

Abstract
Coevolutionary conflict among imprinted genes that influence traits such as offspring growth may arise when maternal and paternal genomes have different evolutionary optima. This conflict is expected in outcrossing taxa with multiple paternity, but not self-fertilizing taxa. MEDEA (MEA) is an imprinted plant gene that influences seed growth. Disagreement exists regarding the type of selection acting on this gene. We present new data and analyses of sequence diversity of MEA in self-fertilizing and outcrossing Arabidopsis and its relatives, to help clarify the form of selection acting on this gene. Codon-based branch analysis among taxa (PAML) suggests that selection on the coding region is changing over time, and nonsynonymous substitution is elevated in at least one outcrossing branch. Codon-based analysis of diversity within outcrossing Arabidopsis lyrata ssp. petraea (OmegaMap) suggests that diversifying selection is acting on a portion of the gene, to cause elevated nonsynonymous polymorphism. Providing further support for balancing selection in A. lyrata, Hudson, Kreitman and Aguadé analysis indicates that diversity/divergence at silent sites in the MEA promoter and genic region is elevated relative to reference genes, and there are deviations from the neutral frequency spectrum. This combination of positive selection as well as balancing and diversifying selection in outcrossing lineages is consistent with other genes influence by evolutionary conflict, such as disease resistance genes. Consistent with predictions that conflict would be eliminated in self-fertilizing taxa, we found no evidence of positive, balancing, or diversifying selection in A. thaliana promoter or genic region.
Keywords: genomic imprinting, Arabidopsis, MEDEA, diversifying selection, antagonistic coevolution
Introduction
Genomic imprinting is an epigenetic phenomenon in which the expression level of a gene depends on whether it was inherited maternally or paternally. Imprinting has evolved in analogous life stages in both plants and mammals. The majority of imprinted mouse genes with a phenotypic effect when mutagenized influence fetal or placental growth (13 of 19 genes, Beechey et al. 2003). In plants, the endosperm of developing seeds is the only tissue where imprinting has been observed (Vinkenoog et al. 2003). Like the mammalian placenta, the endosperm carries the same genes as the embryo (though not the same ploidy) and mediates nutrient transfer between the mother and the developing embryo. Thus, imprinted genes influence maternal provisioning and offspring growth in two widely separated taxonomic groups. This parallel evolution suggests that similar selection pressures may be driving the independent origins of imprinting.
A primary theory for the origin of imprinting is the conflict theory (Moore and Haig 1991). In both plants and mammals, mothers provision their young with resources used in growth and early survival. The general logic is that individual offspring will have higher growth and survival if they receive more maternal resources (Krannitz et al. 1991; Khurana and Singh 2000), but the mother will mature fewer offspring (size–number trade-off) (Alonso-Blanco et al. 1999). If a mother provisions offspring from multiple fathers, the offspring's maternally inherited genomes are 50% related to each other, but paternally inherited genomes are less related. Thus, when kin selection is considered, optimum offspring size from the perspective of the paternal genome is larger than that from the perspective of the maternal genome (Wilkins and Haig 2003), creating a conflict of interest between differently inherited genomes over the level of resources acquired from the mother. Consistent with this hypothesis, imprinted genes promoting growth are frequently silenced when maternally inherited, whereas genes suppressing growth are silenced when paternally inherited (although there are exceptions) (Moore and Haig 1991; Hurst and McVean 1997).
Whether or not the origin of imprinting is due to a conflict of interest, maternally and paternally inherited genes may have different optima for traits such as maternal provisioning and offspring size, so imprinted genes that influence these traits may experience a conflict of interest (Wilkins and Haig 2003). Paternally expressed genes will experience constant selection toward the paternal genome's optimum (larger offspring), whereas maternally expressed genes will be under constant selection toward the maternal optimum (smaller offspring). Although offspring size is expected to reach an evolutionarily stable strategy between the maternal and paternal optima (Wilkins and Haig 2003), there will be continued selection that has the potential to drive ongoing antagonistic coevolution at the level of the gene (McVean and Hurst 1997; Williams et al. 2000).
When a single-sire mating system such as monogamy or self-fertilization evolves from a system with multiple sires, the intergenomic conflict over offspring size should disappear (Mochizuki et al. 1996; Hurst 1999; Wilkins and Haig 2003; de Jong et al. 2005), although imprinting is likely to remain (Moore and Mills 1999; Wilkins and Haig 2003). This is because the offspring competing for maternal resources share just as many paternal alleles as maternal alleles, resulting in identical optima for the two genomes (Wilkins and Haig 2003). Thus, antagonistic coevolution among imprinted genes should disappear when strict monogamy or self-fertilization evolves (McVean and Hurst 1997; Smith and Hurst 1998; Wilkins and Haig 2003).
MEDEA (MEA/FIS1) is a maternally expressed suppressor of seed growth and one of the best-studied imprinted genes in plants. MEA encodes a SET-domain Polycomb group protein, which is homologous to Drosophila Enhancer of Zeste [E(Z)] and has histone methyltransferase activity that catalyzes the addition of methyl groups to histone H3 at lysines 27 (H3K27) (Grossniklaus et al. 1998; Kiyosue et al. 1999; Luo et al. 1999). The maternally inherited MEA allele is activated in the central cell prior to fertilization through a process involving the removal of both DNA methylation and histone methylation (H3K27) (Gehring et al. 2006). The diploid central cell gives rise to the triploid endosperm, where the maternal but not paternal copy of MEA is active. Loss-of-function mutations indicate that MEA functions to prevent replication of the central cell nucleus in the female gametophyte prior to fertilization (Chaudhury et al. 1997; Kiyosue et al. 1999), and restricts endosperm proliferation after fertilization (Kiyosue et al. 1999; Sorensen et al. 2001). Thus, MEA is a maternally expressed imprinted gene that suppresses seed growth, presumably by suppressing the transcription of growth-promoting genes in the developing endosperm. MEA is involved in imprinting of the paternally expressed PHERES1 (PHE1) gene by preventing transcription of the maternally inherited copy (Köhler et al. 2003; Makarevich et al. 2006). MEA is also involved in regulation of MEIDOS (MEO) (Köhler et al. 2003) and in its own imprinting. Protein produced from the maternally inherited MEA maintains the repressed state of the paternally inherited MEA allele (Baroux et al. 2006; Gehring et al. 2006; Jullien et al. 2006). Thus, MEA is both imprinted and an imprintor of itself and other loci.
In this paper, we exploit the difference between mating systems of outcrossing and self-fertilizing Arabidopsis species to investigate the possibility that intergenomic conflict could be driving the evolution of MEDEA. We expect intergenomic conflict to create positive, balancing, and/or diversifying selection in outcrossing taxa with multiple paternity, but not self-fertilizing taxa. We use diversity and divergence data in both selfing and outcrossing taxa to investigate evidence of selection in the MEA genic region, promoter, and 5′ upstream gene. Here, we define positive selection as the rapid fixation of beneficial mutations, balancing selection as the maintenance of polymorphism due to local adaptation, heterozygote advantage, or frequency-dependent selection, where new mutations may or may not be favored (Charlesworth 2006), and diversifying selection as one type of balancing selection where new functional mutations are favored but not necessarily swept to fixation and diversity is maintained (sensu Wilson and McVean 2006), such as that found in self-incompatibility genes. Our results provide the first evidence of diversifying selection in the coding region of an imprinted gene. As predicted, diversifying selection was found in the outcrossing, but not self-fertilizing species. In addition, our results support the possibility of both positive selection in the coding region and balancing selection in the promoter of outcrossers but not selfers.
Materials and Methods
Plant Samples
Arabidopsis thaliana is an annual plant that reproduces primarily through self-fertilization. However, its close relatives, Arabidopsis lyrata ssp. lyrata, A. lyrata ssp. petraea, and Arabidopsis halleri ssp. halleri are perennial self-incompatible outcrossers, and outcrossing is thought to be ancestral in the genus (Tang et al. 2007). Arabidopsis thaliana seeds from 23 different wild ecotypes throughout the world were obtained from the Arabidopsis Biological Resource Center (Columbus, OH). Seeds from five A. l. ssp. lyrata populations from the east coast of North America, six A. l. ssp. petraea populations from western Europe, one A. h. ssp. halleri population from France, and one Boechera stricta (= Arabis drummundii) from Alaska, United States, were obtained through kind donations (supplementary table S1, Supplementary Material online).
Laboratory Methods
Genomic DNA was extracted from leaf tissue with the cetyltrimethylammonium bromide method (Doyle and Doyle 1987) or with the PUREGENE DNA purification kit (Gentra Systems Inc., Minneapolis, MN). Genomic DNA was polymerase chain reaction (PCR) amplified in two fragments, one containing the entire 5′ intergenic spacer, which we refer to as the promoter, and an overlapping fragment containing the entire genic region of MEDEA (MEA; At1g02580), including all introns and exons. The promoter was amplified by placing primers in the last exon of the 5′ upstream gene (At1g02570; mea6f: 5′-AAGATGGATGTTGGTTAGGGT or mea36f: 5′-GGATGTTGGTTAGGGTTTTGTTC) and the beginning of the second exon of MEA (mea26r: 5′-CCATCGTCCTCATGGTTTTC or mea25r: 5′-GATTTAGTTCGGGTGGCAAA). Primers for the genic region were at the beginning of the first exon (mea17f: 5′-GGCGAGTGGTTAATGGAGA) and in the 3′ untranscribed region (UTR; mea16r: 5′-GACTGCTTGAATTGCTGCTTCT) in Arabidopsis. In B. stricta, we were unable to amplify the promoter, so only the genic region was amplified. The B. stricta reverse primer was placed in exon 16 (mea15r: 5′-CTTGGCGTAGCAGTTAGGT), so we are lacking sequence for exon 17 and the 3′ UTR. PCR products were cloned using TOPO-TA and TOPO-XL Cloning Kits (Invitrogen, Carlsbad, CA). Three to six clones per allele were sequenced on an ABI Prism 3100 automatic sequencer using BigDye terminator cycle sequencing ready reaction kit ver. 3.1 (Applied Biosystems, Foster City, CA).
Sequences were base called and assembled using Phred and Phrap (Ewing and Green 1998; Ewing et al. 1998), and manually edited in Consed (Gordon et al. 1998). When there were singleton differences among clones, we used a majority rule. If at least two clones differed from other clones at multiple sites, we assumed that the individual was a heterozygote. In rare cases where an allele was only sequenced from two clones and the two clones had a different base, or in short regions where sequence quality was below a phred score of 20, the sites were scored as missing data. Chimeric clones were identified manually and discarded. If only a single clone was sequenced from an allele, the allele was deemed unreliable due to the potential for PCR mutation or chimeras, and was discarded. However, the individual was considered to be a heterozygote for diversity statistics. For diversity statistics, alleles from homozygotes of outcrossing A. lyrata were duplicated, but not in self-fertilizing A. thaliana where the two alleles were not independent. Multiple sequence alignment was performed using MUSCLE (Edgar 2004) and manually corrected using the SeaView alignment editor (Galtier et al. 1996). Sequences were deposited in GenBank under accessions FJ600407-FJ600481. Summary statistics, including π (Tajima 1983), Tajima's D (Tajima 1989), Fu and Li's D* and F* (Fu and Li 1993), and Fay and Wu's H (Fay and Wu 2000), were estimated using the program package DnaSP ver. 4.10.9 (Rozas J and Rozas R 1999).
