

Abstract
We present a Moran-model approach to modeling general multiallelic selection in a finite population and show how it may be used to develop theoretical models of biological systems of balancing selection such as plant gametophytic self-incompatibility loci. We propose new expressions for the stationary distribution of allele frequencies under selection and use them to show that the continuous-time Markov chain describing allele frequency change with exchangeable selection and Moran-model reproduction is reversible. We then use the reversibility property to derive the expected allele frequency spectrum in a finite population for several general models of multiallelic selection. Using simulations, we show that our approach is valid over a broader range of parameters than previous analyses of balancing selection based on diffusion approximations to the Wright–Fisher model of reproduction. Our results can be applied to any model of multiallelic selection in which fitness is solely a function of allele frequency.
NATURAL selection has long been a topic of interest in population genetics, yet the stochastic theory of genes under selection remains underdeveloped compared to the theory of neutral genes. Due to the interplay of stochastic and deterministic forces, models of selection present analytical challenges beyond those of neutral models, although a great deal of progress has been made with models that use diffusion approximations to a Wright–Fisher model of reproduction. Diffusion approximations with selection are, however, sometimes difficult to employ and always require assumptions about population parameters for tractability. These limitations suggest that there may be value in developing new methods of solving the problem of selection in a finite population, and here we do so using a Moran model of reproduction in place of the familiar Wright–Fisher model. Our approach has two major advantages over previous models: general applicability to a wide variety of selection models and accuracy over a broad range of parameter values. In this work, we propose new expressions for the full stationary distributions of allele frequencies under multiallelic selection, as well as expressions for average allele frequency distributions.
We restrict our attention to exchangeable models of selection, meaning that relabeling the alleles will not change selective outcomes and thus that selection will be a function of allele frequency rather than allele identity. Many models of selection can be transformed into frequency-dependent forms (Denniston and Crow 1990), and some common models of selection have the desired property of exchangeability. For example, symmetric overdominant selection, in which heterozygotes have a selective advantage over homozygotes but the specific genotype of homozygote or heterozygote has no further selective effect, can be expressed as frequency-dependent selection on individual (exchangeable) alleles, although the direct selection is actually on diploid genotypes. Many other proposed models of multiallelic balancing selection, in which substantial variation is maintained by selection, can be viewed in this way. Such models have been of particular interest because of the potential application to highly multiallelic systems found in nature, such as self-incompatibility (SI) loci in plants and the major histocompatibility complex (MHC) loci in vertebrates, and the desire to analyze these systems is a motivation for the present work. We now review some of the population genetic theory related to these systems.
Early in the history of population genetics, Wright (1939) presented a somewhat controversial stochastic model of gametophytic self-incompatibility (GSI) genes, sparking much further theoretical and empirical work. An analytic theory of multiallelic symmetric overdominance was developed along similar lines to this early model (Kimura and Crow 1964; Takahata 1990) and has been used as an approximation to the unknown mode of selection in the MHC (Takahata et al. 1992). Drawing insights from these first two applications, other biological systems where balancing selection was posited, including sex determination in honeybees (Yokoyama and Nei 1979), fungal mating systems (May et al. 1999), and heterokaryon incompatibility in fungi (Muirhead et al. 2002), have also been modeled successfully using closely related approaches. Progress has been made in using these models to address genealogical (Takahata 1990; Vekemans and Slatkin 1994) and demographic (Muirhead 2001) questions, as well as extending the models into more complex modes of selection (Uyenoyama 2003) and reproduction (Vallejo-Marin and Uyenoyama 2008).
Models of genetic variation under balancing selection have traditionally been focused on specific systems, such that extensions require entirely new analyses, and have also included a number of simplifying assumptions in the interest of mathematical tractability. For example, the symmetric overdominance model has been strongly criticized as an unrealistic approximation of MHC evolution (Paterson et al. 1998; Hedrick 2002; Penn et al. 2002; Ilmonen et al. 2007; Stoffels and Spencer 2008), and yet it has proved difficult to make finite-population models of any of the more realistic frequency dependence schemes using the same approaches. A constraint on further progress is the fact that the standard model of stochastic population genetics, the Wright–Fisher model, is in fact quite difficult to analyze.
The Wright–Fisher model of reproduction employs nonoverlapping generations, so that for a diploid population of size N, all 2N allele copies are chosen simultaneously when forming a new generation of individuals. While it is straightforward to describe this reproduction scheme mathematically as a discrete-time Markov chain, that chain unfortunately appears intractable even in simple cases (Ewens 2004). Traditionally, then, diffusion approximations have been used to obtain quantities of interest, such as the equilibrium expected number of alleles, allele frequency spectra, and fixation probabilities and times. Diffusion approximations are derived in the limit 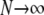 , but are applicable to problems of finite N, provided that the strengths of other forces such as mutation and selection can be assumed to be weak, of O(N−1) (Ewens 2004). Watterson (1977) derived such a diffusion approximation for multiallelic symmetric overdominance using these assumptions. More recently, as interest in population genetics has turned to problems of inference, Grote and Speed (2002) considered sampling probabilities under the diffusion approximation for symmetric overdominance, while Donnelly et al. (2001) and Stephens and Donnelly (2003) proposed computational methods for some asymmetric models.
, but are applicable to problems of finite N, provided that the strengths of other forces such as mutation and selection can be assumed to be weak, of O(N−1) (Ewens 2004). Watterson (1977) derived such a diffusion approximation for multiallelic symmetric overdominance using these assumptions. More recently, as interest in population genetics has turned to problems of inference, Grote and Speed (2002) considered sampling probabilities under the diffusion approximation for symmetric overdominance, while Donnelly et al. (2001) and Stephens and Donnelly (2003) proposed computational methods for some asymmetric models.
Although strong selection can be modeled using diffusion approximations by making the product of the population size and the selection coefficient (Ns) large, the assumption of weak selection is not in fact appropriate for the canonical biological systems of balancing selection. Specifically, selection coefficients are defined by the differences in fitness (the expected number of offspring) among individuals in the population at a given time. These differences may be large in systems such as GSI, where the fitness of a very common allele may be very small while the fitness of other alleles may be greater than one.
In an attempt to deal with the extremely strong selection of gametophytic self-incompatibility, Wright's (1939) original model focused attention on the dynamics of a single representative allele. He collapsed the influence of all other alleles into a single summary statistic: the homozygosity, F, which is a function of the frequencies of all alleles, and which Wright (1939) assumed to be constant. The analysis is essentially that of a two-allele system, using a one-dimensional diffusion analysis. This approach, while shown by simulation to be very effective in the appropriate parameter range (Ewens and Ewens 1966), received substantial criticism on mathematical grounds (Fisher 1958; Moran 1962; Ewens 1964b). Ewens (1964b), in particular, objected to the use of diffusion theory for GSI, pointing out that strong frequency-dependent selection violates the diffusion requirement that both the mean and the variance of the change in allele frequencies be small and of O(N−1). Ewens (1964a) then applied Wright's basic one-dimensional diffusion approach to modeling symmetric overdominance, but assumed that selection was weak and of O(N−1) to stay within the strict limits of the diffusion approximation.
Kimura and Crow (1964) and Wright (1966), on the other hand, presented alternative one-dimensional diffusion approximations to symmetric overdominance, closer in spirit to Wright's original model of GSI, that did not make the weak-selection assumption. Watterson (1977) was concerned about both the inconsistencies of the approximations used in these models and the treatment of F as a constant rather than as a random variable dependent upon allele frequencies. Using his own multiallelic diffusion approximation for symmetric overdominance (Watterson 1977), he derived an alternative (small-Ns) approximation to the frequency of a single representative allele. We consider this approximation, as well as the best-known one-dimensional symmetric overdominance diffusion, the strong-selection approximation of Kimura and Crow (1964), in comparison with our alternative approach to deriving allele frequency spectra under general multiallelic selection with exchangeable alleles.
