

Abstract
The reaction center from Rhodobacter sphaeroides uses light energy for the reduction and protonation of a quinone molecule, QB. This process involves the transfer of two protons from the aqueous solution to the protein-bound QB molecule. The second proton, H+(2), is supplied to QB by Glu-L212, an internal residue protonated in response to formation of QA− and QB−. In this work, the pathway for H+(2) to Glu-L212 was studied by measuring the effects of divalent metal ion binding on the protonation of Glu-L212, which was assayed by two types of processes. One was proton uptake from solution after the one-electron reduction of QA (DQA→D+QA−) and QB (DQB→D+QB−), studied by using pH-sensitive dyes. The other was the electron transfer kAB(1) (QA−QB→QAQB−). At pH 8.5, binding of Zn2+, Cd2+, or Ni2+ reduced the rates of proton uptake upon QA− and QB− formation as well as kAB(1) by ≈an order of magnitude, resulting in similar final values, indicating that there is a common rate-limiting step. Because D+QA− is formed 105-fold faster than the induced proton uptake, the observed rate decrease must be caused by an inhibition of the proton transfer. The Glu-L212→Gln mutant reaction centers displayed greatly reduced amplitudes of proton uptake and exhibited no changes in rates of proton uptake or electron transfer upon Zn2+ binding. Therefore, metal binding specifically decreased the rate of proton transfer to Glu-L212, because the observed rates were decreased only when proton uptake by Glu-L212 was required. The entry point for the second proton H+(2) was thus identified to be the same as for the first proton H+(1), close to the metal binding region Asp-H124, His-H126, and His-H128.
Keywords: Rhodobacter sphaeroides, proton transfer, electron transfer, protein dynamics
The reaction center (RC) from the photosynthetic bacterium Rhodobacter sphaeroides is a membrane-bound protein complex that catalyzes the reduction and protonation of a quinone molecule (Q+2e−+2H+→QH2) by using sunlight as the energy source (1, 2). The protons required for the reduction of the quinone to quinol come exclusively from the cytoplasmic side of the membrane. When the ubiquinol is reoxidized by another membrane protein, the bc1 complex, protons are released on the periplasmic side of the membrane, creating a proton gradient that is used by the bacterium for ATP synthesis (3).
The double reduction of the quinone takes place in two sequential light-induced electron transfer reactions labeled kAB(1) and kAB(2) and involves the uptake of two protons, H+(1) and H+(2) (see Fig. 1). The first proton H+(1) is taken up from solution and transferred to the reduced semiquinone QB− before the second electron transfer (step 5 in Fig. 1). The second proton H+(2) is transferred internally from the protonated residue Glu-L212 (step 6 in Fig. 1), which was protonated from solution after the formation of QA− and QB− (steps 2 and 3 in Fig. 1).
Figure 1.

The catalytic photo-cycle of quinone reduction in the RC. Electron transfer to QB occurs in two sequential reactions, kAB(1) and kAB(2) (steps 3 and 5). The transitions studied in this work are in bold. In transition 1 the donor bacterio-chlorophyll (D+) is excited by light and transfers an electron to QA. D+ is reduced by cytochrome c2. In step 2 a fraction (δ) of a proton is taken up in the D+QA− state by the amino acid (or cluster of amino acids) A. At pH > 8, A is predominantly Glu-L212. In step 3 the electron on QA is transferred to QB (kAB(1)), concomitant with additional fractional proton uptake (ɛ). In step 4 there is a second light-excited electron transfer from D to QA that involves several steps as in step 1. In step 5 the second electron transfer from QA− to QB (kAB(2)) takes place. kAB(2) is coupled to the uptake of H+(1) to the QB headgroup. In step 6, the second proton, H+(2), is transferred internally (from Glu-L212) to QB forming quinol, which in step 7 dissociates from the RC. A second quinone binds, completing the cycle.
Upon light excitation, an electron is transferred from the bacteriochlorophyll primary electron donor (D) to the primary quinone electron acceptor, QA, at a rate of ≈1010 s−1. From QA− the electron is transferred to the secondary quinone electron acceptor, QB (QA−QB→ QAQB−, step 3 in Fig. 1). The rate of the first electron transfer from QA− to QB [kAB(1) ≈3 × 103 s−1 (pH 8.5)] has been shown not to be limited by the intrinsic electron transfer rate but by a conformational change in the protein (4), which was suggested to involve movement of the QB molecule into the binding pocket (5) or structural changes involving protonation events (6). Furthermore, the rate of kAB(1) has been shown to be slowed down in the presence of Zn2+ (7, 8) and Cd2+ (8). This decrease in kAB(1) was attributed to a decrease in the rate-limiting conformationally gated step and/or internal proton rearrangement. These hypotheses concerning the rate-limiting step of kAB(1) in native and Zn2+-bound RCs were re-examined in this work.
