

Summary
Self-renewal of embryonic stem cells (ESCs) is essential for maintenance of pluripotency, which is defined as the ability to differentiate into any specialised cell type comprising the adult organism. Understanding the mechanisms that regulate ESC self-renewal and proliferation is required before ESCs can fulfil their potential in regenerative therapies, and murine ESCs (mESCs) have been widely used as a model. Members of the class-IA phosphoinositide 3-kinase (PI3K) family of lipid kinases regulate a variety of physiological responses, including cell migration, proliferation and survival. PI3Ks have been reported to regulate both proliferation and self-renewal of mESCs. Here we investigate the contribution of specific class-IA PI3K isoforms to the regulation of mESC fate using small-molecule inhibitors with selectivity for particular class-IA PI3K catalytic isoforms, and siRNA-mediated knockdown. Pharmacological inhibition or knockdown of p110β promoted mESC differentiation, accompanied by a decrease in expression of Nanog. By comparison, pharmacological inhibition or siRNA-mediated knockdown of p110α had no effect on mESC self-renewal per se, but instead appeared to reduce proliferation, which was accompanied by inhibition of leukaemia inhibitory factor (LIF) and insulin-induced PI3K signalling. Our results suggest that PI3Ks contribute to the regulation of both mESC pluripotency and proliferation by differential coupling to selected p110 catalytic isoforms.
Keywords: Embryonic stem cells, Pluripotency, Cell proliferation, PI3 kinase, Signal transduction
Introduction
Murine embryonic stem cells (mESCs) are derived from the inner cell mass (ICM) of the preimplantation blastocyst at embryonic day 3.5 (E3.5) (Evans and Kaufman, 1981). When grown in culture in the presence of serum, leukaemia inhibitory factor (LIF) is required to maintain mESCs in an undifferentiated, pluripotent state (Smith, 2001). Pluripotency is defined as the ability to produce all the differentiated cell types that comprise an adult organism. So that pluripotency is maintained, ESCs in culture need to self-renew, which in essence is proliferation accompanied by the suppression of differentiation. LIF promotes an array of intracellular signalling pathways that are important for promoting self-renewal, including the activation of Stat3 (Boeuf et al., 1997; Cartwright et al., 2005; Niwa et al., 1998) and phosphoinositide 3-kinase (PI3K) signalling (Paling et al., 2004). The MAPK pathway is also activated by LIF and FGF4 but, rather than contribute to self-renewal, activation of this pathway promotes differentiation (Burdon et al., 1999; Kunath et al., 2007; Stavridis et al., 2007).
The PI3K family of lipid kinases catalyse the addition of a phosphate group to the D3 position of the inositol ring of phosphoinositides, generating PtdIns(3)P, PtdIns(3,4)P2 and PtdIns(3,4,5)P3, which subsequently act as secondary messengers for signal transduction (Vanhaesebroeck and Waterfield, 1999). Mammalian PI3Ks are categorised into three classes, with the class-IA PI3Ks comprising a 110-kDa catalytic subunit (p110α, p110β or p110δ, encoded by the genes Pik3ca, Pik3cb and Pik3cd, respectively) coupled to a regulatory subunit (p85α, p55α or p50α, encoded by Pik3r1; p85β, encoded by Pik3r2; or p55γ, encoded by Pik3r3) (Cantrell, 2001; Vanhaesebroeck and Waterfield, 1999). The phosphatase PTEN (phosphatase and tensin homologue deleted on chromosome ten) dephosphorylates PtdIns(3,4,5)P3, the key product of class-IA PI3K activity and so acts as a negative regulator of this pathway. In mESCs, PI3Ks were first implicated in the regulation of cell proliferation, initially owing to the fact that PTEN-null mESCs proliferate in reduced levels of serum and show a modestly enhanced growth rate (Sun et al., 1999). Direct inhibition of PI3K signalling with LY294002 (at a dose of 25 μM) was reported to lead to a decrease in mESC proliferation (Jirmanova et al., 2002), whereas deletion or pharmacological inhibition of mammalian target of rapamycin (mTOR), a downstream effector of PI3K signalling, also resulted in proliferative defects (Murakami et al., 2004). Furthermore, deletion of the mESC-specific Ras protein, ERas, leads to a reduction in proliferation of ESCs, which was rescued by expression of an activated form of p110α (Takahashi et al., 2003). By contrast, ESCs lacking the key PI3K target phosphoinositide-dependent kinase-1 exhibit no changes in proliferation compared with wild-type cells (Williams et al., 2000).
We, and others, have also implicated PI3Ks in the regulation of ESC self-renewal. We demonstrated that inhibition of PI3Ks with the broad-selectivity PI3K inhibitor LY294002 (at a dose of 5 μM) leads to a loss of self-renewal, without demonstrating appreciable effects on proliferation, and implicated class-IA PI3Ks in this response (Paling et al., 2004). Furthermore, we have shown that both LIF and BMP4 activate PI3K signalling in mESCs (Welham et al., 2007). Consistent with our observations, expression of a myristoylated, active form of protein kinase B (PKB, also known as Akt) promotes self-renewal of mESCs in the absence of LIF (Watanabe et al., 2006). PKB normally inhibits glycogen-synthase kinase-3 (GSK-3) activity; consequently, it is interesting to note that inhibition of GSK-3 with 6-bromoindirubin-3′-oxime (BIO) (Sato et al., 2004), or a series of bisindolylmaleimides (Bone et al., 2009), enhances mESC self-renewal. In addition, PKB was identified in a screen for activators of self-renewal (Pritsker et al., 2006), whereas a short-interfering RNA (siRNA)-based investigation demonstrated a requirement for the PKB-binding protein Tcl1 in the maintenance of mESC pluripotency (Ivanova et al., 2006). Mechanistically, we have recently reported that PI3K activity appears to be coupled with the regulation of expression of the homeodomain transcription factor Nanog (Storm et al., 2007), which, along with Oct4 and Sox2, forms the core transcriptional circuitry that is involved in the maintenance of pluripotency (Niwa, 2007). A requirement for PI3K signalling in the maintenance of pluripotency of human ESCs has also been reported (Armstrong et al., 2006; Pyle et al., 2006).
Class-IA PI3Ks are expressed from the single-cell stage of mammalian development and their activity is required in order for development to proceed normally (Riley et al., 2005; Riley et al., 2006). Evidence supports the notion that individual isoforms play specific physiological roles in vivo. For example, mouse embryos lacking p110α exhibit proliferative defects and die at E9.5-E10.5 (Bi et al., 1999). Replacement of endogenous p110α, by knock-in of a kinase-dead version of p110α, also results in embryonic lethality of homozygous embryos, whereas heterozygotes are viable and fertile but exhibit disrupted insulin signalling (Foukas et al., 2006). Interestingly, embryos that are deficient in p110β perish at a very early stage of development, with a failure in blastocyst implantation (Bi et al., 2002). By contrast, mice that are deficient in active p110δ are viable and fertile but are subject to impaired immune responses (Okkenhaug et al., 2002). In addition to distinct physiological roles, there is evidence emerging that class-IA PI3K isoforms are also differentially regulated in distinct cellular contexts (Papakonstanti et al., 2008).
To develop a greater understanding of the role of PI3Ks in the regulation of ESC behaviour, we investigated the contribution of individual isoforms to the control of mESC fate. Using inhibitors that are selective for different isoforms, complemented by siRNA-mediated knockdown, our findings suggest that p110β plays the predominant role in the regulation of ESC self-renewal, whereas p110α appears to regulate ESC proliferation.
Results
PI3Ks have previously been implicated in the regulation of both ESC proliferation and self-renewal (see Introduction), and, in this study, we sought to investigate whether individual isoforms of the class-IA PI3K catalytic subunit are responsible for regulating distinct aspects of mESC behaviour. Recently, a number of cell-permeable, small-molecule inhibitors have been characterised regarding their selectivity for the class-I PI3K family (Condliffe et al., 2005; Hayakawa et al., 2006; Hennessy et al., 2005; Jackson et al., 2005; Knight et al., 2006; Sadhu et al., 2003). We used a range of these small molecules, complemented with siRNA-mediated knockdown of individual isoforms, to investigate the roles of different isoforms of the class-IA PI3K catalytic subunit in the regulation of ESC self-renewal.
