

Abstract
The E6 and E7 proteins of high-risk HPVs are both required for the immortalization of primary human keratinocytes and the maintenance of the malignant phenotype of HPV-positive cancer cell lines. Our previous studies have shown that E6 protein binds Myc protein and that both E6 and Myc associate with and cooperatively activate the hTERT promoter, thereby increasing cellular telomerase activity. In this study, we evaluated the role of E7 in the maintenance and activation of telomerase in immortalized and tumorigenic cells. siRNA knockdown of either E6 or E7 (or both) in HPV-immortalized cells or an HPV-positive cancer cell line reduced hTERT transcription and telomerase activity. Since telomerase was inhibited by E7 siRNA in cells that independently expressed the E6 and E7 genes, our results reveal an independent role for E7 in the maintenance of telomerase activity. However, E7 alone was insufficient to increase endogenous hTERT mRNA or telomerase activity, although it significantly augmented E6-induced hTERT transcription and telomerase activity. To further explore this apparent E7-induced promoter augmentation, we analyzed an exogenous hTERT core promoter in transduced keratinocytes. E7 alone induced the wt hTERT promoter and augmented E6-induced hTERT promoter activity. Mutation of the E2F site in the hTERT promoter abrogated the ability of E7 to induce the hTERT promoter or to enhance the ability of E6 to induce the promoter. Correspondingly, keratinocytes expressing E6 and a mutant E7 (defective for binding pRb pocket proteins) showed lower telomerase activity than cells expressing wt E6 and wt E7. Thus, HPV E7 plays a role in the maintenance of telomerase activity in stable cell lines and augments acute, E6-induced hTERT promoter activity.
Keywords: telomerase, papillomaviruses, oncoprotein
Introduction
The high-risk human papillomaviruses (HPV16, 18) are critical etiologic agents in human malignancy, most importantly in cervical cancer (zur Hausen, 2002). These oncogenic viruses encode the E6 and E7 proteins which are uniformly retained and expressed in cervical cancers (Androphy et al., 1987; Banks et al., 1987; Schwarz et al., 1985) and their continued expression is required for the cells to retain the tumorigenic phenotype. Inhibiting expression of these genes leads to inhibition of cell growth (Butz et al., 2003; Fujii et al., 2006; Venturini et al., 1999; Vormwald-Dogan et al., 1992) and the specific ablation of either the E6 or E7 gene (even in HeLa cells which have been in culture for decades) leads to cell apoptosis or senescence (DeFilippis et al., 2003; Hall and Alexander, 2003). The E6 and E7 proteins were first identified as targeting the p53 and pRb tumor suppressor pathways in host cells (Munger and Howley, 2002), thereby leading to disruption of cell cycle controls. The simultaneous expression of E6 and E7 appears to be requisite for cervical cancer and indeed these two genes seem to have evolved complementary functions (zur Hausen, 2002). E7 stimulates the cell cycle via its ability to bind and inactivate the cellular pRb protein (Munger et al., 2001; Munger et al., 1989b), and this stimulatory effect has the potential to enhance apoptosis in primary cells (Stoppler et al., 1998). However, this apoptotic pathway is blocked by the ability of E6 to prevent apoptosis via inhibition of p53 and Bak (Pan and Griep, 1994; Pan and Griep, 1995; Steller et al., 1996; Thomas and Banks, 1998). Reciprocally, the ability of E6 to transform cells appears to be inhibited by the expression of p16 which inhibits cyclin D1-cdk4 or cyclin D1/cdk6 complexes. The E7 protein bypasses this blockade apparently by activating certain cyclins such as cyclin A and E (zur Hausen, 2002). Thus, the efficient immortalization of primary genital keratinocytes by HPV relies upon the cooperative activity of these two viral genes (Hawley-Nelson et al., 1989; Munger et al., 1989a).
E6 displays several activities which might be relevant to its role in cervical neoplasia, including its cooperation with Ras in the transformation of primary rodent cells (Liu et al., 1994; Pim et al., 1994; Storey and Banks, 1993), inhibition of apoptosis (Alfandari et al., 1999; Pan and Griep, 1995; Steller et al., 1996; Thomas and Banks, 1998), interference with cell differentiation (Pan and Griep, 1994; Sherman et al., 2002; Sherman et al., 1997; Sherman and Schlegel, 1996; Thomas and Banks, 1998), immortalization of primary mammary epithelial cells (Band et al., 1991; Liu et al., 1999), cooperation with E7 in the immortalization of human genital keratinocytes (Hawley-Nelson et al., 1989; Munger et al., 1989a), and induction of malignant tumors in transgenic mice (Griep et al., 1993; Song et al., 2000; Song, Pitot, and Lambert, 1999). Corresponding to this multitude of activities, the E6 protein can bind a plethora of cellular proteins (Hebner and Laimins, 2006; Mantovani and Banks, 2001; Munger et al., 2004; Munger and Howley, 2002). Another major function of HPV E6 is its ability to activate the expression of the catalytic subunit of the telomerase, hTERT (Gewin and Galloway, 2001; Klingelhutz, Foster, and McDougall, 1996; Oh, Kyo, and Laimins, 2001; Veldman et al., 2001), which is critical for cell immortalization, thereby maintaining chromosome telomere length and continued cell proliferation (Kiyono et al., 1998). Although this function of E6 is independent of p53 degradation and its PDZ binding ability, it appears to require E6AP (Gewin et al., 2004; James, Lee, and Klingelhutz, 2006; Liu et al., 2005). We and others have shown that E6 appears to activate hTERT transcription through Myc binding sites in the hTERT promoter (Gewin and Galloway, 2001; Oh, Kyo, and Laimins, 2001; Veldman et al., 2001). However, E6 does not induce Myc expression (Galloway et al., 2005; Gewin and Galloway, 2001; Oh, Kyo, and Laimins, 2001; Veldman et al., 2001) nor increase Myc binding to the hTERT promoter (Galloway et al., 2005; Sekaric, Cherry, and Androphy, 2008; Veldman et al., 2003); Myc protein binds to the promoter in the presence or absence of E6 protein in keratinocytes (Galloway et al., 2005; Sekaric, Cherry, and Androphy, 2008; Veldman et al., 2003). Interestingly, E6 associates with Myc in vivo and in vitro and both bind to the core hTERT promoter (Veldman et al., 2003). E6 also increases histone acetylation on the promoter (Galloway et al., 2005, our unpublished data; James, Lee, and Klingelhutz, 2006), suggesting that it might alter the local chromatin structure and enhance promoter activity. The core hTERT promoter is relatively weak and contains multiple potential binding sites for transcription factors, including E2F, Sp1, NF-kB, and Ets (Fig. 3A). Three potential intact E2F sites brought our attention to the possibility that HPV E7 might participate in the transactivation of hTERT.
Fig. 3. E6/E7 contribute to the telomerase activity of E6/E7 immortalized HFKs and HPV-positive tumor cells.
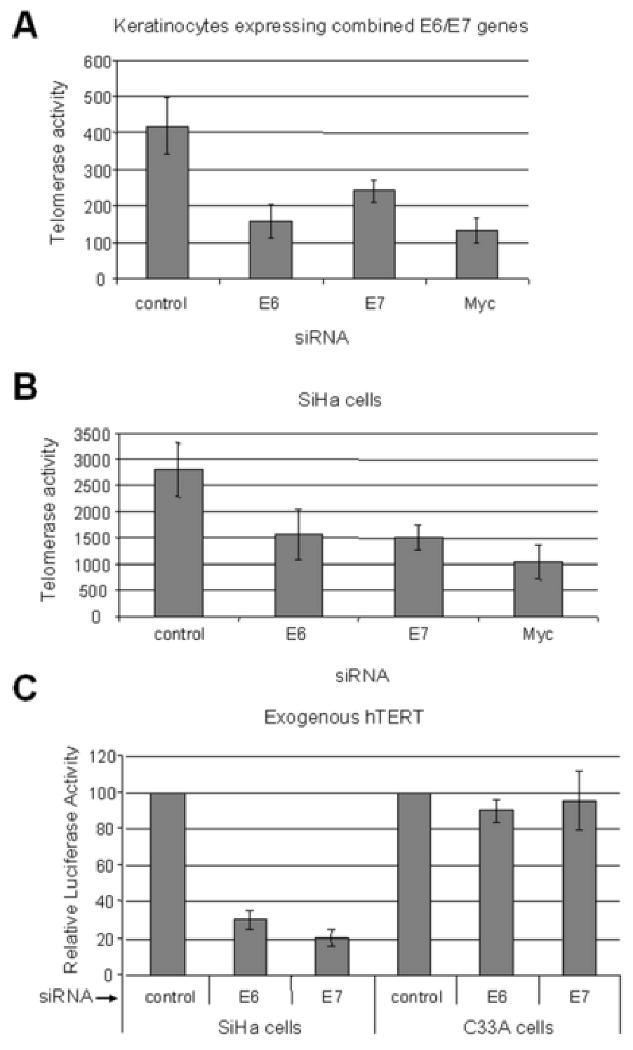
(A) Telomerase activity inhibition by siRNAs in E6/E7 expressing HFKs. Telomerase activity was measured with a quantitative assay in cells treated with the above siRNAs. Consistent with their ability to inhibit E6, the siRNAs for E6 and E7 decreased telomerase activity by approximately 50%. Myc siRNA was used as a positive control since it has been shown to inhibit cellular telomerase activity. (B) Telomerase activity in SiHa cells after siRNA transfection. Telomerase activity was also reduced in SiHa cancer cells by approximately 50% after treatment with either the E6 or E7 siRNA duplexes. However, since the SiHa cells express E6/E7 coordinately, we cannot conclude an independent role for E7 from these experiments. (C) hTERT promoter activity in tumor cells with siRNA treatment. The wild-type hTERT promoter reporter vector was transfected into the indicated cells together with siRNA for either E6, E7, or control. The value of the promoter activity with control siRNA was set to 100. The hTERT activity was reduced in SiHa cells after treatment with either E6 or E7 specific siRNA by approximately 70% and 80% respectively. Importantly, in cervical cancer cells which lack HPV-16 E6/E7 (C33A), the siRNAs showed no significant inhibition of telomerase activity.