Although neither the reduction of QA nor the first electron transfer from QA− to QB involves direct protonation of the quinones, there is a fractional proton uptake from solution to the RC in both the D+QA− and the D+QB− states caused by shifts in the pKas of amino acid residues interacting electrostatically with the quinones (6, 9–11). Reduction of QB is accompanied by a larger proton uptake than reduction of QA (10, 11). The extent of proton uptake in response to reduction of either quinone has a complex pH-titration curve, indicating contribution from several amino acid residues with different pKas (10, 11). At higher pH (pH > 8), Glu-L212 was shown to be primarily responsible for the proton uptake, as evidenced by the much smaller proton uptake in the Glu-L212→Gln mutant RCs (12–16).
The rate of proton uptake to D+QB− is identical to kAB(1), and similar to the rate of proton uptake to D+QA−, suggesting a common rate-limiting step (6).
The second light-induced electron transfer to QB occurs after reduction of D+ by cytochrome c2 (step 4 in Fig. 1). This electron transfer reaction [kAB(2)] leads, in contrast to kAB(1), to direct protonation of the reduced quinone (step 5 in Fig. 1). The kAB(2) reaction (QA−QB− + 2H+→QAQBH2) has been shown (17) to be composed of reversible binding of H+(1), taken up from the cytoplasm, followed by rate-limiting electron transfer, followed by fast binding to QBH− of H+(2) transferred internally from the protonated Glu-L212 (step 6 in Fig. 1) (16, 18).
The QB molecule is located in the interior of the RC without direct contact with the outside solution. Consequently, there is a need for proton-transfer pathway(s) from the cytoplasm to the quinone-binding site. Several putative proton transfer pathways consisting of protonatable amino acids and/or water molecules leading from the cytoplasm to the quinone-binding site have been identified in the x-ray structures of the RC (19–21).
The pathway for H+(1) has been shown by site-directed mutagenesis to involve Asp-L213 (16, 22, 23), Ser-L223 (24), Asp-L210 and Asp-M17 (25). More recently the entry point for H+(1) was identified by using the inhibitory effect on proton transfer by metal ions Zn2+ and Cd2+ that bind to the surface of the RC at His-H126, His-H128, and Asp-H124 (8, 25, 26).
Much less was known about the pathway for H+(2). Site-directed mutagenesis has shown that H+(2) is supplied to reduced QB by Glu-L212. However the pathway to Glu-L212 was not known. To investigate the pathway for H+(2), we measured the effect of metal ion binding on proton uptake to Glu-L212 by using pH-indicator dyes. Proton uptake was measured on both QA− and QB− formation to determine whether the rate-limiting steps for these two different reactions were the same. The involvement of Glu-L212 in the proton uptake was corroborated by using mutant RCs in which Glu-L212 was replaced by Gln (12, 15). In addition, we measured the rate of the first electron transfer from QA− to QB, kAB(1), to determine the rate-limiting step of this reaction in the presence of bound metal ions.
Materials and Methods
Reagents and Quinones.
Coenzyme Q10 was obtained from Sigma-Aldrich, prepared in ethanol, dried under nitrogen, and solubilized in 1% lauryl-dimethylamine-N-oxide. Terbutryn was obtained from Fluka and prepared in ethanol. Phenol red and m-cresol purple were obtained from Sigma-Aldrich.
Preparation of RCs.
RCs from the R. sphaeroides strain R26.1 were purified to a ratio A280/A800 of 1.2 in lauryl-dimethylamine-N-oxide as described (27). The QB site was reconstituted by addition of 3–4 times excess coenzyme Q10 in 1% lauryl-dimethylamine-N-oxide followed by dialysis against 15 mM Tris, 0.1 mM EDTA, and 0.04% β-d-dodecylmaltoside. The RCs were stored at −80°C. Construction and preparation of the Glu-L212→Gln mutant RC were described (18).
Electron Transfer Measurements.
Absorbance changes in response to a laser flash were measured by using a set-up of local design (28). Actinic illumination was provided by a Nd-YAG laser (Opotek, Carlsbad, CA). The RC concentration was determined from the absorbance at 802 nm by using ɛ = 288 mM−1. Charge recombination kBD (D+QB−→DQB) and kAD (D+QA−→DQA) was measured by monitoring the recovery of D at 865 nm. The occupancy of the QB site was determined from the fraction of the slower recombination rate, kBD (28). This fraction, typically 75–80%, was taken into account when calculating the amount of H+/D+QB−. Electron transfer, kAB(1), was measured by monitoring the bacteriopheophytin band-shift at 750 nm (29, 30). The absorption changes caused by D+QA− formation were accounted for by subtracting the optical changes in the presence of terbutryn. The second electron transfer kAB(2) was determined by monitoring the decay of the semiquinone absorption at 450 nm after a second laser flash in the presence of an external reductant (20 μM horse heart cytochrome c) (31).
Proton Uptake Measurements.