PI3K p110β-mediated signalling regulates self-renewal
Mouse embryos homozygous for a partial deletion of Pik3cb, the gene encoding the p110β subunit, exhibit early embryonic lethality (E3.5) and cells cannot be derived from these blastocysts (Bi et al., 2002). mESCs are isolated from blastocysts at E3.5; hence, the role of p110β in mESCs was of particular interest. Treatment of mESCs, cultured in the presence LIF and serum, with the p110β-isoform selective inhibitor TGX-221 (Condliffe et al., 2005; Hennessy et al., 2005; Jackson et al., 2005) generated colonies displaying a more differentiated morphology and reduced staining for alkaline phosphatase, a marker of undifferentiated mESCs, compared with controls (Fig. 1A). Treatment with 25 nM or 50 nM TGX-221 led to significant decreases in the proportion of alkaline-phosphatase-positive colonies, as was observed following incubation with LY294002 (Fig. 1B), whereas higher doses did not lead to further decreases (supplementary material Fig. S1A). No significant change in the total number of colonies generated was observed. A less-potent p110β inhibitor, TGX-121, had similar effects to TGX-221 (supplementary material Fig. S1B). These results, using two different p110β selective inhibitors, suggest that this isoform is involved in the regulation of mESC self-renewal.
Fig. 1.

Pharmacological inhibition of p110β leads to a decrease in self-renewal of murine ESCs. (A) Murine ESCs (mESCs), cultured in the presence of LIF and serum, were treated for 4 days with LY294002 or TGX-221 at the doses shown or with vehicle alone (DMSO). Self-renewal was assessed by staining for alkaline-phosphatase activity and representative colonies are shown. Scale bars: 100 μm. (B) The average percentage of alkaline-phosphatase-positive, self-renewing colonies in each condition are shown with s.e.m. (n=4). ANOVA and Dunnett's post-hoc test were applied; ***P<0.001. (C) Cells were treated with TGX-221 for 30 minutes prior to stimulation with 103 U/ml LIF for 10 minutes. Cell lysates were resolved by SDS-PAGE and immunoblotting carried out using the antibodies indicated to detect PI3K downstream signalling. (D) Cells were cultured for 4 days in the presence or absence of LIF or in the presence of LIF and 100 nM TGX-221 and immunoblotting carried out as above.
To further characterise the involvement of p110β in the regulation of mESC self-renewal, mESCs were pre-treated with TGX-221 prior to acute stimulation with LIF (Fig. 1C). Surprisingly, inhibition of p110β with TGX-221 did not affect basal or LIF-stimulated levels of phosphorylation of Ser473 of PKB (Fig. 1C, pPKB). Similarly, basal levels of both GSK-3α/β phosphorylation and ribosomal protein S6 phosphorylation (sites Ser20/9 and Ser 235/236, respectively) were unchanged by TGX-221 treatment. Whereas LIF-stimulated phosphorylation of the MAPKs Erk1 and Erk2 was unaffected by TGX-221, basal levels of phosphorylated Erk2 were modestly enhanced in unstimulated cells following incubation with TGX-221. Pre-treatment with TGX-121 demonstrated similar effects on basal signalling and negligible consistent changes on LIF-stimulated signalling (supplementary material Fig. S1C). We also examined the longer-term effects of inhibiting p110β on downstream signalling in mESCs cultured in the presence of LIF. As shown in Fig. 1D, treatment for 4 days with TGX-221 led to a decrease in PKB phosphorylation and a small decline in S6 phosphorylation, whereas levels of Erk phosphorylation were unaltered.
p110β is required to maintain optimal mESC self-renewal
To complement the use of TGX-221, we further investigated the role of p110β in mESC self-renewal using siRNAs targeting Pik3cb to knockdown the expression of p110β. Both Smartpool siRNAs (Dharmacon), supplied as a pool of four single siRNAs, and individual Silencer Select siRNAs (Ambion, Applied Biosystems) were used to decrease Pik3cb gene expression. As a positive control, we used Smartpool siRNAs targeting Nanog, a homeodomain transcription factor known to play a key role in controlling ESC fate (Chambers et al., 2003; Chambers et al., 2007; Mitsui et al., 2003) that we have shown is downregulated following inhibition of PI3Ks over a 72-hour timecourse (Storm et al., 2007).
Quantitative reverse transcriptase (RT)-PCR revealed significant knockdown of p110β expression using Smartpool Pik3cb siRNAs (Fig. 2A), with 50 nM being most effective. No effect on expression of Pik3ca or Pik3cd RNAs observed (supplementary material Fig. S2A,B). Owing to antibody-sensitivity issues and low expression levels, we were unable to consistently detect catalytic-isoform protein expression, so cannot entirely rule out compensation at the protein level. However, levels of p85 subunit were either unchanged or slightly decreased following knockdown of p110β (Fig. 2Eii), the latter being consistent with the overall stoichiometry of p85:p110 being preserved. No decrease in the level of mRNA encoding p110β was detected following Nanog knockdown (Fig. 2A), whereas expression of Nanog mRNA was decreased following knockdown of p110β (Fig. 2B), consistent with our previous observation that PI3K inhibition leads to the downregulation of Nanog expression (Storm et al., 2007). On assessment of self-renewal, knockdown of p110β led to a reduction of approximately 50% in the proportion of alkaline-phosphatase-positive colonies (Fig. 2C). Furthermore, a significant reduction in the proportion of pure self-renewing alkaline-phosphatase-positive colonies was observed using 50 nM of p110β-targeting siRNA (Fig. 2D). Importantly, the levels of self-renewal following siRNA-mediated knockdown of p110β were comparable to pharmacological inhibition of p110β with TGX-221 (Fig. 1B) and siRNA-mediated knockdown of Nanog (Fig. 2C). Consistent with a decrease in self-renewal, the level of Nanog protein was also reduced following knockdown of p110β expression (Fig. 2Ei); however, the effect of targeting p110β on Oct4 expression was not as clear, despite cells appearing morphologically differentiated. This is reminiscent of the effects we observed upon inhibition of PI3Ks with LY294002, in which Oct4 levels were maintained even though cells appear differentiated, possibly owing to the slower decline in Oct4 protein expression upon loss of pluripotency (Paling et al., 2004). For subsequent analyses, we used Nanog expression as an indicator of ESC pluripotency.
Fig. 2.

Knockdown of Pik3cb reduces self-renewal and decreases Nanog expression. mESCs were transfected with Pik3cb (p110β), Nanog or non-targeting (NT) Smartpool siRNAs and harvested 72 hours later. (A) Quantitative RT-PCR was conducted on quadruplicate samples to measure knockdown of Pik3cb RNA and levels of RNA encoding p110β were normalised relative to levels of β-actin RNA. Representative data from four independent experiments are shown with s.d.; *P<0.05 following a Student's t-test. (B) Quantitative RT-PCR was conducted to detect knockdown of Nanog RNA. Representative data are shown with s.d.; *P<0.05 and **P<0.01 following a Student's t-test. (C) Following transfection, cells were plated for 4 days and self-renewal assessed by alkaline-phosphatase staining. The average percentage of alkaline-phosphatase-positive, self-renewing colonies in each condition are shown with s.e.m. (n=4); *P<0.05 following a Student's t-test. The number of total colonies formed in each condition did not vary significantly. (D) Assessment of self-renewal by alkaline-phosphatase staining is depicted by the percentage of pure alkaline-phosphatase colonies (compact round red-stained colonies) with s.e.m. (n=4); * P<0.05 following a Student's t-test. (E) Immunoblotting was used to detect (i) Nanog and Oct4 protein levels or (ii) p85 protein levels, with SHP-2 reprobing used to confirm equal loading.
We further verified the role of p110β in the regulation of mESC pluripotency using siRNAs that target different Pi3kcb exons to those targeted by the Smartpool siRNAs. As shown in Fig. 3A, the siRNAs Pik3cb s93107 and Pik3cb s93108 (which reduced p110β RNA expression between 40 and 60%, Fig. 3B, but did not affect levels of Pik3ca or Pik3cd RNAs) (supplementary material Fig. S2C,D) reduced the ability of mESCs to generate alkaline-phosphatase-positive, self-renewing colonies (Fig. 3A). Consistent with the loss of alkaline-phosphatase staining, Nanog RNA (Fig. 3C) and protein (Fig. 3D) expression were also reduced following p110β knockdown, whereas levels of the p85 regulatory subunit were unchanged (Fig. 3D). Taken together, these results indicate that p110β plays a role in the maintenance of pluripotency of mESCs. When expression of p110β is reduced or when p110β is selectively inhibited, mESCs lose their alkaline-phosphatase expression and exhibit a decrease in Nanog RNA and protein expression, features that are consistent with a loss of self-renewal.