To avoid the approximations required to employ Wright–Fisher/diffusion-based methods, we turn to an alternative model of reproduction in a finite population: the overlapping-generations model of Moran (1962). In the Moran model, a single allele copy dies and another reproduces in each time step, rather than all 2N allele copies simultaneously being replaced by offspring each generation. As in the Wright–Fisher model, this reproduction scheme is represented mathematically by a Markov chain. Unlike the Wright–Fisher model, however, the Moran model can sometimes yield tractable, exact solutions to the underlying Markov chain, without the need to resort to diffusion approximations. We exploit this trait to develop a new stochastic theory of multiallelic selection with minimal dependence on assumptions about population parameter values. Our method has the additional benefit of being flexible: it can accommodate any exchangeable model of multiallelic selection and either of two general models of parent-independent mutation, the infinite-alleles and k-allele models of mutation. Our Moran-model predictions agree well with the results of Wright–Fisher simulations.
MODEL
We consider a general model of multiallelic selection, using a continuous-time Moran model of reproduction to incorporate random genetic drift. We explore in detail an infinite-alleles model of mutation, with selection at either death (viability selection) or reproduction (fecundity selection) and where selection depends only on the frequency of an allele, rather than its identity. In appendix b, we present complementary results for a k-allele mutation model, also with two possible modes of exchangeable selection.
Structure of the Moran-model Markov chain:
A single step in the Moran model corresponds to the death of one allele copy and the reproduction of one copy, possibly the same one chosen to die. Mutation may also occur during reproduction. Selection may be introduced at either point, and because it may be more convenient to use one or the other in specific biological situations, we formally develop separate models for selection at death and selection at reproduction. Regardless of when selection acts, we consider one step of the Moran model as consisting of one death event, followed by one reproduction/mutation event. The relative per-copy death rate of an allele in i copies is denoted μi, and the relative per-copy reproduction rate of an i-copy allele is denoted λi. These determine the rates at which allele copies are chosen to die (in the selection-at-death version of the model) and to reproduce (in the selection-at-reproduction version), respectively. For death or reproduction events not affected by selection, allele copies are chosen to die or reproduce with equal probability regardless of type.
We imagine a diploid population of size N, so that there are a total of 2N allele copies in the population. Under the infinite-alleles model of mutation, every new mutation produces a novel allele, and we assume that the probability that an allele copy mutates to a novel allele is u per reproduction event. The state of a population is recorded in a vector, X = {X1, X2,…, X2N}, where Xi denotes the number of alleles present in i copies. Thus, 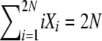 . We are interested in the state of the population at stationarity and consider both the full stationary distribution π(X) = Pr[X1 = x1, X2 = x2,…, X2N = x2N] and the expected values both of the random variables Xi, 1 ≤ i ≤ 2N, and of the products XiXj for all i and j. The vector of expected numbers of alleles in each frequency class corresponds to the average allele frequency spectrum, φ(x)dx, a classic object of population genetic theory usually obtained using diffusion approximations.
. We are interested in the state of the population at stationarity and consider both the full stationary distribution π(X) = Pr[X1 = x1, X2 = x2,…, X2N = x2N] and the expected values both of the random variables Xi, 1 ≤ i ≤ 2N, and of the products XiXj for all i and j. The vector of expected numbers of alleles in each frequency class corresponds to the average allele frequency spectrum, φ(x)dx, a classic object of population genetic theory usually obtained using diffusion approximations.
To analyze evolutionary dynamics, we construct a continuous-time Markov chain on X, the state vector describing the population. A single transition in this Markov chain corresponds to a single step of the Moran model, death followed by reproduction/mutation. The underlying Markov chain is complicated, in part because the number of possible transitions that can be experienced by a particular X vector is very large. However, these transitions can be categorized into 10 distinct types depending on their effects on X (Table 1). To obtain the rate of a particular transition, we consider the two events in a step, death and reproduction, independently. For example, if in one step a copy from a 4-copy allele were picked to die, and a copy from a 10-copy allele were picked to reproduce (without mutation), this would represent a transition of type 5 with i = 4 and j = 10. The population will then have gone from state X = {x1, x2, x3, x4, … , x10, x11, … , x2N} to state X′ = {x1, x2, x3 + 1, x4 − 1,…, x10 − 1, x11 + 1, … , x2N}. In the selection at death model, the rate of such a transition occurring, given X, is
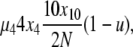 |
and the equivalent rate in the selection at reproduction model is
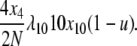 |
With the 10 possible transition types and their effects on X as a complete description of the dynamics of the Markov chain, it is possible to derive equilibrium properties of the system, including stationary distributions and average allele frequency spectra. We are able to obtain exact results for these quantities using the reversibility property of the Moran model with multiallelic selection, which we now prove.
TABLE 1.
Transition types in the infinite-alleles model of mutation
| Event (single time step in a Moran model) | Effect on X | Rate (death selection) | Rate (reproduction selection) | |
|---|---|---|---|---|
| 1 | xi dies, xi−2 reproduces, no mutation | x′i−2 = xi−2 − 1 | 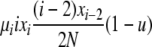 |
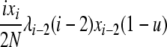 |
| (i > 2) | x′i−1 = xi−1 + 2 | |||
| x′i = xi − 1 | ||||
| 2 | xi dies, xi−1 reproduces, no mutation | — | 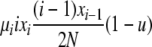 |
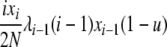 |
| (i > 1) | ||||
| 3 | xi dies, xi (same) reproduces, no mutation | — | 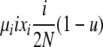 |
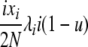 |
| 4 | xi dies, xi (different) reproduces, no mutation | x′i−1 = xi−1 + 1 | 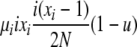 |
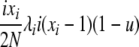 |
| (i > 1) | x′i = xi − 2 | |||
| x′i+1 = xi+1 + 1 | ||||
| 5 | xi dies, xj reproduces, no mutation | x′i−1 = xi−1 + 1 | 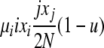 |
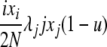 |
| (i > 1, j ≠ i, i − 1, i − 2) | x′i = xi − 1 | |||
| x′j = xj − 1 | ||||
| x′j+1 = xj+1 + 1 | ||||
| 6 | x1 dies, xj reproduces, no mutation | x′1 = x1 − 1 | 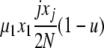 |
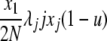 |
| (j ≠ 1) | x′j = xj − 1 | |||
| x′j+1 = xj+1 + 1 | ||||
| 7 | x1 dies, x1 (different) reproduces, no mutation | x′1 = x1 − 2 | 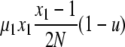 |
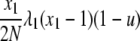 |
| x′2 = x2 + 1 | ||||
| 8 | xi dies, anything reproduces, mutation | x′1 = x1 + 1 | μiixiu | 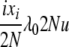 |
| (i > 1) | x′i−1 = xi−1 + 1 | |||
| x′i = xi − 1 | ||||
| 9 | x2 dies, anything reproduces, mutation | x′1 = x1 + 2 | μ22x2u | 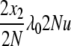 |
| x′2 = x2 − 1 | ||||
| 10 | x1 dies, anything reproduces, mutation | — | μ1x1u | 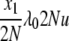 |
The effects of each of the 10 transition types on X = {x1, x2,…, x2N} and their rates under the two modes of selection.
