

Abstract
Alterations in PKC isozyme expression and aberrant induction of cyclin D1 are early events in intestinal tumorigenesis. Previous studies have identified cyclin D1 as a major target in the antiproliferative effects of PKCα in non-transformed intestinal cells; however, a link between PKC signaling and cyclin D1 in colon cancer remained to be established. The current study further characterized PKC isozyme expression in intestinal neoplasms and explored the consequences of restoring PKCα or PKCδ in a panel of colon carcinoma cell lines. Consistent with patterns of PKC expression in primary tumors, PKCα and δ levels were generally reduced in colon carcinoma cell lines, PKCβII was elevated and PKCε showed variable expression, thus establishing the suitability of these models for analysis of PKC signaling. While colon cancer cells were insensitive to the effects of PKC agonists on cyclin D1 levels, restoration of PKCα downregulated cyclin D1 by two independent mechanisms. PKCα expression consistently (a) reduced steady-state levels of cyclin D1 by a novel transcriptional mechanism not previously seen in non-transformed cells, and (b) re-established the ability of PKC agonists to activate the translational repressor 4E-BP1 and inhibit cyclin D1 translation. In contrast, PKCδ had modest and variable effects on cyclin D1 steady state levels and failed to restore responsiveness to PKC agonists. Notably, PKCα expression blocked anchorage-independent growth in colon cancer cells via a mechanism partially dependent on cyclin D1 deficiency, while PKCδ had only minor effects. Loss of PKCα and effects of its re-expression were independent of the status of the APC/β-catenin signaling pathway or known genetic alterations, indicating that they are a general characteristic of colon tumors. Thus, PKCα is a potent negative regulator of cyclin D1 expression and anchorage-independent cell growth in colon tumor cells, findings that offer important perspectives on the frequent loss of this isozyme during intestinal carcinogenesis.
Keywords: Protein Kinase C, Cyclin D1, Transcriptional Control, Colon Cancer, 4E-BP1, β-Catenin, Mouse Models
Introduction
Disruption of the tight control of cell proliferation in self-renewing tissues such as the intestinal epithelium is a key step in tumor development. Increasing evidence points to protein kinase C (PKC) isozymes as important regulators of homeostasis in the intestine [1-3]. PKC is a family of at least 10 serine/threonine kinases that differ in cofactor requirements, tissue distribution, and substrate specificity. These differences likely contribute to isozyme-specific regulation of various cellular processes, including proliferation, differentiation, apoptosis/survival, and transformation [1-5]. The importance of PKC signaling in maintenance of intestinal homeostasis is underscored by frequent deregulation of this enzyme system in intestinal neoplasms [2, 4]. Decreased abundance of PKCα, βI, δ, ζ, and η has been noted in human and rodent intestinal tumors, while levels of PKCβII and ι appear to be elevated during neoplastic transformation [2, 4]. Notably, downregulation of PKCα is an early event in intestinal tumorigenesis, evident in preneoplastic aberrant crypt foci of azoxymethane-treated mice [6] and adenomas of ApcMin/+ mice [7, 8].
The demonstration that PKCα is activated at the point of growth arrest in intestinal crypts in situ [5, 9], and that PKCα signaling promotes cell cycle withdrawal in non-transformed intestinal epithelial cells (IECs) [3], points to a key role of this isozyme in regulating intestinal self-renewal. Consistent with these findings, increased expression of PKCα in APC mutant CaCo-2 colorectal cancer (CRC) cells decreased proliferation, increased differentiation, and attenuated the transformed phenotype, while reduced expression resulted in enhanced growth, decreased differentiation, and a more aggressive tumor phenotype [10, 11]. Strong support for a tumor suppressor role of PKCα in the intestine comes from studies in PKCα knockout mice, which showed increased proliferative activity within intestinal crypts and spontaneous intestinal adenoma formation. Importantly, PKCα-deficient ApcMin/+ mice develop more aggressive tumors and exhibit reduced survival relative to PKCα-expressing littermates [8]. Although PKCα deficiency has been noted in APC/β-catenin mutant intestinal tumors, it remains to be determined if loss of PKCα is directly linked to alterations in β-catenin signaling or is a more general characteristic of intestinal carcinogenesis. Moreover, the molecular basis for loss of PKC signaling during intestinal tumorigenesis has not been defined.
Downregulation of cyclin D1 is one of the earliest consequences of PKCα activation in non-transformed IECs, preceding other hallmark events of cell cycle withdrawal [3, 12]. Thus, cyclin D1 appears to be a critical target of PKCα in intestinal crypt cells. Cyclin D1 is a potent pro-proliferative molecule that links extracellular signaling to the cell cycle machinery [13]. Aberrant expression of cyclin D1 is one of the most common abnormalities in cancer and a key component of tumor development in various tissues [14-16]. Importantly, cyclin D1 deficiency inhibits formation of APC-mutant intestinal tumors in mice [14, 17], pointing to a direct role in intestinal tumorigenesis. However, a link between PKCα deficiency and cyclin D1 in intestinal tumors remains to be established.
To gain further insight into the role of PKCα in intestinal tumorigenesis, this study (a) compared the expression of PKC isozymes and cyclin D1 in a panel of murine and human intestinal tumors and human CRC cell lines, and (b) analyzed the effects of PKCα and PKCδ on cyclin D1 levels and anchorage-independent growth in various CRC cell lines. Our data demonstrate that PKCα is broadly lost in intestinal tumors of diverse genetic backgrounds and point to a specific role for PKCα in regulation of cyclin D1 accumulation in CRC cells, irrespective of β-catenin signaling status. Notably, PKCα suppresses cyclin D1 expression in colon cancer cells by two independent mechanisms: transcriptional repression and blockade of cap-dependent translation. Together, these findings provide a new perspective on the frequent loss of PKCα in intestinal neoplasia.
Experimental Procedures
Materials and Cell Lines
Antibodies were obtained from: Santa Cruz Biotechnology [anti-cyclin D1 (sc-753, sc-450), -PKCα (sc-8393, sc-208), -PKCβII (sc-210), -PKCδ (sc-213 and sc-213P blocking peptide), -PKCε (sc-214), -GAPDH (sc-25778), -phospho-EGFR (Tyr1173) (sc-12351-R), -total EGFR (sc-03), and -actin (sc-7210) antibody]; Lab Vision Corp [anti-cyclin D1 antibody (clone SP4)]; BD Transduction Labs [anti-PKCδ antibody (610397) used for immunohistochemical analysis]; Chemicon [anti-total eIF4E-binding protein 1 (4E-BP1) antibody and peroxidase-conjugated goat anti-rabbit IgG]; Cell Signaling Technology [anti-nonphospho-Thr46-4E-BP1 antibody]; Bio-Rad [peroxidase-conjugated goat anti-mouse IgG]; Epitomics Inc [anti-PKCα rabbit polyclonal antibody (1510-1) used for immunofluorescence]; Jackson ImmunoResearch [TRITC-conjugated donkey anti-rabbit IgG (711-025-152)]; and Sigma-Aldrich [anti-actin (A-2066)]. Phorbol 12-myristate 13-acetate (PMA), propidium iodide, cycloheximide (CHX), actinomycin D (ActD), and p-iodonitrotetrazolium violet were from Sigma-Aldrich. LY294002 was from Calbiochem.
Human CRC cell lines were from Dr. M.G. Brattain, except for DLD-1 and HCT-15 cells (Dr. R.J. Bernacki, RPCI), LS180 cells (Dr. Y. Ionov, RPCI), and Colo205, SW620, and HCT116 cells (ATCC). Normal Fetal Human Colon (FHC) cells and IEC-18 non-transformed rat intestinal crypt cells were from ATCC. Human cell lines were cultured in RPMI 1640, 10% fetal bovine serum (FBS), 2 mmol/L L-glutamine. FHC cells were also cultured in ATCC Complete Culture Medium. IEC-18 cells were cultured as described [3].
Analysis of Protein and RNA Expression
Immunoblot analysis was performed as described [3, 5]. Antibody dilutions were 1:1000 or 1:2000 for anti-cyclin D1; 1:1000 for anti-PKCβII and -PKCε; 1:1000 or 1:5000 for anti-PKCα and -PKCδ; 1:1000 (sc-7210) or 1:10000 (A-2066) for anti-actin; 1:5000 for anti-4E-BP1; 1:2000 for anti-phospho-Thr46-4E-BP1; 1:2000 for anti-GAPDH; and 1:100 for anti-phospho-EGFR (Tyr1173) and anti-total EGFR. In competition experiments, anti-PKCδ was incubated with blocking antigenic peptide at 5:1 (wt/wt) peptide to antibody ratio for 15 min prior to addition to the membrane. Northern blot analysis of total cellular RNA using randomly primed 32P-labeled probe corresponding to mouse cyclin D1 cDNA and quantification of phosphorimaging data with ImageQuant (GE Healthcare) were as described [18].
Human Tissues
CRC tissues and corresponding normal mucosa were collected from 10 patients at RPCI, with written informed consent and approval by the Institutional Review Board.
Adenoviral Transduction and Anchorage-Independent Growth Assays
Adenoviruses expressing LacZ, PKCα, PKCδ, or kinase-dead PKCα (kdPKCα) were from Drs. T. Kuroki (Kobe University, Japan) and M.G. Kazanietz (University of Pennsylvania) [19]. Cyclin D1 adenovirus was from Vector Biolabs. Adenoviruses were amplified in HEK293 packaging cells using standard techniques [20] and titers were determined using the Adeno-X Rapid Titer kit (Clonetech). Cells (2 - 4 × 105), in reduced serum growth medium (2% FBS), were infected with adenovirus in suspension in 6-well plates. After 16 h, cells were rinsed with phosphate buffered saline (PBS), and incubated in complete growth medium for 48 h prior to analysis. LacZ, PKCα, or PKCδ adenoviruses were used at a multiplicity of infection (moi) of 20 infection units/cell (except RKO cells, which required 150 moi to achieve expression comparable with other cell lines). To ensure similar levels of kdPKCα (which is unstable in cells [21]) and wild-type PKCα, kdPKCα adenovirus was added at 500 moi. In PKCα/cyclin D1 co-expression experiments, each adenovirus was added at 20 or 40 moi.
