

Abstract
Mutations in the human gene ALMS1 result in Alström Syndrome, which presents with early childhood obesity and insulin resistance leading to Type 2 diabetes. Previous genomewide scans for selection in the HapMap data based on linkage disequilibrium and population structure suggest that ALMS1 was subject to recent positive selection. Through a detailed population genomic analysis of existing genomewide data sets and new resequencing data obtained in geographically diverse populations, we find that the signature of selection at ALMS1 is considerably more complex than what would be expected for an idealized model of a selective sweep acting on a newly arisen advantageous mutation. Specifically, we observed three highly divergent and globally dispersed haplogroups, two of which carry a set of seven derived nonsynonymous single nucleotide polymorphisms that are nearly fixed in Asian populations. Our data suggest that the interaction of human demographic history and positive selection on standing variation in Eurasian populations approximately 15 thousand years ago parsimoniously explains the spectrum of extant ALMS1 variation. These results provide new insights into the evolutionary history of ALMS1 in humans and suggest that selective events identified in genomewide scans may be more complex than currently appreciated.
Keywords: ALMS1, positive selection, standing variation
Introduction
The recent availability of dense catalogs of human genetic variation such as the HapMap (International HapMap Consortium 2005) and Perlegen (Hinds et al. 2005) data sets has facilitated global inferences of positive selection. Numerous genomewide scans have identified putative targets of positive selection with patterns of variation that show significant deviations from neutral expectations (Sabeti et al. 2002; Kelley et al. 2006; Voight et al. 2006; Wang et al. 2006; Zhang et al. 2006; Kimura et al. 2007; Tang et al. 2007). Although these analyses have provided considerable insight into how often and where in the genome positive selection has shaped extant patterns of human genetic variation, a deeper understanding of human evolutionary history will require in-depth follow-up studies of “outlier loci” identified in genomewide scans (Biswas and Akey 2006).
To this end, we have performed a detailed population genomic analysis of ALMS1, which has been identified as a putative target of recent adaptive evolution in several genomewide scans for selection (International HapMap Consortium 2005; Wang et al. 2006; Kimura et al. 2007; Tang et al. 2007). Mutations in ALMS1 can lead to Alström Syndrome, a rare autosomal recessive disorder with a spectrum of phenotypes including early onset obesity, metabolic disorders, and sensory impairment (Collin et al. 2005; Li et al. 2007). Recent in vitro work demonstrates that ALMS1 is widely expressed and localizes to centrosomes and the base of cilia (Hearn et al. 2005; Arsov et al. 2006), and studies in mice confirm that ALMS1 is involved in cilia formation and function (Li et al. 2007). Alström Syndrome belongs to a growing class of human diseases, referred to as ciliopathies, that includes disorders such as nephronophthisis, Bardet–Biedl syndrome (BBS), and Meckel–Gruber syndrome (MKS) (Badano et al. 2006). Interestingly, several phenotypes, such as childhood obesity and insulin resistance, overlap between Alström Syndrome and BBS (Hildebrandt and Otto 2005), and hypomorphic mutations in MKS causing genes are associated with BBS (Leitch et al. 2008). Thus, distinct genetic perturbations to the network of proteins involved in cilia formation and function can result in overlapping and pleiotropic phenotypic anomalies.
To better understand the evolutionary history of ALMS1, we analyzed ALMS1 genotype, sequence, and haplotype data. These analyses show that ALMS1 has been subjected to recent positive selection in Eurasian populations approximately 15 thousand years ago (kya). However, unexpectedly, the signature of selection at ALMS1 is considerably more complex than what would be expected for an idealized model of selection acting on a newly arisen advantageous mutation. Rather, the interaction of human demography and positive selection on standing variation in Eurasians parsimoniously explains the spectrum of extant ALMS1 variation. In addition, by reanalyzing previously published genomewide association data, we provide evidence that ALMS1 genetic variation contributes to interindividual variation in metabolic phenotypes such as insulin and glucose levels. In summary, our results provide new insights into the evolutionary history of ALMS1 in humans, highlight the need for careful follow-up studies of candidate selection genes identified in genomewide analyses, and suggest that selective events in human populations may be more complex than currently appreciated.
