

Abstract
Herein, we describe the design of high affinity ligands that bind expanded rCUG- and rCAG-repeat RNAs expressed in myotonic dystrophy and spinocerebellar ataxia. These ligands also inhibit, with nanomolar IC50's, the formation of RNA-protein complexes that are implicated in both disorders. The expanded rCUG and rCAG repeats form stable RNA hairpins with regularly repeating internal loops in the stem and have deleterious effects on cell function. The ligands that bind the repeats display a derivative of the bis-benzimidazole Hoechst 33258, which was identified by searching known RNA-ligand interactions. A series of 13 modularly assembled ligands with defined valencies and distances between ligand modules was synthesized to target multiple motifs in these RNAs simultaneously. The most avid binder, a pentamer, binds the rCUG-repeat hairpin with a Kd of 13 nM. As compared to a series of related RNAs, the pentamer binds to rCUG-repeats with 4.4- to >200-fold specificity. Furthermore, the affinity of binding to rCUG-repeats shows incremental gains with increasing valency while the background binding to genomic DNA is correspondingly reduced. Then, it was determined whether the multivalent ligands inhibit the recognition of RNA repeats by Muscleblind-like 1 (MBNL1) protein, the expanded-rCUG binding protein whose sequestration leads to splicing defects in DM1. Among several compounds with nanomolar IC50's, the most potent inhibitor is the pentamer, which also inhibits the formation of rCAG repeat-MBNL1 complexes. Comparison of the binding data of the designed synthetic ligands and MBNL1 to repeating RNAs shows that the synthetic ligand is 23-fold higher affinity and more specific to DM1 RNAs than MBNL1. Further studies show that the designed ligands are cell permeable to mouse myoblasts. Thus, cell permeable ligands that bind repetitive RNAs have been designed that exhibit higher affinity and specificity for binding RNA than natural proteins. These studies suggest a general approach to targeting RNA, including those that cause RNA dominant disease.
Introduction
A wide variety of new and important roles for RNA are being uncovered, particularly for non-coding RNAs such as microRNAs and untranslated regions (UTRs) in mRNAs.1-4 Such studies have expanded the number of RNAs that are potential targets for therapeutics or chemical genetics probes. One interesting RNA target in a non-coding region is the rCUG triplet repeat expansion in the 3′UTR of the dystrophia myotonica protein kinase (DMPK) gene.4,5 The triplet repeat expansion results in a gain-of-function for the RNA and causes myotonic muscular dystrophy type 1 (DM1).
DM1 affects 1 in 6000 individuals and as of 2009 has no known treatment.6,7 The disease is characterized by weakness and wasting of skeletal muscle7 and a wide range of problems in other organ systems.8 The toxic rCUG repeat that causes DM1 folds into a hairpin (Figure 1) that contains regularly repeating UU mismatches flanked by GC pairs (5′CUG/3′GUC) within the stem.5,9,10 These regularly repeating 5′CUG/3′GUC internal loop motifs bind to the alternative splicing regulator Muscleblind-like 1 protein (MBNL1). Formation of the DM1 RNA-MBNL1 complex compromises function of MBNL1, which leads to the misregulation of alternative splicing for a specific set of pre-mRNAs. They include the muscle-specific chloride channel (ClC-1) and the insulin receptor (IR) pre-mRNAs.11,12 Mis-splicing of CIC-1 results in loss of the channel from the surface of muscle cell membranes and explains the altered muscle excitability associated with DM.8 Mis-splicing of the muscle IR may explain why many patients afflicted with DM have insulin insensitivity. This accepted disease model has been established and further cemented by two different mouse models.4,13
Figure 1.

A schematic for the interaction of toxic DM1 rCUG repeats that fold into a hairpin and bind MBNL1. Modularly assembled ligands were used to inhibit the formation of the DM1 hairpin-MBNL1 complex by binding to the RNA.
The disease model suggests that one potential therapeutic avenue for DM would be to displace MBNL1 from rCUG repeats or preclude its binding to the RNA altogether. In support of this strategy, it has been recently shown that over-expression of MBNL1 in DM1 mouse models corrected defects in pre-mRNA splicing associated with this disease.14 Other reports have also indicated that the pathogenic model of expanded RNA repeats interacting with MBNL1 also causes other diseases. For example, the RNAs that cause myotonic muscular dystrophy type 2 (DM2), which has a rCCUG expansion,15 and spinocerebellar ataxia type 3 (SCA3), which has rCAG expansion,16 both interact with Muscleblind proteins. Similar to DM1 RNA, these expanded RNAs fold into a hairpin forming regularly repeating internal loops in the hairpin stem. Thus, design of ligands targeting repeating RNAs to disrupt MBNL1 binding could serve as a general strategy for RNA-mediated diseases.
Herein, we describe the design of cell permeable, modularly assembled ligands that inhibit the formation of the DM1 RNA- and SCA3 RNA-MBNL1 interactions with low nanomolar IC50's. The modularly assembled binder, identified by searching known RNA motif-ligand pairs in the literature,17 is a variant of Hoechst 33258 that binds to single 5′CUG/3′GUC and 5′CAG/3′GAC motifs. By modularly assembling the Hoechst scaffold, a pentameric ligand was designed that is 54-fold specific for the DM1 hairpin (Kd = 13 nM) over herring sperm DNA and inhibits formation of the MBNL1-r(CUG)109 complex with a nanomolar IC50. This ligand also binds SCA3 repeats with a Kd of 130 nM and inhibits the corresponding RNA-protein complex. Furthermore, the designed pentameric ligand binds 23-fold more tightly to DM1 repeats than MBNL1 and is more specific.
Experimental
Instrumentation
NMR spectra were recorded on a Varian NMR operating at 500, 400, or 300 MHz on proton. Chemical shifts were referenced to residual solvent or an internal tetramethylsilane standard. Mass spectra were recorded on a LCQ Advantage Ion Trap LC/MS equipped with a Surveyor HPLC system or on a Bruker Biflex IV MALDI-TOF spectrometer. HPLC was performed on a Waters 1525 Binary HPLC Pump equipped with a Waters 2487 Dual Absorbance Detector system monitoring at 218 and 254 nm. Analytical HPLC separations were performed using a Waters Symmetry C8 or C18 5 μmeter 4.6×150 mm column, and preparative HPLC separations were completed using a Waters Symmetry C8 7 μmeter 19×150 mm column. Sonication was performed using a Branson Bransonic® 5210, 140 watts, 47 kHz sonicator. Resin was agitated by shaking on a Thermolyne Maxi-Mix III™ shaker. All pH measurements were performed at room temperature using a Mettler Toledo SG2 pH meter that was standardized at pH 4.0, 7.0, and 10.0.
Chemicals
The Fmoc-Rink resin and N,N′-diisopropylcarbodiimide (DIC) were from AnaSpec; N,N-dimethylformamide (DMF; 99.8% anhydrous) was from Acros or Baker and was used without further purification; bromoacetic acid was from Sigma Aldrich; 3-bromopropylamine hydrobromide was from TCI or Fluka; all other reagents were from Acros or Alfa Aesar and were used without further purification with the exception of piperidine, which was distilled prior to use. The tris-(benzyltriazolylmethyl)amine (TBTA) catalyst was synthesized as described.18,19 The HPLC solvents used were HPLC grade methanol from Burdick & Jackson or Honeywell and water obtained from a Barnstead NANOpure Diamond Water Purification System operating at 18.2 mΩ-cm. NANOpure water was used to make all buffers and media.
Synthesis of meta-(4-hydroxybutyric acid)-Hoechst
A mixture of ethyl 4-(3-formylphenoxy)butanoate20 (0.37 g, 2.1 mmol) and 4-(5-(4-methylpiperazin-1-yl)-1H-benzo[d]imidazol-2-yl)benzene-1,2-diamine,21 acetate salt (0.8 g, 2.1 mmol) in 45 mL of nitrobenzene was stirred at 140 °C for 36 h under argon. Then the solution was concentrated to dryness in vacuo. The residue was triturated with ethyl ether (50 mL), filtered and washed on the filter with ethyl ether (4×20 mL). The crude product was dried and dissolved in ethanol (15 mL). To the solution, potassium hydroxide (0.47 g, 8 mmol) was added and the mixture was refluxed for 4 h. The reaction was cooled to room temperature, diluted with water (15 mL) and saturated with CO2. In approximately 1 h, crystals of the product started to precipitate. The product was filtered, washed on the filter with ethyl ether (4×20 mL) and dried. Yield 0.9 g (84%). 1H-NMR (DMSO-D6, 500 MHz) δ 13.1 (1H, broad), 12.6 (1H, broad), 8.32 (1H, broad m), 8.03 (2H, d, J=8.6 Hz), 7.79 (2H, m), 7.71 (1H, broad d, J=8.2 Hz), 7.48 (1H, t, J=8 Hz), 7.44 (1H, broad), 7.09 (1H, dd, J=8.2 Hz, J=2.2 Hz), 6.98 (1H, broad), 6.94 (1H, dd, J=8.6 Hz, J=1 Hz), 4.12 (2H, t, J=6.5 Hz), 3.37 (4H, m, overlaps with water), 3.13 (4H, m), 2.44 (2H, t, J=7.4 Hz), 2.25 (3H, s), 2.01 (2H, m). MS-ESI(+) calculated: 511 (M+H+); observed: 511 (M+H+).
meta-(N-(3-azidopropyl)-4-oxybutanamide)-Hoechst (2)
A mixture of meta-(4-hydroxybutyric acid)-Hoechst (0.9 g, 1.76 mmol), PyBOP (1.4 g, 2.64 mmol) and diisopropylethylamine (0.68 g, 5.28 mmol) in DMF (15 mL) was stirred under argon at room temperature for 30 min. Then 3-azidopropylamine (0.27 g, 2.64 mmol) was added. The reaction was stirred at room temperature for 40 h, monitoring the reaction progress by thin layer chromatography (TLC) (16:8:1 ethyl acetate/methanol/triethylamine). After the reaction was complete, the solution was concentrated in vacuo to a thick, gummy residue. The residue was washed with water (3×20 mL) and crystallized from ethanol (10 mL) providing off-white crystals of the product. Yield 0.7 g (54%). 1H-NMR (DMSO-D6, 500 MHz) δ 13.1 (1H, broad), 12.7 (1H, broad), 9.6 (1H, broad), 8.34 (1H, d, J=68.8 Hz), 7.95-8.08 (2H, m), 6.44-7.80 (3H, m), 7.40-7.56 (2H, m), 6.98-7.26 (3H, m), 4.09 (2H, t, J=6.2 Hz), 3.37 (10H, m, overlaps with water), 3.13 (4H, q, J=12.5 Hz, J=6.4 Hz), 2.84 (3H, s), 2.30 (2H, t, J=7.4 Hz), 2.01 (2H, m), 1.65 (2H, m). MS-ESI calculated: 593 (M+H+); observed: MS-ESI(+) 593 (M+H+), MS-ESI(-) 145 (60%, PF6-), 591 (30%, M-), 637 (100%, M+HCO2-).