To observe the pH change that occurs on proton uptake, the buffering capacity of the sample was reduced by passing the sample over a Sephadex column (PD-10, Amersham Pharmacia) equilibrated with 50 mM KCl, 0.04% β-d-dodecylmaltoside. Proton uptake at pH 8.5 in the unbuffered sample supplemented with 40 μM of the pH-sensitive dye m-cresol purple was measured at 580 nm (λmax of the deprotonated dye). At lower pH (pH 7.0–8.0) the dye phenol red (λmax = 560 nm) was used. The signal-to-noise ratio was improved by averaging 5–15 traces. The absorbance changes not caused by the dye response were accounted for by subtracting the signal obtained after addition of 5–10 mM Tris (or Hepes at pH < 8) buffer, which eliminates pH changes. The signal of the dye was calibrated in terms of the change in proton concentration by addition of known amounts of HCl, typically 3 μl of 1.0–2.0 mM HCl. The QB inhibitor terbutryn was used to measure proton uptake in response to forming D+QA−.
Results
Proton Uptake.
Proton uptake in response to formation of D+QA− or D+QB− was measured in isolated RCs from R. sphaeroides by using pH-sensitive dyes. It has been shown that Glu-L212 is the major contributor to this proton uptake at pH > 8 (12, 15). Because we are interested in assessing the transfer of H+(2) to Glu-L212 we measured proton uptake at pH 8.5.
Proton uptake upon formation of D+QA− was measured after addition of the electron transfer inhibitor terbutryn. Reduction of QA after a single laser flash occurs with a rate of ≈1010 s−1, which is several orders of magnitude faster than the rate of proton uptake (≈104 s−1 at pH 8.5) (6). The measured rates of proton uptake at pH 8.5 in the absence and presence of Zn2+ were (see Fig. 2A)
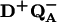 |
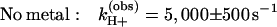 |
1a |
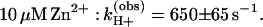 |
1b |
The amplitude,† δH+ (step 2 in Fig. 1) increased upon binding of Zn2+ from 0.13 ± 0.02 H+/D+QA− to 0.23 ± 0.03 H+/D+QA− (Fig. 2A). Slower rates, but similar amplitudes, were observed upon addition of Cd2+ [kH+(obs) ≈500 s−1] and Ni2+ [kH+(obs) ≈450 s−1] (data not shown).
Figure 2.

Effect of Zn2+ binding on proton uptake at pH 8.5 after a single laser flash in native (A and C) and Glu-L212→Gln mutant RCs (B and D) caused by formation of D+QA− (A and B, 120 μM terbutryn added) and D+QB− (C and D). The left ordinate shows the absorbance changes at 580 nm of cresol purple, the right ordinate the corresponding proton uptake. Experimental conditions were: 2 μM RC, 50 mM KCl, 0.04% β-d-dodecyl maltoside, 40 μM cresol purple, pH 8.5. The traces shown are the differences between the traces obtained in the unbuffered solution and the traces obtained after adding 7 mM Tris. Note that the traces with and without Zn2+ overlap in B and D.
In the Glu-L212→Gln mutant RCs, the amplitude of proton uptake at pH 8.5 was decreased ≈4-fold, confirming previous observations (12, 15). No significant changes in rate or amplitude were observed upon Zn2+ binding (Fig. 2B).
Proton uptake upon formation of D+QB− was measured in RCs reconstituted with coenzyme Q10 (see Materials and Methods); the occupancy of the QB site was typically 75–80%. The measured rates of proton uptake at pH 8.5 in the absence and presence of Zn2+ were (see Fig. 2C):
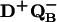 |
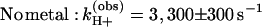 |
2a |
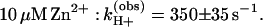 |
2b |
The amplitude,† ɛH+ (step 3 in Fig. 1) increased upon addition of Zn2+ from 0.50 ± 0.05 H+/D+QB− to 0.60 ± 0.06 H+/D+QB− at pH 8.5 (Fig. 2C). As in the case of proton uptake to D+QA−, even slower rates but similar amplitudes were observed upon addition of Cd2+ [kH+(obs) ≈180 s−1] or Ni2+ [kH+(obs) ≈150 s−1] (data not shown).
In the Glu-L212→Gln mutant RCs, the amplitude of proton uptake was decreased ≈8-fold (Fig. 2D), confirming earlier observations (12). No significant changes in rate or amplitude were observed upon Zn2+ binding (Fig. 2D).
At lower pH the effect in native RCs on the rate and amplitude of proton uptake to D+QA− and D+QB− upon binding of Zn2+ became smaller, and for pH < 7 the effect disappeared (data not shown).
The effects of the added metal ions could be reversed at all pHs by addition of EDTA at concentrations above that of the metal ions.
Electron Transfer kAB(1): D+QA−QB → D+QAQB−.
The rate of electron transfer kAB(1) was measured by monitoring the absorbance change at 750 nm of the bacteriopheophytins occurring in response to changing the charged states of the quinones (29, 30). The measured rate constants in the absence and presence of Zn2+ were (Fig. 3A):
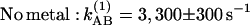 |
3a |
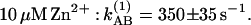 |
3b |
These are the same rate constants as those for proton uptake upon formation of D+QB− (Eq. 2). No effect of Zn2+ on the amplitude of the signal was observed. Slower rates (the same as those for D+QB− proton uptake) were seen upon addition of Cd2+ [kAB(1) ≈180 s−1] or Ni2+ [kAB(1) ≈150 s−1] (data not shown).