The HPV E7 protein is a low-molecular-weight protein of approximately 100 amino acids. Like E6, E7 also appears to be a multifunctional oncoprotein; it interacts with the pRb, p107 and p130 proteins through a conserved LXCXE sequence within the CR2 sequences (Dyson et al., 1992; Dyson et al., 1989). These pocket proteins regulate the activities of the E2F family of transcription factors that control multiple cell cycle transitions as well as other cellular activities (Cam and Dynlacht, 2003). The ability of E7 to destabilize pocket proteins is critical for cellular transformation (Gonzalez et al., 2001; Helt and Galloway, 2001; Jones, Thompson, and Munger, 1997). In addition to binding and degrading pRb and its related pocket proteins (p107, p130), E7 has other cellular targets that are relevant to cellular transformation. HPV E7 can override the growth-inhibitory activities of cyclin-dependent kinase inhibitors, including p21CIP1 (Funk et al., 1997; Jones and Munger, 1997) and p27KIP1 (Zerfass-Thome et al., 1996). Additional E7-interacting proteins, including transcription factors p300, CBP, and pCAF (Avvakumov, Torchia, and Mymryk, 2003; Bernat et al., 2003; Huang and McCance, 2002), cell cycle regulators cyclin A, cyclin E, p21, p27, and metabolic enzymes, have been isolated by various methods, and many of these candidates appear to associate with carboxyl-terminal E7 sequences (Munger et al., 2001). The carboxyl-terminal HPV E7 domain also contributes to association with chromatin-modifying enzymes, particularly histone deacetylases (HDACs) and histone acetyl transferases (HATs) (Brehm et al., 1999). Each of the above E7-interacting proteins might contribute to transforming activities of high-risk HPV E7 proteins. More recently, E7 has been shown to interact with a pRb-associated protein, p600 (DeMasi et al., 2005; Huh et al., 2005), and induce genetic instability, predominantly via its ability to induce abnormal centrosome numbers and multipolar mitotic spindles (Duensing and Munger, 2002; Munger et al., 2004; Munger et al., 2001; Piboonniyom et al., 2003).
In the current study we utilized E6 and E7 siRNAs to demonstrate that E7 contributes to the sustained telomerase activity in E6/E7-immortalized keratinocytes and tumor cells. In addition, real-time quantitative TRAP assays showed that E6/E7-transduced cells had a higher telomerase activity than E6-transduced cells and that the augmentation by E7 required pRb binding and an intact E2F site in the hTERT promoter.
Results
Generation and characterization of keratinocyte cell strains expressing the HPV-16 E6 and E7 oncogenes
Recently, several studies have demonstrated that E2F strongly regulates the hTERT promoter (Alonso et al., 2005; Alonso et al., 2006; Crowe and Nguyen, 2001; Crowe et al., 2001; Won et al., 2004), at least in glioblastoma cancer cells and fibroblasts. Since the HPV-16 E7 protein inactivates the pRb tumor suppressor and releases the repression of E2F target genes, we hypothesized that E7 might also regulate the hTERT promoter in keratinocytes. To address this possibility, we constructed retroviruses with pLXSN vector, pLXSN-16E6, pLXSN-16E7 or pLXSN-16E6E7 and then generated keratinocyte strains expressing these constructs (Stoppler et al., 1997; Veldman et al., 2001). To confirm the appropriate gene expression (E6, E7) in the cell lines, RT-PCR was performed with E6- or E7-specific primers (Fig. 1A). As anticipated, E6 mRNA was expressed in HFKs transduced with the E6 and E6E7 retroviruses, while E7 mRNA was expressed in the E7 and E6E7 transduced cells. HFKs transduced with the pLXSN vector served as the control, and GAPDH was used to normalize gene expression (Fig. 1B). We also performed reactions without reverse transcriptase (-RT) to confirm that PCR products came from mRNA, not DNA (data not shown). To further verify whether the E6 and E7 were functional in the stable cell lines, we assayed the level of tumor suppressor p53 and pRb in these lines by Western Blotting (Fig 1C). The data showed decreased p53 levels in E6-transduced cells and reduction of pRb protein in E7-expressing cells, respectively.
Fig. 1. Generation and characterization of keratinocyte strains expressing the HPV-16 E6 and E7 genes.
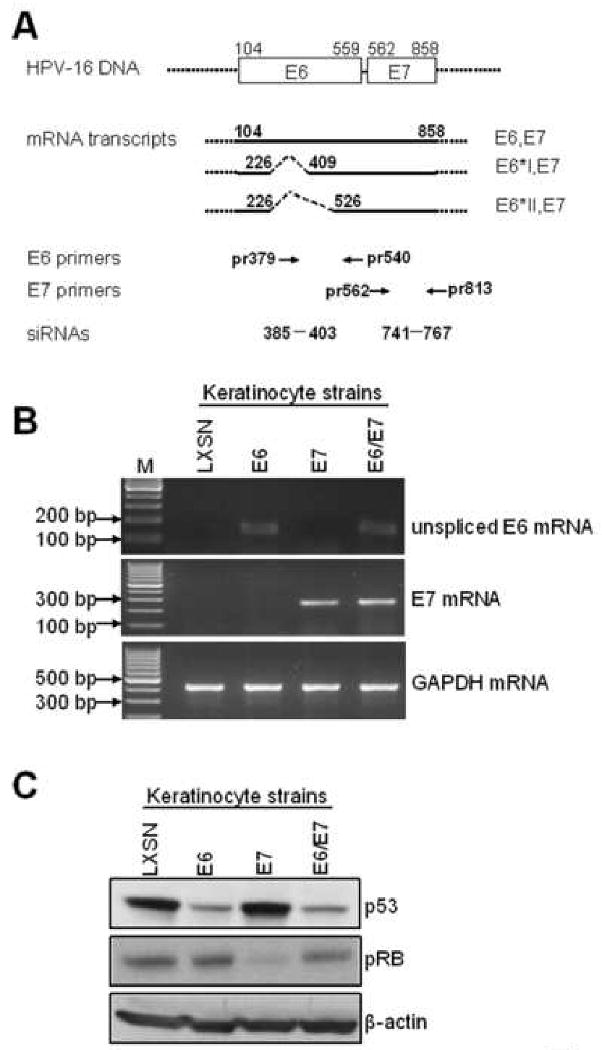
(A): Diagram of HPV-16 E6 and E7 mRNA expression and locations of primers and siRNA targets used in this study. The E6 and E7 open reading frames (ORFs) are shown as open boxes, with the nucleotide positions of the first nucleotide of the start codons and the last nucleotide of the stop codons indicated. The dotted lines flanking the open boxes and the heavy lines represent vector sequences. The heavy lines below the open boxes represent the E6/E7 bicistronic transcripts with alternative splicing sites depicted as dotted lines. The numbers above the full length E6E7 transcript are the nucleotide positions of the first nucleotide of the start codon of E6 and the last nucleotide of the stop codon of E7 in the HPV16 genome. The numbers above the E6*IE7 and E6*IIE7 transcripts are the positions of either 5′ or 3′ splicing sites. The primers used in this study are depicted below the transcript lines as arrows and numbers showing the locations of primers in the genome. The siRNA targets are also depicted as dashes and numbers showing the start and end nucleotides of the targets. (B): confirmation of E6, E7 mRNA expression. Primary human foreskin keratinocytes (HFK) were transduced with pLXSN expressing HPV-16 E6, E7, E6E7 or empty vector as described. Total cellular RNA was isolated from the transduced HFK cell strains and RT-PCR was performed with sets of HPV16 unspliced E6 and E7 specific primers described in Material and Methods, GAPDH mRNA was detected as an internal control. PCR products were analyzed with 2% agarose gel. (C): p53 and pRb proteins. To verify the expression of the E6 and E7 proteins, we screened the above stable cell lines by Western blotting for expression of p53 and pRb, respectively. Western blotting for β-actin also was used as a loading control. As anticipated, a decreased p53 protein was observed in E6 and E6E7 expressing cells, and decreased pRb was only observed in E7-expressing cells.
Specificity of E6 and E7 siRNA duplexes
To study the role of E6 and E7 in the regulation of telomerase, we used the small interfering RNA (siRNA) technique depicted in Figure 1A. In the keratinocyte strain with combined E6/E7 genes, the E6 siRNA (which targets the unspliced E6 mRNA that produces E6 protein but not the spliced mRNAs that produce E7) decreased unspliced E6 mRNA levels but not E7 mRNA levels (Fig. 2A). A similar strategy has been used to knockdown E6 expression without affecting E7 expression (Butz et al., 2003; Kelley et al., 2005; Tang et al., 2006). In contrast, the E7 siRNA decreased both E7 mRNA and unspliced E6 mRNA due to the bicistronic early mRNA in these cells (Fig. 2A). We also used siRNA to knock down Myc expression since this cellular protein is the target by which E6 induces hTERT mRNA. Myc siRNA also serves as a specificity control for the E6 and E7 siRNAs. Neither scrambled nor Myc siRNAs affected E6 or E7 expression. Confirming that both the E6 and E7 siRNAs decreased E6 protein levels, we observed that the amount of p53 in these transfected cells was markedly elevated (Fig. 2B). We applied the same siRNAs to the keratinocyte strain expressing separate E6/E7 genes (Fig. 2C). The E6 and E7 siRNAs specifically decreased E6 and E7 transcription, respectively. Corresponding with the specific inhibition of E6 and E7 expression, only the E6 siRNA (but not the E7 or Myc siRNAs) dramatically restored p53 protein level in these cells (Fig. 2D). While we did not look at the phenotype of cell gowth or apoptosis for 48 hrs treatment with siRNAs, based upon previous studies (DeFilippis et al., 2003; Nishimura et al., 2006; Tang et al., 2006), we anticipate that selective inhibition of E6 or E7 would result in apoptosis or senescence during the course of treatment, respectively.
Fig. 2. siRNA knockdown of E6 and E7 expression in E6E7 transduced keratinocytes.
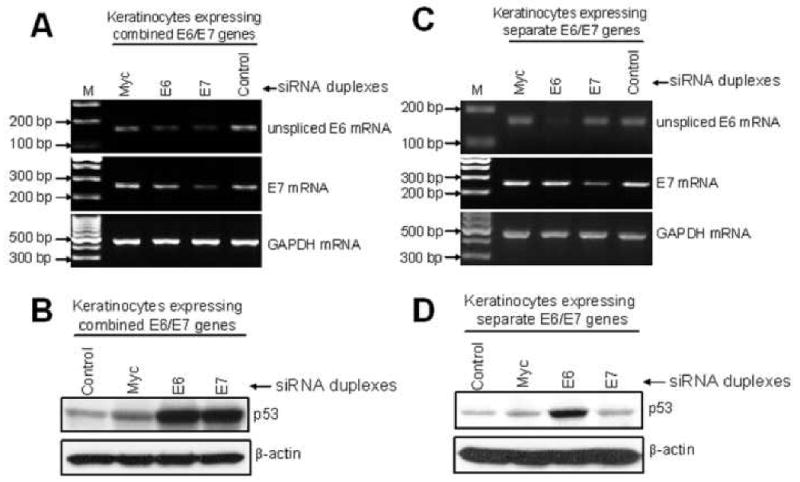
(A) RT-PCR detection of E6 and E7 mRNA (upper and middle panels) and GAPDH mRNA (bottom panel) in keratinocytes expressing the combined E6/E7 genes. The PCR signal for E6 mRNA was reduced in cells treated with siRNA for either E6 or E7. E7 mRNA was decreased in cells treated with siRNA for E7 only. Myc and control siRNA duplexes did not affect the expression of either E6 or E7. The expression of GAPDH mRNA was unaffected by any of these treatments. (B) Western blot detection of p53 protein (upper panel) and β-actin (bottom panel) in HFKs treated with siRNAs. Keratinocytes were treated identically with siRNAs for E6, E7, or Myc as described in (A) but were then analyzed for p53 protein expression by Western blot. p53 protein was increased in cells treated with either E6 or E7 siRNAs, consistent with their postulated inhibition of E6 protein expression. Cells treated with Myc or control siRNA showed lower levels of p53, consistent with continued E6 expression. β-actin protein expression was not altered by any siRNA treatment. (C) RT-PCR detection of E6 and E7 mRNA (upper and middle panels) and GAPDH mRNA (bottom panel) in HFKs that express E6 and E7 from separate vectors. In contrast to experiments with cells expressing the combined E6/E7 genes (panels A-B), cells expressing the E6 and E7 genes from separate vectors showed a specific inhibition by respective siRNAs. E6 mRNA was reduced only when treated with E6 siRNA and E7 mRNA was decreased only in cells treated with E7 siRNA. Myc and control siRNA duplexes had no effect on E6 or E7 expression. The expression of GAPDH mRNA was unaffected by these treatments. (D) Western blot detection of p53 protein (upper panel) and β-actin (bottom panel) in HFKs expressing the E6 and E7 genes from separate vectors. p53 was increased only in cells that were treated with E6 siRNA, indicating specific knockdown of E6 in these cells. These results also verify the results in panel C demonstrating that the E7 siRNA is not inhibiting E6 expression.