Flow cytometry
Cell cycle distribution of adenovirus-infected cells was determined by flow cytometry of propidium iodide-stained cells and analysis with Modfit (Verity Software House) as described [3].
Anchorage-Independent Growth Assays
Cells (5 × 103) were plated in soft agarose as described [22]. After 1-2 weeks, colonies were stained with p-iodonitrotetrazolium violet (1 mg/mL) for 24 h and photographed on a transilluminator using a digital camera.
β-catenin silencing
β-catenin was silenced in HCT116 or DLD-1 cells by transient transfection with 100 nM ON-TARGETplus SMARTpool β-catenin siRNA (Dharmacon) or non-silencing siRNA (Qiagen), according to manufacturer protocols (using Lipofectamine 2000, Invitrogen). β-catenin-silenced cells were infected with 20 moi LacZ or PKCα adenovirus 72 h post-transfection.
Carcinogen Treatment of Mice and Immunohistochemistry
Experiments involving mice were in accordance with institutional and national guidelines/regulations, with appropriate approvals. A/J and KK/HIJ mice (2-4 months old, Jackson Laboratories) received azoxymethane (10 mg/kg) in 4 weekly IP injections, and were euthanized 5-6 months after the first injection. Tissues were fixed in 10% neutral buffered formalin overnight and embedded in paraffin. Paraffin-embedded tissues were sectioned (7 μm), deparaffinized, rehydrated, and incubated in 3% H2O2 (15 min) to block endogenous peroxidase activity. Antigen retrieval involved microwaving (2 × 10 min) in citrate buffer. Sections were “blocked” with 0.03% casein (30 min) and incubated with Santa Cruz Biotechnology sc-208 anti-PKCα antibody [1 μg/mL in PBS/Tween-20 buffer], BD Biosciences anti-PKCδ antibody (10 μg/mL), or Lab Vision Corp SP4 anti-cyclin D1 antibody (1:100) for 1 h, followed by biotinylated anti-rabbit secondary antibody (Vector Labs, 1:250) and streptavidin HRP (1:20, Zymed). Sections were then incubated with Vectastain Elite ABC reagent (Vector Laboratories) and DAKO DAB chromogen solution (K3466), counterstained with hematoxylin, and imaged with an Olympus BPX41 microscope and DP70 digital camera.
Luciferase Reporter Assays
-1745CD1Luc and -163CD1Luc cyclin D1 promoter constructs were from Dr. R. Pestell (Kimmel Cancer Center) [23]. Adenovirus-infected DLD-1 cells were transfected and assayed for luciferase activity as described [18].
Immunofluorescence
Cells plated on glass coverslips were infected with adenovirus and incubated in fresh medium for 24 h prior to fixation in 2% formaldehyde/PBS and permeabilization in 0.2% Triton X-100 (PBS/Triton) as described [5]. Anti-PKCα antibody (Epitomics) and TRITC-conjugated donkey anti-rabbit secondary antibody were applied at 1:1000 and 1:100 dilution in PBS/Triton, respectively. Cells were imaged using a Zeiss Axioskop epifluorescence microscope (x63 Plan Apochromat (1.4 NA) objective lens) and Hamamatsu C7780 digital camera.
Results
Alterations in PKCα expression in neoplastic intestinal cells occur independently of perturbations in APC/β-catenin signaling
To explore further the role of alterations in PKCα signaling in CRC, PKCα protein expression was profiled in a panel of mouse intestinal tumors and human CRC cell lines that reflect the range of genetic changes associated with colon cancer development (Supplementary Table 1) [24]. Immunohistochemical analysis of PKCα was performed in murine intestinal tumors expressing mutant Apc (from ApcMin/+ and Apc1638 mice [25, 26]) or enhanced β-catenin signaling (from azoxymethane-treated KK/HIJ and A/J mice [27]), as well as in tumors which are wild-type for APC and β-catenin but mutant for K-Ras (from pVillin-KRasV12G mice [28]) or deficient in Muc2 (from Muc2-/- mice [29]). Comparison of PKCα immunostaining in tumors and “normal” adjacent mucosa revealed loss of the enzyme in all neoplastic tissues examined, irrespective of genetic background (Figure 1A, A/J mice not shown), thus demonstrating for the first time that PKCα deficiency can occur independently of aberrations in APC/β-catenin signaling.
Figure 1. PKC isozyme expression is altered in neoplastic intestinal tissues and cell lines.
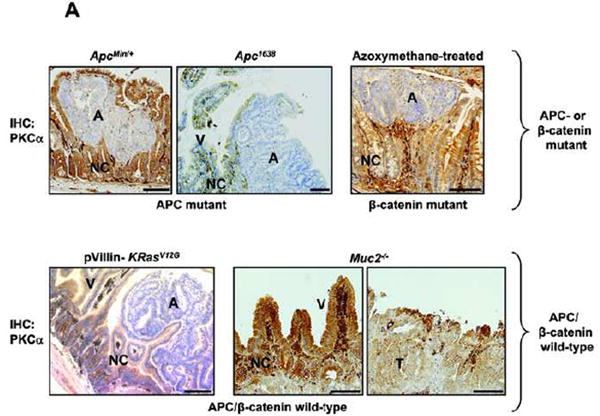

(A) Immunohistochemical analysis of PKCα expression (brown staining) in adenomas (A) or tumors (T) and adjacent normal crypt (NC) and villus (V)/surface mucosa cells from ApcMin/+ and Apc1638 mice (APC mutant), [25], [26] azoxymethane-treated KK/HIJ mice (β-catenin mutant),[27] and Apc/β-catenin wild-type pVillin-KRasV12G mice[28] and Muc2-/- mice[29]. Magnification Bar: 100 μm. PKCα is diffusely distributed throughout the cytoplasm of normal, proliferating intestinal crypt cells (NC) (likely in an inactive conformation), largely at the plasma membrane/active in normal post-mitotic intestinal cells (V), and absent from neoplastic cells.
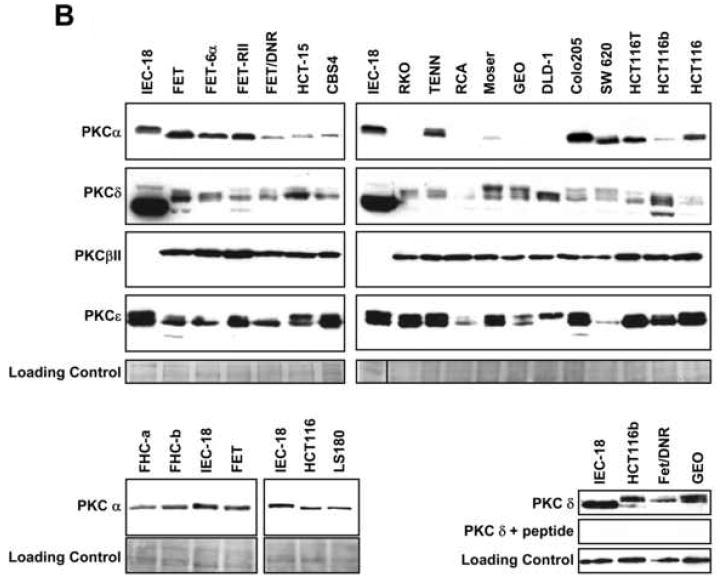
(B) Immunoblot analysis of PKC isozymes in human CRC cells and non-transformed IEC-18 and FHC cells [cultured in the same medium as CRC cells (FHC-a) or in ATCC Complete Culture Medium (FHC-b)]. The faster mobility PKCα species seen in CRC cells likely lacks phosphorylation at Ser657, one of the priming sites on the enzyme (Leontieva and Black, unpublished data). PKCδ consistently showed differential migration patterns in CRC cells. Lower right panel: Peptide competition analysis of the specificity of PKCδ immunoblotting. Samples were probed with anti-PKCδ antibody or anti-PKCδ antibody pre-incubated with blocking peptide. Data represent ≥3 independent experiments.
(C) Top panel: Immunohistochemical analysis of PKCδ expression in adenomas (A) and adjacent normal mucosa (NM)/normal crypt (NC) cells from ApcMin/+ and azoxymethane-treated KK/HIJ mice. Magnification Bar: 100 μm. Lower panel: Immunoblot analysis of PKCδ expression in extracts of colon tumor tissue (T) and adjacent normal colonic mucosa (N) from 10 patients. Equal loading was confirmed by fast green staining (not shown).
Analysis was then extended to a panel of 18 human CRC cell lines. Consistent with expression patterns in mouse intestinal tumors (Figure 1A), a majority of human CRC cell lines express significantly reduced levels of PKCα protein compared with non-transformed IEC-18 cells and FHC human colonic crypt cells (Figure 1B). APC mutant (e.g., FET/DNR, GEO, DLD-1), β-catenin mutant (HCT-116, LS180), and APC/β-catenin wild-type cells (RKO) exhibit decreased abundance of this isozyme, confirming that loss of PKCα occurs independently of APC/β-catenin signaling status and providing further support for the importance of PKCα deficiency in intestinal tumor development.