Hudson, Kreitman and Aguadé Tests
We used Hudson, Kreitman and Aguadé tests (HKA; Hudson et al. 1987) to compare silent diversity-to-divergence ratios in the MEA promoter, MEA genic regions, and other regions of the genome. Comparisons between the MEA promoter and coding region were done using chi-squared tests implemented in DnaSP (Rozas J and Rozas R 1999). To compare MEA with other loci, we used a maximum likelihood version of the HKA test that allows an explicit test of whether a particular locus is different from a set of reference loci (MLHKA, Wright and Charlesworth 2004). Sequences from reference loci were from a similar population sample as the MEA sequences (Wright et al. 2003; Ramos-Onsins et al. 2004) although population substructure (Muller et al. 2008; Ross-Ibarra et al. 2008) can potentially influence the results. Loci used were CAL/CAUL (CAULIFLOWER; At1g26310), ETR1 (“ethylene response 1”; At1g66340), MAM-L (methylthioalkylmalate synthase-like; At5g23020), HAT4 (homeodomain-leucine zipper protein HAT4; At4g16780), CHI (“chalcone isomerase”; At3g55120), PGIC (“glucose-6-phosphate isomerase, cytosolic”; At5g42740), and scADH/SDR (“short chain dehydrogenase/reductase”; At4g05530) for A. l. petraea, CAL, ETR1, PGIC, MAM-L, HAT4, and scADH for A. l. lyrata, and CAL, ETR1, PGIC, scADH, and CHI for A. thaliana. Divergence and diversity estimates for the tests were prepared using DnaSP.
Sliding Window Tests
To visually examine diversity and divergence across the promoter and genic regions of MEA, DnaSP was used to create sliding window plots of silent diversity (πsil), nonsynonymous divergence (Ka), silent divergence (Ksil), and Tajima's D. We statistically assessed whether heterogeneity in the silent polymorphism-to-divergence ratio across the MEA promoter and gene is greater than expected under the neutral coalescent with DNASlider (McDonald 1998). The spatial distribution of the polymorphic and fixed sites was extracted with DnaSP. We used a single haphazardly chosen sequence as the outgroup; A. l. petraea was the outgroup of A. thaliana, and A. thaliana was the outgroup for both A. lyrata subspecies. Following (McDonald 1998), we ran coalescent simulations assuming several different recombination parameter values (e.g., R = 1, 2, 4, 8, 16, 32, 64, 128, and 256), and used the most conservative assumption for each data set (the most conservative values of R were between 4 and 64). Each set of simulations consisted of 10,000 runs. To evaluate the significance of sliding window Tajima's D, we conducted 10,000 replicate simulations in SCANMS (Ardell 2004). The critical values calculated by SCANMS account for the multiple comparisons in a sliding window analysis.
MK Tests
The McDonald–Kreitman test (MK; McDonald and Kreitman 1991) was used both to compare diversity-to-divergence ratios at synonymous and nonsynonymous sites, as well as to compare diversity-to-divergence ratios in promoter sites with synonymous sites (e.g., Fay and Benavides 2005). When comparing synonymous and nonsynonymous sites in the coding region, it was possible to isolate divergence along different lineages of the evolutionary tree by using an estimated ancestral sequence as the outgroup. This was estimated in codeml in PAML ver. 4a using the tree topology in figure 3 and exon sequences from A. thaliana, A. l. lyrata, A. l. petraea, and B. stricta.
FIG. 3.—

Phylogenetic relationship of MEA genes estimated by maximum likelihood from five Brassicaceae species. The numbers above branches are the ratios of nonsynonymous/synonymous substitution (ω) estimated by free-ratio branch model of codeml (Yang 1997). Although branch B had a significantly greater ω than other branches, ω was not significantly greater than 1.
PAML Analysis
To investigate nonneutral evolution in the coding region, and heterogeneity in selection among lineages on the phylogenetic tree, we used codon-based branch models implemented in codeml of PAML 4a (Yang 2007). To avoid the possibility that intragenic recombination could cause false identification of positive selection (Anisimova et al. 2003), we randomly selected a single sequence from each taxonomic group: three Arabidopsis species, B. stricta, and Brassica rapa (GenBank accession: AC189527). Because divergence among species is generally high relative to diversity within species, the particular selection of representative sequences did not strongly influence the overall conclusion. We completely excluded the last 66 codons from the analysis because we were unable to obtain B. stricta sequence for this region. In total, we used 648 codons. The topology of the phylogenetic tree used in this analysis was reconstructed with the maximum likelihood method using PAUP* 4.0b10 (Swofford 2003). The model selected by Modeltest 3.7 (Posada and Crandall 1998) was the general time reversible model with gamma-distributed rate variation. Branch-site models (Yang and Nielsen 2002; Zhang et al. 2005) were attempted, but produced unstable results potentially due to having too many parameters for the amount of data (five species); therefore, only the results of the branch models are presented.
OmegaMap
We investigated the possibility that certain codon sites are under diversifying selection within species (amino acid substitutions are favored among lineages) using OmegaMap (Wilson and McVean 2006). The Bayesian method implemented in OmegaMap incorporates intragenic recombination and does not assume a known fixed genealogy, so that recombination does not inflate the false detection rate of positive sites (Wilson and McVean 2006). For details on OmegaMap analysis, see Supplement A, Supplementary Material online.
Results
For both self-fertilizing A. thaliana, and outcrossing A. lyrata, we obtained sequence data from part of a 5′ flanking gene (At1g02570), the MEA promoter, and the MEA genic region (table 2). The average lengths of sequenced fragments from At1g02570 were 396 bp for A. thaliana (A.t.) and 381 bp for A. lyrata (A.l.). The MEA promoter region was 813 bp (A.t.) and 851 bp (A.l.; excluding the long insertions described below), and the MEA genic region was 4,207 bp (A.t.) and 4,231 bp (A.l.). Although the function of At1g02570 is unknown, the gene is expressed in siliques (genbank accessions BX817913, BX818110, Castelli et al. 2004) and roots (genbank CF651712, Schmid et al. 2003). We did not find any evidence that it is a pseudogene such as premature termination codons or frameshift mutations. Likewise, we found no stop codons or long frameshifts in the MEA coding region. In the MEA promoter region, there were several large indels, including a large insertion in nine out of 10 A. l. lyrata alleles (average size 587 bp; size range 564–599 bp), a 308-bp insertion in one A. l. petraea allele, and an insertion of at least 1,067 bp in another A. l. petraea allele. All indel regions were excluded from analyses. Overall results are summarized in table 1.
Table 2.
Nucleotide Polymorphisms and Summary Statistics for MEA Promoter, Genic Region, and a Gene Flanking the 5′-end of the Promotera
| Taxon | n | Ltotal | La | Lsilent | L4 | Lsyn | Lintr | Stotal | S4 | πtotalb | πab | πsilb | π4b | πsynb | πintrb | DTajcd | FFLc | Fsc | Hc |
| 5’ Gene | |||||||||||||||||||
| Arabidopsis lyrata | 22 | 378 | 291.1 | 86.9 | 46 | 86.9 | 0 | 19 | 4 | 0.010 | 0.010 | 0.011 | 0.008 | 0.011 | — | −1.878* (−0.922) | −2.262* (−1.701) | −3.817** (−0.566) | −1.558 (−3.983) |
| Arabidopsis lyrata lyrata | 10 | 378 | 290.9 | 87.1 | 46 | 87.1 | 0 | 9 | 3 | 0.005 | 0.003 | 0.011 | 0.013 | 0.011 | — | −1.562 (−1.687+) | −1.355 (−0.461) | −1.964+ (−0.653) | −1.244 (−7.20*) |
| Arabidopsis lyrata petraea | 12 | 378 | 291.3 | 86.7 | 46 | 86.7 | 0 | 12 | 1 | 0.005 | 0.004 | 0.010 | 0.004 | 0.010 | — | −1.141 (−2.087*) | −1.567 (−2.53*) | −0.476 (0.924) | 0.152 (−3.636) |
| Arabidopsis thaliana | 18 | 393 | 307.5 | 85.5 | 46 | 85.5 | 0 | 2 | 1 | 0.001 | 0.000 | 0.007 | 0.005 | 0.007 | — | −0.529 (−0.026) | 0.398 (0.74) | −0.011 (−0.027) | 0.183 (0.444) |
| MEA promoter | |||||||||||||||||||
| A. lyrata | 23 | 804 | — | — | — | — | — | 64 | — | 0.022 | — | 0.022 | — | — | — | −0.248 | −1.166 | 1.135 | −1.941 |
| A. l. lyrata | 10 | 814 | — | — | — | — | — | 39 | — | 0.011 | — | 0.011 | — | — | — | −1.795* | −1.865 | 0.606 | −11.556+ |
| A. l. petraea | 13 | 811 | — | — | — | — | — | 50 | — | 0.017 | — | 0.017 | — | — | — | −0.916 | −0.618 | 3.209 | −14.308+ |
| A. thaliana | 18 | 797 | — | — | — | — | — | 16 | — | 0.005 | — | 0.005 | — | — | — | −0.878 | −0.746 | −2.475 | −2.327 |
| MEA gene | |||||||||||||||||||
| A. lyrata | 28 | 3,844 | 1,545.0 | 2,283.0 | 2,075 | 429.0 | 1,854 | 151 | 114 | 0.010 | 0.004 | 0.013 | 0.014 | 0.008 | 0.015 | −0.195 (−0.273) | −0.676 (−0.677) | −1.19 (−0.242) | −0.735 (−1.836) |
| A. l. lyrata | 13 | 4,058 | 1,572.6 | 2,483.4 | 2,271 | 434.4 | 2,049 | 86 | 72 | 0.006 | 0.002 | 0.008 | 0.009 | 0.005 | 0.009 | −0.579 (−0.537) | −0.544 (−0.656) | −0.203 (0.226) | −16.974 (−15.41) |
| A. l. petraea | 15 | 3,927 | 1,544.7 | 2,366.3 | 2,158 | 429.3 | 1,937 | 124 | 94 | 0.008 | 0.004 | 0.012 | 0.012 | 0.008 | 0.012 | −0.596 (−0.659) | −0.461 (−0.443) | 1.637 (2.411) | −8.771 (−12.267) |
| A. thaliana | 13 | 4,202 | 1,611.8 | 2,588.2 | 2,369 | 446.2 | 2,142 | 50 | 37 | 0.005 | 0.001 | 0.007 | 0.007 | 0.005 | 0.007 | 1.381 (0.796) | 1.623+ (0.757) | 3.896 (1.881) | −1.628 (0.462) |
n, number of chromosomes sampled; L, number of sites used in analysis; S, Number of segregating sites; π, average pairwise differences per site; Subscripts for L, S, π: Xtotal, all sites; Xa, nonsynonymous sites; Xsilent, silent sites (= synonymous and noncoding sites); X4, 4-fold degenerate and noncoding sites; Xsyn, synonymous sites; Xintr, intron sites; DTaj, Tajima's D; FFL, Fu and Li's F; Fs, Fu's Fs; H, Fay and Wu's H; Significance levels were determined by 10,000 random coalescent simulations conditioned on the observed number of segregating sites and assuming no recombination, and statistically significant values (P ≤ 0.05) are bold faced: + 0.05 < P ≤ 0.1; * 0.01 < P ≤ 0.05; **P ≤ 0.01.