General Protocol for Peptoid Synthesis
Peptoid oligomers, with the exception of 5H-4, were synthesized at room temperature (22°C) in BioRad Poly-Prep® chromatography columns (0.8×4 cm). These syntheses were based on a previously published synthetic procedure.22 Characterization of the peptoids is available in the Supporting Information. All peptoids were >95% pure.
Fmoc-protected Rink amide polystyrene resin with a substitution level of 0.45 mmol/g (23 mg, 10 μmol) was swollen in dichloromethane (DCM) (1 mL) for 20 min. The solution was drained, and the resin was deprotected with 1 mL of 20% piperidine in DMF for 40 min with shaking (800 rpm). The column was drained, and then the resin was rinsed with DMF (6×3 mL), with mixing between each wash.
Coupling Step
To the resin-bound amine, bromoacetic acid (0.2 mL, 1 M in DMF) and DIC (0.2 mL, 1 M in DMF) were added, and the resin was shaken at 1000 rpm for 20 min. The solution was drained, and then the column was rinsed with DMF (5×2 mL), with mixing between each wash.
Displacement Step
a) Introduction of a click counterpart: Into a solid phase reaction vessel, DMF (0.2 mL) and propargylamine (20 μL) were added sequentially, and the resin was shaken at 1000 rpm for 3 h. After draining the column, the resin was rinsed with DMF (5×2 mL), with mixing between each wash. b) Chain extension with a spacer: Into a solid phase reaction vessel, DMF (0.2 mL) and propylamine (50 μL) were added sequentially, and the resin was shaken at 1000 rpm for 20 min. The column was drained and the resin rinsed with DMF (5×2 mL).
Conjugation of 2 to Peptoids via HDCR
The resin-bound peptoid oligomer was washed with methanol (3×2 mL) and DCM (3×2 mL), and dried under stream of air. A small portion of the resin was cleaved and analyzed by HPLC and MS-ESI prior to conjugation with 2 to confirm formation of the target peptoid. Then into a solid phase reaction vessel containing oligomer-bound resin, 2 (4 equivalents per conjugation site) was added. The vessel was sealed with a rubber septum and purged with argon for 20 min. The vessel was capped and 2 mL of the pre-prepared catalyst solution (0.1 M copper acetate, 1 M diisopropylethylamine, 0.1 M ascorbic acid and 0.01 M TBTA 18 in pyridine/DMF, 3:7) was loaded under argon. The reaction was sonicated in darkness at 40°C with periodic vortexing for 36 h. The click solution was drained, and the resin was rinsed with DMF (5×2 mL), 2% ascorbic acid in pyridine (5×2 mL), and DMF (5×2 mL) with mixing between each wash. After washing with methanol (3×2 mL) and DCM (3×2 mL), the product was cleaved from the resin using a mixture of trifluoracetic acid (TFA)/DCM/water (60:40:2, 2×1 mL) for 1 h at room temperature. The filtrate was concentrated under a stream of air, and the resulting residue was dissolved in water. The product was isolated by preparative HPLC, and fractions were analyzed by MS-ESI. (Please see the Supporting Information for synthetic details and characterization of all compounds.) Combined fractions of the product were concentrated to dryness and the product was resuspended in water and lyophilized.
Synthesis of 5H-4
The alkyne-peptoid was synthesized in a ChemGlass 15 mL solid phase reaction flask using a microwave-based protocol reported previously23 with each submonomer double coupled. Fmoc-Rink amide resin (230 mg, 100 μmole loading) was deprotected as described above. After completion of the synthesis, the sample was cleaved from the resin with TFA/DCM/H2O (60:40:2) for 1 h at room temperature. The sample was then lyophilized to an off-yellow oil and purified by HPLC using a flow rate of 5 mL/min and a gradient of 30% to 70% B in A over 35 min (A: 0.1% TFA in water, B: 0.1% TFA in methanol) (tr = 22.5 min, yield 115 mg, 55 μmoles, 55%). The sample's identity was confirmed by mass spectrometry; ESI-MS+: calculated 1040 (M+2H+)/2; observed: 1040 (M+2H+)/2. This peptoid (22 mg, 9.5 μmol) was dissolved in DMF (0.4 mL) and then 2 (HPF6 salt, 50 mg, 67 μmol) was added. The mixture was sonicated until the solution became clear. Then 70 μL of 1 M aqueous CuSO4 and 105 μL of 1 M aqueous ascorbic acid were added. The reaction was mixed and incubated at 60 °C for 70 h. The sample was purified via the same method described above for 2-functionalized peptoids synthesized by solid-phase methods to yield 48 mg of 5H-4 (7 μmol, 74%; assuming 15×TFA salt, MW 6752).
Synthesis of the Pentatriazolyl Ligand, 5A-4: Conjugation to 3-Azidopropylamine
The corresponding alkyne peptoid (14 mg, 6.4 μmol) was dissolved in 50% aqueous ethanol (0.5 mL) and then 3-azidopropylamine (45 mg, 450 μmol), 10 μL of 1 M aqueous CuSO4, and 20 μL of 1 M aqueous ascorbic acid were added. The reaction was mixed and incubated at room temperature for 10 h. The mixture was then acidified with TFA (50 μL) and purified by HPLC using a flow rate of 4 mL/min and a gradient from 5% to 100% B in A over 95 min. This reaction yielded 5 mg of pure product (1.5 μmol, 24%; assuming 6×TFA salt, MW 3265): MS-ESI(+) calculated: 1290 (MH2)2+; observed: 1290 (MH2)2+
Plasmid Purification and RNA Transcription
The plasmid encoding for r(CUG)1095 was isolated using a Qiagen maxi prep kit. To generate RNA suitable for MBNL1 displacement assays, the plasmid was linearized with XbaI, which affords an RNA transcript with a single stranded region for immobilization into wells containing a complementary DNA tail. RNAs used in binding assays were generated by digestion of the plasmid with BamHI. RNAs were transcribed using a Stratagene RiboMaxx transcription kit and 5 μg of plasmid DNA per the manufacturer's protocol. After incubation at 37 °C, the RNA transcript was purified using a denaturing 5% polyacrylamide gel. RNA was visualized by UV shadowing, the product band was excised, and the RNA extracted into 0.3 M NaCl by tumbling overnight at 4 °C. The resulting solution was concentrated with 2-butanol and ethanol precipitated. The precipitated RNA was resuspended in diethyl pyrocarbonate (DEPC)-treated water and stored at -20 °C until use. Concentrations were determined by UV absorbance at 260 nm using extinction coefficients calculated by the HyTher program.24,25
r(CUG)109-MBNL1 Displacement Assays
The recombinant MBNL1 protein, which is fused to a 25 amino acid sequence encoding the LacZα peptide, was expressed and purified as previously described.26 All steps of the displacement assay were completed at room temperature. For higher loading displacement assays, 25 pmoles of biotinylated DNA capture probe (5′-Biotin - TTTTAATTTTAGGATCCCCCCAG-3′; Integrated DNA Technologies) were prepared in 100 μL of 1× MBNL1 Buffer (50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 50 mM KCl, 1 mM MgCl2, 0.05% Tween-20, and 1 mg/mL BSA) and incubated in a well of a 96-well Reacti-Bind streptavidin coated plate (Pierce) for 3 h. The solution was removed and the wells washed with 2 × 200 μL 1× MBNL1 Buffer. A 25 nM solution of r(CUG)109 transcribed from the plasmid linearized with XbaI was annealed in 1× MBNL1 Buffer without MgCl2 at 60 °C for 1 min and allowed to slowly cool to room temperature. Then, MgCl2, Tween-20, and BSA were added to final concentrations of 1 mM, 0.05%, and 1 mg/mL, respectively. A 100 μL aliquot of the annealed RNA was added per well, and the solution incubated in the plate for 1 h. For lower loadings of RNA, 10 pmoles of the biotinylated DNA capture probe and 1 pmole of r(CUG)109 were used. The average amount of RNA immobilized in the well was determined using SYBR Green II (Invitrogen) and known concentrations of r(CUG)109. On average, when 2.5 pmoles of RNA were delivered to a well, 0.65 pmoles were immobilized; when 1 pmole of RNA was delivered, 0.19 pmole was immobilized. Please see the Supporting Information for details.
After washing the wells with 2 × 200 μL of 1× MBNL1 Buffer, 100 μL of a solution containing 32 pmoles of MBNL1, 3.7 μM tRNA, and the ligand of interest in 1× MBNL1 Buffer was added to each well and incubated for 1 h. For experiments in which 1 pmole of r(CUG)109 (on average 0.19 pmoles is immobilized) was delivered, 1.48 μM bulk yeast tRNA and 13.5 pmoles of MBNL1 were used. The wells were washed with 2 × 200 μL 1× MBNL1 Buffer followed by 1 × 200 μL 1× Phosphate Buffered Saline (PBS). Enzymatic complementation was completed for 3 h by adding 1.2 μL EA reagent (LacZΩ, DiscoverX) in 100 μL of PBS to each well. (Binding of LacZΩ and LacZα results in functional β–galactosidase.) Then, 10 μL of 85 μM resorufin-β-D-galactopyranoside (Invitrogen) was added to each well, and the fluorescence measured on a BioTek FLX-800 fluorescence plate reader (Excitation filter: 530/25; Emission filter 590/35; Sensitivity = 50-80). For the order of addition experiments, when 5H-4 was added first, the ligand was added and the samples were incubated with the RNA for 1 h. Then, MBNL1 was added and the samples allowed to equilibrate for another hour prior to washing and complementation. Analogously, when MBNL1 was added first, the protein was allowed to equilibrate with the RNA for 1 h followed by addition of 5H-4 for 1 h.