CRC cell lines were also profiled for other members of the PKC family that have been linked to intestinal carcinogenesis, including PKCδ, βII, and ε. PKCδ levels were also generally decreased in CRC cell lines, irrespective of APC/β-catenin status (Figure 1B). This expression pattern is consistent with its downregulation in murine and human neoplastic intestinal tissues (Figure 1C and [4]). Interestingly, anti-PKCδ antibody detected several specific bands in tumor cells (confirmed using blocking peptide; Figure 1B, lower right panel), but not in non-transformed cells, indicating that this isozyme may undergo differential post-translational modification as well as downregulation in colon tumors. In contrast to PKCα and δ, PKCβII levels were consistently higher in CRC cells, paralleling findings in experimental models of colon cancer (for reviews, see [2, 4]). PKCε protein showed variability in total levels and in the relative proportion of the two major species detected in CRC cells, consistent with discrepancies in the literature regarding its expression in primary colon tumors [30-32]. Thus, CRC cell lines are similar to neoplastic intestinal tissues in that they generally express reduced levels of PKCα and δ, increased levels of PKCβII, and variable levels of PKCε. These studies, therefore, identify a panel of human CRC cell lines that can be used to address the role of alterations in the PKC enzyme system in intestinal neoplasia.
Expression of PKCα decreases cyclin D1 steady-state levels in CRC cells
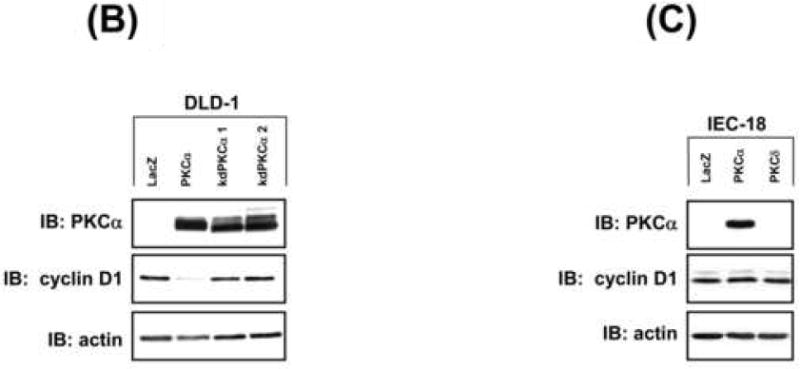
Based on evidence that cyclin D1 is a key target of PKC/PKCα signaling in non-transformed IECs [3, 12, 33], we explored the effect of restoring PKCα expression on cyclin D1 levels in CRC cells. Effects of PKCδ restoration were determined for comparison. Expression of PKCα, but not PKCδ, promoted a marked reduction in steady-state levels of cyclin D1 protein in all cell lines tested, with the exception of RKO cells (Figure 2A; RCA and HCT-15 data not shown). Expression of comparable levels of kdPKCα did not affect cyclin D1 abundance, pointing to a kinase-dependent effect (Figure 2B). Specificity for cyclin D1 was indicated by the failure of PKCα expression to promote consistent alterations in levels of cyclin E (Figure 2A, lower panel). PKCα did not alter cell cycle distribution in the majority of CRC cell lines (Supplementary Figure 1), confirming that cyclin D1 downregulation was not simply a reflection of PKC-induced cell cycle perturbation. Interestingly, increased expression of PKCα led to a reduction in cyclin D1 levels in SW620, FET, HCT116, and LS180 cells (Figure 2A, Supplementary Figure 4B), arguing that levels of PKCα are limiting even in CRC cells that retain expression of the protein (see Figure 1A). The effect appears to be specific to transformed cells since PKCα expression failed to affect steady-state cyclin D1 levels in non-transformed IEC-18 cells (Figure 2C). Together, the data point to PKCα as an important suppressor of cyclin D1 protein accumulation in CRC cells.
Figure 2. PKCα represses cyclin D1 protein expression in CRC cells.
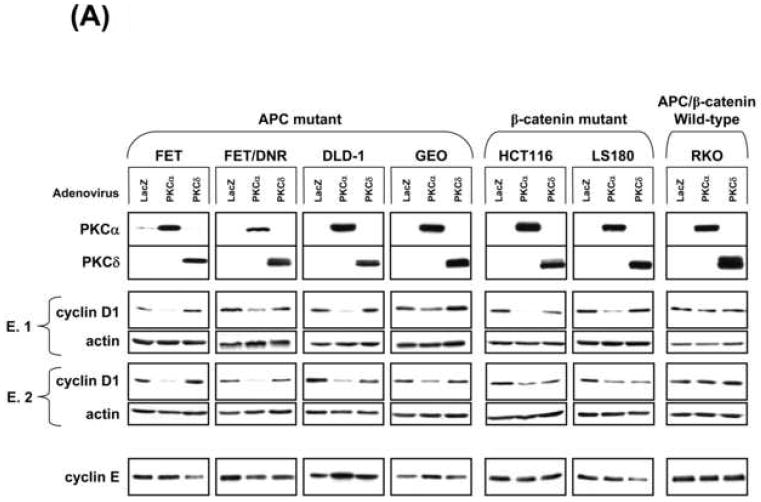
(A) CRC cells infected with 20 moi LacZ, PKCα, or PKCδ adenovirus (150 moi for RKO) were subjected to immunoblot analysis as indicated. Data represent >3 independent experiments; two experiments (E.1 and E.2) are shown to demonstrate consistency of results.
(B) Immunoblot analysis of DLD-1 cells infected with 20 moi LacZ, 20 moi PKCα, 500 moi (kdPKCα1) or 700 moi (kdPKCα2) kdPKCα adenovirus (increased moi compensated for instability of the kinase dead enzyme[21]).
(C) Immunoblot analysis of transduced (20 moi) IEC-18 cells.
PKCα-mediated repression of cyclin D1 in CRC cells does not appear to be dependent on enhanced APC/β-catenin signaling
The inability of PKCα to suppress cyclin D1 steady-state levels in APC/β-catenin wild-type intestinal cells (RKO and IEC-18) suggested that the effect may be dependent on aberrant APC/β-catenin signaling, a characteristic of a majority of CRC cells. To investigate this possibility, β-catenin activity was abrogated in DLD-1 (APC mutant) and HCT116 (β-catenin mutant) cells using siRNA, prior to expression of exogenous PKCα. Despite near-complete silencing of β-catenin expression, PKCα maintained the ability to promote marked downregulation of cyclin D1 steady-state protein levels in both cell lines (Figure 3A), thus excluding a direct link between aberrant APC/β-catenin signaling and PKCα-induced effects. We next compared the subcellular distribution of PKCα in non-responsive RKO and IEC-18 cells with that in responsive HCT116 and DLD-1 cells. Immunofluorescence analysis detected PKCα in the cytoplasm of all PKCα-transduced cells (Figure 3B; IEC-18 and DLD-1 data not shown). Notably, a pool of the enzyme was also detected at the plasma membrane in responsive HCT116 and DLD-1 cells, but not in non-responsive RKO and IEC-18 cells (Figure 3B, arrows). Since membrane association of PKC can reflect enzyme activation [5], these data indicate that repression of cyclin D1 steady-state levels correlates with the presence of an intrinsic mechanism for PKCα stimulation. Responsive cell lines, therefore, appear to have at least two pools of transduced PKCα: a cytoplasmic pool that is available for future activation by exogenous PKC agonists, and a membrane-associated active pool that mediates repression of steady-state levels of cyclin D1. The latter appears to be absent in non-responsive cell lines.
Figure 3. PKCα-induced repression of cyclin D1 occurs independently of aberrant β-catenin signaling and may be regulated by PKCα subcellular distribution.

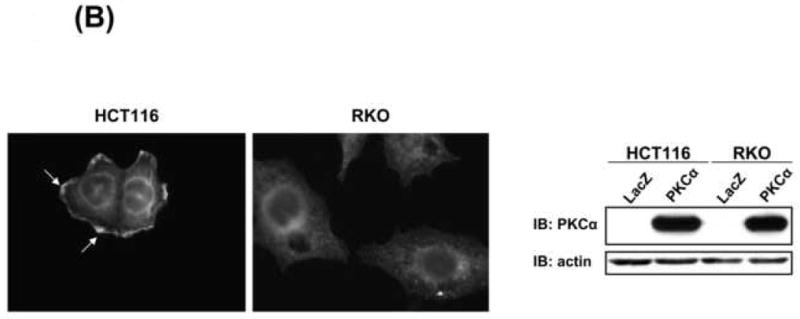
(A) DLD-1 and HCT116 cells were transfected with 100 nM non-silencing or β-catenin siRNA and infected with 20 moi LacZ or PKCα adenovirus 72 h later. Left panel: Immunoblot analysis of HCT116 cells 48 h after adenoviral infection. Right panel: Quantification of cyclin D1 expression (normalized to actin) for 2 (DLD-1) or 3 (HCT116) experiments (average±s.e.).
(B) Immunofluorescence detection of PKCα in PKCα-transduced HCT116 (20 moi) and RKO cells (150 moi). Arrows: membrane staining. Right panel: Immunoblot analysis confirmed similar PKCα expression levels in these cells following adenoviral infection. Data represent ≥2 independent experiments.
PKCα expression decreases cyclin D1 transcription in CRC cells
To investigate mechanism(s) underlying PKCα-induced downregulation of cyclin D1 steady-state levels in CRC cells, cyclin D1 protein stability and mRNA expression were examined. Comparison of cyclin D1 protein decay rate in CHX-treated PKCα- and LacZ-expressing FET, FET/DNR, or DLD-1 cells excluded changes in cyclin D1 protein turnover (Supplementary Figure 2A and not shown). In contrast, Northern blot analysis detected a reduction in cyclin D1 mRNA levels in PKCα-transduced CRC cells (Figure 4A). The extent of inhibition varied among cell lines, ranging from 0.3- to 0.6-fold. Notably, PKCα expression failed to affect cyclin D1 message levels in RKO (Figure 4A) or IEC-18 cells (not shown), consistent with the failure to alter cyclin D1 steady-state protein expression. Northern analysis of kdPKCα-expressing DLD-1 cells confirmed that catalytic activity is required for PKCα-mediated repression of cyclin D1 mRNA (Figure 4A, lower panel).
