

Abstract
Type 2 diabetes is characterized by hyperglycemia, a deficit in β-cells, increased β-cell apoptosis, and islet amyloid derived from islet amyloid polypeptide (IAPP). These characteristics are recapitulated in the human IAPP transgenic (HIP) rat. We developed a mathematical model to quantify β-cell turnover and applied it to nondiabetic wild type (WT) vs. HIP rats from age 2 days to 10 mo to establish 1) whether β-cell formation is derived exclusively from β-cell replication, or whether other sources of β-cells (OSB) are present, and 2) to what extent, if any, there is attempted β-cell regeneration in the HIP rat and if this is through β-cell replication or OSB. We conclude that formation and maintenance of adult β-cells depends largely (∼80%) on formation of β-cells independent from β-cell duplication. Moreover, this source adaptively increases in the HIP rat, implying attempted β-cell regeneration that substantially slows loss of β-cell mass.
Keywords: human islet amyloid polypeptide, diabetes mellitus, mathematical model
glucose concentrations are regulated primarily by insulin secreted from pancreatic β-cells (34). In type 1 and 2 diabetes, there is a near-complete and partial loss, respectively, of β-cells (34). In type 1 diabetes the mechanism of loss is autoimmune-mediated apoptosis (2), whereas in type 2 diabetes several mechanisms have been proposed, including lipotoxicity (30), glucotoxicity (41), and endoplasmic reticulum (ER) stress secondary to misfolding and aggregation of islet amyloid polypeptide, a protein coexpressed and secreted with insulin by pancreatic β-cells (22). As a consequence of the progressive β-cell loss and dysfunction underlying both type 1 and 2 diabetes, insulin daily demand eventually exceeds insulin secretion, leading to diabetes (34). Since the underlying defect in both type 1 and 2 diabetes is β-cell failure, there is increasing interest in the capacity, if any, for β-cell regeneration (14).
Tissue regeneration typically harnesses mechanisms that contribute to cell turnover in health. Thus tissues with relatively high turnover in health (for example, gut epithelium) when subject to increased loss [for example, inflammation in celiac disease (4)] have an excellent capacity to overcome the loss from an underlying stem cell pool, provided that the mechanism provoking the underlying loss is overcome [for example, a gluten-free diet in celiac disease (4)].
β-Cells expand in number after birth in both rodents and humans at least in part through the mechanism of duplication of existing β-cells, i.e., β-cell replication (17, 26, 33, 45). The frequency of β-cell replication declines in adults through epigenetic modification of aging β-cells (12, 26, 32, 33, 47). Whereas there is an impressive capacity to restore β-cell mass in juvenile rodents [following a single dose of streptozotocin (27) or partial pancreatectomy (6, 9)], this capacity is diminished in adult rodents (39, 47).
The extent to which β-cell turnover continues in adult humans or rodents and the extent to which there are potentially new sources of β-cells other than through β-cell replication is controversial (7, 8, 10, 17, 23, 45). In the present study, we first sought to establish a model to quantify β-cell turnover in rats of varying ages. Second, we sought to apply this model to wild-type (WT) Sprague-Dawley rats through their normal life span to address the following questions. 1) Is there ongoing β-cell turnover in adult rats, and 2) what proportion of these cells is derived from duplication of existing β-cells vs. derived from other sources of β-cells?
Moreover, we sought to quantify β-cell turnover in a rodent model of type 2 diabetes, the human islet amyloid polypeptide (IAPP) transgenic (HIP) rat. The HIP rat is transgenic for human IAPP on a Sprague-Dawley background. As described previously, this rat model of type 2 diabetes has ER-stress induced β-cell apoptosis that leads to a progressive deficit in β-cell mass and develops both an anatomical as well as a functional islet phenotype comparable with that in humans with type 2 diabetes mellitus (T2DM) (12, 22, 32).
MATERIALS AND METHODS
Animals.
Most pancreata studied here were obtained from a previously published study that reported the metabolic changes in the HIP rat vs. WT controls from 2 to 10 mo of age (32). With the approval of the University of California Los Angeles (UCLA) Institutional Animal Care and Use Committee, experiments were carried out in 20 WT and 18 HIP rats at 0.07 (i.e., 2 days), 2, 5, and 10 mo of age (3–5 WT and HIP rats/type and age group). The generation of the HIP rats has previously been described in detail (12). Rats were bred and housed at the University of California Los Angeles animal housing facility. Rats were housed in pairs (aged 2–5 mo) or in individual cages (aged 5–10 mo), fed Rodent Diet 8604 (50% carbohydrate, 24% protein, and 4% fat; Harlan Teklad, Madison, WI) ad libitum, and subjected to the standard 12:12-h light-dark cycle.
Morphological techniques.