Stationary distributions and reversibility:
Based on a product form of the stationary distribution of allele counts as in Moran (1962), e.g., see p. 134, we posit that the equilibrium distributions, π(X), of the population state vector under infinite-alleles mutation are of the form
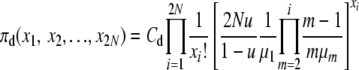 |
(1) |
when selection occurs at death and
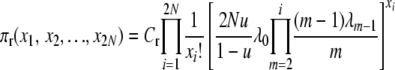 |
(2) |
when selection occurs at reproduction. The normalization constants, Cd and Cr, are unknown functions of selection and population parameters. These probability distributions are difficult to work with directly, in part because of the unknown constants, but they can be verified as correct, and used to show that their respective Markov chains are reversible, by a simple proof. With reversibility, at stationarity we have
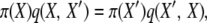 |
(3) |
where q(i, j) is the rate of transition from state i to state j in the chain. If our posited π-distributions satisfy all such relationships, the stationary distributions are verified, and the processes are reversible; see Theorem 1.3 in Kelly (1979).
Using Table 1, we can group transitions into classes that change X in similar ways: type 1 and type 4 alter the counts in three adjacent elements, types 6 and 8 both alter x1 and two adjacent elements, types 7 and 9 alter x1 and x2, and type 5 transitions alter two pairs of (nonoverlapping) adjacent elements, xi−1 and xi, and xj and xj+1. Each one-step transition can be undone by another one-step transition within the same class. For example, a type 1 transition with i = 6 is reversed by a type 4 transition with i = 5, and a type 6 transition with j = 5 is reversed by a type 8 transition with i = 6.
We can use these four transition classes to present a set of detailed balance equations (3) for the process at stationarity, and we use those balance equations to validate our posited stationary distributions and verify reversibility for the infinite-alleles mutation models in appendix a. We also present similar reversibility arguments for a k-allele model of mutation, in appendix b. There we derive both stationary distributions analogous to (1) and (2) and average allele frequency spectra for that mutation model, as we now do for infinite-alleles mutation.
Average allele frequency spectra:
We begin our derivation of average allele frequency spectra with an identity,
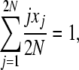 |
which is true for every state vector X = {x1, x2, … , x2N}. Then, we have
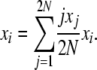 |
Using a stationary distribution π(X) and taking expectations at equilibrium, this becomes
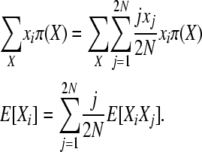 |
(4) |
We derive the equilibrium expected values E[Xi] by first finding the expectations of the products E[XiXj] and then applying the equation above. The approach we use is to consider again the detailed balance equations and take expectations at stationarity, yielding a relatively simple system of equations relating the E[Xi]'s and E[XiXj]'s. This system of equations can then be solved numerically.
Selection at death:
The first balance equation uses transitions of type 1 and type 4. The total expected rate of type 1 transitions at stationarity is
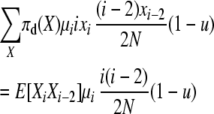 |
and the total reverse rate of corresponding type 4 transitions (that would on average exactly undo the type 1 transitions above) is
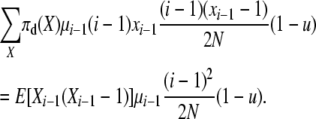 |
With reversibility these total rates are equal so that
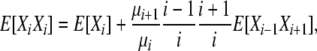 |
(5) |
giving us our first expression for the relationship of the random variables Xi−2, Xi−1, and Xi. Two more such expressions are necessary and are derived from two more balance equations. Balancing two type 5 transitions and taking expectations gives
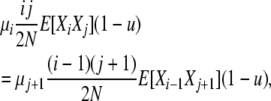 |
where i > 1, j ≠ i, i − 1, i − 2, so that
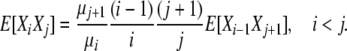 |
(6) |
The last necessary pair required equates a type 6 transition and a type 8 transition for
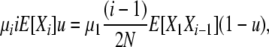 |
for i > 1, and thus
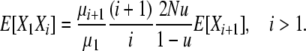 |
(7) |
Combining Equations 5–7, we have
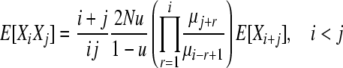 |
(8) |
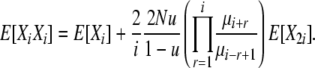 |
(9) |
From the identity (4) and Equations 8 and 9, we arrive finally at an expression for the average allele frequency spectrum under infinite-alleles mutation and selection at death:
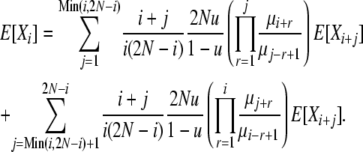 |
(10) |
While not a closed-form solution, this suffices to calculate numerically the vector of E[Xi] values. We first calculate all terms recursively relative to E[X2N] and then normalize by 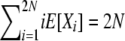 . Note that once we have computed expectations E[Xi], we can also compute Var[Xi] and Cov[XiXj], through use of (8)–(10). In addition, we can use the last element of the vector, E[X2N], to obtain the normalization constant Cd of the stationary distribution πd(X), using E
. Note that once we have computed expectations E[Xi], we can also compute Var[Xi] and Cov[XiXj], through use of (8)–(10). In addition, we can use the last element of the vector, E[X2N], to obtain the normalization constant Cd of the stationary distribution πd(X), using E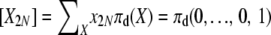 . With this, we now have a fully specified stationary distribution (1) for the Markov chain.
. With this, we now have a fully specified stationary distribution (1) for the Markov chain.
Selection at reproduction:
Analysis for this model proceeds along a very similar line to that for the previous model. The rates of the 10 types of transitions in this model are listed in the last column of Table 1, and reversibility of the process is formally verified in appendix a. We can use the same fundamental relation (4) for the expectations and again focus on using the detailed balance equations to derive expressions for the cross-product expectations. Using the same informative pairs of transitions as before and the transition rates for this model (Table 1), we find
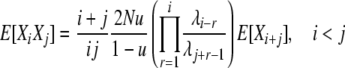 |
(11) |
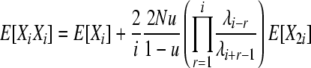 |
(12) |
and
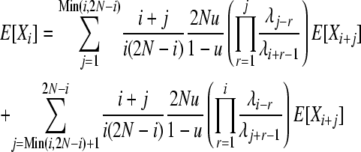 |
(13) |
so that we may calculate E[Xi] values as before.
RESULTS
Comparison to results from diffusion theory and simulations:
We compare results from our approach to those from existing stochastic models of balancing selection, beginning with the well-studied case of symmetric overdominance. Watterson (1977) assumed very weak selection to derive his multidimensional diffusion approximation to symmetric overdominance. In this case, weak means that the fitness advantage of heterozygotes, s, is O(N−1). Analysis of the full diffusion model is impractical, and Watterson (1977) provided the following approximation for the expected one-dimensional allele frequency spectrum under an infinite-alleles model of mutation,
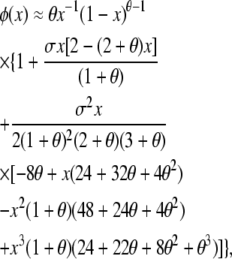 |
which is valid for small σ = 2Nes, where Ne is the effective population size, and θ = 4Neu. φ(x)dx is defined as the equilibrium number of alleles expected in the frequency class (x, x + dx) and is equivalent to our vector of E[Xi] values with an appropriate dx. We have kept multiple terms in the Taylor expansion above to improve the accuracy of the approximation.