E6/E7 expression is required for the maintenance of telomerase activity in HPV-immortalized and tumorigenic cervical cells
E6 and E7 are required for the efficient immortalization of primary HFKs as well as the maintenance of the malignant phenotype of HPV-positive cancer cells. Similarly, telomerase activity is also critical for immortalization and the maintenance of a malignant phenotype. We therefore speculated that the HPV oncoproteins played an important role in the continued expression of telomerase in HPV positive cancers and E6/E7 immortalized cells. To test this hypothesis, we selectively knocked down expression of E6 (using the E6 siRNA construct, Fig 1A) or E6/E7 (using the E7 siRNA construct, Fig. 1A) in HPV positive cervical cancer cells (SiHa cells) and E6/E7 immortalized HFKs with siRNA duplexes. The level of telomerase activity in these cells was reduced 50-70% by the E6, E7 and Myc siRNAs, indicating the dependence of this enzyme activity on continued viral oncoprotein expression as well as on Myc expression (Fig. 3A). Thus, these data indicate that E6 and Myc are required for maintenance of telomerase activity. Whether E7 protein expression is required cannot be evaluated in these experiments due to the dual effect of the E7 siRNA on both E6 and E7 mRNA. The potential role of E7 is considered in the next section. These data are consistent with previous studies of HeLa cells in which E2-repressed HPV-18 E6/E7 genes were replaced by the HPV-16 E6 and E7 genes (DeFilippis et al., 2003; Nishimura et al., 2006), as well as studies of HPV positive cancer lines (HeLa and SiHa cells) in which E6 or E6/E7 siRNAs were used (Tang et al., 2006).
The E6, E7 and Myc siRNAs were also able to significantly inhibit telomerase activity in SiHa cervical cancer cells (Fig. 3B), suggesting a similar dependence upon the viral oncogenes and Myc. However, similar to the limitations in Fig 3A, we cannot conclude an independent role for E7 in these cells since the E7 siRNA also affects E6 mRNA levels.
Finally, to evaluate the presumed dependence of hTERT promoter activity on E6/E7 expression in cervical cancer cells, we compared the activity of a transfected hTERT promoter (expressing luciferase) in HPV-16 positive and HPV-negative cervical cancer cells. While both E6 and E7 siRNAs decreased hTERT promoter activity in SiHa cells, neither siRNA affected hTERT promoter activity in the HPV-negative C33A cell line (Fig. 3C).
E7 is also required for the maintenance of telomerase and is independent of E6
To demonstrate that telomerase activity was also dependent upon E7 protein, we generated keratinocyte cell lines that were immortalized by the E6 and E7 genes expressed separately by different vectors. This would allow us to bypass the complex alternative splicing of the bicistronic E6/E7 genes and regulate E6 and E7 gene expression individually. In these cells, siRNAs to the E6 and E7 were able to differentially and selectively inhibit the expression of the corresponding mRNAs (Fig. 2C). This selective inhibition was verified by the ability of the E6 siRNA, but not the E7 siRNA, to inhibit E6 protein and thereby increase cellular p53 levels (Fig. 2D). Similar to the results in Fig 3A, the siRNA for E6, E7 and Myc inihibited telomerase activity by 50-70% (Fig. 4A). However, the experiments in Fig. 4A now allow us to conclude that E7 is independently responsible for significant telomerase activity in E6/E7 immortalized cells. As expected, we also observed an siRNA-dependent decrease in promoter activity in the two keratinocyte lines expressing the E6/E7 genes together or separately (Fig. 4B).
Fig. 4. E7 contributes to the telomerase activity of E6/E7 immortalized cells.
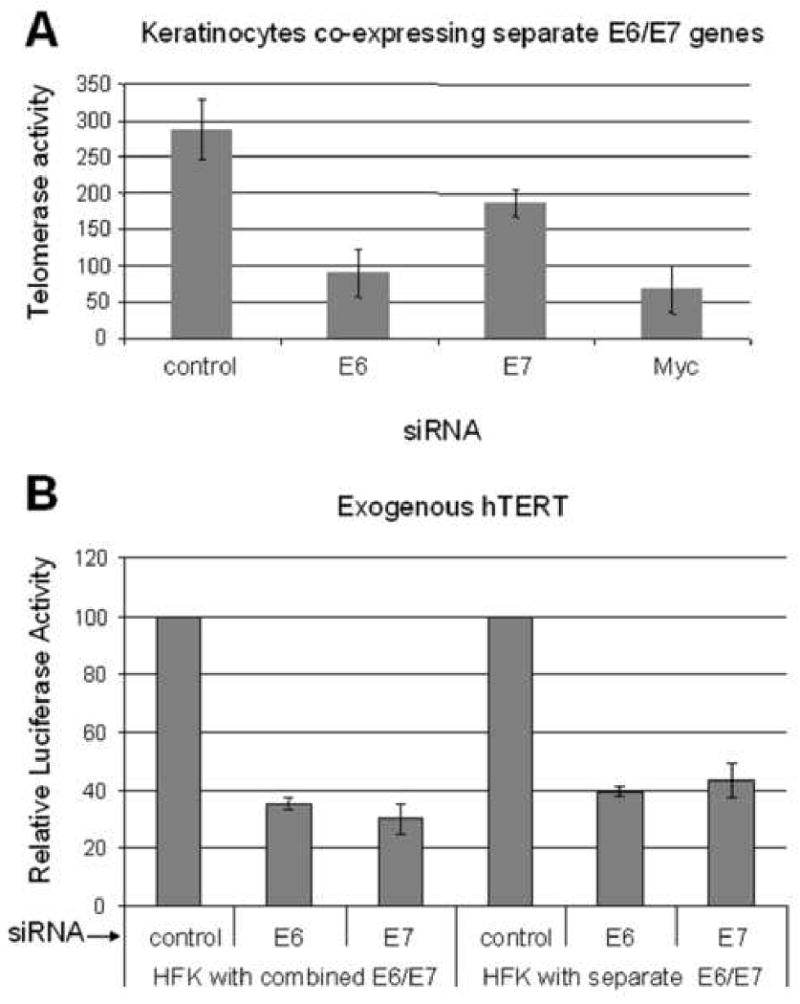
(A) Telomerase activity is dependent upon E7. HFKs expressing the separate E6 and E7 genes were transfected with either the E6 and E7 siRNAs and quantified for telomerase activity as described. Both siRNAs decreased telomerase activity when transfected into recipient cells. Thus, E7 is required for full telomerase activity and its effect on the hTERT promoter is independent of E6. As shown previously in panel C, Myc siRNA also inhibited telomerase activity. (B) hTERT promoter activity is also dependent upon E7. An hTERT promoter reporter construct was transfected into the indicated cells together with siRNA for either E6, E7, or control siRNA. The value of the promoter activity with control siRNA was set to 100. hTERT promoter activity was reduced dramatically in both types of HFKs after treatment with either E6 or E7 specific siRNA. However, only the experiment with HFKs expressing the separate E6 and E7 genes conclusively demonstrates the specific requirement for E7 in maintaining full telomerase activity.
Qualitative assays indicate that HPV E6, but not E7, increases hTERT mRNA and telomerase activity in primary keratinocytes
In support of previous findings, (Kiyono et al., 1998; Klingelhutz, Foster, and McDougall, 1996; Sherman et al., 1997; Veldman et al., 2001), qualitative assays demonstrated that telomerase activity was detectable in E6- or E6E7-expressing cells but not in vector- or E7-expressing cells (Fig. 5B). Correlating with this increase in enzymatic activity, hTERT mRNA was expressed selectively in E6- or E6E7-expressing cells (Fig. 5A). In contrast, there was no increase in hTERC mRNA levels (Fig. 5A), consistent with the hypothesis that hTERT is a limiting factor for telomerase activity. GAPDH was used as an internal control for RT-PCR (Fig. 5A). While these results seem straight-forward, we noted variations in several independent qualitative assays, which suggested at times that E7 might enhance hTERT promoter activity. We therefore utilized quantitative assays to re-evaluate these findings.
Fig. 5. E7 augments E6-induced hTERT transcription and telomerase activity.
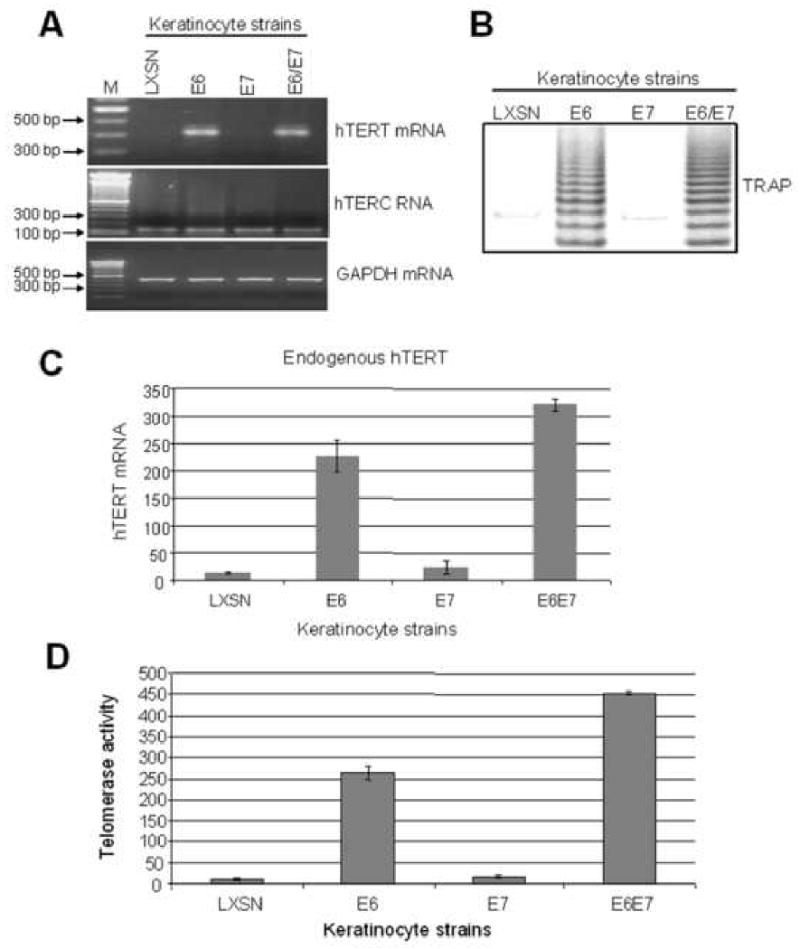
(A) hTERT and hTERC mRNA expression in stable keratinocyte cell lines. hTERT mRNA and hTERC RNA were detected with sets of the primers described in Materials and Methods. Detectable hTERT mRNA was observed only in cells expressing the E6 gene. hTERC RNA was expressed constitutively in all cell strains. (B) Telomerase activity. A qualitative TRAP assay was performed as described in Materials and Methods. A typical DNA ladder (6-base pair difference per band) was generated by cellular telomerase in the cell extract of keratinocytes transduced with the E6 or E6E7 genes. (C) Quantitative RT-PCR detection of hTERT mRNA. A real time quantitative RT-PCR was performed to quantify hTERT mRNA levels shown in panel A. These quantitative data demonstrated reproducible, higher hTERT mRNA levels in E6/E7 expressing cells than in E6 expressing cells. (D) Quantitative TRAP detection of telomerase activity. A quantitative-TRAP assay (Materials and Methods) was also performed to quantify the changes observed in panel B. The E6/E7 cells showed higher telomerase activity than E6 cells, correlating with the hTERT mRNA levels shown in panel C.