The complete pancreas was rapidly resected from euthanized rats, all fat and nonpancreatic tissue were trimmed, and the pancreas was weighed. A longitudinal section of the pancreas (tail through head in the flat plane of the pancreas) was fixed in formaldehyde and then embedded in paraffin. Sections of pancreas were then taken through the fixed tissue in the plane of embedding so that a section of pancreas (head, body, and tail) through its maximal width was obtained with each section. These sections were stained for hematoxylin-eosin and insulin, as described before (15, 24). Sections were double immunostained for the marker of replication Ki67 (rat anti-murine Ki67 monoclonal antibody TEC-3, 1/45; Dako, Carpinteria, CA) and insulin (guinea pig anti-insulin, 1:100; Zymed, Carlsbad, CA) and for the the terminal deoxynucleotidyl (TdT)-mediated dUTP nick-end labeling (TUNEL) method for apoptosis using the TdT-Frag El Kit from Oncogene Research Products (Cambridge, MA) and insulin (guinea pig anti-insulin, 1:100; Zymed, Carlsbad, CA), as described previously (13). We selected Ki67 and TUNEL to quantify replication and apoptosis, since we have previously developed conversion factors (frequency to rate) for these methods (43). If other methods were to be selected for use with the present model, it would be necessary to establish conversion factors for those methods. Other methods commonly used to detect replication include 5-bromo-2′-deoxyuridine (BrdU) incorporation into β-cells and PCNA staining. The advantages of Ki67 over these have been addressed elsewhere (14). In brief, BrdU and PCNA detect both DNA repair and replication and are therefore prone to overestimate replication. Alternative approaches to quantify apoptosis include detection of cleaved caspase-3 and annexin V. We selected TUNEL because in our hands the available antibodies for TUNEL have shown the most reproducibility, with those for caspase-3 being the least reproducible.
The β-cell mass for each rat was measured by first obtaining the fraction of the cross-sectional area of pancreatic tissue (exocrine and endocrine) positive for insulin staining and then multiplying this by the pancreatic weight. All β-cells per pancreatic section (2 sections/animal) were examined in detail at ×400 magnification (×40 objective, ×10 ocular) for the frequency of β-cell replication as fractional Ki67 and insulin-positive cells. The frequency of β-cell apoptosis for each rat was similarly computed by examination of the TUNEL-positive β-cells. These analyses were meanly performed in a mean of 70 islets/rat.
To measure the mean radius of β-cells, insulin-stained sections of pancreas (peroxidase) counterstained with hematoxylin-eosin were used. The sections were scanned by ScanScope XT System (Aperio, Vista, CA) at ×20 magnification, and 10 islets/case were selected. Each of these islets was then examined with ImageScope software (Aperio) to identify 10 representative distances between the centers of adjacent β-cell nuclei. Selection criteria included clear presence of the nucleus within a β-cell, the ability to clearly visualize nuclear boundaries, circular shape (similar dimensions in all directions), and the appearance to the observer that the nucleus had been sectioned through its maximum diameter. Having satisfied all of these requirements, measurement of 100 distances was taken in each rat and the mean volume of an individual β-cell (μm3) was calculated, considering it reasonable to approximate the irregular polyhedral shape of a cell to a sphere (48). Then, on the assumption that in aqueous organs, such as pancreas, 1 g of weight equals 1 cm3 of volume, in each rat the total number of β-cells was established as the ratio between β-cell mass (converted in μm3) and β-cell volume.
Model design.
The model of β-cell turnover illustrated in Fig. 1 describes β-cell mass as the balance between β-cell formation and loss. β-cells are added either by replication of existing β-cells or by other sources of β-cells. β-cells are lost through β-cell apoptosis. The model equation is thus
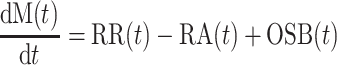 |
(1) |
where dM(t)/dt (mg/mo) is the mass derivative, i.e., rate of change in β-cell mass, M (mg); RR (mg/mo) and RA (mg/mo) are, respectively, rates of β-cell replication and apoptosis derived from frequencies of β-cell replication and apoptosis [fraction of replicated (apoptotic) β-cells], each multiplied by the respective conversion factors estimated in Ref. 43 and by β-cell mass; and OSB (mg/mo) indicates the contribution of other sources of β-cells, i.e., from sources different from β-cell replication, to β-cell mass.
Fig. 1.

Model of β-cell turnover. The β-cell mass depends on the balance of β-cell formation either by replication of existing β-cells (left tap) or, eventually, by other sources (right tap) and β-cell loss (black drain) through β-cell apoptosis. OSB, other sources of β-cells.
In Eq. 1, the term OSB(t) includes change in β-cell mass due to both the change in number of β-cells and changes in the mean β-cell size. Since our primary interest is the turnover of β-cells, we further refined Eq. 1 to focus on turnover of β-cell number, as in the following equation:
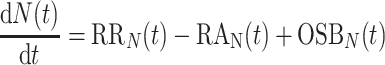 |
(2) |
where dN(t)/dt (106/mo) is the rate of change in β-cell number, N (106); RRN (106/mo) and RAN (106/mo) are, respectively, rates of β-cell replication and apoptosis referred to N and derived from frequencies of β-cell replication and apoptosis [fraction of replicated (apoptotic) β-cells], each multiplied by the respective conversion factors estimated in Ref. 43 and by β-cell number; and OSBN (106/mo) indicates the contribution of other sources to changes in β-cell number.
Quantification of β-cell turnover by Eqs. 1 and 2 requires the following assumptions. First, we assume that β-cell turnover is homogeneously distributed; this implies that duration of β-cell replication (apoptosis) is the same among individual β-cells and that all β-cells have the same probability to replicate (11, 45) or die. Second, we assume that changes in β-cell volume are homogeneous. Third, we assume that the conversion factors for β-cell replication (αR) and apoptosis (αA) are applicable across the population of β-cells and are not changed by age or development of diabetes. Macrophage function declines with hyperglycemia in diabetes (31), which may reduce the clearance rate of apoptotic cells and theoretically alter the conversion factor from the frequency to the rate of β-cell apoptosis. However, by design we only count cells as β-cells undergoing apoptosis if the cell is positive for TUNEL, the cytoplasm is positive for insulin, and the cell is intact. Macrophage clearance removes cellular debris (apoptotic bodies) that is not included in our evaluation of β-cell apoptosis. Fourth, since it is impossible to obtain data on β-cell turnover from an individual animal at multiple time points, we have to assume that the behavior of a group of individuals examined at a variety of time points can be used to predict β-cell turnover over time in the population from which those individuals were sampled (i.e., Sprague-Dawley rats or HIP rats).