Kimura and Crow (1964) assumed strong selection to obtain a different, one-dimensional, diffusion approximation for multiallelic symmetric overdominance and infinite-alleles mutation:
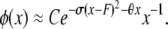 |
θ and σ are as before, except that in this model s represents the decrease in fitness of homozygotes. The random variable F is the homozygosity of the population (the sum of squared allele frequencies), which Kimura and Crow (1964) assumed to be a constant at equilibrium under strong selection. When evaluating Kimura and Crow's (1964) diffusion, instead of using one of the available closed-form approximations for the two unknown constants C and F (requiring additional assumptions), we used Mathematica 7.0 (Wolfram 2008) to numerically integrate the equations
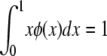 |
and
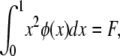 |
solving simultaneously for C and F. The intent was to provide a solution that was minimally dependent on approximations beyond the assumptions inherent in the strong-selection, one-dimensional diffusion approach itself.
To assess the accuracy of the Moran model approach developed here, relative to the Wright–Fisher model diffusions above, we performed extensive individual-based simulations of Wright–Fisher populations with symmetric overdominant selection and infinite-alleles mutation. The simulations use a discrete-time, nonoverlapping generation framework, with reproduction and selection implemented as follows. In each generation, tentative new diploid individuals are created by drawing two allele copies in a multinomial draw from the previous generation's gamete pool (with each allele copy given a chance to mutate to a novel allele with probability u). The individual created is given a fitness value according to its genotype and is accepted or rejected as a member of the next generation according to the value of a random number drawn and compared to the fitness value. The process is repeated for all N individuals in each generation. Simulations were begun with triallelic populations and run for at least 100,000 generations, at which point allele frequencies were recorded. To ensure that the data were being sampled at stationarity, allele number and homozygosity were recorded at regular time points during the run. All sets of simulations appeared to have reached stationarity for average allele number and homozygosity by generation 20,000.
Comparisons between the Moran model and the Wright–Fisher model must account for the factor of 2 difference in timescale that results from differences in the distributions of the numbers of offspring per individual between these two modes of reproduction (Feldman 1966). All comparisons among the two Wright–Fisher diffusions, the Moran model prediction, and the simulation results adjust for this difference, as well as for the slight differences in the selection model between the two diffusion approximations. In all cases the N reported is the N of the Moran model, so that, for example, the corresponding Wright–Fisher simulation would be of a population of size N/2. The s reported is the s of the following Moran model implementation of symmetric overdominance.
Symmetric overdominant selection in the Moran model is easily imposed through death-step selection. Allele copies in heterozygotes die at rate 1, and allele copies in homozygotes die at rate 1 + s. Assuming Hardy–Weinberg proportions, a copy of an allele whose population frequency is i/2N will find itself in a homozygote with probability i/2N and in a heterozygote with probability 1 − i/2N. This leads to
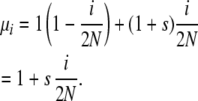 |
The s used here is equivalent to the s of the Kimura–Crow diffusion model.
Figure 1 shows three comparisons between the simulations and the various analytic predictions for average allele frequency spectra. In Figure 1A (where N = 2000, s = 0.001, and u = 10−5) selection is relatively weak (σ = 2) and Watterson's weak-selection diffusion (dotted line) tracks closely with the simulations (solid line, average of 10,000 iterations). The Moran model solution (thick dots) also tracks the simulations, while the Kimura–Crow strong-selection diffusion (dashed line) does not. In Figure 1C (where N = 2000, s = 0.01, and u = 10−5), selection is strong (σ = 20) and the Watterson diffusion no longer performs well, while the Kimura–Crow diffusion does. Again, the Moran model also accurately predicts the outcome of simulations. Figure 1B shows an intermediate situation (N = 2000, s = 0.005, u = 10−5) in which neither diffusion approximation is able to predict the results of the simulations, but the Moran model predicts simulation behavior very accurately.
Figure 1.—

Correspondence between Moran and diffusion predictions and accuracy of predictions as assessed by simulations (averaged over 10,000 iterations). Dotted line, Watterson diffusion; dashed line, Kimura–Crow diffusion; thick dots, Moran solution; solid line, simulations. Symmetric overdominant selection is shown. Moran model parameters: (A) N = 2000, s = 0.001, u = 10−5; (B) N = 2000, s = 0.005, u = 10−5; (C) N = 2000, s = 0.01, u = 10−5.
Application to two specific types of selection:
Plant self-incompatibility:
GSI is a genetic system by which some plant species ensure outcrossing. A compatible mating can result when there are no alleles in common at the incompatibility locus (S locus) between haploid pollen (male gamete) and diploid stigma (female reproductive structure). For example, if a plant has diploid genotype AiAj at the S locus, its stigmas will express that diploid genotype and the ovules of the plant may be fertilized only by pollen bearing a different allele Ak, k ≠ i, j. Pollen expressing Ai or Aj landing on an AiAj stigma will trigger an incompatibility reaction, so that no zygote will be formed.
This type of mechanism, in a Moran model framework, naturally suggests selection at the reproduction step. We approximate GSI by assuming that selection occurs only through the male part and that the reproductive success of an allele copy in pollen (relative to the success of a copy in a female gamete) is directly proportional to the frequency of diploid plants not containing that allele. Because all plants are heterozygotes under GSI, the frequency of plants containing an allele in frequency i/2N is simply i/N. Thus, we can approximate the reproductive rate of an allele copy in frequency i/2N by
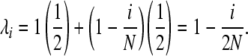 |
(14) |
This approximation lacks some of the notable features of GSI. In particular, it does not require an overall number of alleles ≥3 (as required for a functional GSI system), and it does not restrict the allele frequencies to be ≤0.5 (which follows directly from the observation that all individuals in a GSI system must be heterozygotes). Nevertheless, this simple formulation performs well as judged by simulations.
The simulations shown in Figures 2 and 3 are again of Wright–Fisher (nonoverlapping generations) reproduction with an appropriate population size conversion and in this case are individual-based simulations of gametophytic plant self-incompatibility under an infinite-alleles model of mutation. GSI was simulated by the following scheme: a diploid female parent and haploid pollen were picked randomly from the population and tested for compatibility. If they were compatible, a new zygote was generated from the possible gamete combinations; if incompatible, the pollen was discarded and a new pollen gamete picked until a compatible mating was achieved. Mutation to novel alleles occurred with probability u per gamete per generation.
Figure 2.—

Accuracy of self-incompatibility approximation assessed by simulation: average allele frequency spectrum. Solid line, Moran solution using (14) for N = 2000, u = 10−6; dashed line, diffusion approximations for gametophytic self-incompatibility, Ne = 1000, u = 10−6; dots, individual-based simulations of a population (averaged over 10,000 independent iterations) with Wright–Fisher reproduction (nonoverlapping generations) and gametophytic self-incompatibility, Ne = 1000, u = 10−6.
Figure 3.—

Accuracy of self-incompatibility approximation assessed by simulation: allele number. Shaded triangles, expected total number of alleles from the Moran solution for N = 1000 and mutation rates u = 10−6, 10−5, 10−4, and 10−3; shaded stars, expected total number of alleles from the Wright–Fisher model diffusion approximation with Ne = 500 and the appropriate mutation rates. Simulation results, from individual-based forward simulations of a Wright–Fisher population (Ne = 500) with gametophytic self-incompatibility, are represented by box-and-whisker diagrams.