Materials and Methods
Samples
We sequenced approximately 6 kb of ALMS1 in DNA samples from 91 individuals representing six populations that were obtained from the Coriell Institute for Medical Research Cell Repositories (Camden, NJ). Coriell repository numbers for these samples are as follows: CEPH (n = 21: NA06990, NA07019, NA07348–9, NA10830–1, NA10842–5, NA10848, NA10850–4, NA10857–8, NA10860–1, and NA17201), Han Chinese of L.A. (n = 21: NA17733–NA17749, NA17752–56), Middle East (n = 10: NA17041–50), Pygmy (n = 10: NA10469–73, NA10492–96), South Africa (n = 9: NA17341–49), South America (n = 10: NA17301–10) and South East Asia (n = 10: NA17081–90). In addition, we sequenced the same regions in four nonhuman primate DNA samples from the Coriell Institute for Medical Research Cell Repositories with the following repository numbers: gorilla (Gorilla gorilla; AG05251), bonobo (Pan paniscus; AG05253), chimpanzee (Pan troglodytes; AG06939), and orangutan (Pongo pygmaeus; AG12256).
DNA Sequencing
Sequencing primers were designed from published human sequence (NM_015120) with primer3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) for coding and noncoding regions of ALMS1: upstream, intron 2, exon 5, intron 7, exon 8, intron 8, exon 10, and downstream (primer sequences are available upon request). We used standard polymerase chain reaction–based sequencing reactions using Applied Biosystem's Big Dye sequencing protocol on an ABI 3130xl. Sequence data were assembled using Phred/Phrap (Ewing and Green 1998; Ewing et al. 1998), and the alignments were inspected for accuracy with Consed (Gordon et al. 1998, 2001). Polymorphisms were identified with PolyPhred 4.0 (Bhangale et al. 2006). All polymorphic sites were manually verified and confirmed by sequencing the opposite strand. Genotype data from 210 unrelated individuals were obtained from the HapMap project (Release 22 NCBI Build 36) (International HapMap Consortium 2005).
Linkage Disequilibrium (LD)
We calculated r2 between all pairwise combinations (Hill 1968) of markers in ALMS1 and approximately 1 Mb of flanking sequences (both 5′ and 3′) using HapMap genotype data. Estimates of r2 were obtained from Haploview (Barrett et al. 2005) for all markers with a minor allele frequency ≥5% and used in subsequent analyses. To evaluate and compare the distribution of LD within and between the HapMap CEU, YRI, and ASN samples, and how LD decays as a function of distance from ALMS1, we calculated a statistic related to ZnS (Kelly 1997). Specifically, we calculated the average r2 between all pairwise comparisons of single nucleotide polymorphisms (SNPs) in bin 1 and bin 2:
 |
where n1 is the number of SNPs in bin 1 and n2 is the number of SNPs in bin 2. Here, n2 represents the number of SNPs in ALMS1 and n1 the number of SNPs in nonoverlapping 50-kb windows up and downstream of ALMS1.
Haplotype Analysis
Haplotypes were reconstructed in the HapMap and sequence data with Phase 2.1.1 (Stephens et al. 2001; Stephens and Scheet 2005) using 10 iterations to confirm consistency among runs, and the run with the best average goodness-of-fit was used. We defined Haplogroup A (ancestral) and Haplogroup D (derived) based on the allelic state of seven nonsynonymous SNPs (nsSNPs) (rs3813227, rs6546837, rs6546838, rs6724782, rs6546839, rs2056486, and rs10193972) and Haplogroup D1 and Haplogroup D2 based on the allelic state of two additional SNPs (rs6730785 and rs7598901). The ancestral allele was determined by the chimpanzee sequence.
We used Neighbor from the software package PHYLIP 3.6 (Felsenstein 1989, 2005) to construct unrooted phylogenetic trees on phased sequence (Dnadist was used to calculate the pairwise distance matrix) and HapMap data (average pairwise distances were calculated for the distance matrix). In both cases, we removed recombinant haplotypes occurring among the seven aforementioned nsSNPs (three unique haplotypes/four total haplotypes from the sequence data and two unique haplotypes/three total haplotypes from the HapMap data). We visualized the Neighbor-Joining trees with the APE package in R (http://cran.r-project.org/web/packages/ape/).