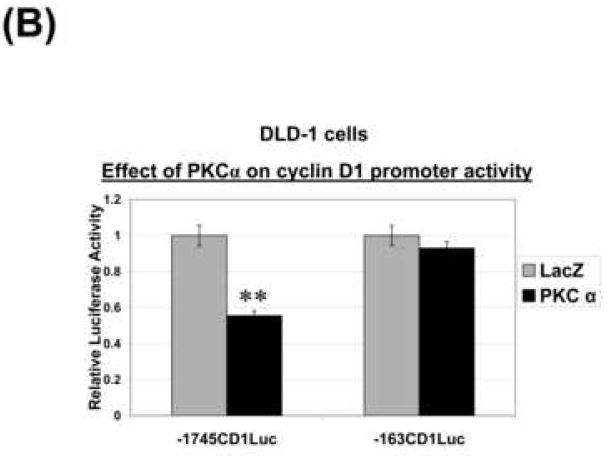
Figure 4. PKCα reduces cyclin D1 steady-state levels through transcriptional repression.
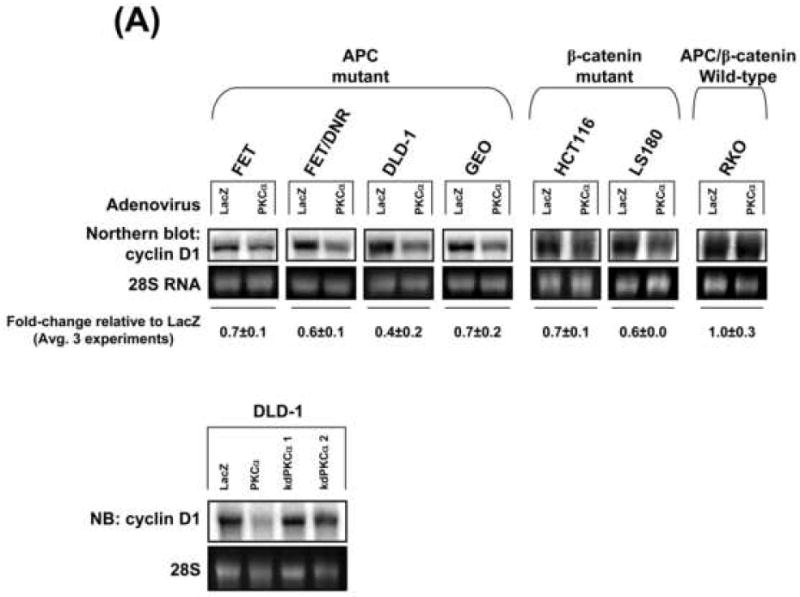
(A) PKCα suppresses cyclin D1 mRNA accumulation. Upper panel: Northern blot analysis of cyclin D1 mRNA in adenovirus-transduced cells. Numbers show levels of cyclin D1 mRNA (normalized to 28S rRNA) in PKCα-expressing cells relative to LacZ-transduced cells (averages±s.e.). Lower panel: Northern blot analysis of DLD-1 cells infected as in Figure 2B.
(B) LacZ- or PKCα-transduced DLD-1 cells were transfected with the indicated cyclin D1 promoter-luciferase reporter constructs, and luciferase activity (expressed relative to LacZ-transduced cells) was determined 24 h later. ** Significantly different from LacZ control ( P<0.005).
Data represent ≥3 independent experiments.
Analysis of ActD-treated cells demonstrated that PKCα does not alter the stability of cyclin D1 message in CRC cells (Supplementary Figure 2B), pointing to a transcriptional effect. To explore this possibility, LacZ- and PKCα-expressing DLD-1 cells were transfected with a construct in which luciferase expression is driven by 1745 bp of the cyclin D1 promoter (-1745CD1Luc) or by a promoter truncated at -163 bp (-163CD1Luc) [23]. PKCα expression reduced the activity of the -1745CD1 promoter by ~50%, relative to the LacZ control. In contrast, PKCα had no significant effect on the -163CD1 promoter, excluding non-specific effects of PKCα expression on transfection efficiency or general transcription, and pointing to the involvement of promoter elements between -1745 and -163. The extent of PKCα-induced repression of cyclin D1 promoter activity was comparable with the decrease in cyclin D1 mRNA accumulation, arguing that transcriptional modulation is the predominant mechanism underlying the reduction in cyclin D1 mRNA levels. Thus, these studies identify a novel mechanism of cyclin D1 regulation by PKCα in colon cancer cells.
Exogenous PKCα restores the ability of PKC agonists to downregulate cyclin D1 and inhibit cap-dependent translation
The data presented above demonstrate that PKCα expression reduces cyclin D1 steady-state levels in CRC cells by kinase-dependent transcriptional repression. Based on evidence that full activation of PKC/PKCα in non-transformed IEC-18 cells promotes near depletion of cyclin D1 via translational suppression ([12, 33], Figure 5A, left panel), we examined the ability of exogenous PKC agonists to further suppress cyclin D1 expression in CRC cells. PMA failed to affect cyclin D1 levels in all untransduced (not shown) or LacZ-transduced CRC cell lines tested, including CRC cells that retain PKCα expression (e.g., HCT116, LS180), further supporting the notion that PKCα levels are limiting in these cells (Figure 5A, right panel; similar results were seen in FET, FET/DNR, HCT-15, and RCA cells). Notably, however, expression of exogenous PKCα, but not kdPKCα or PKCδ, restored the ability of PMA to promote near depletion of cyclin D1 protein in all CRC cells tested (Figure 5B,D).
Figure 5. PKCα is required for PMA-induced downregulation of cyclin D1 and inhibition of cap-dependent translation in CRC cells.
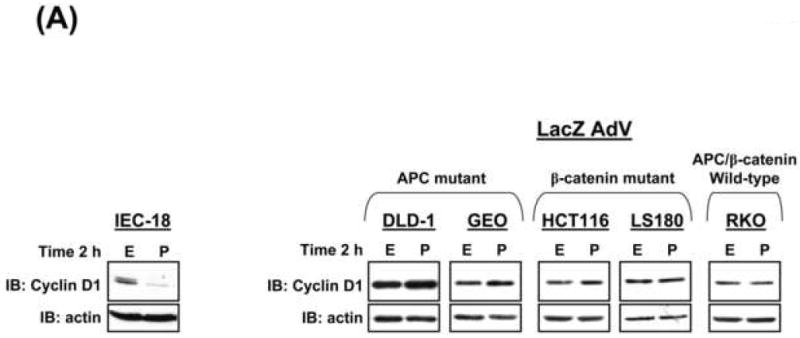
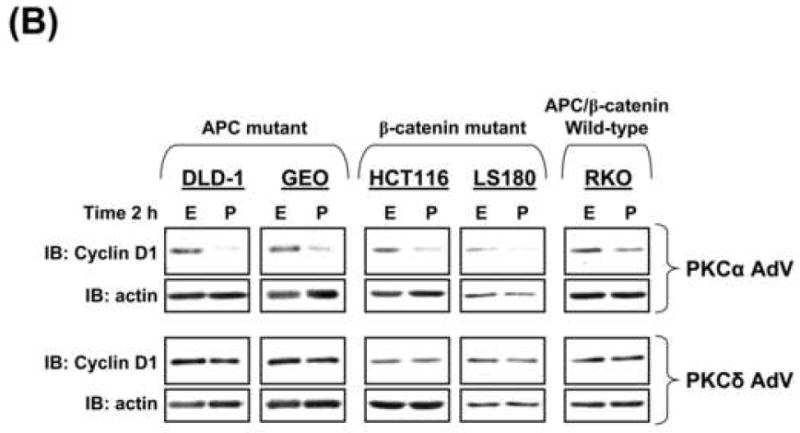
(A) Immunoblot analysis of cyclin D1 in IEC-18 cells (left panel) or LacZ-transduced CRC cells (right panel) treated with 100 nmol/L PMA (P) or vehicle (EtOH, E).
(B) Immunoblot analysis of CRC cells transduced with PKCα or PKCδ adenovirus and treated with PMA or ethanol for 2 h.
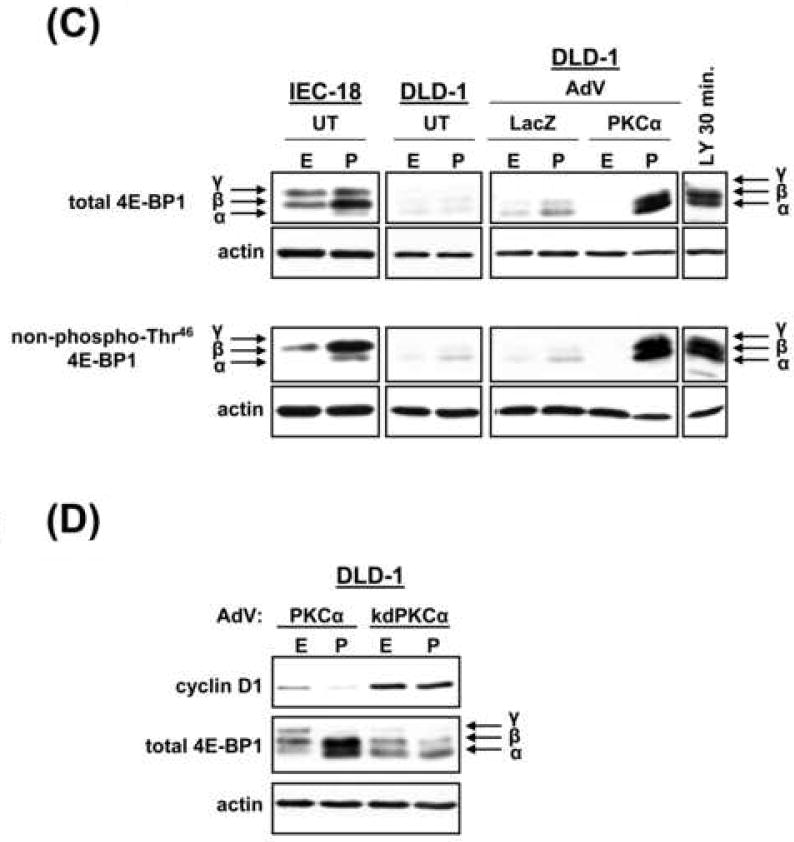
(C) Untransduced (UT) IEC-18 and DLD-1 cells, and LacZ- or PKCα-transduced DLD-1 cells were treated with PMA or vehicle for 2 h and subjected to immunoblotting for total and non-phospho-Thr46 4E-BP1. DLD-1 cells treated with LY294002 (LY) (50 μmol/L, 30 min) are included as a positive control for 4E-BP1 activation. Arrows indicate 4E-BP1 phosphoforms.