Figure 3.

Effect of Zn2+ binding on electron transfer kAB(1) at pH 8.5, measured as the bandshift of the pheophytin cofactor at 750 nm. (A) Native RCs. (B) Glu-L212→Gln mutant RCs. Experimental conditions were as in Fig. 2. Note that the traces with and without Zn2+ overlap in B.
In the Glu-L212→Gln mutant RCs, the rate constant for kAB(1) was 3,300 s−1 at pH 8.5, the same as in native RCs (Fig. 3B) as reported (16, 18). In the mutant RCs kAB(1) was unaffected by the binding of Zn2+ (Fig. 3B).
As the pH was lowered below pH 8.5 in native Zn2+-bound RCs, the kinetics became biphasic, with the metal-sensitive fraction decreasing with decreasing pH (data not shown). At pH 6.5, the kinetics were essentially unaffected by the binding of Zn2+ (data not shown). The rate of the metal-sensitive fraction increases with decreasing pH, in contrast to native Zn2+-free RCs where kAB(1) is essentially pH-independent below pH 8.5 (data not shown).
As in the case of the proton uptake measurements, the effects of the added metal ions could be reversed by addition of EDTA at concentrations above that of the metal ions.
Electron Transfer kAB(2): DQA−QB− + H+ → DQAQBH−.
To confirm that the metal ions were bound to the native RCs at low pH and to the Glu-L212→Gln mutant RCs for which no metal effect on kAB(1) was observed, kAB(2) was measured in the presence of Zn2+. It was found to have decreased in agreement with previous observations (8), indicating the presence of a bound metal ion.
Charge Recombination kAD: D+QA− → DQA and kBD: D+QB− → DQB.
Charge recombination rates kAD and kBD, measured at pH 8.5, remained essentially unchanged upon addition of Zn2+ and Cd2+, confirming previous observations (8).
Discussion
The main motivation for this study was to determine the entry point for transfer of the second proton H+(2) to QB, the entry point for the first proton having been determined earlier (8, 26).
It has been shown by site-directed mutagenesis that Glu-L212 supplies the second proton, H+(2), to QB (12, 16, 18). Consequently, a study of the protonation of Glu-L212 should shed light on the pathway and kinetics of the uptake of H+(2). Two types of processes were used to assay the protonation of Glu-L212. One was proton uptake from solution after the one-electron reduction of either QA or QB, and the other was the electron transfer kAB(1) (QA−QB→QAQB−). All of these processes are inhibited by divalent metal ions such as Zn2+, Cd2+, and Ni2+.
Protonation of Glu-L212 upon Reduction of QA and QB: Reduced Protonation Rate by Binding of Metal Ions.
After a single laser flash, the creation of D+QA− or D+QB− results in proton uptake as shown in Fig. 2 A and C. This uptake is associated with the protonation of Glu-L212 as shown by the greatly reduced amplitude of proton uptake in mutant RCs in which Glu-L212 had been replaced by Gln (Fig. 2 B and D and see refs. 12 and 15). The origin of the proton uptake is a shift in the pKa of Glu-L212 caused by the negative charges on QA− and QB−, respectively. Because Glu-L212 is closer to QB (≈5 Å) than to QA (≈15 Å), the electrostatic interaction and hence the pKa shift is larger, resulting in a larger proton uptake upon D+QB− formation.
The rates of proton uptake upon formation of both D+QA− and D+QB− are decreased by metal binding as shown in Fig. 2 A and C. Because the rate of formation of D+QA− (≈1010 s−1) is much faster than the rate of the induced proton uptake [kH+obs(+Zn2+) = 650 s−1], the observed decrease in rate upon metal ion binding must be caused by an inhibition of proton transfer and not electron transfer. In the Zn2+-bound RCs, the proton uptake rate upon forming D+QB− is similar to that upon D+QA− formation, suggesting that there is a common rate-limiting step, which is the intrinsic rate of protonation. However, the observed rates of proton uptake in the Zn2+-bound RCs upon forming D+QA− (650 s−1, Eq. 1b) and D+QB− (350 s−1, Eq. 2b) are not identical. This can be explained by the fact that the magnitudes of the proton uptake, and thus the final equilibria of Glu-L212, are different (see Fig. 2 A and C). The observed rate of protonation, kH+(obs), is given by the sum of the rate of protonation kH+ and the rate of deprotonation k-H+ [kH+(obs) = kH+ + k-H+]. For QB− k-H+ is smaller than for QA− because of the larger electrostatic interaction with Glu-L212. This accounts for the differences in kH+(obs), although kH+ is the same for both processes. Evidence that kH+ is the same for D+QA− and D+QB− proton uptake in Zn2+-bound RCs comes from the observation that the initial rates (which have no contribution from k-H+) are the same (see Fig. 2 A and C).