Jukes–Cantor correction was used.
Four-fold degenerate and noncoding sites were used. Values in parentheses are for all sites.
All significant values of Tajima's D are more extreme than the range of values calculated from multiple other genes in the same taxa (Wright and Gaut 2005, table 2).
Table 1.
Summary of Results
| Promoter |
Genic |
|||||||
| Neutrality tests | Elevated diversity | Heterogeneity in diversity/divergencea | Neutrality tests | Elevated silent diversity | Heterogeneity in silent diversity/divergencea | Positive selection | Diversifying selectionb | |
| Arabidopsis lyratac | n.s.d | Yesef | Yes | n.s. | n.s. | Yes | NA | n.s. |
| Arabidopsis lyrata lyrata | Deviationg | Yesde | n.s. | 5’-end | Yes | Yes | NA | n.s. |
| Arabidopsis lyrata petraea | n.s. | Yese | Yes | n.s. | n.s. | Yes | Nearly significant | Yes |
| Arabidopsis thaliana | n.s. | n.s. | n.s. | n.s.h | n.s. | Yes | n.s. | n.s. |
Analyzed by DNASlider.
Analyzed by OmegaMap.
Pooled sequences from A. l. lyrata and A. l. petraea.
Not significant.
MLHKA test against reference genes.
MK test against synonymous sites and against introns 3–16 of MEA gene.
Negative Tajima's D.
SCANMS detect significantly positive Tajima's D in 3’-end, but the resampling tests from empirical distribution was not significant.
Promoter
Diversity in the Promoter
In A. thaliana, no deviations from neutrality were detected in the promoter even though the sample size was larger than in A. lyrata. The only significant deviation from neutrality observed in the promoter region is a negative value of Tajima's D in A. l. lyrata (table 2). This deviation appears to be due to a single allele (one of two alleles of a heterozygous individual from Wisconsin) that is very distinct from the other alleles. Our result is consistent with Kawabe et al.’s (2007) finding of two distinct clades in the promoter of samples from some populations, which was attributed to balancing selection. Although they found a positive value of Tajima's D in this region, the opposite sign of Tajima's D in our data can be attributed to sampling differences producing a different number of samples from each clade. In the following section, we provide additional support for balancing selection by showing elevated genetic diversity of the A. l. lyrata promoter region relative to divergence observed in our data set.
Comparison of the Promoter to Other Genomic Regions
Balancing selection should elevate diversity at this locus relative to other loci. To further test the hypothesis of balancing selection in the promoter, we used variations of the HKA and MK tests to compare the relative ratios of diversity-to-divergence in the MEA promoter with the neighboring MEA genic region and with putatively neutral, unlinked reference loci.
Following the approach used by Fay and Benavides (2005), we compared diversity and divergence in the MEA promoter with silent (synonymous and intron) sites in the MEA genic region (table 3). The more conservative HKA tests did not detect any significant differences between the gene and promoter in any of the taxa. However, MK tests suggest that the diversity-to-divergence ratio is higher in the promoter of A. l. lyrata and A. l. petraea, than in synonymous sites. Diversity/divergence is also elevated in introns 1–2, which were not significantly different from the promoter. However, the promoter has elevated diversity/divergence relative to the other introns (3–16). There were no significant differences between the promoter and any genic sites in A. thaliana.
Table 3.
HKA and MK Tests Comparing the MEA Promoter to Silent Sites within the MEA Gene and the 5′ Flanking Gene
|
Nsequencesb |
Test | # Polymorphic/#Differencesc |
MEA promoter versusd |
|||||||||
| Taxona | 5′Gene + Promoter | MEA Gene | 5’ Gene syn | MEA prom. | MEA syn. | Introns 1–2 | Introns 3–16 | 5’ Gene syn | MEA synonymous | MEA introns 1–2 | MEA introns 3–16 | |
| Arabidopsis lyrata | 23 | 28 | MK | 9/6 | 67/57 | 18/39 | 48/31 | 57/116 | P = 0.786 | P =0.006 | P = 0.385 | P = 0.0003 |
| HKA | 9/9.2 | 67/74 | 18/38 | 48/44 | 57/128 | χ2 = 0.02, ns | χ2 = 1.36, ns | χ2 = 0.05, ns | χ2 = 2.00, ns | |||
| Arabidopsis lyrata lyrata | 10 | 13 | MK | 4/8 | 40/63 | 6/40 | 33/42 | 30/123 | P = 0.766 | P = 0.002 | P = 0.538 | P = 0.0009 |
| HKA | 4/9.3 | 40/76 | 6/38 | 33/57 | 30/131 | χ2 = 0.06, ns | χ2 = 2.83, P = 0.09 | χ2 = 0, ns | χ2 = 2.46, ns | |||
| Arabidopsis lyrata petraea | 13 | 15 | MK | 5/7 | 49/57 | 13/39 | 46/33 | 40/122 | P = 1.000 | P = 0.014 | P = 0.137 | P = 0.0003 |
| HKA | 5/9.0 | 49/75 | 13/39 | 46/46 | 40/134 | χ2 = 0.05, ns | χ2 = 1.14, ns | χ2 = 0.31, ns | χ2 = 2.16, ns | |||
| Arabidopsis thaliana | 18 | 13 | MK | 2/9 | 15/71 | 5/43 | 7/70 | 24/122 | P = 1.000 | P = 0.322 | P = 0.167 | P = 0.857 |
| HKA | 2/9.3 | 15/75 | 5/41 | 7/72 | 24/131 | χ2 = 0.01, ns | χ2 = 0.22, ns | χ2 = 0.97, ns | χ2 = 0, ns | |||
One A. thaliana sequence was used as the outgroup for A. lyrata diversity tests, and one A. l. petraea sequence was used as the outgroup for A. thaliana.
Number of sites used in the analysis are 80–87 (5′ gene), 747–756 (MEA promoter), 429–436 (MEA synonymous), 480–721 (MEA introns 1–2), and 1208–1269 (MEA introns 3–16).
The two tests use different methods to calculate the number of polymorphic sites and differences; MK test uses fixed differences, whereas HKA test uses average pairwise differences.
Fisher's exact test was used to calculate significance for the MK test. Chi-square tests based on coalescent simulations implemented in the HKA software were used for HKA tests.
Using a maximum likelihood based variant of the HKA test (MLHKA, Wright and Charlesworth 2004), we compared the MEA promoter with putatively neutral reference genes in other parts of the genome. We found that diversity relative to divergence of the A. l. lyrata promoter was more than 5-fold higher than silent sites in the reference genes (table 4). This was due to both elevated diversity (θW was more than 4-fold higher than reference genes) and also slightly reduced divergence between species. Thus, the deviations from neutrality in the A. l. lyrata promoter are probably not due to an incomplete selective sweep but may be due to balancing selection. Diversity/divergence in the A. l. petraea promoter was not significantly different from other genes, but it did show the same trend as A. l. lyrata: Diversity/divergence was generally higher in MEA than other genes. When the two A. lyrata subspecies are combined, MEA diversity–divergence ratio is significantly greater than other genes. Diversity/divergence in the A. thaliana promoter is not different from the reference genes (table 4).
Table 4.
MLHKA Tests Comparing MEA Silent Sites in the Promoter and Genic Region to Silent Sites in Reference Genes
| MEA |
θW/Ksilent ± SD |
||||||
| Taxona | Region | MEA | Ref. Genes | 2Δlb | d.f. | P | kc |
| Arabidopsis lyrata | Promoter | 0.205 | — | 4.859 | 1 | 0.027 | 2.60 |
| Gene | 0.14 | 0.101 ± 0.049 | 1.192 | 1 | 0.275 | — | |
| Arabidopsis lyrata lyrata | Promoter | 0.165 | — | 13.906 | 1 | 0.0002 | 6.65 |
| Gene | 0.093 | 0.031 ± 0.03 | 7.011 | 1 | 0.008 | 3.57 | |
| Arabidopsis lyrata petrea | Promoter | 0.164 | — | 2.521 | 1 | 0.112 | — |
| Gene | 0.130 | 0.102 ± 0.058 | 0.45 | 1 | 0.503 | — | |
| Arabidopsis thaliana | Promoter | 0.045 | — | 0.007 | 1 | 0.933 | — |
| Gene | 0.065 | 0.059 ± 0.027 | 0.042 | 1 | 0.837 | — | |
One A. thaliana sequence was used as the outgroup for A. lyrata diversity tests, and one A. l. petraea sequence was used as the outgroup for A. thaliana.
2Δl is two times the difference in log-likelihood values of a model with selection (k is allowed to vary) and without selection (k = 1) (Wright and Charlesworth 2004).
k is a scaling parameter that measures the degree to which diversity is increased or decreased by the action of selection at the target locus.
Regional Heterogeneity in the Diversity–Divergence Ratio across the Promoter Region
Different portions of the promoter region are likely to differ in their functionality and be subject to different types of selection, so neutrality tests that average over the entire promoter region may fail to detect localized deviations from neutrality. We investigated heterogeneity in the divergence–diversity pattern across the promoter using sliding window plots (fig. 1) and statistical tests of heterogeneity implemented in DNASlider (table 5). There was significant heterogeneity across the promoter in A. l. petraea, but not in the other taxa (table 5). The region approximately 200 bp upstream of the initiation codon showed a high peak in the diversity-to-divergence ratio relative to the rest of the promoter in both A. l. petraea and A. l. lyrata (fig. 1C). However, this peak seems to be caused by unusually low divergence between A. lyrata and A. thaliana rather than elevated diversity (fig. 1). This disparity between diversity and divergence in A. lyrata is unexpected. Interestingly, no polymorphism was observed in the corresponding region of A. thaliana. Although we know of no identified function in this region, low divergence is consistent with strong purifying selection, as is the lack of diversity in A. thaliana, suggesting that this region may be functionally important for gene regulation. The elevated diversity in A. lyrata relative to divergence suggests that there was a recent change in the selection regime in the A. lyrata lineage due to relaxation of purifying selection or possibly balancing selection.