Figure 2 shows the average allele frequency spectrum predicted using the Moran approximation (14) for a population size of N = 2000 and mutation rate of u = 10−6 and compares it to the average spectrum obtained from forward simulations (25,000 iterations) and to the Wright–Fisher model diffusion prediction. The diffusion prediction was calculated following Uyenoyama's (2003) implementation of Wright's model. Specifically, the effective number of common alleles (n) was obtained by solving
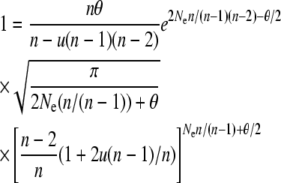 |
numerically and then substituting the resulting value of n into the appropriate expression for φ(x),
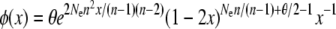 |
(Uyenoyama 2003; Vallejo-Marin and Uyenoyama 2008).
Figure 3 compares the total number of alleles predicted by both the Moran approach and the diffusion approximation to simulation results. To compute the total number of alleles predicted by the diffusion approximation, we integrate φ(x)dx over the allowable allele frequency range. Figure 3 shows that, in general, the Moran approximation tends to slightly underestimate the total number of alleles produced by simulations (100 iterations for each point, box-and-whisker plot) unless the mutational input is extremely high, while the diffusion prediction follows a reverse pattern. Both estimates, however, fall within the ranges commonly seen in simulations for all parameter sets examined.
A model of threshold frequency dependence:
The Moran treatment of multiallelic selection is versatile and allows the study of any model in which the mode of selection is solely due to the frequency of an allele. Figure 4 shows the expected allele frequency spectrum for a very simple model of frequency dependence, in which selection depends on a critical threshold frequency. In this example, selection is applied at the reproduction step, such that
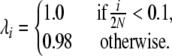 |
(15) |
The expected allele frequency spectrum is shown for this selection model (Figure 4, shaded line; see right-hand axis), with N = 1000 and infinite-alleles mutation rate u = 10−5. The analytic prediction from the Moran model approach is represented by the solid line, and forward simulations (average of 25,000 iterations) are shown by dots. Simulations in this case were of a discrete-time Moran model of reproduction, where in each time step one allele copy was picked to die and one to reproduce and possibly mutate, as in the preceding analytic model. To run discrete-time simulations, we converted the reproduction rates given by (15) into per-time step probabilities, dividing by the total reproduction rate 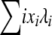 . The analytic prediction provides an accurate description of the consequences of this simple selection model. Analysis of additional complex selection models is equally straightforward with the analytic approach described above, limited only by the requirement that it must be possible to express an allele's fitness as a function of its frequency, independent of allele identity.
. The analytic prediction provides an accurate description of the consequences of this simple selection model. Analysis of additional complex selection models is equally straightforward with the analytic approach described above, limited only by the requirement that it must be possible to express an allele's fitness as a function of its frequency, independent of allele identity.
Figure 4.—

“Threshold” frequency-dependent selection. The fitness function (through differential rate of reproduction) is plotted on the right axis (shaded line). On the left axis is the Moran model prediction for average allele frequency spectrum at equilibrium (solid line) and simulation results (dots, average of 25,000 independent runs). N = 1000, u = 10−5. The simulations are of a population reproducing according to a Moran model.
DISCUSSION
The Moran approach to modeling multiallelic selection has some immediate advantages over previous methods based on diffusion analyses of Wright–Fisher models. Moran models can sometimes yield exact solutions where Wright–Fisher models cannot; in this case, the solution obtained is exact for a continuous-time model and appears to be an excellent approximation for discrete-time models such as those implemented in the Wright–Fisher model simulations. The exact solutions accurately portray the expected state of a population at equilibrium under selection across a range of parameters and modes of selection. The diffusion approximations based on the Wright–Fisher model, on the other hand, can falter when parameter values are outside their applicable ranges and have been developed only for a few well-studied modes of multiallelic selection. Thus, although the diffusion approximations perform very well within their allowable parameter values, and with careful attention should yield accurate results, the Moran approach may be generally preferable. Additional simulations (not shown) demonstrate that the k-allele version of the Moran model with multiallelic selection (appendix b) performs as well as the infinite-alleles version that is the focus of the diffusion comparisons, representing another extension of the applicability of the approach.
The traditional diffusion approximations have one significant practical advantage over the Moran approach, in that the solutions, once obtained, tend to be easier to work with analytically. For example, while the expressions for the expected allele frequency spectra are exact in the continuous-time multiallelic Moran model, they are expressed as a system of equations, rather than closed-form expressions as is possible for some of the one-dimensional diffusion approximations. Moreover, the number of calculations required to obtain results from the Moran solution grows rapidly with population size N, making study of very large population sizes tedious and potentially vulnerable to inaccuracies due to machine precision limits. Nevertheless, despite these difficulties resulting from the large size of the state space in an exact model, we believe that the disadvantages are outweighed by the advantages of having a more fully described stochastic model.
One biological system that could benefit from having a more complete stochastic description of allele dynamics is plant self-incompatibility, in which analysis has been hindered by the complexity of the selection mode. The good fit between the GSI simulations and our simple Moran model parameterization may be initially surprising, particularly in light of the realization that our parameterization (14) is algebraically equivalent to a simple model of overdominance with complete homozygote infertility. It has been pointed out (Vekemans and Slatkin 1994) that such a model underestimates the strength of selection in an SI system, and Wright's more complicated analytic model of relative pollen success (Wright 1939) has thus been favored in the study of GSI. Cases with low mutational input (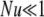 ) show the expected apparently weaker selection of the Moran model approximation compared to full GSI simulations, through a smaller total number of alleles maintained (Figure 3) at higher frequency (Figure 2). The weakness of mutation is also evident in the lack of a low-frequency class in Figure 2. At higher mutational inputs (Nu > 1), however, the Moran model actually overestimates the total number of alleles maintained by selection, apparently overestimating selection. A closer examination reveals that the discrepancy is due to the substantial number of alleles in the lowest (1-copy) frequency class in the Moran model, a frequency class that does not exist in simulations due to the transformation N = 2Ne. Overall, however, the difference between simulation and either of the two analytic predictions is small. In fact, the average allele frequency spectrum obtained via the Moran approach seems to provide as good a match to simulations as does the diffusion prediction based on the more complicated (and biologically accurate) GSI approximation of Wright.
) show the expected apparently weaker selection of the Moran model approximation compared to full GSI simulations, through a smaller total number of alleles maintained (Figure 3) at higher frequency (Figure 2). The weakness of mutation is also evident in the lack of a low-frequency class in Figure 2. At higher mutational inputs (Nu > 1), however, the Moran model actually overestimates the total number of alleles maintained by selection, apparently overestimating selection. A closer examination reveals that the discrepancy is due to the substantial number of alleles in the lowest (1-copy) frequency class in the Moran model, a frequency class that does not exist in simulations due to the transformation N = 2Ne. Overall, however, the difference between simulation and either of the two analytic predictions is small. In fact, the average allele frequency spectrum obtained via the Moran approach seems to provide as good a match to simulations as does the diffusion prediction based on the more complicated (and biologically accurate) GSI approximation of Wright.