Quantitative assays demonstrate that E7, either alone or in combination with E6, does augment telomerase activity and hTERT mRNA expression
Using a quantitative real time TRAP technique described in Materials and Methods, we assayed cell extracts from keratinocytes transduced with pLXSN, E6, E7 and E6E7. Supporting the qualitative data in Fig. 5B, our quantitative data demonstrated a significant activation of telomerase in cells expressing either E6 or E6E7 (Fig. 5D). However, we consistently noted a higher telomerase activity in E6E7 expressing cells (relative activity was 452 ± 4.36, Fig. 5D) compared to those expressing E6 alone (relative activity was 263 ± 15.5, Fig. 5D). In addition, we noted a small but reproducible increase of telomerase activity in E7 cells (relative telomerase activity of 16.3±2.9) compared to LXSN control cells (relative activity of 11.6±2.6) (Fig. 5D). To determine if this E7-dependent increase might reflect enhanced transcription of the hTERT promoter, we measured hTERT mRNA levels in stable cell lines with quantitative RT-PCR (Fig 5C). Our results demonstrated that hTERT mRNA levels paralleled the quantitative telomerase findings. Thus, E7 does enhance hTERT mRNA levels, presumably by increasing the activity of the hTERT promoter. Direct tests of promoter activation were pursued.
E7 induces and further augments E6-induced exogenous hTERT promoter activity
To determine if the E7-augmented expression of hTERT mRNA was related to promoter activity, we used an exogenous hTERT reporter construct (luciferase). We cotransfected empty vector, E6, E7 or E6E7 simultaneously with an hTERT core promoter (pGL3B-hTERT) into primary HFKs and measured the relative promoter activity. We observed a higher promoter activity (9-12 fold) with E6E7 compared to that with E6 alone (5-8 fold) (Fig. 6B). This is consistent with our quantitative telomerase activity and hTERT mRNA levels (Fig. 5C, 5D). We also observed a 2-fold induction of the core hTERT promoter with E7 alone (Fig. 6B). Thus, the ability of E7 to induce detectable increases in telomerase and hTERT mRNA is most likely due to its parallel ability of E7 to induce the hTERT promoter.
Fig. 6. E7 induces the hTERT promoter through an intact E2F site.
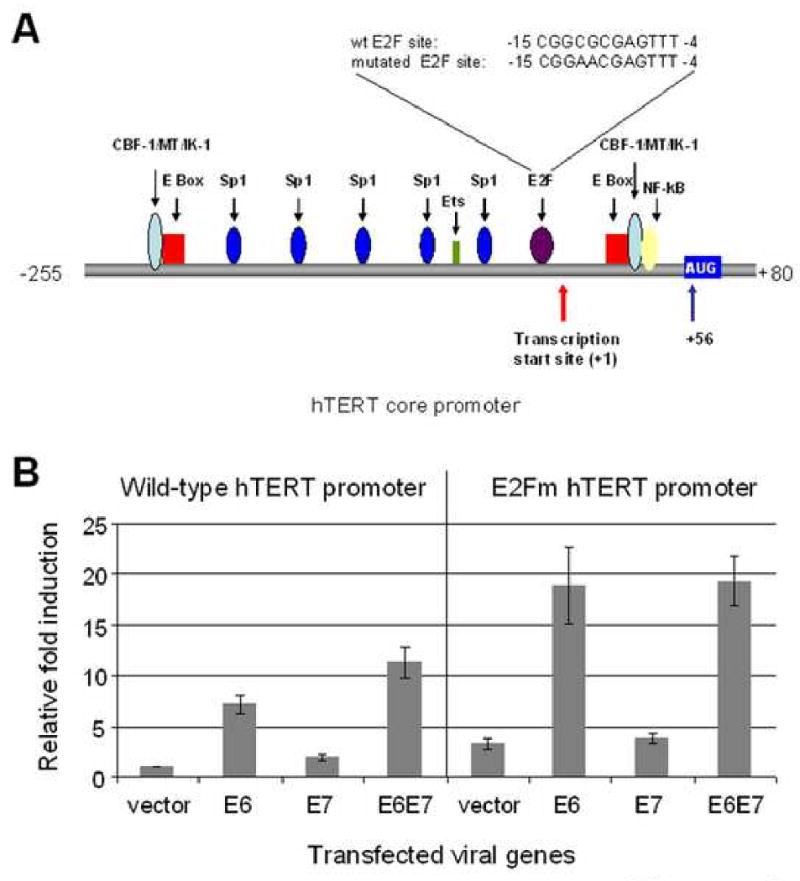
(A) Diagram of the hTERT core promoter. Binding sites for transcription factors are indicated as well as the transcription and translation start sites. The sequences and positions of wt and mutated E2F are also depicted. (B) E7 augments E6-induced hTERT promoter activity and this augmentation requires an intact E2F binding site. The wild-type hTERT core promoter (pGL3B-hTERT) was mutated at the E2F binding site using an overlapping PCR protocol described in Materials and Methods. Keratinocytes were transfected with either wt hTERT core promoter (pGL3B-hTERT) or the E2Fm mutant (pGL3B-hTERT-E2Fm) and either E6, E7 or both. The pRL-CMV R. reniformis reporter plasmid was also transfected into the cells to standardize for transfection efficiency. Relative fold activation reflects the normalized luciferase activity induced by E6 and E7 compared to the normalized activity of vector control. The value of pGL3B-hTERT activity with empty vector was set to 1. Error bars show the standard deviation for at least three independent experiments. The mutated E2F binding site in the core hTERT promoter led to an increased basal activity of the promoter and abrogated E7 induction and augmentation of the promoter. Mutation of the E2F binding site did not affect E6 induction of the promoter compared to vector.
E7 augmentation of hTERT promoter activity requires an intact E2F site in the core hTERT promoter
Since the above experiments indicate that E7 appears to exert its effect on telomerase activity via the hTERT promoter, we pursued a mutagenic analysis of the core promoter. Interestingly, three potential E2F binding sites in the hTERT core promoter have been reported (Alonso et al., 2005; Alonso et al., 2006; Crowe and Nguyen, 2001; Crowe et al., 2001; Won et al., 2004), although only one of these (position nt -15 to nt -4) is an optimal binding site and can regulate the hTERT promoter in fibroblasts (Fig. 6A). To test whether the proximal E2F site regulated E7 activity in keratinocytes, we mutated the site (CGGCGCGAGTTT to CGGAACGATTT) with overlapping PCR and performed luciferase assays using both wt and mutated hTERT promoters (E2Fm). Compared to wt hTERT promoter (wt E2F), the E2Fm promoter exhibited a higher basal activity (3.25 ± 0.48 vs 1 ± 0, p<0.01, Fig. 6B), suggesting that the E2F site was functioning as an inhibitory cis-element in primary keratinocytes. While E6 appeared to induce the mutant promoter (15-24 fold vs vector control with wt promoter) more efficiently than the wt promoter (6-8 fold), the degree of induction [about 6 fold (18.9/3.2)] was actually the same as observed with the wt promoter after the data was normalized to the vector. Importantly, however, E7 did not further induce the mutant hTERT promoter compared to vector control and could not further augment induction of the mutant promoter by E6. This demonstrates that the release of hTERT promoter repression by mutation of the E2F site negates the ability of E7 to induce the promoter and suggests that E7/pRb interactions are critical for this induction.
E7 augmentation of the hTERT promoter activity requires binding to pRb pocket proteins
To further determine whether the effects of E7 on the hTERT promoter reflected its ability to bind pRb pocket proteins and inactivate the ability of E2F to repress the promoter, we made an E7 mutant (Del 21-24 aa) that was defective for pRb binding. We speculated that a pRb-binding defective mutant of E7 would be unable to alter the interaction of pRb with the E2F site and therefore would be defective for relieving repression of the hTERT promoter. Our results confirmed this hypothesis. The E7 mutant (E7m) could not induce either the wt or mutant hTERT promoter (Fig. 7A and 7B). The E7m protein also could not augment E6 induction of wt hTERT promoter (Fig. 7A).
Fig. 7. Augmentation of E6-induced hTERT expression and telomerase activity by E7 requires RB binding.
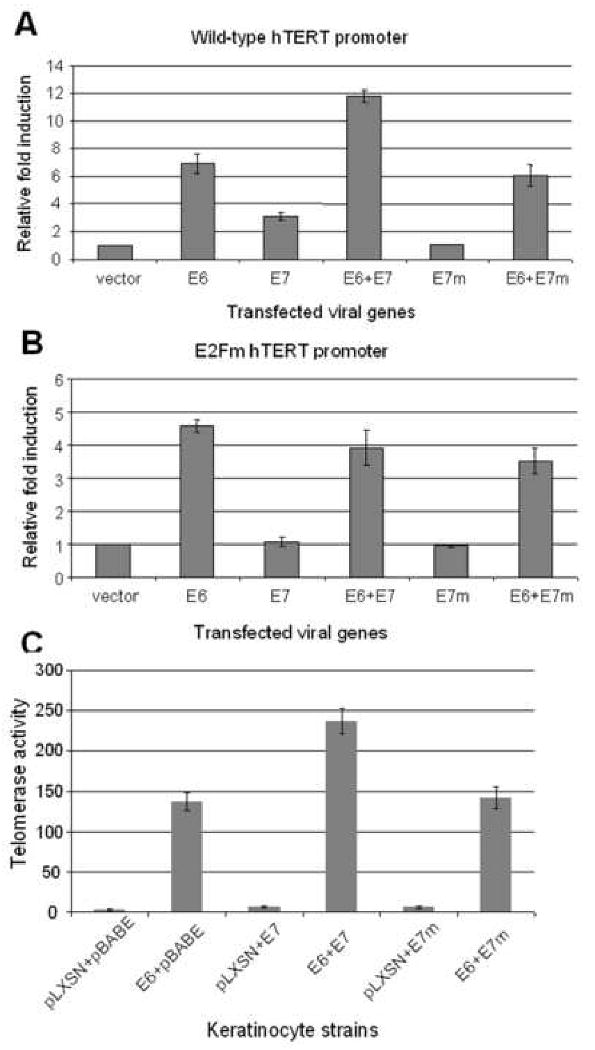
(A) Mutation of the E2F binding site in E7 abolishes the enhancement of hTERT transcription. Wild type and E2Fm promoters (expressing luciferase) were transfected into keratinocytes along with either, E6, E7 or an E7 mutant (E7m), which is unable to bind and degrade pRb. In contrast to wild-type E7, E7m failed to induce the core hTERT promoter and augment E6-induced promoter activity. (B) Neither wt E7 nor E7m induce the hTERT promoter containing an mutated E2F binding site. The mutated hTERT prompter (E2Fm) was transfected into keratinocytes with expression vectors described in panel A. The activity of vector control was set to 1. Both the wt and mutant E7 proteins were unable to induce the mutated hTERT promoter or augment E6-induced promoter activity. (C) Wild-type E7, but not E7m, augments E6-induced telomerase activity in HFKs. Primary HFKs were doubly transduced with two types of retroviruses as described in Materials and Methods. The cell strains were analyzed using a quantitative telomerase assay. Paralleling the results observed with transcription of the hTERT promoter (panel A), we observed that the E7m protein was unable to enhance E6-induced telomerase activity.