Time to the Most Recent Common Ancestor (TMRCA) Estimates
We used the method described by Thomson et al. (2000) to estimate the TMRCA on our phased sequenced data as this method does not utilize any particular population model. Analyses were performed both on all haplotypes as well as on only haplotypes with no recombination among the seven nsSNPs, and we found minimal effects on the estimated TMRCA (data not shown). We used the average divergence between chimpanzee and human sequences divided by two times the estimated divergence time of 6 million years, which we calculated to be 36/(2*60,00,000), or 3 × 10−6 for our sequence mutation rate. Briefly, to estimate the TMRCA, we used the simple estimate of T, the time since the MRCA (Thomson et al. 2000; Mekel-Bobrov et al. 2005):
where  is the unbiased estimator of T, xi is the number of mutational differences between the ith sequence and the MRCA, n is the total number of sequences in the sample, and μ is the mutation rate. In addition, we used three additional methods to estimate the ALMS1 TMRCA (McPeek and Strahs 1999; Bahlo and Griffiths 2000; Templeton 2002), all of which yielded similarly old dates and were not significantly different from one another (data not shown).
is the unbiased estimator of T, xi is the number of mutational differences between the ith sequence and the MRCA, n is the total number of sequences in the sample, and μ is the mutation rate. In addition, we used three additional methods to estimate the ALMS1 TMRCA (McPeek and Strahs 1999; Bahlo and Griffiths 2000; Templeton 2002), all of which yielded similarly old dates and were not significantly different from one another (data not shown).
Coalescent Simulations
We calculated three standard neutrality tests of the site frequency spectrum: Tajima's D (Tajima 1989), Fu and Li's F test (Fu and Li 1993), and Fay and Wu's H test (Fay and Wu 2000). We used the nonhuman primate sequence to establish the ancestral allele for Fay and Wu's H test. To interpret summary statistics derived from the resequencing data, we performed additional coalescent simulations with the program ms (Hudson 2002) using previously inferred demographic parameters that were found to best fit genomic patterns of variation in the HapMap YRI, CEU, and ASN samples (Schaffner et al. 2005). The exact parameters can be found in table 1 of Schaffner et al. (2005), and involve multiple bottlenecks, population expansions, population splitting, recombination, and gene conversion. The only exception is that we did not include migration following population splitting as Schaffner et al. (2005) found these parameters resulted in only slightly worse fitting models, but the modest increase in levels of population differentiation resulted in more accepted simulation replicates to analyze. The ms command line argument for this model is available upon request. We used a rejection sampling method (Beaumont et al. 2002) to account for the a priori observation of ALMS1 population structure and a total of 1 × 107 simulations were performed. Initially, we attempted to accept data sets if they matched observed levels of differentiation in our resequencing data (five or more SNPs with an FST ≥ 0.80 between African and Han Chinese samples and two or more SNPs with an FST ≥ 0.52 between African and CEPH samples). However, none of the 10 million simulation replicates met these criteria, indicating that such levels of structure are incompatible with a neutral demographic model that is consistent with major features of human genomic variation (Schaffner et al. 2005). Thus, for computational tractability, we relaxed the acceptance criteria to one or more SNPs with a pairwise FST ≥ 0.80 and 0.52 between African and Han Chinese samples and African and CEPH samples, respectively. Using these thresholds, 1,405 data sets of the 10 million simulations were accepted and analyzed further. In particular, we evaluated the probability of observing divergent haplotype lineages, TMRCA, and Tajima's D as or more extreme than that observed for ALMS1. In accepted data sets, we calculated TMRCA as described above, Tajima's D (Tajima 1989), and the average number of nucleotide differences between haplogroups carrying the derived allele at the highly differentiated SNP:
where D1 and D2 denote the set of haplotypes belonging to derived haplogroup lineages 1 and 2, respectively. In the simulated data sets, D1 and D2 were chosen so as to maximize dxy.
Table 1.
Summary of Coalescent Simulation Results Conditional on Ascertainment
| Samplea | dxyb | P(dxy|FST)c | TMRCAb (kya) | P(TMRCA|FST)c | Tajima's D | P(D|FST)c |
| Han Chinese (n = 42) | 6.01 | 0.046 | 810 | 0.934 | −0.12 | 0.827 |
| CEPH (n = 42) | 4.98 | 0.349 | 2301 | 0.081 | 1.34 | 0.065 |
Number of chromosomes for each sample is indicated in parentheses.