Data analysis.
By integrating Eq. 1 from 0.07 to 10 mo, the overall OSB over the same period can be expressed as:
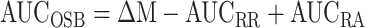 |
(3) |
where AUCOSB, AUCRR, and AUCRA (mg) are the areas under the OSB, RR, and RA curves, respectively, and ΔM (mg) is the change in β-cell mass from 0.07 to 10 mo of age. Eq. 3 was applied to mean data of WT and HIP rats with AUCRR and AUCRA calculated by the trapezoid method to estimate the overall contribution of OSB to β-cell mass balance.
The time course of this contribution can also be determined from Eq. 1, rewritten as Eq. 1′:
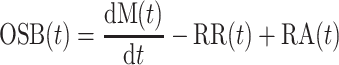 |
(1′) |
However, this equation requires that the rate of change in β-cell mass is calculated. For this purpose, a new algorithm based on a stochastic regularization method (16a) was applied. Traditional methods evaluate the derivative using a two-step process, i.e., the data are first smoothed, followed by numerical estimation of the derivative. In contrast, the new algorithm, by simultaneously performing both data regularization and calculation of the derivative, provides an estimate of the derivative on a uniform, arbitrarily defined grid. To obtain OSB on the same fine grid, smoothed mean profiles for RR and RA were evaluated using the same algorithm.
To express β-cell mass and turnover in terms of N vs. M, we divided M by the average individual β-cell volume. To derive the overall contribution of OSBN to the balance of β-cell number as well as its time course, we performed an analysis similar to that undertaken to calculate the contribution of OSB toward turnover and the time course of β-cell mass (that is, Eqs. 3 and 1′).
Statistical analysis.
All data are represented as means ± SE. The deconvolution method was implemented by MATLAB software (The Mathworks, Natick, MA). Student's t-test (P < 0.05) was performed to compare WT and HIP rats and to verify the difference between OSB and zero.
Supplemental data.
The Supplemental data include three supplemental figures and one supplemental table (Supplemental Material for this article is available at the AJP-Endocrinology and Metabolism web site).
RESULTS
Model of β-cell turnover.
Ideally, to establish a model for β-cell turnover, β-cell mass and the rates of β-cell formation and loss would be measured in the same individual at several time points. However, it is currently not possible to measure β-cell mass, β-cell formation, or β-cell loss in vivo. Therefore, the best approximation to this ideal currently plausible is to obtain pancreas from representative individuals in a relatively homogenous population at different time points. On this basis, we selected the inbred Spague-Dawley rat and Sprague-Dawley rat transgenic for human IAPP (the HIP rat) for these studies. Pancreas was obtained at several time points, 2 days (0.07 mo) after birth and then at ages 2, 5, and 10 mo. The β-cell mass and frequency of β-cell apoptosis in the 2-, 5-, and 10-mo-of-age rats were included in a prior report (32). No data from the 0.07-mo rats and no β-cell replication data from any of the rats were included in that report. In the present report, we employed newly developed conversion factors (43) to quantify the rate of β-cell apoptosis and rate of β-cell replication from the respective frequency of apoptosis and frequency of replication to establish a new model to quantify β-cell turnover.
The model parameters are shown in Fig. 1. The model is based on the assumption that the net balance of β-cells (β-cell mass or number) at any given time is a function of the rate of β-cell formation and β-cell loss. A second assumption is that β-cell formation can be considered as the sum of β-cell formation from the duplication of existing β-cells and the formation of β-cells from all other sources of β-cells. A third assumption is that in vivo β-cell loss is through β-cell apoptosis. Theoretically, β-cells may be lost through β-cell apoptosis and/or necrosis. Because necrosis is relatively rare compared with apoptosis under physiological conditions (18), we use β-cell apoptosis as a synonym of β-cell death where not specified otherwise.
Since all model parameters in Fig. 1 can be quantified with the exception of the formation of β-cells from sources other than duplication from existing β-cells, we could solve for this unknown. It is important to stress that the term “other sources of β-cells” (OSB) is a mathematical term, not a term that identifies the source (or sources) of these cells.
Impaired fasting glucose by 5 mo of age and diabetes by 10 mo of age in HIP rats.
The blood glucose values in the 2-, 5-, and 10-mo-old rats studied here have been reported previously (32). Blood glucose was comparable in 2-day- and 2-mo-old HIP and WT rats (102 ± 2 vs. 109 ± 3 mg/dl in WT vs. HIP rats). Whereas fasting blood glucose concentrations remained unchanged in WT rats at 5 and 10 mo of age (103 ± 2 and 100 ± 2 mg/dl, respectively), by 5 mo of age HIP rats had impaired fasting glucose (128 ± 6 mg/dl), and by 10 mo of age they had diabetes (185 ± 25 mg/dl).
Decreased β-cell mass and increased β-cell apoptosis by 2 mo of age in HIP rats.