We explain this initially puzzling result by pointing out that the simulation method used (repeated pollen trials against a chosen female plant) is intuitively quite close to the Moran formulation of selection (females reproduce at rate one, males reproduce according to their probability of finding a compatible female plant). This is the standard simulation method for GSI (Mayo 1966; Yokoyama and Nei 1979; Vekemans and Slatkin 1994; Uyenoyama 1997; Schierup 1998; Schierup et al. 2000; Muirhead 2001), chosen in part via biological intuition (Mayo 1966). The standard simulation method has been an accepted way to model the complex selection in GSI; our results suggest that the simple Moran approach (or the algebraic equivalent, complete infertility of homozygotes) is an equally acceptable analytic model, at least for considering the quantities derived here, allele number and average allele frequency spectrum. The relationships among simulations, the Moran model analytic predictions, and the Wright–Fisher diffusion analytic predictions are not constant across all population parameters, however, indicating a need for caution in the use of any analytic approach. Here again, however, the Moran model approach may be preferred because, with its limited set of approximations, it is relatively easy to isolate the causes of any discrepancies from simulations and to predict and account for other discrepancies in other parameter ranges.
The exact Moran approach for multiallelic selection may be helpful in studying other difficult systems of selection. Through its generality, it opens up a large number of selection models that were inaccessible (or, at least, unaccessed), using traditional diffusion methods. An example is the fitness function shown in Figure 4, which while itself is quite simple (a threshold model with two alternative reproduction rates) yields a surprisingly complex expected allele frequency distribution. As simple as this fitness model is, the work required to develop an appropriate diffusion approximation to its dynamics would be considerable, and simple adjustments to the model would require additional analysis. With the Moran model approach, due to reversibility, we have immediate access to the stationary distribution of allele frequencies, the equilibrium allele frequency spectrum, and related quantities for any exchangeable selection model, i.e., in which fitnesses depend only on allele frequencies.
The Moran model approach has been shown to provide a useful means of modeling a wide variety of fitness schemes, in particular those relevant to highly multiallelic systems of balancing selection. The approach is minimally dependent on approximations and thus, unlike alternative methods, can be applied regardless of the values of population parameters such as the strength of selection, mutation rate, and effective population size. In addition to generating accurate expressions for the average allele frequency spectrum, using the Moran model yields fully specified expressions for π(X), the joint allele frequency spectrum, independent of the diffusion assumptions used to derive the standard results for multiallelic selection (first developed by Wright 1949). The expressions are general for any model of exchangeable multiallelic selection with parent-independent mutation, as are our proofs of reversibility of the underlying processes. We also obtained exact transition probabilities for all possible one-step transitions from a given population state X under any model of exchangeable selection. It is hoped that these results, by giving us a reasonably detailed picture of this class of complex stochastic processes, may prove useful in extending the analysis of multiallelic selection to problems of ancestral inference, which has previously been restricted to a limited number of models of selection.
Acknowledgments
We are greatly indebted to Rick Durrett for many helpful comments and for suggesting a product form of the stationary distribution of allele frequencies that improved the work immensely. This work was supported by a Career Award (DEB-0133760) to J.W. from the National Science Foundation.
APPENDIX A: REVERSIBILITY, INFINITE-ALLELES MODEL
To simplify notation, let
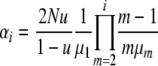 |
(A1) |
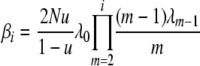 |
(A2) |
so that for our posited stationary distributions we have
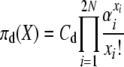 |
(A3) |
and
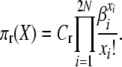 |
(A4) |
We begin by considering the selection at death stationary distribution. If the stationary distribution is correct, and the process is reversible, then
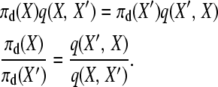 |
(A5) |
Let ei be the unit vector with xi = 1 and all other elements 0. From the transition types in Table 1, there are four possible detailed balances, with new population vectors X′ given by
 |
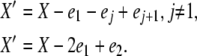 |
To further simplify notation in evaluating the detailed balances, let 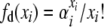 . Then, for the first detailed balance in the selection at death model, we have
. Then, for the first detailed balance in the selection at death model, we have
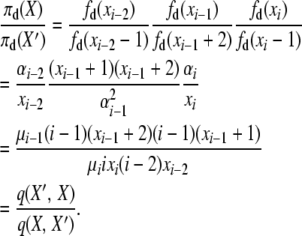 |
(A6) |
The last line holds because this balance equation equates two events, “death of an i-copy allele and reproduction of an (i − 2)-copy allele (no mutation),” which has transition rate q(X, X′) = μiixi((i − 2)xi−2)(1 − u), and “death of an (i − 1)-copy allele and reproduction of a different (i − 1)-copy allele (no mutation),” with rate q(X′, X) = μi−1(i − 1)(xi−1 + 2)((i − 1)(xi−1 + 1)/2N)(1 − u).
We have three remaining detailed balances to verify for the selection at death model. Substituting as before into (A5), we have for the second balance equation, for i ≠ 1, and j ≠ i, i − 1, i − 2, X′ = X + ei−1 − ei − ej + ej+1,
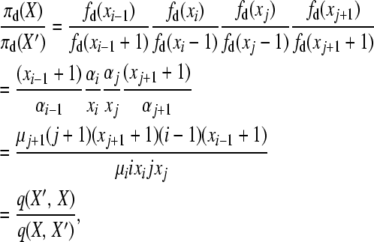 |
(A7) |
where the two events are “i-copy dies, j-copy reproduces, no mutation,” with rate q(X, X′) = μiixi(jxj/2N)(1 − u), and “(j + 1)-copy dies, (i − 1)-copy reproduces, no mutation,” with rate q(X′, X) = μj+1(j + 1)(xj+1 + 1)((i − 1)(xi−1 + 1)/2N)(1 − u).
The third balance uses, with X′ = X − e1 − ej + ej+1 and j ≠ 1,
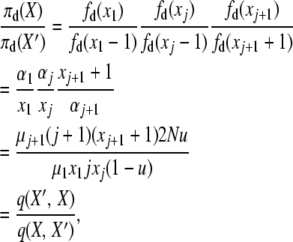 |
(A8) |
with the two events, “1-copy dies, j-copy reproduces, no mutation,” having rate q(X, X′) = μ1x1(jxj/2N)(1 − u), and “(j + 1)-copy dies, mutation,” with rate q(X′, X) = μj+1(j + 1)(xj+1 + 1)u.
The fourth detailed balance has X′ = X − 2e1 + e2, yielding
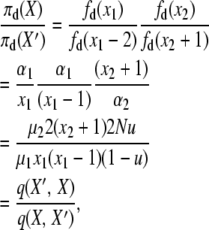 |
(A9) |
where the two events are “1-copy dies, different 1-copy reproduces, no mutation,” with rate q(X, X′) = μ1x1((x1 − 1)/2N)(1 − u), and “2-copy dies, mutation,” with rate q(X′, X) = μ22(x2 + 1)u. And
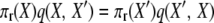 |
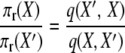 |
(A10) |
to verify the stationary distribution and reversibility of the process.
For the first balance equation, we have for selection at reproduction,
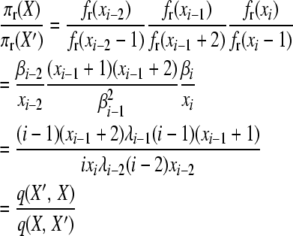 |
(A11) |
with q(X′, X) and q(X, X′) as before, with transition rates appropriate for selection at reproduction.