TMRCA and dxy (the average number of pairwise differences between derived haplogroups) were calculated as described in Methods.
Probability of observed summary given the ascertainment based on strong levels of population structure as assessed by FST. One-sided tests were used for dxy and TMRCA, and a two-sided test was used for Tajima's D.
Human Genome Diversity Project–Centre d'Etude du Polymorphisme Humain (HGDP–CEPH) Analysis
We defined haplogroups in the HGDP–CEPH data set (Li et al. 2008) with six SNPs, Haplogroups A and D were defined based on alleles of four genotyped nsSNPs (rs3813227, rs6546838, rs2056486, and rs10193972) and Haplogroups D1 and D2 were further defined by two additional genotyped SNPs (rs2037814 and rs3820700). Recombinant haplotypes among Haplogroups A and D were excluded from the haplotype frequency map, whereas recombinants between Haplogroups D1 and D2 were included and defined by the allelic status of rs10193972. In order to avoid any single population sample falling below a sample size of 10, we combined the Bantu SE and SW individuals into one Bantu South population.
We developed a simple heuristic statistic to determine how unusual the geographic distribution of ALMS1 genetic variation is relative to the rest of the genome using all autosomal HGDP–CEPH data that had less than 10% missing data. Specifically, for the ith SNP, we define the global deviance score, GDi, as follows:
where FSTi12, FSTi13, and FSTi23is the unbiased pairwise FST (Weir 1996) between East Asian and African samples, East Asian and American samples, and African and American samples, respectively, for the ith SNP, and 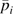 is the average allele frequency across samples weighted by sample size. In words, the global deviance score is large when levels of differentiation between Asian and African and Asian and American samples are greater than the genomewide average and levels of differentiation between African and American samples is less than the genomewide average. We included the Bantu (North and South), Biaka, Mbuti, Mandenka, Yoruba, and San in the African sample; the Colombian, Karitiana, Maya, Pima, and Surui in the American sample; and the Cambodian, Dai, Daur, Han (North and South), Hezhen, Japanese, Lahu, Miaozu, Mongola, Naxi, Oroqen, She, Tu, Tujia, Xibo, Yakut, and Yizu in the East Asian sample.
is the average allele frequency across samples weighted by sample size. In words, the global deviance score is large when levels of differentiation between Asian and African and Asian and American samples are greater than the genomewide average and levels of differentiation between African and American samples is less than the genomewide average. We included the Bantu (North and South), Biaka, Mbuti, Mandenka, Yoruba, and San in the African sample; the Colombian, Karitiana, Maya, Pima, and Surui in the American sample; and the Cambodian, Dai, Daur, Han (North and South), Hezhen, Japanese, Lahu, Miaozu, Mongola, Naxi, Oroqen, She, Tu, Tujia, Xibo, Yakut, and Yizu in the East Asian sample.
We used the expression analysis tool (Thomas et al. 2006) to identify enriched PANTHER Pathways, Biological Processes, and Molecular Functions (Thomas et al. 2003) among genes in the top 0.1% of the distribution of GD scores. Pathways and terms with less than five genes were excluded from further analysis, and Bonferroni corrections were used to correct for multiple testing.
Estimating the Time of the Selective Sweep
We estimated the time since the selective sweep for the derived class of ALMS1 lineages by analyzing the amount of nucleotide diversity that has accumulated on the selected haplotypes as described in Akey et al. (2004) where the time back to the selective sweep, t, can be estimated by S/(nμ), where S is the number of segregating sites, n is the number of haplotypes included and μ is the neutral mutation rate of the locus. For ALMS1 derived haplogroups, n = 120, S = 13, and μ = 1.75 × 10−4. Note that this calculation should be treated as a rough approximation because it assumes a starlike phylogeny, which ALMS1 violates.