Consistent with these changes in hTERT promoter activity, quantitative TRAP assays revealed that E7m could not independently induce telomerase or augment E6-induction of telomerase in stable cell strains (Fig. 7C). Six cell strains were generated from primary keratinocytes via double retroviral transductions (pLXSN and pBABE, pLXSN-16E6 and pBABE, pLXSN and pBABE-16E7, pLXSN-16E6 and pBABE-16E7, pLXSN and pBABE-16E7, pLXSN-16E6 and pBABE-16E7m). All of the cells expressed the appropriate E6, E7 or E7m genes and the corresponding expression of p53 and pRb proteins was verified by Western blotting (data not shown). The quantitative TRAP data showed that the telomerase activity of these cells paralleled those that were generated with single transduction (Fig. 7C). However, these double transduction experiments allowed us to conclude that pRb-binding was essential for E7 to induce telomerase activity in cells.
Discussion
We have previously shown that the E6 oncoprotein of HPV-16 induces hTERT transcription in primary HFK cells through Myc binding sites (Gewin and Galloway, 2001; McMurray and McCance, 2003; Oh, Kyo, and Laimins, 2001; Veldman et al., 2001). However, E6 does not induce Myc expression (Gewin and Galloway, 2001; Oh, Kyo, and Laimins, 2001; Veldman et al., 2001) or Myc binding to the hTERT promoter (Galloway et al., 2005; Liu et al., 2005; Sekaric, Cherry, and Androphy, 2008; Veldman et al., 2003). Although the exact mechanisms by which E6 induces hTERT transcription still remains unknown, E6 associates with Myc in vitro and in vivo and both E6 and Myc bind to the core promoter. More importantly, E6AP is required for the transactivation of hTERT by E6 (Galloway et al., 2005; Gewin et al., 2004; James, Lee, and Klingelhutz, 2006; Kelley et al., 2005; Liu et al., 2005) and for E6 association with the promoter (Liu et al., 2005). In contrast to the above studies, a single recent report suggests that E6AP is not required for hTERT promoter induction (Sekaric, Cherry, and Androphy, 2008). The etiology of these experimental differences is currently unclear but may derive from the characteristics of the E6 mutants employed. Clearly the specific knockdown of E6AP interferes with promoter induction.
It is possible that E6AP directs the ubiquitination of regulatory proteins present on the hTERT promoter. Ubiquitination (with or without subsequent protein degradation) plays a critical role in the control of transcription factor activity (Conaway, Brower, and Conaway, 2002; Muratani and Tansey, 2003), and we postulate that proteins at or nearby the proximal E box are potential targets. For example, Galloway et al. have shown the NFX1-91 contributes to the repression of hTERT in primary cells and is ubiquitinated by E6/E6AP (Galloway et al., 2005; Gewin et al., 2004). In addition, the BRCA1 tumor suppressor protein has been shown to bind at the hTERT E box and to suppress expression (Xiong et al., 2003), and E6 is capable of binding BRCA1 and relieving this repression (Zhang et al., 2005). It is possible that this relief of suppression is mediated by the ubiquitination/degradation of BRCA1. Myc is another potential target for ubiquitination and degradation by E6AP. Indeed, the stability and transcriptional activity of Myc is already known to be regulated by ubiquitination via the Skp2 and/or Fbw7 ubiquitin ligases (Amati, 2004; Kim et al., 2003; von der Lehr et al., 2003; Welcker et al., 2004; Yada et al., 2004).
In this study, we observed a significant decrease in telomerase activity in E6/E7 transduced HFKs or tumor cells after the cells were treated with Myc-specific siRNA (Fig. 3A and B, and 4A), suggesting a important role for Myc in maintaining telomerase activity in HPV-positive cells. However, ChIP experiments by us and others have demonstrated that Myc binds to the hTERT promoter in the absence or presence of E6 (Galloway et al., 2005; Liu et al., 2005; Sekaric, Cherry, and Androphy, 2008; Veldman et al., 2003), suggesting that unlike overexpression of exogenous Myc (Liu et al., 2007), endogenous Myc is insufficient to activate telomerase. Potentially Myc could be modified by E6 or E6/E6AP, thereby altering local chromatin structure and permitting active gene transcription. E6 or E6/E6AP-dependent changes in the acetylation of histones at the hTERT promoter (Galloway et al., 2005; James, Lee, and Klingelhutz, 2006) (our unpublished data) have been observed. Interesting, E6 appears to function in both p53-dependent and –independent pathways and can either positively (in the case of the hTERT promoter) or negatively (p21 promoter) regulate promoter activity (Liu et al., 2005; Sekaric, Cherry, and Androphy, 2008; Thomas and Chiang, 2005; Veldman et al., 2003).
As mentioned previously, the hTERT promoter has no TATA box and is relatively weak. The core promoter contains many known transcription factor binding sites, including those for Myc (E boxes), E2F, Sp1, Ets-1, CBF1, NF-kB sites. The Myc (USF also binds to same site) and Sp1 binding sites contribute to promoter transactivation by E6 (Galloway et al., 2005; Gewin and Galloway, 2001; McMurray and McCance, 2003; Oh, Kyo, and Laimins, 2001; Veldman et al., 2001; Veldman et al., 2003). In addition to these known activators, we have shown in the current study that E7, pRb pocket proteins and the E2F binding site regulate the hTERT promoter. These findings were dependent upon the development of a more precise, quantitative technique for measuring hTERT mRNA and telomerase activity (Fig. 5A, 5B). During the course of this study, other laboratories have shown that the pRb/E2F pathway is involved in the regulation of telomerase in gliomas and human fibroblasts (Alonso et al., 2005; Alonso et al., 2006; Won et al., 2004).
In brief, our studies demonstrate that E7 alone induces the wt hTERT two-fold, and augments E6-induced hTERT promoter activity (Fig. 6B and 7A). Mutation of the E2F site in the hTERT promoter abrogated the ability of E7 to induce the hTERT promoter or to enhance the ability of E6 to induce the promoter (Fig. 6B and 7B). The basal activity of the hTERT promoter was also increased by mutation of the E2F site (Fig. 6B). These data indicate that the E2F site in the hTERT promter functions as an inhibitory element in normal keratinocytes, and responds to changes in pRb/E2F pathway. Indeed, recent data from other laboratories demonstrate that pRb and E2F proteins associate with the hTERT promoter and regulate the activity in glioma cells and fibroblasts. Our studies have also shown that the ability of E7 to bind pRb is essential for E7 promoter enhancement (Fig. 7A, 7B), as well as an augmentation of the telomerase activity in cells (Fig. 7C). However, E7 alone is insufficient to initiate the transcription of the endogenous hTERT in primary keratinocytes (Fig. 5A, 5C), suggesting that the pRb/E2F pathway is not the primary reason for repression of the hTERT promoter in keratinocytes.
Telomerase activity is one of the major hallmarks of malignancy and many cellular factors contribute the activation and maintenance of this activity. While it is clear that the E6 gene of the high-risk HPVs induces hTERT transcription during cell immortalization, it also appears that the E6 gene is required to maintain this expression even in cancer cells. Our current results indicate that the HPV E7 oncogene also contributes to the induction and maintenance of telomerase activity and that the pRb pathway is critical for regulating this effect.
Materials and Methods
Plasmids
Expression plasmids (pJS55E6, pJS55E7) coding E6 and E7 of HPV-16 in a modified pSG5 vector (Strategene) for transient transfection in this study, retrovirus vectors expressing E6, E7, or E6E7 (pLSXN-E6, pLXSN-E7, pLXSN-E6E7) for stable cell lines, and hTERT promoter reporter plasmid (pGL3B-hTERT core promoter, defined as pGL3B-255 previously) were described previously (Sherman and Schlegel, 1996; Veldman et al., 2001; Veldman et al., 2003). The hTERT expression vector (pCI-hTERT) was a gift from Dr. Robert Weinberg (MIT). The E7 mutant defective for pRb binding in pCMV was a gift from Dr. Karl Munger (Harvard University), we performed PCR to amplify this E7 mutant using the following primers 5′-ATCGAATTCATGCATGGAGATACACCTAC-3′ with EcoR I site at 5′end and 5′-TAGCTCGAGTTATGGTTTCTGAGAACAGA-3′ with Xho I site at its 5′ end, the double digestion products with EcoR I and Xho I were ligated with the EcoR I and Xho I fragment of pJS55, resulting a new construct, pJS55-16E7m, defective for pRb binding. To generate wt 16 E7 and its pRb binding defective E7 mutant in pBABE-puro (pBABE-16E7 and pBABE-16E7m), PCR products from the following primers 5′-ATCGGATCCATGCATGGAGATACACCTAC-3′ with BamH I site at its 5′ end and 5′-TACGAATTCTTATGGTTTCTGAGAACAGA-3′ with EcoR I site at its 5′ end were digested with BamH I and EcoR I, and then ligated with the BamH I and EcoR I fragment of pBABE-puro vector. To obtain a pGL3B-hTERT promoter with mutated E2F site (pGL3B-hTERT E2Fm, CGGCGCGAGTTT –CGGAACGATTT), overlapping PCR was using to build in a mutated E2F site in pGL3B-hTERT core promoter (Veldman et al., 2001; Veldman et al., 2003). Briefly, using pGL3B-hTERT as a DNA template, the first round PCR was done with primer sets oXL006 5′-AGTACCGGAATGCCAAGCTTAC-3′ and oXL067 5′-CTCTCCTCGCGGAACGAGTTTCAGGCAGCGCTGCG-3′, and oXL022 5′-CTTGGAGCGGCCGCAATAAAATATC-3′ and oXL068 5′-CTGCCTGAAACTCGTTCCGCGAGGAGAGGGCGGG-3′, respectively; the second PCR was done with primers oXL006 and oXL022 using mixture of the 1st round PCR products as template, the double digestion of the 2nd PCR product with Hind III and Xho I was ligated with the Hind III and Xho I fragment of pGL3-basic vector. The expected mutation for E2F binding site was confirmed by sequencing.