The mechanism of the reduced proton uptake rates by the metal ions could be either through the introduction of an electrostatic barrier, an inhibition of protein dynamics important for proton transfer [see section on kAB(1) below], or the elimination of potential proton donors like His-H126 and His-H128, to which the metal is known to bind (26). Evidence for the latter comes from experiments on RCs in which both His residues were replaced by nonprotonatable residues (Ala).‡ The proton uptake rate at pH 8.5 in these mutant RCs was similar to the rate in native Zn2+-bound RCs (unpublished results). Thus, the binding of a metal ion to the His residues removes their capacity to donate protons, as does their replacement by Ala.
Metal binding causes, in addition to the reduced rate of proton uptake, an increase in its amplitude (see Fig. 2 A and C). A simple explanation for this increase would be a decrease in the pKa of Glu-L212 caused by metal binding. However, we rule out this interpretation as discussed in the next section. The increase in proton uptake from solution can be understood in terms of interactions between the metal ion and a pool of protons bound to internal acid residues such as Asp-L210, Asp-M17, Asp-L213, and Glu-H173, as well as the metal ligands His-H126, His-H128, and Asp-H124. These residues are partially protonated and provide an internal source of protons to Glu-L212. The bound metal interacts electrostatically with these residues, thereby reducing their pKas. Consequently, fewer residues are protonated, reducing the source of internal protons to Glu-L212. This loss is compensated for by an increased uptake from solution. Calculations have shown that in particular the protonation state of Asp-L210 is affected by changes in its vicinity (32).
Electron Transfer, kAB(1) (QA−QB→QAQA−): Metal Ion Binding Slows Protonation of Glu-L212, which Limits Electron Transfer.
An alternate way to assess the protonation of Glu-L212 is to measure the electron transfer rate kAB(1), because this process occurs only when Glu-L212 is protonated. This conclusion was arrived at from the observation that in Glu-L212→Gln mutant RCs, kAB(1) is pH-independent and has the same value as in native RCs at pH << 8 (16, 18). Subsequent calculations have corroborated that conclusion (32, 33). In native RCs at pH > 8, when Glu-L212 is deprotonated before electron transfer, kAB(1) decreases drastically (9, 28). It is in this region that kAB(1) is inhibited by binding of metal ions (Fig. 3 and Eq. 4). We can represent the kAB(1) reaction, in analogy to kAB(2) (8, 17), as a two-step process:§
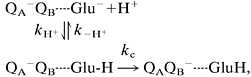 |
4 |
where kc is the rate of protein dynamics preceding electron transfer, and Glu is Glu-L212. From Eq. 4, it follows that when protein dynamics, kc, is the rate-limiting step, kAB(1) = kc × f(Glu-H), where f(Glu-H) is the fraction of RCs having Glu-L212 protonated. When proton uptake is rate-limiting, kAB(1) = kH+.
In native RCs at pH << 8, where f(Glu-H) is 1 (as well as in Glu-L212→Gln mutant RCs), kAB(1) is pH-independent and protein dynamics, kc, is the rate-limiting step, i.e., (kH+ + k-H+) >> kc. A detailed mechanism accounting for protein dynamics to affect kAB(1) is not known, but could involve reorientation of water molecules or other polar groups. The movement of QB from its distal to proximal position (5), which was proposed earlier to represent the rate-limiting step (4, 5), can be ruled out by recent experiments that showed that kAB(1) was unchanged in mutant RCs (34), in which QB was already in the proximal position before reduction (35). Metal binding does not affect protein dynamics, kc, as shown by the lack of metal effect on kAB(1) in the Glu-L212→Gln mutant RCs (see Fig. 3B), as well as the lack of effect on kAB(1) in native RCs at lower pH. This is contrary to previous proposals that the metal affects protein dynamics associated with electron transfer or quinone movement (7, 8).
Above pH > 8, kAB(1) decreases in native RCs. This could be due either to a reduction in f(Glu-H), or to proton transfer kH+ becoming rate-limiting. In the presence of metal ions a further drastic reduction in kAB(1) is observed (Fig. 3A). What is the mechanism for this reduction in kAB(1)? The metal does not affect kc as discussed above. In principle, metal binding could affect f(Glu-H) by decreasing the pKa of Glu-L212. However, we rule this possibility out for two reasons: First, the rate and pH dependence of kBD (D+QB−→DQB), which is sensitive to the pKa of Glu-L212 (16, 18), has been found to be essentially unaffected by the metal (see Results and ref. 8). Second, the rates of kAB(1) and proton uptake are even slower with Cd2+ than with Zn2+ bound, whereas the amplitude of proton uptake is the same. The slower rate with Cd2+ would imply an even larger decrease in the pKa of Glu-L212, which would result in a larger extent of proton uptake. Because this is not observed, we exclude this possibility. We therefore conclude that the rate-limiting step of Eq. 4 in the presence of a bound metal ion is the rate of proton uptake from solution kH+, i.e., kH+ << kc.