Estimating the Strength of Selection
We used the following simple deterministic formula to estimate the selection coefficient, s (Gillespie 1998):
where , p is the frequency of the selected allele, q is the frequency of the nonselected allele, and h is the heterozygous effect. We assumed an initial frequency of 10% (a conservatively high estimate based on current frequencies in African samples) and a final frequency of 95% (a conservatively low estimate based on current frequencies in East Asian samples) for the putatively selected allele. The range of s reported in the main text is based on varying the age of the selective event (from 500 to 1,000 generations) and heterozygous effects (h = 0, 0.5, and 1).
Results
ALMS1 Genetic Variation Is Highly Structured among Populations and Localizes the Signature of Selection
ALMS1 was initially identified as a potential target of positive selection based on large allele frequency differences among populations for six nonsynonymous SNPs (nsSNPs) (International HapMap Consortium 2005). In order to better understand how unusual levels of population structure are in the ALMS1 region relative to the rest of the genome and fine-scale map the signature of selection, we first performed a genomewide analysis of allele frequency differences in the HapMap data. Specifically, we calculated the average pairwise FST (Weir 1996) in nonoverlapping 100-kb windows using HapMap Phase II data (autosomal regions only) among the Yoruba (YRI) individuals from Ibadan, Nigeria (n = 60), CEPH (CEU) individuals with ancestry from northern and western Europe (n = 60), Japanese (JPT) individuals from Tokyo, Japan (n = 45), and Han Chinese (CHB) individuals from Beijing, China (n = 45). In all of the analyses, we combined the JPT and CHB individuals into a single Asian sample (ASN). Windows containing ALMS1 were in the extreme 99th percentile of the empirical distribution for both the ASN and YRI and CEU and YRI comparisons, and only 12 of the 28,652 windows were more differentiated than ALMS1 in the ASN and YRI comparisons.
We performed three additional analyses to determine how robust the signature of strong population structure at ALMS1 is to potential confounding variables. First, we repeated the genomewide analysis of FST on the HapMap data with window sizes in units of genetic distance (0.1 cM) estimated from fine-scale recombination rates. Second, we adjusted each window specific estimate of FST for a larger set of potential confounding variables (number of SNPs, recombination rate, GC%, and heterozygosity per 100-kb bin) by multiple regression. Finally, we performed a genomewide analysis of FST on Class A Perlegen SNPs as described above, which were discovered more uniformly and manifest less ascertainment bias relative to the HapMap SNPs (Hinds et al. 2005; Kelley et al. 2006). In all three cases, ALMS1 remained one of the most differentiated regions in the genome (results not shown), indicating that our results are robust to ascertainment bias, recombination rate heterogeneity, and additional confounding variables.
The distribution of pairwise FST among the ASN, CEU, and YRI samples for all SNPs across an approximately 800-kb region centered on ALMS1 is shown in figure 1. The largest values of FST across the region are coincident with the location of ALMS1 (fig. 1), and SNPs located immediately up and downstream of ALMS1 show markedly lower FST values. Extreme levels of population structure are found throughout ALMS1; specifically, 45 and 35 SNPs have FST values greater than the 99th percentile for the ASN and YRI and CEU and YRI samples, respectively. These highly differentiated SNPs include seven nsSNPs (fig. 2), six of which have been previously described (International HapMap Consortium 2005), and are located in exons 5, 8, and 10 of ALMS1. The derived alleles at each of the seven nsSNPs are found at 99%, 80%, and 8–9% frequency in the ASN, CEU, and YRI samples, respectively. Levels of differentiation between the ASN and CEU samples are not unusual compared with the genome at large (fig. 1).
FIG. 1.—

Distribution of FST across the ALMS1 region. The top panel shows the ALMS1 gene structure (with exons shaded in black) and locations of previously estimated recombination hotspots (International HapMap Consortium, 2007). Pairwise FST between each HapMap sample is shown below, and the dashed line in each panel indicates the 99% threshold for each pairwise FST comparison. The seven highly differentiated ALMS1 nsSNPs are highlighted in red.
FIG. 2.—

Patterns of LD in ALMS1 and flanking regions. The location of ALMS1 is marked by a gray rectangle and additional genes located in the region are shown as white rectangles. Previously inferred recombination hotspots are denoted by black rectangles. For each HapMap sample, r2 is plotted individually. The points centered on ALMS1 denote the average r2 among all pairwise combinations of SNPs in ALMS1. Points up and downstream indicate the average r2 among all ALMS1 SNPs and SNPs in nonoverlapping 50-kb windows. For example, the points at −25 kb correspond to the average r2 between all ALMS1 SNPs and SNPs located between 0 and 50 kb upstream of ALMS1. The point centered on ALMS1 represents the average r2 for all SNP pairs within ALMS1.