FIG. 1.—
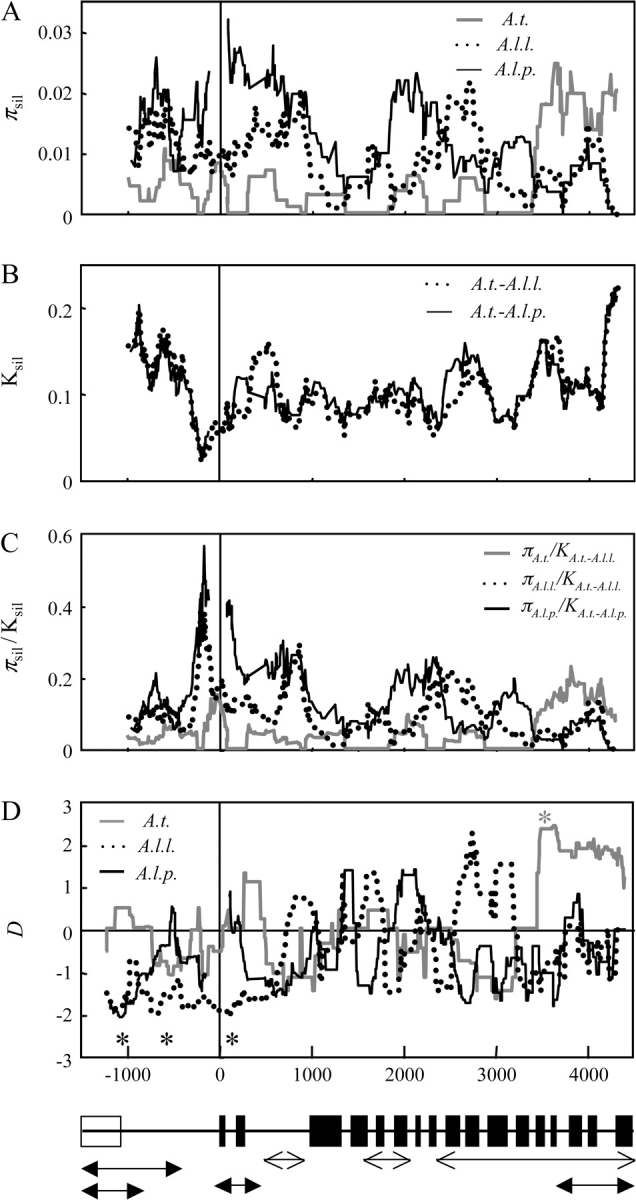
Sliding window plots (window size = 150 bp, step size = 5 bp) for (A) silent nucleotide diversity (πsil), (B) divergence (Ksil), (C) diversity-to-divergence ratio (πsil/Ksil) in Arabidopsis thaliana (A.t.), Arabidopsis lyrata lyrata (A.l.l.), and Arabidopsis lyrata petraea (A.l.p.). (D) Tajima's D (D) was calculated with all sites and a window size of 200 bp (step size = 5 bp). *Regions where Tajima's D is significantly different from zero (P < 0.05) using SCANMS (Ardell 2004). (E) The black boxes correspond with MEA exons and the white box indicates the 5′ gene (At1g02570). Arrows with closed ends indicate regions that may be important for MEA cis regulation (Baroux et al. 2006; Gehring et al. 2006; Jullien et al. 2006). Arrows with open ends indicate regions with high H3K27 methylation in 10-day old seedlings, where MEA is silent (Zhang et al. 2007).
Table 5.
Tests of Heterogeneity among Sites in Polymorphism and Divergence from DNASlidera
| Taxon | Nsequences | Npoly | Nfixed | Runs Test |
Kolmogorov–Smirnovb |
Mean sliding G |
|||
| Test Statisticc | P | Test Statisticd | P | Test statisticd | P | ||||
| Promoter | |||||||||
| Arabidopsis lyrata | 23 | 67 | 57 | 56 | 0.2293 | 0.0801 | 0.0334 | 2.9725 | 0.2899 |
| Arabidopsis lyrata lyrata | 10 | 40 | 63 | 48 | 0.5293 | 0.0508 | 0.3949 | 2.2286 | 0.4418 |
| Arabidopsis lyrata petraea | 13 | 49 | 57 | 44 | 0.0846 | 0.0919 | 0.0257 | 3.5192 | 0.2071 |
| Arabidopsis thaliana | 18 | 15 | 71 | 27 | 0.795 | 0.0381 | 0.3685 | 1.1409 | 0.557 |
| Gene (silent sites) | |||||||||
| A. lyrata | 28 | 123 | 188 | 126 | 0.0122 | 0.0066 | 0.0136 | 6.8013 | 0.0692 |
| A. l. lyrata | 13 | 69 | 207 | 89 | 0.035 | 0.0562 | 0.025 | 5.6133 | 0.1015 |
| A. l. petraea | 15 | 99 | 196 | 112 | 0.24 | 0.0722 | 0.0089 | 9.2926 | 0.0294 |
| A. thaliana | 13 | 37 | 241 | 66 | 0.6776 | 0.0406 | 0.0136 | 5.2926 | 0.0591 |
For each test, we used the value of the recombination rate parameter (R) that produced the most conservative test statistic.
The test using Kolmogorov–Smirnov statistics is most powerful when the ratios at the far ends of the gene differ (McDonald 1998), and this test appears to be the most sensitive to the heterogeneity in our data set.
A smaller value indicates greater heterogeneity.
A larger value indicates greater heterogeneity.
Table 6.
Likelihood and Estimated Parameter Values of Models from Codon-Based Branch Model (Codeml)
| Model | κa | ω0 | ω1 | lnLb | npc |
| M0 | 1.932 | 0.546 | — | −5,343.21 | 9 |
| M1neutrald | 1.916 | 0.5077 | (1) | −5,339.97 | 9 |
| M1positivee | 1.932 | 0.505 | 2.755 | −5,338.51 | 10 |
| Free ratio | 1.939 | — | — | −5,336.63 | 15 |
Transition–transversion ratio.
Log-likelihood of the fitted model.
Number of parameters estimated in the model.
Branch B of figure 3 was constrained to have ω1 = 1, and the rest of branches were assumed to have ω0.
Branch B of figure 3 was assumed to have ω1, different from the rest of branches with ω0.
5′ Flanking Gene
In order to understand whether deviations from neutrality in the promoter are likely due to selection in the promoter, or due to selection on a nearby coding region, we investigated diversity in the last exon of the gene flanking the 5′-end of the promoter region (At1g02570). This is a region not previously investigated. We detected no significant deviations from neutrality in the 4-fold degenerate sites of the 5′-gene in A. l. petraea (table 2); however, the number of sites was small. When all sites were used, Tajima's D in A. l. petraea was significantly negative due to a single highly divergent allele (fig. 1D). In the A. l. lyrata 5′ gene, Tajima's D is negative, but not significant. Like the A. l. lyrata promoter, it has one very divergent allele, and the region of negative Tajima's D stretches from the 5′ end of the MEA genic region, through the promoter to the 5′ gene, but Tajima's D is not significant in the 5′ gene (table 2). The diversity–divergence ratio is not elevated in the 5′ gene relative to the promoter as one would expect if balancing selection were acting on the 5′ gene rather than the promoter (table 3). However, because of low recombination between the two regions (supplementary table S2, Supplementary Material online), we cannot reliably distinguish between selection acting on the MEA promoter and the 5′ flanking gene. Further obscuring the target of selection, the MEA protein appears to interact with DNA in the last exon of At1g02570 (Baroux et al. 2006), and histone (H3K27) methylation in this region is associated with the silencing of transcription in MEA (Jullien et al. 2006). Therefore, if balancing selection is acting on the 5′ flanking gene, it is difficult to determine whether selection is acting on MEA expression or the protein of this gene of unknown function.
Genic Region
Silent Diversity in the MEA Genic Region
Standard tests of neutrality on silent sites (synonymous and noncoding sites) failed to detect any deviations from neutral expectations in the genic region of any of the three taxa (table 2). However, the diversity–divergence ratio in MEA silent sites of A. l. lyrata was more than three times higher than the reference genes, a significant difference (table 4). The elevated diversity could be due to balancing selection in the promoter and hitchhiking in the genic region, because diversity was higher in the promoter than the genic region (table 3). However, DNASlider detected significant heterogeneity in the silent polymorphism-to-divergence ratios across the length of the gene (table 5), and the location of peaks in diversity (one in intron 2 and one around introns 9–10; fig. 1) argues that there could be some nonneutral evolution within the gene as well. Silent sites in A. l. petraea were not significantly different from other genes (table 4), but there was significant heterogeneity across the gene (table 5), with peaks around introns 1–2 and exons 6–9 (fig. 1).
In A. thaliana, diversity/divergence in MEA silent sites did not differ from the reference genes (table 4). There was significant heterogeneity across the gene (table 5), but the pattern of diversity-to-divergence was quite different in A. thaliana compared with A. lyrata. Diversity in A. thaliana was low throughout most of the gene, but was elevated in the last four introns and 3′ untranslated region (fig. 1A). Further, SCANMS revealed that Tajima's D is significantly positive (P < 0.01) in five consecutive windows in this region (fig. 1D), caused by the presence of two distinct haplotype groups. There were no nonsynonymous polymorphisms around the region, suggesting that there might be some balancing selection on silent sites at or beyond the 3′ end of the gene in A. thaliana. Histone (H3K27) methylation in this region is correlated with MEA expression in vegetative tissue, and is likely to be involved in the control of paternal silencing in the endosperm (Jullien et al. 2006), so DNA sequence variation in this region may influence MEA expression.
Population subdivision and other demographic factors have the potential to increase the variance in Tajima's D (Wright and Gaut 2005). In a sample of 876 gene fragments from A. thaliana samples with a similar geographic distribution as ours, Tajima's D is generally negative (mean = −0.8) throughout the genome (Nordborg et al. 2005). However, the variance in Tajima's D among genes was higher than expected under neutral coalescent models such that standard significance cutoff values for Tajima's D have the potential to be anticonservative in both ends (Nordborg et al. 2005). To compare the sliding window results from the MEA gene, with the genomewide distribution of Tajima's D, we created an empirical sampling distribution of maximum Tajima's D values observed from sliding window analyses of the Nordborg et al. (2005) data set. The procedure is analogous to how SCANMS determines the critical value, except it uses empirical distributions, rather than distributions from neutral coalescent simulations. For each repetition of resampling, we randomly selected the same number of haplotypes as our data set (13 haplotypes, each from a different population). Because the length of each sequence fragment in the Nordborg et al. (2005) data was shorter than MEA coding region, we concatenated randomly selected fragments to create the length equivalent to our data set (4,202 bp). On each 4,202-bp fragment, we conducted the sliding window analysis of Tajima's D and recorded the maximum Tajima's D among windows. We repeated this procedure 10,000 times to create the sampling distribution of maximum Tajima's D for a sliding window analysis of given size. In contrast to the results of SCANMS, this analysis suggests that the high Tajima's D in 3′-end of the MEA gene is not statistically significant. Although we should note that this procedure is likely to be conservative because some genes in the genomic data are under selection and not neutral, we conclude that the high Tajima's D in the 3′ end of this A. thaliana gene is probably not due to balancing selection, but may be due to some genomewide factor such as population subdivision.
Nonsynonymous and Synonymous Divergence in the MEA Coding Region
If evolution of the MEA coding region in outcrossers is driven by conflict, we may expect outcrossers, but not selfers, to have experienced repeated selective sweeps and to show an excess of nonsynonymous/synonymous substitutions. The rate of nonsynonymous/synonymous substitution (Ka/Ks) between A. lyrata and A. thaliana is 7-fold higher in MEA than our seven reference genes (MEA Ka/Ks = 0.64, reference genes Ka/Ks average ± SD = 0.09 ± 0.07) and more than 4-fold higher than the average between Arabidopsis and Brassica genes (Ka/Ks = 0.14; Tiffin and Hahn 2002). MK tests do not reveal evidence of positive selection causing excessive fixation of nonsynonymous mutations in any of the taxa studied (A. lyrata P = 0.11, A. thaliana P = 1.0). However, codon-based models using codeml of the PAML package (Yang 1997) suggest the possibility of positive selection on at least one branch. We analyzed MEA evolution in a tree containing five self-compatible (SC) and (SI) species in the mustard family, including A. thaliana (SC), A. lyrata (SI), A. halleri (SI), B. stricta (= Arabis drummondii; SC) and B. rapa subsp. pekinensis (SI; GenBank Acc#: AC189527). We used the free-ratio branch model, which allows a different nonsynonymous/synonymous substitution rate (ω) on each branch of a phylogenetic tree (fig. 3). The free-ratio model had a significantly better fit to the data than a model assuming no variation in ω among lineages (M0; table 6; 2ΔlnL = 13.18, df = 6, P = 0.040 from χ2 approximation). This suggests that there is heterogeneity in ω among lineages, and selection has been changing over time.
