

Abstract
Plasminogen activator inhibitor-1 (PAI1) can promote cancer progression, and its protein expression in tumors is an independent indicator of poor prognosis in many forms of cancer. Here, we show that high PAI1 mRNA levels also predict for shorter overall survival in two independent breast cancer data sets, highlighting the importance of its transcriptional regulation. The −675insG (4G/5G) single-nucleotide polymorphism in the PAI1 gene promoter has been shown to influence PAI1 transcription, with the 4G allele eliciting higher reporter gene expression in vitro and higher levels of circulating PAI1 in vivo. Nevertheless, its genotypic distribution in 2,539 British women with invasive breast cancer was virtually identical to that seen in 1,832 matched controls (P = 0.72), and annual mortality rates for 4G4G, 4G5G, and 5G5G cases were 2.6%, 2.8%, and 3.1% per year, respectively (P = 0.10). Thus, there was no association with breast cancer incidence or outcome, and in a separate set of breast cancers, the 4G/5G single-nucleotide polymorphism showed no association with PAI1 mRNA expression (P = 0.85). By contrast, connective tissue growth factor (CTGF), which can regulate PAI1 expression in culture, was associated with PAI1 expression in three independent cohorts (P ≪ 0.0001). In addition, PAI1 gene copy number differences in the tumors were correlated with PAI1 mRNA expression (P = 0.0005) and seemed to affect expression independently of CTGF. Thus, local factors, such as CTGF and genomic amplification, seem to be more important than germ line genetic variation in influencing PAI1 expression and its untoward effects in breast cancer.
Introduction
Considerable evidence indicates that PAI1, a major physiologic inhibitor of urokinase-type and tissue-type plasminogen activators, promotes cancer progression. High tumoral PAI1 protein expression is associated with poor survival in several forms of cancer (1-3) and is a strong independent prognostic factor in breast cancer, with elevated levels forecasting shorter recurrence-free and overall survival (4-8). In addition, prospective data suggest that high tumoral PAI1 levels can identify those lymph node–negative (LN−) breast cancer patients who are most likely to benefit from adjuvant chemotherapy (9, 10). Nevertheless, PAI1 expression is rarely used in clinical decision making, as the protein-based assays used to establish its prognostic and predictive value are poorly adapted to the limited amounts of tissue that are commonly available after routine screening and early cancer detection.
Experimental data also indicate that PAI1 promotes cancer progression. In cancer transplantation models, tumor growth, invasion, and angiogenesis are essentially abolished in PAI1-deficient mice and are rescued by adenoviral PAI1 replacement (11, 12). In addition, data show that PAI1 promotes tumor growth in a dose-dependent and stage-dependent manner and suggest that stromal PAI1 is more important than tumor cell–derived PAI1 (13, 14). These effects seem to depend on the proteinase inhibitory activity of PAI1 (15, 16), although its ability to disrupt integrin-mediated adhesion and promote cell motility independent of its inhibitory function may also contribute (17, 18). Thus, PAI1 is not simply an indicator of progression but an active participant. Consequently, factors that affect its expression should also affect progression.
One means of regulating PAI1 is at the level of transcription. Notably, the PAI1 (SERPINE1) promoter contains a common transcription-altering insertion/deletion single-nucleotide polymorphism (SNP; rs1799889) with four or five guanine nucleotides 675 bp upstream of the transcription start site (19). Gel shift, methylation interference, and DNase footprinting assays show that at least one extra protein binds the 5G allele, and reporter assays show that the 4G promoter has enhanced basal activity and is more responsive to transforming growth factor-β (TGFβ) and interleukin-1 (IL-1) induction than the 5G variant (19-21). In addition, induction of PAI1 by tumor necrosis factor-α is mediated by interaction of the transcription factor nuclear factor-κB with a promoter element that includes the 4G/5G site, although it is not known whether the 4G/5G SNP alters its responsiveness to nuclear factor-κB (22). Moreover, at least 37 separate studies have detected significant allelic dose-dependent correlations between carriage of the 4G allele and PAI1 protein levels in vivo, such that 4G4G homozygotes have the highest levels of circulating PAI1, 5G5G homozygotes have the lowest levels, and heterozygous individuals have intermediate levels of circulating PAI1 (Supplementary Table S1). Indeed, multiple studies and meta-analyses have found significant associations between 4G/5G genotypes and various vascular and thrombotic diseases (23-27); yet, its role in cancer remains unclear.
Clearly, many factors influence PAI1 promoter activity. These include TGFβ, tumor necrosis factor-α, platelet-derived growth factor (PDGF), IL-1, IL-6, insulin, insulin-like growth factor-1, and glucose (21, 22, 28). TGFβ, a known regulator of cancer initiation and progression, is a particularly potent inducer of PAI1 (21), whereas PAI1 suppresses the plasmin-mediated activation of TGFβ by inhibiting plasmin formation (29). Conversely, TGFβ elicits the expression of connective tissue growth factor (CTGF), which enhances the TGFβ-mediated induction of various TGFβ-responsive genes, including PAI1 and CTGF itself (30, 31). Nevertheless, it is unclear which factors affect tumoral PAI1 expression in vivo. Thus, we evaluated the role of germ line genetic variability and local microenvironmental and cellular factors in determining the level of PAI1 expression in breast cancer.
Materials and Methods
Study Populations
Global mRNA expression levels were analyzed for three breast cancer data sets: Miller et al. (32), van de Vijver et al. (33), and a set7 from the University of California San Francisco (UCSF; Supplementary Table S2). Data sets that used a Research Genetics cDNA platform were excluded based on evidence that the PAI1 clone in this set does not match the PAI1 sequence.8 The UCSF data set contained expression profiles from Affymetrix HG-U133A arrays run on 118 breast cancers as well as genomic copy number information obtained by array-based comparative genomic hybridization (CGH). The Miller data set was obtained through the Gene Expression Omnibus (accession no. GSE3494). It contains comprehensive expression profiles for 251 Swedish breast cancers that were analyzed using Affymetrix U133A and B arrays covering >30,000 genes. Data from the B array were excluded because PAI1 was absent from this array and because some genes represented on both arrays were not correlated across arrays. The van de Vijver data set was obtained from Rosetta Inpharmatics. Expression profiles in this set of 295 Dutch breast cancers were obtained by mixing Cy-labeled cRNA from individual tumors with an equal amount of a pool of reverse color–labeled cRNA representing all 295 tumors equally. These were hybridized to Agilent Technologies microarrays with 24,479 biological features.
Invasive breast cancer cases for SNP analysis were drawn from SEARCH (breast), an on-going population-based study of breast cancer cases ascertained through the East Anglian Cancer Registry (34). Incident cases were diagnosed at ≤65 years of age after the study began on July 1, 1996, and prevalent cases were diagnosed at ≤55 years of age between January 1, 1991 and initiation of the study. Participants who died before initiation of the study were not included. Controls were randomly selected from the Norfolk component of the European Prospective Investigation of Cancer (EPIC) from the same geographic area as the SEARCH study (34). Vital status was available for 2,524 genotyped SEARCH participants. Controls were not matched to cases but were broadly similar in age (median = 63 years; range = 42-81) and ethnicity (>98% White). Tumor and matched normal tissue DNAs were also isolated from 129 breast cancer cases provided by the UCSF Cancer Center Breast Oncology Program Tissue Core. Human research approval was obtained from the UCSF Committee on Human Research and the Eastern Region Multicentre Research Ethics Committee. Informed, written consent was obtained from each participant.