In summary, the patterns of FST in the HapMap and Perlegen data suggest ALMS1 was a target of recent positive selection in East Asian and European populations, and indicate several plausible sites (i.e., one or more of the seven highly differentiated nsSNPs) conferring a fitness advantage. Consistent with this interpretation, previous genomewide scans of selection based on LD have also identified ALMS1 as an outlier in the ASN and CEU samples (Wang et al. 2006; Kimura et al. 2007; Tang et al. 2007). A graphical summary of the distribution of LD across the ALMS1 region in the HapMap samples is shown in figure 2.
Unusual Patterns of ALMS1 Haplotype Variation Are Inconsistent with Simple Models of a Selective Sweep
Under simple models of genetic hitchhiking (Maynard-Smith and Haigh 1974), we would expect to find a single haplotype carrying an advantageous allele at high frequency. To test this prediction, we reconstructed haplotypes (Stephens et al. 2001; Stephens and Scheet 2005) in the HapMap samples. A visual representation of haplotypes shows a striking departure from predictions of a simple hitchhiking model, where haplotypes carrying the derived alleles at the seven highly differentiated nsSNPs exist on two distinct backgrounds (fig. 3). In addition, haplotypes carrying the ancestral allele at each of the seven highly differentiated nsSNPs exist on a background that is distinct relative to the two derived classes (fig. 3). Similar results were observed in visual representations of genotypes (results not shown), demonstrating these patterns are not an artifact of haplotype inference.
FIG. 3.—

ALMS1 haplotypes are organized into three divergent haplogroup lineages. On the left, a visual representation of ALMS1 haplotypes derived from the HapMap samples is shown, where rows correspond to individual phased haplotypes and columns represent SNPs. Black and gray rectangles indicate ancestral and derived alleles, respectively. Haplotypes are sorted according to ancestral state at the seven highly differentiated nsSNPs. White horizontal lines separate haplotypes carrying the ancestral alleles at the nsSNPs (labeled A) and the two divergent haplogroup lineages carrying the derived alleles at the nsSNPs (labeled D1 and D2). Black rectangles above the visual haplotype indicate SNPs with an allele frequency difference ≥90% between the ancestral and derived haplogroups (A/D) and haplogroups D1 and D2 (D1/D2). Red arrows at the bottom and red rectangles at the top of the visual haplotype indicate positions of the seven nsSNPs. A Neighbor-Joining tree of haplotypes is shown on the top right. Haplogroup A, D1, and D2 branches are colored in magenta, green, and blue, respectively. The length of the scale bar corresponds to 1% of sites differing between haplotypes. The splits between haplogroups A and D and D1 and D2 were assessed by bootstrapping and were supported 94% and 54% of the time in 1,000 replicates, respectively. Finally, haplogroup frequencies in the HapMap ASN, CEU, and YRI samples are shown in the bottom right.
To more quantitatively assess haplotype structure at ALMS1, we constructed a Neighbor-Joining tree based on pairwise distances among haplotypes. The Neighbor-Joining tree shows three distinct haplogroups (fig. 3), formed by an initial split between haplotypes carrying the seven ancestral nsSNPs from those containing the derived nsSNPs (which we will refer to as Haplogroup A and Haplogroup D, respectively). Furthermore, there is an additional deep split among the derived haplotypes forming two distinct haplogroups (which we will refer to as Haplogroup D1 and Haplogroup D2).
The average pairwise difference (based on all 242 HapMap SNPs spanning ALMS1) between Haplogroups A and D and D1 and D2 are 104.2 and 57.6, respectively. Thus, on average, Haplogroups A and D possess alternative alleles at approximately 43% of sites and Haplogroups D1 and D2 differ at approximately 24% of sites. In contrast, the average number of pairwise differences within Haplogroups A, D, D1, and D2 are 24.0, 15.5, 7.3, and 1.5, respectively. Although marked differences exist in Haplogroup D frequency between African and non-African HapMap samples (0.99, 0.80, and 0.08 in the ASN, CEU, and YRI samples, respectively) both derived lineages are found in Africa (Haplogroups D1 and D2 exist in the YRI sample at a frequency of 6% and 2%, respectively; fig. 3). Furthermore, haplotype heterozygosities of the YRI, ASN, and CEU samples are 0.953, 0.837, and 0.700, respectively.
