

Abstract
The 90 kDa heal shock proteins (Hsp90), which are integrally involved in cell signaling, proliferation, and survival, are ubiquitously expressed in cells. Many proteins in tumor cells are dependent upon the Hsp90 protein folding machinery for their stability, refolding, and maturation. Inhibition of Hsp90 uniquely targets client proteins associated with all six hallmarks of cancer. Thus, Hsp90 has emerged as a promising target for the treatment of cancer.
Hsp90 exists as a homodimer, which contains three domains. The N-terminal domain contains an ATP-binding site that binds the natural products geldanamycin and radicicol. The middle domain is highly charged and has high affinity for co-chaperones and client proteins. Initial studies by Csermely and co-workers suggested a second ATP-binding site in the C-terminus of Hsp90. This C-terminal nucleotide binding pocket has been shown to not only bind ATP, but cisplatin, novobiocin, epilgallocatechin-3-gallate (EGCG) and taxol.
The coumarin antibiotics novobiocin, clorobiocin, and coumermycin A1 were isolated from several streptomyces strains and exhibit potent activity against Gram-positive bacteria. These compounds bind type II topoisomerases, including DNA gyrase, and inhibit the enzyme-catalyzed hydrolysis of ATP. As a result, novobiocin analogues have garnered the attention of numerous researchers as an attractive agent for the treatment of bacterial infection. Novobiocin was reported to bind weakly to the newly discovered Hsp90 C-terminal ATP binding site (~700 M in SkBr3 cells) and induce degradation of Hsp90 client proteins. Structural modification of this compound has led to an increase of 1000-fold in activity in anti-proliferative assays. Recent studies of structure-activity relationship (SAR) by Renoir and co-workers highlighted the crucial role of the C-4 and/or C-7 positions of the coumarin and removal of the noviose moiety, which appeared to be essential for degradation of Hsp90 client proteins. Unlike the N-terminal ATP binding site, there is no reported co-crystal structure of Hsp90 C-terminus bound to any inhibitor. The Hsp90 C-terminal domain, however, is known to contain a conserved pentapeptide sequence (MEEVD) which is recognized by co-chaperones.
Cisplatin is a platinum-containing chemotherapeutic used to treat various types of cancers, including testicular, ovarian, bladder, and small cell lung cancer. Most notably, cisplatin coordinates to DNA bases, resulting in cross-linked DNA, which prohibits rapidly dividing cells from duplicating DNA for mitosis. Itoh and co-workers reported that cisplatin decreases the chaperone activity of Hsp90. This group applied bovine brain cytosol to a cisplatin affinity column, eluted with cisplatin and detected Hsp90 in the eluent. Subsequent experiments indicated that cisplatin exhibits high affinity for Hsp90. Moreover Csermely and co-workers determined that the cisplatin binding site is located proximal to the C-terminal ATP binding site.
EGCG is one of the active ingredients found in green tea EGCG is known to inhibit the activity of many Hsp90-dependent client proteins, including telomerase, several kinases, and the aryl hydrocarbon receptor (AhR). Recently Gasiewicz and co-workers reported that EGCG manifests its antagonistic activity against AhR through binding Hsp90. Similar to novobiocin, EGCG was shown to bind the C-terminus of Hsp90. Unlike previously identified N-terminal Hsp90 inhibitors, EGCG does not appear to prevent Hsp90 from forming multiprotein complexes. Studies are currently underway to determine whether EGCG competes with novobiocin or cisplatin binding.
Taxol, a well-known drug for the treatment of cancer, is responsible for the stabilization of microtubules and the inhibition of mitosis. Previous studies have shown that taxol induces the activation of kinases and transcription factors, and mimies the effect of bacterial lipopolysaccharide (LPS), an attribute unrelated to its tubulin-binding properties. Rosen and co-workers prepared a biotinylated taxol derivative and performed affinity chromatography experiments with lysates from both mouse brain and macrophage cell lines. These studies led to identification of two chaperones. Hsp70 and Hsp90, by mass spectrometry. In contrast to typical Hsp90-binding drugs, taxol exhibits a stimulatory response. Recently it was reported that the geldanamycin derivative 17-AAG behaves synergistically with taxol-induced apoptosis.
This review describes the different C-terminal inhibitors of Hsp90, with specific emphasis on structure-activity relationship studies of novobiocin and their effects on anti-proliferative activity.
INTRODUCTION
The goal of many research groups, internationally, has been to better understand the ubiquitously expressed 90 kDa heat shock proteins (Hsp90), Many studies I have been published, revealing these proteins to be integrally involved in cell signaling, proliferation and survival. This family of proteins plays an essential role as molecular chaperones and is responsible for the conformational maturation of nascent polypeptides and the refolding of denatured proteins [1]. More than 100 Hsp90-dependent client proteins have been identified [2,3]. Many of these proteins are associated with cellular signaling networks such as steroid hormone receptors, transcription factors and protein kinases, which represent individually sought after targets for the development of cancer chemotherapeutics [1,4–8]. Hsp90 is an abundant molecular chaperone and is constitutively expressed in eukaryotic cells. Under homeostatic conditions, this protein accounts for nearly 1% of the total cellular protein in eukaryotic cells [9]. Cells exposed to heat shock and other stressed conditions, such as in the case of cancer, overexpress Hsp90 [10]. Many proteins in tumor cells are dependent upon the Hsp90 protein folding machinery for their maturation, stability and activation [2,3]. Hsp90 inhibition uniquely targets client proteins associated with all six hallmarks of cancer [11]. Thus, Hsp90 has emerged as a promising target for cancer chemotherapy [12]. Moreover, this molecular chaperone has exhibited exceptional neuroprotective properties due to its ability to refold aggregated proteins associated with several neurodegenerative diseases [13].
In humans, Hsp90 exists as a homodimer, which contains three highly conserved domains. These regions consist of a 25 kDa N-terminal ATP-binding domain, a 35 kDa middle domain, and a 12 kDa C-terminal dimerization domain (Fig. 1). Inhibitors block the ability of Hsp90 to stabilize and/or fold client proteins, leading to an unproductive heteroprotein complex that is degraded by the ubiquitin-proteasome pathway [14–19]. The N-terminal domain contains an ATP-binding site that also binds the natural products, geldanamycin (GDA) and radicicol (Fig. 2) [20–23]. Hsp90 function depends upon the ability of the N-terminal domain to bind and hydrolyze ATP [24–26]. While the solution structure of Hsp90 exists as a continuum of C-terminally dimerized conformations, the ATP-bound state is a highly constrained structure [27]. The formation of this structure involves coupled conformational switches to position the catalytic apparatus for ATP hydrolysis [24]. An unstructured, highly charged linker joins the N-terminus to the middle domain.
Fig. 1.

Structure of Hsp90 in open state.
Fig. 2.
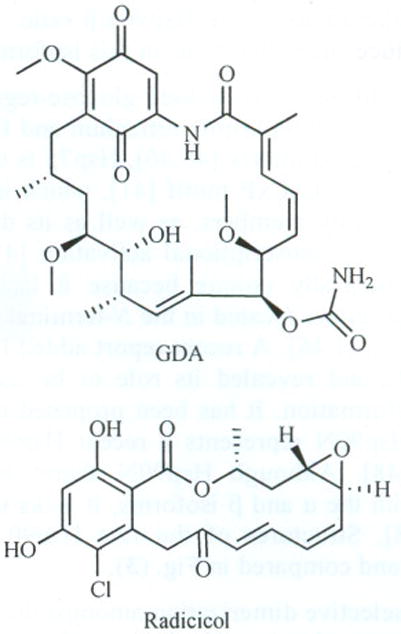
Structure of GDA and radicicol.
The middle domain exhibits high affinity for co-chaperones as well as client proteins [28–34]. Structural and functional analyses have demonstrated that the middle domain of Hsp90 contains a catalytic loop which may serve as an acceptor for the γ-phosphate of ATP, when it is bound to the N-terminus [32]. The structure of the C-terminus of Hsp90 contains the MEEVD sequence, which is known to bind co-chaperones that contain multiple copies of the tetratricopeptide repeat (TPR), a 34 amino acid sequence. Chadli and co-workers recently identified a second, novel site near the N-terminus that also binds TPR proteins. This second site is either within or near the N-terminal ATP-binding pocket and is strongly regulated by nucleotide binding [35].
Initial studies by Csermely and co-workers suggested a second ATP-binding site located in the Hsp90 C-terminus [36]. This C-terminal nucleotide binding pocket has been shown to not only bind ATP, but also novobiocin, cisplatin, epilgallocatechin-3-gallate (EGCG) and Taxol [20]. This review examines the different classes of Hsp90 C-terminal inhibitors, with specific emphasis on structure–activity relationships for novobiocin and their implications for neuroprotection and/or anti-cancer activity.
THE HSP90 FAMILY
Heat shock proteins consist of several subfamilies of molecular chaperones classified by their molecular weights. These subfamilies include Hsp90, Hsp70, Hsp60 and the small Hsps [37]. Each of these subfamilies plays critical cellular roles, such as the prevention of protein aggregation and the direction of misfolded and transient proteins to proteasomal degradation [38]. Hsp90 proteins are highly conserved and four human isoforms have been identified. The two cytosolic isoforms include the major, inducible Hsp90α and the minor, constitutively expressed, Hsp90β [39–42]. Although there is a relatively high conservation within these two isoforms (85% sequence identity), it is suggested that they may display altering chaperone activity [43]. Due to slight perturbations in amino acid sequence, it has been proposed that the α and β forms may exhibit differential binding to client protein substrates [39].
Millson and co-workers used yeast to show that activation of certain Hsp90 clients, such as heat shock transcription factor and v-src were more efficient with Hsp90α, rather than Hsp90β,. In contrast, activation of other clients, such as glucocorticoid receptor and extracellular signal-regulated kinase-5 mitogen-activated protein kinase, demonstrated less dependence on the human Hsp90 isoform expressed. Differential expression patterns were observed when inhibitors, such as radicicol, were selectively introduced to each isoform. It was concluded that in yeast and mammalian systems, cellular susceptibility to Hsp90 inhibitors may be dependent on alterations of the Hsp90α/β ratio. Heat shock is known to induce such alterations in this isoform ratio [44].
Other Hsp90 isoforms include glucose-regulated protein 94 (Grp94) in the endoplasmic reticulum and Hsp75/TRAP1 in the mitochondrial matrix [45,46]. Hsp75 is unique in both its expression of a LxCxE motif [41], which is absent in all other Hsp90 family members, as well as its dependence on stress kinases for transcriptional activation [47]. Moreover, Hsp75 is structurally unique because it lacks the highly charged hinge region located in the N-terminal domain of the other isoforms [41,46]. A recent report added Hsp90N to the Hsp90 family and revealed its role to be associated with cellular transformation. It has been proposed that the newly discovered Hsp90N represents a recent Hsp90α gene rearrangement [48]. Although Hsp90N shares high sequence homology with the α and β isoforms, it lacks the 25 kDa N-terminus [48]. Structures of the five Hsp90 isoforms are summarized and compared in Fig. (3).