(D) Immunoblot analysis of cyclin D1 and total 4E-BP1 in PKCα (20 moi)- or kdPKCα (500 moi)-transduced DLD-1 cells treated with PMA or vehicle for 2 h.
Data represent ≥3 independent experiments.
PMA-induced downregulation of cyclin D1 in IEC-18 cells involves inhibition of cap-dependent translation initiation via PKCα-dependent dephosphorylation/activation of 4E-BP1 [12, 33]. To determine if activation of exogenous PKCα affects translation in CRC cells, LacZ-and PKCα-transduced cells were treated with PMA for 2 h and analyzed for the presence of translation-repressive hypophosphorylated α/β forms of 4E-BP1, which migrate faster than the inactive hyperphosphorylated γ form in SDS-PAGE gels (Figure 5C, HCT116 and RKO data not shown). Samples were also directly examined for alterations in Thr46 phosphorylation, using an antibody specific for the unphosphorylated residue. PKC activation did not significantly affect 4E-BP1 phosphorylation/activity in untransduced or LacZ-transduced cells (Figure 5C). In contrast, PMA promoted marked accumulation of translation-inhibitory α/β forms of 4E-BP1 in PKCα-expressing cells, but not in kdPKCα-expressing cells (Figure 5C,D). Thus, consistent with the mechanism of cyclin D1 downregulation identified in non-transformed IECs [12, 33], expression of exogenous PKCα in CRC cells restores the ability of PKC agonists to regulate cyclin D1 protein expression via translational repression.
PKCα broadly inhibits anchorage-independent growth of CRC cells via a mechanism that is partially dependent on reduced cyclin D1 expression
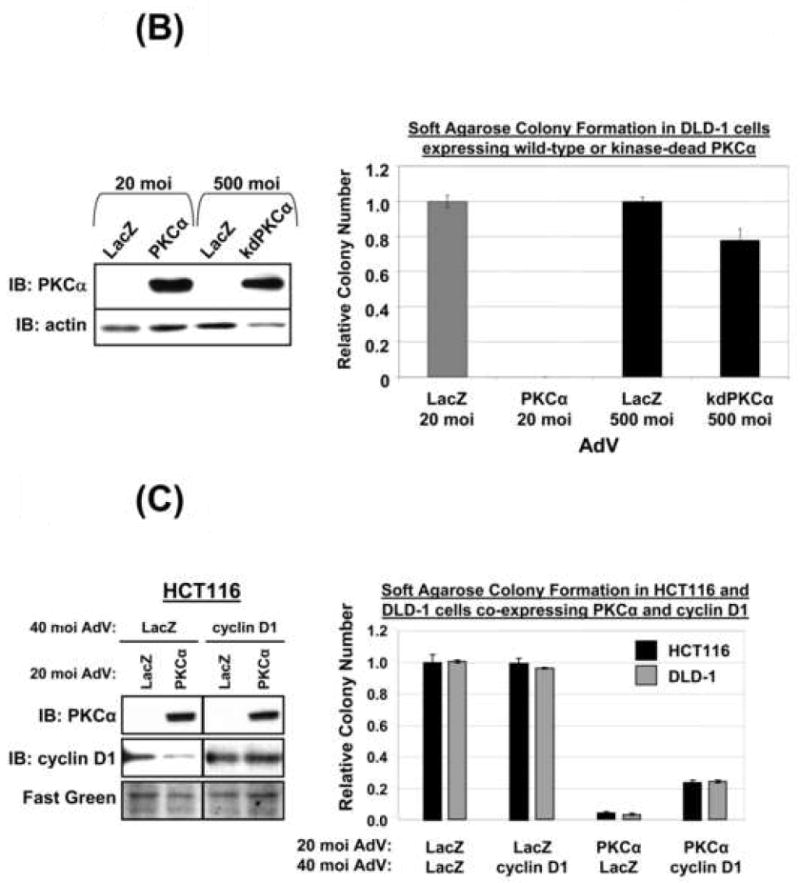
PKCα reduces the growth and in vitro tumorigenicity of APC mutant CaCo-2 and HT-29 CRC cells [10, 11, 34]. To determine if PKCα is broadly tumor suppressive in CRC cells with different genetic alterations and degrees of differentiation, the ability of PKCα to inhibit soft agarose colony formation was tested in a panel of CRC cell lines. Effects of PKCα were compared with those of PKCδ. PKCα expression generally promoted near complete inhibition of anchorage-independent growth in CRC cell lines (Figure 6A, Supplementary Figure 3), consistent with a strong tumor suppressive activity of this isozyme in colon cancer [10, 11, 34]. The failure of kdPKCα to significantly affect colony formation pointed to a kinase-dependent effect (Figure 6B). Interestingly, restoration of PKCα was unable to affect anchorage-independent growth of RKO cells (Figure 6A), which are also resistant to the suppressive effects of the enzyme on cyclin D1 steady-state levels (Figure 2A). To further explore the contribution of cyclin D1 deficiency to the growth inhibitory actions of PKCα, DLD-1 and HCT116 cells were co-transduced with cyclin D1 or LacZ adenovirus and PKCα adenovirus, and plated in soft agar. Notably, co-transduction with cyclin D1 led to a ~10-fold increase in colony number in PKCα expressing cells, while having no effect in LacZ control cells (Figure 6C). This partial rescue indicates that cyclin D1 is limiting for anchorage-independent growth of PKCα-expressing CRC cells, while pointing to the existence of additional mediators of the powerful tumor suppressive effects of PKCα in CRC cells (e.g., p21waf1) [10].
Figure 6. PKCα and PKCδ differentially suppress anchorage-independent growth of CRC cells.
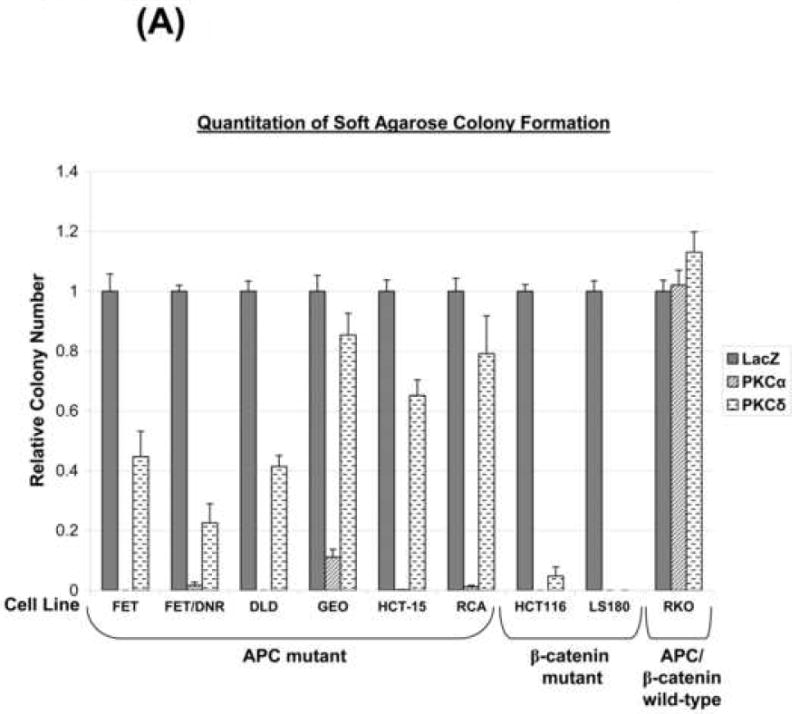
(A) CRC cells were infected with LacZ, PKCα, or PKCδ adenovirus as indicated and plated in soft agarose. Quantification (average of ≥2 independent experiments ± s.e.) of colony formation is presented relative to LacZ control. All samples transduced with PKCα were significantly different from LacZ-transduced controls (P< 0.05) except for RKO cells.
(B) DLD-1 cells were infected with LacZ, PKCα or kdPKCα adenovirus as indicated and subjected to immunoblot analysis (left panel) or soft agarose colony formation assays (right panel). Data represent the average of two independent experiments (± s.e.).
(C) DLD-1 and HCT116 cells were infected with indicated combinations of PKCα, LacZ and cyclin D1 adenovirus and analyzed by immunoblotting (Left panel) or colony formation in soft agarose (quantified as in A, Right panel). Data represent the average of 3 independent experiments ± s.e. Similar results were obtained using 20 moi cyclin D1 adenovirus.
PKCδ expression also reduced anchorage-independent growth of CRC cells (Figure 6A, Supplementary Figure 3), although this suppression was more modest and variable (ranging from little effect in RKO and GEO cells to near complete inhibition in HCT116 and LS180 cells). Thus, consistent with its effects on cyclin D1 regulation, PKCα appears to be more effective than PKCδ in suppressing the transformed phenotype of CRC cells.