In order to examine a data set not limited by the ascertainment biases inherent in the HapMap data set, we sequenced approximately 6 kb of coding and noncoding ALMS1 in 91 globally dispersed individuals (see Methods). As shown in supplementary figure S1, Supplementary Material online, the Neighbor-Joining tree of ALMS1 sequence variation recapitulates the topology of the three divergent haplogroups consisting of Haplogroups A, D1, and D2, and derived haplogroups are present in low frequencies in African samples (0.13). Consistent with the patterns of divergence among haplogroups in the HapMap data, our estimate of the TMRCA of ALMS1 is 2,158 ± 848 kya (see Methods), which is among the oldest reported autosomal TMRCAs (Kreitman and Di Rienzo 2004).
Thus, both the HapMap and resequencing data demonstrate that the origins of Haplogroups D1 and D2 can be traced back to Africa, and these haplogroups have dramatically increased in frequency in Eurasian populations sometime after the dispersal of humans out of Africa. The large divergence among haplogroups and their global occurrence strongly argue for a model of selection acting upon standing variation, rather than on a newly arisen advantageous mutation.
ALMS1 DNA Sequence Variation
Summary and standard neutrality test statistics for the resequencing data are shown in supplementary table S1, Supplementary Material online. Typically, patterns of DNA sequence variation are evaluated by determining how unusual observed values are under neutral expectations. However, this canonical approach fails to properly account for the fact that ALMS1 was not chosen at random, but rather was ascertained based on its high level of population structure. Such ascertainment biases need to be taken into account when interpreting patterns of DNA sequence variation in subsequent analyses of outlier loci (Kreitman and Di Rienzo 2004; Thornton and Jensen 2007). To this end, we used a rejection sampling approach (Beaumont et al. 2002) to explicitly control for the a priori observation of strong population structure when evaluating the probability of observing additional aspects of ALMS1 genetic variation under neutrality as described in the Methods section. For simplicity, we will focus on results from the Han Chinese and CEPH, as the calibrated model of Schaffner et al. (2005) is most appropriate for these samples.
Two interesting points emerge from the simulations. First, when ascertainment is taken into account, values of haplogroup divergence, Tajima's D, and TMRCA are either marginally significant or not significant at all in both the Han Chinese and CEPH samples. At least for Tajima's D, this result is unsurprising, as theoretical analyses have shown that tests of the site frequency spectrum have low power to detect deviations from neutrality under models of selection from standing variation (Hermisson and Pennings 2005; Przeworski et al. 2005; Barrett and Schluter 2008).
Second, table 1 illustrates the contrasting patterns of sequence characteristics between the CEPH and the Han Chinese samples. Specifically, the average pairwise difference between derived lineages is marginally significant in the Han Chinese, but not in the CEPH (table 1). In contrast, Tajima's D and the TMRCA are marginally significant in the CEPH, but not the Han Chinese. This result is due to the fact that the ancestral haplogroup is absent in the Han Chinese, but its frequency is 26% in the CEPH, raising the TMRCA of the latter. Furthermore, the presence of three common and divergent haplogroups in the CEPH leads to a modestly positive Tajima's D.