All branches of the MEA tree showed a high ω (fig. 3). However, we observed ω > 1 only on branch B, after the divergence of A. lyrata and A. halleri from A. thaliana (node A; fig. 3). In order to identify which branches differed from the others, we followed the multiple hypothesis testing approach of Anisimova et al. (2007). For each branch, we estimated the likelihood of a two-ratio model assuming that the ratio for that branch (ω1) was different from the rest of the branches (ω0) and compared the likelihood with a one-ratio model (M0). To avoid false positives due to multiple hypothesis testing, Rom's (1990) correction procedure was employed (Anisimova et al. 2007). This analysis revealed that branch B (fig. 3) was significantly different from the rest of the branches (M1positive vs. M0; 2ΔlnL = 9.410, df = 1, P = 0.002; table 6).
In order to test whether ω1 = 2.50 of branch B was significantly higher than 1, and therefore evidence of positive selection, we compared the model where ω1 of branch B was free to vary (model M1positive) with a model that constrained the ω value of branch B to neutrality (ω1 = 1; model M1neutral; table 6). The model M1positive, which allowed ω1 > 1, had a better fit to the data; however, the improvement was only marginally significant (2ΔlnL = 2.92, df = 1, P = 0.087). To test whether the particular sequences chosen for this analysis influenced the result, we repeated the analyses with all combinations of each A. lyrata and A. thaliana sequence. In the comparison of model M1positive to M1neutral, the average P value was 0.10 (0.069–0.15). Statistical significance of M1positive versus M0 was not influenced by the choice of sequences, and average P value was 0.003 (0.001–0.006). Thus, although it is possible that positive selection occurred in some region(s) of the gene along this branch, we cannot statistically exclude the possibility the elevated ω is simply due to relaxed purifying selection and the gene was evolving neutrally. Note this gene is unlikely to be completely neutral; it has an essential function and mutations in the gene have been shown to be lethal (Chaudhury et al. 1997; Kiyosue et al. 1999). Nonetheless, because there was significant heterogeneity among branches, we can conclude that selection pressure on the MEA coding region has been variable over time.
Although none of the above statistical tests, which average across the entire coding region, provide strong evidence for positive selection, the pattern of substitutions along the coding region appears to be highly heterogeneous. A sliding window plot of the ratio of nonsynonymous to synonymous substitutions (Ka/Ks) shows different patterns in comparisons among taxa with different mating systems (fig. 2). The comparison of A. thaliana and B. stricta (both SI) shows no high peaks in Ka/Ks. However, when outcrossers are included in the comparison, there are several regions where Ka/Ks >>1. Two of the highest peaks (b and f; fig. 2) are regions with very low Ks (Ks ≈ 0), which might be caused by purifying selection on synonymous sites perhaps due to a role in intron splicing (Parmley et al. 2006). However, there are also some peaks with elevated Ka (a, b, c). All of exon 3 shows very high nonsynonymous divergence (Ka/Ks > 1), and comparison among species shows several large indels in exon 3, such that amino acid divergence in this region is even higher than that indicated by Ka/Ks. At least some of this region is important in MEA-FIE protein interactions (Spillane et al. 2000), indicating that the region should not be neutral. According to Chamary et al. (2006), very few genes (6 of 148 surveyed) contain peaks of Ka/Ks > 1 that are due to elevated Ka, so these peaks could indicate nonneutral evolution. We were unable to statistically test for localized regions of positive selection with branch-site models in PAML because results of different runs did not converge, perhaps due to the small number of taxa in our study. A site model was not significant, presumably because selection was not acting uniformly on all branches. Nonetheless, the large number of Ka/Ks > 1 peaks, in conjunction with the PAML branch analysis showing ω ≥ 1 in part of the tree suggest that there may be some positive selection, or at least that selection on different portions of the gene is changing through time.
FIG. 2.—
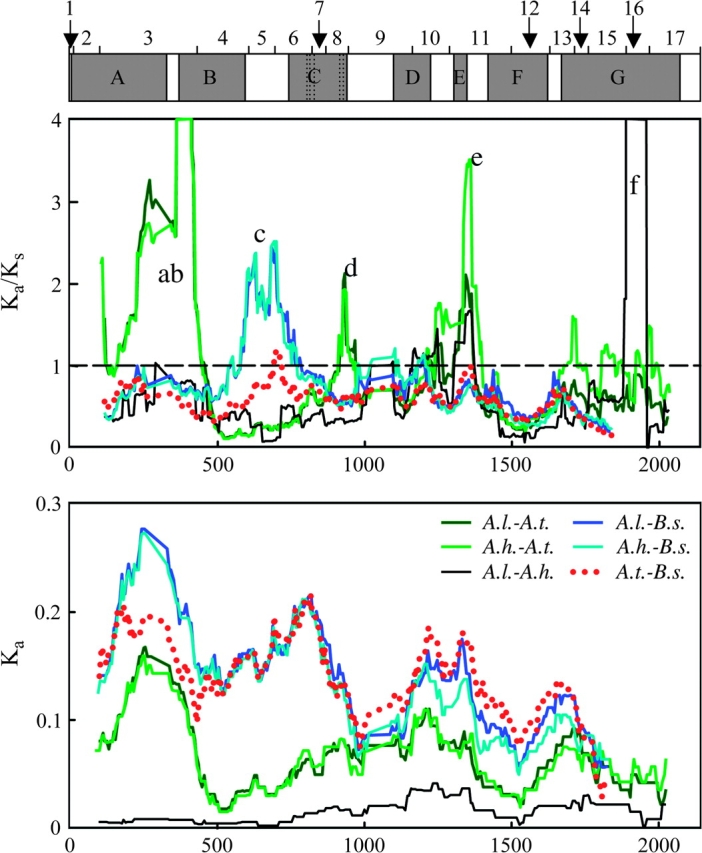
Sliding window plots for Ka/Ks (window size = 200 bp, step size = 5 bp) and Ka (window size = 150 bp, step size = 5 bp) of comparisons between species pairs: Arabidopsis thaliana (A.t.), Arabidopsis lyrata (A.l.), Arabidopsis halleri (A.h.), and Boechera stricta (B.s.). Color coding is the same in both plots. The small letters in the upper plot indicate regions with elevated Ka/Ks discussed in the text. At the top of the figure is a map of exons (1–17) and functional domains (gray boxes): (A) an important region for the interaction between MEA and FIE (Spillane et al. 2000; Yadegari et al. 2000); (B) EZD1 and (C) EZD2 are found in E(Z) homologs but their function is unknown; (D) a SANT domain, which may be involved in histone binding (Boyer et al. 2004); (E) a putative nuclear localization signal (NLS); (F) a cysteine-rich region (CXC), which may be important in protein folding (Sardiu et al. 2007). (G) a SET domain, which has H3K27 methylation activity (Springer et al. 2002; Ng et al. 2007).
Nonsynonymous and Synonymous Diversity across MEA Coding Region
We found potential evidence for recent diversifying selection in a portion of the A. l. petraea coding region with elevated nonsynonymous/synonymous diversity. Using a codon model with recombination in OmegaMap (Wilson and McVean 2006) to analyze heterogeneity in the ratio of nonsynonymous and synonymous diversity (ω) within taxa, we found one area in exon 9 with elevated nonsynonymous diversity (ω > 1) suggesting that selection favors new and rare amino acids (figs. 2 and 4). This region corresponds to a region surrounding codon 330 in A. thaliana Col-0 (Genbank NM_100139). We conducted this analysis with three sets of prior assumptions (A, B, and C; Supplement A, supplementary table S2, Supplementary Material online), and in the area of codon 330, the posterior probability of ω > 1 was greater than 0.95 under the prior sets A (estimated ω = 2.66; with 95% highest posterior density, HPD, interval of 1.44–19.13) and C (estimated ω = 2.45; 1.05–5.98), but not in set B (estimated ω = 1.64; 0.7–3.95). Although these results were not sensitive to the shape of prior distributions (compare sets A and C), the results are sensitive to the prior mean of ω. Prior set B assumes the prior mean ω < 0.5, whereas the other prior sets use higher prior means of ω = 1. Set B therefore produces the most conservative test of diversifying selection. When the results of a Bayesian analysis are influenced by the prior assumptions, the data may not contain a sufficiently strong signal for a reliable conclusion, and additional research on the function of this region is needed. Nonetheless, our analysis suggests the possibility of recent diversifying selection favoring amino acid diversity around codon 330 in A. l. petraea. Arabidopsis thaliana and A. l. lyrata did not have any regions where ω was statistically greater than 1, and therefore, show no evidence of diversifying selection within the coding region.
FIG. 4.—

Results of OmegaMap analysis for Arabidopsis lyrata petraea coding region under Prior set A of supplementary table S2, Supplementary Material online. (a) The sitewise mean ω (solid line) and the 95% HPD interval (shaded area) of ω along the gene. The dotted line corresponds to neutrality (ω = 1). (b) Sitewise posterior probability of diversifying selection (ω > 1). The two vertical dotted lines correspond to 95% and 99% probabilities. The map of exon boundaries and conserved functional domains at the top of the figure is the same as in figure 2.
Discussion
We have examined the population genetic structure of an imprinted gene, MEDEA, and its promoter, in both self-fertilizing and outcrossing Arabidopsis species, as well as divergence among Arabidopsis, Boechera, and Brassica. We expected to find some evidence of conflict in outcrossing taxa (A. lyrata), but not the self-fertilizing species (A. thaliana). We find nearly significant evidence of positive selection for amino acid substitution on one outcrossing branch of the gene tree (fig. 3). We also found possible evidence of diversifying selection in a portion of the coding region in A. l. petraea (fig. 4), and we add support for Kawabe et al.’s (2007) finding of balancing selection in the promoter and first two introns in A. l. lyrata, using a different sampling scheme, and a different class of analyses (fig. 1, tables 2–4). Selection in selfers as well as outcrossers would shed doubt on the conflict hypothesis and suggest that some other force acting on both selfers and outcrossers may be driving evolution in this gene, such as maternal–offspring coadaptation (Agrawal et al. 2001; Wolf and Hager 2006). Importantly, we do not find evidence for selection in self-fertlizing A. thaliana, which was not investigated extensively in the previous studies.