Fig. 3.

Summary of various Hsp90 isoforms [39].
There is selective dimerization amongst the two cytosolic isoforms. The two dominant isoforms differ in their ability to dimerize, with the α form doing so readily and the β with much less efficiency [39]. Upon dimerization, Hsp90 exists mainly as a constitutive dimer (αα or ββ), but monomers (α or β), heterodimers (αβ) and higher oligomers of both isoforms may also exist [39]. Dimerization is dependent upon the last 190 amino acids in the C-terminus [46,48]. This C-terminal dimerization is essential for efficient ATP hydrolysis and dependent on both intra- and inter-domain interactions for its formation and stability [49]. 16 amino acids located in the 561–685 amino acid region of the C-terminal dimerization domain were suggested to be responsible for dimerization of Hsp90β [50]. Kobayakawa and co-workers defined which specific amino acid substitutions impeded dimerization and explained the results in terms of the differences between the two major isoforms [51].
In addition to structures of the isolated domains published previously, the full length crystal structure of the closed chaperone complex of yeast Hsp90 bound to nucleotide-p23/Sba1 was published in 2006 by Ali and associates. The crystal structure was solved in complex with a non-hydrolyzable ATP analogue and the Hsp90 co-chaperone p23/Sba1. This crystal structure revealed several novel aspects of Hsp90 which were previously unknown. Firstly, the complex architecture of the ‘closed’ state of Hsp90 was elucidated. In addition, the experimentally described interactions between Hsp90 and partner proteins were confirmed. Moreover, a detailed conformational change in the N-terminal domain was demonstrated to result from ATP binding in the closed, ATP-bound state. Finally, the structural and stabilizing role of co-chaperone p23/Sba1 in the ATP-bound closed dimer state was clarified. This closed Hsp90 state was shown to provide a binding surface for protein substrates rather than enclosing them. Formation and disruption of this surface was found to be directly coupled to the Hsp90 ATPase cycle. The full-length crystal structure verified the widely accepted ATPase-coupled molecular clamp mechanism and structurally elucidated the ATP-dependent activation of Hsp90 client proteins. In contrast to the closed ATP-bound state, the relaxed, structurally unconstrained structure was described as a continuum between C-terminally dimerized conformations [24].
In addition to the crystal structure of Hsp90 bound to p23/Sba1, the structures of other Hsp90 complexes have also been published. The structure of the Hsp90-Cdc37-Cdk4 complex was published by Vaughan and co-workers in 2005. The 3D structure of this complex was determined by single-particle electron microscopy. This study helped elucidate the locations to which Cdc37 and Cdk4 bind in the complex as well as the link between conformational changes in the kinase and ATPase cycle [52]. Another important structure, solved by Meyer and co-workers, was that of Aha1 bound to Hsp90. Aha1 plays an important role in stimulating the ATPase activity of Hsp90. Through a crystal structure of the N-terminal domain of Aha1 in complex with the middle segment of Hsp90, it was confirmed that this activity is mediated through an interaction between Aha1 and the central segment of Hsp90. This binding promotes a conformational switch in the middle-segment catalytic loop and enables the interaction of the N-terminus with ATP [32].
More recently, Bron and co-workers reported the solution structures of two open conformational states of eukaryotic Hsp90. This group presented the first nucleotide-free structure of the full-length chaperone and confirmed that, in solution, apo-Hsp90 is in conformational equilibrium between two states. Switching between the two Hsp90 conformations was described to require movement of the N-terminal and middle domains around two flexible hinge regions. Due to the intrinsic flexibility and dynamic nature of the Hsp90 dimer observed, Bron and associates challenged the accepted ATPase cycle and proposed an alternative mechanism for chaperone activity [53].
HSP90 AS A MOLECULAR CHAPERONE
Eukaryotic Hsp90 is constitutively expressed under normal conditions [54], and significantly overexpressed upon exposure to stress. Stress to the cell, including elevated temperature, non-physiological pH, nutrient deprivation and malignancy, results in the accumulation of misfolded proteins and increased translation of new proteins [20]. Heat shock proteins are overexpressed to refold both denatured and newly synthesized polypeptides into their native conformation [54–56].
Induction of the molecular chaperones Hsp27, Hsp40 and Hsp70 depends on Hsp90 [4]. Release of heat-shock transcription factor 1 (HSF1) is responsible for this Hsp upregulation. While Hsp90 is bound to HSF1 in resting cells, it dissociates upon cellular stress and is translocated to the cell nucleus. Once in the nucleus, HSF1 is phosphorylated and undergoes trimerization [57,58]. The activated HSF1 trimer binds to heat shock response elements (HSE), the consensus sequence for Hsp promoters [59], and elicits transcription of Hsps.
After the synthesis of single-stranded polypeptides by the ribosome, nascent polypeptides have the propensity to aggregate via interactions between amino acid side chains. This aggregation is prevented through the action of molecular chaperones. Because of their role in the transformation of linear polypeptides into tertiary and quaternary structures, chaperones are considered essential for the second half of the genetic code [60]. Linear polypeptides released from the ribosome are subsequently bound by the promiscuous molecular chaperone, Hsp70, in complex with ATP and Hsp40. The bound ATP is then hydrolyzed to stabilize and prevent aggregation of these proteins [60].
Hsp70 interacting protein (HIP), subsequently, binds to and stabilizes the Hsp70-ADP-client complex. BAG (Bcl2-associated athanogene) homologues, on the contrary, cause dissociation of this Hsp70-protein complex by stimulating exchange of ATP for ADP and polypeptide release [20]. Hsp90-Hsp70 organizing protein (HOP), which contains highly conserved tetratricopeptide repeats (TPRs), unites the Hsp70-protein complex with Hsp90, thus forming a multiprotein complex [61]. HOP has been shown to interact with the Hsp90 C-terminus through its TPR domain as well as at other locations in the middle domain of Hsp90. Onuoha and co-workers biophysically analyzed the structure and binding of HOP to Hsp90 using a variety of truncation mutants of both the client and chaperone. Their results confirmed that while the primary binding site of HOP is the C-terminal MEEVD peptide, binding also occurs at additional sites in the C-terminal and middle domains [62]. Immunophilins, co-chaperones, and partner proteins bind to the newly formed heteroprotein complex and facilitate the transfer of client proteins from Hsp70 to Hsp90. Simultaneously, Hsp70, HIP and HOP are released from the complex [63]. The action of immunophilins, such as FKBP51 FKBP52 and CyP-40, or PP5 enable cis/trans peptidylprolylisomerase activity and form a heteroprotein complex that represents the activated Hsp90 protein folding machine [63–65]. Fig. (4) summarizes essential co-chaperones and partner proteins involved in the protein folding mechanism of Hsp90 and highlights those participants that contain a TPR domain.
Fig. 4.
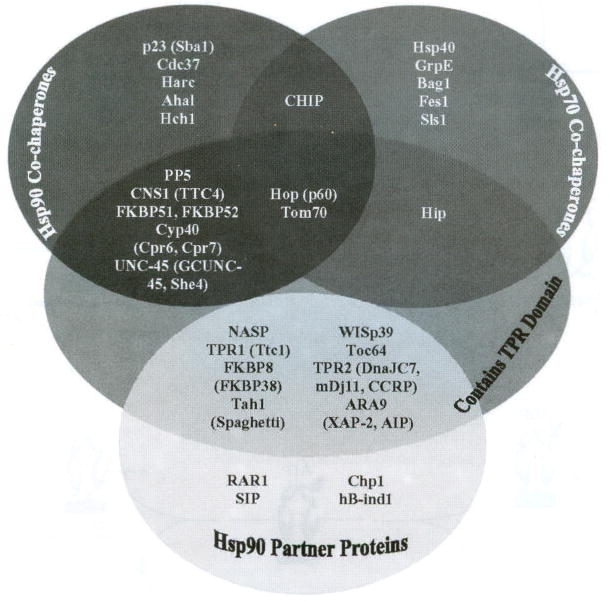
Co-chaperones and partner proteins involved in the Hsp90 protein folding mechanism [66,67].
At this point of the folding process, ATP binds to the open conformation and Hsp90 clamps around the client protein substrate resulting in a closed clamp [68]. It is at this stage of the folding process that an inhibitor, instead of ATP, can bind competitively to the multiprotein complex and cause destabilization of the heteroprotein complex which will transform it into a substrate for the ubiquitin-proteasome pathway. During the protein folding cycle, ATP hydrolysis occurs through the binding co-chaperone p23 to the ATP-bound Hsp90 multiprotein complex. Co-chaperone p23 is also responsible for stabilizing this clamped, high-affinity client-bound Hsp90 conformation. The bound client protein is then folded into its three-dimensional structure by this complex. Release of the folded protein has not been fully characterized, but is thought to also be stimulated by p23 [2]. Once the mature protein is released, Hsp90 can reenter the next catalytic protein folding cycle. The schematic diagram shown in Fig. (5) represents a simplified Hsp90-rnediated protein folding mechanism. This process has been shown to utilize more than 20 associated proteins for the maturation of various clients [1,20,69–75].
Fig. 5.
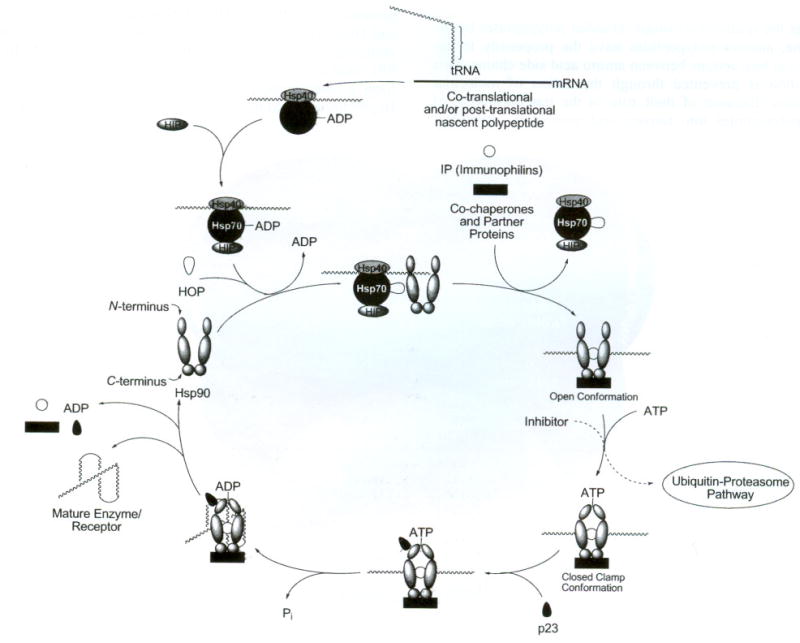
Proposed Hsp90-mediated protein folding mechanism.