In the intermediate pH region (pH 7–8) in which Glu-L212 is partially protonated, we observe biphasic kinetics in Zn2+-bound RCs. This is predicted by Eq. 4 when proton transfer is rate-limiting. The fast phase with rate constant kc is caused by the fraction of the RCs that have Glu-L212 protonated, and the slow phase is caused by the fraction of RCs that have Glu-L212 ionized, requiring rate-limiting proton transfer kH+.
Proton Transfer Pathway.
The results presented above show that with Zn2+ bound to the RC, the rate of transfer of H+(2) to Glu-L212 from solution is greatly decreased. Zn2+ (and Cd2+) binds to His-H126, His-H128, and Asp-H124 (see Fig. 4), defining the entry point for the transfer of H+(2) to be near the suggested P3 pathway (21). This is the same entry point as that found for H+(1) (8), making it the common, unique entry point for both protons used for the reduction of quinone to quinol. The involvement of this surface region in proton transfer is further supported by the experiments in RCs lacking the His-H126 and His-H128 mentioned above.
Figure 4.

Part of the structure of the RC from R. sphaeroides in the charge-separated state (coordinates are from ref. 5). Shown are the two quinones with their bridging amino acid residues, His-M219 and His-L190, and the nonheme Fe2+. The amino acid residues Asp-M17, Asp-L210, Asp-L213, Ser-L223, and Glu-L212, involved in proton transfer to QB are shown, with the transfer of H+(2) (bold) to Glu-L212 (boxed). The pathway (thin arrow) for H+(1) has recently been determined (8) and the pathway for H+(2) (bold arrow) was studied in this work. H+(2) is taken up to Glu-L212 in response to reduction of the quinones and then is transferred to QB after kAB(2) (see text and Fig. 1). Filled circles represent water molecules, and dashed lines potential hydrogen bonds. Also shown are Asp-H124, His-H126, and His-H128 that bind Zn2+ and Cd2+ at the circled star. Ni2+ binds to His-H126 and Asp-M17 at the boxed star (26). In the absence of metal ions, the positions of the circled and boxed stars are occupied by water molecules. The illustration was made by using the programs molscript (36) and raster3d (37).
The transfer of H+(1), as determined from the value of kAB(2), also involves Asp-M17 and Asp-L210 (Fig. 4) (25). This was shown by a decrease in kAB(2) in the presence of Cd2+ when either Asp-M17 or Asp-L210 was replaced with Asn. The same study showed that in Cd2+-inhibited RCs, kAB(1) was also decreased in those mutant RCs. This supports our conclusion that in the metal-inhibited RCs proton transfer becomes rate-limiting for kAB(1) as well as kAB(2), and further suggests that the pathway for the transfer of H+(2) from solution to Glu-L212 also involves Asp-M17 and Asp-L210. Whether Asp-L210 or Asp-L213, the amino acid closest to Glu-L212 (Fig. 4) serves as the branch point for H+(2) remains to be determined. In either case, the transfer to Glu-L212 likely involves water molecules, because the distance from either Asp-L213 (≈7 Å) or Asp-L210 (≈8 Å) is too large for direct transfer.
The Rate of Proton Transfer Through the H+(1) and H+(2) Pathways.
It is interesting to note that the relative effects of the different metals on the rate of H+(1) and H+(2) uptake are the same, with Zn2+ having the smallest and Ni2+ the largest effect (8). However, the absolute rates for the two proton uptake steps differ. In the presence of Zn2+ at pH 8.5 the measured rates are ≈350 s−1 for H+(2) and ≈50 s−1 for H+(1) (8). This suggests that the rate-limiting step of the proton uptake is an intraprotein transfer, because the rate of a diffusion-controlled reaction should be the same for the two processes. The observed difference in the rates could be explained by the Marcus theory for proton transfer (38), which shows that there are two important parameters for the rate of transfer. The first is the activation energy, which involves a reorganization energy and the pKa difference between the proton donor and acceptor. The second is the work term, which describes the energy cost to align the components of the proton transfer chain to facilitate the proton transfer. For our situation, both the activation energy and the work term are likely to be different for the transfer of H+(1) and H+(2) because the pKas of QB− and Glu-L212 are quite different [ΔpKa = 4–5 (16, 18, 39)] and parts of the pathways follow structurally different routes (Fig. 4). Which of these terms accounts for the observed difference in rates remains to be determined.
The above argument also can be used to explain why the rates depend on the identity of the bound metal ion if we consider the possibility that the metal-bound water/hydroxide could be the proton donor in the rate-limiting step. As the pKas of these metal-bound hydroxides differ, the rates are expected to be different.
To understand the details of proton transfer reactions in proteins one needs to identify the proton donor and acceptor components of the pathway. In this study, we identified components of the transfer pathway for H+(2). We are, therefore, in a position now to systematically investigate the parameters that characterize each step in the proton transfer reaction, e.g., by using site-directed mutagenesis to vary the pKa (and hence the driving force) of the amino acid residues in the pathway or by the use of external proton donors with different pKas, such as imidazoles. Such studies should help elucidate the relative importance of the activation energy and the work term. Experiments along these lines have been performed on carbonic anhydrase (refs. 40 and 41, reviewed in ref. 42).