Comparison to Previous Research
There has been little empirical research on the population genetics of imprinted genes, and results have been mixed. Comparison of mouse and rat sequences in seven imprinted genes did not reveal an elevated rate of nonsynonymous mutation relative to synonymous mutation (Ka/Ks < 1, McVean and Hurst 1997). Likewise, a gene that is imprinted in Maize endosperm (Mez1) failed to reveal strong evidence of elevated nonsynonymous mutation (Ka/Ks < 1, Haun et al. 2007). However, none of these imprinted genes has been subject to the more powerful analyses of diversity that have the potential to expose either balancing selection or selection that is localized to a portion of the gene.
Conflicting reports exist over whether MEA has been subject to positive selection in outcrossing Arabidopsis. Using phylogeny-based models in PAML, Spillane et al. (2007) found that the outcrossing A. lyrata branch had a much higher rate of nonsynonymous:synonymous change (ω = 2.9) than the selfing A. thaliana branch (ω = 0.1), suggesting that there was positive selection in outcrossers but not selfers. However, because MEA was rooted to paralagous genes in this analysis, it is difficult to disentangle the effects of low sequence homology, positive selection due to neofunctionalization and positive selection due to conflict (Kawabe et al. 2007). Further, Kawabe et al. (2007) argue that because MK tests are not significant, there is no compelling evidence for positive selection on the MEA coding region.
We found nearly significant evidence of positive selection on one outcrossing branch after the divergence of A. lyrata and A. thaliana (ω = 2.5, a nearly significant difference from ω = 1). In contrast to Spillane et al. (2007), we did not root MEA to its paralogs, but to the MEA gene in Brassica, so neofunctionalization and long branch lengths should have less influence on our results. Thus, it is possible that a portion of the gene was influenced by positive selection independent of neofunctionalization. However, the effect of selection is variable across lineages and not all outcrossing branches showed elevated (ω ≥ 1) amino acid change in contrast to the expectation that all outcrossing lineages should be under positive selection. This does not appear to be simply due to the lack of statistical power because the other outcrossing branches outside of genus Arabidopsis were longer with more substitutions. Thus, if positive selection is acting on portions of this gene, it is probably sporadic.
Although positive selection is often seen in conjunction with gene duplication (He and Zhang 2005), balancing and diversifying selection are not. Kawabe et al. (2007) showed evidence of balancing selection on the promoter and possibly the first two introns within some populations of outcrossing A. lyrata; Tajima's D, Fu's Fs, and Strobeck's S were significantly positive due to the presence of three highly divergent haplotypes. We provide further support for balancing selection in the promoter by showing that the ratio of diversity-to-divergence is elevated in the promoter relative to the coding region (MK test; table 3), and relative to silent sites in other genes (MLHKA test; table 4) with a different sampling scheme (i.e., specieswide). Although both frequency spectrum tests and comparisons among loci may be influenced by nonequilibrium demography, the bias should be different in the two types of tests. Taken together, these results support balancing selection on the promoter or a nearby genomic region in outcrossers.
The possibility of balancing and/or diversifying selection in self-fertilizing A. thaliana has not been previously investigated, yet it is critical to know whether selection in selfers, where conflict should be eliminated, is different from outcrossers. The promoter and first two introns showed no evidence of balancing selection in A. thaliana (tables 2–4). Introns at the 3’ end of the gene showed elevated diversity-to-divergence and Tajima's D was positive due to the presence of two divergent haplotype groups, and deviations were significant when compared with neutral expectations (fig. 1, table 5), but not when compared with an empirical distribution from A. thaliana. Further, no codons showed evidence of diversifying selection with the OmegaMap analysis. Thus, positive, balancing, and diversifying selection in MEA gene appear to be unique to the outcrossing taxa, suggesting the possibility that selection may be due to conflict among imprinted genes.
Nonequilibrium demographic processes appear to have played a role in shaping the diversity of both A. lyrata (Ross-Ibarra et al. 2008) and A. thaliana (Nordborg et al. 2005). Population substructure and nonequilibrium demography can cause difficulties in any studies of selection on sequence evolution (e.g., Wright and Gaut 2005). For example, the HKA test was originally considered to be robust against systematic bias due to population bottlenecks (Hudson et al. 1987), but simulations with some bottleneck scenarios show that biases in the HKA test can occur (Hammer et al. 2004) although this effect has not been thoroughly studied. Population substructure is high in A. lyrata, especially European A. l. petraea (Muller et al. 2008; Ross-Ibarra et al. 2008). When A. lyrata populations are analyzed at the species level, two north American A. l. lyrata are closely related and clustered together (Ross-Ibarra et al. 2008). We have analyzed the two A. lyrata subspecies separately in the hope of reducing this effect. Although the potential for bias applies to both A. lyrata and A. thaliana, we found that only the outcrossing species show the signature of selection using multiple analyses with different types of bias. Balancing selection should be easier to detect in self-fertilizing species than outcrossers (Innan 2006) because of lower effective recombination rate in selfers (Hagenblad and Nordborg 2002; Wright et al. 2003), so we believe that our finding of balancing and diversifying selection in outcrossers but not selfers is robust.
Conflict and Population Genetics
Imprinted genes have been suggested to experience antagonistic coevolution: Paternally expressed imprinted genes should be under continuous directional selection for increased offspring size, whereas maternally expressed genes should be under continuous directional selection for decreased offspring size (McVean and Hurst 1997). Thus, one might expect to find continuous selective turnover of alleles, evidenced by an increased rate of nonsynonymous fixation and reduced diversity (Maynard Smith and Haigh 1974). However, if we look at population genetics data from other genes influenced by coevolutionary conflict, we see that the findings are more complex.
The interaction between pathogens and disease resistance genes is clearly known to exhibit conflict, and many, but not all, disease resistance genes show evidence of selection (Hughes and Nei 1988; Bakker et al. 2006). In those that are evolving under selection, there are often regions with elevated nonsynonymous substitution rates, suggesting that new mutations are favored (Hughes and Nei 1988). However, these new mutations do not always sweep to fixation (Bakker et al. 2006). Rather, diversity is often elevated (Parham and Ohta 1996; Tan et al. 2005), with long coalescence times among alleles (Takahata and Nei 1990), suggesting that balancing selection is important. One explanation is that rare mutations are favored because they are not targeted by pathogens, favoring both new mutations as well as retaining old alleles when they become rare (Dybdahl and Lively 1998; Stahl et al. 1999; Borghans et al. 2004). Different disease resistance genes from the same species show substantial differences in genealogical shape and depth, suggesting that balancing selection may persist for highly variable lengths of time (Bakker et al. 2006). Further, genes that show balancing selection in some taxa, show evidence of a recent selective sweep, and low diversity, in closely related taxa (de Groot et al. 2008). Finally, selection appears to be episodic, with differences in selection pressure among lineages (Abi-Rached et al. 2007). Mating-related genes influenced by sexual conflict (such as seminal fluid proteins) show patterns similar to disease resistance genes. New mutations are favored by positive selection (Tsaur et al. 1998; Swanson et al. 2001; Andrés et al. 2006), and amino acid changes are fixed more often than expected (Tsaur et al. 1998), but selection pressure is variable among lineages (Clark and Swanson 2005), and high levels of nonsynonymous polymorphism indicate that balancing selection is widespread (Begun et al. 2000). Thus, it appears that coevolutionary conflict is likely to create a combination of positive selection favoring new mutations that may or may not sweep to fixation, as well as balancing selection maintaining old alleles (i.e., diversifying selection).
Population genetic data from MEA show some of the same patterns seen in genes known to be influenced by conflict, including variable selection among lineages, the possibility of sporadic positive selection in the coding region (figs. 2 and 3), as well as elevated diversity in both the promoter and coding region (figs. 1 and 4). Thus, the evolution of MEA may well be influenced by selection driven by coevolutionary conflict.
Imprinting and Balancing Selection
An evolutionary mechanism to promote balancing or diversifying selection in imprinted genes has not been previously proposed. How can balancing selection arise in imprinted genes? One potential mechanism can be derived by analogy to the coevolution of a disease and a disease resistance gene. Consider a system with a growth-promoting gene that is expressed when paternally inherited, P, and a growth-suppressing gene that is expressed when maternally inherited, S. Assume that gene S suppresses growth by interfering with the action of P in some way, for instance by binding either to the P protein or binding to the promoter of the P gene to suppress transcription. A mutation in P (from allele P1 to P2) that reduces the ability of the S1 protein to bind should be selectively favored if it allows greater offspring growth. If P2 becomes common, a mutation in S (from S1 to S2) should be favored if S2 suppresses P2 more effectively. S1 may bind stably to P1 but not to P2, and S2 may bind stably to P2 but not P1. Like in a host–pathogen system (Borghans et al. 2004), allele frequencies may oscillate due to negative frequency–dependent selection where allele P1 is favored when S2 is common, but as P1 increases in frequency, so does S1. As S1 becomes common, P2 will be favored, creating long-term balancing selection. Alternatively, if new mutations altering binding are constantly arising and being swept to fixation, one may observe positive selection and a reduction in diversity (McVean and Hurst 1997). In reality, balancing selection and positive selection favoring new mutations are likely to be acting simultaneously, creating diversifying selection.
Diversity can only be maintained if the mutation from S1 to S2 occurs before P2 can sweep to fixation, eliminating polymorphism. Population substructure is one factor that could slow or prevent complete fixation, such that some populations may be fixed for S1 and P1 whereas others may be fixed for S2 and P2. Positive selection within each population could cause elevated nonsynonymous differentiation among lineages, as observed in our analysis by OmegaMap. Secondary gene flow among populations may reintroduce diversity to a population, and subsequent negative frequency selection can preserve the segregating alleles. Thus, the high level of population substructure, observed in A. lyrata (Muller et al. 2008), may promote the occurrence of this hypothetical scenario. Indeed Kawabe et al. (2007) observed that highly divergent haplotypes are maintained within some populations but not others, and a similar pattern was seen in our data.
Our proposed mechanism of balancing selection is not inconsistent with known molecular mechanisms of MEA. MEA is likely to interact with other genes in a multifactorial network. MEA is a polycomb protein that binds DNA in a complex with other proteins: MEA forms a complex with Fertilization Independent Seed2 (FIS2), Fertilization Independent Endosperm (FIE), and Multicopy Suppressor of Ira1 (MSI1) (Köhler et al. 2003; Chanvivattana et al. 2004). This complex is likely to suppress transcription at multiple downstream target genes including MEIDOS (MEO), the maternally inherited copy of the imprinted gene PHERES1 (Köhler et al. 2003), as well as the paternally inherited copy of MEA (Gehring et al. 2006; Jullien et al. 2006). There could be coevolutionary conflict between the MEA protein and the proteins it binds to, or the DNA regions to which it binds (e.g., regulatory regions of PHERES1), or other proteins that activate transcription in genes that MEA suppresses (e.g., a trithorax homolog). There could also be conflict with proteins that regulate the expression of MEA. Antagonistic coevolution among these interacting factors could create a combination of positive and balancing selection.