Fig. 3.
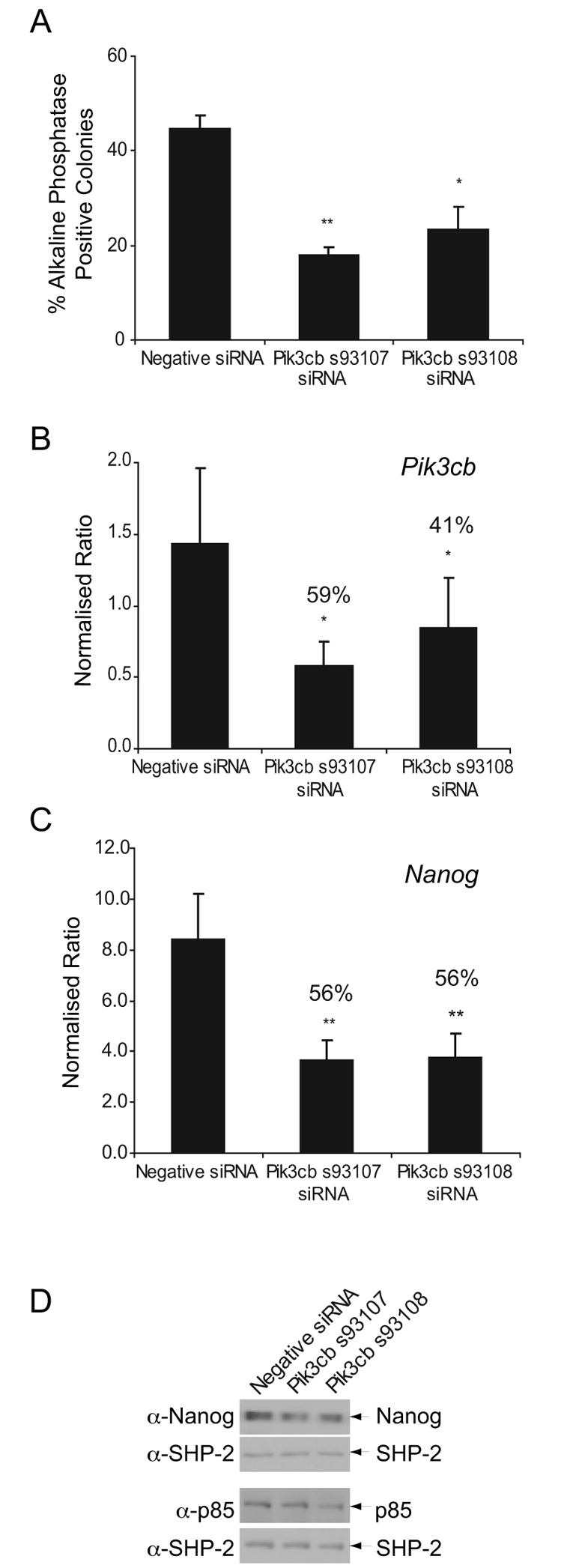
siRNA-mediated knockdown of p110β expression reduces self-renewal. mESCs were transfected with Pik3cb-targeting siRNAs or with Negative Control Silencer Select siRNA (Ambion). (A) Following transfection, cells were plated for 4 days and self-renewal assessed by alkaline-phosphatase staining. The average percentage of alkaline-phosphatase-positive colonies are shown, plus s.e.m. (n=4); *P<0.05 and **P<0.01 following a Student's t-test. The number of total colonies formed in each condition did not vary significantly. (B,C) Following transfection, cells were re-plated for a further 4 days then harvested for RNA analysis. Quantitative RT-PCR was conducted to detect knockdown of Pik3cb RNA (B) or expression of Nanog (C). Representative data, with s.d., are shown; *P<0.05 and **P<0.01 following a Student's t-test. The percentage of knockdowns, compared with the negative control, are indicated. (D) Immunoblots detecting expression of Nanog and p85 proteins in mESCs after transfection and 4 days of culture are shown and the position of proteins indicated. Blots were stripped and reprobed with antibodies detecting SHP-2 to assess loading.
p110δ and the control of mESC fate
p110δ plays a predominant role in cells of the lymphohaemopoietic system owing largely to its restricted expression in cells of the immune system (Okkenhaug et al., 2002; Vanhaesebroeck et al., 2004; Vanhaesebroeck et al., 1997). Although expressed at low levels by mESCs, given the importance of low levels of p110γ in cardiomyocytes (Patrucco et al., 2004), we investigated whether p110δ contributes to the regulation of ESC fate. Treatment of mESCs with the p110δ selective inhibitor IC87114 (Knight et al., 2006; Sadhu et al., 2003) only led to a significant decrease in self-renewal at a dose of 10 μM (Fig. 4A), whereas siRNA-mediated knockdown of p110δ (Fig. 4B) had little effect on the levels of alkaline-phosphatase-positive colonies (Fig. 4C). Furthermore, expression of Nanog RNA was not reduced following p110δ knockdown (Fig. 4D) and levels of p85 regulatory-subunit protein were unaltered (Fig. 4E). These data suggest that, if p110δ is playing a role in ESCs, it is minor in comparison to p110β.
Fig. 4.

A minor role for p110δ in mESC self-renewal. (A) mESCs were treated with a p110δ inhibitor, IC87114, for 4 days and self-renewal assessed by staining for alkaline-phosphatase activity. The average percentage of alkaline-phosphatase-positive, self-renewing colonies in each condition are shown with s.e.m. (n=4); *P<0.05 following a Student's t-test. The number of total colonies formed in each condition did not vary significantly. (B) Quantitative RT-PCR was conducted to detect knockdown of Pik3cd RNA following transfection with Pik3cd-targeting siRNA or with non-targeting siRNA. Representative data is shown with s.d. and the percentage of knockdowns, compared with control, are indicated. (C) Following transfection, cells were plated for 4 days and self-renewal assessed by alkaline-phosphatase staining. The average percentage of alkaline-phosphatase-positive, self-renewing colonies in each condition are shown with s.e.m. The number of total colonies formed in each condition did not vary significantly. (D) Expression of Nanog was assessed by quantitative RT-PCR following knockdown of Pik3cd. Representative data is shown with s.d. (E) Immunoblotting was carried out to detect p85 protein levels, with SHP-2 reprobing used to confirm equal loading.
p110α does not regulate pluripotency in mESCs
p110α has been widely implicated in the regulation of cell proliferation (Bi et al., 1999), particularly in response to insulin signalling (Foukas et al., 2006; Knight et al., 2006). To investigate whether p110α plays a selective role in the control of mESC fate, we first assessed its importance using siRNA-mediated knockdown. Smartpool Pik3ca-targeting siRNAs effected an 88% knockdown of mRNA encoding p110α (Fig. 5A), but had no significant effect on the expression of Pik3cb or Pik3cd RNAs (supplementary material Fig. S2G,H) or protein levels of the p85 regulatory subunit (Fig. 5Cii). Despite achieving a high level of knockdown of p110α, we observed no change in the proportion of alkaline-phosphatase-positive, self-renewing colonies (Fig. 5B). Consistent with this, we observed no alteration in the expression of the pluripotency markers Nanog or Rex1 (Fig. 5C,D). These studies suggest that p110α might not be required for the maintenance of mESCs in an undifferentiated state.
Fig. 5.

p110α does not regulate mESC self-renewal. mESCs were transfected with Pik3ca-targeting (p110α) or with non-targeting (NT) siRNAs. (A) Quantitative RT-PCR of quadruplicate samples was conducted to detect knockdown of Pik3ca RNA. The averages, with s.d., are shown and are representative of four independent experiments. *P<0.05 following a Student's t-test. (B) Following transfection, cells were plated for 4 days and self-renewal assessed by alkaline-phosphatase staining. The average percentage of alkaline-phosphatase-positive, self-renewing colonies in each condition are shown with s.e.m. (n=4). The total number of colonies formed in each condition did not vary significantly. (Ci) Semi-quantitative RT-PCR was used to assess knockdown of RNA encoding p110α, Nanog or Rex1 following transfection with the indicated siRNAs. (Cii) Immunoblotting was used to detect Nanog, p85 and SHP-2 protein levels. (D) Nanog expression was also assessed by quantitative RT-PCR as in B. (E,F) mESCs were treated for 4 days with the indicated concentrations of LY294002, compound 15e (E), PIK-75 (F) or with vehicle alone (DMSO). The average number of self-renewing alkaline-phosphatase-positive and non-self-renewing colonies in each condition is shown with s.e.m. (15e, n=5; PIK-75, n=3); *P<0.05 following a Student's t-test. (G) Following treatment with 15e or PIK-75, colonies were fixed and stained for alkaline phosphatase. Representative colonies are depicted. Scale bars: 100 μm.