The resulting data were then fit to a four parameter logistic curve to determine the IC50's when the percentage of MBNL1 bound ranged from 0-100%. Each IC50 was the average of at least two measurements, and the error is the standard deviation in those measurements. The values for the multivalent effects were computed using the two equations below:
| (1) |
| (2) |
Fluorescence Binding Assays
Nucleic acids were annealed in Assay Buffer (8 mM sodium phosphate, pH 7.0, 185 mM NaCl, and 1 mM EDTA) at 60 °C for 1 min (RNA) or 90 °C for 3 min (herring sperm DNA), followed by slow cooling to room temperature. Binding assays were completed by titrating the annealed nucleic acid into 1 μM of the corresponding compound in 1× Assay Buffer, with the exception of determination of the affinity of r(CUG)109 for 2 (5 μM). After a 5 min incubation, the fluorescence intensity was measured using a BioTek FLX-800 fluorescence plate reader (Excitation: 360/40; Emission: 460/40; Sensitivity = 90). Two types of plots were constructed: Δ fluorescence vs. [Nucleic Acid]/[Ligand] to determine stoichiometry; and Fraction Bound / [Nucleic Acid] vs. Fraction Bound to determine binding constants. Stoichiometries were determined from the former plots by fitting each of the two slopes (pre-saturated and saturated portions of the curves) to a line. The two resulting equations were solved simultaneously to afford the stoichiometry.27 For Herring Sperm DNA, the latter plots were fit to a straight line. For RNA-ligand interactions, statistical effects had to be taken into account. Therefore, the interaction was treated as a large ligand binding to a lattice-like chain as described.28,29 As such, the resulting curves were fit to Equation 3:
| (3) |
where v is the moles of ligand per moles of RNA lattice, [L] is the concentration of ligand, N is the number of repeating units on the RNA, l is the number of consecutive lattice units occupied by the ligand, and k is the microscopic dissociation constant. Interestingly, if l is treated as a variable, the resulting value is consistent with ligand valency and the stoichiometries determined from Δ fluorescence vs. [Nucleic Acid]/[Ligand] plots (The ratio N/l is the stoichiometry.) Please see the Supporting Information for representative binding curves and a summary of all data.
Cell Culture, Uptake, and Microscopy
The C2C12 (mouse myoblast) cell line was maintained as a monolayer in 1× DMEM supplemented with 10% FBS and 0.5% penicillin/streptomycin. For uptake experiments, cells were added to a well of a 6-well plate containing a sterile glass cover slip and 1.5 mL of fresh medium. The cells were grown for 24 h at 37 °C and 5% CO2. The medium was removed and replaced with fresh medium. Then compound was added to a final concentration of 5 μM and incubated for 14 h. The medium containing the compound of interest was removed and the cells were washed with 1× DPBS (Invitrogen). The cover slip was mounted in 5 μL of 1× DPBS + 50% glycerol, and the cells were imaged using a Zeiss photomicroscope equipped with a Princeton Micromax CCD and Scanalytics IPLab software.
Flow Cytometry Analysis for Uptake and Toxicity
In order to quantify cell uptake of modularly assembled ligands and assess toxicity, flow cytometry analyses were completed. Uptake assays were completed as described above except the ligand of interest was incubated with the cells for 14 h or 48 h. The cells were then trypsinized, pelleted and washed with ice-cold 1× DPBS. After pelleting the cells, they were resuspended in ice-cold 1× DPBS and placed on ice. Then, 1 μL of 1.5 mM propidium iodide was incubated with the cells (on ice) in the dark for 20-30 min. Analysis of 30,000 events was completed using a BD LSR II System Flow Cytometer.
Results
Buoyed by earlier results of targeting of the rCCUG repeats that cause DM2 with aminoglycoside modules displayed on a peptoid backbone,26 we searched the literature to identify lead modules that are known to bind to the 5′CUG/3′GUC motif present in the DM1 hairpin (Figure 1). Searches were constrained to identify ligands that have been successfully applied in mammalian cells and mice. Gratifyingly, Hoechst 33258 (1, Figure 2), which is well tolerated by and non-toxic to mice,30 was identified to bind 5′CUG/3′GUC with nanomolar affinity during studies on the binding of 1 to the 5′UTR of thymidylate synthetase messenger RNA.17
Figure 2.

The structures of Hoechst 33258 (1) and a Hoechst derivative that contains an azide handle (2) to anchor the module on a peptoid backbone.
Validation of 2 as a Lead Ligand for Disruption of the r(CUG)109-MBNL1 Complex
In order to multivalently display 1 to bind multiple copies of the 5′CUG/3′GUC motif present in the DM1 hairpin, an azide chemical handle was installed in the 1 scaffold to afford compound 2 (Figure 2). The azide handle allows 2 to be multivalently displayed on an alkyne-functionalized peptoid backbone via a Huisgen dipolar cycloaddition reaction (HDCR), a variant of “click” chemistry.22 Prior to synthesis of the multivalent compounds, studies were undertaken to determine if 2 binds selectively to RNAs that display a single copy of the DM1 motif, or 5′CUG/3′GUC. The 5′CUG/3′GUC motif was inserted into a hairpin cassette (RNA1) to afford RNA2 (Figure 3). In good agreement with the previous report that piqued our interest in this ligand,17 2 binds RNA2 13-fold more tightly than RNA1 with dissociation constants of 130 ± 25 nM and 1700 ± 70 nM, respectively. The affinities of 1 and 2 for a DNA hairpin, DNA1 (Figure 3) that contains the Hoechst binding motif 5′AATT/3′TTAA was also determined (Table 1). Results show that 1 and 2 bind to DNA1 with Kd's of 280 and 250 nM, respectively. These results indicate that functionalization of 1 to install a chemical handle to enable modular assembly does not impair binding to nucleic acids or alter its specificity. Furthermore, monomer 2 is only ∼2-fold specific for RNA2 over DNA1.
Figure 3.

The nucleic acids used to study RNA-ligand interactions. Boxed nucleotides shown to the right were inserted into RNA 1. RNA2-RNA10 contain single copies of an internal loop motif. RNA11-RNA17 contain 12 copies of a motif. RNA2 and RNA11 contain the DM1 motif; RNA9 and RNA12 contain the SCA3 motif; RNA14 contains the DM2 motif; RNA6 and RNA13 contain 1×1 CC internal loops; RNA15 and RNA16 contain 2×2 CC or UU internal loops, respectively; RNA17 is a fully paired RNA control. DNA1 contains a single copy of the consensus Hoechst 33258 binding site and DNA2 is the DNA analogue of DM1 RNA11.
Table 1. Binding of Nucleic Acids to 2a.
| Nucleic Acid | Kd (nM) | Specificity to RNA2 |
|---|---|---|
| RNA1 | 1700±70 | 13.1 |
| RNA2 | 130±25 | - |
| RNA3 | 1050±212 | 8.0 |
| RNA4 | 370±107 | 2.8 |
| RNA5 | 810±150 | 6.2 |
| RNA6 | 290±50 | 2.2 |
| RNA7 | ∼1000 | ∼7.7 |
| RNA8 | 520±18 | 4.0 |
| RNA9 | 450±100 | 3.5 |
| RNA10 | 820±160 | 6.3 |
| DNA1 | 250±63 (280±37)b | 1.9 |
All 2-RNA complexes occur with a 1:1 stoichiometry.
Binding of 1 to DNA1.
Ligand 2 was also studied for binding to a series of RNAs containing 1×1 nucleotide internal loops to determine features in the RNA that are important for molecular recognition. First a series of 1×1 nucleotide UU loops were studied in which the loop closing base pairs were changed, or RNA3-RNA5 (Figure 3). Approximately 3-fold and 6-fold weaker binding is observed when loop GC pairs (RNA2) are changed to AU (RNA4) or GU (RNA5), respectively. Binding is even more significantly affected when the orientation of the closing pairs are changed. For example, compound 2 binds 5′CUG/3′GUC (RNA2) >8-fold more tightly than 5′CUC/3′GUG (RNA3), which has a similar affinity as the RNA that is fully paired (RNA1). Similar diminished affinities related to the orientation of the loop closing pairs are observed with 1×1 CC (RNA6 and RNA7) and 1×1 AA loops (RNA9 and RNA10). Thus, the identity and orientation of the loop closing base pairs is an important factor in molecular recognition of RNA by 2. These results point to appropriate display of RNA functional groups in the grooves as an important determinant in molecular recognition, as altering the orientation of the loop closing pairs would affect display of these groups.
Next, 2 was studied for binding to r(CUG)109 hairpin, which has 109 rCUG repeats or 54 DM1 motifs (Table 2). Results showed that 2 has a similar affinity to r(CUG)109 and RNA2; the Kd for binding to r(CUG)109 is 150 ± 25 nM while the Kd for binding RNA2 is 130 ± 25 nM. In addition, the stoichiometry of 2 binding r(CUG)109 is 54 ± 3 ligands per RNA, indicating that 2 binds every 5′CUG/3′GUC motif (Table 2). These studies provide two important results: (1) 2 binds to every copy of the DM1 motif in r(CUG)109; and, (2) since the affinity of 2 to RNAs containing single or multiple 5′CUG/3′GUC motif(s) are similar, there is no cooperativity between recognition of adjacent 5′CUG/3′GUC modules. Similarly, no cooperativity is observed in studies of MBNL1 binding to DM1 motifs.31 Compound 2 was then tested for inhibiting the formation of the toxic r(CUG)109-MBNL1 complex using a microtiter plate displacement assay with a MBNL1-β galactosidase fusion protein. In these assays, 0.65 pmoles of r(CUG)109 were immobilized in a well of a 96-well plate and incubated simultaneously with the ligand of interest, 32 pmoles of MBNL1, and 3.7 μM competing yeast bulk tRNA (∼570-fold higher concentration than r(CUG)109). Results show that 2 inhibits the formation of the r(CUG)109-MBNL1 complex with an IC50 of 110 μM.