The β-cell mass and the frequency of β-cell replication and apoptosis are shown in Fig. 2. β-cell mass in WT rats (Fig. 2A, left) increased from 0.9 ± 0.2 mg at 0.07 mo to 14.9 ± 4.0 mg by 2 mo. Thereafter, it continued to increase: 19.2 ± 3.5 mg at 5 mo and 34.0 ± 6.9 mg by 10 mo. In HIP rats (Fig. 2A, right), β-cell mass increased similarly to WT rats until 2 mo (1.9 ± 0.6 mg at 0.07 mo, 13.6 ± 3.6 mg at 2 mo), but thereafter it decreased compared with that in WT rats: 9.9 ± 0.8 mg (P < 0.04) at 5 mo and to 8.3 ± 1.5 mg by 10 mo (P < 0.01).
Fig. 2.

β-Cell mass and the frequency of β-cell replication and apoptosis. The mean β-cell mass (A), frequencies of β-cell replication (B), and β-cell apoptosis (C) in 20 wild-type (WT; A–C, left) and 18 human islet amyloid polypeptide transgenic rats (HIP; A–C, right) at 0.07, 2, 5, and 10 mo of age. *HIP vs. WT, P < 0.05. Bars represent SE.
The frequency of β-cell replication in WT rats (Fig. 2B, left) declined from a highest value at 0.07 mo of age (1.98 ± 0.41%) to 0.25 ± 0.04% at 2 mo, 0.24 ± 0.05% at 5 mo, and 0.13 ± 0.07% by 10 mo. The pattern was similar in HIP rats (Fig. 2B, right): 2.0 ± 0.9% at 0.07 mo, 0.2 ± 0.1% at 2 mo, 0.5 ± 0.1% at 5 mo, and 0.2 ± 0.0% at 10 mo.
The frequency of β-cell apoptosis in WT rats (Fig. 2C, left) was 0.21 ± 0.08% at 0.07 mo of age, decreasing to 0.03 ± 0.02% by 2 mo and thereafter remaining relatively low (0.07 ± 0.02% at 5 mo and 0.07 ± 0.03% by 10 mo). The frequency of β-cell apoptosis was already increased in HIP vs. WT rats by 0.07 mo (0.40 ± 0.20% vs. at 0.07 mo) and further increased by 2, 5, and 10 mo (0.18 ± 0.04, 0.36 ± 0.07, and 0.37 ± 0.10% at 2, 5, and 10 mo, respectively, all P < 0.01 vs. WT) (Fig. 2C, right).
Total β-cell number showed a pattern similar to β-cell mass.
The mean individual β-cell volume and the total β-cell number are shown in Fig. 3. The mean individual β-cell volume in WT rats (Fig. 3A, left) increased from 247 ± 19 μm3 at 0.07 mo to 448 ± 18 μm3 by 2 mo. Thereafter, it remained almost constant: 416 ± 4 μm3 at 5 mo and 443 ± 20 μm3 by 10 mo. In HIP rats (Fig. 3A, right), the mean volume of a β-cell increased similarly to that in WT rats until 2 mo (311 ± 35 μm3 at 0.07 mo and 574 ± 67 μm3 at 2 mo); it then decreased slightly to 486 ± 22 μm3 by 5 and 10 mo (460 ± 6 μm3).
Fig. 3.

Mean β-cell volume and total β-cell number. The mean individual β-cell volume (A) and β-cell number (B) in 20 WT (A and B, left) and 18 HIP rats (A and B, right) at 0.07, 2, 5, and 10 mo of age. *HIP vs. WT, P < 0.05. Bars represent SE.
The β-cell number, obtained by dividing β-cell mass by the average β-cell volume, in WT rats (Fig. 3B, left) increased from (3.5 ± 1.0) × 106 at 0.07 mo to (32.3 ± 7.6) × 106 by 2 mo, and thereafter it continued to increase: (46.2 ± 8.8) × 106 at 5 mo and (78.2 ± 13.3) × 106 by 10 mo. In HIP rats (Fig. 3B, right), the number of β-cells increased similarly to that in WT rats until 2 mo [(5.8 ± 1.4) × 106 at 0.07 mo and (24.7 ± 6.4) × 106 at 2 mo] but thereafter decreased compared with WT rats to (20.9 ± 2.8) × 106 (P < 0.03) at 5 mo and to (18.1 ± 3.0) × 106 by 10 mo (P < 0.02).
β-Cell mass turnover: a major role from other sources of β-cells.
A graph to depict overall contributions to β-cell mass turnover (Eq. 3; see materials and methods) in WT and HIP rats is shown in Fig. 4. In WT rats, to account for the net increase in β-cell mass (ΔM = 33 mg) from 0.07 to 10 mo of age, a contribution of OSB (AUCOSB = 61 mg) was required, with only a smaller contribution of β-cell formation from β-cell replication (AUCRR = 7 mg). Formation of 68 mg of β-cells total (90% from OSB and 10% from β-cell replication) was required to permit the net increase in β-cell mass in the face of 35 mg of β-cell apoptosis (AUCRA = 7 mg) during the same period. In HIP rats, the net increase in β-cell mass from age 0.07 to 10 mo (ΔM = 7 mg) was more modest. However, given the much greater loss of β-cells from apoptosis (AUCRA = 88 mg) and the relatively minor contribution of new β-cells from replication of existing β-cells (AUCRR = 6 mg, 7% of β-cell formation), we computed that the contribution of new β-cells from OSB in the HIP rat was 89 mg (AUCOSB, 93% of new β-cells formed).
Fig. 4.