Inverse relationship between PKCα and cyclin D1 expression in neoplastic intestinal tissues and cells with and without perturbations in APC/β-catenin signaling
To determine if there is a relationship between expression of PKCα and cyclin D1 in intestinal tumors with different genetic backgrounds, immunohistochemical analysis was performed in murine intestinal tumors that are Apc mutant, β-catenin-mutant, or wild-type for APC/β-catenin but mutant for K-Ras or deficient in Muc2. Markedly increased levels of cyclin D1 were consistently observed in neoplasms arising in all of these mice (Figure 7A and not shown). In contrast, levels of PKCα expression were consistently reduced in these neoplastic tissues (Figure 7A). Thus, reciprocal changes in the expression of cyclin D1 and PKCα are seen at early stages of tumor development in a range of mouse models of intestinal cancer, consistent with a potential link between these events.
Figure 7. PKCα deficiency and increased cyclin D1 expression are characteristics of intestinal tumors and CRC cell lines independent of APC/β-catenin mutation status.
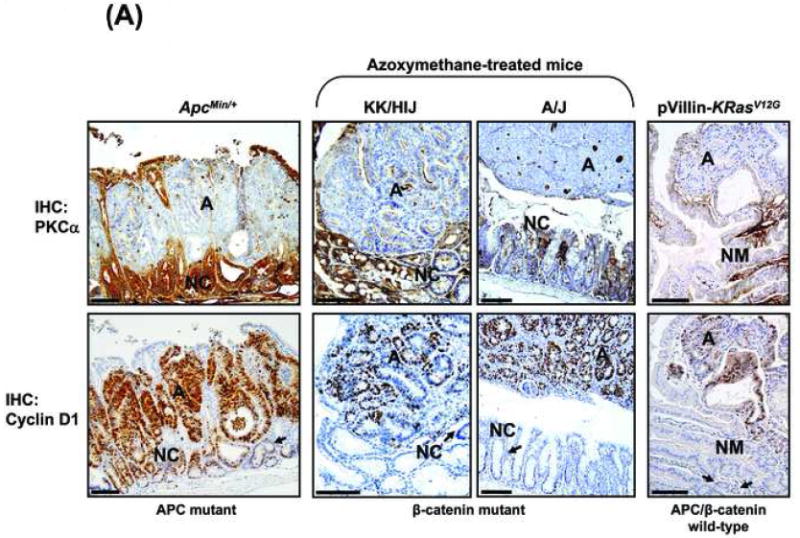
(A) Immunohistochemical analysis of PKCα and cyclin D1 in adenomas (A) and adjacent “normal” mucosa/crypts (NM; NC) from ApcMin/+ mice, azoxymethane-treated KK/HIJ and A/J mice, and pVillin-KRasV12G mice. Arrows: cyclin D1 staining in “normal” crypt cells. Magnification Bar: 100 μm.
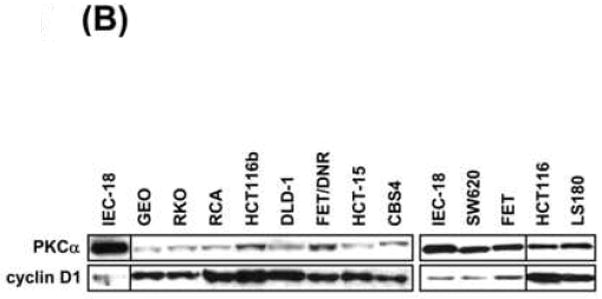
(B) Western blot analysis of PKCα and cyclin D1 in CRC cells and non-transformed IEC-18 cells. Lines indicate regrouping of lanes from the same gel.
Cyclin D1 and PKCα protein levels were also surveyed in a panel of 12 human CRC cell lines used in this study. Notably, all of these cell lines (except for SW620) express increased levels of cyclin D1 relative to non-transformed IEC-18 cells (Figure 7B). While a majority of these cell lines showed a concomitant reduction in PKCα levels, some lines (e.g., HCT116 and LS180) showed upregulation of cyclin D1 in the context of significant levels of PKCα. Our findings (Figure 5A) indicate that this expression pattern may reflect limiting levels of PKCα signaling in these cells, a defect that can be overcome by increased expression of wild-type enzyme.
Discussion
PKCα is growth inhibitory in non-transformed IECs and has been implicated in tumor suppression in APC/β-catenin mutant intestinal neoplasms [3, 5-11, 30, 34, 35]. This study extends previous findings by providing the first demonstration that the tumor suppressive effects of PKCα signaling, as well as early loss of PKCα protein, occur independently of specific genetic alterations or degree of differentiation. The study further demonstrates that an important target of PKCα regulation in CRC cells is the potent mitogen cyclin D1. PKCα is shown to suppress cyclin D1 accumulation in CRC cells independently of β-catenin signaling, and cyclin D1 deficiency is implicated in PKCα-induced inhibition of anchorage-independent growth. Two distinct mechanisms of PKCα-mediated cyclin D1 control are identified: transcriptional repression and blockade of cap-dependent translation. Finally, to our knowledge, this study is the first to report a comprehensive analysis of PKC isozyme expression in a panel of CRC cells, and to validate these cells as models for studies on the PKC enzyme system in intestinal tumors.
PKCα is downregulated in murine intestinal tumors and in human CRC cells that are APC mutant (e.g., ApcMin/+, Apc1638 and KRas/Apc+/1638N mice, and FET/DNR, DLD-1, GEO, HCT-15, and RCA cells) or β-catenin-mutant (e.g., azoxymethane-treated mice and HCT116 and LS180 cells), as well as in neoplastic intestinal tissues and cells that are APC/β-catenin wild-type (i.e., pVillin-KRasV12G and Muc2-/- mice, and RKO cells). PKCα signaling appears to be limiting even in CRC cells that retain expression of the enzyme (e.g., FET, HCT116 and LS180), as indicated by their resistance to the effects of PKC agonists on cyclin D1 expression, a deficiency that can be overcome by exogenous expression of the enzyme. Although the underlying mechanisms remain to be delineated, defective PKCα signaling appears to be a general characteristic of early intestinal tumorigenesis, and alterations in this pathway are likely related to its negative growth effects rather than a secondary consequence of specific alterations in individual tumors.
Previous studies in CaCo-2 [10, 11] and HT-29 [34] cells, which are capable of enterocytic differentiation in vitro and form moderately well- or well-differentiated tumors in mice [36], demonstrated the ability of PKCα signaling to inhibit anchorage-independent growth. The finding that PKCα expression inhibits colony formation in soft agarose of highly aggressive, poorly differentiated cells (DLD-1, HCT116, and FET/DNR), intermediately aggressive, moderately differentiated cells (RCA), as well as non-aggressive, well-differentiated cells (GEO), argues that PKCα retains its tumor suppressive effects even in advanced tumors. This notion is consistent with evidence that PKCα deficiency not only increases the number of early lesions (i.e., aberrant crypt foci and adenomas) in the murine intestine, but also promotes the development of aggressive intestinal adenocarcinomas in ApcMin/+ mice [8].
Loss of PKCα and upregulation of cyclin D1 are both early events in intestinal tumorigenesis (Figure 7 and [6, 7, 37]). Evidence in support of a direct relationship between these events includes (a) reciprocal changes in the abundance of PKCα and cyclin D1 in diverse mouse models of intestinal neoplasia and human CRC cell lines; (b) rapid downregulation of cyclin D1 protein in non-transformed IECs in response to PKCα activation [12, 33]; (c) the failure of PKC agonists to downregulate cyclin D1 in colon tumor cells, which have limiting levels of PKCα signaling; (d) the ability of PKCα re-expression to reduce steady-state levels of cyclin D1 in CRC cells and to re-establish PKC agonist-mediated downregulation of the protein; and (e) the involvement of cyclin D1 deficiency in PKCα-induced suppression of CRC cell growth in soft agar.
Analysis of the effects of PKCα re-expression in CRC cells has identified two distinct mechanisms of PKCα-induced cyclin D1 repression. In addition to agonist-induced translational effects mediated by activation of 4E-BP1, PKCα re-expression inhibits steady state levels of cyclin D1 by a novel transcriptional mechanism not previously identified in non-transformed IECs. Notably, both of these mechanisms appear to be defective in tumor cells, even in those that retain appreciable levels of PKCα. The fact that the defect could be rescued by expression of exogenous PKCα likely reflects reduced sensitivity of downstream signaling components to levels of PKCα activation or defects in activation of the endogenous enzyme. In regard to the latter possibility, it is interesting to note that the migration of PKCα in SDS-PAGE gels often differed between CRC cells and non-transformed IECs (Figure 1A), pointing to a potential difference in post-translational modification. The precise defects in CRC cells are currently under investigation. Our data also indicate that the transcriptional and translational effects on cyclin D1 can occur independently: while endogenous levels of PKCα agonist activity in CRC cells were sufficient to elicit transcriptional effects following PKCα re-expression, further activation with pharmacological agonists was required for the translational effects. Whether this reflects differential sensitivity to levels of PKC activation or unique effects of different stimuli remains to be determined.
