

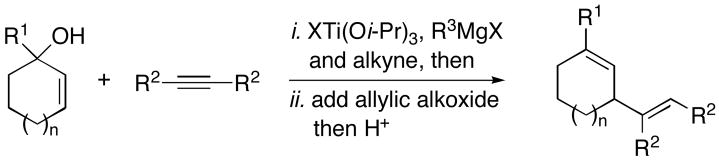
Convergent C–C bond formation is a central theme in modern synthetic organic chemistry. Among the many strategies to accomplish this type of bond construction, allylic alkylation represents an emerging and powerful reaction class. Generally, the chemistry of allyl electrophiles dominates this area, and major challenges within the field reside in the control of regio- and stereoselection in the bimolecular C–C bond forming event (i.e. 1 → 2–5; Figure 1).1 Here, we describe a metal-mediated alkylation of unactivated allylic alcohols with internal alkynes2 that proceeds with net allylic transposition and delivers stereodefined 1,4-dienes in a regioselective manner.3
Figure 1.

Issues of selectivity in modern allylic alkylation.
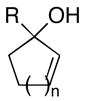
Treatment of substituted allylic alkoxides with preformed titanium–π complexes, generated in situ from the corresponding alkyne and Ti(Oi-Pr)4 or ClTi(Oi-Pr)3,4 results in efficient allylic alkylation (Table 1). As depicted in entries 1 and 2, cross-coupling of the cyclic allylic alcohol 6 or 9 with the symmetric alkyne 7 provides stereodefined 1,4-dienes 8 or 10 in 65 and 68% yields respectively. Entris 3–6 demonstrate that this convergent C–C bond forming reaction occurs with allylic transposition, providing the stereodefined 1,4-diene products 12, 14, 16 and 18 as single isomers.
Table 1.
 | ||||
|---|---|---|---|---|
| entry | allylic alcohol | alkyne | yield (%) | 1,4-dienea |
|
|
|
||
| 1 | 6; R = H, n=1 | 7 | 65 | 8; n=1 |
| 2 | 9; R = H, n=2 | 68 | 10; n=2 | |
| 3 | 11; R = Me, n=1 | 61 | 12; R = Me, n=1 | |
| 4 | 13; R = Ph, n=1 | 79 | 14; R = Ph, n=1 | |
| 5 | 15; R = c-C5H9, n=1 | 40 | 16; R = c-C5H9, n=1 | |
| 6 |
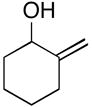
17 |
7 | 57 |
18 |
| 7b |
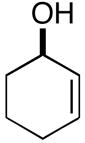
19(er = 97:3) |
7 | 54 |
20c (er = 96:4) |
| 8 |
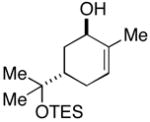
21 |
7 | 50 |
22(dr ≥ 20:1) |
Reaction conditions for cross coupling: alkyne (1.0 eq), ClTi(Oi-Pr)3, PhMe, C5H9MgCl, −78 to −35 °C, then recool to −78 °C, add Li-alkoxide of allylic alcohol (1.0 eq) (−78 to 0 °C).
ClTi(Oi-Pr)3 was replaced with Ti(Oi-Pr)4 in this experiment.
Absolute stereochemistry not determined.
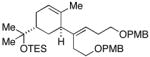
In addition to being regioselective, this cross-coupling reaction proceeds with a high degree of stereochemical control. As depicted in entry 7, coupling of a stereodefined allylic alcohol 19 (er = 97:3) with alkyne 7 provides the functionalized 1,4-diene 20 with negligible erosion of stereochemistry (er = 96:4). The absolute stereochemistry of 20 was assigned based on the stereoselection observed in the coupling of 21 with alkyne 7 (entry 8). This process provides the trans-trisubstituted cyclohexene 22 in 50% yield (dr ≥20:1), demonstrating that C–C bond formation occurs in a suprafacial manner across the allyl system.
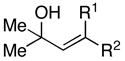
Whereas simple primay allylic alcohols can be employed in coupling reactions with internal alkynes (Table 2, entry 1), increased efficiency is observed with more substituted coupling partners (entry 2). Tertiary acyclic allylic alcohols are also effective in this reaction, and provide highly substituted 1,4-dienes when coupled with internal alkynes. For example, coupling of 2-methyl-3-buten-2-ol (27) with alkyne 7 furnishes the prenylated product 28 in 53% yield (entry 3). Similarly, both (E)- and (Z)-2-methyl-3-penten-2-ol (29 and 31) can be coupled to an internal alkyne to furnish a 3-alkyl-1,4-diene-containing product (30) (entries 4 and 5). As depicted in entry 6, even tetrasubstituted olefins can be preparred with this cross coupling reaction.