β-Cell turnover: overall contributions in WT and HIP rats from birth to age 10 mo. β-Cell mass balance in WT (left) and HIP rats (right) aged 0.07–10 mo. Hatched bars represent the net change in β-cell mass (ΔM) from age 0.07 to 10 mo. Dark gray and light gray bars show, respectively, the overall contributions to β-cell mass formation from replication of existing β-cells (AUCRR) and from other sources (AUCOSB). Black bars represent the loss due to β-cell apoptosis (AUCRA).
A similar overall pattern of findings was obtained considering β-cell number instead of β-cell mass (Supplemental Fig. S1). In WT rats, the net increase in β-cell number (ΔN = 75 × 106) during the study period was mostly dependent on the contribution from OSB (AUCOSBN = 141 × 106, 89% of new cells formed), with a much smaller contribution from β-cell replication (AUCRRN = 18 × 106, 11% of new cells formed). These newly formed β-cells permitted a net expansion of β-cell number despite the concurrent loss of 84 × 106 cells through apoptosis (AUCRAN). In contrast, in HIP rats, whereas the overall variation in β-cell number (ΔN = 12 × 106) over the 10-mo observation period was less than that in WT rats, because of a much higher number of β-cells lost through apoptosis (AUCRAN = 180 × 106) and again a quite modest (7%) contribution of newly forming cells from β-cell replication (AUCRRN = 13 × 106), the contribution from OSB was 179] × 106 (AUCOSBN), or 93.0% of newly formed cells. Clearly, in the absence of OSB, HIP rats would have had a much more marked deficit in β-cells at an earlier age.
Estimation of other sources of β-cell profile.
In Fig. 5, the time profiles of rate of change in β-cell mass (dM/dt), rate of β-cell replication (RR), rate of β-cell apoptosis (RA), and the calculated OSB (Eq. 1′; see materials and methods) are shown in WT (Fig. 5, left) and HIP rats (Fig. 5, right). In WT rats, OSB plays an important role in permitting the relatively high rate of change of β-cell mass during the first month (7 mg/mo at 0.07 mo). OSB decreases in 2- to 4-mo period when the rate of expansion of β-cell mass (dM/dt) slows. Finally, OSB again increases by 10 mo of age to 9 mg/mo, counterbalancing a gradual increase in the rate of β-cell apoptosis in the older animals. In HIP rats, OSB was comparatively even more important than in WT rats. If it was not for the high rate of OSB, the mass (or number) of β-cells would have rapidly declined to nearly zero by 5 mo of age. The overall conclusions were essentially the same when β-cell number rather than β-cell mass was considered (Supplemental Fig. S2).
Fig. 5.

β-Cell turnover: time profiles. Time profiles of rate of change in β-cell mass dM/dt (dotted line), rate of β-cell replication RR (○, data; solid thin line, smoothed profile), OSB (solid thick line), and rate of β-cell apoptosis RA (⧫, data; dashed line, smoothed profile) in WT (left) and HIP rats (right) aged 0.07–10 mo.
To verify the consistency between the overall OSB over the period of the study, i.e., AUCOSB (Eq. 3), and the OSB profile obtained by Eq. 1′, we evaluated the AUC of the latter by the trapezoid method and compared it to AUCOSB. In both WT and HIP rats, the percent relative differences between the two approaches are <4%, confirming that the applied smoothing procedure does not introduce bias into the results.
DISCUSSION
Our first objective was to establish a model to permit insights into the regulation and dysregulation of β-cell turnover in vivo. We then applied this model in nondiabetic rats from birth to adult life to gain insights into β-cell turnover during growth and maintenance of β-cell mass in health. Specifically, we tested the hypothesis that β-cell formation can be accounted for by duplication of existing β-cells. This hypothesis was rejected by the modeled data, and thus we concluded that formation and maintenance of β-cell mass depends on sources of β-cells independent of duplication of existing β-cells. Moreover, we concluded that the rate of formation of β-cells from this source (or sources) increases in the setting of increased β-cell apoptosis in the HIP rat model of T2DM. The effect of the increased formation of β-cells in the HIP rat was to slow the progression of β-cell loss, implying that there is attempted β-cell regeneration in this model of T2DM. Having established that changes in β-cell mass in vivo could be explained only by a source (or sources) of newly forming β-cells in addition to those arising from duplication of existing β-cells, we introduced the term other sources of β-cells, abbreviated to OSB.
The term OSB was carefully chosen to not offer any prejudice as to the origins of these β-cells, since the experimental approach here does not seek to identify their source. It is plausible that they arise from a single source, such as differentiation from a pancreatic stem cell pool (5, 37, 50), or from several sources. Bone marrow-derived stem cells (44), transdifferentiation of pancreatic acinar cells (28) and ductal cells (7, 8), and dedifferentiation (42) and then redifferentiation of pancreatic β-cells (21) have all been proposed. Although there has been controversy as to whether there is any source of β-cells other than duplication of existing β-cells, there is accumulating evidence in favor of such a pool. For example, lineage studies reveal that pancreatic ductal cells can serve as precursors for subsequent formation of pancreatic islets or acinar cells, implying that duct cells include cells that are pluripotential (23, 49).