For the second balance equation,
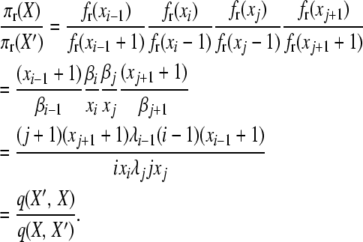 |
(A12) |
The third balance equation gives
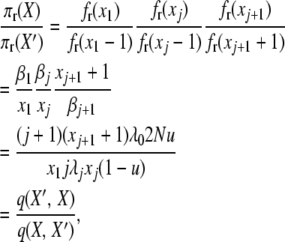 |
(A13) |
and for the final detailed balance, we have
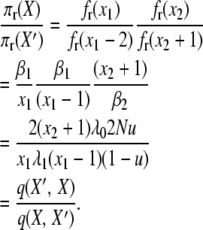 |
(A14) |
This completes the reversibility proof for the infinite-alleles model with selection at reproduction and validates the posited stationary distribution (2).
APPENDIX B: k-ALLELE MUTATION
Stationary distributions and reversibility:
We now consider the case of k-allele mutation. In this model, there are k possible allelic types, and the probability of mutation to a particular type is u/k, independent of the “parental” type, for each reproduction event. Analysis of the k-allele model is similar to that of the infinite-alleles model, with some important departures. The population state vector X now includes a term x0, representing the number of the k possible alleles not present in the population (i.e., those having zero copies). There are, as in the infinite-alleles models, 10 basic types of transitions possible from a given X vector, but because of the different role of mutation, these transitions are not the same as in the previous model. Because of the more consistent role of mutation in affecting alleles in different frequency classes, there are fewer special cases among the transitions that change the population state. We need to consider only two detailed balances: one in which death and reproduction occur to copies in nonoverlapping frequency classes (affecting i, i − 1, j, and j + 1, i ≠ 0 and j ≠ i, i − 1, i − 2) and one in which reproduction and death affect a common frequency class, similar to paired transitions 1 and 4 in the infinite-alleles model. Formally, the two X′ vectors we must consider are
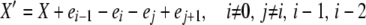 |
and
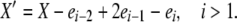 |
We again suggest expressions for the stationary distributions,
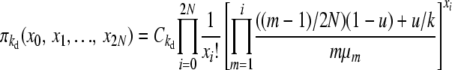 |
(B1) |
for selection at death and
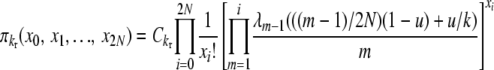 |
(B2) |
for selection at reproduction, where Ckd and Ckr again represent normalization constants. Reversibility can be verified by the same methods as before, using these stationary distributions and appropriate transition rates. Our simplifying notation is, for the k-allele model,
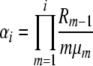 |
(B3) |
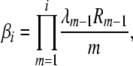 |
(B4) |
where we have defined Ri as
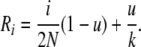 |
(B5) |
We also use fd(xi) and fr(xi) as before in the infinite-alleles model.
For the first balance equation we have, with X′ = X + ei−1 − ei − ej + ej+1,
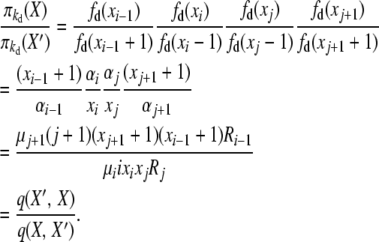 |
(B6) |
Here the forward event is “an i-copy allele dies, and a new copy of a j-copy allele is created (through reproduction or mutation),” with rate q(X, X′) = μiixixjRj, and the reverse event is “a (j + 1)-copy allele dies, and a new copy of an (i − 1)-copy allele is created (through reproduction or mutation),” with rate q(X′, X) = μj+1(j + 1)(xj+1 + 1)(xi−1 + 1)Ri−1.
For the second balance, X′ = X − ei−2 + 2ei−1 − ei, and with selection at death,
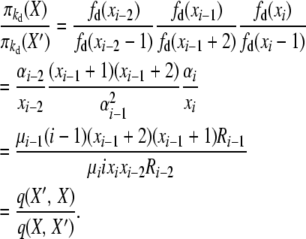 |
(B7) |
The events here are “i-copy dies, and an (i − 2)-copy is created (through reproduction or mutation),” with rate q(X, X′) = μiixixi−2Ri−2, and “(i − 1)-copy dies, and a (different) (i − 1)-copy is created (through reproduction or mutation),” with rate q(X′, X) = μi−1(i − 1)(xi−1 + 2)(xi−1 + 1)Ri−1.
This completes the reversibility proof for selection at death with k-allele mutation. The proof for the selection at reproduction case is similar, using the same balance equations. We have, for the first balance equation,
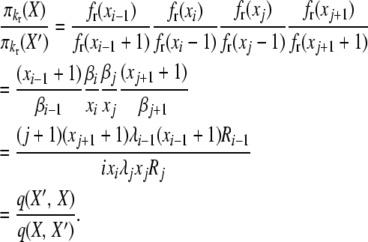 |
(B8) |
For the second balance equation we have
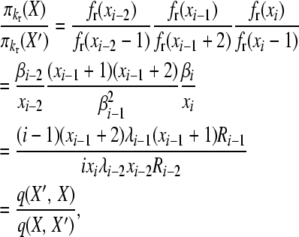 |
(B9) |
verifying the posited stationary distributions and reversibility in the Moran model of selection at reproduction under a k-allele model of mutation.
Average allele frequency spectra:
To obtain the equilibrium expectations for X, we begin again with the identity (4),
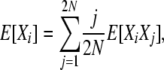 |
and focus on finding expressions for the cross-products E[XiXj]. Using the transitions in the first detailed balance above and taking expectations gives us, for the selection at death case,
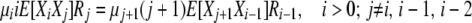 |
so that, for i < j
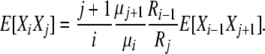 |
(B10) |
We also have from the other set of detailed balances that
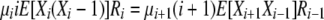 |
for i > 1 or
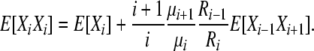 |
(B11) |
Combining (B10) and (B11), we have
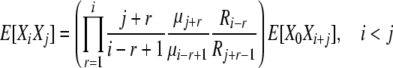 |
(B12) |
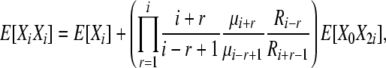 |
(B13) |
leading to
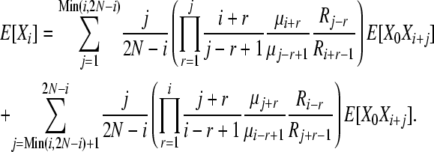 |
(B14) |
These expressions are more complex than in the infinite-alleles case, and it appears that in addition to the problem of finding the set of 2N + 1 E[Xi] elements, we have also set ourselves the task of first finding the set of E[X0Xi] elements. These tasks, however, can be accomplished simultaneously. We first note that
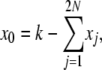 |
and therefore
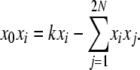 |
Then, taking the usual equilibrium expectations,
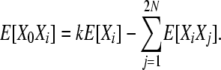 |
(B15) |
We then have
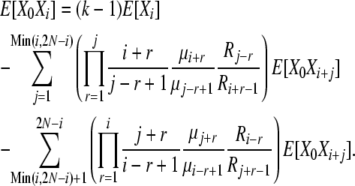 |
(B16) |
We have now expressed E[X0Xi] in terms of the “higher” vector elements E[X0Xi+j], just as we were able to express E[Xi] in terms of the higher E[Xi+j] in the infinite-alleles model. In that case, we obtained results by calculating all E[Xi] terms beginning with E[X2N] and moving down the vector, relative to E[X2N], and then normalizing to obtain E[X2N]. Here, our procedure is very similar, but with an added layer of calculation as we move down the vector to calculate the E[X0Xi]'s relative to E[X0X2N]. The procedure is straightforward because E[X0X2N] = (k − 1)E[X2N], and thus we can calculate all the E[X0Xi]'s, as well as E[Xi], relative to E[X2N], and normalize at the end as before. The full expression for E[Xi] obtained in this manner is cumbersome, but a computer program can calculate the values readily. As in the infinite-alleles model, once we have the expectations E[Xi], we can immediately calculate the normalization constant Ckd, as well as Var[Xi] and Cov[XiXj] for the k-allele model.