To further probe the role of p110α in mESCs, we used some of the recently described small-molecule inhibitors that exhibit selectivity for p110α. Compound 15e has been characterised as a selective inhibitor of p110α with, as yet, no reported off-target inhibition of mTOR (Hayakawa et al., 2006). PIK-75 has been used more widely and is a selective p110α inhibitor with a half-maximal inhibitory concentration (IC50) value in the range of 5-10 nM (Chaussade et al., 2007; Knight et al., 2006). Importantly, unlike the broad-selectivity PI3K inhibitors LY294002 and PI-103, at concentrations at which PIK-75 is selective for p110α (5-10 nM) it does not inhibit mTOR, which is reported to occur at concentrations of 1 μM or higher (Knight et al., 2006). Treatment of mESCs with either 15e (Fig. 5E) or PIK-75 (Fig. 5F) did not lead to a decrease in the proportion of self-renewing colonies, in agreement with our siRNA studies. However, we noted a decrease in the total number of colonies formed in these cultures (Fig. 5E,F) and observed that colonies were smaller than those with vehicle treatment alone (Fig. 5G), suggesting that p110α plays a role in regulating the proliferation of mESCs.
p110α-mediated signalling appears to regulate mESC proliferation
In light of earlier reports (Jirmanova et al., 2002; Murakami et al., 2004; Sun et al., 1999; Takahashi et al., 2003), and our results demonstrating that inhibition of p110α had no discernable effect on self-renewal but rather led to a reduction in colony size and number, we considered that p110α might couple to regulation of ESC proliferation, distinct from a role in the maintenance of pluripotency. We found that both 15e and PIK-75 reduced mESC growth (Fig. 6A), which appears to predominantly be the result of reduced cell division, as levels of apoptosis were similar in control and treated samples, with the exception of cells treated with 800 nM 15e, in which levels of apoptosis were slightly elevated (supplementary material Fig. S3). p110α has been linked to the regulation of insulin signalling in many cell types (Foukas et al., 2006; Knight et al., 2006), including ESCs (Welham et al., 2007). We found that basal and insulin-stimulated phosphorylation of Ser473 on PKB were inhibited in a dose-dependent manner by both 15e (Fig. 6Bi) and PIK-75 (Fig. 6Bii), whereas S6 phosphorylation was also reduced (Fig. 6Bi,ii). In contrast to the p110β selective inhibitor TGX-221, 15e and PIK-75 also reduced LIF-stimulated PKB Ser473 and S6 Ser235/236 phosphorylation (Fig. 6Ci,ii).
Fig. 6.

p110α appears to be coupled to proliferation, insulin signalling and LIF signalling in mESCs. (A) To assess cell proliferation, viable cell numbers were determined in triplicate at 24-hour intervals for up to 4 days. The average number of cells per dish, ± s.d., are shown for each treatment. (B,C) Following 30 minutes pre-treatment with either (i) 15e or (ii) PIK-75 at the doses indicated, mESCs were stimulated with 10 μg/ml insulin for 5 minutes (B) or 103 U/ml LIF for 10 minutes (C). Signalling downstream of PI3K was assessed by SDS-PAGE and immunoblotting with the antibodies indicated. Data, representative of three independent experiments, are shown.
Inhibition of p110α activity does not prevent differentiation
Inhibition of p110α activity or knockdown of p110α do not appear to result in loss of pluripotency, but rather our data with PIK-75 and compound 15e suggest a role in cell division and proliferation. Alternative interpretations of our data are that inhibition or knockdown of p110α could either enhance self-renewal, block ESC differentiation or lead to selective apoptosis of spontaneously differentiating ESCs. To distinguish between these possibilities, we examined the effects of PIK-75 and 15e on mESCs cultured in the absence of LIF but in the presence of serum. The results reveal that treatment with neither PIK-75 nor 15e is sufficient to maintain self-renewal in the absence of LIF, demonstrated by the very low numbers of alkaline-phosphatase-positive colonies that were generated (Fig. 7A). However, colonies did form in the absence of LIF, arguing against selective apoptosis of differentiating cells, and these colonies had a differentiated morphology, suggesting differentiation was not blocked to any significant extent (Fig. 7B). We also investigated what effect inhibition of p110α would have on the loss of self-renewal observed following p110β selective inhibition. Interestingly, a greater loss of self-renewal was observed when mESCs were treated with both TGX-221 and 15e, compared with TGX-221 alone (Fig. 7C). These data suggest that, although effects on mESC self-renewal and proliferation seem to be coupled to different isoforms of the class-IA PI3K catalytic subunit, there is crosstalk between the isoforms.
Fig. 7.

p110α inhibition does not prevent loss of self-renewal or block mESC differentiation. (A) mESCs were treated with the p110α inhibitors 15e or PIK-75 for 4 days, at the doses indicated, in the presence or absence of LIF. The average percentage of alkaline-phosphatase-positive colonies, with s.e.m. (n=3), is shown, ***P<0.001 following a Student's t-test. (B) The average number of self-renewing alkaline-phosphatase-positive and non-self-renewing colonies in each condition is shown with s.e.m. (n=3). (C) mESCs cultured in the presence of LIF were treated with TGX-221 for 4 days in the presence or absence of 600 nM 15e. Self-renewal was assessed by staining for alkaline-phosphatase activity and the average percentage of alkaline-phosphatase-positive colonies, with s.e.m., are shown (n=4). ANOVA (P=0.002) and Dunnett's post-hoc test were applied; **P<0.01.
Discussion
ESC pluripotency is regulated by the coordinated actions of a network of extrinsic factors, signalling pathways and core transcriptional components (Boiani and Schoeler, 2005; Niwa, 2007). Self-renewal of ESCs, which is widely defined as proliferation accompanied by suppression of differentiation (Burdon et al., 2002), is required in order that pluripotency is retained. Thus, a comprehensive understanding of the regulatory mechanisms that control self-renewal is essential. The PI3K family of lipid kinases has been implicated in the regulation of ESC proliferation (Jirmanova et al., 2002; Murakami et al., 2004; Sun et al., 1999; Takahashi et al., 2003) and also in the control of self-renewal (Armstrong et al., 2006; Paling et al., 2004; Pritsker et al., 2006; Pyle et al., 2006; Watanabe et al., 2006). We rationalised that specific class-IA PI3K catalytic subunits might couple to particular physiological responses in mESCs, providing an explanation for these findings. We tested this hypothesis using isoform-selective pharmacological inhibitors and siRNA-mediated knockdown. Our data clearly demonstrate that p110β is the main isoform that contributes to the regulation of mESC pluripotency. By contrast, our data suggest that p110α plays a role in regulating mESC proliferation.
The p110β-specific inhibitors TGX-221 and TGX-121 caused a loss of mESC self-renewal, which was exemplified by significant decreases in the proportion of alkaline-phosphatase-positive colonies formed and a decline in expression of the master regulator of pluripotency, Nanog. Importantly, we observed very similar results using three different siRNAs, all of which selectively knocked down expression of p110β. In view of the lack of effect of knockdown of p110δ, it is likely that the reduction in self-renewal that was observed with the p110δ selective inhibitor IC87114 is also due to inhibition of p110β, which would be predicted at the dose of this inhibitor at which an effect was observed (Knight et al., 2006). It has been known for some time that complete knockout of p110β results in very early embryonic lethality, at the blastocyst stage of development (Bi et al., 2002), which corresponds to the exact stage of development that the ICM, from which ESCs are derived, is present (Evans and Kaufman, 1981). Our results are consistent with a key requirement for PI3K signalling, mediated at least in part via p110β, for the maintenance of cells of the blastocyst. More recently, mice have been generated that contain a knock-in of a catalytically inactive form of p110β, which retain p110β protein expression. These mice demonstrate lethality, but with incomplete penetrance, the reasons for which are not currently clear (Ciraolo et al., 2008; Guillermet-Guibert et al., 2008). One proposal to rationalise the different phenotypes observed in mice that completely lack p110β (Bi et al., 2002) compared with those with knock-in of a kinase-inactive form (Ciraolo et al., 2008; Guillermet-Guibert et al., 2008), is that p110β has non-catalytic functions (Ciraolo et al., 2008). For example, the small GTPase Rab5, located mainly on endosomes, associates with p110β (Cristoforidis et al., 1999) and, when p110β expression is reduced, endocytosis of epidermal growth factor receptor is impaired (Ciraolo et al., 2008). In ESCs, we have demonstrated that inhibition of p110β activity or knockdown of p110β expression both result in decreased self-renewal, suggesting that the catalytic functions of p110β are functionally important. However, at this stage we cannot rule out the possibility that p110β also has non-catalytic functions in mESCs or that compensatory changes in expression of other PI3Ks are involved.