Before accepting the principal conclusion of the present study, that there is indeed a quantitatively significant source of new β-cells that arise from a source distinct from duplication of existing β-cells, it is important to question the limitations of the approach used. In short, are the potential errors of sufficient magnitude that they could have permitted us to falsely conclude that β-cells arise from sources other than β-cell replication? To address this, we used the approach of propagation of errors to establish the variance in the computed OSB and then taking this variance into account tested the hypothesis that OSB is different from zero. Even when taking into account the sum of the variance of individual measurements required to compute OSB, the hypothesis that OSB is different from zero was consistently proven (Fig. 6), implying that OSB is not simply a result of sample variance. A second plausible source of error that might account for the conclusion that OSB exists could be errors in the factors used to convert the frequency of β-cell replication and apoptosis to rates of replication and apoptosis. To address this we examined the range of conversion factors that would be required to permit the observed changes in β-cell mass over time if OSB was equal to zero. The required conversion factors to conclude that OSB is zero (Supplemental Fig. S3) are implausible based on the published literature (1, 3, 25, 35, 38, 40). Although these analyses suggest that OSB does indeed exist, as in most complex biological systems, it is impossible to exclude all possible sources of systematic bias. Some of the limitations in developing the model are listed in the description of model development. In brief, these include the possible confounding effects of inhomogeneity in behavior of the β-cell pool with regard to turnover. It should also be emphasized that if β-cells defined here by detectable insulin by immunohistochemistry were to dedifferentiate to the extent that insulin was no longer detectable before undergoing replication, this would be assigned to OSB by the present approach.
Fig. 6.

Uncertainty of the calculated other sources of β-cell mass. Time profile of OSB in WT (left) and HIP rats (right). Error bars are SE of OSB obtained by propogating the interrat variability. OSB was significantly different from 0 at all time points except at 10 mo of age in HIP rats, when variance was high (*OSB vs. 0, P < 0.05).
One prior approach to evaluate β-cell mass balance was reported (19, 46). Finegood et al. (19) and Topp et al. (46) estimated the rate of β-cell replication by BrdU incorporation into β-cells, whereas we employed Ki67 and insulin double staining and then applied the conversion factors developed previously by use of time-lapse video microscopy (43). There are some advantages of use of Ki67 over BrdU that have been discussed previously (14). A second difference between the new model and that of Finegood is the inclusion of β-cell apoptosis (43). This permits quantification of the contribution of β-cell formation arising from β-cell replication vs. other sources of β-cells and not just a “net balance” that included both apoptosis and OSB (called net neogenesis in Refs. 19 and 46).
The current study also examines the role of changes in individual β-cell size (volume) in involution and expansion of β-cell mass (16, 29). For this purpose we examined β-cell turnover by two models, the first considering β-cell mass (Eq. 1; see materials and methods), where OSB includes changes in β-cell mass due to both the change in number of β-cells by other sources and changes in the mean β-cell size, and the second based on β-cell number (Eq. 2; see materials and methods). The small percent relative differences between the two models shown in Supplemental Table S1 (<12%) reveal that the cell size does not much influence the estimation of β-cell turnover with β-cell mass data in the WT or HIP rats with aging.
The application of the model to evaluate β-cell turnover provided several interesting insights. First, the model implies that OSB provides an important contribution to the postnatal expansion of β-cell mass in the rat. This result differs with the conclusions of studies in mice that imply that the postnatal expansion of β-cell mass is derived exclusively by duplication of existing β-cells (11, 17, 20, 45). One possible explanation is that rats differ from mice, although this seems unlikely. An alternative explanation is that the cells that give rise to pancreatic β-cells identified as OSBs by this model are labeled by the same lineage markers as differentiated β-cells. Recent data support the concept that ductal progenitor cells have the capacity to form new β-cells both during postnatal growth and in response to injury (7, 23).
An insight that emerged by modeling β-cell turnover in the HIP rat is that the decline in β-cell mass in this model of type 2 diabetes is delayed by a compensatory increase in the rate of formation of β-cells from OSB. Indeed, it appears that OSB might be regulated and the principal mechanism for maintaining β-cell mass. In older wild-type rats the rate of OSB increased as β-cell apoptosis increased with aging, and in the HIP rat the rate of increase in OSB was impressive albeit still insufficient to maintain β-cell mass in the face of a progressive increase in β-cell apoptosis. The apparently relatively slow decline in β-cell function, and presumably mass, in humans with either type 1 or 2 diabetes might be due to ongoing β-cell regeneration that, as in the HIP rat, is eventually overcome by ongoing increased β-cell apoptosis.
In summary, we report a new modeling approach for the evaluation of changes in β-cell mass and β-cell turnover applied here first to the HIP rat model of type 2 diabetes and its wild-type counterpart, the Sprague-Dawley rat. The novel insights that emerge from these studies include affirmation that there is ongoing β-cell turnover in wild-type rats through adult life and also that the replication of existing β-cells provides only a relatively minor contribution to this turnover, the most important contribution arising from mechanisms independent of duplication of existing β-cells (termed other sources of β-cells in this model). Moreover, the rate of formation of new β-cells from these other sources of β-cells increases substantially in the face of the increased rate of β-cell apoptosis in the HIP rat model of type 2 diabetes, delaying the decline in β-cell mass in this model.
GRANTS
This study was partially supported by the National Institute of Diabetes and Digestive and Kidney Diseases (no. DK-077967), Juvenile Diabetes Research Foundation, Larry L. Hillblom Foundation, and Ministero dell'Istruzione, dell'Università e della Ricerca.
Supplementary Material
Acknowledgments
We thank Giovanni Sparacino and Chiara Dalla Man (Department of Information Engineering, University of Padua) for help with deconvolution techniques, Ryan Galasso, Heather Gerber, and Inderroop Singh (Larry Hillblom Islet Research Center, UCLA) for technical assistance, and our colleagues in the Larry Hillblom Islet Research Center at UCLA for their useful comments.
REFERENCES
- 1.Al-Rubeai M, Fussenegger M. Cell Engineering: Apoptosis. Dordrecht, The Netherlands: Kluwer Academic, 2004.
