where A is the absorbance of the specimen at that pixel, Ar is the absorbance of other things in the beam-path (e.g., mounting tape, air, windows), and the constant ki accounts for different ion chamber responses. Simple subtraction of an energy-dependent background absorbance (Ab = Ar + ki) of everything except the specimen yields the specimen absorbance, A, at each pixel. This may also be given by A = σρt, where σ is the energy-dependent absorption cross section, ρ is the density, and t is the effective sample thickness. Because the experiment is in the dilute limit, the element of interest (Se) contributes negligibly to the absorbance. The reasonable approximation is also made that the plant material has ρ and σ (15) of water. Hence, from the absorbance image a map of the effective sample thickness is determined using:
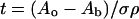 |
2 |
and is shown for our example in Fig. 2.
Calibration of Molar Quantities.
The amount of Se per pixel was determined from the fluorescence intensity by using a calibration raster scan. Filter paper wetted with selenate solution (0.2 μl, 10 mM) was scanned as for a plant sample. The fluorescence was corrected using Eq. 1 and FT, and the total corrected fluorescence integrated over all pixels was calculated. A quantitation constant kq was then determined as kq = FT/MIcc, where M is the number of moles deposited and Icc is the intensity of the calibrant species at the calibrant energy, normalized by the spline (see below).
Determination of Concentrations of Chemical Species.
At each pixel we wish to determine mA and mB, the molar quantities, respectively, of species A and B. The true fluorescence per pixel at energy j, Fj (from Eq. 1) may be written as Fj = mAkqIAj + mBkqIBj, where Iij is the normalized intensity of component i (i is A or B) at energy j. Iij is determined independently from spectra of the pure components by using the spline normalization (see below). Solving for mi, we obtain mA = (F1IB2 − F2IB1)/K and mB = (F2IA1 − F1IA2)/K, where K = kq(IA1IB2 − IA2IB1). The concentrations per pixel of species A and B, cA and cB, can be simply determined from the molar quantities mi, the effective thickness t (Eq. 2), and the pixel area, a:
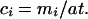 |
3 |
The concentration maps of organoselenium, selenate, and total Se are shown in Fig. 2. To extend the two-component method to more complex mixtures would require measurement of additional energies where the contributions of the different species were distinct.
Near-Edge Spectra Data Reduction.
X-ray absorption spectra (both bulk and spatially resolved) were collected to k ≥ 6 Å−1 to ensure proper spline normalization. Spectra were processed by using exafspak (http://ssrl.slac.stanford.edu/exafspak.html). After calibration and averaging of independent reproducible sweeps, a pre-edge background was subtracted, a low-order spline function was fitted to the post-edge region, and the edge was normalized to the spline value at 12695 eV. Near-edge spectra were quantitatively curve-fit by using exafspak the program datfit. Here, the spectrum of a plant sample containing unknown Se species is fit to a linear combination of the spectra of standard Se compounds by using a least-squares minimization procedure (12). As all of the spectra have been normalized to the spline, the fractional contribution of a standard spectrum to the fit is equivalent to the fractional abundance of Se in that chemical species in the plant sample.
Results
X-Ray Absorption Spectroscopy on Bulk Tissues.
A representative x-ray absorption near-edge spectrum of root tissue from A. bisulcatus plants initially grown for 3 weeks in the absence of selenate and then exposed to 25 μM selenate in the hydroponic medium for 4 days is shown in Fig. 1. The result of fitting to the spectra of aqueous selenate and selenomethionine is also shown. Although A. bisulcatus is known to accumulate Se-methylselenocysteine (6), selenomethionine was used as a commercially available surrogate—because the local Se environments are very similar (both R-CH2-Se-CH3), the near-edge spectra are also expected to be very similar. The spectrum of Fig. 1 is well modeled by linear combinations of selenate and selenomethionine, as were all of the plant spectra; other forms such as selenite were not detected. Extended x-ray absorption fine structure analyses (data not shown) gave complementary results, with data well modeled by mixtures of Se–C and Se–O interactions, in proportions agreeing very well (within 5%) with the near-edge fitting results. Se–Se interactions (which would indicate the substantial presence of diselenides) were not detected. It was observed that longer times of exposure to selenate yielded greater proportions of organoselenium in both root and shoot tissue (data not shown).