Somewhat surprisingly, we discovered that neither TGX-221 nor TGX-121 affected acute LIF-induced activation of PI3K signalling, although, following a 4-day treatment, TGX-221 did lead to a reduction in PKB phosphorylation in cells cultured in LIF, perhaps reflecting a role for p110β in long-term LIF action, which requires further investigation. The results of our acute LIF-stimulation experiments are very reminiscent of reports that suggest that p110β is not a major effector of tyrosine-kinase-coupled signalling pathways, but rather is linked to G-protein-coupled receptors (GPCRs) (Ciraolo et al., 2008; Guillermet-Guibert et al., 2008; Roche et al., 1998). p110γ is expressed at extremely low levels, if at all, by mESCs (M. Storm and M.W., unpublished data), and treatment with the p110γ selective inhibitor AS605240 has no measurable effect on mESC self-renewal (our unpublished data). Thus, p110β is likely to be the sole PI3K isoform present in mESCs that is able to couple to GPCRs. There is little information relating to signalling via GPCRs in mESCs, although, in human ESCs, sphingosine-1-phosphate, in conjunction with platelet-derived growth factor, has been reported to promote pluripotency (Pebay et al., 2005). Components in serum, media supplements or autocrine factors might be capable of activating GPCRs, which could contribute to what might be termed `basal' levels of signal generation. In relation to this, it is interesting to note that inhibition of p110β with TGX-221 or TGX-121 led to an increase in phosphorylation of Erk2 in unstimulated mESCs, similar to effects we have previously observed following treatment with LY294002 (Paling et al., 2004). Erk1/2 MAP-kinase signalling promotes differentiation of ESCs (Burdon et al., 1999; Kunath et al., 2007; Stavridis et al., 2007); hence, the increase we observe here could favour differentiation. A modest enhancement in basal Erk2 phosphorylation is also apparent in MEFs derived from p110β kinase-dead knock-in mice (Ciraolo et al., 2008). The mechanism underlying this enhancement in Erk2 phosphorylation is unclear, but could be related to the ability of PKB to control Raf1 phosphorylation (Rommel et al., 1999). Further studies will be required to unravel the mechanisms regulating p110β activity in ESCs.
Despite highly effective siRNA-mediated knockdown of p110α, mESCs retained their undifferentiated phenotype. Consistent with these effects, we found that two structurally distinct p110α selective inhibitors, 15e and PIK-75, did not perturb the proportion of self-renewing mESCs. Rather, our results show that treatment with 15e and PIK-75 lead to a reduction in ESC proliferation, which is exemplified by the direct measurement of cell growth and indirectly via a reduction in the size of colonies observed in self-renewal assays, similar to that reported upon knockdown of Pim-1 and Pim-3 (Aksoy et al., 2007). We also observed a decrease in the total number of colonies generated at higher doses of PIK-75, which could be due to the fact that cells are dividing more slowly and that, below a certain size, colonies will not be scored. Overall, these findings suggest that p110α is not playing a major role in regulating ESC pluripotency, but instead might regulate mESC proliferation. We also considered alternative explanations of our results. A common issue relating to PI3K inhibitors has been their tendency to inhibit mTOR, a protein that has previously been shown to regulate ESC proliferation (Murakami et al., 2004). Although much higher concentrations of PIK-75 than those we used here are required to inhibit mTOR (1 μM, compared with 6 nM to inhibit p110α) (Knight et al., 2006), no data have been reported on the possible effect of compound 15e on mTOR activity, so there remains a formal possibility that the reduction in proliferation could be due to inhibition of mTOR directly, rather than directly via p110α, although for PIK-75 we believe this is unlikely. Other alternatives include the possibility that inhibition of p110α potentiates self-renewal, blocks differentiation or causes apoptosis of spontaneously differentiating mESCs, but our data did not support these alternatives. There is extensive evidence for a role for p110α in the control of proliferation in many cell types (Bi et al., 1999; Foukas et al., 2006; Hooshmand-Rad et al., 2000; Kang et al., 2006; Knight et al., 2006; Roche et al., 1994; Vanhaesebroeck et al., 1999). Our data suggest that p110α might play a similar role in mESCs, although definitive proof requires conditional deletion of p110α in mESCs by either siRNA or gene-targeting approaches, as well as confirmation that there are no compensatory changes in expression of other PI3K isoforms. While bearing these caveats in mind, our findings now provide a rationale for the reported role of PI3Ks in the regulation of ESC proliferation (Jirmanova et al., 2002; Murakami et al., 2004; Sun et al., 1999; Takahashi et al., 2003). Most of these previous studies had not distinguished between the isoforms of the p110 catalytic subunit, although, interestingly, Takahashi et al., did show that expression of p110α could rescue the defect in proliferation that arises from a lack of ERas (Takahashi et al., 2003).
The signals that integrate the self-renewal machinery of ESC and their control of proliferation remain enigmatic. Self-renewal is widely defined as proliferation with the suppression of differentiation (Burdon et al., 2002), but it is evident that the rate of ESC proliferation can be altered while ESCs retain their undifferentiated status (Murakami et al., 2004; Takahashi et al., 2003). Although our data implicate the coupling of p110β to self-renewal and p110α to proliferation, we also present evidence that there is crosstalk between these two pathways. We found that inhibition of p110α concomitantly with p110β led to a greater decline in ESC self-renewal compared with inhibition of p110β alone. Evidence from other systems suggests that one PI3K isoform can regulate signalling via another isoform, e.g. p110β sets a threshold for p110α in insulin signalling (Chaussade et al., 2007; Knight et al., 2006) and, in priming the neutrophil respiratory burst, p110γ activation in the first phase is required for activation of p110δ in the second phase (Condliffe et al., 2005). In ESCs, p110α could set a threshold linked to ESC proliferation, acting potentially via LIF or insulin signalling. Inhibition of p110α would reduce the rate of proliferation and at the same time make self-renewal more sensitive to levels of p110β activation. Thus, PI3K signalling could potentially link the machinery controlling pluripotency with that regulating proliferation. Further detailed analyses will be required to test this hypothesis.
Materials and Methods
ESC culture
E14tg2a mESCs were routinely cultured as previously described (Paling et al., 2004). E14tg2a mESCs were also cultured in N2B27-defined media containing a 1:1 ratio of Neurobasal media to DMEM F-12 media, supplemented with N2 (Bottenstein and Sato, 1979) and B27 (Brewer and Cotman, 1989; Brewer et al., 1993) supplements (Invitrogen), 2 mM glutamine, 50 mg/ml bovine serum albumin (BSA), 0.0125% (v/v) Monothioglycerol (Sigma), 1000 U/ml LIF (Chemicon) and 10 ng/ml BMP4 (R&D Systems), as described previously (Ying et al., 2003a; Ying et al., 2003b). Stimulation experiments were conducted using cells cultured for at least 2 days in N2B27-defined media. Cells were washed and incubated in 1:1 Neurobasal media to DMEM F-12 media alone for 4 hours prior to stimulation.
Inhibitors
LY294002 (Calbiochem), compound 15e (Alexis Biochemicals), PI-103 (Patent WO01083456), TGX-121 (Patent WO0153266), IC87114 (Patent WO0181346) (all generous gifts from Tom Crabbe, UCB, Slough, UK) or PIK-75 (Patent WO2001083481), TGX-221 (Patent WO2004016607) [generous gifts from Peter Shepherd, University of Auckland, New Zealand (Chaussade et al., 2007)] were used in this study at the concentrations indicated. To verify their activity and cell permeability, the effects of these small molecules on IL-3-induced signalling in the mouse pro-B cell line BaF/3, which expresses all class-IA catalytic isoforms (Vanhaesebroeck et al., 1997), were examined (supplementary material Fig. S4). All inhibitors used in this study perturbed IL-3-induced signalling in BaF/3, confirming their activity.