- 2.Atkinson MA ADA Outstanding Scientific Achievement Lecture 2004. Thirty years of investigating the autoimmune basis for type 1 diabetes: why can't we prevent or reverse this disease? Diabetes 54: 1253–1263, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Baker AJ, Mooney A, Hughes J, Lombardi D, Johnson RJ, Savill J. Mesangial cell apoptosis: the major mechanism for resolution of glomerular hypercellularity in experimental mesangial proliferative nephritis. J Clin Invest 94: 2105–2116, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barker JM, Liu E. Celiac disease: pathophysiology, clinical manifestations, and associated autoimmune conditions. Adv Pediatr 55: 349–365, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernard C, Berthault MF, Saulnier C, Ktorza A. Neogenesis vs. apoptosis As main components of pancreatic β cell mass changes in glucose-infused normal and mildly diabetic adult rats. FASEB J 13: 1195–1205, 1999. [DOI] [PubMed] [Google Scholar]
- 6.Bonner-Weir S Regulation of pancreatic beta-cell mass in vivo. Recent Prog Horm Res 49: 91–104, 1994. [DOI] [PubMed] [Google Scholar]
- 7.Bonner-Weir S, Inada A, Yatoh S, Li WC, Aye T, Toschi E, Sharma A. Transdifferentiation of pancreatic ductal cells to endocrine beta-cells. Biochem Soc Trans 36: 353–356, 2008. [DOI] [PubMed] [Google Scholar]
- 8.Bonner-Weir S, Taneja M, Weir GC, Tatarkiewicz K, Song KH, Sharma A, O'Neil JJ. In vitro cultivation of human islets from expanded ductal tissue. Proc Natl Acad Sci USA 97: 7999–8004, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonner-Weir S, Trent DF, Weir GC. Partial pancreatectomy in the rat and subsequent defect in glucose-induced insulin release. J Clin Invest 71: 1544–1553, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bouwens L, Pipeleers DG. Extra-insular beta cells associated with ductules are frequent in adult human pancreas. Diabetologia 41: 629–633, 1998. [DOI] [PubMed] [Google Scholar]
- 11.Brennand K, Huangfu D, Melton D. All beta cells contribute equally to islet growth and maintenance. PLoS Biol 5: e163, 2007. [DOI] [PMC free article] [PubMed]
- 12.Butler AE, Jang J, Gurlo T, Carty MD, Soeller WC, Butler PC. Diabetes due to a progressive defect in beta-cell mass in rats transgenic for human islet amyloid polypeptide (HIP Rat): a new model for type 2 diabetes. Diabetes 53: 1509–1516, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52: 102–110, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Butler PC, Meier JJ, Butler AE, Bhushan A. The replication of beta cells in normal physiology, in disease and for therapy. Nat Clin Pract Endocrinol Metab 3: 758–768, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Couce M, Kane LA, O'Brien TD, Charlesworth J, Soeller W, McNeish J, Kreutter D, Roche P, Butler PC. Treatment with growth hormone and dexamethasone in mice transgenic for human islet amyloid polypeptide causes islet amyloidosis and beta-cell dysfunction. Diabetes 45: 1094–1101, 1996. [DOI] [PubMed] [Google Scholar]
- 16.Del Prato S, Wishner WJ, Gromada J, Schluchter BJ. Beta-cell mass plasticity in type 2 diabetes. Diabetes Obes Metab 6: 319–331, 2004. [DOI] [PubMed] [Google Scholar]
- 16a.De Nicolao G, Sparacino G, Cobelli C. Nonparametric input estimation in physiological systems: problems, methods, and case studies. Automatica 33: 851–870, 1997. [Google Scholar]
- 17.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429: 41–46, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol 16: 663–669, 2004. [DOI] [PubMed] [Google Scholar]
- 19.Finegood DT, McArthur MD, Kojwang D, Thomas MJ, Topp BG, Leonard T, Buckingham RE. Beta-cell mass dynamics in Zucker diabetic fatty rats. Rosiglitazone prevents the rise in net cell death. Diabetes 50: 1021–1029, 2001. [DOI] [PubMed] [Google Scholar]
- 20.Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest 114: 963–968, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanley S, Rosenberg L. Islet-derived progenitors as a source of in vitro islet regeneration. Methods Mol Biol 482: 371–385, 2009. [DOI] [PubMed] [Google Scholar]
- 22.Huang CJ, Lin CY, Haataja L, Gurlo T, Butler AE, Rizza RA, Butler PC. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 56: 2016–2027, 2007. [DOI] [PubMed] [Google Scholar]
- 23.Inada A, Nienaber C, Katsuta H, Fujitani Y, Levine J, Morita R, Sharma A, Bonner-Weir S. Carbonic anhydrase II-positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc Natl Acad Sci USA 105: 19915–19919, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janson J, Soeller WC, Roche PC, Nelson RT, Torchia AJ, Kreutter DK, Butler PC. Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proc Natl Acad Sci USA 93: 7283–7288, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalashnik L, Bridgeman CJ, King AR, Francis SE, Mikhalovsky S, Wallis C, Denyer SP, Crossman D, Faragher RG. A cell kinetic analysis of human umbilical vein endothelial cells. Mech Ageing Dev 120: 23–32, 2000. [DOI] [PubMed] [Google Scholar]