Table 2. Binding constants and stoichiometry of monovalent and modularly assembled ligands to nucleic acids.
| Ligand | Kd (nM) | Stoichiometry |
|---|---|---|
| r(CUG)109 | ||
| 2 | 150 ± 25 | 54 ± 3 (54)a |
| 2H-4 | 100 ± 13 | 18 ± 2 (27) a |
| 3H-4 | 65 ± 8 | 16 ± 2 (18) a |
| 4H-4 | 35 ± 4 | 11 ± 2 (13.5) a |
| 5H-4 | 13 ± 3 | 8 ± 1 (10.8) a |
| Herring Sperm DNA | ||
| 2 | 110 ± 19 | 9 ± 1.3 |
| 2H-4 | 60 ± 7 | 9 ± 0.4 |
| 3H-4 | 430 ± 17 | 2 ± 0.1 |
| 4H-4 | 460 ± 31 | 2 ± 0.1 |
| 5H-4 | 700 ± 13 | 1 ± 0.04 |
| Bulk Yeast tRNA | ||
| 5H-4 | 1300 ± 250 | 1.0 ±0.1 |
Values in parentheses are the numbers of 5′CUG/3′GUC motifs present in r(CUG)109 divided by the number of bis-benzimidazole modules displayed by the compounds.
Multivalent Display of 2 Increases Potency for Disruption of the r(CUG)109-MBNL1 Complex
These initial studies validated 2 as a lead ligand for binding to the DM1 hairpin and for inhibition of the r(CUG)109-MBNL1 complex. In order to increase affinity and potency of the lead, a modular assembly approach was used in which the azide handle present in 2 was conjugated to alkyne-displaying peptoids using a HDCR (Figure 4). A small library of nine alkyne-displaying dimeric peptoids was synthesized with varying distances between ligand modules afforded by coupling different numbers of propylamines (1-6, 8, 12, or 16) between alkyne submonomers (propargylamine). The most potent dimer spacing was then used as a basis to synthesize trimeric, tetrameric, and pentameric ligands. Each peptoid is named using the nomenclature described in Figure 4. Representative structures are shown in Figure 5. The general format for peptoid nomenclature is as follows: nL-m where n is the ligand valency (c + 2), L is the ligand module, and m is the number of propylamine submonomers between ligand modules (a & b). The ligand modules (L) that were conjugated to the peptoid are: H which indicates the Hoechst derivative 2 (Figure 2) or A which refers to 3-azidopropylamine. Thus, 2H-4 describes a peptoid that displays two 2 modules separated by four spacing modules (a dimer) while 3H-4 describes a peptoid that displays three 2 modules each separated by four spacing modules (a trimer), etc.
Figure 4.

Anchoring of 2 to peptoids displaying alkyne units with various valencies and distances between ligand modules using a HDCR and the nomenclature used to describe the multivalent peptoid ligands. The general format for peptoid nomenclature is as follows: nL-m where n is the ligand valency (c + 2), L is the ligand module, and m is the number of propylamine submonomers between ligand modules (a & b). The ligand modules that were conjugated to the peptoid are: H, which indicates Hoechst derivative 2 (Figure 2), or A, which refers to 3-azidopropylamine. Thus, 2H-4 describes a peptoid that displays two 2 modules separated by four spacing modules (a dimer) while 3H-4 describes a peptoid that displays three 2 modules each separated by four spacing modules (a trimer), etc. The structures of the most potent dimer, trimer, tetramer, and pentamer are shown in Figure 5.
Figure 5.
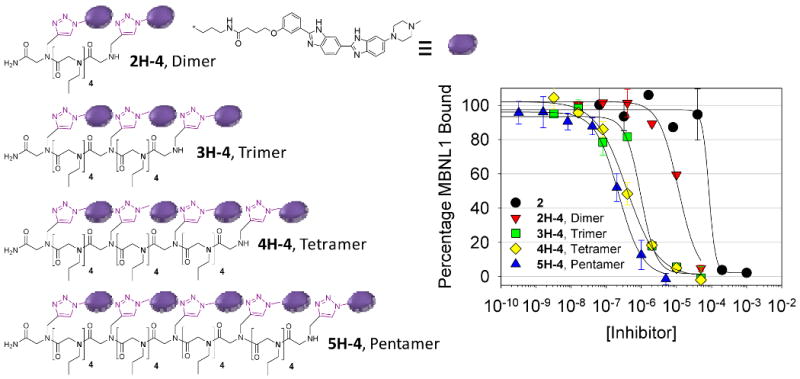
Left, structures of the most potent dimer, trimer, tetramer, and pentamer identified in these studies. Right, plots for inhibition of the r(CUG)109-MBNL1 interaction via monovalent and multivalent ligands.
Initial studies were then completed on the library of dimers to identify the spacing that gave the most potent inhibitory activity. Results showed that the dimer with the highest potency contained four propylamine spacers between the 2 modules (2H-4) and has an IC50 of 11 μM (Figure 5 and Table 4). Thus, an appropriately spaced dimer is an ∼10-fold more potent inhibitor than monomer 2.
Table 4. Inhibition of the r(CUG)109-MBNL1 complex with monovalent and multivalent ligands a.
| Compound | IC50, μM | Multivalent Effect b | Compound | IC50, μM | Multivalent Effect b |
|---|---|---|---|---|---|
| 2, Monomer | 110 ± 20 | - | 3H-4, Trimer | 0.960 ± 0.100 | 38 |
| 2H-1, Dimer | 25 ± 6 | 2.2 | 4H-4, Tetramer | 0.390 ± 0.110 | 71 |
| 2H-2, Dimer | 22 ± 5 | 2.5 | 5H-4, Pentamer | 0.220 ± 0.010 | 100 |
| 2H-3, Dimer | 18 ± 4 | 3.0 | 3H-4, Trimer c | 0.410 ± 0.023 | - |
| 2H-4, Dimer | 11 ± 2 | 5.0 | 4H-4, Tetramer c | 0.210 ± 0.085 | - |
| 2H-5, Dimer | 22 ± 4 | 2.5 | 5H-4, Pentamer c | 0.077 ± 0.015 | - |
| 2H-6, Dimer | >30 | <1.8 | 5H-4, Pentamer d | 0.140 ± 0.040 | - |
| 2H-8 Dimer | >30 | <1.8 |
5H-4, Pentamer e (no competitor; 5H-4 added before MBNL1) |
0.038 ± 0.021 | - |
| 2H-12, Dimer | >30 | <1.8 |
5H-4, Pentamer e (no competitor,;5H-4 and MBNL1 added together) |
0.086 ± 0.005 | - |
| 2H-16, Dimer | >30 | <1.8 |
5H-4, Pentamer e (no competitor; MBNL1 added before 5H-4) |
0.95 ± 0.08 | - |
|
5A-4, Control Pentamer (no 2 module) f |
>50 | - |
Nomenclature describing spacing of modules and valency is described in Figure 4. Unless otherwise noted, experiments were completed in the presence of 3.7 μM bulk yeast tRNA (0.1 mg/mL or 290 μM nucleotides) with 0.65 pmoles of r(CUG)109 immobilized in a well of a 96-well plate.
Values for the multivalent effect were calculated as described in the Experimental.
Experiments were completed with 0.19 pmole of r(CUG)109 immobilized in a well and 1.48 μM bulk yeast tRNA.
These experiments were completed in the presence of 1.48 μM herring sperm DNA (0.65 mM nucleotides; molecular weight, 220 base pairs).
Order of addition experiments were completed without competing nucleic acid with 0.19 pmole of r(CUG)109 immobilized.
Control pentamer, 5A-4, is the pentameric peptoid functionalized with 3-azidopropylamine via HDCR. These experiments were completed with 0.19 pmole of r(CUG)109 immobilized in a well and 1.48 μM bulk yeast tRNA.
Based on the results of the dimers, a series of compounds with increasing valency displaying the optimal four propylamine spacing was synthesized and tested for disruption of the r(CUG)109-MBNL1 complex. The resulting IC50's for inhibition of the r(CUG)109-MBNL1 complex in the presence of tRNA competitor for the trimer (3H-4), tetramer (4H-4), and pentamer (5H-4) are 960, 390 and 220 nM, respectively (Figure 5 and Table 4). Thus, for each increase in valency there is, on average, an ∼2-fold increase in the potency of the ligands.
Inhibition assays were also completed at lower RNA loadings by immobilizing 0.19 pmole of r(CUG)109 in the MBNL1 displacement assay (Table 4). As expected, the multivalent compounds had improved IC50's at the lower RNA loadings of 410, 210, and 77 nM for 3H-4, 4H-4, and 5H-4, respectively. The IC50 for 5H-4 was also determined in the presence of herring sperm DNA and is 140 nM; the IC50 in the absence of competitor is 86 nM (which is within error of the IC50 for the pentamer in the presence of tRNA). A 3-azidopropylamine-functionalized pentameric peptoid, 5A-4, that has the same spacing as 5H-4, was also tested in order to determine if the peptoid itself contributes to inhibition of the RNA-protein interaction. As expected, this compound does not inhibit formation of the DM1 hairpin-MBNL1 interaction up to the highest concentration tested, 50 μM, unambiguously showing that inhibition is due to the RNA-binding modules not the peptoid or the 5 amines displayed on this control.
The effect of the order in which MBNL1 and 5H-4 were added on potency was also determined in the absence of competing nucleic acids with 0.19 pmole of r(CUG)109 immobilized (Table 4). When MBNL1 and 5H-4 are added at the same time, the IC50 is 86 nM. When 5H-4 is added first followed by addition of MBNL1, an IC50 of 40 nM is obtained. A higher IC50 of 950 nM is obtained if MBNL1 is pre-incubated with r(CUG)109 and then 5H-4 added. Since it is unclear which order of addition experiment would more closely mimic the cellular interaction, it is encouraging that sub-micromolar IC50's are obtained in each case.