Summary
We have determined the entry point of the pathway for H+(2) to the reduced quinone, QB, as the region around His-H126, His128, and Asp-H124 (Fig. 4). The logic of the approach was as follows:
(i) The second proton, H+(2), is supplied to reduced QB by Glu-L212 (12, 16, 18).
(ii) We measured the rate of protonation of Glu-L212 after the reduction of QA and QB and the electron transfer rate kAB(1) (QA−QB→QAQB−), which depends on the protonation of Glu-L212.
(iii) The binding of divalent metal ions (Zn2+, Cd2+, Ni2+) decreases the rate of proton transfer, which is the rate-limiting step. (The binding of the metal ions do not affect intrinsic rates of electron transfer or protein dynamics associated with the movement of QB.)
(iv) Metal binding decreases the rate of transfer of both the first [H+(1)] and second [H+(2)] proton, showing that both protons involved in the formation of QBH2 share a common entry point. This entry point is defined by the location of the bound metal ions (26). The pathways for H+(1) and H+(2) also share the involvement of Asp-M17 and Asp-L210 (Fig. 4).
Acknowledgments
We thank E. Abresch for technical assistance, H. Axelrod for help in preparing Fig. 4, and T. Beatty for the mutant His-H126→Ala/His-H128→Ala RCs. This work was supported by the National Science Foundation (Grant MCB 99–82186), the National Institutes of Health (Grants GM-41637 and GM-13191), and a postdoctoral fellowship from the Swedish Foundation for International Cooperation in Research and Higher Education (STINT) to P.Ä.
Abbreviations
- D
primary electron donor
- QA
primary quinone electron acceptor
- QB
secondary quinone electron acceptor
- RC
reaction center
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.230439597.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.230439597
The difference in amplitudes of proton uptake observed in this study compared with those published in ref. 10 are attributed to the presence of terbutryn in the QB site and to the use of different detergents.
Beatty, J. T., Paddock, M. L., Feher, G. & Okamura, M. Y. (2000) Biophys. J. 78, 339A.
It should be noted that proton transfer to Glu-L212 from the internal pool of protons discussed in the previous section is likely to have a much faster rate than kH+ and may be important in understanding the details of the kAB(1) reaction in native RCs.
References
- 1.Okamura M Y, Paddock M L, Graige M S, Feher G. Biochim Biophys Acta. 2000;1458:148–163. doi: 10.1016/s0005-2728(00)00065-7. [DOI] [PubMed] [Google Scholar]
- 2.Blankenship R E, Madigan M T, Bauer C E. Anoxygenic Photosynthetic Bacteria. Dordrecht, The Netherlands: Kluwer; 1995. [Google Scholar]
- 3.Cramer W A, Knaff D B. Energy Transduction in Biological Membranes. New York: Springer; 1990. [Google Scholar]
- 4.Graige M S, Feher G, Okamura M Y. Proc Natl Acad Sci USA. 1998;95:11679–11684. doi: 10.1073/pnas.95.20.11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stowell M H, McPhillips T M, Rees D C, Soltis S M, Abresch E, Feher G. Science. 1997;276:812–816. doi: 10.1126/science.276.5313.812. [DOI] [PubMed] [Google Scholar]
- 6.Maróti P, Wraight C A. Biophys J. 1997;73:367–381. doi: 10.1016/S0006-3495(97)78077-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Utschig L M, Ohigashi Y, Thurnauer M C, Tiede D M. Biochemistry. 1998;37:8278–8281. doi: 10.1021/bi980395n. [DOI] [PubMed] [Google Scholar]
- 8.Paddock M L, Graige M S, Feher G, Okamura M Y. Proc Natl Acad Sci USA. 1999;96:6183–6188. doi: 10.1073/pnas.96.11.6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wraight C A. Biochim Biophys Acta. 1979;548:309–327. doi: 10.1016/0005-2728(79)90138-5. [DOI] [PubMed] [Google Scholar]
- 10.McPherson P H, Okamura M Y, Feher G. Biochim Biophys Acta. 1988;934:348–368. doi: 10.1016/0005-2728(93)90116-w. [DOI] [PubMed] [Google Scholar]
- 11.Maróti P, Wraight C A. Biochim Biophys Acta. 1988;934:329–347. [Google Scholar]
- 12.McPherson P H, Schönfeld M, Paddock M L, Okamura M Y, Feher G. Biochemistry. 1994;33:1181–1193. doi: 10.1021/bi00171a018. [DOI] [PubMed] [Google Scholar]