Self-renewal assays
Self-renewal assays were performed as previously described (Paling et al., 2004). mESCs were allowed to adhere for a minimum of 4 hours before the addition of inhibitors. The number of alkaline-phosphatase-positive colonies (total self-renewing), the number of pure, compact, highly self-renewing colonies (pure self-renewing) and the total number of colonies per dish were determined. The mean numbers are shown for each treatment with s.e.m. Statistical significance of the results was assessed using ANOVA or t-test where appropriate; *P<0.05, **P<0.01 and ***P<0.001.
Cell lysates and immunoblotting
ESCs were washed with phosphate-buffered saline (PBS) and incubated for 4 hours in media lacking supplements and cytokines. Inhibitors were added 0.5-1.0 hour prior to a 10-minute stimulation with 103 U/ml LIF or a 5-minute stimulation with 10 μg/ml insulin. Culture plates were placed on ice, washed three times with ice-cold PBS and lysed with solubilisation buffer (Welham et al., 1994). Insoluble cell debris was removed by centrifugation for 2 minutes at 20,800 g at 4°C. Protein concentration was determined with a Bradford Assay using the Bio-Rad protein assay kit according to the manufacturer's instructions. A total of 10-20 μg of protein was separated by SDS-PAGE and transferred onto nitrocellulose. Immunoblotting was performed as previously described (Welham et al., 1994) using the following primary antibodies at 1:1000 dilution: rabbit polyclonal antibodies against phosphorylated p44/42 MAPK (anti-pERK-1/2; Santa Cruz Biotechnology, sc93), p44/42 MAPK [anti-panERK-1/2; Cell Signalling Technologies (CST), 9102], phosphorylated Ser473 PKB (anti-pPKB; CST, 4058), PKB (anti-panPKB; CST, 9272), phosphorylated ribosomal protein S6 (anti-pS6; CST, 2211), SH2-domain tyrosine phosphatase (anti-SHP-2; Santa Cruz Biotechnology, sc280), phosphorylated Ser 20/9 GSK-3 (anti-pGSK-3α/β; CST, 9331), GSK-3β (anti-panGSKβ; CST, 9315) and Nanog (anti-Nanog; Abcam, 21603). Horseradish-peroxidase-conjugated anti-rabbit secondary antibodies (Dako, UK) were used at 1:20,000 dilution and blots were developed using ECL or ECL Advance (Amersham Biosciences, UK). Blots were stripped and re-probed as described previously (Welham et al., 1994).
Analysis of apoptosis using DiOC6 staining
Mitochondrial-membrane-permeability transition, a hallmark of and an irreversible point in programmed cell death (Rottenberg and Wu, 1998), was assessed by DiOC6 (3,3′-dihexyloxacarbocyanide iodide) staining. Cells were pre-treated with compound 15e, PIK-75 or DMSO for 18 hours, then media and cells were collected, washed in PBS and incubated with 1 nM DiOC6 for 30 minutes at room temperature. Samples of unstained cells were retained. An aliquot of DMSO-treated cells were pre-treated with 150 μM carbonyl cyanide m-cholorphenyl hydrazone (CCCP) for 20 minutes. After staining, cells were washed and fluorescence measured by flow cytometry.
Growth-curve analyses
Cells were plated at 2×104 cells/5-cm plate and incubated with DMSO or p110α inhibitors for 1-5 days. Cells were counted each day, in triplicate, using Trypan Blue vital dye.
Gene silencing with siRNAs
Cultures of mESCs were transfected with doses of Pik3ca-, Pik3cb-, Pik3cd- or Nanog-targeting, or non-targeting, siRNAs (Dharmacon) as indicated, using Lipofectamine 2000 transfection reagent (Invitrogen). After 48 hours, cells were re-transfected and incubated for a further 24 hours. Transfected cells were plated into a self-renewal assay, or harvested for protein or RNA expression analysis. Cultures of mESCs transfected with 10 nM Pik3cb-targeting or non-targeting negative siRNA (Ambion, Applied Biosystems) were plated into self-renewal assays after 48 hours, harvested for protein or RNA expression analyses at the same time or re-plated for a further 4 days before protein and RNA were harvested.
Gene-expression analyses
RNA was isolated using TRIzol (Invitrogen) according to the manufacturer's instructions. DNase treatment, reverse transcription and quantitative RT-PCR (qRT-PCR) were conducted as described previously (Storm et al., 2007). Briefly, qRT-PCR was conducted using LightCycler FastStart DNA Master SYBR Green (Roche Applied Science, East Sussex, UK) according to the manufacturer's instructions. The primers used in this study were: Nanog Forward 5′-CTCTTCAAGGCAGCCCTGAT-3′; Nanog Reverse 5′-CCATTGCTAGTCTTCAACCAC-3′; Rex1 Forward 5′-CGTGTAACATACACCATCCG-3′; Rex1 Reverse 5′-GAAATCCTCTTCCAGAATGG-3′; p110α (Pik3ca) Forward 5′-AAATGGCGACGACTTACG-3′; p110α (Pik3ca) Reverse 5′-TTGTTCTTGTCCTTGAGC-3′; p110β (Pik3cb) Forward 5′-TAATGTGTCAAGTCGTGG-3′; p110β (Pik3cb) Reverse 5′-CAGCCTACAGCGTATTCC-3′; p110δ (Pik3cd) Forward 5′-CTGGACCTGAGGATGACG-3′; p110δ (Pik3cd) Reverse 5′-GGCTCAAGTCCAAGAACC-3′; β-Actin Forward 5′-TAGGCACCAGGGTGTGATGG-3′; β-Actin Reverse 5′-CATGGCTGGGGTGTTGAAGG-3′.
Representative experiments, showing expression of quadruplicate samples normalised to β-actin, are presented with standard deviations. Expression following targeted siRNA transfection was compared with expression following transfection with non-targeting siRNA controls and statistical significance assessed using a Student's t-test, where *P<0.05 and **P<0.01.
Supplementary Material
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/122/13/2311/DC1
This work was supported by an MRC Capacity-Building studentship and a BBSRC Research Development Fellowship to M.W. The authors are grateful to Tom Crabbe (UCB) and Peter Shepherd (Auckland, New Zealand) for the gifts of isoform-selective PI3K inhibitors. Deposited in PMC for release after 6 months.