For each inhibitor, the effect of multivalency on increasing potency was calculated (Equations 1 and 2 and Table 4). Values were computed for experiments completed with 0.65 pmoles of immobilized r(CUG)109 in the presence of 3.7 μM bulk yeast tRNA. The range of the multivalent effects were from <1.8- to 100-fold. Dimers ranged from <1.8 to 5, with 2H-4 having the largest value. The trimer 3H-4, tetramer 4H-4, and pentamer 5H-4 had values of 38-, 71-, and 100-fold, respectively. Thus, increases in the valency of the inhibitors increases their potency beyond the value expected if only the number of modules displayed on the chain is considered.
The Affinities of Monovalent and Modularly Assembled Ligands for Nucleic Acids
The binding affinities and stoichiometries of the ligands to r(CUG)109, herring sperm DNA and yeast tRNAs were determined (Figure 6 and Table 2). The goals of these experiments were two-fold. The first objective is to understand the effect that affinity has on the potency of r(CUG)109-MBNL1 inhibition and how these results correlate with multivalent effects. The second objective is to understand the effect that multivalent display of the bis-benzimidazole 2 has on specificity between recognition of DNA and RNA.
Figure 6.

A, Titration of r(CUG)109, top, or herring sperm DNA, bottom, into a solution of 2H-4. B, Titration of r(CUG)109, top, or herring sperm DNA, bottom, into a solution of 5H-4.
The affinities of the ligands for r(CUG)109 scales with ligand valency and the Kd's range from 150 nM for 2 to 13 nM for 5H-4 (Table 2). The same trend does not hold for binding to herring sperm DNA, however. While the binding affinities of monomeric 2 and the dimer 2H-4 were 110 and 60 nM, respectively, affinity for herring sperm DNA decreased with higher valencies; trimer 3H-4, tetramer 4H-4, and pentamer 5H-4 bound herring sperm DNA with Kd's of 430, 460, and 700 nM, respectively. Therefore, both the monomer and dimer are slightly specific for herring sperm DNA while the trimer, tetramer, and pentamer are specific for r(CUG)109. The tetramer and pentamer are 13- and 54-fold specific, respectively (Table 5). Additionally, 5H-4 was tested for binding to bulk yeast tRNA. The Kd of the tRNA-5H-4 interaction is 1300 ± 300 nM with a 1:1 stoichiometry. Thus, 5H-4 is 100-fold specific for r(CUG)109 over bulk yeast tRNA. These results show that appropriate display of the ligand module can convert a monomeric DNA binder (2) into a modularly assembled ligand that has high specificity for the target r(CUG)109 hairpin over DNA. There was no change in fluorescence of 5H-4 when up to equimolar concentrations of MBNL1 were added, indicating that it does not bind the protein.
Table 5. Selectivity of ligands for r(CUG)109 over herring sperm DNAa.
| Ligand | Selectivity |
|---|---|
| 2 | 0.73 |
| 2H-4 | 0.60 |
| 3H-4 | 7 |
| 4H-4 | 13 |
| 5H-4 | 54 |
Selectivity was calculated by dividing the Kd for the ligand binding to herring sperm DNA by the Kd for binding to r(CUG)109.
Stoichiometries were also determined for each ligand-herring sperm DNA and ligand-r(CUG)109 complex. For herring sperm DNA, the stoichiometries for the monomer 2, dimer 2H-4, trimer 3H-4, tetramer 4H-4, and pentamer 5H-4 were 9, 9, 2, 2, and 1 ligand(s) per DNA, respectively. For r(CUG)109, the stoichiometries for the monomer 2, dimer 2H-4, trimer 3H-4, tetramer 4H-4, and pentamer 5H-4 were 54, 18, 16, 11, and 8 ligands per RNA, respectively (Table 2). (These stoichiometries are expected when statistical effects are taken into account.28,29) Since there are 54 copies of 5′CUG/3′GUC in the r(CUG)109 hairpin, these results indicate that each ligand module, whether a monomer or part of a modularly assembled ligand, interacts with one 5′CUG/3′GUC motif present in the RNA. More specifically, these results suggest that 5H-4 interacts with five 5′CUG/3′GUC motifs, 4H-4 interacts with four 5′CUG/3′GUC motifs, etc.
The 5H-4 pentamer was also tested for binding to related RNAs that contain 12 copies of a motif to determine features in RNA repeats that govern molecular recognition (RNA11-RNA17, Table 3 and Figure 3). The repeating RNAs include: RNA11 that has the DM1 motif; RNA12 that contains the 5′CAG/3′GAC repeat present in SCA3 and polyQ disorders;16 RNA13 that has a 5′CCG/3′GCC, or polypyrimidine, repeat; RNA14 that contains the 5′CCUG/3′GUCC tetranucleotide repeat that is present in DM2;15 RNA15 that contains a 5′CUUG/3′GUUC repeat with the 2×2 all-U loop; RNA16 that has a 5′CCCG/3′GCCC repeat with a 2×2 all-C loop; and, RNA17 that contains 12 copies of 5′CAG/3′GUC that forms a fully Watson-Crick paired region. For all RNAs studied, the pentamer binds DM1 RNA11 with the highest affinity with a Kd of 25 nM. The next tightest binder is RNA13, and it binds pentamer 4.4-fold weaker than RNA11. Binding is the weakest to fully paired RNA17, which binds with a Kd of >5000 nM. Interestingly, the pentamer binds to the DM2 RNA (RNA14) 32-fold more weakly than DM1 RNA11. Previously, we designed a modularly assembled aminoglycoside for binding DM2 RNA14, and that ligand was 30-fold specific for RNA14 over RNA11.26 Thus, specificity can be controlled by modular display of an appropriate ligand module.
Table 3. Binding of 5H-4 and MBNL1 to a variety of nucleic acids.
| Nucleic Acid | 5H-4 | MBNL1 | |||
|---|---|---|---|---|---|
| Kd (nM) | Stoichiometry | Specificity to DM1 RNA11 | Kd (nM)a | Specificity to DM1 RNA11 | |
| r(CUG)109 | 13 ± 3 | 8 ± 1 | - | 300±93b | - |
| RNA11 | 25 ± 6 | 1.9 ± 0.2 | - | 250±10b | - |
| RNA12 | 130 ± 35 | 1.7 ± 0.2 | 5.2 | 630±70 b | 2.5 |
| RNA13 | 110 ± 22 | 2.0 ± 0.3 | 4.4 | 200±30 b | 0.8 |
| RNA14 | 790 ± 31 | 1.3 ± 0.04 | 32 | 120±40 b | 0.5 |
| RNA15 | 1150 ± 80 | 1.2 ± 0.03 | 46 | >2500 b | 10 |
| RNA16 | 650 ± 74 | 1.1 ± 0.2 | 26 | 920±150 b | 3.7 |
| RNA17 | >5000 | N.D. | >200 | >1000 | >4 |
| DNA1 | 580 ± 30 | 1.0 ± 0.1 | 23 | NDc | NDc |
| DNA2 | 1200 ± 250 | 1.5 ± 0.2 | 48 | NDc | NDc |
Binding of 5H-4 to synthetic DNAs was also completed. DNA1 binds pentamer with a Kd of 580 nM, which is 2-fold weaker than the binding of DNA1 to 2; these results mirror those with herring sperm DNA in that modularly assembled ligands bind more weakly to DNA than RNA. Binding of 5H-4 to DNA2, which is the DNA analogue of RNA11, occurs with a Kd of 1200 nM. Thus, the 5H-4 pentamer is 48-fold specific for RNA over DNA even when the sequence is similar.
Binding of RNAs to MBNL1
The affinity and specificity of the modularly assembled ligands for RNA were then compared to that of MBNL1 (Table 3). We previously determined the affinity of MBNL1 for RNA11-RNA16 by gel retardation.26 These values are in good agreement with a previous report.31 In summary, MBNL1 binds r(CUG)109 with a Kd of 300 nM and to RNA11 with a Kd of 250 nM. The similar affinities to these RNAs despite the difference in the number of DM1 motifs indicate a lack of cooperativity in the binding of MBNL1 to RNA as observed previously.31 The highest affinity complex, formed between MBNL1 and DM2 RNA14, has a Kd of 120 nM, which is 2-fold higher affinity than the DM1-MBNL1 complex. The RNA13-MBNL1 (1×1 CC loop) complex has a slightly higher affinity than the DM1-MBNL1 complexes. The rCAG (SCA3)-MBNL1 16 repeat, or RNA12, complex is 2.5-fold weaker than the DM1-MBNL1 complex. MBNL1 binds more weakly to the RNAs displaying 2×2 nucleotide loops that form tandem UU or CC mismatches (RNA15 and RNA16). The dissociation constants for these interactions are >2000 and 920 nM, respectively. As expected, MBNL1 also binds weakly to the fully paired RNA17 with a Kd of >1000 nM.
Uptake of the Modularly Assembled Ligands into Mouse Myoblasts
If the modularly assembled ligands described herein are to have any use as therapeutics, then they should be cell permeable. Ideally no transfection agent should be required for uptake. One potential issue with using modularly assembled ligands is that there could be a point at which the molecular weight becomes so large that ligands will not be cell permeable. To qualitatively probe the effects of ligand valency on uptake, we studied the uptake of 3H-4, 4H-4, and 5H-4 into mouse myoblasts, which serve as a model for human muscle cells into which DM1 therapeutics must gain entry. Compounds were simply added to the medium with serum and incubated at 37 °C overnight. Microscopy of unfixed cells shows the trimer, tetramer, and pentamer all enter cells and localize to the nuclei (Figure 7A). Fortuitously, this is where the toxic DM1- MBNL1 hairpin interaction occurs in DM-affected cells.4,13,32,33
Figure 7.

A, Microscopy of mouse myoblasts (C2C12 cell line) incubated with 3H-4 (top), 4H-4 (middle), and 5H-4 (bottom) by simply adding the ligand to the culture medium. Left, phase contrast image of C2C12 cells; middle, fluorescence image; right, overlay of the phase contrast and fluorescence images. The compounds localize in the cells' nuclei where the toxic rCUG-MBNL1 interaction occurs. B, Flow cytometry histograms of C2C12 cells to determine uptake of 3H-4, 4H-4 and 5H-4 at various incubation times. The percentage of cells that are fluorescent increases with valency.