- 26.Kassem SA, Ariel I, Thornton PS, Scheimberg I, Glaser B. Beta-cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes 49: 1325–1333, 2000. [DOI] [PubMed] [Google Scholar]
- 27.Kim BM, Kim SY, Lee S, Shin YJ, Min BH, Bendayan M, Park IS. Clusterin induces differentiation of pancreatic duct cells into insulin-secreting cells. Diabetologia 49: 311–320, 2006. [DOI] [PubMed] [Google Scholar]
- 28.Lipsett M, Finegood DT. Beta-cell neogenesis during prolonged hyperglycemia in rats. Diabetes 51: 1834–1841, 2002. [DOI] [PubMed] [Google Scholar]
- 29.Lupi R, Del Prato S. Beta-cell apoptosis in type 2 diabetes: quantitative and functional consequences. Diabetes Metab 34, Suppl 2: S56–S64, 2008. [DOI] [PubMed] [Google Scholar]
- 30.Lupi R, Dotta F, Marselli L, Del Guerra S, Masini M, Santangelo C, Patane G, Boggi U, Piro S, Anello M, Bergamini E, Mosca F, Di Mario U, Del Prato S, Marchetti P. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 51: 1437–1442, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Marée AF, Komba M, Finegood DT, Edelstein-Keshet L. A quantitative comparison of rates of phagocytosis and digestion of apoptotic cells by macrophages from normal (BALB/c) and diabetes-prone (NOD) mice. J Appl Physiol 104: 157–169, 2008. [DOI] [PubMed] [Google Scholar]
- 32.Matveyenko AV, Butler PC. Beta-cell deficit due to increased apoptosis in the human islet amyloid polypeptide transgenic (HIP) rat recapitulates the metabolic defects present in type 2 diabetes. Diabetes 55: 2106–2114, 2006. [DOI] [PubMed] [Google Scholar]
- 33.Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, Butler PC. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 57: 1584–1594, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meier JJ, Butler PC. Insulin secretion. In: Endocrinology, edited by DeGroot LJ and Jameson JL. Philadelphia, PA: Elsevier Saunders, 2006, p. 961–973.
- 35.Meier JJ, Ritzel RA, Maedler K, Gurlo T, Butler PC. Increased vulnerability of newly forming beta cells to cytokine-induced cell death. Diabetologia 49: 83–89, 2006. [DOI] [PubMed] [Google Scholar]
- 37.Noguchi H, Xu G, Matsumoto S, Kaneto H, Kobayashi N, Bonner-Weir S, Hayashi S. Induction of pancreatic stem/progenitor cells into insulin-producing cells by adenoviral-mediated gene transfer technology. Cell Transplant 15: 929–938, 2006. [DOI] [PubMed] [Google Scholar]
- 38.Potten C, Wilson J. Apoptosis: The Life and Death of Cells. New York: Cambridge University, 2004.
- 39.Rankin MM, Kushner JA. Adaptive beta-cell proliferation is severely restricted with advanced age. Diabetes 58: 1365–1372, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ritzel RA, Butler PC. Replication increases beta-cell vulnerability to human islet amyloid polypeptide-induced apoptosis. Diabetes 52: 1701–1708, 2003. [DOI] [PubMed] [Google Scholar]
- 41.Robertson RP Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem 279: 42351–42354, 2004. [DOI] [PubMed] [Google Scholar]
- 42.Russ HA, Bar Y, Ravassard P, Efrat S. In vitro proliferation of cells derived from adult human beta-cells revealed by cell-lineage tracing. Diabetes 57: 1575–1583, 2008. [DOI] [PubMed] [Google Scholar]
- 43.Saisho Y, Manesso E, Gurlo T, Huang CJ, Toffolo GM, Cobelli C, Butler PC. Development of factors to convert frequency to rate for β-cell replication and apoptosis quantified by time-lapse video microscopy and immunohistochemistry. Am J Physiol Endocrinol Metab 296: E89–E96, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sapir T, Shternhall K, Meivar-Levy I, Blumenfeld T, Cohen H, Skutelsky E, Eventov-Friedman S, Barshack I, Goldberg I, Pri-Chen S, Ben-Dor L, Polak-Charcon S, Karasik A, Shimon I, Mor E, Ferber S. Cell-replacement therapy for diabetes: Generating functional insulin-producing tissue from adult human liver cells. Proc Natl Acad Sci USA 102: 7964–7969, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell 12: 817–826, 2007. [DOI] [PubMed] [Google Scholar]
- 46.Topp BG, Atkinson LL, Finegood DT. Dynamics of insulin sensitivity, β-cell function, and β-cell mass during the development of diabetes in fa/fa rats. Am J Physiol Endocrinol Metab 293: E1730–E1735, 2007. [DOI] [PubMed] [Google Scholar]
- 47.Tschen SI, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes 58: 1312–1320, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weibel ER Stereological principles for morphometry in electron microscopic cytology. Int Rev Cytol 26: 235–302, 1969. [DOI] [PubMed] [Google Scholar]
- 49.Xu X, D'Hoker J, Stangé G, Bonné S, De Leu N, Xiao X, Van de Casteele M, Mellitzer G, Ling Z, Pipeleers D, Bouwens L, Scharfmann R, Gradwohl G, Heimberg H. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 132: 197–207, 2008. [DOI] [PubMed] [Google Scholar]
- 50.Zulewski H, Abraham EJ, Gerlach MJ, Daniel PB, Moritz W, Muller B, Vallejo M, Thomas MK, Habener JF. Multipotential nestin-positive stem cells isolated from adult pancreatic islets differentiate ex vivo into pancreatic endocrine, exocrine, and hepatic phenotypes. Diabetes 50: 521–533, 2001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.