Conclusions
A combination of balancing, diversifying, and positive selection are likely to have influenced the evolution of MEA in both regulatory and coding regions. Codon-based branch analysis among taxa (PAML) suggests that selection on the coding region is changing over time and that nonsynonymous substitution may be elevated in at least one outcrossing branch. Codon-based analysis of outcrossing A. lyrata ssp. petraea (OmegaMap) indicates that nonsynonymous polymorphism is elevated in a portion of the gene, suggesting the possibility of diversifying selection. HKA analysis of outcrossers indicates that diversity/divergence at silent sites in the MEA promoter and genic region is elevated relative to reference genes, and there are deviations from the neutral frequency spectrum, providing support for balancing selection in outcrossers. This combination of positive selection as well as balancing and diversifying selection in outcrossing lineages is consistent with other genes influenced by evolutionary conflict such as disease resistance genes. Supporting predictions that conflict would be eliminated in self-fertilizing taxa, we found no strong evidence of positive, balancing, or diversifying selection in A. thaliana promoter or genic regions.
Supplementary Material
Supplementary tables S1 and S2 and Supplement A are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We are grateful to Dane Salter, Lacie Jo Westbrook, Jim Warner, Carrie Topp, and Leif Vick for assistance with laboratory work. Mark Rausher provided the use of his laboratory in the early part of this study. Stephen Wright, Deborah Charlesworth, Mark MacNair, Matt Olson, Tom Mitchell-Olds, and ARBC provided seeds. We thank Peter Morrell, Marcy Uyenoyama, and anonymous reviewers for constructive comments on an earlier draft of the manuscript. T.M. and N.T. were supported by Alaska INBRE Grant Number RR016466 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH). DEW was supported by National Science Foundation (NSF) Experimental Program to Stimulate Competitive Research (Alaska EPSCoR) grant 0346770. N.T. and D.E.W. are supported by National Science Foundation (DEB-0640520). This project was initiated through funding from a Duke University Postdoctoral fellowship in Molecular Evolution to D.E.W.
References
- Abi-Rached L, Dorighi K, Norman PJ, Yawata M, Parham P. Episodes of natural selection shaped the interactions of IgA-Fc with FcαRI and bacterial decoy proteins. J Immunol. 2007;178:7943–7954. doi: 10.4049/jimmunol.178.12.7943. [DOI] [PubMed] [Google Scholar]
- Agrawal AF, Brodie ED, Brown J. Parent–offspring coadaptation and the dual genetic control of maternal care. Science. 2001;292:1710–1712. doi: 10.1126/science.1059910. [DOI] [PubMed] [Google Scholar]
- Alonso-Blanco C, Blankestijn-de Vries H, Hanhart CJ, Koornneef M. Natural allelic variation at seed size loci in relation to other life history traits of Arabidopsis thaliana. Proc Natl Acad Sci USA. 1999;96:4710–4717. doi: 10.1073/pnas.96.8.4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrés JA, Maroja LS, Bogdanowicz SM, Swanson WJ, Harrison RG. Molecular evolution of seminal proteins in field crickets. Mol Biol Evol. 2006;23:1574–1584. doi: 10.1093/molbev/msl020. [DOI] [PubMed] [Google Scholar]
- Anisimova M, Nielsen R, Yang Z. Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics. 2003;164:1229–1236. doi: 10.1093/genetics/164.3.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimova M, Nielsen R, Yang Z. Multiple hypothesis testing to detect lineages under positive selection that affects only a few sites. Mol Biol Evol. 2007;24:1219–1228. doi: 10.1093/molbev/msm042. [DOI] [PubMed] [Google Scholar]
- Ardell DH. SCANMS: adjusting for multiple comparisons in sliding window neutrality tests. Bioinformatics. 2004;20:1986–1988. doi: 10.1093/bioinformatics/bth187. [DOI] [PubMed] [Google Scholar]
- Bakker EG, Toomajian C, Kreitman M, Bergelson J. A genome-wide survey of R gene polymorphisms in Arabidopsis. Plant Cell. 2006;18:1803–1818. doi: 10.1105/tpc.106.042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroux U, Gagliardini V, Page DR, Grossniklaus U. Dynamic regulatory interactions of Polycomb group genes: MEDEA autoregulation is required for imprinted gene expression in Arabidopsis. Genes Dev. 2006;20:1081–1086. doi: 10.1101/gad.378106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beechey CV, Cattanach BM, Blake A, Peters J. 2003. MRC Mammalian Genetics Unit Harwell, Oxfordshire World Wide Web site – mouse imprinting data and references. [Google Scholar]
- Begun DJ, Whitley P, Todd BL, Waldrip-Dail HM, Clark AG. Molecular population genetics of male accessory gland proteins in Drosophila. Genetics. 2000;156:1879–1888. doi: 10.1093/genetics/156.4.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghans JAM, Beltman JB, De Boer RJ. MHC polymorphism under host–pathogen coevolution. Immunogenetics. 2004;55:732–739. doi: 10.1007/s00251-003-0630-5. [DOI] [PubMed] [Google Scholar]
- Boyer LA, Latek RR, Peterson CL. The SANT domain: a unique histone-tail-binding module? Nat Rev Mol Cell Biol. 2004;5:158–163. doi: 10.1038/nrm1314. [DOI] [PubMed] [Google Scholar]
- Castelli V, Aury J-M, Jaillon O, et al. (13 co-authors) Whole genome sequence comparisons and “full-length” cDNA sequences: a combined approach to evaluate and improve Arabidopsis genome annotation. Genome Res. 2004;14:406–413. doi: 10.1101/gr.1515604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamary JV, Parmley JL, Hurst LD. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat Rev Genet. 2006;7:98–108. doi: 10.1038/nrg1770. [DOI] [PubMed] [Google Scholar]
- Chanvivattana Y, Bishopp A, Schubert D, Stock C, Moon Y-H, Sung ZR, Goodrich J. Interaction of Polycomb-group proteins controlling flowering in Arabidopsis. Development. 2004;131:5263–5276. doi: 10.1242/dev.01400. [DOI] [PubMed] [Google Scholar]
- Charlesworth D. Balancing selection and its effects on sequences in nearby genome regions. PLoS Genet. 2006;2:e64. doi: 10.1371/journal.pgen.0020064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury AM, Ming L, Miller C, Craig S, Dennis ES, Peacock WJ. Fertilization-independent seed development in Arabidopsis thaliana. Proc Natl Acad Sci USA. 1997;94:4223–4228. doi: 10.1073/pnas.94.8.4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark NL, Swanson WJ. Pervasive adaptive evolution in primate seminal proteins. PLoS Genet. 2005;1:e35. doi: 10.1371/journal.pgen.0010035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot NG, Heijmans CMC, de Groot N, Otting N, de Vos-Rouweler AJM, Remarque EJ, Bonhomme M, Doxiadis GGM, Crouau-Roy B, Bontrop RE. Pinpointing a selective sweep to the chimpanzee MHC class I region by comparative genomics. Mol Ecol. 2008;17:2074–2088. doi: 10.1111/j.1365-294X.2008.03716.x. [DOI] [PubMed] [Google Scholar]
- de Jong TJ, Van Dijk H, Klinkhamer PGL. Hamilton's rule, imprinting and parent–offspring conflict over seed mass in partially selfing plants. J Evol Biol. 2005;18:676–682. doi: 10.1111/j.1420-9101.2004.00856.x. [DOI] [PubMed] [Google Scholar]
- Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–15. [Google Scholar]
- Dybdahl MF, Lively CM. Host–parasite coevolution: evidence for rare advantage and time-lagged selection in a natural population. Evolution. 1998;52:1057–1066. doi: 10.1111/j.1558-5646.1998.tb01833.x. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Fay JC, Benavides JA. Hypervariable noncoding sequences in Saccharomyces cerevisiae. Genetics. 2005;170:1575–1587. doi: 10.1534/genetics.105.042283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wu C-I. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N, Gouy M, Gautier C. SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci. 1996;12:543–548. doi: 10.1093/bioinformatics/12.6.543. [DOI] [PubMed] [Google Scholar]
- Gehring M, Huh JH, Hsieh T-F, Penterman J, Choi Y, Harada JJ, Goldberg RB, Fischer RL. DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell. 2006;124:495–506. doi: 10.1016/j.cell.2005.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Grossniklaus U, Vielle-Calzada J-P, Hoeppner MA, Gagliano WB. Maternal control of embryogenesis by MEDEA, a polycomb group gene in Arabidopsis. Science. 1998;280:446–450. doi: 10.1126/science.280.5362.446. [DOI] [PubMed] [Google Scholar]
- Hagenblad J, Nordborg M. Sequence variation and haplotype structure surrounding the flowering time locus FRI in Arabidopsis thaliana. Genetics. 2002;61:289–298. doi: 10.1093/genetics/161.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer MF, Garrigan D, Wood E, Wilder JA, Mobasher Z, Bigham A, Krenz JG, Nachman MW. Heterogeneous patterns of variation among multiple human X-linked loci: the possible role of diversity-reducing selection in non-Africans. Genetics. 2004;167:1841–1853. doi: 10.1534/genetics.103.025361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haun WJ, Laoueille-Duprat S, O'Connell MJ, Spillane C, Grossniklaus O, Phillips AR, Kaeppler SM, Springer NM. Genomic imprinting, methylation and molecular evolution of maize Enhancer of zeste (Mez) homologs. Plant J. 2007;49:325–337. doi: 10.1111/j.1365-313X.2006.02965.x. [DOI] [PubMed] [Google Scholar]
- He X, Zhang J. Rapid subfunctionalization accompanied by prolonged and substantial neofunctionalization in duplicate gene evolution. Genetics. 2005;169:1157–1164. doi: 10.1534/genetics.104.037051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Kreitman M, Aguadé M. A test of neutral molecular evolution based on nucleotide data. Genetics. 1987;116:153–159. doi: 10.1093/genetics/116.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Nei M. Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature. 1988;335:167–170. doi: 10.1038/335167a0. [DOI] [PubMed] [Google Scholar]
- Hurst LD. Is multiple paternity necessary for the evolution of genomic imprinting? Genetics. 1999;153:509–512. doi: 10.1093/genetics/153.1.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst LD, McVean GT. Growth effects of uniparental disomies and the conflict theory of genomic imprinting. Trends Genet. 1997;13:436–443. doi: 10.1016/s0168-9525(97)01273-0. [DOI] [PubMed] [Google Scholar]
- Innan H. Modified Hudson–Kreitman–Aguadé test and two-dimensional evaluation of neutrality tests. Genetics. 2006;173:1725–1733. doi: 10.1534/genetics.106.056242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jullien PE, Katz A, Oliva M, Ohad N, Berger F. Polycomb group complexes self-regulate imprinting of the polycomb group gene MEDEA in Arabidopsis. Curr Biol. 2006;16:486–492. doi: 10.1016/j.cub.2006.01.020. [DOI] [PubMed] [Google Scholar]
- Kawabe A, Fujimoto R, Charlesworth D. High diversity due to balancing selection in the promoter region of the Medea gene in Arabidopsis lyrata. Curr Biol. 2007;17:1885–1889. doi: 10.1016/j.cub.2007.09.051. [DOI] [PubMed] [Google Scholar]