DESCRIPTION OF THE C-TERMINAL ATP BINDING POCKET
The existence of a second ATP binding site on the Hsp90 carboxyl terminus, separate from the well-documented N-terminal ATP binding site, has only recently been reported [9,76,77]. Since no co-crystal structure of the Hsp90 C-terminus bound to inhibitors has been published, knowledge of the binding pocket is limited, but many hypotheses have been proposed to account for its function. The Hsp90 C-terminal domain is known to display chaperone activity independent of the N-terminus, as well as mediate dimerization and oligimerization of Hsp90 monomeric species. Structurally, the C-terminus of Hsp90 contains a conserved pentapeptide sequence (MEEVD) that is recognized by co-chaperones [9,78]. The co-chaperones that recognize this sequence all contain multiple copies of the tetratricopeptide repeat (TPR), a 34 amino acid sequence that elicits specific binding to Hsp90 [64,79–81]. This sequence, though conserved, has been reported as dispensable for activity [82].
Many groups have used novobiocin to study Hsp90 C-terminal binding, as it was the first and remains the most studied inhibitor of the Hsp90 carboxyl terminus [83]. Neckers and co-workers revealed via truncation studies that the novobiocin binding site resides in the Hsp90 C-terminus, in a region that is proximal to the carboxyl-terminal dimerization domain. Several amino-terminal point mutations known to disrupt binding of geldanamycin and radicicol were tested for their ability to perturb binding to immobilized novobiocin. These mutants bound novobiocin-Sepharose as well as or better than did wild type Hsp90, Moreover, the N-terminal Hsp90 fragment containing the ATP-binding site of GDA and radicicol failed to bind. Upon demonstration that the amino-terminus did not bind novobiocin, several C-terminal fragments were analyzed for novobiocin binding. These fragments revealed that novobiocin binds to a carboxyl-terminal Hsp90 fragment containing amino acids 538–728. Moreover, this group demonstrated that novobiocin competes with ATP for binding and that association of the co-chaperones Hsc70 and p23 with Hsp90 is disrupted by novobiocin. Through immobilization of ATP, the same fragments that demonstrated binding to immobilized novobiocin were tested for ATP binding. The same fragments that bound novobiocin were also shown to bind ATP, demonstrating a competitive nature between the two small molecules. Finally, novobiocin preincubation with rabbit reticulocyte lysate, which contains Hsc70- and p23-Hsp90-multichaperone complexes, caused a marked decrease in the amounts of both p23 and Hsc70 when co-precipitated with Hsp90. This study confirmed the disruption of co-chaperone binding by novobiocin [76].
Csermely and co-workers reported that the C-terminal site becomes available for binding only after occupancy of the N-terminal site. Moreover, through oxidative nucleotide affinity cleavage, this group characterized that while the N-terminal binding site is fairly specific for adenine nucleotides, the C-terminus binds both purines and pyrimidines (GTP and UTP preferentially) [36]. Garnier and co-workers utilized isothermal titration calorimetry, scanning differential calorimetry and fluorescence spectroscopy to study the interaction of ATP with native Hsp90 and its recombinant C-terminal domain. Results clearly demonstrated that a second ATP-binding site is present in the carboxyl terminus and that the secondary structure of this site may resemble a Rossmann fold [9,84]. Garnier and co-workers concluded that the nucleotide-binding site overlaps with the dimerization domain, which explains the close relationship between ATP binding, dimerization, and magnesium-dependent oligomerization [9].
Although the Hsp90 C-terminus does not exhibit ATPase activity, it is involved in the conformational rearrangement of Hsp90 upon ATP binding [78,85]. The ATPase activity of Hsp90, which leads to a conformational change of the entire homodimer, is dependent upon the Hsp90 C-terminal region to trap the nucleotide during the ATPase cycle [9]. Yun and co-workers suggested that the conformational switch upon novobiocin binding causes changes to Hsp90/cochaperone/client interactions and may be responsible for the observed biological activities [85]. Complex interactions between the N- and C-terminus are a critical regulatory component of chaperone function. Garnier and co-workers have alluded to the cross-talk observed as allosteric interactions between the two termini [9,36,86–89]. Garnier also observed that when a nucleotide is bound to the N-terminus, the molecule exhibits a strong negative impact on the binding of nucleotides to the C-terminus. Furthermore, affinity of the truncated C-terminus for ATP was higher than that of the entire protein. This result confirms that the presence of the amino-terminus negatively affects binding to the carboxyl-terminus and that interdomain cross-talk occurs. The development of improved analogues should further refine the Hsp90 C-terminal nucleotide-binding pocket and provide insight into the unique mechanism exhibited by Hsp90 during the protein folding process [78].
INHIBITORS OF THE C-TERMINAL ATP BINDING POCKET
Novobiocin and Analogues
The coumarin antibiotics novobiocin, chlorobiocin, and coumermycin A1 (Fig. 6) have been isolated from several streptomyces strains and all exhibit potent activity against Gram-positive bacteria. These compounds bind type II topoisomerases, including DNA gyrase, and inhibit the enzyme-catalyzed hydrolysis of ATP [78,90–93]. As a result, novobiocin analogues have garnered the attention of numerous researchers as attractive agents for the treatment of bacterial infections. In addition, novobiocin was reported to bind weakly to the newly discovered Hsp90 C-terminal nucleotide-binding site (~700 μM in SKBr3 cells) and induce degradation of Hsp90 client proteins. Structural modification of this compound has led to analogues with 1000-fold greater efficacy in anti-proliferative assays against various cancer cell lines.
Fig. 6.
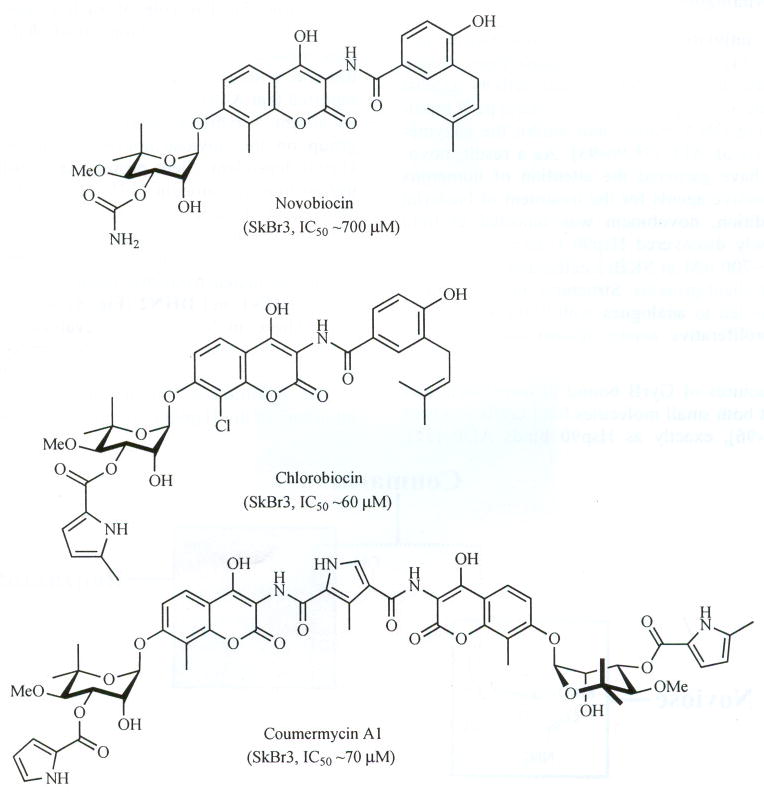
Structures of novobiocin, chlorobiocin and coumermycin A1.
Co-crystal structures of GyrB bound to novobiocin and ADP revealed that both small molecules bind GyrB in a bent conformation [94–96], exactly as Hsp90 binds ADP [22]. With the observations that novobiocin binds in this bent conformation and also exhibits cytotoxicity [93,97–100], Neckers and co-workers hypothesized and subsequently proved that it also binds Hsp90. It is through binding to Hsp90 that novobiocin exerts its anti-tumor activity against breast cancer cells. Using SkBr3 breast cancer cells, Neckers and co-workers demonstrated that 16-hour exposure to novobiocin induces degradation of Hsp90-dependent clients ErbB2, mutant p53 and Raf-1 in a concentration-dependent manner. The same laboratory eluted truncated Hsp90 from an immobilized novobiocin solid-support and concluded that, in contrast to all other Hsp90 inhibitors that bind to the well-established N-terminal ATP-binding site, only the Hsp90 C-terminus is capable of binding novobiocin [83]. Binding of novobiocin to the C-terminus was found to displace inhibitors bound to the Hsp90 N-terminus, a phenomenon that is not reciprocal with N-terminal inhibitors [76,83]. Allan and co-workers proposed that novobiocin may lead to substrate release by inducing a conformational change that results in separation of the homodimeric C-terminal domains [78].
Two research laboratories have synthesized analogues of novobiocin in an attempt to improve upon its poor Hsp90 inhibitory activity [83]. Novobiocin is composed of three distinct parts upon which modifications can be made: the benzamide side chain, the coumarin core and the noviose sugar (Fig. 7). The role of each contributing part can be studied through the development of analogues to probe specific structure–activity relationships for this molecule. To this end, a library of novobiocin analogues published in 2005 reported that A4 (Fig. 8), with a shortened N-acyl side chain, an absent 4-hydroxy substituent and a missing carbamoyl group on the noviose appendage, induced degradation of Hsp90-dependent client proteins at ~70-fold lower concentration than novobiocin [14]. This study demonstrated that attachment of the noviose appendage to the 7-position and an amide linker at the 3-position of the coumarin ring are essential for Hsp 90 inhibition [14]. To confirm the observed SAR trends elucidated from this library, the natural product analogues DHN1 and DHN2 (Fig. 8) were prepared and evaluated. These molecules were evaluated in several assays, which confirmed that while the 4-hydroxyl and 3′-carbamate are essential for DNA gyrase inhibition, they are detrimental to Hsp90 inhibitory activity [17]. Thus, the first selective inhibitors of the Hsp90 C-terminus were born.
Fig. 7.

Structural analysis of novobiocin.
Fig. 8.

Structures of A4, DHN1 and DHN2.
Compound A4 was found to exhibit unique properties unbenounced previously. A4 induced Hsp90 levels at concentrations 1000–10000-fold lower than that required for client protein degradation and was thus evaluated for neuroprotective activity. In addition to outstanding neuroprotective properties, A4 exhibited no toxicity at all concentrations tested [101]. In contrast to the monomeric species, dimers of A4 (based on the structure of cournermycin A1, Fig. 9) were found to manifest anti-proliferative activity. This study sought to fully investigate variants of A4 through preparation of dimers that were linked through meta- and para-phthalic acids and others that contained methylene spacers in the tether. The phthalic acid derivatives were proposed to represent steric mimics of the pyrrole linker found in the natural product, coumermycin A1. Unfortunately, the phthalic acid-linked derivatives manifested no activity against cell cultures.
Fig. 9.