References
- Aksoy, I., Sakabedoyan, C., Bourillot, P. Y., Malashicheva, A. B., Mancip, J., Knoblauch, K., Afanassieff, M. and Savatier, P. (2007). Self-renewal of murine embryonic stem cells is supported by the serine/threonine kinases pim-1 and pim-3. Stem Cells 25, 2996-3004. [DOI] [PubMed] [Google Scholar]
- Armstrong, L., Hughes, O., Yung, S., Hyslop, L., Stewart, R., Wappler, I., Peters, H., Walter, T., Stojkovic, P., Evans, J. et al. (2006). The role of PI3K/AKT, MAPK/ERK and NFkappabeta signalling in the maintenance of human embryonic stem cell pluripotency and viability highlighted by transcriptional profiling and functional analysis. Hum. Mol. Gen. 15, 1894-1913. [DOI] [PubMed] [Google Scholar]
- Bi, L., Okabe, I., Bernard, D. J., Wynshaw-Boris, A. and Nussbaum, R. L. (1999). Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3-kinase. J. Biol. Chem. 274, 10963-10968. [DOI] [PubMed] [Google Scholar]
- Bi, L., Okabe, I., Bernard, D. J. and Nussbaum, R. L. (2002). Early embryonic lethality in mice deficient in the p110beta catalytic subunit of PI 3-kinase. Mamm. Genome 13, 169-172. [DOI] [PubMed] [Google Scholar]
- Boeuf, H., Hauss, C., DeGraeve, F., Baran, N. and Kedinger, C. (1997). Leukemia inhibitory factor-dependent transcriptional activation in embryonic stem cells. J. Cell Biol. 138, 1207-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiani, M. and Schoeler, H. R. (2005). Regulatory networks in embryo-derived pluripotent stem cells. Nat. Rev. Mol. Cell. Biol. 6, 872-881. [DOI] [PubMed] [Google Scholar]
- Bone, H. K., Damiano, T., Bartlett, S., Perry, A., Letchford, J., Sanchez Ripoll, Y., Nelson, A. S. and Welham, M. J. (2008). Involvement of glycogen synthase kinase-3 in regulation of murine embryonic stem cell self-renewal revealed by a series of bisindolylmaleimides. Chem. Biol. 16, 15-27. [DOI] [PubMed] [Google Scholar]
- Bottenstein, J. E. and Sato, G. H. (1979). Growth of a rat neuroblastoma cell line in serum-free supplemented medium. Proc. Natl. Acad. Sci. USA 76, 514-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer, G. J. and Cotman, C. W. (1989). Survival and growth of hippocampal neurons in defined medium at low density: advantages of a sandwich culture technique or low oxygen. Brain Res. 494, 65-74. [DOI] [PubMed] [Google Scholar]
- Brewer, G. J., Torricelli, J. R., Evege, E. K. and Prive, P. J. (1993). Optimized survival of hippocampal neurons in B27-Supplemented neurobasal, a new serum-free medium combination. J. Neurosci. Res. 35, 567-576. [DOI] [PubMed] [Google Scholar]
- Burdon, T., Stracey, C., Chambers, I., Nichols, J. and Smith, A. (1999). Suppression of SHP-2 and ERK signalling promotes self-renewal of mouse embryonic stem cells. Dev. Biol. 210, 30-43. [DOI] [PubMed] [Google Scholar]
- Burdon, T., Smith, A. and Savatier, P. (2002). Signalling, cell cycle and pluripotency in embryonic stem cells. Trends Cell Biol. 12, 432-438. [DOI] [PubMed] [Google Scholar]
- Cantrell, D. A. (2001). Phosphoinositide 3-kinase signalling pathways. J. Cell Sci 114, 1439-1445. [DOI] [PubMed] [Google Scholar]
- Cartwright, P., McLean, C., Sheppard, A., Rivett, D., Jones, K. and Dalton, S. (2005). LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 132, 885-896. [DOI] [PubMed] [Google Scholar]
- Chambers, I., Colby, D., Robertson, M., Nichols, J., Lee, S., Tweedie, S. and Smith, A. (2003). Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell 113, 643-655. [DOI] [PubMed] [Google Scholar]
- Chambers, I., Silva, J., Colby, D., Nichols, J., Nijmeijer, B., Robertson, M., Vrana, J., Jones, K., Grotewold, L. and Smith, A. (2007). Nanog safeguards pluripotency and mediates germline development. Nature 450, 1230-1234. [DOI] [PubMed] [Google Scholar]
- Chaussade, C., Rewcastle, G. W., Kendall, J. D., Denny, W. A., Cho, K., Gronning, L. M., Chong, M. L., Anagnostou, S. H., Jackson, S. P., Daniele, N. et al. (2007). Evidence for functional redundancy of class IA PI3K isoforms in insulin signalling. Biochem. J. 404, 449-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciraolo, E., Iezzi, M., Marone, R., Marengo, S., Curcio, C., Costa, C., Azzolino, O., Gonella, C., Rubinetto, C., Wu, H. et al. (2008). Phosphoinositide 3-Kinase p110β activity: key role in metabolism and mammary gland cancer but not development. Sci. Signal. 1, 1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condliffe, A., Davidson, K., Anderson, K., Ellson, C., Crabbe, T., Okkenhaug, K., Vanhaesebroeck, B., Turner, M., Webb, L., Wymann, M. et al. (2005). Sequential activation of class IB and class IA PI3K is important for the primed respiratory burst of human but not murine neutrophils. Blood 106, 1432-1440. [DOI] [PubMed] [Google Scholar]
- Cristoforidis, S., Miaczynska, M., Ashman, K., Wilm, M., Zhao, L., Yip, S. C., Waterfield, M. D., Backer, J. M. and Zerial, M. (1999). Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat. Cell Biol. 1, 249-252. [DOI] [PubMed] [Google Scholar]
- Evans, M. J. and Kaufman, M. H. (1981). Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154-156. [DOI] [PubMed] [Google Scholar]
- Foukas, L. C., Claret, M., Pearce, W., Okkenhaug, K., Meek, S., Peskett, E., Sancho, S., Smith, A. J. H., Withers, D. J. and Vanhaesebroeck, B. (2006). Critical role for the p110 alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature 441, 366-370. [DOI] [PubMed] [Google Scholar]
- Guillermet-Guibert, J., Bjorklof, K., Salpekar, A., Gonella, C., Ramadani, F., Bilancio, A., Meek, S., Smith, A. J. H., Okkenhaug, K. and Vanhaesebroeck, B. (2008). The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc. Natl. Acad. Sci. USA 105, 8292-8297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa, M., Kaizawa, H., Moritomo, H., Koizumi, T., Ohishi, T., Okada, M., Ohta, M., Tsukamoto, S. i., Parker, P., Workman, P. et al. (2006). Synthesis and biological evaluation of 4-morpholino-2-phenylquinazolines and related derivatives as novel PI3 kinase p110[alpha] inhibitors. Bioorg. Med. Chem. 14, 6847-6858. [DOI] [PubMed] [Google Scholar]
- Hennessy, B., Smith, D., Ram, P., Lu, Y. and Mills, G. (2005). Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 4, 988-1004. [DOI] [PubMed] [Google Scholar]
- Hooshmand-Rad, R., Hajkova, L., Klint, P., Karlsson, R., Vanhaesebroeck, B., Claesson-Welsh, L. and Heldin, C. H. (2000). The PI 3-kinase isoforms p110 alpha and p110 beta have differential roles in PDGF- and insulin-mediated signaling. J. Cell Sci. 113, 207-214. [DOI] [PubMed] [Google Scholar]
- Ivanova, N., Dobrin, R., Lu, R., Kotenko, I., Lervorse, J., DeCoste, C., Schafer, X., Lun, Y. and Lemischka, I. R. (2006). Dissecting self-renewal in stem cells with RNA interference. Nature 442, 533-538. [DOI] [PubMed] [Google Scholar]
- Jackson, S., Schoenwaelder, S., Goncalves, I., Nesbitt, W., Yap, C., Wright, C., Kenche, V., Anderson, K., Dopheide, S., Yuan, Y. et al. (2005). PI 3-kinase p110beta a new target for antithrombotic therapy. Nat. Med. 11, 507-514. [DOI] [PubMed] [Google Scholar]
- Jirmanova, L., Afanassieff, M., Gobert-Gosse, S., Markossian, S. and Savatier, P. (2002). Differential contributions of ERK and PI3-kinase to the regulation of cyclin D1 expression and to the control of the G1/S transition in mouse embryonic stem cells. Oncogene 21, 5515-5528. [DOI] [PubMed] [Google Scholar]
- Kang, S., Denley, A., Vanhaesebroeck, B. and Vogt, P. K. (2006). Oncogenic transformation induced by the p110 beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc. Natl. Acad. Sci. USA 103, 1289-1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight, Z. A., Gonzalez, B., Feldman, M. E., Zunder, E. R., Goldenberg, D. D., Williams, O., Loewith, R., Stokoe, D., Balla, A. and Toth, B. (2006). A pharmacological map of the PI3-K family defines a role for p110[alpha] in insulin signaling. Cell 125, 733-747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunath, T., Saba-El-Leil, M. K., Almousailleakh, M., Wray, J., Meloche, S. and Smith, A. (2007). FGF stimulation of the Erk1/2 signalling cascade triggers transition of pluripotent embryonic stem cells from self-renewal to lineage committment. Development 134, 2895-2902. [DOI] [PubMed] [Google Scholar]