The ability of PKCα to downregulate cyclin D1 in CRC cell lines did not require activated β-catenin signaling and was independent of known genetic changes. While our studies in non-transformed IECs point to a role for PP2A in the translational effects of PKCα [33], downstream effectors that mediate the reduced steady-state levels of cyclin D1 mRNA are not immediately apparent. A potential target is the epidermal growth factor receptor (EGFR) which can promote cyclin D1 expression through various signaling intermediates [38]. Inhibition of EGFR activation by PKCα has been noted in a number of systems (e.g., [39-41]) and was also seen following re-expression of the isozyme in CRC cells (Supplementary Figure 4A). However, while this receptor is likely to play a role in the regulation of cyclin D1 under some circumstances, our data point to the involvement of additional factors. PKCα repressed cyclin D1 (both at the steady state level and in response to PKC agonist treatment) in SW620 cells which lack EGFR expression [42, 43]. Indeed, EGFR-independent mechanisms appear to play a significant role even in cells that express the receptor since (a) PKCα is downregulated in tumors and CRC cell lines with activating mutations in proteins downstream of EGFR (e.g., KRas mutant mice and KRas/PI3K mutant DLD1, HCT116 and HCT-15 cells [44]), mutations that correlate with insensitivity to EGFR inhibition [44, 45], and (b) effects of PKCα on cyclin D1 mRNA levels did not correlate with sensitivity of CRC cells to EGFR inhibitors [44] (e.g., the effect seen in resistant HCT116 and HCT-15 cells was comparable with that in sensitive GEO cells (Figure 4A and data not shown). These EGFR-independent mechanisms are currently under investigation.
A role for downregulation of cyclin D1 in the tumor suppressive effects of PKCα is consistent with known functions of this cyclin in tumorigenesis [13, 14, 16, 17]. Overexpression of cyclin D1 mRNA and protein is a common characteristic of various cancers, including those of the intestine, where it appears to be caused by trans-acting influences as opposed to genetic mutation/rearrangement [46]. A direct role in intestinal tumorigenesis is supported by evidence that cyclin D1-deficiency (a) reduces the number and size of tumors in ApcMin/+ mice [14], (b) inhibits intestinal tumor progression following conditional loss of APC in vivo [17], and (c) reverses the transformed phenotype of SW480 CRC cells [15]. The finding that PKCα-induced downregulation of cyclin D1 in CRC cells does not affect the cell cycle (Supplementary Figure 2) suggests that levels of this cyclin are not limiting for growth of tumor cells under ‘optimal’ conditions (as seen in SW480 cells [15]). The high levels of cyclin D1 in CRC cells may, thus, be required to maintain cell growth in the less favorable environments found in tumors.
Previous studies also support a role for PKCδ in tumor suppression in the intestine [47]. Expression of this isozyme is also reduced/lost in rodent and human intestinal neoplasms and CRC cell lines (this study and [4, 7, 30, 32, 48]). Our comparative analysis demonstrates that, although PKCδ variably affects CRC cells of different genetic background, PKCα is generally more potent in blocking soft agar colony formation, suppressing cyclin D1 steady-state levels, re-establishing downregulation of cyclin D1 in response to PKC agonists, and mediating translational inhibition in response to PMA. Thus, these two isozymes are likely to mediate their tumor suppressive effects through different mechanisms.
Taken together, our findings point to cyclin D1 repression as an important component of PKCα tumor suppression in intestinal cells. Since PKCα has been shown to have tumor suppressive properties in other tissues (e.g., epidermis [49, 50], pituitary and thyroid [51]), these findings may be relevant to multiple tumor types.
Supplementary Material
Acknowledgments
We thank Dr. M. Brattain, J. Hauser, and A. Byrd for their help with analysis of human CRC cells/tissues, and D. Oleszek and J. Marinaro for expert technical assistance.
Grant Support: This work was supported by NIH grants DK60632, DK54909, and CA16056, and a grant from the Mae Stone Goode/Roswell Park Alliance Foundation. MAP is supported by NIH Postdoctoral Fellowship CA113048.
Abbreviations
- ActD
actinomycin D
- APC
adenomatous polyposis coli
- CHX
cycloheximide
- CRC
colorectal cancer
- 4E-BP1
eIF4E-binding protein 1
- FBS
fetal bovine serum
- FHC
fetal human colon
- IEC
intestinal epithelial cell
- ifu
infection units
- kdPKCα
kinase-dead PKCα
- moi
multiplicity of infection
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
- RPCI
Roswell Park Cancer Institute
- TGF
transforming growth factor
Footnotes
There are no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Black JD. Protein kinase C-mediated regulation of the cell cycle. Front Biosci. 2000;5:D406–423. doi: 10.2741/black. [DOI] [PubMed] [Google Scholar]
- 2.Di Mari JF, Mifflin RC, Powell DW. The role of protein kinase C in gastrointestinal function and disease. Gastroenterology. 2005;128:2131–2146. doi: 10.1053/j.gastro.2004.09.078. [DOI] [PubMed] [Google Scholar]
- 3.Frey MR, Clark JA, Leontieva O, Uronis JM, Black AR, Black JD. Protein kinase C signaling mediates a program of cell cycle withdrawal in the intestinal epithelium. J Cell Biol. 2000;151:763–778. doi: 10.1083/jcb.151.4.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Black JD. Protein kinase C isozymes in colon carcinogenesis: guilt by omission. Gastroenterology. 2001;120:1868–1872. doi: 10.1053/gast.2001.25287. [DOI] [PubMed] [Google Scholar]
- 5.Saxon ML, Zhao X, Black JD. Activation of protein kinase C isozymes is associated with post-mitotic events in intestinal epithelial cells in situ. J Cell Biol. 1994;126:747–763. doi: 10.1083/jcb.126.3.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gokmen-Polar Y, Murray NR, Velasco MA, Gatalica Z, Fields AP. Elevated protein kinase C betaII is an early promotive event in colon carcinogenesis. Cancer Res. 2001;61:1375–1381. [PubMed] [Google Scholar]
- 7.Klein IK, Ritland SR, Burgart LJ, Ziesmer SC, Roche PC, Gendler SJ, Karnes WE., Jr Adenoma-specific alterations of protein kinase C isozyme expression in Apc(MIN) mice. Cancer Res. 2000;60:2077–2080. [PubMed] [Google Scholar]
- 8.Oster H, Leitges M. Protein kinase C alpha but not PKCzeta suppresses intestinal tumor formation in ApcMin/+ mice. Cancer Res. 2006;66:6955–6963. doi: 10.1158/0008-5472.CAN-06-0268. [DOI] [PubMed] [Google Scholar]
- 9.Verstovsek G, Byrd A, Frey MR, Petrelli NJ, Black JD. Colonocyte differentiation is associated with increased expression and altered distribution of protein kinase C isozymes. Gastroenterology. 1998;115:75–85. doi: 10.1016/s0016-5085(98)70367-1. [DOI] [PubMed] [Google Scholar]
- 10.Abraham C, Scaglione-Sewell B, Skarosi SF, Qin W, Bissonnette M, Brasitus TA. Protein kinase C alpha modulates growth and differentiation in Caco-2 cells. Gastroenterology. 1998;114:503–509. doi: 10.1016/s0016-5085(98)70533-5. [DOI] [PubMed] [Google Scholar]
- 11.Scaglione-Sewell B, Abraham C, Bissonnette M, Skarosi SF, Hart J, Davidson NO, Wali RK, Davis BH, Sitrin M, Brasitus TA. Decreased PKC-alpha expression increases cellular proliferation, decreases differentiation, and enhances the transformed phenotype of CaCo-2 cells. Cancer Res. 1998;58:1074–1081. [PubMed] [Google Scholar]
- 12.Hizli AA, Black AR, Pysz MA, Black JD. Protein kinase C alpha signaling inhibits cyclin D1 translation in intestinal epithelial cells. J Biol Chem. 2006;281:14596–14603. doi: 10.1074/jbc.M601959200. [DOI] [PubMed] [Google Scholar]
- 13.Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: Cyclin D1: normal and abnormal functions. Endocrinology. 2004;145:5439–5447. doi: 10.1210/en.2004-0959. [DOI] [PubMed] [Google Scholar]
- 14.Hulit J, Wang C, Li Z, Albanese C, Rao M, Di Vizio D, Shah S, Byers SW, Mahmood R, Augenlicht LH, Russell R, Pestell RG. Cyclin D1 genetic heterozygosity regulates colonic epithelial cell differentiation and tumor number in ApcMin mice. Mol Cell Biol. 2004;24:7598–7611. doi: 10.1128/MCB.24.17.7598-7611.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weinstein IB. Disorders in cell circuitry during multistage carcinogenesis: the role of homeostasis. Carcinogenesis. 2000;21:857–864. doi: 10.1093/carcin/21.5.857. [DOI] [PubMed] [Google Scholar]
- 16.Diehl JA. Cycling to cancer with cyclin D1. Cancer Biol Ther. 2002;1:226–231. doi: 10.4161/cbt.72. [DOI] [PubMed] [Google Scholar]
- 17.Sansom OJ, Reed KR, van de Wetering M, Muncan V, Winton DJ, Clevers H, Clarke AR. Cyclin D1 is not an immediate target of beta -catenin following Apc loss in the intestine. J Biol Chem. 2005 doi: 10.1074/jbc.M500191200. [DOI] [PubMed] [Google Scholar]