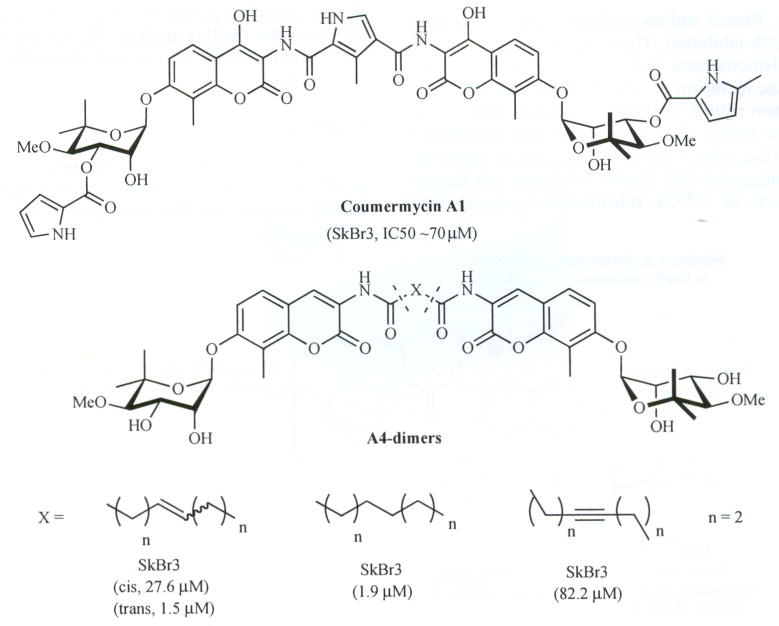
Structures of coumermycin A1 and A4-dimcrs.
Through the synthesis and biological evaluation of methylene linked dimers, the optimal tether length and degree of unsaturation were determined. The saturated A4-dimer with a tether length of eight carbons was found to be the most potent compound in this series and exhibited anti-proliferative activity against two different breast cancer cell lines at low micromolar concentrations. The fact that the dimeric species exhibited anti-proliferative activity led to the hypothesis that conversion of a nontoxic molecule into a potent anti-proliferative agent was accomplished through modification of the amide side chain [102). Consequently, a series of monomeric species based on A4 were synthesized and evaluated against a series of cancer cell lines. Both biaryl and heterocyclic amide derivatives were prepared to explore potential hydrogen-bonding interactions with the putative novobiocin binding pocket that is responsible for binding the prenylated benzamide of the natural product. This study led to the first SAR for the amide side chain and led to identification of the biaryl and the 2-indolyl side chains (Fig. 10), both of which exhibited anti-proliferative activity [101].
Fig. 10.
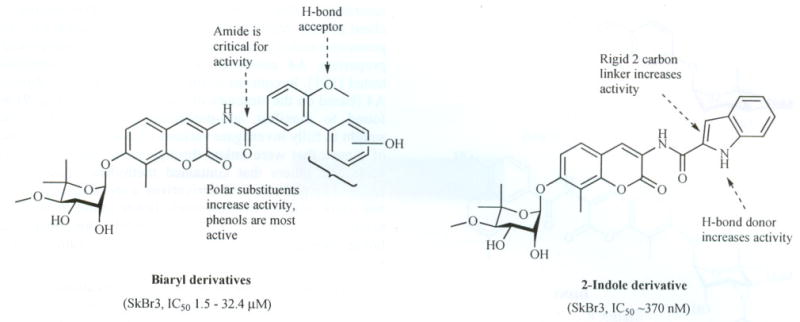
SAR elucidated for novobiocin.
A paper published by Huang and co-workers in 2007 represented the first combinatorial library of coumarin analogues aimed at Hsp90 inhibition. The library was designed to probe hydrophobic and hydrogen bonding interactions produced by the binding pocket. The analogues incorporated previously determined SAR trends as well as strategically placed H-bond donors/acceptors [103].
More recently, Renoir and co-workers examined the role of noviose in Hsp90 inhibition. Their structure–activity relationship studies demonstrated that when analogues lack the noviose moiety, the inclusion of a tosyl substituent at C-4 or C-7 of the coumarin results in Hsp90 inhibition. These analogues were more potent than novobiocin and manifested mid-micromolar IC50 values [104]. A subsequent paper by the same group suggested that Hsp90 inhibition can be enhanced by removal of C7/C8 substituents in desnoviose analogues bearing a tosyl group at the 4-position. These studies produced inhibitors with simplified coumarins that also exhibited mid-micromolar IC50 values [105]. A summary of the novobiocin SAR determined thus far is summarized in Fig. (11).
Fig. 11.

Summary of SAR between novobiocin and Hsp90.
To extend SAR for novobiocin, modified coumarin derivatives of A4 were designed to complement and probe interactions typically manifested by the purine ring of ATP. These coumarin-modified ring systems possess hydrogen bonding capabilities that mimic those of the nucleotide bases, adenine and guanine. In addition, these derivatives contain strategically placed hydrogen bond acceptors and donors and alkyl groups of variable size to probe the size and nature of the binding pocket. The results from such studies are currently under review and should be available in the near future.
Cisplatin
Cisplatin (Fig. 12) is a platinum-containing chemotherapeutic used to treat various types of cancers, including testicular, ovarian, bladder, and small cell lung cancer [106]. Most notably, cisplatin coordinates to DNA bases, resulting in cross-linked DNA, which prohibits rapidly dividing cells from duplicating DNA during mitosis [107–109]. In addition to its DNA-mediated effects, Sreedhar and co-workers reported that cisplatin binds the Hsp90 C-terminal domain and interferes with nucleotide binding [110]. Rosenhagen points to physiological effects as indicators of the interaction between cisplatin and Hsp90. The hyperactive Hsp90-androgen receptor (AR) in prostate cancer is treated with cisplatin through Hsp90 inhibition. Likewise, Hsp 90 inhibitors can be used to treat cisplatin-resistance in cells transfected with the Hsp90-dependent protein kinase v-src [111].
Fig. 12.

Structure of cisplatin.
Itoh and co-workers reported that cisplatin decreases Msp90 chaperone activity. They applied bovine brain cytosol to a cisplatin-affinity column, eluted with cisplatin and detected Hsp90 in the eluent. The results of this study indicated that cisplatin has a high affinity for Hsp90. Moreover, through the use of proteolyzed Hsp90 fragments and affinity purification, it was demonstrated that cisplatin binds near the C-terminus [112]. Upon treatment of neuroblastoma cells with cisplatin, Rosenhagen and co-workers observed degradation of the androgen and glucocorticoid steroid receptors but not other Hsp90 clients, such as raf-1. Ick and c-src. The steroid-receptor-specific proteolysis induced by cisplatin suggests that the compound does not complex Hsp90 and other client proteins, but rather it specifically inhibits steroid receptor-Hsp90 complexes [111]. Csermely and co-workers determined that the cisplatin binding site is located proximal to the C-terminal nucleotide binding site. This study concluded that cisplatin can be used to inhibit the in vitro chaperone activity of Hsp90 as well as to efficiently and selectively block C-terminal nucleotide binding [77].
Acquired resistance to cisplatin can limit its therapeutic potential and many resistance mechanisms have been reported [113]. These pathways include decreased intracellular drug accumulation, enhanced cellular detoxification by glutathione and metallothionein, altered DNA repair and inhibition of apoptosis [113,114]. The observed in vivo unresponsiveness of certain tumors to cisplatin cannot be explained by these mechanisms, however, pointing to novel pathways mediating cisplatin resistance [115,116]. Genomic screening in vivo has helped to elucidate the mechanisms of both cisplatin toxicity and acquired resistance [117].
Epigallocatechin-3-Gallate
EGCG (Fig. 13) is one of the active polyphenolic components found in green tea. EGCG is known to inhibit the activity of many Hsp90 client proteins, including telomerase, multiple kinases, and the aryl hydrocarbon receptor (AhR). EGCG is also involved in growth factor signaling, which involves epidermal and vascular endothelial growth factors as well as transcription factors such as AP-1 and NF-κB. Recently Palermo and co-workers demonstrated via affinity chromatography that EGCG manifests its antagonistic activity against AhR through Hsp90 binding [118]. Affinity purification of Hsp90 fragments from immobilized EGCG revealed that EGCG binds to the Hsp90 C-terminus. This interaction was reported to occur specifically with amino acids 538–728, suggesting that binding takes place at the C-terminal ATP-binding site. Unlike previously identified N-terminal Hsp90 inhibitors, EGCG does not appear to prevent Hsp90 from forming heteroprotein complexes, Studies are currently underway to determine whether EGCG competes with novobiocin or cisplatin binding [118].
Fig. 13.

Structure of EGCG.
Taxol
Taxol (Fig. 14), a well-known drug for the treatment of cancer, is responsible for the stabilization of microtubules and inhibition of mitosis [119]. Previous studies have shown that Taxol induces transcription factors and kinase activation, mimicking the effect of bacterial lipopolysaccharide (LPS), an attribute unrelated to its tubulin-binding properties [120]. A significant amount of evidence suggests that the LPS-mimetic activity of Taxol is independent of (β-tubulin binding. Thus, Rosen and co-workers prepared a biotinylated Taxol derivative and performed affinity chromatography experiments with lysates from both mouse brain and macrophage cell lines. These studies led to affinity purification of two chaperones, Hsp70 and Hsp90, by mass spectrometry from the mouse brain. In contrast to typical Hsp90-binding drugs, Taxol exhibits a stimulatory response, mediating the activation of macrophages and exerting the LPS-mimetic effects observed [121].
Fig. 14.

Structure of Taxol.
Recently it was reported that the geldanamycin derivative 17-AAG (Fig. 15) behaves synergistically with Taxol-induced apoptosis. The mechanism by which these two interact is best explained as sensitization of tumor cells to Taxol-induced apoptosis by 17-AAG through suppression of Akt kinase [122]. The use of Hsp90 inhibitors in combination with proapoptotic therapies represents an exciting new strategy for chemotherapy.
Fig. 15.
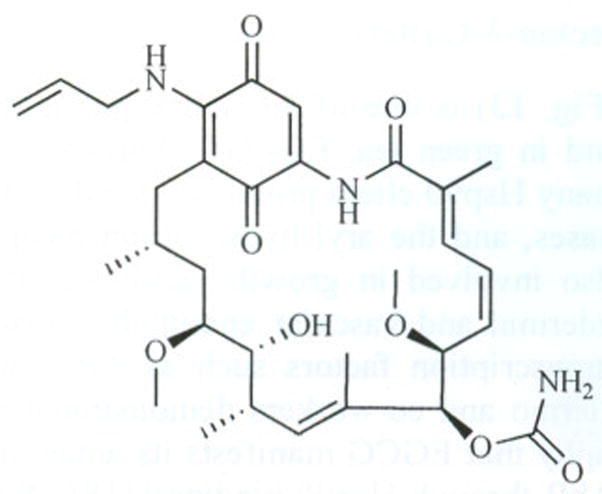
Structure of 17-AAG.
IMPLICATIONS FOR HSP90 INHIBITORS
Key roles for Hsp90 involve the folding of client proteins and the refolding of aggregated or misfolded proteins. These functions make Hsp90 an attractive target for the development of potential therapeutics. By taking advantage of these roles, Hsp90 can be transformed from a mechanism for protein folding to a means of therapy delivery. Moreover, the divergence of these functions makes Hsp90 amenable to the treatment of various disease states (Fig. 16).