Retroviruses
Retrovirus packaging cells, SD3443, were transfected with pLXSN, pBABE-puro vectors with either E6 or E7, or both E6 and E7 as described above using LipofectAmine 2000 (Invitrogen) as suggested by the manufacturer. Culture supernatants containing retrovirus were collected 24 hours after transfections.
Cell Culture and stable lines selection
Primary human foreskin keratinocytes (HFKs) were cultured from neonatal foreskins as described (Schlegel et al., 1988) and maintained in keratinocyte growth media (Gibco-BRL), supplemented with gentamycin (50 μg/ml). Primary HFKs P2 were infected with amphotropic LXSN or pBABE-puro retroviruses expressing HPV-16E6, E7, E7m or both E6 and E7 (or E7m) described above. Retrovirus-infected cells were selected in G418 (100 μg/ml) or puromycin (200 ng/ml) for 5 days. Resistant colonies were pooled and passaged every 3-4 days (1 to 4 ratio). SiHa (HPV16 positive cervical cancer cell line), C33A (HPV negative cervical cancer cell line) cells were maintained in complete DMEM medium. All cells were cultured on non-coated, plastic tissue culture dishes or flasks.
Reverse Transcription and Polymerase Chain Reaction (RT-PCR)
Total cellular RNA was isolated with TRIZol reagent (Invitrogen) and treated with DNA-free kit (Ambion) according to the manufacturer's instructions. First-strand cDNA was synthesized with some modifications using 2 μg of total cellular RNA following the instructions of Superscript First-Strand Synthesis System for RT-PCR (Invitrogen). Five percent of the RNase H-treated cDNA products were subjected to PCR amplification in a total volume of 50 μl containing 10 μmol/L sense primer and 10 μmol/L anti-sense primer as described in the manufacturer's protocol. Initial denaturation for 3 minutes at 94°C was followed by 30 cycles of PCR amplification (94°C for 30 seconds, 52°C for 30 seconds, 72°C for 60 seconds). PCR products were analyzed for E6, E7, hTERC, hTERT, and GAPDH mRNA with 1.5% agaraose gel. The primers used for RT-PCR were: 5′-CAACAAACCGTTGTGTGAT-3′ and 5′-CGTGTTCTTGATGATCTGC-3′ for unspliced E6; 5′-ATGCATGGAGATACACCTAC-3′ and 5′-CATTAACAGGTCTTCCAAAG-3′ for E7; 5′-GGCTCTTTTTCTACCGGAAG-3′ and 5′-ACAAAGTACAGCTCAGGCGG-3′ for hTERT mRNA; 5′-TCTAACCCTAACTGAGAAGGGCGTAG-3′ and 5′-GTTTGCTCTAGAATGAACGGTGGAAG-3′ for hTERC; and 5′-CTCAGACACCATGGGGAAGGTGA-3′ and 5′-ATGATCTTGAGGCTGTTGTCATA -3′ for GAPDH mRNA.
Real-time quantitative RT-PCR (QRT-PCR)
Taqman real-time QRT-PCR was performed on the Bio-Rad iCycler MyiQ for quantitation of hTERT mRNA using primers and probes (sense primer, 5′-TGACACCTCACCTCACCCAC-3′, anti-sense primer, 5-CACTGTCTTCCGCAAGTTCAC-3′, and Tagman probe, 5′-ACCCTGGTCCGAGGTGTCCCTGAG-3′) as previously reported (Fu, Quintero, and Baker, 2003). All samples were run in triplicate and each reaction contained the cDNA from 50 ng of RNA. Standard curves were created for each run using 10-fold serial dilutions of pCI-hTERT cDNA expression vector. The concentration of each mRNA was expressed as fg of the respective cDNA vector.
Western blot
Stable cell lines in 10-mm dishes or cells with siRNA duplex treatment in 6-well plate were washed once with PBS, lysed in 2× SDS gel electrophoresis sample buffer. Proteins were separated on a 4 to 20% Tris-glycine gradient gel (Invitrogen) and then were electrophoretically transferred to an Immobilon-P polyvinylidane difluorid (PVDF) membrane (Millipore). The membranes were blocked in 5% milk-PBST or 5% BSA-PBST and incubated with the primary antibodies, mouse anti-p53 monoclonal antibody (Santa Cruz, mouse monoclonal, 1:1000 dilution), pRb (Cell Signaling, rabbit polyclonal, 1:500 dilution) or β-actin (Sigma, mouse monoclonal, 1:5000 dilution). The secondary antibodies, HRP-conjugated goat anti-mouse IgG and anti-rabbit IgG antibodies (Santa Cruz), were used at a dilution of 1:10,000. The membranes were visualized by using Western Blotting Chemiluminescence Luminol Reagent (Santa Cruz).
siRNA transfection
The siRNA target sequences were as follows (Kelley et al., 2005): for 16E6 siRNA duplex target (nt385-403), 5′-ACCGTTGTGTGATTTGTTA-3′; for 18E6 siRNA duplex target (nt231-249), 5′-GAGGTATTTGAATTTGCAT-3′; for 16E7 siRNA duplex target (nt741-767), 5′-GTGTGACTCTACGCTTCGGTTGTGCGT-3′; for c-myc siRNA duplexes targeting four different region in myc (Smartpool from Dharmacon), the targets are nt437-455 5′-CAGAGAAGCTGGCCTCCTA-3′, nt360-378 5′-CGACGAGACCTTCATCAAA-3, nt1263-1281 5′-GAAACGACGAGAACAGTTG-3′ and nt908-926 5′-CCACACATCAGCACAACTA-3′. 60-70% confluency of C33A cells or 70-80% confluency of SiHa cells, pLXSN-16E6E7 or pLXSN-16E6E6/pBABE-16E7 transduced HFKs were tranfected with a final concentration of 40 nM of negative control siRNA, E6-specific siRNA (Dharmacon), or E7-specific siRNA (IDT), or Myc specific siRNA duplex mixture (Dharmacon, Smartpool) with LipofectAmine 2000 (Invitrogen) following the manufacturer's instructions. Cells were harvested for Luciferase assay 24 hrs posttransfection, and for RT-PCR, western blot, and TRAP assay 48 hrs posttransfection, respectively. For Luciferase assay, SiHa, C33A, pLXSN-16E6E7, and pLXSN-16E6/pBABE-16E7 transduced HFK cells were co-tranfected with siRNA duplexes plus pGL3B-hTERT and Renilla reniformis luciferase gene as described below.
Luciferase Assay
1 × 105 telomerase-negative HFKs, pLXSN-16E6E7 transduced HFKs, pLXSN16E6/pBABE-16E7 transduced HFKs, SiHa, or C33Acells were seeded onto 24-well plates and grown overnight. Transient transfections were performed using LipofectAmine 2000 reagent (Invitrogen) according to the protocol provided by the manufacturer. Cotransfections were performed using 0.5 ug of a core hTERT reporter plasmid (pGL3B-hTERT or its mutant pGL3B-hTERT E2Fm) and 20 ng of each expression vector as indicated (pJS55-16E6, pJS55-16E7 or both) or empty vectors as control for basal promoter activity. Cells also were cotransfected with 2 ng of the pRL-CMV plasmid (Promega), which contains the Renilla reniformis luciferase gene as a transfection control. Firefly and Renilla luciferase activities were measured 24 hr after transfection using the Dual luciferase reporter assay system (Promega).
Telomeric repeat amplification protocol (TRAP) and Real-time quantitative TRAP (Q-TRAP)
Human keratinocytes were grown in 100-mm tissue-culture dishes to 80% confluence, harvested by trypsinization, washed in cold PBS, and transferred to a microfuge tube. Cell pellets were lysed for 30 minutes on ice in 400 μl of telomeric repeat amplification protocol (TRAP) buffer (0.5% Chaps, 10 mmol/L Tris, pH 7.5, 1 mmol/L MgCl2, 1 mmol/L EGTA, 5 mmol/L β-ME, 10% glycerol, 0.1 mmol/L 4-(2-amino-ethyl)benzene-sulfonyl fluoride hydrochloride (AEBSF). Lysates were centrifuged at 14,000 × g for 5 minutes at 4°C, the supernatant was transferred to a new tube, and protein concentration was determined (Bio-Rad, Richmond, CA). A TRAP assay was performed on 1 μg of protein lysates as described (Baege et al., 2002; Veldman et al., 2001). Twenty percent of the PCR products were separated on 10% nondenaturing polyacrylamide gels and visualized using the Gelcode color silver-staining kit (Pierce, Rockford, IL).
Quantitative TRAP assay was performed as described before (Fu, Quintero, and Baker, 2003) with SYBR Green Supermixure (Bio-Rad) A standard curve was produced for the real-time Q-TRAP assay using serially diluted 293 cell extracts. All samples were run in triplicate. This assay is linear over at least a 500-fold range (0.008 to 4 μg of 293 cell protein input).
Fig. 8. A working model for regulation of the hTERT promoter by E6 and E7.
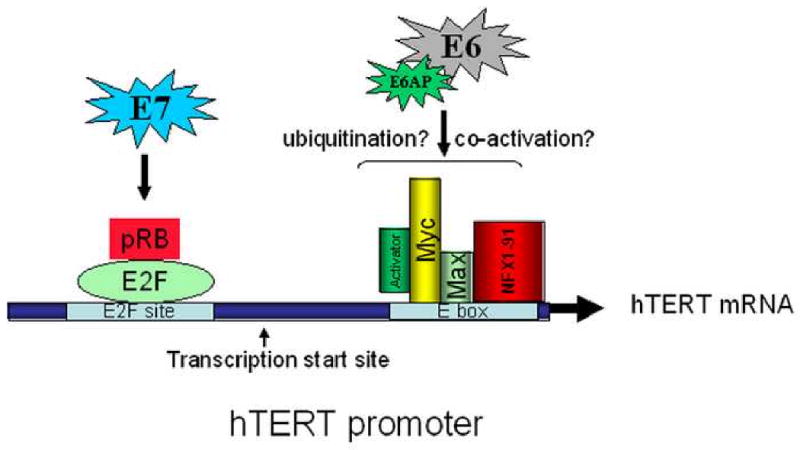
Results from the current and previous studies are summarized to illustrate the possible mechanism for regulation of the hTERT promoter by the HPV E6 and E7 oncoproteins. A critical regulator of hTERT transcription is the binding of Myc/Max to the promoter E box, as well as the physical interaction of E6/E6AP with this E box site. In addition, the interaction of E7 with the pRb/E2F site is also involved in regulating promoter activity. The transcription start site for hTERT mRNA is indicated.