- Mitsui, K., Tokuzawa, Y., Itoh, H., Segawa, K., Murakami, M., Takahashi, K., Maruyama, M., Maeda, M. and Yamanaka, S. (2003). The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 113, 631-642. [DOI] [PubMed] [Google Scholar]
- Murakami, M., Ichisaka, T., Maeda, M., Oshiro, N., Hara, K., Edenhofer, F., Kiyama, H., Yonezawa, K. and Yamanaka, S. (2004). mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol. Cell. Biol. 24, 6710-6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa, H. (2007). How is pluripotency determined and maintained? Development 134, 635-646. [DOI] [PubMed] [Google Scholar]
- Niwa, H., Burdon, T., Chambers, I. and Smith, A. (1998). Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 12, 2048-2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okkenhaug, K., Bilancio, A., Farjot, G., Priddle, H., Sancho, S., Peskett, E., Pearce, W., Meek, S. E., Salpekar, A., Waterfield, M. D. et al. (2002). Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science 297, 1031-1034. [DOI] [PubMed] [Google Scholar]
- Paling, N. R., Wheadon, H., Bone, H. K. and Welham, M. J. (2004). Regulation of embryonic stem cell self-renewal by phosphoinositide 3-kinase-dependent signaling. J. Biol. Chem. 279, 48063-48070. [DOI] [PubMed] [Google Scholar]
- Papakonstanti, E. A., Zwaenepoel, O., Bilancio, A., Burns, E., Nock, G. E., Houseman, B., Shokat, K., Ridley, A. J. and Vanhaesebroeck, B. (2008). Distinct roles of class IA PI3K isoforms in primary and immortalised macrophages J. Cell Sci. 121, 4124-4133. [DOI] [PubMed] [Google Scholar]
- Patrucco, E., Notte, A., Barberis, L., Selvetella, G., Maffei, A., Brancaccio, M., Marengo, S., Russo, G., Azzolino, O., Rybalkin, S. D. et al. (2004). PI3Kgamma modulates the cardiac response to chronic pressure overlaod by distinct kinase-dependent and -independent effects. Cell 118, 375-387. [DOI] [PubMed] [Google Scholar]
- Pebay, A., Wong, R. C., Pitson, S. M., Wolvetang, E. J., Peh, G. S., Filipczyk, A., Koh, K. L., Tellis, I., Nguyen, L. T. and Pera, M. F. (2005). Essential roles of sphongosine-1-phosphate and platelet-derived growth factor in the maintenance of human embryonic stem cells. Stem Cells 23, 1541-1548. [DOI] [PubMed] [Google Scholar]
- Pritsker, M., Ford, N., Jenq, H. and Lemischka, I. (2006). Genomewide gain-of-function genetic screen identifies functionally active genes in mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA 103, 6946-6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle, A., Lock, L. and Donovan, P. (2006). Neurotrophins mediate human embryonic stem cell survival. Nat. Biotech. 24, 344-350. [DOI] [PubMed] [Google Scholar]
- Riley, J. K., Carayannopoulos, M. O., Wyman, A. H., Chi, M., Ratajczak, C. K. and Moley, K. H. (2005). The PI3K/Akt pathway is present and functional in the preimplantation mouse embryo. Dev. Biol. 284, 377-386. [DOI] [PubMed] [Google Scholar]
- Riley, J. K., Carayannopoulos, M. O., Wyman, A. H., Chi, M. and Moley, K. H. (2006). Phosphatidylinositol 3-Kinase activity is critical for glucose metabolism and embryo survival in murine blastocysts. J. Biol. Chem. 281, 6010-6019. [DOI] [PubMed] [Google Scholar]
- Roche, S., Koegl, M. and Courtneidge, S. A. (1994). The phosphatidylinositol 3-kinase-Alpha is required for DNA-synthesis induced by some, but not all, growth-factors. Proc. Natl. Acad. Sci. USA 91, 9185-9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche, S., Downward, J., Raynal, P. and Courtneidge, S. A. (1998). A function for phosphatidylinositol 3-kinase beta (p85 alpha-p110 beta) in fibroblasts during mitogenesis: Requirement for insulin- and lysophosphatidic acid-mediated signal transduction. Mol. Cell. Biol. 18, 7119-7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommel, C., Clarke, B. A., Zimermann, S., Nunez, L., Rossman, R., Reid, K., Moelling, K., Yancopoulos, G. D. and Glass, D. J. (1999). Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science 286, 1738-1741. [DOI] [PubMed] [Google Scholar]
- Rottenberg, H. and Wu, S. L. (1998). Quantitative assay by flow cytometry of the mitochondrial membrane potential in intact cells. BBA-Mol. Cell Res. 1404, 393-404. [DOI] [PubMed] [Google Scholar]
- Sadhu, C., Masinovsky, B., Dick, K., Sowell, C. G. and Staunton, D. E. (2003). Essential role of phosphoinositide 3-kinase {delta} in Neutrophil directional movement. J. Immunol. 170, 2647-2654. [DOI] [PubMed] [Google Scholar]
- Sato, N., Meijer, L., Skaltsounis, L., Greengard, P. and Brivanlou, A. H. (2004). Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat. Med. 10, 55-63. [DOI] [PubMed] [Google Scholar]
- Smith, A. G. (2001). Embryo-derived stem cells: of mice and men. Ann. Rev. Cell Dev. Biol. 17, 435-462. [DOI] [PubMed] [Google Scholar]
- Stavridis, M., Lunn, J. S., Collins, B. J. and Storey, K. G. (2007). A discrete period of FGF-induced Erk1/2 signalling is required fro vertebrate neural specification. Development 134, 2889-2894. [DOI] [PubMed] [Google Scholar]
- Storm, M., Bone, H. K., Beck, C. G., Bourillot, P. Y., Schreiber, V., Damiano, T., Nelson, A., Savatier, P. and Welham, M. J. (2007). Regulation of Nanog expression by phosphoinositide 3-kinase -dependent signalling in murine embryonic stem cells. J. Biol. Chem. 282, 6265-6273. [DOI] [PubMed] [Google Scholar]
- Sun, H., Lesche, R., Li, D. M., Liliental, J., Zhang, H., Gao, J., Gavrilova, N., Mueller, B., Liu, X. and Wu, H. (1999). PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5-trisphosphate and Akt/protein kinase B signaling pathway. Proc. Natl. Acad. Sci. USA 96, 6199-6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, K., Mitsui, K. and Yamanaka, S. (2003). Role of ERas in promoting tumour-like properties in mouse embryonic stem cells. Nature 423, 541-545. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck, B. and Waterfield, M. D. (1999). Signaling by distinct classes of phosphoinositide 3-kinases. Exp. Cell Res. 253, 239-254. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck, B., Welham, M. J., Kotani, K., Stein, R., Warne, P. H., Zvelebil, M. J., Higashi, K., Volinia, S., Downward, J. and Waterfield, M. D. (1997). P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc. Natl. Acad. Sci. USA 94, 4330-4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck, B., Jones, G. E., Allen, W. E., Zicha, D., Hooshmand-Rad, R., Sawyer, C., Wells, C., Waterfield, M. D. and Ridley, A. J. (1999). Distinct PI(3)Ks mediate mitogenic signalling and cell migration in macrophages. Nat. Cell Biol. 1, 69-71. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck, B., Rohn, J. L. and Waterfield, M. D. (2004). Gene targeting: attention to detail. Cell 118, 274-276. [DOI] [PubMed] [Google Scholar]
- Watanabe, S., Umehara, H., Murayama, K., Okabe, M., Kimura, T. and Nakano, K. (2006). Activation of Akt signalling is sufficient to maintain pluripotency in mouse and primate embryonic stem cells. Oncogene 25, 2697-2707. [DOI] [PubMed] [Google Scholar]
- Welham, M. J., Dechert, U., Leslie, K. B., Jirik, F. and Schrader, J. W. (1994). Interleukin (IL)-3 and granulocyte/macrophage colony-stimulating factor, but not IL-4, induce tyrosine phosphorylation, activation, and association of SHPTP2 with Grb2 and phosphatidylinositol 3′-kinase. J. Biol. Chem. 269, 23764-23768. [PubMed] [Google Scholar]
- Welham, M. J., Storm, M. P., Kingham, E. and Bone, H. K. (2007). Phosphoinositide 3-kinases and regulation of embryonic stem cell fate. Biochem. Soc. Trans. 35, 225-228. [DOI] [PubMed] [Google Scholar]
- Williams, M. R., Arthur, J. S., Balendran, A., van der Kaay, J., Poli, V., Cohen, P. and Alessi, D. R. (2000). The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr. Biol. 10, 439-448. [DOI] [PubMed] [Google Scholar]
- Ying, Q. L., Stavridis, M., Griffiths, D., Li, M. and Smith, A. (2003a). Conversion of embryonic stem cell into neuroectoderma precursos in adherent monoculture. Nat. Biotech. 21, 183-186. [DOI] [PubMed] [Google Scholar]
- Ying, Q. L., Nichols, J., Chambers, I. and Smith, A. (2003b). BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell 115, 281-292. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.