Fig. 16.
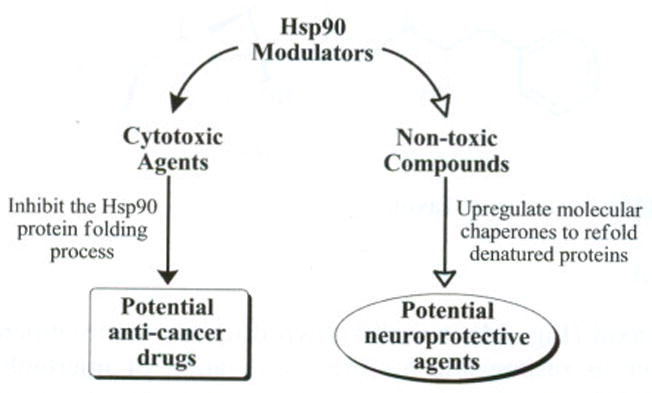
Bidirectional approach to Hsp90 modulation.
Cancer
Many proteins responsible for malignant progression within tumor cells are Hsp90-dependent and more than 40 oncogenic substrates have been identified to date [123]. Therefore, targeting Hsp90 can simultaneously disrupt all six hallmarks of cancer (Fig. 18) and offer a unified mechanism for chemotherapy [11]. Moreover, Hsp90 is overexpressed in malignant cells, and its expression correlates directly with the proliferation of these cells [40,124–126]. Hsp90 inhibitors represent a unique class of compounds that demonstrate high differential selectivity for malignant versus normal cells [10] at concentrations that are well tolerated by humans [127,128]. There are currently more than 20 Hsp90-targeted clinical trials in progress and many more inhibitors are in preclinical development.
Fig. 18.

Hallmarks of cancer and corresponding Hsp90 client proteins [15, 130–133].
The mechanism by which Hsp90 inhibitors exert their anti-cancer effect is by competitively binding to the nucleotide binding site. Upon inhibitor binding, the heteroprotein complex becomes unable to fold or stabilize client proteins. This unproductive complex becomes ubiquitinylated and degradation by the ubiquitin-proteasome pathway ensues. Fig. (17) demonstrates at which step disruption of the protein folding process occurs.
Fig. 17.

Hsp90 inhibition by anti-cancer agents.
Fig. (18) lists the six hallmarks of cancer as defined by Weinberg as well as associated client proteins for each [11]. A cell is defined as cancerous only if all six of these hallmarks are present. The manifestation of each is mediated through a number of proteins, many of which are Hsp90-dependent. The proteins associated with the first two hallmarks are those which facilitate and/or inhibit mitogenic signaling. Those listed with the third hallmark inhibit programmed cell death by preventing normal apoptotic pathways from killing the transformed cell. Telomerase enables DNA replication without harming valuable genetic material and thus provides immortality [129]. Hallmark five is related to the recruitment of vasculature, a process which is directly regulated by several Hsp90 clients. Finally, several Hsp90-dependent clients enable metastasis, leading to the spread of cancer from the initial site of the tumor to other parts of the body.
While many of these proteins are associated with only one hallmark of cancer, other Hsp90-dependent proteins fall into many. The Hsp90 client HDAC6 is involved with the control of gene expression through deacetylation of histones, many of which are Hsp90 clients as well. Deacetylation of histones causes DNA to become too tightly wound, inhibiting gene expression. Therefore, inhibition of this deacetylation may lead to the increased expression of genes responsible for tumor suppression. Moreover, HDAC6 inhibition can inhibit cancer cell proliferation, induce apoptosis and block tumor angiogenesis [134]. Similarly, the SMYD family of proteins also contains several Hsp90-dependent clients which are directly related to modification of histones. SMYD1, SMYD2 and SMYD3 are histone methyltransferases, which act much like HDACs in their ability to alter gene expression [135,136]. Inhibition of these Hsp90-dependent proteins lead to the same anti-cancer effects as observed through histone deacetylase inhibition.
Akt is another Hsp90-dependent protein that is associated with several hallmarks of cancer. Akt (protein kinase B) is a serine/threonine kinase involved in signal transduction pathways that has implications in self-sufficiency of growth signals, evasion of apoptosis and sustained angiogenesis. Inhibition of apoptosis by Akt is accomplished through inhibition of a number of proapoptotic proteins, such as kinase ASK1 [137], glycogen synthase kinase 3, BAD, caspase 9 and Forkhead transcription factors [138–141]. Moreover, through interaction with phosphatidylinositol-3 kinase (PI3K), Akt signaling regulates many angiogenic growth factors involved with recruitment of vasculature [142]. The PI3K-Akt interaction is responsible for an important cell survival signal pathway that is targeted by many anti-cancer drugs [143]. The ability of Hsp90 inhibitors to disrupt the many associations of Akt with oncogenic pathways has generated much interest in studying the interaction between Akt and Hsp90.
The receptor tyrosine kinase c-Met is another example of an Hsp90-client that is involved with several of the hallmarks defined by Weinberg. This kinase plays important roles in cell growth, apoptosis, angiogenesis and apoptosis [144] and thus can be categorized as fitting into several hallmarks. c-Met is overexpressed and mutated in a variety of cancers, and its function and stability depend on Hsp90. Hsp90 inhibitors, such as geldanamycin, have been shown to block c-Met oncogenic signaling [145–147]. Clinical trials are under way to study the effects of such inhibitors in a variety of cancers that demonstrate an overactive c-Met pathway [148].
Although the anti-cancer drug effects of many Hsp90 inhibitors have been previously ascribed to specific inhibition of growth-related (tyrosine) protein kinases, a more complicated mechanism has been recently suggested. MAP kinases, which play an important role in cellular signaling, have been known to be activated by various stresses and growth stimuli [149–156]. The recently identified and cloned member of the MAP kinase superfamily, MOK, was found to be Hsp90-dependent for its intracellular stability and solubility. The Hsp90/Cdc37 complex that binds MOK specifically binds closely related protein kinases MAK and MRK, but not conventional MAP kinases, such as ERK, p38 and JNK [157]. With the knowledge that Hsp90 inhibition leads to degradation of certain other kinases, such as MOK, it was concluded that molecular chaperones play an essential role in the stability of signal transuding protein kinases. This role may be directly related to the anti-cancer effects observed upon introduction of Hsp90 inhibitors.
The more than 40 client proteins associated with oncogenesis include proteins from the classes of transcription factors, kinases, and other proteins [20,46,124–126]. Many of these proteins are individually sought after anti-cancer targets for which therapies have been developed. While many of these proteins can be associated with a specific hallmark of cancer as defined by Weinberg, other examples exist that regulate factors upstream from cancer development. Oncogenic proteins like Mdm2 and SV40 large T-antigen are associated with tumor suppressor genes. These Hsp90-dependent proteins play essential roles in regulating p53, a tumor suppressor which is commonly mutated in many cancers. Ral-binding protein I is another example of an oncogenic Hsp90-dependent protein. This protein interacts with RalA and RalB, both or which are associated with Ras and many signaling pathways directly related to the malignant phenotype. Hsp90 inhibitors, therefore, offer the opportunity to treat cancer through disrupting many targets at different stages as it advances, further increasing their utility to treat a variety of cancers. The possibility to disrupt many targets is also what gives Hsp90 inhibition its seemingly divergent role in the treatment of neurodegenerative diseases.
Neurodegenerative Diseases
The accrual of misfolded proteins that result in plaque formation causes neurodegenerative diseases including Alzheimer’s, Parkinson’s, Huntington’s, and prion disease [13]. Hsp90 is a major molecular chaperone responsible for the rematuration, disaggregation, and resolubilization of these misfolded proteins and their aggregates. Hsp90 inhibitors can lead to Hsp induction, refolding of aggregated proteins and provide neuroprotective activities via this mechanism [158]. Fig. (19) summarizes this role of Hsp90 as it fits into the generally accepted protein folding scheme.
Fig. 19.

Hsp90 inhibition by anti-cancer agents.
There are several Hsp90-dependent proteins with roles within the central nervous system related to disease states. Tau proteins are associated with microtubules and are abundant in neurons within the central nervous system. These proteins promote tubulin assembly into microtubules and the different Tau isoforms stabilize these microtubules, often after phosphorylation by a series of kinases. Hyperphosphorylation of the Tau protein results in the self-assembly of filament tangles, which are involved in the pathogenesis of Alzheimer’s disease [159]. This aggregation of Tau proteins into neurofibrillary tangles has also been associated with diseases such as progressive supranuclear palsy, corticobasal degeneration, and Pick’s disease [160].
Soluble protein levels correlate well with high levels of Hsp90. In contrast, high levels of granular Tau oligimers (Tau filaments and intermediates) have been observed when Hsp levels are low. Although it has been suggested that Hsp90 functions to regulate levels of soluble Tau, the chaperone system can become saturated [161]. Chiosis and co-workers studied Tau hyperphosphorylation as the direct result of the aberrant activation of several kinases, such as cyclin-dependent protein kinase 5 (Cdk5) and glycogen synthase kinase-3β). The group specifically studied the Cdk5/p35 kinase complex, demonstrating in mice that Cdk5 inhibitors reduce Tau hyperphosphorylation and apoptosis in neurons [162,163]. In addition to abnormal phosphorylation of Tau by kinases, the accumulation of aggregated Tau in several tauopathies has been linked to mutations in human Tau isoforms on chromosome 17 [163–165]. Chiosis and co-workers demonstrated that the expression of the most common mutation, TauP301L, can be suppressed to inhibit neuronal loss, leading to function improvement in mice. Both Cdk5/p35 and TauP301L were cited as clients that require Hsp90 assistance for their stability and proper function [166].
Another Hsp90-dependent client associated with neurological disease is alpha-synuclein. This protein is found predominantly at presynaptic terminals in neural tissue, but its primary function remains unknown. Although it is usually a soluble protein, alpha-synuclein can aggregate to form insoluble fibrils in diseases characterized by Lewy bodies, such as Parkinson’s disease, dementia with Lewy bodies and multiple system atrophy [167]. An alpha-synuclein fragment, the non-Aβ component (NAC), is also found in the amyloid plaques associated with Alzheimer’s disease [168].
Hsp90 offers a new range of therapies for treating neurodegenerative diseases. Whether through the induction of Hsp90 to allow refolding of denatured or aggregated proteins or through directly inhibiting clients related to neurodegeneration, Hsp90 offers a unique target for therapy. Hsp90 inhibitors are a future direction for the development of clinical drugs to slow the progression of and cure these debilitating diseases.
CONCLUSION AND FUTURE DIRECTIONS IN THE FIELD OF C-TERMINAL INHIBITION
Mechanistic implications for targeting the Hsp90 protein folding machinery continue to evolve at a high rate. Newly identified client proteins and co-chaperones have led to additional biological targets that can be modulated by small molecules that bind Hsp90. Crystal and co-crystal structures of nearly the entire Hsp90 scaffold have provided significant advancements in the field. These structures allow for a more precise understanding of the protein and provide a scaffold upon which rationally-designed inhibitors can be developed. Biochemical and spectroscopic techniques, molecular modeling and inhibitor design have indirectly revealed much about the C-terminus, but most remains speculative without confirmation through co-crystal structures. The mechanism of action for C-terminal inhibitors will finally be clarified through further understanding of the C-terminal structure and its nucleotide binding site. Thus, although considerable advancements have been made, continued efforts that focus on the Hsp90 C-terminus are required to fully understand the Hsp90 protein folding machine and its potential role against various diseases.