Genotyping
The PAI1 −675insG SNP and SNPs in five other genes were genotyped by multiplex capillary electrophoresis-based minisequencing. Six regions of interest, including the PAI1 region from −745 to −646, were amplified by multiplex PCR in a 25-μL volume containing 1.4× PCR Buffer-II (Applied Biosystems, Foster City, CA), 4.5 mmol/L MgCl2, 0.4 mmol/L of each deoxynucleotide, 0.3 μmol/L of each primer, 1.25 units of AmpliTaq Gold DNA Polymerase (Applied Biosystems), and 20 ng of DNA from whole blood. To facilitate even primer annealing, each forward primer had the nonspecific sequence 5′-GCGGTCCCAAAAGGGTCAGT-3′ from bacteriophage M13mp18 added to its 5′ end, and each reverse primer contained the T7 RNA promoter sequence 5′-TTCTAATACGACTCACTATAG-3′ at its 5′ end. The −675insG region was amplified using a −745/−726 forward primer (5′-TCCAACCTCAGCCAGACAAG-3′) and a −646/−663 reverse primer (5′-CCGCCTCCGATGATACAC-3′). Thermal cycling conditions were 95°C for 10 minutes, 42 cycles of 95°C for 30 seconds, 66°C for 45 seconds, and 72°C for 45 seconds and a final 7 minutes elongation step at 72°C. Residual primers and deoxynucleotide triphosphates were then removed by treating 5 μL of PCR product with 2 units of shrimp alkaline phosphatase (U.S. Biochemical, Cleveland, OH) and 10 units of exonuclease I (U.S. Biochemical) at 37°C for 1 hour followed by a 15-minute enzyme inactivation step at 80°C. Single-base extension reactions were then done in a 10-μL volume containing ∼0.15 pmol of amplified target DNA (purified multiplex PCR product), 2.5 μl of SNaPshot Ready Reaction Premix (Applied Biosystems) with fluorescent dideoxynucleotides (R6G-ddATP, ROX-ddTTP, TAMRA-ddCTP, and R110-ddGTP), and Taq DNA polymerase and a cocktail of sense- and antisense-oriented SNP-specific primers (0.15 pmol each). The six sense-oriented and six antisense-oriented primers had free 3′ ends that annealed immediately 5′ to the six polymorphic sites of interest and 5′ poly-T tails of various lengths. For the PAI1 −675insG site, a 46-nucleotide sense-oriented −693/ −672 oligonucleotide (5′-T24-GAGAGAGTCTGGACACGTGGGG-3′) and a 30-nucleotide antisense −655/−675 oligonucleotide (5′-T9-ATGATACACGGCTGACTCCCC-3′) were used. Single-base extension thermal cycling conditions were 25 cycles of 96°C for 10 seconds, 50°C for 5 seconds, and 60°C for 30 seconds. Unincorporated dideoxynucleotide triphosphates were removed by incubating single-base extension products with 0.5 unit of shrimp alkaline phosphatase at 37°C for 1 hour followed by a 15-minute 72°C inactivation step. One microliter each of shrimp alkaline phosphatase-treated single-base extension product and LIZ-120 size standard (Applied Biosystems) were added to 25 μL of deionized water, and the 12 single-base extension products were resolved on an ABI-PRISM 3700 DNA Analyzer.
Because the 12 sense and antisense primers had different lengths, all six SNPs were simultaneously sequenced from both directions for internal validation. We also validated the accuracy of our method using at least one alternative method for each site, including direct sequencing and a restriction fragment-based method designed for the 4G/5G SNP (35). Our minisequencing results and the results of these other approaches were 100% concordant for the PAI1 site in 21 comparisons and >99% concordant for all six loci in >3,600 comparisons. The sense and antisense primers also yielded identical results for >99.8% of >27,000 separate genotypes (5,065 PAI1 genotypes), and replicate samples and matched normal and tumor tissue pairs yielded identical results for 100% of >2,400 genotypes (447 PAI1 genotypes). Only one SEARCH case and one EPIC control had discordant sense and antisense PAI1 results and were thus excluded from our analyses. Thus, our method was >99% accurate, reproducible, and internally consistent.
Statistical Analysis
Expression-based univariate and multivariate survival analyses were done by Cox proportional hazards regression analysis using Stata-9.1. To adjust for platform differences and provide comparable meaning to the hazard ratios across all data sets, PAI1 expression values were normalized by subtracting the mean PAI1 expression for all cases, dividing the difference by the SD, and adding the constant five to avoid negative values. Survival curves were constructed for the highest versus lowest 50% of PAI1-expressing tumors and were compared using the log-rank test. Pearson correlations between PAI1 expression and the expression of all other genes were calculated, and their corresponding two-tailed parametric Ps were adjusted for multiple comparisons using the false discovery rate procedure of Benjamini and Hochberg (36). Because each platform contained duplicate PAI1 probes that yielded correlated expression values (r = 0.83-0.97), all analyses were done using the average value from both probes. Average values were also used for all other genes with replicate probes. Genes were considered significantly correlated with PAI1 if the false discovery rate-adjusted P < 0.0001.
For SNP analyses, deviations of the observed genotypic frequencies from those expected under Hardy-Weinberg equilibrium were assessed by χ2 tests. Allelic and genotypic frequencies in cases and controls were compared by one degree and two degrees of freedom χ2 tests, respectively. Genotype-specific risks were estimated as odds ratios by unconditional logistic regression, and 95% confidence intervals were obtained using the floating absolute risk method (34). Attributable risks were estimated as g(r − 1) / [1 + g(r − 1)], where g is the genotypic frequency in the control population and r is the associated relative risk estimate. For survival analysis, time at risk was defined from the date of blood sample receipt until death due to any cause or until March 21, 2003 for surviving participants. Genotype-specific hazard ratios and 95% confidence intervals were estimated by Cox regression analysis. The proportional hazards assumption was examined and tested by adding a time × genotype term to the model. The level of significance was determined using an overall test for heterogeneity among all three genotypes.
Results
Prognostic Value of PAI1 mRNA Expression in Breast Cancer
As predicted from protein-based studies, high PAI1 mRNA expression was associated with shorter overall survival in two breast cancer data sets (Table 1; Fig. 1). In the UCSF data set, PAI1 expression was prognostic by univariate analysis but failed to provide significant additional prognostic information when tumor size and LN status (which were the only independent prognostic variables in this cohort) were accounted for in the Cox model. In the van de Vijver data set, LN status was not prognostic, whereas estrogen receptor (ER) status and a 70-gene expression signature derived from this set were (33). PAI1 expression was also prognostic in this set when considered alone. In addition, it was an independent indicator of poor prognosis in the 151 LN− cases after adjusting for ER status (P = 0.014) and/or the 70-gene signature (P < 0.05) but was not prognostic in the remaining LN+ cases. PAI1 was also prognostic in the subset of 180 high-risk cases with a poor 70-gene expression signature.