- 18.Bateman NW, Tan D, Pestell RG, Black JD, Black AR. Intestinal tumor progression is associated with altered function of KLF5. J Biol Chem. 2004;279:12093–12101. doi: 10.1074/jbc.M311532200. [DOI] [PubMed] [Google Scholar]
- 19.Garcia-Bermejo ML, Leskow FC, Fujii T, Wang Q, Blumberg PM, Ohba M, Kuroki T, Han KC, Lee J, Marquez VE, Kazanietz MG. Diacylglycerol (DAG)-lactones, a new class of protein kinase C (PKC) agonists, induce apoptosis in LNCaP prostate cancer cells by selective activation of PKCalpha. J Biol Chem. 2002;277:645–655. doi: 10.1074/jbc.M107639200. [DOI] [PubMed] [Google Scholar]
- 20.Berkner KL. Expression of heterologous sequences in adenoviral vectors. Curr Top Microbiol Immunol. 1992;158:39–66. doi: 10.1007/978-3-642-75608-5_3. [DOI] [PubMed] [Google Scholar]
- 21.Carpenter L, Mitchell CJ, Xu ZZ, Poronnik P, Both GW, Biden TJ. PKC alpha is activated but not required during glucose-induced insulin secretion from rat pancreatic islets. Diabetes. 2004;53:53–60. doi: 10.2337/diabetes.53.1.53. [DOI] [PubMed] [Google Scholar]
- 22.Brattain MG, Levine AE, Chakrabarty S, Yeoman LC, Willson JK, Long B. Heterogeneity of human colon carcinoma. Cancer Metastasis Rev. 1984;3:177–191. doi: 10.1007/BF00048384. [DOI] [PubMed] [Google Scholar]
- 23.Watanabe G, Howe A, Lee RJ, Albanese C, Shu IW, Karnezis AN, Zon L, Kyriakis J, Rundell K, Pestell RG. Induction of cyclin D1 by simian virus 40 small tumor antigen. Proc Natl Acad Sci U S A. 1996;93:12861–12866. doi: 10.1073/pnas.93.23.12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 25.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 26.Fodde R, Edelmann W, Yang K, van Leeuwen C, Carlson C, Renault B, Breukel C, Alt E, Lipkin M, Khan PM, et al. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci U S A. 1994;91:8969–8973. doi: 10.1073/pnas.91.19.8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takahashi M, Nakatsugi S, Sugimura T, Wakabayashi K. Frequent mutations of the beta-catenin gene in mouse colon tumors induced by azoxymethane. Carcinogenesis. 2000;21:1117–1120. [PubMed] [Google Scholar]
- 28.Janssen KP, el-Marjou F, Pinto D, Sastre X, Rouillard D, Fouquet C, Soussi T, Louvard D, Robine S. Targeted expression of oncogenic K-ras in intestinal epithelium causes spontaneous tumorigenesis in mice. Gastroenterology. 2002;123:492–504. doi: 10.1053/gast.2002.34786. [DOI] [PubMed] [Google Scholar]
- 29.Velcich A, Yang W, Heyer J, Fragale A, Nicholas C, Viani S, Kucherlapati R, Lipkin M, Yang K, Augenlicht L. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science. 2002;295:1726–1729. doi: 10.1126/science.1069094. [DOI] [PubMed] [Google Scholar]
- 30.Kahl-Rainer P, Karner-Hanusch J, Weiss W, Marian B. Five of six protein kinase C isoenzymes present in normal mucosa show reduced protein levels during tumor development in the human colon. Carcinogenesis. 1994;15:779–782. doi: 10.1093/carcin/15.4.779. [DOI] [PubMed] [Google Scholar]
- 31.Pongracz J, Clark P, Neoptolemos JP, Lord JM. Expression of protein kinase C isoenzymes in colorectal cancer tissue and their differential activation by different bile acids. Int J Cancer. 1995;61:35–39. doi: 10.1002/ijc.2910610107. [DOI] [PubMed] [Google Scholar]
- 32.Wali RK, Bissonnette M, Khare S, Aquino B, Niedziela S, Sitrin M, Brasitus TA. Protein kinase C isoforms in the chemopreventive effects of a novel vitamin D3 analogue in rat colonic tumorigenesis. Gastroenterology. 1996;111:118–126. doi: 10.1053/gast.1996.v111.pm8698190. [DOI] [PubMed] [Google Scholar]
- 33.Guan L, Song K, Pysz MA, Curry KJ, Hizli AA, Danielpour D, Black AR, Black JD. Protein kinase C-mediated down-regulation of cyclin D1 involves activation of the translational repressor 4E-BP1 via a phosphoinositide 3-kinase/Akt-independent, protein phosphatase 2A-dependent mechanism in intestinal epithelial cells. J Biol Chem. 2007;282:14213–14225. doi: 10.1074/jbc.M610513200. [DOI] [PubMed] [Google Scholar]
- 34.Batlle E, Verdu J, Dominguez D, del Mont Llosas M, Diaz V, Loukili N, Paciucci R, Alameda F, de Herreros AG. Protein kinase C-alpha activity inversely modulates invasion and growth of intestinal cells. J Biol Chem. 1998;273:15091–15098. doi: 10.1074/jbc.273.24.15091. [DOI] [PubMed] [Google Scholar]
- 35.Frey MR, Saxon ML, Zhao X, Rollins A, Evans SS, Black JD. Protein kinase C isozyme-mediated cell cycle arrest involves induction of p21(waf1/cip1) and p27(kip1) and hypophosphorylation of the retinoblastoma protein in intestinal epithelial cells. J Biol Chem. 1997;272:9424–9435. doi: 10.1074/jbc.272.14.9424. [DOI] [PubMed] [Google Scholar]
- 36.Rousset M. The human colon carcinoma cell lines HT-29 and Caco-2: two in vitro models for the study of intestinal differentiation. Biochimie. 1986;68:1035–1040. doi: 10.1016/s0300-9084(86)80177-8. [DOI] [PubMed] [Google Scholar]
- 37.Otori K, Sugiyama K, Fukushima S, Esumi H. Expression of the cyclin D1 gene in rat colorectal aberrant crypt foci and tumors induced by azoxymethane. Cancer Lett. 1999;140:99–104. doi: 10.1016/s0304-3835(99)00058-0. [DOI] [PubMed] [Google Scholar]
- 38.Waugh MG, Hsuan JJ. EGF receptors as transcription factors: ridiculous or sublime? Nature cell biology. 2001;3:E209–211. doi: 10.1038/ncb0901-e209. [DOI] [PubMed] [Google Scholar]
- 39.Santiskulvong C, Rozengurt E. Protein kinase C[alpha] mediates feedback inhibition of EGF receptor transactivation induced by Gq-coupled receptor agonists. Cellular Signalling. 2007;19:1348–1357. doi: 10.1016/j.cellsig.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 40.Wang X-q, Yan Q, Sun P, Liu J-W, Go L, McDaniel SM, Paller AS. Suppression of Epidermal Growth Factor Receptor Signaling by Protein Kinase C-{alpha} Activation Requires CD82, Caveolin-1, and Ganglioside. 2007;67:9986–9995. doi: 10.1158/0008-5472.CAN-07-1300. [DOI] [PubMed] [Google Scholar]
- 41.Hornia A, Lu Z, Sukezane T, Zhong M, Joseph T, Frankel P, Foster DA. Antagonistic Effects of Protein Kinase C alpha and delta on Both Transformation and Phospholipase D Activity Mediated by the Epidermal Growth Factor Receptor. 1999;19:7672–7680. doi: 10.1128/mcb.19.11.7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manning HC, Merchant NB, Foutch AC, Virostko JM, Wyatt SK, Shah C, McKinley ET, Xie J, Mutic NJ, Washington MK, LaFleur B, Tantawy MN, Peterson TE, Ansari MS, Baldwin RM, Rothenberg ML, Bornhop DJ, Gore JC, Coffey RJ. Molecular Imaging of Therapeutic Response to Epidermal Growth Factor Receptor Blockade in Colorectal Cancer. 2008;14:7413–7422. doi: 10.1158/1078-0432.CCR-08-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang JL, Qu XJ, Russell PJ, Goldstein D. Interferon-alpha promotes the anti-proliferative effect of gefitinib (ZD 1839) on human colon cancer cell lines. Oncology. 2005;69:224–238. doi: 10.1159/000088070. [DOI] [PubMed] [Google Scholar]
- 44.Jhawer M, Goel S, Wilson AJ, Montagna C, Ling YH, Byun DS, Nasser S, Arango D, Shin J, Klampfer L, Augenlicht LH, Perez-Soler R, Mariadason JM. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008;68:1953–1961. doi: 10.1158/0008-5472.CAN-07-5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheridan C. EGFR inhibitors embrace KRAS. Nat Biotech. 2008;26:839–840. doi: 10.1038/nbt0808-839. [DOI] [PubMed] [Google Scholar]
- 46.Hosokawa Y, Arnold A. Mechanism of cyclin D1 (CCND1, PRAD1) overexpression in human cancer cells: analysis of allele-specific expression. Genes Chromosomes Cancer. 1998;22:66–71. doi: 10.1002/(sici)1098-2264(199805)22:1<66::aid-gcc9>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 47.Cerda SR, Mustafi R, Little H, Cohen G, Khare S, Moore C, Majumder P, Bissonnette M. Protein kinase C delta inhibits Caco-2 cell proliferation by selective changes in cell cycle and cell death regulators. Oncogene. 2006;25:3123–3138. doi: 10.1038/sj.onc.1209360. [DOI] [PubMed] [Google Scholar]
- 48.Wali RK, Baum CL, Bolt MJ, Dudeja PK, Sitrin MD, Brasitus TA. Down-regulation of protein kinase C activity in 1,2-dimethylhydrazine-induced rat colonic tumors. Biochim Biophys Acta. 1991;1092:119–123. doi: 10.1016/0167-4889(91)90185-z. [DOI] [PubMed] [Google Scholar]
- 49.Neill GW, Ghali LR, Green JL, Ikram MS, Philpott MP, Quinn AG. Loss of protein kinase Calpha expression may enhance the tumorigenic potential of Gli1 in basal cell carcinoma. Cancer Res. 2003;63:4692–4697. [PubMed] [Google Scholar]
- 50.Tibudan SS, Wang Y, Denning MF. Activation of protein kinase C triggers irreversible cell cycle withdrawal in human keratinocytes. J Invest Dermatol. 2002;119:1282–1289. doi: 10.1046/j.1523-1747.2002.19625.x. [DOI] [PubMed] [Google Scholar]
- 51.Zhu Y, Dong Q, Tan BJ, Lim WG, Zhou S, Duan W. The PKCalpha-D294G mutant found in pituitary and thyroid tumors fails to transduce extracellular signals. Cancer Res. 2005;65:4520–4524. doi: 10.1158/0008-5472.CAN-04-4506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.