Chemically Specific Imaging.
The montage in Fig. 3 compares quantitative, chemically specific images of leaves and roots from two A. bisulcatus plants of the same age exposed to gradually increased levels of selenate (see Materials and Methods). The figure shows the optical micrograph, the effective thickness, and the concentration of selenate and organoselenium. Leaves of three different ages are shown: mature leaves, dating from exposure to low selenate levels; intermediate leaves, which were small (and still expanding) at the time of peak selenate exposure; and young leaves, which had developed during exposure to 5 μM selenate.
Figure 3.

Chemically specific concentration images of different parts of A. bisulcatus. From left to right: optical micrograph; effective tissue thickness; concentration of organoselenium; concentration of selenate. Top to bottom: mature, intermediate and young leaves, and roots. The mature leaves, young leaves, and roots were taken from the same plant, the mature leaves from the lowest shoot and the young leaves from the highest shoot of the same plant branch.
The mature leaves show some shape distortion (see optical micrograph) and slight discoloration. They show a much greater proportion of selenate than any other specimen (approaching 100% in the leaf blade), and the total concentration of Se in this sample was lowest among the leaves studied (<0.6 mM in the leaf blade). Selenate was most concentrated in the leaf blade (0.3–0.6 mM), especially toward the outer edges, less concentrated in the midvein (0.15–0.25 mM) and stem (0.04–0.12 mM), and apparently absent in the petiole. The concentration of organoselenium was low (<0.1 mM) in all areas except the base and periphery of the leaf and the petiole, where it was 0.2–0.4 mM.
The intermediate leaves were rounded, large, and dark green; they contained more Se than the mature leaves (0.5–1.1 mM in the leaf blade). Selenate was notably more concentrated in the leaf blade (0.15–0.30 mM), where it accounted for some 20–40% of the total, than it was in the stem, petiole, or midvein (<0.1 mM, <10% of total). Organoselenium was present at 0.4–0.8 mM in the leaf blade, with higher concentrations along the periphery of the leaf, the edge of the stem, and at the petiole (typically up to 2 mM, although the highest may be as much as 3.5 mM).
The youngest leaves were paler green, recently expanded, and pointed. They had the highest Se content (3–4 mM in the leaf blade), which was almost exclusively organoselenium. The concentration appeared to be higher toward the periphery of the leaves. Selenate concentrations were also highest in the leaf blade but nowhere exceeded 0.25 mM, or 8% of the total. The concentration of organoselenium was a little less in the midvein (1.8–2.2 mM) and in the stem and petiole (0.8–1.8 mM). The fraction of selenate is largely uniform except in the petiole, where it is very close to zero.
The roots of the plant from which the mature and young leaves were harvested showed a background total Se level of only 0.2–0.3 mM, which was essentially all organic. However, they also exhibited small regions, typically <400 μm across, where the Se concentration, again all organic, was substantially higher (1–4 mM), with the highest concentration at the periphery. These areas may correspond to root meristems, the area of cell division. Note that the highest concentration of Se in the hydroponic solution was 5 μM.
Spatially Resolved Spectroscopy.
The identity of Se species and the fraction of selenate at selected locations on the specimens were confirmed by recording x-ray absorption near-edge spectra by using a 100 × 100 μm2 beam. The spectra and their corresponding locations on the leaf samples are shown in Fig. 4. They are in excellent agreement with the chemically specific imaging data, and are all accurately modeled by linear combinations of selenate and selenomethionine alone.
Figure 4.

Spatially resolved x-ray absorption near-edge spectra recorded by using a 100 × 100 μm2 beam from selected portions of mature (Top), intermediate (Middle), and young (Bottom) leaves. The spectra of selenate and selenomethionine (org. Se) are also included. The Insets show maps of the percentage of total Se as organoselenium (balance selenate) and indicate the spatial location for the spectra. The percentages of selenate in each leaf specimen as determined by near-edge fitting for a (stem), b (petiole), c (mid-vein), and d (leaf blade) respectively are: mature 36, 0, 69, 88; intermediate 3, 0, 7, 28; young 3, 0, 1, 1. Spectra from two locations on the root (main part of the root, and a high-concentration spot, data not shown) both yielded 0% selenate.