Table 1. PAI1 expression and overall survival in three breast cancer cohorts.
| Cohort | n | Hazard ratio (95% confidence interval) |
P |
|---|---|---|---|
| UCSF | |||
| All cases | 117 | 1.46 (1.07-1.98) | 0.016 |
| Subgroup | |||
| LN− cases | 50 | 1.14 (0.52-2.47) | 0.746 |
| LN+ cases | 67 | 1.53 (1.06-2.20) | 0.023 |
| ER− cases | 43 | 1.65 (1.06-2.58) | 0.026 |
| ER+ cases | 74 | 1.33 (0.86-2.07) | 0.202 |
| Multivariate* | |||
| All cases | 114 | 1.37 (0.98-1.93) | 0.068 |
| van de Vijver | |||
| All cases | 295 | 1.31 (1.09-1.58) | 0.003 |
| Subgroup | |||
| LN− cases | 151 | 1.55 (1.21-1.99) | 0.001 |
| LN+ cases | 144 | 1.13 (0.84-1.52) | 0.427 |
| ER− cases | 69 | 1.08 (0.85-1.38) | 0.508 |
| ER+ cases | 226 | 1.34 (0.99-1.83) | 0.062 |
| High risk † | 180 | 1.21 (1.01-1.45) | 0.042 |
| Multivariate ‡ | |||
| All cases | 295 | 1.18 (0.98-1.41) | 0.083 |
| Miller | |||
| All cases | 234 | 0.95 (0.72-1.24) | 0.699 |
| Subgroup | |||
| LN− cases | 149 | 0.83 (0.55-1.26) | 0.381 |
| LN+ cases | 77 | 0.99 (0.70-1.39) | 0.957 |
| ER− cases | 30 | 1.93 (0.63-5.88) | 0.250 |
| ER+ cases | 200 | 0.90 (0.68-1.19) | 0.454 |
| Multivariate§ | |||
| All cases | 223 | 0.95 (0.72-1.24) | 0.708 |
NOTE: Cox proportional hazards analysis was performed for overall survival using PAI1 expression as a normalized continuous variable. Hazard ratios and 95% confidence intervals are for 1-unit increase in the standard deviation of the log 2 expression of PAI1 (range = 1.11-8.33).
Multivariate analyses were adjusted for LN status, tumor size, age at diagnosis, and ER status.
Cases with a poor 70-gene prognostic signature (33).
Multivariate analyses were adjusted for LN status and ER status.
Multivariate analyses were adjusted for LN status, age at diagnosis, tumor size, and ER status.
Figure 1.
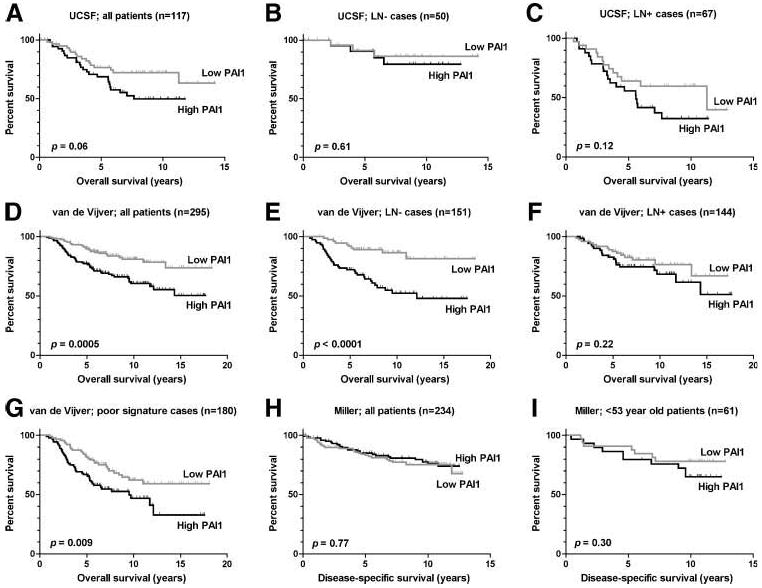
Kaplan-Meier plots for overall or disease-specific survival for the highest (black) versus lowest (gray) 50% of tumors in terms of their PAI1 expression. Survival curves for all outcome-informative patients (A, D, and H), LN− cases (B and E) and LN+ cases (C and F) for the indicated cohorts, as well as for the subset of cases from the van de Vijver study with a poor 70-gene prognostic signature (G) and the subset of Miller cases younger than 53 years of age (I). Log-rank Ps are provided.
Conversely, PAI1 had no prognostic power in the Miller data set, suggesting that key differences between this cohort and the others may modify the biological effects and prognostic use of PAI1. Indeed, substantial differences were seen between the patient sets (Supplementary Table S2). The proportion of ER− cases in the Miller data set was significantly lower than in the van de Vijver (P = 0.004) and UCSF (P < 0.0001) data sets, and the proportion of LN− cases in the Miller data set was significantly greater than in the van de Vijver (P = 0.0009) and UCSF (P < 0.0001) data sets. In addition, none of the van de Vijver cases were older than 52 years of age at diagnosis (i.e., most were premenopausal), whereas 48% of UCSF cases and 73% of Miller cases were over 52 years old. Thus, the UCSF cohort had an older age distribution than the van de Vijver cohort (P < 0.0001), and the Miller cohort had a far older distribution than either of the other cohorts (P < 0.0001). Nevertheless, PAI1 expression continued to lack prognostic significance in the 61 Miller cases diagnosed before age 53 or in cases stratified by LN or ER status. Thus, although these differences may contribute to the presence or absence of an association between PAI1 and survival, they are not entirely responsible and are probably indicative of other important differences that we were unable to ascertain from the data. Nevertheless, mRNA associations in the van de Vijver and UCSF cohorts are consistent with the proven prognostic value of PAI1 and thus indicate that transcriptional controls are important in regulating tumoral PAI1 expression.
PAI1 4G/5G SNP Is Not Associated with Breast Cancer Incidence, Outcome, or PAI1 Expression
PAI1 4G/5G genotypes were obtained for 2,539 SEARCH cases and 1,832 EPIC controls. The cases and controls had virtually identical allelic (P = 0.44) and genotypic (P = 0.72) distributions (Table 2). Similar 4G4G, 4G5G, and 5G5G genotypic frequencies (0.32, 0.43, and 0.25, respectively) and 4G/5G allelic frequencies (0.53/0.47) were also seen in the Caucasian subset of UCSF breast cancer cases, and no British or U.S. cohort deviated significantly from Hardy-Weinberg equilibrium (P ≥ 0.29). Thus, the 4G/5G SNP was not associated with the incidence of invasive breast cancer.
Table 2. Genotypic distributions and allelic frequencies in East Anglian women with and without invasive breast cancer.
| Genotype | Cases (n = 2,539) |
Controls (n = 1,832) |
Odds ratio (95% confidence interval) |
|---|---|---|---|
| 5G5G | 520 (0.205) | 386 (0.211) | 1.00 (reference) |
| 4G5G | 1,229 (0.484) | 896 (0.489) | 1.02 (0.87-1.19) |
| 4G4G | 790 (0.311) | 550 (0.300) | 1.07 (0.90-1.26) |
| 4G allele | 0.553 | 0.545 | |
| 5G allele | 0.447 | 0.455 |
Notably, the 4G/5G SNP sits within one of six TGFβ response elements in the PAI1 promoter and affects PAI1 induction by TGFβ (21), whereas PAI1 can indirectly suppress TGFβ activation and the release of matrix-sequestered TGFβ (29). In addition, the P (proline) allele of the nonsynonymous TGFB1 L10P (T+29C) SNP has been associated with elevated TGFβ1 secretion in culture and an increased incidence of invasive breast cancer in three case-control sets, including the SEARCH-EPIC set (34). Therefore, we explored the possibility that the PAI1 and TGFB1 SNPs might interact to influence cancer risk. L10P and 4G/5G genotypes were available for 2,074 SEARCH cases and 1,766 EPIC controls. However, neither SNP was associated with breast cancer incidence when analyzed separately (odds ratio, 1.16; 95% confidence interval, 0.95-1.43 for PP versus LL homozygotes; odds ratio, 1.06; 95% confidence interval, 0.88-1.28 for 4G4G versus 5G5G homozygotes). When they were analyzed together, the distribution of the nine possible genotypic combinations in the controls was virtually identical to the expected pattern based on the observed allele frequencies and an assumption of independent inheritance (P = 0.98). However, a marginally significant difference between observed and expected distributions was detected among the cases (P = 0.055).