It appeared from microscopy studies that a higher percentage of cells were fluorescent when dosed with 5H-4 than with 3H-4. In order to quantify how valency affects uptake and to assess toxicity, flow cytometry analyses were completed (Figure 7B and Table 7). As also observed by microscopy, the compound that affords the highest percentage of fluorescent cells after a 14 h incubation is the pentamer, 5H-4 (83%). In contrast, only 69% of the cells treated with 4H-4 and 44% of the cells treated with 3H-4 have taken up modularly assembled ligand. After 48 h, the percentages of cells that are fluorescent are similar for 4H-4 and 5H-4, 90% and 88%, respectively, but slightly lower for 3H-4 (71%). The cells were also stained with propidium iodide after treatment with the modularly assembled ligands in order to assess toxicity. These experiments confirmed that the modularly assembled ligands are non-toxic at concentrations ≤5 μM. Specifically, a small percentage of cells had fluorescence from both the modularly assembled ligand and propidium iodide at 14 h or 48 h (<6% in all cases), indicating that uptake is not due to leaky cell membranes present in apoptotic cells. In addition, few cells were only stained with propidium iodide (<5% in all cases), or were apoptotic.
Table 7. Uptake and Toxicity of Inhibitor (Percentage of Cells that take up a Fluorescently Labeled Ligand)a.
| Compound | 48 h | 14 h |
|---|---|---|
| Cells with Fluorescence Only from nH-4 Ligand (%) | ||
| None | 3.2 | 0.55 |
| 3H-4 | 71 | 44 |
| 4H-4 | 90 | 69 |
| 5H-4 | 88 | 83 |
| Cells with Fluorescence from nH-4 Ligand and Propidium Iodide (%) | ||
| None | 0.69 | 0.11 |
| 3H-4 | 4.1 | 5.8 |
| 4H-4 | 1.3 | 13 |
| 5H-4 | 2.8 | 4.4 |
| Cells with Fluorescence Only from Propidium Iodide (%) | ||
| None | 0.02 | 0.00 |
| 3H-4 | 0.24 | 4.7 |
| 4H-4 | 0.03 | 1.4 |
| 5H-4 | 0.04 | 0.37 |
| Cells with No Fluorescence (%) | ||
| None | 96 | 99 |
| 3H-4 | 24 | 46 |
| 4H-4 | 8.9 | 17 |
| 5H-4 | 8.9 | 13 |
Analysis was performed by flow cytometry. Errors in each measurement are, on average, ± 5%.
Pentameric 5H-4 Potently Inhibits Formation of the SCA3 RNA-MBNL1 Interaction
As mentioned above, both MBNL1 and 5H-4 bind to RNA12, which contains 12 copies of the 5′CAG/3′GAC motif that is present in toxic SCA3 repeats.16 Recently, the interaction of SCA3 RNAs with Muscleblind proteins has been implicated in the disease pathology of spinocerebellar ataxia type 3.16 Therefore, we sought to study the ability of 5H-4 to inhibit SCA3 RNA-MBNL1 interactions.
In order to make the best comparison of ligand potency for DM1 and SCA3, RNA11 was used as a mimic of DM1 RNAs rather than r(CUG)109. Monomer 2 inhibits the interaction of MBNL1 with both RNAs with micromolar IC50's (Table 6). The IC50's for 5H-4 are 130 nM for disruption of both the RNA11- and RNA12-MBNL1 complexes when 2.0 pmoles of RNA are loaded into each well. Similar IC50's are observed despite 5H-4 being 5-fold more avid for RNA11 than RNA12. These results are explained by the fact that MBNL1 binds RNA11 with a 2.5-fold higher affinity than RNA12. Multivalent effects of 30- and 75-fold for inhibition of MBNL1 interactions with RNA11 and RNA13, respectively, were observed. These results demonstrate that modular assembly of ligands can allow for the potent design of inhibitors for a different triplet repeating RNA-MBNL1 interactions.
Table 6. Inhibition of MBNL1-RNA11 (DM1) and RNA12(SCA3) Interactions with 5H-4. The IC50's are reported in μM.
| Ligand | Condition Aa | Condition Ba | ||
|---|---|---|---|---|
| RNA 11 | RNA 12 | RNA 11 | RNA 12 | |
| 2 | 30.2 ± 6.8 | 48.4 ± 9.9 | 38.1 ± 6.9 | 21.1 ± 0.5 |
| 5H-4 | 0.25 ± 0.01 | 0.16 ± 0.03 | 0.13 ± 0.01 | 0.13 ± 0.06 |
| Multivalent Enhancements b | ||||
| 5H-4 | 24 | 60.5 | 58 | 32 |
In condition A, 7.5 pmoles of MBNL1 are added to each well; in condition B, 2.0 pmoles of MBNL1 were added per well.
Calculated as described in the Experimental.
Discussion
Molecular Recognition of rCUG Repeats by Ligands
A crystal structure of r(CUG)6 has been reported.9 Overall, the oligonucleotide adopts a structure similar to A-form RNA. Folding is stabilized by optimal base-stacking interactions of GC/CG base steps. In contrast, the CU/UG and UG/CU base steps have poor intrastrand overlap. The UU mismatches themselves stack within the helix and do not distort the helical backbone. In order to do so, the mismatches do not hydrogen bond to each other; rather they hydrogen bond to water. This results in a repeating pattern of alternating electrostatic potentials in the minor groove that is distinct from Watson-Crick paired A-form RNA. The alternating electrostatic potential may also provide a good binding pocket in the minor groove for 2 and may explain the 13-fold selectivity of 2 for RNA2 over RNA1.
An additional factor that could explain the selectivity of 2 for RNA2 is the difference in the shape of the major and minor grooves when mismatches are present. Such differences may not be observed in crystal structures because the structure in solution may be dynamic. Indeed, studies by Weeks and Crothers have shown through chemical modification that mismatches can affect RNA groove size.34 Interestingly, studies from a previous report of 1 binding to various RNAs showed that it recognizes many RNAs with 1×1 nucleotide loops with similar affinities.17 In addition, the DNA groove binders distamycin and DAPI (2-(4-amidinophenyl)-1H-indole-6-carboxamidine) both bind RNAs containing a 1×1 nucleotide CC internal loop with nanomolar affinities.17 Herein, we have found that the nature of the loop closing pairs affects binding of 2. For example, RNA2 with 5′CUG/3′GUC motif and RNA3 with a 5′CUC/3′GUG motif bind 2 with Kd's of 130 and 1050 nM, respectively. These effects are also observed with other 1×1 loops. Collectively, these results point to the importance of the loop closing base pairs in the recognition of RNA by 2.
An advantage of the bis-benzimidazole scaffold is that it can be easily diversified.35-37 Therefore, other related modules can be synthesized and tested to identify ones with improved selectivity and affinity to r(CUG)109. Detailed insights into molecular recognition of the RNA-ligand complexes will have to wait for high resolution structures, which will undoubtedly facilitate the rational design of improved ligands to both this and other targets.
As a regulator of alternative splicing, MBNL1 must interact specifically with RNA and does so via four zinc finger (ZnF) domains. In order to gain insight into RNA-MBNL1 interactions and how this translates into regulation of alternative splicing, crystal structures of the ZnF3/4 domain with and without single stranded r(CGCUGU) were solved and described by Teplova and Patel.38 Both zinc fingers interact with one molecule of RNA with ZnF3 forming contacts to the 5′GC step and ZnF4 forming contacts to 5′GCU. The RNA molecules are oriented anti-parallel to each other. This study also investigated how the distance between 5′GCU elements affects MBNL1 binding affinity. If ten or 15 nucleotides separate two 5′GCU's, then the binding affinities are similar; however, if only five nucleotides separate two elements, an ∼3-fold decrease in affinity is observed. Taken together, these results suggest that MBNL1 binding induces a chain-reversal trajectory in the bound RNA that requires a certain length between 5′GCU's. Interestingly, this chain reversal and separation of 5′GCU elements are already present in long r(CUG) repeats that fold into a hairpin. Perhaps MBNL1 ordinarily opens the r(CUG) hairpin stem to afford two single stranded regions and the presence of our multivalent ligands prevents this unzipping. MBNL1 binds more weakly to fully base-paired RNAs, 31 which also supports this hypothesis.
Advantages of the Modular Assembly Approach
The DM1-MBNL1 interaction is unique because multiple proteins bind to a single RNA. Studies have shown that a single MBNL1 molecule interacts with ≤ 6 base pairs 31 or two 5′CUG/3′GUC motifs. Therefore each copy of the r(CUG)109 hairpin, which has 54 DM1 motifs, can interact with at most 27 molecules of MBNL1. Because of the high number of proteins that are bound to this RNA and the surface area of the RNA-protein complex, it could be very difficult for a traditional small molecule to potently disrupt this interaction. Therefore it is likely that surface area effects in addition to the relative affinities of the ligand-RNA and MBNL1-RNA complexes are important factors governing inhibition. Monomer 2 binds to r(CUG)109 with a Kd of 150 nM while binding of MBNL1 to r(CUG)109 is 300 nM.26 Despite the observation that 2 binds more tightly to the RNA than MBNL1, 2 is a weak inhibitor of the r(CUG)109-MBNL1 interaction with an IC50 of 110 μM. Such results may further suggest that surface area effects are an important factor in identifying potent inhibitors.
The multivalent effect quantifies the increased potency that multivalent ligands have over monovalent ones. Values can then be compared with binding constants to determine if multivalent enhancements are accounted for by an increase in affinity alone. Table 4 summarizes the data for inhibition of the r(CUG)109-MBNL1 interaction including multivalent effects, and Table 2 summarizes the binding affinities of the ligands for r(CUG)109. Comparison of the data in these two tables shows that multivalent effects increase from 5- to 100-fold as the valency increases from the dimer, 2H-4, to the pentamer, 5H-4. Enhancements in potency of the ligands are not totally accounted for by increased affinity, however. For example, 5H-4 binds 12-fold more tightly to r(CUG)109 than 2.