Acknowledgments
This work was supported by NIH grants R01CA106440 and R01CA53371 to R.S. We thank Karl Munger for providing the construct pCMV-E7, Carl Baker and Jesse Quintero for assistance with quantitative PCR, and Hang Yuan, Frank Suprynowicz, and Gary Disbrow for their advice and suggestions during this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alfandari J, Shnitman Magal S, Jackman A, Schlegel R, Gonen P, Sherman L. HPV16 E6 oncoprotein inhibits apoptosis induced during serum-calcium differentiation of foreskin human keratinocytes. Virology. 1999;257(2):383–96. doi: 10.1006/viro.1999.9675. [DOI] [PubMed] [Google Scholar]
- Alonso MM, Fueyo J, Shay JW, Aldape KD, Jiang H, Lee OH, Johnson DG, Xu J, Kondo Y, Kanzawa T, Kyo S, Bekele BN, Zhou X, Nigro J, McDonald JM, Yung WK, Gomez-Manzano C. Expression of transcription factor E2F1 and telomerase in glioblastomas: mechanistic linkage and prognostic significance. J Natl Cancer Inst. 2005;97(21):1589–600. doi: 10.1093/jnci/dji340. [DOI] [PubMed] [Google Scholar]
- Alonso MM, Fueyo J, Yung WK, Gomez-Manzano C. E2F1 and telomerase: alliance in the dark side. Cell Cycle. 2006;5(9):930–5. doi: 10.4161/cc.5.9.2698. [DOI] [PubMed] [Google Scholar]
- Amati B. Myc degradation: dancing with ubiquitin ligases. Proc Natl Acad Sci U S A. 2004;101(24):8843–4. doi: 10.1073/pnas.0403046101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Androphy EJ, Hubbert NL, Schiller JT, Lowy DR. Identification of the HPV-16 E6 protein from transformed mouse cells and human cervical carcinoma cell lines. Embo J. 1987;6(4):989–92. doi: 10.1002/j.1460-2075.1987.tb04849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avvakumov N, Torchia J, Mymryk JS. Interaction of the HPV E7 proteins with the pCAF acetyltransferase. Oncogene. 2003;22(25):3833–41. doi: 10.1038/sj.onc.1206562. [DOI] [PubMed] [Google Scholar]
- Baege AC, Berger A, Schlegel R, Veldman T. Cervical epithelial cells transduced with the papillomavirus E6/E7 oncogenes maintain stable levels of oncoprotein expression but exhibit progressive, major increases in hTERT gene expression and telomerase activity. Am J Pathol. 2002;160(4):1251–7. doi: 10.1016/S0002-9440(10)62552-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Band V, De Caprio JA, Delmolino L, Kulesa V, Sager R. Loss of p53 protein in human papillomavirus type 16 E6-immortalized human mammary epithelial cells. J Virol. 1991;65(12):6671–6. doi: 10.1128/jvi.65.12.6671-6676.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks L, Spence P, Androphy E, Hubbert N, Matlashewski G, Murray A, Crawford L. Identification of human papillomavirus type 18 E6 polypeptide in cells derived from human cervical carcinomas. J Gen Virol. 1987;68(Pt 5):1351–9. doi: 10.1099/0022-1317-68-5-1351. [DOI] [PubMed] [Google Scholar]
- Bernat A, Avvakumov N, Mymryk JS, Banks L. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene. 2003;22(39):7871–81. doi: 10.1038/sj.onc.1206896. [DOI] [PubMed] [Google Scholar]
- Brehm A, Nielsen SJ, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. Embo J. 1999;18(9):2449–58. doi: 10.1093/emboj/18.9.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butz K, Ristriani T, Hengstermann A, Denk C, Scheffner M, Hoppe-Seyler F. siRNA targeting of the viral E6 oncogene efficiently kills human papillomavirus-positive cancer cells. Oncogene. 2003;22(38):5938–45. doi: 10.1038/sj.onc.1206894. [DOI] [PubMed] [Google Scholar]
- Cam H, Dynlacht BD. Emerging roles for E2F: beyond the G1/S transition and DNA replication. Cancer Cell. 2003;3(4):311–6. doi: 10.1016/s1535-6108(03)00080-1. [DOI] [PubMed] [Google Scholar]
- Conaway RC, Brower CS, Conaway JW. Emerging roles of ubiquitin in transcription regulation. Science. 2002;296(5571):1254–8. doi: 10.1126/science.1067466. [DOI] [PubMed] [Google Scholar]
- Crowe DL, Nguyen DC. Rb and E2F-1 regulate telomerase activity in human cancer cells. Biochim Biophys Acta. 2001;1518(12):1–6. doi: 10.1016/s0167-4781(00)00296-7. [DOI] [PubMed] [Google Scholar]
- Crowe DL, Nguyen DC, Tsang KJ, Kyo S. E2F-1 represses transcription of the human telomerase reverse transcriptase gene. Nucleic Acids Res. 2001;29(13):2789–94. doi: 10.1093/nar/29.13.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFilippis RA, Goodwin EC, Wu L, DiMaio D. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells. J Virol. 2003;77(2):1551–63. doi: 10.1128/JVI.77.2.1551-1563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMasi J, Huh KW, Nakatani Y, Munger K, Howley PM. Bovine papillomavirus E7 transformation function correlates with cellular p600 protein binding. Proc Natl Acad Sci U S A. 2005;102(32):11486–91. doi: 10.1073/pnas.0505322102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing S, Munger K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002;62(23):7075–82. [PubMed] [Google Scholar]
- Dyson N, Guida P, Munger K, Harlow E. Homologous sequences in adenovirus E1A and human papillomavirus E7 proteins mediate interaction with the same set of cellular proteins. J Virol. 1992;66(12):6893–902. doi: 10.1128/jvi.66.12.6893-6902.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243(4893):934–7. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- Fu B, Quintero J, Baker CC. Keratinocyte growth conditions modulate telomerase expression, senescence, and immortalization by human papillomavirus type 16 E6 and E7 oncogenes. Cancer Res. 2003;63(22):7815–24. [PubMed] [Google Scholar]
- Fujii T, Saito M, Iwasaki E, Ochiya T, Takei Y, Hayashi S, Ono A, Hirao N, Nakamura M, Kubushiro K, Tsukazaki K, Aoki D. Intratumor injection of small interfering RNA-targeting human papillomavirus 18 E6 and E7 successfully inhibits the growth of cervical cancer. Int J Oncol. 2006;29(3):541–8. [PubMed] [Google Scholar]
- Funk JO, Waga S, Harry JB, Espling E, Stillman B, Galloway DA. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev. 1997;11(16):2090–100. doi: 10.1101/gad.11.16.2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway DA, Gewin LC, Myers H, Luo W, Grandori C, Katzenellenbogen RA, McDougall JK. Regulation of telomerase by human papillomaviruses. Cold Spring Harb Symp Quant Biol. 2005;70:209–15. doi: 10.1101/sqb.2005.70.041. [DOI] [PubMed] [Google Scholar]
- Gewin L, Galloway DA. E box-dependent activation of telomerase by human papillomavirus type 16 E6 does not require induction of c-myc. J Virol. 2001;75(15):7198–201. doi: 10.1128/JVI.75.15.7198-7201.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewin L, Myers H, Kiyono T, Galloway DA. Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 E6/E6-AP complex. Genes Dev. 2004;18(18):2269–82. doi: 10.1101/gad.1214704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez SL, Stremlau M, He X, Basile JR, Munger K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J Virol. 2001;75(16):7583–91. doi: 10.1128/JVI.75.16.7583-7591.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griep AE, Herber R, Jeon S, Lohse JK, Dubielzig RR, Lambert PF. Tumorigenicity by human papillomavirus type 16 E6 and E7 in transgenic mice correlates with alterations in epithelial cell growth and differentiation. J Virol. 1993;67(3):1373–84. doi: 10.1128/jvi.67.3.1373-1384.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AH, Alexander KA. RNA interference of human papillomavirus type 18 E6 and E7 induces senescence in HeLa cells. J Virol. 2003;77(10):6066–9. doi: 10.1128/JVI.77.10.6066-6069.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley-Nelson P, Vousden KH, Hubbert NL, Lowy DR, Schiller JT. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. Embo J. 1989;8(12):3905–10. doi: 10.1002/j.1460-2075.1989.tb08570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebner CM, Laimins LA. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity. Rev Med Virol. 2006;16(2):83–97. doi: 10.1002/rmv.488. [DOI] [PubMed] [Google Scholar]
- Helt AM, Galloway DA. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J Virol. 2001;75(15):6737–47. doi: 10.1128/JVI.75.15.6737-6747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, McCance DJ. Down regulation of the interleukin-8 promoter by human papillomavirus type 16 E6 and E7 through effects on CREB binding protein/p300 and P/CAF. J Virol. 2002;76(17):8710–21. doi: 10.1128/JVI.76.17.8710-8721.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh KW, DeMasi J, Ogawa H, Nakatani Y, Howley PM, Munger K. Association of the human papillomavirus type 16 E7 oncoprotein with the 600-kDa retinoblastoma protein-associated factor, p600. Proc Natl Acad Sci U S A. 2005;102(32):11492–7. doi: 10.1073/pnas.0505337102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James MA, Lee JH, Klingelhutz AJ. HPV16-E6 associated hTERT promoter acetylation is E6AP dependent, increased in later passage cells and enhanced by loss of p300. Int J Cancer. 2006;119(8):1878–85. doi: 10.1002/ijc.22064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DL, Munger K. Analysis of the p53-mediated G1 growth arrest pathway in cells expressing the human papillomavirus type 16 E7 oncoprotein. J Virol. 1997;71(4):2905–12. doi: 10.1128/jvi.71.4.2905-2912.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DL, Thompson DA, Munger K. Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology. 1997;239(1):97–107. doi: 10.1006/viro.1997.8851. [DOI] [PubMed] [Google Scholar]
- Kelley ML, Keiger KE, Lee CJ, Huibregtse JM. The global transcriptional effects of the human papillomavirus E6 protein in cervical carcinoma cell lines are mediated by the E6AP ubiquitin ligase. J Virol. 2005;79(6):3737–47. doi: 10.1128/JVI.79.6.3737-3747.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Herbst A, Tworkowski KA, Salghetti SE, Tansey WP. Skp2 regulates Myc protein stability and activity. Mol Cell. 2003;11(5):1177–88. doi: 10.1016/s1097-2765(03)00173-4. [DOI] [PubMed] [Google Scholar]
- Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998;396(6706):84–8. doi: 10.1038/23962. [DOI] [PubMed] [Google Scholar]
- Klingelhutz AJ, Foster SA, McDougall JK. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature. 1996;380(6569):79–82. doi: 10.1038/380079a0. [DOI] [PubMed] [Google Scholar]