Discussion
Chemically specific imaging and spatially resolved spectroscopy have revealed some of the complexity of Se transformations in the hyperaccumulator A. bisulcatus. Our approach combines the quantitative x-ray absorption spectroscopy speciation of Se, which has been proven in environmental (12, 16, 17) and biological (5, 11) studies, with spatial resolution to provide two-dimensional maps of the location of chemical species. Although two-dimensional elemental mapping can be achieved through other methods, chemically specific mapping of two or more species requires monochromatic x-rays and has been applied in only a few cases (13, 14, 18, 19). A more common method is elemental mapping in combination with micro-x-ray absorption spectroscopy to provide speciation at specific locations of interest (e.g., ref. 20). However, none of these studies quantitates the speciation in terms of absolute concentrations, and, to our knowledge, this work provides the first example of this approach.
Selenium was provided to the plants as selenate, and may have been taken up by the sulfate-uptake system (21, 22), although a more specific uptake mechanism cannot be excluded. Our results indicate that the fate of this selenate is quite different in different tissues of the plant, and indeed in different parts of those tissues. Selenate is clearly transported throughout the plant (Figs. 3 and 4); however, the distribution between selenate and organic forms varies depending on the tissue and its developmental stage. This suggests that in planta chemical reduction of selenate to organic forms must be tissue-specific, inducible, or developmentally dependent. The absence of intermediate oxidation states such as selenite suggests that selenate reduction is rate-limiting in the conversion to organic forms.
A critical step in the biotransformation of selenate is the initial two-electron reduction to selenite. Hyperaccumulating plants might achieve this in at least three different ways: by substitution for sulfate within the sulfate reduction pathway [reduction by ATP sulfurylase/adenosine 5′-phosphosulfate (APS) reductase (23, 24)], by substitution for nitrate in the nitrate uptake pathway [microbial nitrate reductases can reduce selenate (25)], or by a specific selenate reductase. For nonhyperaccumulating plants there is good evidence that selenate reduction occurs via substitution for sulfate in the ATP sulfurylase/APS reductase system, and that this is the rate-limiting step in selenate transformation (11, 23, 26, 27). Although it is often assumed that analogous biochemistry accounts for selenate reduction in hyperaccumulating plants, the evidence presented here (that the ability to reduce selenate is inducible or developmentally dependent) suggests a different mechanism, as significant ATP sulfurylase/APS reductase activity for sulfate must always be present for healthy plant growth. The presence of a specific inducible selenate reductase system seems a plausible explanation, either a modified ATP sulfurylase/APS reductase couple with greater selenate specificity or a previously unidentified selenate reductase. A highly specific respiratory selenate reductase, having negligible activity for sulfate, has recently been purified from the proteobacterium Thauera selenatis (28).
We can also make further, more tentative, deductions about the physiology of Se detoxification. Transport within the xylem depends on the rate of transpiration, and all tissues receive whatever the xylem is transporting. Because the mature leaves have predominantly selenate in the leaf blade and a relatively low total Se content, it seems unlikely that the roots transform selenate to organic forms before transport in the xylem. Older leaves would be expected to have lower rates of transpiration than younger ones, resulting in reduced transport of Se and the observed lower Se content. The intermediate-age leaves had more total Se than the oldest, and the majority of this Se was in the organic form, hence, the selenate-reduction pathway must have been present in these younger tissues. The significant fraction of selenate suggests either that the inorganic form is somehow partitioned (instead of converted) to reduce its toxic effects, or that some, but not all, cells in the intermediate leaves have the pathway induced. In either case, after the selenate-reduction pathway is induced, subsequent selenate is converted to Se-methylselenocysteine and stored, resulting in the observed mixed composition. The highest concentration of Se in the youngest leaves is consistent with the highest rates of transpiration, and thus of Se delivery. The young leaves developed during exposure of the plant to high levels of selenate, and their selenate-reduction pathway appears to be well developed; they show almost exclusively organic Se.