Table 2.
 | |||||
|---|---|---|---|---|---|
| entry | allylic alcohol | alkyne | yield (%) | E:Z | 1,4-dienea, b, c |
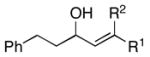
|
|
|
|||
| 1 | 23; R = H | 7 | 41 | – | 24; R = H |
| 2 | 25; R = Me | 66 | – | 26; R = Me | |
| 3 |
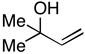
27 |
7 | 53 | – |
28 |
|
|
||||
| 4 | 29; R1 =H, R2 =Me | 7 | 59 | – | 30 |
| 5 | 31; R1 =Me, R2 =H | 67 | – | ||
| 6 |
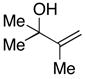
32 |
7 | 57 | – |
33 |
|
|
||||
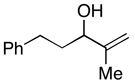
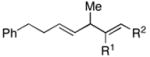
| 7 | 34; R=Ph | 7 | 78 | 1:1 | 35; R=Ph (E/Z=1:1) |
| 8 | 36; R=Et | 67 | 1:1 | 37; R=Et (E/Z=1:1) | |
| 9 |
38 |
7 | 72 | ≥20:1 |
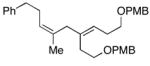
39 |
| 10 | 38 |
40 |
77 | ≥20:1 |
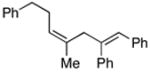
41d |
| 11 | 38 |
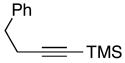
42 |
59 | ≥20:1 |
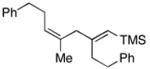
43e |
|
|
||||
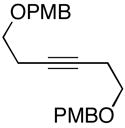
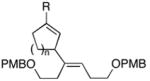
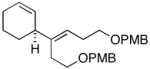
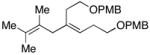
| 12 | 44; R1=Me, R2=H | 7 | 67 | 1:1 | 45; R1, R2=(CH2)2OPMB |
| 13 | 46; R1=H, R2=Me | 42 | 59 | 8:1 | 47e; R1=(CH2)2Ph, R2=TMS |
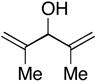
| 14 |
48 |
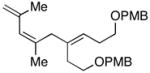
7 | 59 | ≥20:1 |
49 |
Reaction conditions for cross coupling: alkyne (1.0 eq), ClTi(Oi-Pr)3, PhMe, C5H9-MgCl, −78 to −35 °C, then recool to −78 °C, add Li-alkoxide of allylic alcohol (1.0 eq) (−78 to 0 °C).
All 1,4 diene products were isolated as a single olefin isomers.
No evidence was found for the production of regioisomeric products.
In the formation of the titanium–alkyne complex, the temperature was kept under −55 °C (see supporting information for details).
rr ≥ 20:1.
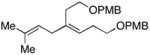
Coupling of acyclic secondary allylic alcohols with internal alkynes is also possible, yet these processes are more complex due to the generation of an additional stereodefined double bond. Whereas secondary allylic alcohols containing mono-substituted olefins (34 and 36) can be coupled to internal alkynes in an efficient manner, these processes proceed without stereoselection (E/Z ca. 1:1) (entries 7 and 8). In contrast, secondary allylic alcohols bearing 1,1-disubstituted olefins can be coupled with internal alkynes in a highly stereoselective manner. In these cases, 1,4-dienes bearing a stereodefined (Z)-trisubstituted olefin are produced with high selectivity (entries 9 and 10).
As depicted in entry 11, this stereoselective cross-coupling reaction can be employed with unsymmetrical alkynes. In this case, cross coupling of allylic alcohol 38 with the TMS-substituted alkyne 42 provides the stereodefined 1,4-diene 43 in 59% yield. In accord with known preferences for titanium alkoxide-mediated functionalization of silyl-substituted alkynes, C–C bond formation occurs selectively at the site distal to the TMS-substituent of alkyne 42.5
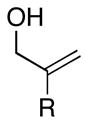
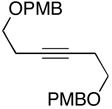
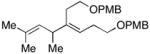
As depicted in entries 12 and 13, allylic alcohols bearing (Z)-disubstituted olefins provide 1,4-diene products with superior selectivity in comparison to the (E)-disubstituted olefin isomers. Whereas cross-coupling of (E)-44 with alkyne 7 affords the 1,4-diene 45 as a 1:1 mixture of olefin isomers (entry 12), the corresponding cross-coupling of (Z)-46 with 42 provides 1,4-diene 47 as an 8:1 mixture favoring the formation of a product containing an (E)-disubstituted olefin (entry 13).