- Liu X, Disbrow GL, Yuan H, Tomaic V, Schlegel R. Myc and human papillomavirus type 16 E7 genes cooperate to immortalize human keratinocytes. J Virol. 2007;81(22):12689–95. doi: 10.1128/JVI.00669-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Yuan H, Fu B, Disbrow GL, Apolinario T, Tomaic V, Kelley ML, Baker CC, Huibregtse J, Schlegel R. The E6AP ubiquitin ligase is required for transactivation of the hTERT promoter by the human papillomavirus E6 oncoprotein. J Biol Chem. 2005;280(11):10807–16. doi: 10.1074/jbc.M410343200. [DOI] [PubMed] [Google Scholar]
- Liu Y, Chen JJ, Gao Q, Dalal S, Hong Y, Mansur CP, Band V, Androphy EJ. Multiple functions of human papillomavirus type 16 E6 contribute to the immortalization of mammary epithelial cells. J Virol. 1999;73(9):7297–307. doi: 10.1128/jvi.73.9.7297-7307.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Ghai J, Ostrow RS, McGlennen RC, Faras AJ. The E6 gene of human papillomavirus type 16 is sufficient for transformation of baby rat kidney cells in cotransfection with activated Ha-ras. Virology. 1994;201(2):388–96. doi: 10.1006/viro.1994.1306. [DOI] [PubMed] [Google Scholar]
- Mantovani F, Banks L. The human papillomavirus E6 protein and its contribution to malignant progression. Oncogene. 2001;20(54):7874–87. doi: 10.1038/sj.onc.1204869. [DOI] [PubMed] [Google Scholar]
- McMurray HR, McCance DJ. Human papillomavirus type 16 E6 activates TERT gene transcription through induction of c-Myc and release of USF-mediated repression. J Virol. 2003;77(18):9852–61. doi: 10.1128/JVI.77.18.9852-9861.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 2004;78(21):11451–60. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger K, Basile JR, Duensing S, Eichten A, Gonzalez SL, Grace M, Zacny VL. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene. 2001;20(54):7888–98. doi: 10.1038/sj.onc.1204860. [DOI] [PubMed] [Google Scholar]
- Munger K, Howley PM. Human papillomavirus immortalization and transformation functions. Virus Res. 2002;89(2):213–28. doi: 10.1016/s0168-1702(02)00190-9. [DOI] [PubMed] [Google Scholar]
- Munger K, Phelps WC, Bubb V, Howley PM, Schlegel R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol. 1989a;63(10):4417–21. doi: 10.1128/jvi.63.10.4417-4421.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. Embo J. 1989b;8(13):4099–105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratani M, Tansey WP. How the ubiquitin-proteasome system controls transcription. Nat Rev Mol Cell Biol. 2003;4(3):192–201. doi: 10.1038/nrm1049. [DOI] [PubMed] [Google Scholar]
- Nishimura A, Nakahara T, Ueno T, Sasaki K, Yoshida S, Kyo S, Howley PM, Sakai H. Requirement of E7 oncoprotein for viability of HeLa cells. Microbes Infect. 2006;8(4):984–93. doi: 10.1016/j.micinf.2005.10.015. [DOI] [PubMed] [Google Scholar]
- Oh ST, Kyo S, Laimins LA. Telomerase activation by human papillomavirus type 16 E6 protein: induction of human telomerase reverse transcriptase expression through Myc and GC-rich Sp1 binding sites. J Virol. 2001;75(12):5559–66. doi: 10.1128/JVI.75.12.5559-5566.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Griep AE. Altered cell cycle regulation in the lens of HPV-16 E6 or E7 transgenic mice: implications for tumor suppressor gene function in development. Genes Dev. 1994;8(11):1285–99. doi: 10.1101/gad.8.11.1285. [DOI] [PubMed] [Google Scholar]
- Pan H, Griep AE. Temporally distinct patterns of p53-dependent and p53-independent apoptosis during mouse lens development. Genes Dev. 1995;9(17):2157–69. doi: 10.1101/gad.9.17.2157. [DOI] [PubMed] [Google Scholar]
- Piboonniyom SO, Duensing S, Swilling NW, Hasskarl J, Hinds PW, Munger K. Abrogation of the retinoblastoma tumor suppressor checkpoint during keratinocyte immortalization is not sufficient for induction of centrosome-mediated genomic instability. Cancer Res. 2003;63(2):476–83. [PubMed] [Google Scholar]
- Pim D, Storey A, Thomas M, Massimi P, Banks L. Mutational analysis of HPV-18 E6 identifies domains required for p53 degradation in vitro, abolition of p53 transactivation in vivo and immortalisation of primary BMK cells. Oncogene. 1994;9(7):1869–76. [PubMed] [Google Scholar]
- Schlegel R, Phelps WC, Zhang YL, Barbosa M. Quantitative keratinocyte assay detects two biological activities of human papillomavirus DNA and identifies viral types associated with cervical carcinoma. Embo J. 1988;7(10):3181–7. doi: 10.1002/j.1460-2075.1988.tb03185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz E, Freese UK, Gissmann L, Mayer W, Roggenbuck B, Stremlau A, zur Hausen H. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature. 1985;314(6006):111–4. doi: 10.1038/314111a0. [DOI] [PubMed] [Google Scholar]
- Sekaric P, Cherry JJ, Androphy EJ. Binding of human papillomavirus type 16 E6 to E6AP is not required for activation of hTERT. J Virol. 2008;82(1):71–6. doi: 10.1128/JVI.01776-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman L, Itzhaki H, Jackman A, Chen JJ, Koval D, Schlegel R. Inhibition of serum- and calcium-induced terminal differentiation of human keratinocytes by HPV 16 E6: study of the association with p53 degradation, inhibition of p53 transactivation, and binding to E6BP. Virology. 2002;292(2):309–20. doi: 10.1006/viro.2001.1263. [DOI] [PubMed] [Google Scholar]
- Sherman L, Jackman A, Itzhaki H, Stoppler MC, Koval D, Schlegel R. Inhibition of serum- and calcium-induced differentiation of human keratinocytes by HPV16 E6 oncoprotein: role of p53 inactivation. Virology. 1997;237(2):296–306. doi: 10.1006/viro.1997.8778. [DOI] [PubMed] [Google Scholar]
- Sherman L, Schlegel R. Serum- and calcium-induced differentiation of human keratinocytes is inhibited by the E6 oncoprotein of human papillomavirus type 16. J Virol. 1996;70(5):3269–79. doi: 10.1128/jvi.70.5.3269-3279.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Liem A, Miller JA, Lambert PF. Human papillomavirus types 16 E6 and E7 contribute differently to carcinogenesis. Virology. 2000;267(2):141–50. doi: 10.1006/viro.1999.0106. [DOI] [PubMed] [Google Scholar]
- Song S, Pitot HC, Lambert PF. The human papillomavirus type 16 E6 gene alone is sufficient to induce carcinomas in transgenic animals. J Virol. 1999;73(7):5887–93. doi: 10.1128/jvi.73.7.5887-5893.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steller MA, Zou Z, Schiller JT, Baserga R. Transformation by human papillomavirus 16 E6 and E7: role of the insulin-like growth factor 1 receptor. Cancer Res. 1996;56(21):5087–91. [PubMed] [Google Scholar]
- Stoppler H, Hartmann DP, Sherman L, Schlegel R. The human papillomavirus type 16 E6 and E7 oncoproteins dissociate cellular telomerase activity from the maintenance of telomere length. J Biol Chem. 1997;272(20):13332–7. doi: 10.1074/jbc.272.20.13332. [DOI] [PubMed] [Google Scholar]
- Stoppler H, Stoppler MC, Johnson E, Simbulan-Rosenthal CM, Smulson ME, Iyer S, Rosenthal DS, Schlegel R. The E7 protein of human papillomavirus type 16 sensitizes primary human keratinocytes to apoptosis. Oncogene. 1998;17(10):1207–14. doi: 10.1038/sj.onc.1202053. [DOI] [PubMed] [Google Scholar]
- Storey A, Banks L. Human papillomavirus type 16 E6 gene cooperates with EJ-ras to immortalize primary mouse cells. Oncogene. 1993;8(4):919–24. [PubMed] [Google Scholar]
- Tang S, Tao M, McCoy JP, Jr, Zheng ZM. The E7 oncoprotein is translated from spliced E6*I transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation. J Virol. 2006;80(9):4249–63. doi: 10.1128/JVI.80.9.4249-4263.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, Banks L. Inhibition of Bak-induced apoptosis by HPV-18 E6. Oncogene. 1998;17(23):2943–54. doi: 10.1038/sj.onc.1202223. [DOI] [PubMed] [Google Scholar]
- Thomas MC, Chiang CM. E6 oncoprotein represses p53-dependent gene activation via inhibition of protein acetylation independently of inducing p53 degradation. Mol Cell. 2005;17(2):251–64. doi: 10.1016/j.molcel.2004.12.016. [DOI] [PubMed] [Google Scholar]
- Veldman T, Horikawa I, Barrett JC, Schlegel R. Transcriptional activation of the telomerase hTERT gene by human papillomavirus type 16 E6 oncoprotein. J Virol. 2001;75(9):4467–72. doi: 10.1128/JVI.75.9.4467-4472.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldman T, Liu X, Yuan H, Schlegel R. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc Natl Acad Sci U S A. 2003;100(14):8211–6. doi: 10.1073/pnas.1435900100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturini F, Braspenning J, Homann M, Gissmann L, Sczakiel G. Kinetic selection of HPV 16 E6/E7-directed antisense nucleic acids: anti-proliferative effects on HPV 16-transformed cells. Nucleic Acids Res. 1999;27(7):1585–92. doi: 10.1093/nar/27.7.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Lehr N, Johansson S, Wu S, Bahram F, Castell A, Cetinkaya C, Hydbring P, Weidung I, Nakayama K, Nakayama KI, Soderberg O, Kerppola TK, Larsson LG. The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol Cell. 2003;11(5):1189–200. doi: 10.1016/s1097-2765(03)00193-x. [DOI] [PubMed] [Google Scholar]
- Vormwald-Dogan V, Fischer B, Bludau H, Freese UK, Gissmann L, Glitz D, Schwartz E, Durst M. Sense and antisense transcripts of human papillomavirus type 16 in cervical cancers. J Gen Virol. 1992;73(Pt 7):1833–8. doi: 10.1099/0022-1317-73-7-1833. [DOI] [PubMed] [Google Scholar]
- Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, Clurman BE. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A. 2004;101(24):9085–90. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won J, Chang S, Oh S, Kim TK. Small-molecule-based identification of dynamic assembly of E2F-pocket protein-histone deacetylase complex for telomerase regulation in human cells. Proc Natl Acad Sci U S A. 2004;101(31):11328–33. doi: 10.1073/pnas.0401801101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J, Fan S, Meng Q, Schramm L, Wang C, Bouzahza B, Zhou J, Zafonte B, Goldberg ID, Haddad BR, Pestell RG, Rosen EM. BRCA1 inhibition of telomerase activity in cultured cells. Mol Cell Biol. 2003;23(23):8668–90. doi: 10.1128/MCB.23.23.8668-8690.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. Embo J. 2004;23(10):2116–25. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerfass-Thome K, Zwerschke W, Mannhardt B, Tindle R, Botz JW, Jansen-Durr P. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene. 1996;13(11):2323–30. [PubMed] [Google Scholar]
- Zhang Y, Fan S, Meng Q, Ma Y, Katiyar P, Schlegel R, Rosen EM. BRCA1 interaction with human papillomavirus oncoproteins. J Biol Chem. 2005;280(39):33165–77. doi: 10.1074/jbc.M505124200. [DOI] [PubMed] [Google Scholar]
- zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2(5):342–50. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]