Selenate is remarkably absent in all of the petioles investigated—even those in the old leaves—and the stems of all three leaves show very similar (low) levels of selenate. The organic Se concentrations in the midvein, relative to the blade, are comparable for the intermediate leaves but substantially higher for the young leaves. The increased concentrations of Se at the periphery of the leaves suggests the possibility of Se storage in the trichomes (leaf hairs), as has been observed for cadmium accumulated by Brassica juncea (9). Higher resolution experiments (in progress) will assist our understanding of all these phenomena.
The roots had relatively low levels of Se, but almost all of it was present in the organic form. Possibly, organic forms of Se produced in the roots by reduction of selenate are compartmentalized within the root tissue for storage, whereas unmodified selenate passes through the root into the xylem for transport to the leaves. Alternatively, the root cells may receive organic Se via phloem transport from the younger leaves. Although we saw no direct evidence for this, it would be equivalent to the recent finding that S-methylmethionine is a phloem-transportable form of sulfur in plants (29). In either case it is possible that Se-methylselenocysteine localized in these areas of rapid cell division deters herbivory, protecting the sensitive growing tissues of the root.
We anticipate that the quantitative, chemically specific imaging described here will be developed into a broadly useful new tool of general applicability. It should be generally applicable to materials for which a matrix density and absorption cross section can be approximated, to materials in which the element of interest is in the dilute limit, and to materials which are sufficiently thin that both the incident and fluorescent photon beams can penetrate without substantial attenuation. The latter depends both on the absorption edge energy and on the specimen thickness, so that whereas the method could in principle be applied to imaging any element, in practice it will be easier to image those with Z > 25. It should be possible to extend the approach to more complex mixtures if the spectra of the components are sufficiently distinct. Selenium, sulfur, and arsenic are particularly favorable elements for this approach, because different chemical forms of these elements display a broad diversity of spectra. While other elements may display less dramatic differences for different chemical forms, in many cases there will be a suite of compounds that will be different enough in their spectroscopy that the technique should be valuable.
Acknowledgments
We are grateful to the SSRL staff and V. F. Pickering for their assistance and encouragement, and to C. Skinner for collecting the Astragalus seeds in Big Hollow, Wyoming. SSRL is funded by the Department of Energy, Offices of Basic Energy Sciences and Biological and Environmental Research; the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program; and the National Institute of General Medical Sciences. D.E.S. was supported by the Department of Energy Environmental Management Science program (Grant DE-FG07–98ER20295).
Abbreviation
- SSRL
Stanford Synchrotron Radiation Laboratory
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.200244597.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.200244597
References
- 1.Stadtman T C. Annu Rev Biochem. 1996;65:83–100. doi: 10.1146/annurev.bi.65.070196.000503. [DOI] [PubMed] [Google Scholar]
- 2.Beath O A, Draize J H, Eppson H F, Gilbert C S, McCreary O C. J Am Pharm Assoc. 1934;23:94–97. [Google Scholar]