The relative risks for the nine possible PAI1-TGFB1 genotypes showed an inconsistent trend in which the high-expressing PAI1 4G and TGFB1 P alleles each seemed to confer an increased risk of breast cancer in an additive, allelic dose-dependent manner (Table 3). According to this semidominant model, double-homozygous 4G4G/PP individuals with four putative high-risk alleles should show a greater risk of developing cancer than individuals with three high-risk alleles, and so on, with double-homozygous 5G5G/LL individuals having the lowest relative risk. Approximately 4% of British Caucasians are 4G4G/PP double homozygotes. Thus, if the relative risk of 1.24 is correct, then ∼1% of all breast cancers would be attributable to this genotype. If we instead assume that the L10P SNP is recessive for increased risk, as prior data suggest (34), and that 4G/5G is dominant, then the odds ratio for the high-risk 4G-carrier/PP group (∼ 11% of the population) remains at 1.24 versus the 5G5G/L-carrier group; the 95% confidence interval improves to 0.97 to 1.59; and the attributable fraction increases to ∼ 2.6%. Either way, the risks associated with a 4G/5G-L10P interaction are weak, and the population attributable risk decreases below the 3% level estimated for the TGFB1 PP genotype alone (34), further suggesting that the 4G/5G SNP does not contribute to the risk of developing breast cancer.
Table 3. PAI1 4G/5G and TGFB1 L10P genotypic distributions in East Anglian women with and without invasive breast cancer.
| D | PAI1genotype | TGFB1Genotype | Cases | Controls | Genotypic odds ratio (95% confidence interval) |
Additive odds ratio (95% confidence interval) |
|
|---|---|---|---|---|---|---|---|
| 0 | 5G5G | LL | 161 | 127 | 1.00 (reference) |  |
1.00 (reference) |
| 1 | 5G5G | LP | 204 | 153 | 1.05 (0.77-1.44) | 1.02 (0.78-1.33) | |
| 1 | 4G5G | LL | 356 | 281 | 1.00 (0.76-1.32) | ||
| 2 | 5G5G | PP | 58 | 42 | 1.09 (0.69-1.73) | 1.11 (0.86-1.43) | |
| 2 | 4G5G | LP | 487 | 362 | 1.06 (0.81-1.39) | ||
| 2 | 4G4G | LL | 266 | 174 | 1.21 (0.89-1.63) | ||
| 3 | 4G5G | PP | 170 | 104 | 1.29 (0.92-1.81) | 1.07 (0.81-1.41) | |
| 3 | 4G4G | LP | 271 | 221 | 0.97 (0.72-1.30) | ||
| 4 | 4G4G | PP | 101 | 64 | 1.24 (0.84-1.84) | 1.24 (0.84-1.84) | |
NOTE: D = dose of putative high-risk alleles. Additive odds ratios were determined for each combined allelic dosage group relative to the reference 5G5G/LL participants who had no putative high-risk alleles.
Among the 2,524 genotyped SEARCH cases for which survival data were available, 291 deaths occurred over 10,517 person-years, for an overall mortality rate of 2.8% per year. Significant heterogeneity was not observed among the three genotypes; yet, annual mortality tended to decrease in an allelic dose-dependent manner from 3.1% for 5G5G cases and 2.8% for 4G5G cases to 2.6% for 4G4G cases. This was reflected in a nonsignificant increase in the unadjusted hazard ratio when using homozygous 4G4G cases as the reference group (Table 4; Fig. 2A). Thus, if anything, the high-expressing 4G allele tended to provide a slightly better, rather than worse, prognosis (proportional hazards P = 0.10). Likewise, there was no association with overall survival in 121 informative UCSF cases (P = 0.74; Fig. 2B) or in the smaller subset of 79 Caucasian patients (P = 0.34). Nor was the 4G/5G SNP associated with tumor stage, LN status, ER status, pathologic grade, or ductal versus lobular histology in this subset (P ≥ 0.30).
Table 4. Genotypic mortality rates and relative hazards.
| Genotype | n | Time at risk (y) |
Annual %mortality | Hazard ratio (95% confidence interval) |
|---|---|---|---|---|
| 4G4G | 784 | 3,269.6 | 2.57 | 1.00 (reference) |
| 4G5G | 1,222 | 5,106.8 | 2.76 | 1.07 (0.82-1.40) |
| 5G5G | 518 | 2,141.1 | 3.08 | 1.20 (0.87-1.66) |
Figure 2.
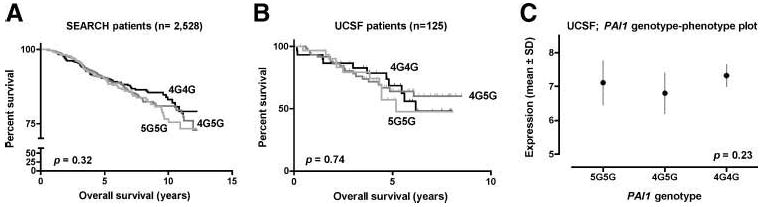
The PAI1 4G/5G SNP is not associated with overall survival or tumoral PAI1 mRNA expression in breast cancer. Kaplan-Meier survival curves for the putative high expression 4G4G (black), intermediate expression 4G5G (dark gray), and low expression 5G5G (light gray) genotypes for all outcome-informative SEARCH (A) and UCSF (B) patients. Mean tumoral PAI1 mRNA expression levels obtained by microarray expression profiling in a subset of UCSF cases for the corresponding PAI1 genotypes (C). Two-tailed log-rank test for trend (A and B) and ANOVA (C) Ps are provided.
Array-based PAI1 mRNA expression data were available for 27 UCSF cases for which 4G/5G genotypic data were also available. Among these tumors, no genotype exhibited a significantly altered level of PAI1 expression (ANOVA, P = 0.23; Fig. 2C), nor was there an allelic dose-dependent trend in terms of expression (P = 0.85), suggesting that the 4G/5G SNP does not have a substantial effect on PAI1 expression in breast cancer.