References
- 1.Terasawa K, Minami M, Minami Y. J Biochem. 2005;137:443–447. doi: 10.1093/jb/mvi056. [DOI] [PubMed] [Google Scholar]
- 2.Pratt WB, Toft DO. Exp Biol Med. 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 3.Issacs JS, Xu W, Neckers L. Cancer Cell. 2003;3:213–217. doi: 10.1016/s1535-6108(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 4.Buchner J. Trends Biochem Sci. 1999;24:136–141. doi: 10.1016/s0968-0004(99)01373-0. [DOI] [PubMed] [Google Scholar]
- 5.Picard D. Cell Mol Life Sci. 2002;59:1640–1648. doi: 10.1007/PL00012491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yonehara M, Minami Y, Kawata Y, Nagai J, Yahara I. J Biol Chem. 1996;271:2641–2645. doi: 10.1074/jbc.271.5.2641. [DOI] [PubMed] [Google Scholar]
- 7.Xiao L, Lu X, Ruden DM. Mini Rev Med Chem. 2006;6:1137–1143. doi: 10.2174/138955706778560166. [DOI] [PubMed] [Google Scholar]
- 8.Zhao R, Houry WA. Biochem Cell Biol. 2005;83:703. doi: 10.1139/o05-158. [DOI] [PubMed] [Google Scholar]
- 9.Garnier C, Lafitte D, Tsvetkov PO, Barbier P, Leclerc-Devin J, Millot JM, Briand C, Makarov AA, Catelli MG, Peyrot V. J Biol Chem. 2002;277:12208–12214. doi: 10.1074/jbc.M111874200. [DOI] [PubMed] [Google Scholar]
- 10.Chiosis G, Huezo H, Rosen N, Mimnaugh E, Whitesell L, Neckers L. Mol Cancer Ther. 2003;2:123–129. [PubMed] [Google Scholar]
- 11.Hanahan D, Weinberg RA. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 12.Neckers L, Ivy SP. Curr Opin Oncol. 2003;15:419–424. doi: 10.1097/00001622-200311000-00003. [DOI] [PubMed] [Google Scholar]
- 13.Warrick JM, Chan HY, Chai GBY, Paulson HL, Bonin NM. Nat Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- 14.Yu XM, Shen G, Neckers L, Blake H, Holzbeierlein J, Cronk B, Blagg BSJ. J Am Chem Soc. 2005;127:12778–12779. doi: 10.1021/ja0535864. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H, Burrows F. J Mol Med. 2004;82:488. doi: 10.1007/s00109-004-0549-9. [DOI] [PubMed] [Google Scholar]
- 16.Chiosis G, Vilenchik M, Kim J, Solit D. Drug Discuss Today. 2004;9:881. doi: 10.1016/S1359-6446(04)03245-3. [DOI] [PubMed] [Google Scholar]
- 17.Burlison JA, Neckers L, Smith AB, Maxwell A, Blagg BSJ. J Am Chem Soc. 2006;128:15529–15536. doi: 10.1021/ja065793p. [DOI] [PubMed] [Google Scholar]
- 18.Toft DO. Trends Endocrin Metab. 1998;9:238–243. doi: 10.1016/s1043-2760(98)00060-5. [DOI] [PubMed] [Google Scholar]
- 19.Walter S, Buchner J. J Agnew Chem Int Ed. 2002;41:1098–1113. doi: 10.1002/1521-3773(20020402)41:7<1098::aid-anie1098>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 20.Chaudhury S, Welch TR, Blagg BSJ. ChemMedChem. 2006;1:1331–1340. doi: 10.1002/cmdc.200600112. [DOI] [PubMed] [Google Scholar]
- 21.Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, Pearl LH. Cell. 1997;90:65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- 22.Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH. J Med Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- 23.Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Proc Natl Acad Sci USA. 1994;91:8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ali MMU, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, Pearl LH. Nature. 2006;440:1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Panaretou B, Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, Pearl LH. EMBO J. 1998;17:4829–4836. doi: 10.1093/emboj/17.16.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Obermann WMJ, Sondermann H, Russo AA, Pavletich NP, Hartl FUJ. Cell Biol. 1998;143:901–910. doi: 10.1083/jcb.143.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang W, Hirshberg M, McLaughlin SH, Lazar GA, Grossmann JG, Nielsen PR, Sobott F, Robinson CV, Jackson SE, Laue ED. J Mol Biol. 2005;340:891–907. doi: 10.1016/j.jmb.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 28.Hawl P, Siepmann M, Harst A, Siderius M, Reusch HP, Obermann WMJ. Mol Cell Biol. 2006;26:8385–8395. doi: 10.1128/MCB.02188-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harst A, Lin H, Obermann WMJ. Biochem J. 2005;387:789–796. doi: 10.1042/BJ20041283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lotz GP, Lin H, Harst A, Obermann WMJ. J Biol Chem. 2003;278:17228–7235. doi: 10.1074/jbc.M212761200. [DOI] [PubMed] [Google Scholar]
- 31.Matsumoto S, Tanaka E, Nemoto TK, Ono T, Takagi T, Imai J, Kimura Y, Yahara I, Kobayakawa T, Ayuse T, Oi K, Mizuno A. J Biol Chem. 2002;277:32959–34966. doi: 10.1074/jbc.M203038200. [DOI] [PubMed] [Google Scholar]
- 32.Meyer P, Prodromou C, Hu B, Vaughan C, Roe SM, Panaretou B, Piper PW, Pearl LH. Mol Cell. 2003;11:647–658. doi: 10.1016/s1097-2765(03)00065-0. [DOI] [PubMed] [Google Scholar]
- 33.Panaretou B, Siligardi G, Meyer P, Maloney A, Sullivan JK, Singh S, Millson SH, Clarke PA, Naaby-Hansen S, Stein R, Cramer R, Mollapour M, Workman P, Piper PW, Pearl LH, Prodromou C. Mol Cell. 2002;10:1307–1318. doi: 10.1016/s1097-2765(02)00785-2. [DOI] [PubMed] [Google Scholar]
- 34.Sato S, Fujita N, Tsuruo T. Proc Natl Acad Sci USA. 2000;97:10832–10837. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chadli A, Bruinsma ES, Stensqard B, Toft D. Biochemistry. 2008;47:2850–2857. doi: 10.1021/bi7023332. [DOI] [PubMed] [Google Scholar]
- 36.Soti C, Vermes A, Haystead TAJ, Csermely P. Eur J Biochem. 2003;270:2421–2428. doi: 10.1046/j.1432-1033.2003.03610.x. [DOI] [PubMed] [Google Scholar]
- 37.Welch WJ. Curr Opin Cell Biol. 1991;3:1033–1038. doi: 10.1016/0955-0674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- 38.Ferris DK, Harel-Bellan A, Morimoto RI, Welch WJ, Farrar WL. Proc Natl Acad Sci USA. 1988;85:3850–3854. doi: 10.1073/pnas.85.11.3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sreedhar AS, Kalmar E, Csermely P, Shen YF. FEBS Lett. 2004:562,11–15. doi: 10.1016/s0014-5793(04)00229-7. [DOI] [PubMed] [Google Scholar]
- 40.Csermly P, Schnaider T, Soiti C, Prohaszka Z, Nardi G. Pharmacol Ther. 1998;79:129–168. doi: 10.1016/s0163-7258(98)00013-8. [DOI] [PubMed] [Google Scholar]
- 41.Felts SJ, Owen BAL, Nguyen P, Trepel J, Donner DB, Toft DO. J Biol Chem. 2000;275:3305–3312. doi: 10.1074/jbc.275.5.3305. [DOI] [PubMed] [Google Scholar]
- 42.Krishna P, Gloor G. Cell Stress Chaperones. 2001;6:238–246. doi: 10.1379/1466-1268(2001)006<0238:thfopi>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pepin K, Momose F, Ishida N, Nagata K. J Vet Med Sci. 2001;63:115–124. doi: 10.1292/jvms.63.115. [DOI] [PubMed] [Google Scholar]
- 44.Millson SH, Truman AW, Racz A, Hu B, Panaretou B, Nuttall J, Mollapour M, Soti C, Piper PW. FEBS J. 2007;274:4453–4463. doi: 10.1111/j.1742-4658.2007.05974.x. [DOI] [PubMed] [Google Scholar]
- 45.Sorger PK, Pelham HRB. J Mol Biol. 1987;194:341–344. doi: 10.1016/0022-2836(87)90380-9. [DOI] [PubMed] [Google Scholar]
- 46.Csermely P, Schnaider T, Soti C, Prohaszka Z, Nardi G. Pharmacol Ther. 1998;79:129–168. doi: 10.1016/s0163-7258(98)00013-8. [DOI] [PubMed] [Google Scholar]
- 47.Kim SH, Kim D, Jung GS, Um JH, Chung BS, Kang CD. Biochem Biophys Res Commun. 1999;262:516–522. doi: 10.1006/bbrc.1999.1229. [DOI] [PubMed] [Google Scholar]
- 48.Grammatikakis N, Vultur A, Ramana CV, Siganou A, Schweinfest CW, Watson DK, Raptis L. J Biol Chem. 2002;277:8312–8320. doi: 10.1074/jbc.M109200200. [DOI] [PubMed] [Google Scholar]
- 49.Cunningham CN, Krukenberg KA, Agard DA. J Biol Chem. 2008 doi: 10.1074/jbc.M800046200. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nemoto T, Ohara-Nemoto Y, Ota M, Takagi T, Yokoyama K. Eur J Biochem. 1995;233:1–8. doi: 10.1111/j.1432-1033.1995.001_1.x. [DOI] [PubMed] [Google Scholar]
- 51.Kobayakawa T, Yamada S, Mizuno A, Nemoto TK. Cell Stress Chaperones. 2008;13:97–104. doi: 10.1007/s12192-008-0017-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vaughan CK, Gohlke U, Sobott F, Good VM, Ali MMU, Prodromou C, Robinson CV, Saibil HR, Pearl LH. Mol Cell. 2006;23:697–707. doi: 10.1016/j.molcel.2006.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bron P, Giudice E, Rolland J-P, Buey RM, Barbier P, Diaz JF, Peyrot V, Thomas D, Garnier C. Biol Cell. 2008 doi: 10.1042/BC20070149. in press. [DOI] [PubMed] [Google Scholar]