Analysis of k-allele mutation and reproduction-step selection is very similar, using the familiar balance equations and the appropriate transition rates. The expression for the cross-product expectations in the case of selection at reproduction is
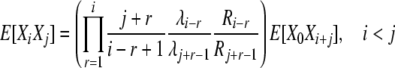 |
(B17) |
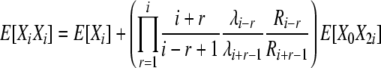 |
(B18) |
and we again use (B15) to obtain
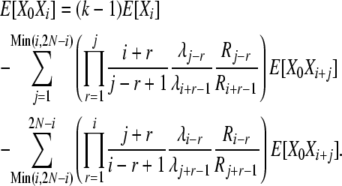 |
(B19) |
Again, a full expression for E[Xi] is not useful to write down, but a computer program can readily calculate the values.
References
- Denniston, C., and J. F. Crow, 1990. Alternative fitness models with the same allele frequency dynamics. Genetics 125 201–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly, P., M. Nordborg and P. Joyce, 2001. Likelihoods and simulation methods for a class of nonneutral population genetics models. Genetics 159 853–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewens, W. J., 1964. a The maintenance of alleles by mutation. Genetics 50 891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewens, W. J., 1964. b On the problem of self-sterility alleles. Genetics 50 1433–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewens, W. J., 2004. Mathematical Population Genetics I. Theoretical Introduction, Ed. 2. Springer-Verlag, New York.
- Ewens, W. J., and P. M. Ewens, 1966. The maintenance of alleles by mutation–Monte Carlo results for normal and self-sterility populations. Heredity 21 371–378. [DOI] [PubMed] [Google Scholar]
- Feldman, M. W., 1966. On the offspring number distribution in a genetic population. J. Appl. Probab. 3 129–141. [Google Scholar]
- Fisher, R. A., 1958. The Genetical Theory of Natural Selection. Dover, New York.
- Grote, M. N., and T. P. Speed, 2002. Approximate Ewens formulae for symmetric overdominance selection. Ann. Appl. Probab. 12 637–663. [Google Scholar]
- Hedrick, P. W., 2002. Pathogen resistance and genetic variation at MHC loci. Evolution 56 1902–1908. [DOI] [PubMed] [Google Scholar]
- Ilmonen, P., D. Penn, K. Damjanovich, L. Morrison, L. Ghotbi et al., 2007. Major histocompatibility complex heterozygosity reduces fitness in experimentally infected mice. Genetics 176 2501–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, F. P., 1979. Reversibility and Stochastic Networks. Wiley, Chichester, UK.
- Kimura, M., and J. F. Crow, 1964. The number of alleles that can be maintained in a finite population. Genetics 49 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May, G., F. Shaw, H. Badrane and X. Vekemans, 1999. The signature of balancing selection: fungal mating compatibility gene evolution. Proc. Natl. Acad. Sci. USA 96 9172–9177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo, O., 1966. On the problem of self-incompatibility alleles. Biometrics 22 111–120. [Google Scholar]
- Moran, P. A. P., 1962. The Statistical Processes of Evolutionary Theory. Clarendon Press, Oxford.
- Muirhead, C. A., 2001. Consequences of population structure on genes under balancing selection. Evolution 55 1532–1541. [DOI] [PubMed] [Google Scholar]
- Muirhead, C. A., N. L. Glass and M. Slatkin, 2002. Multilocus self-recognition systems in fungi as a cause of trans-species polymorphism. Genetics 161 633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson, S., K. Wilson and J. M. Pemberton, 1998. Major histocompatibility complex variation associated with juvenile survival and parasite resistance in a large unmanaged ungulate population (Ovis aries L.). Proc. Natl. Acad. Sci. USA 95 3714–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn, D. J., K. Damjanovich and W. K. Potts, 2002. MHC heterozygosity confers a selective advantage against multiple-strain infections. Proc. Natl. Acad. Sci. USA 99 11260–11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierup, M. H., 1998. The number of self-incompatibility alleles in a finite, subdivided population. Genetics 149 1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierup, M. H., X. Vekemans and D. Charlesworth, 2000. The effect of subdivision on variation at multi-allelic loci under balancing selection. Genet. Res. 76 51–62. [DOI] [PubMed] [Google Scholar]
- Stephens, M., and P. Donnelly, 2003. Ancestral inference in population genetics models with selection. Aust. N. Z. J. Stat. 45 395–430. [Google Scholar]
- Stoffels, R. J., and H. G. Spencer, 2008. An asymmetric model of heterozygote advantage at major histocompatibility complex genes: degenerate pathogen recognition and intersection advantage. Genetics 178 1473–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata, N., 1990. A simple genealogical structure of strongly balanced allelic lines and trans-species evolution of polymorphism. Proc. Natl. Acad. Sci. USA 87 2419–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata, N., Y. Satta and Y. Klein, 1992. Polymorphism and balancing selection at major histocompatibility complex loci. Genetics 130 925–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyenoyama, M. K., 1997. Genealogical structure among alleles regulating self-incompatibility in natural populations of flowering plants. Genetics 147 1389–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyenoyama, M. K., 2003. Genealogy-dependent variation in viability among self-incompatibility genotypes. Theor. Popul. Biol. 63 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo-Marin, M., and M. K. Uyenoyama, 2008. On the Evolutionary Modification of Self-Incompatibility: Implications of Partial Clonality for Allelic Diversity and Genealogical Structure, pp. 53–71 in Self-Incompatibility in Flowering Plants: Evolution, Diversity, and Mechanisms, edited by V. E. Franklin-tong. Springer-Verlag, Berlin/Heidelberg, Germany.
- Vekemans, X., and M. Slatkin, 1994. Gene and allelic genealogies at a gametophytic self-incompatibility locus. Genetics 137 1157–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G. A., 1977. Heterosis or neutrality? Genetics 85 789–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfram, S., 2008. Mathematica, Version 7.0. Wolfram Research, Champaign, IL.
- Wright, S., 1939. The distribution of self-sterility alleles in populations. Genetics 24 538–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1949. Adaptation and Selection, pp. 365–389 in Genetics, Paleontology, and Evolution, edited by G. L. Jepsen, E. Mayr and G. G. Simpson. Princeton University Press, Princeton, NJ.
- Wright, S., 1966. Polyallelic random drift in relation to evolution. Proc. Natl. Acad. Sci. USA 55 1074–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama, S., and M. Nei, 1979. Population dynamics of sex-determining alleles in honey bees and self-incompatibility alleles in plants. Genetics 91 609–626. [DOI] [PMC free article] [PubMed] [Google Scholar]