Correlations between PAI1 mRNA Expression and Other Factors
Three global gene expression data sets were used to identify genes that might regulate PAI1 expression, or that might be influenced by PAI1 or regulated in common with it. Of ∼ 24,000 arrayed genes, 532 were significantly correlated with PAI1 at a false discovery rate–adjusted P < 0.0001 (Supplementary Table S3). Of these, 68 were correlated with PAI1 in two data sets, and 32 were correlated in all three data sets. Thus, not only were these 32 genes significantly correlated within each data set, but the probability that any one would be significantly correlated in all three data sets by chance was ∼ 1 × 10−6. Furthermore, only five genes were among the 40 most highly correlated genes in all three data sets: CTGF, the facilitated glucose transporter SLC2A3, the cysteine-rich angiogenic inducer CYR61, the bone morphogenetic protein antagonist gremlin (GREM1), and the extracellular matrix protein fibronectin (FN1). CTGF was by far the most highly correlated gene in the UCSF (r = 0.68) and Miller (r = 0.66) data sets and ranked ninth in the van de Vijver data set (r = 0.45). Likewise, SLC2A3 ranked first, second, and ninth among PAI1-correlated genes in the van de Vijver (r = 0.57), Miller (r = 0.59), and UCSF (r = 0.54) data sets, respectively. The probability of CTGF or SLC2A3 being so highly ranked in all three sets by chance was <4 × 10−12. Thus, PAI1 was more highly and consistently correlated with CTGF and SLC2A3 than with any other gene.
Other consistently correlated genes included angiopoietin-like 2 (ANGPTL2), mitogen-inducible 2 (MIG2), growth-arrest and DNA-damage-inducible β (GADD45B), tristetraprolin (ZFP36), urokinase-type plasminogen activator and its receptor (PLAU and PLAUR), inhibin-βA (INHBA), IL6, laminin-β1 (LAMB1), nidogen (NID), versican (CSPG2), PDGF receptor-β (PDGFRB), and TGFβ-inducible early growth response (TIEG/KLF10). Notably, PAI1 and TGFβ were not correlated with one another in any data set (r = 0.10-0.29, P = 0.02-0.38). However, the mRNA expression of TGFβ does not necessarily reflect its activity, as TGFβ is extensively regulated post-transcriptionally (37), whereas PAI1 is a robust indicator of TGFβ activity (38). Moreover, at least 15 (47%) of the 32 genes that were correlated with PAI1 in all data sets respond to TGFβ, as do many of the less consistently correlated genes, such as TGFBI (TGFβ-induced, 68 kDa) and TGFB1I1 (TGFβ1-induced transcript 1). Thus, although TGFβ and PAI1 were not correlated at the mRNA level, local TGFβ activity probably affects tumoral PAI1 expression.
Genomic and epigenetic changes in PAI1 may also affect its overall expression in tumors. Notably, a P1-derived artificial chromosome (RP4-747G18) that contains the entire PAI1 gene plus 24.4 kb of upstream DNA was among the 4,325 array comparative genomic hybridization probes in the UCSF data set. Comparison of DNA copy number results for this probe and expression estimates for PAI1 revealed that this region was amplified in 21 (30%) of the 70 cases for which genomic and expression data were available, and that the copy number ratios for all 70 cases were positively correlated with average PAI1 expression (r = 0.40, P = 0.0005). Thus, PAI1 copy number changes also seem to influence overall PAI1 mRNA levels in breast cancer. Moreover, gene copy number differences (which act in cis) and CTGF (which acts in trans) should affect PAI1 expression independently. That is, tumors with high CTGF expression and PAI1 amplification should have the highest levels of PAI1 expression. Indeed, PAI1 expression was more highly correlated with the product of CTGF expression × the PAI1 copy number ratio (r = 0.73, P < 0.0001) than with either variable alone. Furthermore, partial correlations for PAI1 expression versus CTGF (r = 0.74, P < 0.001) and copy number (r = 0.33, P = 0.005) indicate that the interaction between CTGF and PAI1 is even stronger when gene amplification is taken into account.
Discussion
Gene ablation and reconstitution data show that PAI1 can promote cancer progression (11-14), and correlative data consistently indicate that its elevated expression in breast tumors is an independent indicator of poor prognosis (4-8). Here, we found that high PAI1 mRNA expression is also associated with shorter overall survival in breast cancer. Nevertheless, the transcription-altering PAI1 4G/5G SNP was not associated with breast cancer incidence, outcome, or tumoral PAI1 mRNA expression. Thus, local factors seem to be more important than germ line genetic variability in determining the level of PAI1 expression in breast cancer. Furthermore, our data show that PAI1 mRNA levels correlate with CTGF and the level of PAI1 gene amplification, suggesting that they play an important role in regulating the expression and adverse effects of PAI1 in breast cancer.
Because PAI1 mRNA expression correlates with its protein expression in breast cancer (39), we reasoned that its mRNA expression should also be prognostic. Indeed, elevated PAI1 mRNA expression has been associated with shorter disease-free survival in 130 breast cancers (39). Here, we show that tumoral PAI1 mRNA expression is also an independent or borderline independent prognostic indicator for overall survival in two breast cancer cohorts. Furthermore, the absence of an association in a third cohort suggests that there are demographic and molecular factors that modify the effects of PAI1 and might aid in identifying cancers for which PAI1 has the greatest prognostic value. Indeed, the cohorts had significantly different age, ER status, and LN status distributions; however, none of these seemed to explain the between-cohort differences in the prognostic power of PAI1. In one cohort, PAI1 was prognostic in LN− cases, whereas in another, it was prognostic in the LN+ subset. Thus, it seems critical to determine what dictates the differential activity and prognostic use of PAI1. Nonetheless, our mRNA data were consistent with protein-based observations and support the hypothesis that transcriptional regulation is an important factor in controlling the tumoral levels of PAI1.
One factor that can affect PAI1 transcription is the 4G/5G SNP; yet, our data indicate that it is not associated with breast cancer incidence or survival. Given the common nature of this SNP and the size of our cohort, we had >95% power to detect an odds ratio of 1.5 at P < 0.001. By comparison, the five other studies that have examined this SNP in cancer had an average of only 92 cases and 122 controls per study and were thus far less apt to detect a true association (40-44). Despite or due to their limited size, three of these studies detected borderline associations between the high-expressing 4G allele and breast (40, 41) or general cancer incidence (42), whereas no associations were seen in ovarian (43) or colorectal cancer (44). In both breast cancer studies, the 4G allele was more common among cases than controls, and 4G carriers were more apt to have cancer than 5G homozygotes, particularly when the studies were combined. However, these associations disappear in a meta-analysis that includes SEARCH-EPIC data (Supplementary Table S4). To our knowledge, no functional PAI1 polymorphism other than the 4G/5G SNP has been described, although a nonsynonymous PAI1 signal sequence SNP of unknown functional significance (rs6092, Ala15Thr) has been associated with colorectal cancer risk (45). However, the genotypic distribution of the cases in this study was disconcertingly far from Hardy-Weinberg equilibrium, and this same SNP was not associated with risk in a small ovarian cancer study (43).
The high-expressing 4G4G genotype has also been associated with more advanced colorectal cancers (44) as well as larger diameter and higher-grade breast cancers (40), whereas no such associations were seen in another breast cancer cohort (41). Indeed, these few significant associations are outnumbered by multiple null comparisons and were seen in subsets with an average of only 38 cases (range = 17-55), thus increasing the likelihood that they represent false-positive associations. By comparison, our far larger study revealed no association with outcome and, if anything, showed an unexpected tendency for patients with higher expressing genotypes to have prolonged rather than diminished survival. Thus, our data support the conclusion that cancer-associated signals are more important than germ line genetic variability in determining the expression and ill effects of PAI1 in breast cancer.