- 54.Lindquist S, Craig EA. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 55.Gallo KA. J Chem Biol. 2006;13:115–116. doi: 10.1016/j.chembiol.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 56.Frydman J. Annu Rev Biochem. 2001;70:603–649. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- 57.Kim SA, Yoon JH, Lee SH, Ahn SG. J Biol Chem. 2005;280:12653–12657. doi: 10.1074/jbc.M411908200. [DOI] [PubMed] [Google Scholar]
- 58.Guettouche T, Boellmann F, Lane WS, Voellmy R. BMC Biochem. 2005;6:4. doi: 10.1186/1471-2091-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shamovsky I, Ivannikov M, Kandel ES, Gershon D, Nudler E. Nature. 2006;440:556–560. doi: 10.1038/nature04518. [DOI] [PubMed] [Google Scholar]
- 60.Soti C, Nagy E, Giricz Z, Vigh L, Csermely P, Ferdinandy P. Br J Pharmacol. 2005;146:769–780. doi: 10.1038/sj.bjp.0706396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murphy PJM, Kanelakis KC, Galigniana MD, Morishima Y, Pratt WB. J Biol Chem. 2001;276:30092–30098. doi: 10.1074/jbc.M103773200. [DOI] [PubMed] [Google Scholar]
- 62.Onuoha SC, Coulstock ET, Grossmann JG, Jackson SE. J Mol Biol. 2008;379:732–744. doi: 10.1016/j.jmb.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 63.Kosano H, Stensgard B, Charlesworth MC, McMahon N, Toft D. J Biol Chem. 1998;273:32973–32979. doi: 10.1074/jbc.273.49.32973. [DOI] [PubMed] [Google Scholar]
- 64.Chen S, Sullivan WP, Toft DO, Smith DF. Cell Stress Chaperones. 1998;3:1118–129. doi: 10.1379/1466-1268(1998)003<0118:diopat>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Forsythe HL, Jarvis JL, Turner JW, Elmore LW, Holt SE. J Biol Chem. 2001;276:15571–15574. doi: 10.1074/jbc.C100055200. [DOI] [PubMed] [Google Scholar]
- 66.Caplan AJ. Cell Stress Chaperones. 2003;8:105–107. doi: 10.1379/1466-1268(2003)008<0105:wiac>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blagg BSJ, Kerr TD. Med Res Rev. 2006;26:310–338. doi: 10.1002/med.20052. [DOI] [PubMed] [Google Scholar]
- 68.Prodromou C, Panaretou B, Chohan S, Siligardi G, O’Brien R, Ladbury JE, Roe SM, Piper PW, Pearl LH. EMBO J. 2000;19:4383–4392. doi: 10.1093/emboj/19.16.4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu Z, Horwich AL, Sigler PB. Nature (London) 1997;388:741–750. doi: 10.1038/41944. [DOI] [PubMed] [Google Scholar]
- 70.Kimura Y, Matsumoto S, Yahara I. Mol Gen Genet. 1994;242:517–527. doi: 10.1007/BF00285275. [DOI] [PubMed] [Google Scholar]
- 71.Imai J, Maruya M, Yashiroda H, Yahara I, Tanaka K. EMBO J. 2003;22:3557–3567. doi: 10.1093/emboj/cdg349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Correia MA, Sadeghi S, Mundo-Paredes E. Annu Rev Pharmacol Toxicol. 2005;45:439–464. doi: 10.1146/annurev.pharmtox.45.120403.100127. [DOI] [PubMed] [Google Scholar]
- 73.Goetz MP, Toft DO, Ames MM, Erlichman C. Ann Oncol. 2003;14:1169–1176. doi: 10.1093/annonc/mdg316. [DOI] [PubMed] [Google Scholar]
- 74.Xu W, Mimnaugh EG, Kim JS, Trepel JB, Neekers LM. Cell Stress Chaperones. 2002;7:91–96. doi: 10.1379/1466-1268(2002)007<0091:hngrti>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. J Biol Chem. 2002;277:39858–39866. doi: 10.1074/jbc.M206322200. [DOI] [PubMed] [Google Scholar]
- 76.Marcu MG, Chadli A, Bohouche I, Catelli B, Neekers LM. J Biol Chem. 2001;276:37181–37186. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- 77.Soti C, Racz A, Csermely P. J Biol Chem. 2002;277:7066–7075. doi: 10.1074/jbc.M105568200. [DOI] [PubMed] [Google Scholar]
- 78.Allan RK, Mok D, Ward BK, Ratajczak T. J Biol Chem. 2006;281:7161–7171. doi: 10.1074/jbc.M512406200. [DOI] [PubMed] [Google Scholar]
- 79.Young JC, Obermann WMJ, Hartl FU. J Biol Chem. 1998;273:18001–18010. doi: 10.1074/jbc.273.29.18007. [DOI] [PubMed] [Google Scholar]
- 80.Prodromou C, Siligardi G, O’Brien R, Woolfson DN, Regan L, Panaretou B, Ladbury JE, Piper PW, Peral LH. EMBO J. 1999;18:754–762. doi: 10.1093/emboj/18.3.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Harl FU, Moarefi I. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- 82.Louvion JF, Warth R, Picard D. Proc Natl Acad Sci USA. 1996;93:13937–13942. doi: 10.1073/pnas.93.24.13937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marcu MG, Schulte TW, Neekers L. J Natl Cancer Inst. 2000;92:242–248. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- 84.Callebaut I, Catelli MG, Portetelle D, Meng X, Cadepond F, Burny A, Baulieu EE, Momon JP. C R Acad Sci III (Paris) 1994:317. [PubMed] [Google Scholar]
- 85.Yun BG, Huang W, Leach N, Hartson SD, Matts RL. Biochemistry. 2004;43:8217–8229. doi: 10.1021/bi0497998. [DOI] [PubMed] [Google Scholar]
- 86.Scheibel T, Weikl T, Buchner J. Proc Natl Acad Sci USA. 1988;95:1495–1499. doi: 10.1073/pnas.95.4.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hartson SD, Thulasiraman V, Huang W, Whitesell L, Matts RL. Biochemistry. 1999;38:3837–3849. doi: 10.1021/bi983027s. [DOI] [PubMed] [Google Scholar]
- 88.Jibard N, Meng X, Leclerc P, Rajkowski K, Fortin D, Schweizer-Groyer G, Catelli MG, Baulieu EE, Cadepond F. Exp Cell Res. 1999;247:461–474. doi: 10.1006/excr.1998.4375. [DOI] [PubMed] [Google Scholar]
- 89.Scheibel T, Siegmund HI, Jaenicke R, Ganz P, Lilie H, Buchner J. Proc Natl Acad Sci USA. 1999;96:1297–1302. doi: 10.1073/pnas.96.4.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Reece RJ, Maxwell A. Crit Rev Biochem Mol Biol. 1991;26:335–375. doi: 10.3109/10409239109114072. [DOI] [PubMed] [Google Scholar]
- 91.Laurin P, Ferroud D, Schio L, Klich M, Dupuis-Hamelin C, Mauvais P, Lassaigne P, Bonnefoy A, Musicki B. Bioorg Med Chem Lett. 1999;9:2875–2880. doi: 10.1016/s0960-894x(99)00492-8. [DOI] [PubMed] [Google Scholar]
- 92.Lewis RJ, Tsai FT, Wigley DB. BioEssays. 1996;18:661–671. doi: 10.1002/bies.950180810. [DOI] [PubMed] [Google Scholar]
- 93.Ali JA, Jackson AP, Howells AJ, Maxwell A. Biochemistry. 1993:32. doi: 10.1021/bi00061a033. [DOI] [PubMed] [Google Scholar]
- 94.Holdgate GA, Tunnicliffe A, Ward WHJ, Weston SA, Rosenbrock G, Barth PT, Taylor IWF, Paupit RA, Timms D. Biochemistry. 1997;36:9663–9673. doi: 10.1021/bi970294+. [DOI] [PubMed] [Google Scholar]
- 95.Lewis RJ, Singh OMP, Smith CV, Skarzyknski T, Maxwell A, Wonacott AJ, Wigley DB. EMBO J. 1996;15:1412–1420. [PMC free article] [PubMed] [Google Scholar]
- 96.Tsai FTF, Singh OMP, Skarzynski T, Wonacott AJ, Weston S, Tucker A, Pauptit RA, Breeze AL, Poyser JP, O’Brien R, Ladbury JE, Wigley DB. Proteins Struct Funct Genet. 1997;28:41–52. [PubMed] [Google Scholar]
- 97.Gobernado M, Canton E, Santos M. J Clin Microbiol. 1984;3:371. doi: 10.1007/BF01977500. [DOI] [PubMed] [Google Scholar]
- 98.Schwartz GN, Teicher BA, Eder JP, Jr, Korbut T, Holden SA, Ara G, Herman TS. Cancer Chemother Pharmacol. 1993;32:455–462. doi: 10.1007/BF00685890. [DOI] [PubMed] [Google Scholar]
- 99.Nordenberg J, Albukrek D, Hadar T, Fux A, Wasserman L, Novogrodsky A, Sidid Y. Br J Cancer. 1992;65:183–188. doi: 10.1038/bjc.1992.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hombrouck C, Capmau M, Moreau N. Cell Mol Biol. 1999;45:347–352. [PubMed] [Google Scholar]
- 101.Burlison JA, Avila C, Vielhauer G, Lubbers DJ, Holzbeierlein J, Blagg BSJ. J Org Chem. 2008;73:2130–2137. doi: 10.1021/jo702191a. [DOI] [PubMed] [Google Scholar]
- 102.Burlison JA, Blagg BS. J Org Lett. 2006;8:4855–4858. doi: 10.1021/ol061918j. [DOI] [PubMed] [Google Scholar]
- 103.Huang YT, Blagg BS. J Org Chem. 2007;72:3609–3613. doi: 10.1021/jo062083t. [DOI] [PubMed] [Google Scholar]
- 104.Le Bras G, Radanyi C, Peyrat JF, Brio JD, Alami M, Marsaud V, Stella B, Renoir JM. J Med Chem. 2007;50:6189–6200. doi: 10.1021/jm0707774. [DOI] [PubMed] [Google Scholar]
- 105.Radanyi C, Le Bras G, Messaoudi S, Bouclier C, Peyrat J-F, Brion J-D, Marsaud V, Renoir J-M, Alami M. Bioorg Med Chem Lett. 2008;18:2495–2498. doi: 10.1016/j.bmcl.2008.01.128. [DOI] [PubMed] [Google Scholar]
- 106.Galanski M. Recent Patents Anticancer Drug Discov. 2006;1:285–295. doi: 10.2174/157489206777442287. [DOI] [PubMed] [Google Scholar]
- 107.Goodisman J, Hagrman D, Tacka KA, Souid AK. Cancer Chemother Pharmacol. 2006;57:257. doi: 10.1007/s00280-005-0041-4. [DOI] [PubMed] [Google Scholar]
- 108.Brabec V, Kasparkova J. Drug Resist Updat. 2005;8:131. doi: 10.1016/j.drup.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 109.Frankenberg-Schwager M, Kirchermeier D, Greif G, Baer K, Beeker M, Frankenberg D. Toxicology. 2005;212:175. doi: 10.1016/j.tox.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 110.Sreedhar AS, Soti C, Csermely P. Biochim Biophys Acta. 2004;1697:233–242. doi: 10.1016/j.bbapap.2003.11.027. [DOI] [PubMed] [Google Scholar]