- Khurana E, Singh JS. Influence of seed size on seedling growth of Albizia procera under different soil water levels. Ann Bot. 2000;86:1185–1192. [Google Scholar]
- Kiyosue T, Ohad N, Yadegari R, et al. (10 co-authors) Control of fertilization-independent endosperm development by the MEDEA polycomb gene in Arabidopsis. Proc Natl Acad Sci USA. 1999;96:4186–4191. doi: 10.1073/pnas.96.7.4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler C, Hennig L, Spillane C, Pien S, Gruissem W, Grossniklaus U. The Polycomb-group protein MEDEA regulates seed development by controlling expression of the MADS-box gene PHERES1. Genes Dev. 2003;17:1540–1553. doi: 10.1101/gad.257403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krannitz PG, Aarsen LW, Dow JM. The effect of genetically based differences in seed size on seedling survival in Arabidopsis thaliana (Brassicaceae) Am J Bot. 1991;78:446–450. [Google Scholar]
- Luo M, Bilodeau P, Koltunow A, Dennis ES, Peacock WJ, Chaudhury AM. Genes controlling fertilization-independent seed development in Arabidopsis thaliana. Proc Natl Acad Sci USA. 1999;96:296–301. doi: 10.1073/pnas.96.1.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarevich G, Leroy O, Akinci U, Schubert D, Clarenz O, Goodrich J, Grossniklaus U, Köhler C. Different Polycomb group complexes regulate common target genes in Arabidopsis. EMBO Rep. 2006;7:947–952. doi: 10.1038/sj.embor.7400760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith J, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res. 1974;23:23–35. [PubMed] [Google Scholar]
- McDonald JH. Improved tests for heterogeneity across a region of DNA sequence in the ratio of polymorphism to divergence. Mol Biol Evol. 1998;15:377–384. doi: 10.1093/oxfordjournals.molbev.a025934. [DOI] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- McVean GT, Hurst LD. Molecular evolution of imprinted genes, no evidence for antagonistic coevolution. Proc R Soc Lond B Biol Sci. 1997;264:739–746. doi: 10.1098/rspb.1997.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki A, Takeda Y, Iwasa Y. The evolution of genomic imprinting. Genetics. 1996;144:1283–1295. doi: 10.1093/genetics/144.3.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore T, Haig D. Genomic imiprinting in mammalian development – a parental tug-of-war. Trends Genet. 1991;7:45–49. doi: 10.1016/0168-9525(91)90230-N. [DOI] [PubMed] [Google Scholar]
- Moore T, Mills W. Imprinting and monogamy. Nat Genet. 1999;22:130–131. doi: 10.1038/9639. [DOI] [PubMed] [Google Scholar]
- Muller M-H, Leppälä J, Savolainen O. Genome-wide effects of postglacial colonization in Arabidopsis lyrata. Heredity. 2008;100:47–58. doi: 10.1038/sj.hdy.6801057. [DOI] [PubMed] [Google Scholar]
- Ng DWK, Wang T, Chandrasekharan MB, Aramayo R, Kertbundit S, Hall TC. Plant SET domain-containing proteins: structure, function and regulation. Biochim Biophys Acta. 2007;1769:316–329. doi: 10.1016/j.bbaexp.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg M, Hu TT, Ishino Y, et al. (23 co-authors) The pattern of polymorphism in Arabidopsis thaliana. PLoS Biol. 2005;3:e196. doi: 10.1371/journal.pbio.0030196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham P, Ohta T. Population biology of antigen presentation by MHC class I molecules. Science. 1996;272:67–74. doi: 10.1126/science.272.5258.67. [DOI] [PubMed] [Google Scholar]
- Parmley JL, Chamary JV, Hurst LD. Evidence for purifying selection against synonymous mutations in mammalian exonic splicing enhancers. Mol Biol Evol. 2006;23:301–309. doi: 10.1093/molbev/msj035. [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- Ramos-Onsins SE, Stranger BE, Mitchell-Olds T, Aguade M. Multilocus analysis of variation and speciation in the closely related species Arabidopsis halleri and A. lyrata. Genetics. 2004;166:373–388. doi: 10.1534/genetics.166.1.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rom DM. A sequentially rejective test procedure based on a modified Bonferroni inequality. Biometrika. 1990;77:663–665. [Google Scholar]
- Ross-Ibarra J, Wright SI, Foxe JP, Kawabe A, Derose-Wilson L, Gos G, Charlesworth D, Gaut BS. Patterns of polymorphism and demographic history in natural populations of Arabidopsis lyrata. PLoS One. 2008;3:e2411. doi: 10.1371/journal.pone.0002411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J, Rozas R. DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics. 1999;15:174–175. doi: 10.1093/bioinformatics/15.2.174. [DOI] [PubMed] [Google Scholar]
- Sardiu ME, Cheung MS, Yu YK. Cysteine–cysteine contact preference leads to target-focusing in protein folding. Biophys J. 2007;93:938–951. doi: 10.1529/biophysj.106.097824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid KJ, Sorensen TR, Stracke R, Torjek O, Altmann T, Mitchell-Olds T, Weisshaar B. Large-scale identification and analysis of genome-wide single-nucleotide polymorphisms for mapping in Arabidopsis thaliana. Genome Res. 2003;13:1250–1257. doi: 10.1101/gr.728603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NGC, Hurst LD. Molecular evolution of an imprinted gene: repeatability of patterns of evolution within the mammalian insulin-like growth factor type II receptor. Genetics. 1998;150:823–833. doi: 10.1093/genetics/150.2.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen MB, Chaudhury AM, Robert H, Bancharel E, Berger F. Polycomb group genes control pattern formation in plant seed. Curr Biol. 2001;11:277–281. doi: 10.1016/s0960-9822(01)00072-0. [DOI] [PubMed] [Google Scholar]
- Spillane C, MacDougall C, Stock C, Kohler C, Vielle-Calzada JP, Nunes SM, Grossniklaus U, Goodrich J. Interaction of the Arabidopsis Polycomb group proteins FIE and MEA mediates their common phenotypes. Curr Biol. 2000;10:1535–1538. doi: 10.1016/s0960-9822(00)00839-3. [DOI] [PubMed] [Google Scholar]
- Spillane C, Schmid KJ, Laoueille-Duprat S, Pien S, Escobar-Restrepo JM, Baroux C, Gagliardini V, Page DR, Wolfe KH, Grossniklaus U. Positive darwinian selection at the imprinted MEDEA locus in plants. Nature. 2007;448:U349–U348. doi: 10.1038/nature05984. [DOI] [PubMed] [Google Scholar]
- Springer NM, Danilevskaya ON, Hermon P, Helentjaris TG, Phillips RL, Kaeppler HF, Kaeppler SM. Sequence relationships, conserved domains and expression patterns for maize homologs of the polycomb group genes E(z), esc, and E(Pc) Plant Physiol. 2002;128:1332–1345. doi: 10.1104/pp.010742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl EA, Dwyer G, Mauricio R, Kreitman M, Bergelson J. Dynamics of disease resistance polymorphism at the Rpm1 locus of Arabidopsis. Nature. 1999;400:667–671. doi: 10.1038/23260. [DOI] [PubMed] [Google Scholar]
- Swanson WJ, Clark AG, Waldrip-Dail HM, Wolfner MF, Aquadro CF. Evolutionary EST analysis identifies rapidly evolving male reproductive proteins in Drosophila. Proc Natl Acad Sci USA. 2001;98:7375–7379. doi: 10.1073/pnas.131568198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford DL. Phylogenetic analysis using parsimony (and other methods) Sunderland (MA): Sinauer Associates; 2003. Version 4. PAUP*. [Google Scholar]
- Tajima F. Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983;105:437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical-method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata N, Nei M. Allelic genealogy under overdominant and frequency-dependent selection and polymorphism of major histocompatibility complex loci. Genetics. 1990;124:967–978. doi: 10.1093/genetics/124.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z, Shon AM, Ober C. Evidence of balancing selection at the HLA-G promoter region. Hum Mol Genet. 2005;14:3619–3628. doi: 10.1093/hmg/ddi389. [DOI] [PubMed] [Google Scholar]
- Tang C, Toomajian C, Sherman-Broyles S, Plagnol V, Guo YL, Hu TT, Clark RM, Nasrallah JB, Weigel D, Nordborg M. The evolution of selfing in Arabidopsis thaliana. Science. 2007;317:1070–1072. doi: 10.1126/science.1143153. [DOI] [PubMed] [Google Scholar]
- Tiffin P, Hahn MW. Coding sequence divergence between two closely related plant species: Arabidopsis thaliana and Brassica rapa ssp pekinensis. J Mol Evol. 2002;54:746–753. doi: 10.1007/s00239-001-0074-1. [DOI] [PubMed] [Google Scholar]
- Tsaur SC, Ting CT, Wu CI. Positive selection driving the evolution of a gene of male reproduction, Acp26Aa, of Drosophila: II. Divergence versus polymorphism. Mol Biol Evol. 1998;15:1040–1046. doi: 10.1093/oxfordjournals.molbev.a026002. [DOI] [PubMed] [Google Scholar]
- Vinkenoog R, Bushell C, Spielman M, Adams S, Dickinson HG, Scott RJ. Genomic imprinting and endosperm development in flowering plants. Mol Biotechnol. 2003;25:149–184. doi: 10.1385/MB:25:2:149. [DOI] [PubMed] [Google Scholar]
- Wilkins JF, Haig D. What good is genomic imprinting: the function of parent-specific gene expression. Nat Rev Genet. 2003;4:1–10. doi: 10.1038/nrg1062. [DOI] [PubMed] [Google Scholar]
- Williams EJB, Pal C, Hurst LD. The molecular evolution of signal peptides. Gene. 2000;253:313–322. doi: 10.1016/s0378-1119(00)00233-x. [DOI] [PubMed] [Google Scholar]
- Wilson DJ, McVean G. Estimating diversifying selection and functional constraint in the presence of recombination. Genetics. 2006;172:1411–1425. doi: 10.1534/genetics.105.044917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf JB, Hager R. A maternal–offspring coadaptation theory for the evolution of genomic imprinting. PLoS Biol. 2006;4:e380. doi: 10.1371/journal.pbio.0040380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SI, Charlesworth B. The HKA test revisited: a maximum-likelihood-ratio test of the standard neutral model. Genetics. 2004;168:1071–1076. doi: 10.1534/genetics.104.026500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SI, Gaut BS. Molecular population genetics and the search for adaptive evolution in plants. Mol Biol Evol. 2005;22:506–519. doi: 10.1093/molbev/msi035. [DOI] [PubMed] [Google Scholar]
- Wright SI, Lauga B, Charlesworth D. Subdivision and haplotype structure in natural populations of Arabidopsis lyrata. Mol Ecol. 2003;12:1247–1263. doi: 10.1046/j.1365-294x.2003.01743.x. [DOI] [PubMed] [Google Scholar]
- Yadegari R, Kinoshita T, Lotan O, et al. (11 co-authors) Mutations in the FIE and MEA genes that encode interacting polycomb proteins cause parent-of-origin effects on seed development by distinct mechanisms. Plant Cell. 2000;12:2367–2381. doi: 10.1105/tpc.12.12.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19:908–917. doi: 10.1093/oxfordjournals.molbev.a004148. [DOI] [PubMed] [Google Scholar]
- Zhang JZ, Nielsen R, Yang ZH. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- Zhang X, Clarenz O, Cokus S, Bernatavichute YV, Pellegrini M, Goodrich J, Jacobsen SE. Whole-genome analysis of histone H3 Lysine 27 trimethylation in Arabidopsis. PLoS Biol. 2007;5:e129. doi: 10.1371/journal.pbio.0050129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.