The selectivity of the ligands for r(CUG)109 is also enhanced by modular assembly. Specificity for binding to r(CUG)109 versus herring sperm DNA increased from 0.7-fold for 2 to 54-fold for 5H-4 (Table 6). Binding of 5H-4 is also 100-fold specific for r(CUG)109 over bulk yeast tRNA. Thus, appropriate multivalent display has converted a ligand that was slightly specific for herring sperm DNA to being very specific for the DM1 hairpin.
Modular assembly has been used as a strategy to design inhibitors for a variety of targets.39-42 For example, STARFISH ligands have been developed to inhibit the Shiga-like toxins.43 These compounds have multivalent enhancements as high as 1,000,000, which are some of the highest observed. Crystallographic analysis of the ligand-Shiga-like toxin complex revealed that the STARFISH scaffold pre-organizes the multivalent ligands for binding to the target protein. More commonly, multivalent enhancements range from 100 to 1000-fold. 28
Fragment-based assembly of small molecules has also been used in SAR (structure-activity relationships) by NMR,42 in screening of small chemical libraries,44,45 and in the modular design of polyamides targeting the DNA minor groove.46 In each of these cases, pre-organization of the ligand modules for binding to the target is critical for constructing potent ligands. Our future studies will focus on optimizing the display of ligand modules in order to pre-organize them to bind the DM1 hairpin. These studies can yield higher potency, lower molecular weight inhibitors. However, the modular assembly strategy described herein has provided cell permeable, designed ligands that bind with higher affinity and specificity to DM1 RNAs than Muscleblind-like 1 protein (Table 3).
Comparison to Other Studies Targeting DM1- and DM2-repeating RNA Hairpins
During the course of this work, another study identified compounds that disrupt the r(CUG)109-MBNL1 interaction.47 Compounds were identified by screening a resin-based dynamic combinatorial library containing, in theory, 11,325 members. The most potent compound found in these studies exhibited a Ki value of ∼3 μM in the absence of bulk tRNA. It must be noted that these compounds only inhibited ≤50% of the formation of the r(CUG)109-MBNL1 complex at the highest concentrations of ligand used, ∼20 μM. Furthermore, the ligands are linked by disulfides that are not likely to be stable in vivo, which is a reducing environment.
The most potent modularly assembled ligand (5H-4) described herein and monomeric 2 inhibit ≥95% of the MBNL1-r(CUG)109 interaction in the presence of bulk yeast tRNA (Figure 5). Furthermore, three inhibitors with submicromolar IC50's in the presence of bulk yeast tRNA were identified by testing only 13 compounds. The pentamer 5H-4 has an IC50 of 86 nM in the absence of competitor (Table 4). Thus, it may not be necessary to screen chemical libraries to identify lead molecules targeting the DM1 hairpin; rather, they can be designed once appropriate modules are identified that bind the 5′CUG/3′GUC motif. Furthermore, designed ligands are greater than two orders of magnitude more potent inhibitors compared to the ligands identified by screening and are higher affinity and more specific DM1 RNA binders than MBNL1 (Table 3).
Modularly assembled aminoglycoside ligands have been used to target the DM2 repeat-MBNL1 interaction by displaying the 5′CCUG/3′GUCC-binding module 6′-N-5-hexynoate kanamycin on a peptoid backbone.26 A series of nanomolar inhibitors of the DM2-MBNL1 interaction were identified. A trimeric azide-displaying peptoid, similar to 3H-4 but conjugated to 6′-N-5-hexynoate kanamycin (K), was the most potent inhibitor with an IC50 of 1.6 nM. The multivalent effect for this ligand was 20,000, which is greater than the value for 5H-4. This difference may be due to the number of repeats from which MBNL1 was displaced (r(CUG)109 versus r(CCUG)24). It could also be due to differences in recognition of the RNA by the two ligand modules or the pre-organization of the modules on the peptoid backbone.
Interestingly, the peptoid trimer functionalized with 6′-N-5-hexynoate kanamycin A (3K-4) is 30-fold specific for DM2 RNA over DM1 RNA.26 In the present study, we found that 5H-4 binds 32-fold more tightly to RNA11 (DM1) than RNA14 (DM2). Thus, the specificity for RNA targets can be precisely controlled by the module that is displayed on the scaffold. These collective observations bode well for the use of a modular assembly approach to provide ligands that are specific for a given RNA target.
In both studies, the most potent modularly assembled compounds displayed ligand modules separated by four propylamine spacers on a peptoid chain. Thus, the spacing provided by four propylamine spacers is sufficient to allow RNA binding modules to interact with two (or more) internal loops simultaneously when separated by two canonical pairs. Additional studies must be completed to determine how many spacing modules are needed to span other lengths between RNA secondary structures. This spacing could be general or the number of spacing modules required may depend on the RNA-binding module displayed. These studies are critical if the full potential of a developing RNA motif-ligand database 19,48,49 to target RNA is to be harnessed. Fine tuning may be necessary for each target; however, the ability to increase and decrease the spacing between ligands in a modular manner via solid-phase peptoid synthesis can allow for these molecules to be quickly synthesized and tested.
Uptake and Localization of Peptoids into Mouse Myoblasts
In a previous study, we reported that a fluorescently labeled peptoid trimer functionalized with 6′-N-5-hexynoate kanamycin A (3K-4) is cell permeable to mouse myoblasts.26 In contrast to 3H-4 which localizes in the nucleus, the kanamycin-functionalized trimer localizes mainly in the cytoplasm and the perinuclear region. We therefore synthesized a trimeric peptoid conjugated to propargylamine (3P-4) and studied its cellular uptake by microscopy. Interestingly, as observed for 3K-4, 3P-4 is cytoplasmic and perinuclear (Supporting Information). Taken together, this suggests that the ligand module conjugated to the peptoid backbone affects cellular localization. Flow cytometry analysis of cells incubated with 3H-4, 4H-4, and 5H-4 indicate that the size of the peptoid and/or the number of ligand modules correlates with uptake. That is, the higher the valency of the modularly assembled ligand, the greater the percentage of cells that are fluorescent.
Although uptake was not an issue with this cell line-ligand combination (Figure 7), this may not be the case for other combinations. Indeed, studies by the Dervan group have shown that uptake patterns can be idiosyncratic for different cell line-polyamide combinations.50-53 If uptake or cellular localization proves to be problematic, these issues potentially could be overcome by changing the identity of the spacing module. Several studies have shown that appropriately functionalized peptoids can improve uptake properties of cargo to which they are attached.54,55
It remains to be seen if a ligand that binds DM repeats and displaces MBNL1 would be effective at curing myotonic dystrophies or SCA3. Several factors are likely to be important for ligand efficacy based on the disease pathogenesis. For example, the toxicity of DM repeats has been associated with both the decreased translation of DM1-affected RNAs due to nuclear retention56 and sequestration of MBNL1 which affects pre-mRNA splicing.7,14,57,58 Splicing defects associated with DM1, however, have been corrected when MBNL1 is over-expressed in a DM1 mouse model.14 Thus, increasing the free amount of MBNL1 can allow for correction of splicing defects. It is possible, therefore, that a ligand disrupting the DM1 RNA-MBNL1 complex could increase the free concentration of MBNL1 leading to correction of splicing defects. On the other hand, the ligands could also prevent cytoplasmic transport of transcripts and not correct the translational defect. To correct both defects, it may be advantageous for a small molecule ligand to partition between the nucleus and cytoplasm. Such questions can only be answered when ligands are tested in cellular systems.
The modular assembly approach described herein may allow for the development of ligands that display different cellular localization properties by changing the spacing module.54,55,59 By synthesizing a library of small molecules with different spacing modules, ligands that are nuclear, cytoplasmic, and partition between the two could be identified. The effects of ligand localization on correction of splicing and translational defects could then be investigated more thoroughly.
Summary and Outlook
A modular assembly strategy was used to design nanomolar inhibitors of the toxic RNA-protein interactions that cause DM1 and SCA3. The most potent designed ligands are higher affinity and more specific binders for DM1 RNA than MBNL1 (Table 3). Since these diseases currently (2009) have no treatment, these studies may provide insights to develop therapies. Especially encouraging in this regard is the observation that modularly assembled ligands are cell permeable. Perhaps, this approach can be applied towards targeting other toxic repeating RNAs 16,60 and other RNA drug targets identified through genomic sequencing and biochemical investigations.3,61-64 One potential limitation for applying this approach to other RNAs is that only limited information on RNA motif-ligand partners is currently available, especially compared to the diversity of RNA loops in genomic RNA structures. The development of Two-Dimensional Combinatorial Screening (2DCS) to probe both RNA and chemical space simultaneously may expand this information, however.19,48,49,65 Perhaps, modular assembly strategies like those described herein will allow ligands targeting RNA to be designed quickly using computational mining of genomic sequences 66-68 rather than having to subject each new RNA target to which a binder is desired to a high throughput screening assay.
Supplementary Material
Acknowledgments
The authors thank Mr. Alan J. Siegel and the University at Buffalo's Microscopic Imaging Facility, Department of Biological Sciences for obtaining all microscopy images. Financial support by a NYSTAR JD Watson Award, The Research Corporation by a Cottrell Scholar Award, The New York State Center of Excellence in Bioinformatics and Life Sciences, the National Institutes of Health (R01-GM079235 to MDD), and from the Wellstone Center (08U54NS048843-05 and AR046806) are gratefully acknowledged.