- 111.Rosenhagen MC, Soti C, Schmidt U, Wochnik GM, Hartl FU, Hosboer F, Young JC, Rein T. Mol Endocrinol. 2003;17:1991. doi: 10.1210/me.2003-0141. [DOI] [PubMed] [Google Scholar]
- 112.Itoh H, Ogura M, Komatsuda A, Wakui H, Miura AB, Tashima Y. Biochem J. 1999;343:697–703. [PMC free article] [PubMed] [Google Scholar]
- 113.Perez RP. Eur J Cancer. 1998;34:1535–1542. doi: 10.1016/s0959-8049(98)00227-5. [DOI] [PubMed] [Google Scholar]
- 114.Huang RY, Eddy M, Vujcic M, Kowalski D. Cancer Res. 2005;65:5890–5897. doi: 10.1158/0008-5472.CAN-04-4093. [DOI] [PubMed] [Google Scholar]
- 115.Schenk PW, Brok M, Boersma AWM, Brandsma JA, Kulk HD, Brok M, Burger H, Stolen G, Brouwer J, Nooter K. Mol Pharmacol. 2003;64:259–268. doi: 10.1124/mol.64.2.259. [DOI] [PubMed] [Google Scholar]
- 116.Niedner H, Christen R, Lin X, Kondo A, Howell SB. Mol Pharmacol. 2001;60:1153–1160. [PubMed] [Google Scholar]
- 117.Liao C, Hu B, Arno MJ, Panarelou B. Mol Pharmacol. 2007;71:416–425. doi: 10.1124/mol.106.030494. [DOI] [PubMed] [Google Scholar]
- 118.Palermo CM, Westlake CA, Gasiewicz TA. Biochemistry. 2005;44:5041–5052. doi: 10.1021/bi047433p. [DOI] [PubMed] [Google Scholar]
- 119.Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. J Am Chem Soc. 1971;93:2325–2327. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- 120.Ding AH, Porteu F, Sanchez E, Nathan CF. Science. 1990;248:370–372. doi: 10.1126/science.1970196. [DOI] [PubMed] [Google Scholar]
- 121.Byrd CA, Bornmann W, Erdjument-Bromage H, Tempst P, Pavletich N, Rosen N, Nathan CF, Ding A. Proc Natl Acad Sci USA. 1999;96:5645–5650. doi: 10.1073/pnas.96.10.5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Solit DB, Basso AD, Olshen AB, Scher HI, Rosen N. Cancer Res. 2003;63:2139–2144. [PubMed] [Google Scholar]
- 123.Adams J, Elliot PJ. Oncogene. 2000;19:6687–6692. doi: 10.1038/sj.onc.1204088. [DOI] [PubMed] [Google Scholar]
- 124.Yufu Y, Nishimura J, Nawata H. Leuk Res. 1992;16:597–605. doi: 10.1016/0145-2126(92)90008-u. [DOI] [PubMed] [Google Scholar]
- 125.Franzen B, Linder S, Alaiya AA, Eriksson E, Fujioka K, Bergman AC. Electrophoresis. 1997;18:582–587. doi: 10.1002/elps.1150180341. [DOI] [PubMed] [Google Scholar]
- 126.Luparello C, Noel A, Pucci-Minafra I. DNA Cell Biol. 1997;16:1231–1236. doi: 10.1089/dna.1997.16.1231. [DOI] [PubMed] [Google Scholar]
- 127.Banerji U. Proc Am Assoc Cancer Ther. 2003;44:677. [Google Scholar]
- 128.Sausville EA. Curr Cancer Drug Targets. 2003;3:337–383. doi: 10.2174/1568009033481831. [DOI] [PubMed] [Google Scholar]
- 129.Aubert G, Lansdorp PM. Physiol Rev. 2008;88:557–579. doi: 10.1152/physrev.00026.2007. [DOI] [PubMed] [Google Scholar]
- 130.Hurst DR, Mehta A, Moore BP, Phadke PA, Meehan WJ, Accavitti MA, Shevde LA, Hopper JE, Xie Y, Welch DR, Samant RS. Biochem Biophys Res Commun. 2006;348:1429–1435. doi: 10.1016/j.bbrc.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Neckers L. J Biosci. 2007;32:517–530. doi: 10.1007/s12038-007-0051-y. [DOI] [PubMed] [Google Scholar]
- 132.Eustace BK, Sakurai T, Stewart JK, Yimlamai D, Unger C, Zehetmeier C, Lain B, Torella C, Henning SW, Beste G, Scroggins BT, Neckers L, Ilag LL, Jay DG. Nat Cell Biol. 2004;6:507–510. doi: 10.1038/ncb1131. [DOI] [PubMed] [Google Scholar]
- 133.Whitesell L, Lindquist SL. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 134.Monneret C. Anticancer Drugs. 2007;4:363–370. doi: 10.1097/CAD.0b013e328012a5db. [DOI] [PubMed] [Google Scholar]
- 135.Hamamoto R, Furukawa Y, Morita M, Iimura Y, Piettella Silva F, Li M, Ragyu R, Nakamura Y. Nat Cell Biol. 2004;6:731–740. doi: 10.1038/ncb1151. [DOI] [PubMed] [Google Scholar]
- 136.Brown MA, Sims RJ, Gottlieb PD, Tucker PW. Mol Cancer. 2006;5:26. doi: 10.1186/1476-4598-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang R, Luo D, Miao R, Vai L, Ge Q, Sessa WC, Min W. Oncogene. 2005;24:3954–3963. doi: 10.1038/sj.onc.1208548. [DOI] [PubMed] [Google Scholar]
- 138.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 139.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 140.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 141.Kops GJ, de Ruiter ND, De Vries-Smits AM, Powell DR, Bos JL, Burgering BM. Nature. 1999;398:630–634. doi: 10.1038/19328. [DOI] [PubMed] [Google Scholar]
- 142.Shiojima I, Walsh K. Circ Res. 2002;90:1243–1250. doi: 10.1161/01.res.0000022200.71892.9f. [DOI] [PubMed] [Google Scholar]
- 143.Tsuruo T, Naito M, Tomida A, Fujita N, Mashima T, Sakamoto H, Haga N. Cancer Sci. 2003;94:15–21. doi: 10.1111/j.1349-7006.2003.tb01345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sattler M, Salgia R. Curr Oncol Rep. 2007;9:102–108. doi: 10.1007/s11912-007-0005-4. [DOI] [PubMed] [Google Scholar]
- 145.Neckers L. Trends Mol Med. 2002;8:S55–61. doi: 10.1016/s1471-4914(02)02316-x. [DOI] [PubMed] [Google Scholar]
- 146.Webb CP, Hose CD, Koochekpour S, Jeffers M, Oskarsson M, Sausville E, Monks A, Vande Woude GF. Cancer Res. 2000;60:342–349. [PubMed] [Google Scholar]
- 147.Xie Q, Gao CF, Shinomiya N, Sausville E, Hay R, Gustafson M, Shen Y, Wenkert D, Vande Woude GF. Oncogene. 2005;24:3697–3707. doi: 10.1038/sj.onc.1208499. [DOI] [PubMed] [Google Scholar]
- 148.Peruzzi B, Bottaro DP. Clin Cancer Res. 2006;12:3657–3660. doi: 10.1158/1078-0432.CCR-06-0818. [DOI] [PubMed] [Google Scholar]
- 149.Sturgill TW, Wu J. Biochim Biophys Acta. 1991;1092:350–357. doi: 10.1016/s0167-4889(97)90012-4. [DOI] [PubMed] [Google Scholar]
- 150.Ahn NG, Seger R, Krebs EG. Curr Opin Cell Biol. 1992:4. doi: 10.1016/0955-0674(92)90131-u. [DOI] [PubMed] [Google Scholar]
- 151.Nishida E, Gotoh Y. Trends Biochem Sci. 1993;18:128–131. doi: 10.1016/0968-0004(93)90019-j. [DOI] [PubMed] [Google Scholar]
- 152.Davis RJ. Trends Biochem Sci. 1994;19:470–473. doi: 10.1016/0968-0004(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 153.Marshall CJ. Curr Opin Genet Dev. 1994;4:82–89. doi: 10.1016/0959-437x(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 154.Cobb MH, Goldsmith EJ. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- 155.Kyriakis JM, Avruch J. BioEssays. 1996;18:567–577. doi: 10.1002/bies.950180708. [DOI] [PubMed] [Google Scholar]
- 156.Lavoie JN, Rivard N, L’Allemain G, Poyssegur J. Prog Cell Cycle Res. 1996;2:49–58. doi: 10.1007/978-1-4615-5873-6_5. [DOI] [PubMed] [Google Scholar]
- 157.Miyata Y, Ikawa Y, Shibuya M, Nishida E. J Biol Chem. 2001;276:21841–21848. doi: 10.1074/jbc.M010944200. [DOI] [PubMed] [Google Scholar]
- 158.Kim HR, Kang HS, Kim HD. IUBMB Life. 1999;48:429–433. doi: 10.1080/713803536. [DOI] [PubMed] [Google Scholar]
- 159.Alonso AdC, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Proc Natl Acad Sci USA. 2001;98:6923–6928. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Delacourte A. Folia Neuropathol. 2005;43:244–257. [PubMed] [Google Scholar]
- 161.Sahara N, Maeda S, Yoshiike Y, Mizoroki T, Yamashita S, Murayama M, Park JM, Saito Y, Murayama S, Takashima A. J Neurosci Res. 2007;85:3098–3108. doi: 10.1002/jnr.21417. [DOI] [PubMed] [Google Scholar]
- 162.Lau LF, Schachter JB, Seymour PA, Sanner MA. Curr Top Med Chem. 2002;4:395–415. doi: 10.2174/1568026024607526. [DOI] [PubMed] [Google Scholar]
- 163.Lee VM, Goedert M, Trojanowski JQ. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 164.Goedert M, Jakes R. Biochim Biophys Acta. 2005;1739:240–250. doi: 10.1016/j.bbadis.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 165.Zheng YL, Kesavapany S, Gravell M, Hamilton RS, Schubert M, Amin N, Albers W, Grant P, Pant HC. EMBO J. 2005;24:209–220. doi: 10.1038/sj.emboj.7600441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Luo W, Dou F, Rodina A, Chip S, Kim J, Zhao Q, Moulick K, Aquirre J, Wu N, Greengard P, Chiosis G. Proc Natl Acad Sci USA. 2007;104:9511–9516. doi: 10.1073/pnas.0701055104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kim HY, Heise H, Fernandez CO, Baldus M, Zweckstetter M. ChemBioChem. 2007;8:1671–1674. doi: 10.1002/cbic.200700366. [DOI] [PubMed] [Google Scholar]
- 168.Culvenor JG, McLean CA, Cutt S, Campbell BCV, Maher F, Jakala P, Hartmann T, Beyreuther K, Masters CL, Li QX. Am J Pathol. 1999;255:1173–1181. doi: 10.1016/s0002-9440(10)65220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]