- 3.Baker A J M, Brooks R R. Biorecovery. 1989;1:81–126. [Google Scholar]
- 4.Salt D E, Smith R D, Raskin I. Annu Rev Plant Physiol Plant Mol Biol. 1998;49:643–668. doi: 10.1146/annurev.arplant.49.1.643. [DOI] [PubMed] [Google Scholar]
- 5.Orser C S, Salt D E, Pickering I J, Prince R C, Epstein A, Ensley B D. J Medicinal Food. 1999;1:253–261. [Google Scholar]
- 6.Trelease S F, Di Somma A A, Jacobs A L. Science. 1960;132:618. doi: 10.1126/science.132.3427.618. [DOI] [PubMed] [Google Scholar]
- 7.Nigam S N, McConnell W B. Biochim Biophys Acta. 1969;192:185–190. doi: 10.1016/0304-4165(69)90354-7. [DOI] [PubMed] [Google Scholar]
- 8.Neuhierl B, Thanbichler T, Lottspeich F, Böck A. J Biol Chem. 1999;274:5407–5414. doi: 10.1074/jbc.274.9.5407. [DOI] [PubMed] [Google Scholar]
- 9.Salt D E, Prince R C, Pickering I J, Raskin I. Plant Physiol. 1995;109:1427–1433. doi: 10.1104/pp.109.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kramer U, Cotter-Howells J D, Charnock J M, Baker A J M, Smith J A C. Nature (London) 1996;379:635–638. [Google Scholar]
- 11.Pilon-Smits E A H, Hwang S, Lytle C M, Zhu Y, Tai J C, Bravo R C, Chen Y, Leustek T, Terry N. Plant Physiol. 1999;119:123–132. doi: 10.1104/pp.119.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pickering I J, Brown G E, Tokunaga T K. Environ Sci Technol. 1995;29:2456–2459. doi: 10.1021/es00009a043. [DOI] [PubMed] [Google Scholar]
- 13.Sutton S R, Bajt S, Delaney J, Schulze D, Tokunaga T. Rev Sci Instrum. 1995;66:1464–1467. [Google Scholar]
- 14.Nakai I, Numako C, Hatakawa S, Tsuchiyama A. J Trace Microprobe Tech. 1998;16:87–98. [Google Scholar]
- 15.McMaster W H, Kerr Del Grande N, Mallet J H, Hubell J H. Compilation of X-ray Cross Sections. Springfield, VA: National Technical Information Service; 1969. [Google Scholar]
- 16.Tokunaga T K, Pickering I J, Brown G E., Jr Soil Sci. 1996;60:781–790. [Google Scholar]
- 17.Tokunaga T K, Sutton S R, Bajt S, Nuessle P, Shea-McCarthy G. Environ Sci Technol. 1998;32:1092–1098. [Google Scholar]
- 18.Kinney J, Johnson Q, Nichols M C, Bonse U, Nusshardt R. Appl Opt. 1986;25:4583–4585. doi: 10.1364/ao.25.004583. [DOI] [PubMed] [Google Scholar]
- 19.Schulze D G, McCay-Buis T, Sutton S R, Huber D M. Phytopathology. 1995;85:990–994. [Google Scholar]
- 20.Yun W, Pratt S T, Miller R M, Cai Z, Hunter D B, Jarstfer A G, Kemner K M, Lai B, Lee H-R, Legnini D G, et al. J Synchrotron Radiat. 1998;5:1390–1395. doi: 10.1107/S0909049598007225. [DOI] [PubMed] [Google Scholar]
- 21.Barak P, Goldman I L. J Agric Food Chem. 1997;45:1290–1294. [Google Scholar]
- 22.Mikkeksen R L, Wan H F. Plant Soil. 1990;121:151–153. [Google Scholar]
- 23.Shrift A. Ann Rev Plant Physiol. 1969;20:475–495. [Google Scholar]
- 24.Setya A, Murillo M, Leustek T. Proc Natl Acad Sci USA. 1996;93:13383–13388. doi: 10.1073/pnas.93.23.13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Avazeri C, Turner R J, Pommier J, Weiner J H, Giordano G, Vermeglio A. Microbiology. 1997;143:1181–1189. doi: 10.1099/00221287-143-4-1181. [DOI] [PubMed] [Google Scholar]
- 26.Burnell J N. Plant Physiol. 1981;67:316–324. doi: 10.1104/pp.67.2.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw W H, Anderson J W. Biochem J. 1974;139:37–42. doi: 10.1042/bj1390037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schroder I, Rech S, Krafft T, Macy J M. J Biol Chem. 1997;272:23765–23768. doi: 10.1074/jbc.272.38.23765. [DOI] [PubMed] [Google Scholar]
- 29.Bourgis F, Roje S, Nuccio M L, Fisher D B, Tarczynski M C, Li C, Herschbach C, Rennenberg H, Pimenta M J, Shen T L, et al. Plant Cell. 1999;11:1485–1497. doi: 10.1105/tpc.11.8.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]