PAI1 protein levels tend to be 7- to 10-fold higher in tumors than normal tissues and can vary >1,000-fold between tumors (2-4). Notably, this is far wider than the 2- to 6-fold differences in reporter gene expression seen for 4G/5G alleles in culture (19, 20). Furthermore, although the 4G/5G SNP contributes to the regulation of humoral PAI1 in vivo, it only accounts for 2% to 3% of the variability in circulating levels (46-49), and mean plasma levels are only 1.2- to 2.5-fold higher in 4G4G homozygotes than in 5G5G homozygotes (Supplementary Table S1). Likewise, it is unclear whether the 4G/5G SNP has any appreciable affect on tumoral PAI1 levels or whether it is overpowered by other factors. One study found no association between the 4G/5G SNP and PAI protein levels in 40 colorectal cancers (50), whereas another found that the high-expressing 4G allele was associated with increased tumoral PAI1 expression in 104 breast cancers (40). Our own analyses revealed no such association in a subset of UCSF breast cancers, which is consistent with the lack of association between the 4G/5G SNP and outcome in the SEARCH and UCSF cohorts. Thus, although subtle genotype-driven differences in PAI1 expression could exert profound effects over an entire lifetime or during the prolonged evolution of a cancer, most data suggest that the 4G/5G SNP has little effect on tumoral PAI1 expression, which raises the question of what factors are important.
To identify potential upstream regulators of PAI1 in breast cancer, we examined three comprehensive mRNA expression data sets for genes that were consistently associated with PAI1. Notably, CTGF was far more strongly correlated with PAI1 than any of several thousand other genes in two data sets and was the ninth most highly correlated gene in another. Thus, not only were these associations highly significant within each set, but the likelihood that CTGF would be so highly ranked in all three sets by chance was minuscule. Moreover, CTGF has been shown to enhance TGFβ-mediated induction of PAI1 by stimulating expression of the transcription factor TIEG, which in turn suppresses transcription of the TGFβ antagonist SMAD7 (Fig. 3; refs. 30, 31). However, SMAD7 was positively rather than inversely correlated with PAI1 in two data sets, suggesting that an alternative mechanism may be involved. By contrast, TIEG, which has been shown to enhance PAI1 induction (38), was directly correlated with PAI1 in all data sets, as was FN1, which is also induced by TGFβ and CTGF (31). Thus, TIEG probably coregulates PAI1 and FN1 in response to local TGFβ/CTGF activity. Moreover, not only does CTGF enhance the effects of TGFβ, but its own expression is induced by TGFβ (30, 31). Thus, the involvement of CTGF suggests that TGFβ is also involved. Indeed, 47% of the genes associated with PAI1 in all three data sets are induced by TGFβ. Furthermore, although TGFβ and PAI1 were not correlated in any data set, the expression of TGFβ does not necessarily reflect its actual activity (37), whereas PAI1 is an established indicator of TGFβ activity (38). Alternatively, other TGFβ super-family members may be involved. Indeed, activins have been shown to induce PAI1 expression through the same TGFβ response elements (51). Moreover, the inhibin-βA subunit that homodimerizes to form activin-A was significantly correlated with PAI1 in all three data sets, and the type I activin receptor (ACVR1) and type II TGFβ receptor (TGFBR2) were significantly associated with PAI1 in one data set. Thus, our data support the notion that PAI1 levels in breast cancer are strongly influenced by TGFβ and/or activin-A acting in concert with CTGF and TIEG.
Figure 3.
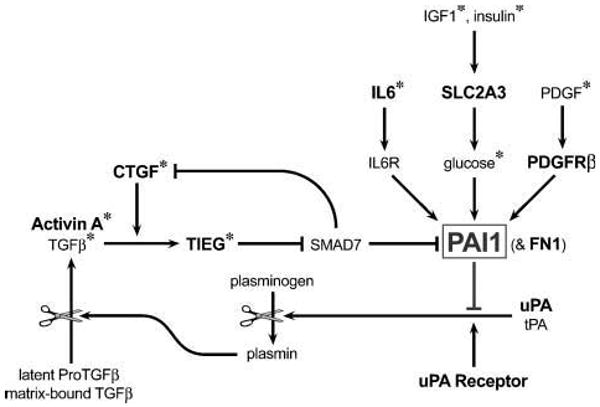
Model depicting the signaling pathways and feedback mechanisms that regulate PAI1 expression. Factors that were correlated with PAI1 expression in all three breast cancer data sets are highlighted in bold. FN1 is included because it is coregulated in parallel with PAI1 by TGFβ and CTGF (31). *, factors that have been shown to promote PAI1 expression in culture (21, 22, 28, 30).
Another highly correlated gene was the glucose transporter SLC2A3. Notably, SLC2A3 was one of three PAI1-correlated genes (including COL4A2 and AKAP2) in the prognostic 70-gene set identified by van de Vijver et al. (33). Thus, its presence in this set may reflect the functional and prognostic importance of PAI1, and PAI1 may add value to the 70-gene signature, as its prognostic power was independent of the 70-gene profile. Moreover, as glucose has been shown to regulate PAI1 expression (22), SLC2A3 may be intimately involved in the glucose-mediated regulation of PAI1 in breast tumors. Likewise, IL-6 was consistently correlated with PAI1 and can enhance PAI1 expression in culture (28), suggesting that it too may influence PAI1 expression in breast cancer. In addition, PDGF may contribute, as its receptor was associated with PAI1 in all data sets and because it can also influence PAI1 expression (22). Thus, although PAI1 expression will depend on the combined effects of many inputs, CTGF and SLC2A3 may be particularly important in breast cancer, given their consistency and level of correlation.
A better understanding of the regulation and role of PAI1 in breast cancer will undoubtedly require integration of many complementary approaches and will almost certainly enhance its use as a prognostic and predictive tool or therapeutic target. Indeed, some breast cancers may be more sensitive to the effects of PAI1 than others and thus more informative when PAI1 is used to predict their prognosis or therapeutic responsiveness. Thus, determining the basis for these differences could prove particularly beneficial. In addition, our data suggest that transcriptional controls play an important role in the overall regulation of PAI1; yet, the transcription-altering 4G/5G SNP is apparently unimportant. Thus, inter-individual genetic differences in PAI1 expression are probably overcome by more profound cancer-associated signals that are responsible for the up-regulation and ill effects of PAI1 in cancer. Indeed, our data suggest that amplification of the PAI1 gene may contribute to its elevated expression in some cancers, and that microenvironmental and cellular factors, such as CTGF and SLC2A3, are key regulators of PAI1 expression in breast cancer and thus ripe for further study.
Supplementary Material
Acknowledgments
We thank K. Redman, J. Gregory, and J. Lipscombe for case sample management; N.E. Day, K-T. Khaw, S. Oakes, S. Bingham, R. Luben, and J. Russell for access to control DNAs; C. Luccarini, J. Weng, A. Sayegh, S.S.F. Leung, S. Dhamija, P. Smith, and J. Long for technical assistance; and J. Fridlyand for technical advice.
Grant support: National Cancer Institute grants CA58207 and CA72006, University of California San Francisco Research Evaluation and Allocation Committee/Simon E. Memorial Fund, and Cancer Research UK.
Footnotes
K. Chin, Devries S, Fridlynd J, et al. Associations of genomic and transcriptional aberrations with human breast cancer pathophysiologies. Cancer Cell, in press.
J.E. Korkola, personal communication.
Note: Supplementary data for this article are available at Cancer Epidemiology Biomakers and Prevention Online (http://cebp.aacrjournals.org/).
P.D.P. Pharoah is a Senior Clinical Research Fellow.
B.A.J. Ponder is a Gibb Fellow of Cancer Research UK.