References
- 1.Yang PK, Kuroda MI. Cell. 2007;128:777–86. doi: 10.1016/j.cell.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 2.Calin GA, Croce CM. Oncogene. 2006;25:6202–10. doi: 10.1038/sj.onc.1209910. [DOI] [PubMed] [Google Scholar]
- 3.Nahvi A, Sudarsan N, Ebert MS, Zou X, Brown KL, Breaker RR. Chem Biol. 2002;9:1043–9. doi: 10.1016/s1074-5521(02)00224-7. [DOI] [PubMed] [Google Scholar]
- 4.Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton CA. Science. 2000;289:1769–73. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 5.Tian B, White RJ, Xia T, Welle S, Turner DH, Mathews MB, Thornton CA. RNA. 2000;6:79–87. doi: 10.1017/s1355838200991544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Savkur RS, Philips AV, Cooper TA, Dalton JC, Moseley ML, Ranum LP, Day JW. Am J Hum Genet. 2004;74:1309–13. doi: 10.1086/421528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Day JW, Ranum LP. Neuromuscul Disord. 2005;15:5–16. doi: 10.1016/j.nmd.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 8.Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT, Cannon SC, Thornton CA. Mol Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- 9.Mooers BH, Logue JS, Berglund JA. Proc Natl Acad Sci U S A. 2005;102:16626–31. doi: 10.1073/pnas.0505873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Napierala M, Krzyzosiak WJ. J Biol Chem. 1997;272:31079–85. doi: 10.1074/jbc.272.49.31079. [DOI] [PubMed] [Google Scholar]
- 11.Kaliman P, Catalucci D, Lam JT, Kondo R, Gutierrez JC, Reddy S, Palacin M, Zorzano A, Chien KR, Ruiz-Lozano P. J Biol Chem. 2005;280:8016–21. doi: 10.1074/jbc.M412845200. [DOI] [PubMed] [Google Scholar]
- 12.Dansithong W, Paul S, Comai L, Reddy S. J Biol Chem. 2005;280:5773–80. doi: 10.1074/jbc.M410781200. [DOI] [PubMed] [Google Scholar]
- 13.Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS. Science. 2003;302:1978–80. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 14.Kanadia RN, Shin J, Yuan Y, Beattie SG, Wheeler TM, Thornton CA, Swanson MS. Proc Natl Acad Sci U S A. 2006;103:11748–53. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP. Science. 2001;293:864–7. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 16.Li LB, Yu Z, Teng X, Bonini NM. Nature. 2008;453:1107–11. doi: 10.1038/nature06909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho J, Rando RR. Nucleic Acids Res. 2000;28:2158–63. doi: 10.1093/nar/28.10.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Org Lett. 2004;6:2853–5. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 19.Disney MD, Childs-Disney JL. Chembiochem. 2007;8:649–56. doi: 10.1002/cbic.200600569. [DOI] [PubMed] [Google Scholar]
- 20.PerreeFauvet M, VerchereBeaur C, Tarnaud E, AnneheimHerbelin G, Bone N, Gaudemer A. Tetrahedron. 1996;52:13569–88. [Google Scholar]
- 21.Satz AL, Bruice TC. Bioorg Med Chem. 2000;8:1871–80. doi: 10.1016/s0968-0896(00)00116-4. [DOI] [PubMed] [Google Scholar]
- 22.Jang H, Fafarman A, Holub JM, Kirshenbaum K. Org Lett. 2005;7:1951–4. doi: 10.1021/ol050371q. [DOI] [PubMed] [Google Scholar]
- 23.Olivos HJ, Alluri PG, Reddy MM, Salony D, Kodadek T. Org Lett. 2002;4:4057–9. doi: 10.1021/ol0267578. [DOI] [PubMed] [Google Scholar]
- 24.Peyret N, Seneviratne PA, Allawi HT, SantaLucia J. Biochemistry. 1999;38:3468–77. doi: 10.1021/bi9825091. [DOI] [PubMed] [Google Scholar]
- 25.SantaLucia J. Proc Natl Acad Sci U S A. 1998;95:1460–5. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee MM, Pushechnikov A, Disney MD. ACS Chem Biol. 2009;4:345–355. doi: 10.1021/cb900025w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tse WC, Boger DL. Acc Chem Res. 2004;37:61–9. doi: 10.1021/ar030113y. [DOI] [PubMed] [Google Scholar]
- 28.McGhee JD, von Hippel PH. J Mol Biol. 1974;86:469–89. doi: 10.1016/0022-2836(74)90031-x. [DOI] [PubMed] [Google Scholar]
- 29.Cantor CR, Schimmel PR. Biophysical Chemistry. Vol. 3. W.H Freeman and Company; San Francisco: 1980. pp. 849–86. [Google Scholar]
- 30.Disney MD, Stephenson R, Wright TW, Haidaris CG, Turner DH, Gigliotti F. Antimicrob Agents Chemother. 2005;49:1326–30. doi: 10.1128/AAC.49.4.1326-1330.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Warf MB, Berglund JA. RNA. 2007;13:2238–51. doi: 10.1261/rna.610607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, Krym M, Henderson D, Schalling M, Swanson MS, Thornton CA. Hum Mol Genet. 2001;10:2165–70. doi: 10.1093/hmg/10.19.2165. [DOI] [PubMed] [Google Scholar]
- 33.Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Hum Mol Genet. 2004;13:3079–88. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- 34.Weeks KM, Crothers DM. Science. 1993;261:1574–7. doi: 10.1126/science.7690496. [DOI] [PubMed] [Google Scholar]
- 35.Wu CH, Sun CM. Tetrahedron Lett. 2006;47:2601–4. [Google Scholar]
- 36.Carpenter RD, DeBerdt PB, Lam KS, Kurth MJ. J Comb Chem. 2006;8:907–14. doi: 10.1021/cc060106b. [DOI] [PubMed] [Google Scholar]
- 37.Smith JM, Gard J, Cummings W, Kanizsai A, Krchnak V. J Comb Chem. 1999;1:368–70. [Google Scholar]
- 38.Teplova M, Patel DJ. Nat Struct Mol Biol. 2008;15:1343–51. doi: 10.1038/nsmb.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mammen M, Choi SK, Whitesides GM. Angew Chem Int Ed Engl. 1998;37:2755–94. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 40.Gestwicki JE, Cairo CW, Strong LE, Oetjen KA, Kiessling LL. J Am Chem Soc. 2002;124:14922–33. doi: 10.1021/ja027184x. [DOI] [PubMed] [Google Scholar]
- 41.Gordon EJ, Sanders WJ, Kiessling LL. Nature. 1998;392:30–1. doi: 10.1038/32073. [DOI] [PubMed] [Google Scholar]
- 42.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Science. 1996;274:1531–4. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 43.Kitov PI, Sadowska JM, Mulvey G, Armstrong GD, Ling H, Pannu NS, Read RJ, Bundle DR. Nature. 2000;403:669–72. doi: 10.1038/35001095. [DOI] [PubMed] [Google Scholar]
- 44.Maly DJ, Choong IC, Ellman JA. Proc Natl Acad Sci U S A. 2000;97:2419–24. doi: 10.1073/pnas.97.6.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Erlanson DA, Braisted AC, Raphael DR, Randal M, Stroud RM, Gordon EM, Wells JA. Proc Natl Acad Sci U S A. 2000;97:9367–72. doi: 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dervan PB. Bioorg Med Chem. 2001;9:2215–35. doi: 10.1016/s0968-0896(01)00262-0. [DOI] [PubMed] [Google Scholar]
- 47.Gareiss PC, Sobczak K, McNaughton BR, Palde PB, Thornton CA, Miller BL. J Am Chem Soc. 2008;130:16254–61. doi: 10.1021/ja804398y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Childs-Disney JL, Wu M, Pushechnikov A, Aminova O, Disney MD. ACS Chem Biol. 2007;2:745–54. doi: 10.1021/cb700174r. [DOI] [PubMed] [Google Scholar]
- 49.Disney MD, Labuda LP, Paul DJ, Poplawski SG, Pushechnikov A, Tran T, Velagapudi SP, Wu M, Childs-Disney JL. J Am Chem Soc. 2008;130:11185–94. doi: 10.1021/ja803234t. [DOI] [PubMed] [Google Scholar]
- 50.Nickols NG, Jacobs CS, Farkas ME, Dervan PB. Nucleic Acids Res. 2007;35:363–70. doi: 10.1093/nar/gkl1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Edelson BS, Best TP, Olenyuk B, Nickols NG, Doss RM, Foister S, Heckel A, Dervan PB. Nucleic Acids Res. 2004;32:2802–18. doi: 10.1093/nar/gkh609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Belitsky JM, Leslie SJ, Arora PS, Beerman TA, Dervan PB. Bioorg Med Chem. 2002;10:3313–8. doi: 10.1016/s0968-0896(02)00204-3. [DOI] [PubMed] [Google Scholar]
- 53.Best TP, Edelson BS, Nickols NG, Dervan PB. Proc Natl Acad Sci U S A. 2003;100:12063–8. doi: 10.1073/pnas.2035074100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goun EA, Pillow TH, Jones LR, Rothbard JB, Wender PA. Chembiochem. 2006;7:1497–515. doi: 10.1002/cbic.200600171. [DOI] [PubMed] [Google Scholar]
- 55.Goun EA, Shinde R, Dehnert KW, Adams-Bond A, Wender PA, Contag CH, Franc BL. Bioconjug Chem. 2006;17:787–96. doi: 10.1021/bc0503216. [DOI] [PubMed] [Google Scholar]
- 56.Mastroyiannopoulos NP, Feldman ML, Uney JB, Mahadevan MS, Phylactou LA. EMBO Rep. 2005;6:458–63. doi: 10.1038/sj.embor.7400390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Philips AV, Timchenko LT, Cooper TA. Science. 1998;280:737–41. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- 58.Savkur RS, Philips AV, Cooper TA. Nat Genet. 2001;29:40–7. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 59.Yu P, Liu B, Kodadek T. Nat Biotechnol. 2005;23:746–51. doi: 10.1038/nbt1099. [DOI] [PubMed] [Google Scholar]
- 60.Orr HT, Zoghbi HY. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 61.Lau NC, Lim LP, Weinstein EG, Bartel DP. Science. 2001;294:858–62. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- 62.Lee RC, Ambros V. Science. 2001;294:862–4. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- 63.Venter JC, et al. Science. 2001;291:1304–51. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 64.Lander ES, et al. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 65.Aminova O, Paul DJ, Childs-Disney JL, Disney MD. Biochemistry. 2008;47:12670–9. doi: 10.1021/bi8012615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Proc Natl Acad Sci U S A. 2004;101:7287–92. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Uzilov AV, Keegan JM, Mathews DH. BMC Bioinformatics. 2006;7:173. doi: 10.1186/1471-2105-7-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Macke TJ, Ecker DJ, Gutell RR, Gautheret D, Case DA, Sampath R. Nucleic Acids Res. 2001;29:4724–35. doi: 10.1093/nar/29.22.4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.