Finally, this new C–C bond forming reaction is tolerant of neighboring π-unsaturation in the allylic alcohol coupling partner. As illustrated in entry 14, cross-coupling of allylic alcohol 48 with alkyne 7 provides the stereodefined triene 49 in 59% yield.
Overall, we have described a new stereoselective cross-coupling reaction between allylic alcohols and alkynes for the synthesis of 1,4-dienes. While occuring with allylic transposition, high stereoselectivity in the generation of substituted olefins is observed in coupling reactions with cyclic, as well as acyclic allylic alcohols. In general, the stereochemical results from this cross-coupling are consistent with an empirical model whereby C–C bond formation occurs through a boat-like geometry of a transient mixed titanate ester (i.e. A and B; Figure 2).6 Further study of the mechanism and scope of this, and related coupling reactions is underway.
Figure 2.

Empirical model for this direct allylic alkylation reaction.
Supplementary Material
Experimental procedures and tabulated spectroscopic data for new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We gratefully acknowledge financial support of this work by the American Cancer Society (RSG-06-117-01), the American Chemical Society (PRF-45334-G1), the Arnold and Mabel Beckman Foundation, Boehringer Ingelheim, Eli Lilly & Co., and the National Institutes of Health – NIGMS (GM80266). The authors also thank Dr. Gorka Peris for the determination of er in entry 7 of Table 1.
References
- 1.For a review of nucleophilic addition reactions to allylic electrophiles, see: Magid RM. Tetrahedron. 1980;36:1901–1930. For a review cross-coupling reactions via π-allyl intermediates, see: Kazmaier U, Pohlman M. In: Metal Catalyzed Cross-Coupling Reactions. De Meijere A, editor. Wiley-VCH; Weinheim: 2004. pp. 531–583. For a recent review of asymmetric allylic substitution catalyzed by copper complexes, see: Yorimitsu H, Oshima K. Angew Chem Int Ed. 2005;44:4435–4439. doi: 10.1002/anie.200500653. For recent examples, see: Zheng W, Zheng B, Zhang Y, Hou X. J Am Chem Soc. 2007;129:7718–7719. doi: 10.1021/ja071098l.Weix DJ, Hartwig JF. J Am Chem Soc. 2007;129:7720–7721. doi: 10.1021/ja071455s.
- 2.For examples of metal-mediated coupling reactions of allylic alcohols with alkynes, see: Trost BM, Kulawiec RJ. J Am Chem Soc. 1992;114:5579–5584.Trost BM, Martinez JA, Kulawiec RJ, Indolese AF. J Am Chem Soc. 1993;115:10402–10403.Trost BM, Indolese AF, Müller TJJ, Treptow B. J Am Chem Soc. 1995;117:615–623.Dérien S, Jan D, Dixneuf PH. Tetrahedron. 1996;52:5511–5524.
- 3.For copper-mediated alkylation of allylic alcohols, see: Tanigawa Y, Ohta H, Sonoda A, Murahashi SI. J Am Chem Soc. 1978;100:4610–4612.Yamamoto Y, Maruyama K. J Organomet Chem. 1978;156:C9–C11.Goering HL, Kantner SS. J Org Chem. 1981;46:2144–2148.
- 4.Harada K, Urabe H, Sato F. Tetrahedron Lett. 1995;36:3203–3206. [Google Scholar]
- 5.Sato F, Urabe H. In: Titanium and Zirconium in Organic Synthesis. Marek I, editor. Wiley-VCH; Weinheim: 2002. pp. 319–354. [Google Scholar]
- 6.The empirical model presented for understanding selectivity in these cross-coupling reactions invokes a formal metallo-[3,3]-rearrangement; we are aware that a plausible mechanistic proposal for these reactions can be based on directed formation of intermediate bicyclic metallacyclopentenes, followed by syn-elimination: Ryan J, Micalizio GC. J Am Chem Soc. 2006;128:2764–2765. doi: 10.1021/ja057352w.Reichard HA, Micalizio GC. Angew Chem Int Ed. 2007;46:1440–1443. doi: 10.1002/anie.200603515.McLaughlin M, Takahashi M, Micalizio GC. Angew Chem Int Ed. 2007;46:3912–3914. doi: 10.1002/anie.200605060.Takahashi M, Micalizio GC. J Am Chem Soc. 2007;129:7514–7516. doi: 10.1021/ja071974v. An analysis of these mechanistic hypotheses is the subject of ongoing studies in our laboratory.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and tabulated spectroscopic data for new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.