References
- 1.Nielsen HJ, Pappot H, Christensen IJ, et al. Association between plasma concentrations of plasminogen activator inhibitor-1 and survival in patients with colorectal cancer. BMJ. 1998;316:829–30. doi: 10.1136/bmj.316.7134.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fredstorp-Lidebring M, Bendahl PO, Brunner N, et al. Urokinase plasminogen activator and its inhibitor, PAI-1, in association with progression-free survival in early stage endometrial cancer. Eur J Cancer. 2001;37:2339–48. doi: 10.1016/s0959-8049(01)00306-9. [DOI] [PubMed] [Google Scholar]
- 3.Nekarda H, Schmitt M, Ulm K, et al. Prognostic impact of urokinase-type plasminogen activator and its inhibitor PAI-1 in completely resected gastric cancer. Cancer Res. 1994;54:2900–7. [PubMed] [Google Scholar]
- 4.Foekens JA, Peters HA, Look MP, et al. The urokinase system of plasminogen activation and prognosis in 2780 breast cancer patients. Cancer Res. 2000;60:636–43. [PubMed] [Google Scholar]
- 5.Pedersen AN, Christensen IJ, Stephens RW, et al. The complex between urokinase and its type-1 inhibitor in primary breast cancer. Cancer Res. 2000;60:6927–34. [PubMed] [Google Scholar]
- 6.Harbeck N, Schmitt M, Kates RE, et al. Clinical utility of urokinase-type plasminogen activator and plasminogen activator inhibitor-1 determination in primary breast cancer tissue for individualized therapy concepts. Clin Breast Cancer. 2002;3:196–200. doi: 10.3816/CBC.2002.n.023. [DOI] [PubMed] [Google Scholar]
- 7.Look MP, van Putten WL, Duffy MJ, et al. Pooled analysis of prognostic impact of urokinase-type plasminogen activator and its inhibitor PAI-1 in 8377 breast cancer patients. J Natl Cancer Inst. 2002;94:116–28. doi: 10.1093/jnci/94.2.116. [DOI] [PubMed] [Google Scholar]
- 8.Zemzoum I, Kates RE, Ross JS, et al. Invasion factors uPA/PAI-1 and HER2 status provide independent and complementary information on patient outcome in node-negative breast cancer. J Clin Oncol. 2003;21:1022–8. doi: 10.1200/JCO.2003.04.170. [DOI] [PubMed] [Google Scholar]
- 9.Harbeck N, Kates RE, Look MP, et al. Enhanced benefit from adjuvant chemotherapy in breast cancer patients classified high-risk according to urokinase-type plasminogen activator (uPA) and plasminogen activator inhibitor type 1 (n = 3424) Cancer Res. 2002;62:4617–22. [PubMed] [Google Scholar]
- 10.Janicke F, Prechtl A, Thomssen C, et al. Randomized adjuvant chemotherapy trial in high-risk, lymph node-negative breast cancer patients identified by urokinase-type plasminogen activator and plasminogen activator inhibitor type 1. J Natl Cancer Inst. 2001;93:913–20. doi: 10.1093/jnci/93.12.913. [DOI] [PubMed] [Google Scholar]
- 11.Bajou K, Noel A, Gerard RD, et al. Absence of host plasminogen activator inhibitor 1 prevents cancer invasion and vascularization. Nat Med. 1998;4:923–8. doi: 10.1038/nm0898-923. [DOI] [PubMed] [Google Scholar]
- 12.Gutierrez LS, Schulman A, Brito-Robinson T, Noria F, Ploplis VA, Castellino FJ. Tumor development is retarded in mice lacking the gene for urokinase-type plasminogen activator or its inhibitor, plasminogen activator inhibitor-1. Cancer Res. 2000;60:5839–47. [PubMed] [Google Scholar]
- 13.Bajou K, Maillard C, Jost M, et al. Host-derived plasminogen activator inhibitor-1 (PAI-1) concentration is critical for in vivo tumoral angiogenesis and growth. Oncogene. 2004;23:6986–90. doi: 10.1038/sj.onc.1207859. [DOI] [PubMed] [Google Scholar]
- 14.Maillard C, Jost M, Romer MU, et al. Host plasminogen activator inhibitor-1 promotes human skin carcinoma progression in a stage-dependent manner. Neoplasia. 2005;7:57–66. doi: 10.1593/neo.04406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bajou K, Masson V, Gerard RD, et al. The plasminogen activator inhibitor PAI-1 controls in vivo tumor vascularization by interaction with proteases, not vitronectin. Implications for antiangiogenic strategies. J Cell Biol. 2001;152:777–84. doi: 10.1083/jcb.152.4.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Devy L, Blacher S, Grignet-Debrus C, et al. The pro- or antiangiogenic effect of plasminogen activator inhibitor 1 is dose dependent. Faseb J. 2002;16:147–54. doi: 10.1096/fj.01-0552com. [DOI] [PubMed] [Google Scholar]
- 17.Isogai C, Laug WE, Shimada H, et al. Plasminogen activator inhibitor-1 promotes angiogenesis by stimulating endothelial cell migration toward fibronectin. Cancer Res. 2001;61:5587–94. [PubMed] [Google Scholar]
- 18.Czekay RP, Loskutoff DJ. Unexpected role of plasminogen activator inhibitor 1 in cell adhesion and detachment. Exp Biol Med (Maywood) 2004;229:1090–6. doi: 10.1177/153537020422901102. [DOI] [PubMed] [Google Scholar]
- 19.Dawson SJ, Wiman B, Hamsten A, Green F, Humphries S, Henney AM. The two allele sequences of a common polymorphism in the promoter of the plasminogen activator inhibitor-1 (PAI-1) gene respond differently to interleukin-1 in HepG2 cells. J Biol Chem. 1993;268:10739–45. [PubMed] [Google Scholar]
- 20.Eriksson P, Kallin B, van't Hooft FM, Bavenholm P, Hamsten A. Allele-specific increase in basal transcription of the plasminogen-activator inhibitor 1 gene is associated with myocardial infarction. Proc Natl Acad Sci U S A. 1995;92:1851–5. doi: 10.1073/pnas.92.6.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen YQ, Sloan-Lancaster J, Berg DT, Richardson MA, Grinnell B, Tseng-Crank J. Differential mechanisms of plasminogen activator inhibitor-1 gene activation by transforming growth factor-β and tumor necrosis factor-α in endothelial cells. Thromb Haemost. 2001;86:1563–72. [PubMed] [Google Scholar]
- 22.Swiatkowska M, Szemraj J, Cierniewski CS. Induction of PAI-1 expression by tumor necrosis factor α in endothelial cells is mediated by its responsive element located in the 4G/5G site. Febs J. 2005;272:5821–31. doi: 10.1111/j.1742-4658.2005.04979.x. [DOI] [PubMed] [Google Scholar]
- 23.Boekholdt SM, Bijsterveld NR, Moons AH, Levi M, Buller HR, Peters RJ. Genetic variation in coagulation and fibrinolytic proteins and their relation with acute myocardial infarction: a systematic review. Circulation. 2001;104:3063–8. doi: 10.1161/hc5001.100793. [DOI] [PubMed] [Google Scholar]
- 24.Iacoviello L, Burzotta F, Di Castelnuovo A, Zito F, Marchioli R, Donati MB. The 4G/5G polymorphism of PAI-1 promoter gene and the risk of myocardial infarction: a meta-analysis. Thromb Haemost. 1998;80:1029–30. [PubMed] [Google Scholar]