- 13.Hienerwadel R, Grzybek S, Fogel C, Kreutz W, Okamura M Y, Paddock M L, Breton J, Nabedryk E, Mäntele W. Biochemistry. 1995;34:2832–2843. doi: 10.1021/bi00009a013. [DOI] [PubMed] [Google Scholar]
- 14.Nabedryk E, Breton J, Hienerwadel R, Fogel C, Mäntele W, Paddock M L, Okamura M Y. Biochemistry. 1995;34:14722–14732. doi: 10.1021/bi00045a013. [DOI] [PubMed] [Google Scholar]
- 15.Miksovska J, Maróti P, Tandori J, Schiffer M, Hanson D K, Sebban P. Biochemistry. 1996;35:15411–15417. doi: 10.1021/bi961299u. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi E, Wraight C A. Biochemistry. 1992;31:855–866. doi: 10.1021/bi00118a031. [DOI] [PubMed] [Google Scholar]
- 17.Graige M S, Paddock M L, Bruce J M, Feher G, Okamura M Y. J Am Chem Soc. 1996;118:9005–9016. [Google Scholar]
- 18.Paddock M L, Rongey S H, Feher G, Okamura M Y. Proc Natl Acad Sci USA. 1989;86:6602–6606. doi: 10.1073/pnas.86.17.6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fritzsch G, Kampmann L, Kapaun G, Michel H. Photosynth Res. 1998;55:127–132. [Google Scholar]
- 20.Baciou L, Michel H. Biochemistry. 1995;34:7967–7972. doi: 10.1021/bi00025a001. [DOI] [PubMed] [Google Scholar]
- 21.Abresch E C, Paddock M L, Stowell M H B, McPhillips T M, Axelrod H L, Soltis S M, Rees D C, Okamura M Y, Feher G. Photosynth Res. 1998;55:119–125. [Google Scholar]
- 22.Paddock M L, Rongey S H, McPherson P H, Juth A, Feher G, Okamura M Y. Biochemistry. 1994;33:734–745. doi: 10.1021/bi00169a015. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi E, Wraight C A. Biochim Biophys Acta. 1990;1020:107–111. [Google Scholar]
- 24.Paddock M L, McPherson P H, Feher G, Okamura M Y. Proc Natl Acad Sci USA. 1990;87:6803–6807. doi: 10.1073/pnas.87.17.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paddock M L, Feher G, Okamura M Y. Proc Natl Acad Sci USA. 2000;97:1548–1553. doi: 10.1073/pnas.97.4.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Axelrod H L, Abresch E C, Paddock M L, Okamura M Y, Feher G. Proc Natl Acad Sci USA. 2000;97:1542–1547. doi: 10.1073/pnas.97.4.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Isaacson R A, Lendzian E, Abresch E C, Lubitz W, Feher G. Biophys J. 1995;69:311–322. doi: 10.1016/S0006-3495(95)79936-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleinfeld D, Okamura M Y, Feher G. Biochim Biophys Acta. 1984;766:126–140. doi: 10.1016/0005-2728(84)90224-x. [DOI] [PubMed] [Google Scholar]
- 29.Verméglio A, Clayton R K. Biochim Biophys Acta. 1977;461:159–165. doi: 10.1016/0005-2728(77)90078-0. [DOI] [PubMed] [Google Scholar]
- 30.Tiede D M, Vazquez J, Cordova J, Marone P A. Biochemistry. 1996;35:10763–10775. doi: 10.1021/bi9605907. [DOI] [PubMed] [Google Scholar]
- 31.Kleinfeld D, Okamura M Y, Feher G. Biochim Biophys Acta. 1985;809:291–310. doi: 10.1016/0005-2728(85)90179-3. [DOI] [PubMed] [Google Scholar]
- 32.Alexov E G, Gunner M R. Biochemistry. 1999;38:8253–8270. doi: 10.1021/bi982700a. [DOI] [PubMed] [Google Scholar]
- 33.Grafton A K, Wheeler R A. J Phys Chem. 1999;103:5380–5387. [Google Scholar]
- 34.Tandori J, Sebban P, Michel H, Baciou L. Biochemistry. 1999;38:13179–13187. doi: 10.1021/bi990192e. [DOI] [PubMed] [Google Scholar]
- 35.Kuglstatter, A., Ermler, U., Michel, H., Baciou, L. & Fritzsch, G. (2000) Biochemistry, in press. [DOI] [PubMed]
- 36.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 37.Merritt E A, Bacon J D. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 38.Marcus R A. J Phys Chem. 1968;72:891–899. [Google Scholar]
- 39.Graige M S, Paddock M L, Feher G, Okamura M Y. Biochemistry. 1999;38:11465–11473. doi: 10.1021/bi990708u. [DOI] [PubMed] [Google Scholar]
- 40.Tu C K, Qian M Z, Earnhardt J N, Laipis P J, Silverman D N. Biophys J. 1998;74:3182–3189. doi: 10.1016/S0006-3495(98)78024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Earnhardt J N, Tu C, Silverman D N. Can J Chem-Rev Canadienne Chimie. 1999;77:726–732. [Google Scholar]
- 42.Silverman D N. Biochim Biophys Acta. 2000;1458:88–103. doi: 10.1016/s0005-2728(00)00061-x. [DOI] [PubMed] [Google Scholar]