- 25.Mansfield MW, Stickland MH, Grant PJ. Plasminogen activator inhibitor-1 (PAI-1) promoter polymorphism and coronary artery disease in non-insulin-dependent diabetes. Thromb Haemost. 1995;74:1032–4. [PubMed] [Google Scholar]
- 26.Iwai N, Shimoike H, Nakamura Y, Tamaki S, Kinoshita M. The 4G/5G polymorphism of the plasminogen activator inhibitor gene is associated with the time course of progression to acute coronary syndromes. Atherosclerosis. 1998;136:109–14. doi: 10.1016/s0021-9150(97)00191-3. [DOI] [PubMed] [Google Scholar]
- 27.Gardemann A, Lohre J, Katz N, Tillmanns H, Hehrlein FW, Haberbosch W. The 4G4G genotype of the plasminogen activator inhibitor 4G/5G gene polymorphism is associated with coronary atherosclerosis in patients at high risk for this disease. Thromb Haemost. 1999;82:1121–6. [PubMed] [Google Scholar]
- 28.Dong J, Fujii S, Li H, et al. Interleukin-6 and mevastatin regulate plasminogen activator inhibitor-1 through CCAAT/enhancer-binding protein-δ. Arterioscler Thromb Vasc Biol. 2005;25:1078–84. doi: 10.1161/01.ATV.0000159701.24372.49. [DOI] [PubMed] [Google Scholar]
- 29.Lyons RM, Gentry LE, Purchio AF, Moses HL. Mechanism of activation of latent recombinant transforming growth factor β1 by plasmin. J Cell Biol. 1990;110:1361–7. doi: 10.1083/jcb.110.4.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wahab NA, Weston BS, Mason RM. Modulation of the TGFβ/Smad signaling pathway in mesangial cells by CTGF/CCN2. Exp Cell Res. 2005;307:305–14. doi: 10.1016/j.yexcr.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 31.Zhang C, Meng XF, Zhu ZH, Yang X, Deng AG. Role of connective growth factor in plasminogen activator inhibitor-1 and fibronectin expression induced by transforming growth factor β1 in renal tubular cells. Chin Med J (Engl) 2004;117:990–6. [PubMed] [Google Scholar]
- 32.Miller LD, Smeds J, George J, et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci U S A. 2005;102:13550–5. doi: 10.1073/pnas.0506230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van de Vijver MJ, He YD, van't Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 34.Dunning AM, Ellis PD, McBride S, et al. A transforming growth factor β1 signal peptide variant increases secretion in vitro and is associated with increased incidence of invasive breast cancer. Cancer Res. 2003;63:2610–5. [PubMed] [Google Scholar]
- 35.Margaglione M, Grandone E, Cappucci G, et al. An alternative method for PAI-1 promoter polymorphism (4G/5G) typing. Thromb Haemost. 1997;77:605–6. [PubMed] [Google Scholar]
- 36.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc Ser B. 1995;57:289–300. [Google Scholar]
- 37.Kim SJ, Park K, Koeller D, et al. Post-transcriptional regulation of the human transforming growth factor-β 1 gene. J Biol Chem. 1992;267:13702–7. [PubMed] [Google Scholar]
- 38.Johnsen SA, Subramaniam M, Janknecht R, Spelsberg TC. TGFβ inducible early gene enhances TGFβ/Smad-dependent transcriptional responses. Oncogene. 2002;21:5783–90. doi: 10.1038/sj.onc.1205681. [DOI] [PubMed] [Google Scholar]
- 39.Spyratos F, Bouchet C, Tozlu S, et al. Prognostic value of uPA, PAI-1 and PAI-2 mRNA expression in primary breast cancer. Anticancer Res. 2002;22:2997–3003. [PubMed] [Google Scholar]
- 40.Castello R, Espana F, Vazquez C, et al. Plasminogen activator inhibitor-1 4G/5G polymorphism in breast cancer patients and its association with tissue PAI-1 levels and tumor severity. Thromb Res. 2006;117:487–92. doi: 10.1016/j.thromres.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 41.Blasiak J, Smolarz B. Plasminogen activator inhibitor-1 (PAI-1) gene 4G/5G promoter polymorphism is not associated with breast cancer. Acta Biochim Pol. 2000;47:191–9. [PubMed] [Google Scholar]
- 42.Smolarz B, Blasiak J, Piestrzeniewicz D, Pytel J. Plasminogen activator inhibitor-1 (PAI-1) gene 4G/5G promoter polymorphism in subjects with cancer. Cell Mol Biol Lett. 1998;3:49–56. [Google Scholar]
- 43.Turkmen B, Schmitt M, Schmalfeldt B, et al. Mutational analysis of the genes encoding urokinase-type plasminogen activator (uPA) and its inhibitor PAI-1 in advanced ovarian cancer. Electrophoresis. 1997;18:686–9. doi: 10.1002/elps.1150180505. [DOI] [PubMed] [Google Scholar]
- 44.Loktionov A, Watson MA, Stebbings WS, Speakman CT, Bingham SA. Plasminogen activator inhibitor-1 gene polymorphism and colorectal cancer risk and prognosis. Cancer Lett. 2003;189:189–96. doi: 10.1016/s0304-3835(02)00556-6. [DOI] [PubMed] [Google Scholar]
- 45.Smolarz B, Romanowicz-Makowska H, Kulig A. Plasminogen activator inhibitor 1 (PAI-1) 1334G/A genetic polymorphism in colorectal cancer. Acta Biochim Pol. 2003;50:489–95. [PubMed] [Google Scholar]
- 46.Henry M, Tregouet DA, Alessi MC, et al. Metabolic determinants are much more important than genetic polymorphisms in determining the PAI-1 activity and antigen plasma concentrations: a family study with part of the Stanislas Cohort. Arterioscler Thromb Vasc Biol. 1998;18:84–91. doi: 10.1161/01.atv.18.1.84. [DOI] [PubMed] [Google Scholar]
- 47.Balasa VV, Gruppo RA, Glueck CJ, et al. The relationship of mutations in the MTHFR, prothrombin, and PAI-1 genes to plasma levels of homocysteine, prothrombin, and PAI-1 in children and adults. Thromb Haemost. 1999;81:739–44. [PubMed] [Google Scholar]
- 48.Kathiresan S, Gabriel SB, Yang Q, et al. Comprehensive survey of common genetic variation at the plasminogen activator inhibitor-1 locus and relations to circulating plasminogen activator inhibitor-1 levels. Circulation. 2005;112:1728–35. doi: 10.1161/CIRCULATIONAHA.105.547836. [DOI] [PubMed] [Google Scholar]
- 49.Freeman MS, Mansfield MW, Barrett JH, Grant PJ. Genetic contribution to circulating levels of hemostatic factors in healthy families with effects of known genetic polymorphisms on heritability. Arterioscler Thromb Vasc Biol. 2002;22:506–10. doi: 10.1161/hq0302.104906. [DOI] [PubMed] [Google Scholar]
- 50.Smolarz B, Blasiak J, Kulig A, et al. Plasminogen activator inhibitor 1 (PAI-1) levels and gene promoter polymorphisms in subjects with colorectal cancer. J Exp Clin Cancer Res. 2001;20:247–52. [PubMed] [Google Scholar]
- 51.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF β-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. Embo J. 1998;17:3091–100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.