Unusual Worldwide Distribution of ALMS1 Haplogroups
Recently, over 650,000 SNPs were genotyped in the HGDP–CEPH samples (Li et al. 2008), which consist of over 1,000 individuals from 52 populations (see Methods). In the HGDP–CEPH data, four of the ALMS1 nsSNPs described above (rs3813227, rs6546838, rs2056486, and rs10193972) were genotyped, and we used two additional genotyped SNPs (rs3820700 and rs1052161) to distinguish between Haplogroups D1 and D2. The worldwide distribution of ALMS1 haplogroups (fig. 4) reveals a particularly interesting pattern where Haplogroup D is nearly fixed in East Asian samples (98.9%), but is at considerably lower frequency in the American samples (43.0%). Similarly, the frequency of Haplogroup D1 in the American samples is extremely low (0.8%) compared with East Asian samples (24.6%). Conversely, Haplogroup A is common in the Americas (57.03%) but nearly absent in East Asia (0.01%). This geographic distribution is peculiar given that Asia was the likely source population of the Americas (Karafet et al. 1997; Mulligan et al. 2004; Goebel et al. 2008; Volodko et al. 2008). The simplest explanation for these data is that Haplogroups A and D were both present in Asia before the founding of the Americas, but Haplogroup D dramatically increased in frequency in East Asia sometime after the colonization of the Americas 15–20 kya (Karafet et al. 1997; Mulligan et al. 2004; Goebel et al. 2008; Volodko et al. 2008). The caveats to this interpretation are that the HGDP–CEPH samples are not ideally suited to test models for the peopling of the Americas, and the SNPs typed in these samples have difficult to account for ascertainment bias.
FIG. 4.—

Distribution of ALMS1 haplogroups in 52 populations. Haplogroup frequencies are indicated with pie charts. Haplogroups A, D1, and D2 are shown in magenta, green, and blue, respectively.
To evaluate how unusual the worldwide distribution of ALMS1 allele frequency variation is relative to the rest of the genome, which would provide insight into whether purely neutral processes such as genetic drift and serial founder effects (Edmonds et al. 2004; Klopfstein et al. 2006; Hallatschek and Nelson 2008) can account for patterns of ALMS1 variation, we analyzed 643,884 SNPs (see Methods) genotyped in the HGDP–CEPH panel (Li et al. 2008). Specifically, we defined a simple heuristic statistic, which we refer to as the global deviance score (see Methods), to capture the worldwide frequency distribution of ALMS1. Seven ALMS1 SNPs rank in the top 50 SNPs (99.99th percentile). Interestingly, 27 of the top 50 SNPs are located in regions of the genome that have previously been implicated as targets of adaptive evolution (supplementary table S2, Supplementary Material online; see also Wang et al. 2006; Frazer et al. 2007; Kimura et al. 2007; Tang et al. 2007). In addition, genes in the 99.9th percentile of the empirical distribution of global deviance scores are significantly enriched (Bonforroni corrected P < 0.05) for particular PANTHER Pathways, Biological Processes, and Molecular Functions (supplementary table S3, Supplementary Material online). Of particular interest is the observation that genes involved in carbohydrate metabolism (including ALMS1) are significantly enriched among the top 0.1% of loci (supplementary table S3, Supplementary Material online), consistent with previous genomewide scans for selection (Kelley and Swanson 2008), indicating this class of genes has been particularly important in the recent evolutionary history of East Asian populations.
In short, the geographic distribution of ALMS1 haplogroup frequencies in the HGDP–CEPH samples further supports a model of selection from standing variation. In particular, the presence of Haplogroup A at high frequency in the American samples combined with its extremely low frequency in East Asia, suggests that the ancestral haplogroup was present at an appreciable frequency in Asia prior to the colonization of the Americas, and subsequently driven to near extinction as selection promoted the rapid increase in Haplogroup D frequency.
Discussion
ALMS1 possesses many anomalous patterns of genetic variation such as extensive population structure, including a cadre of seven nsSNPs, three divergent haplogroup lineages, and a peculiar spatial distribution in geographically diverse populations. We have shown that these characteristics are inconsistent with purely neutral explanations. However, our data are equally inconsistent with simple models of positive selection acting on a newly arisen advantageous mutation (Maynard-Smith and Haigh 1974). Rather, our results support a model of positive selection acting on standing variation in Eurasia populations. In this model, one or more polymorphisms on Haplogroups D1 and D2, which are found at low frequency in the African samples we analyzed, became adaptive following their dispersal out of Africa and rapidly increased in frequency in Eurasians. Furthermore, by considering the geographic distribution of haplogroup frequencies, we are able to narrow down the likely time frame of selection to be either concurrent with or subsequent to the colonization of the Americas 15–20 kya. This interpretation is consistent with our estimate of the time since the selective sweep on the derived lineages of 15.5 kya (see Methods).
A particularly interesting feature of ALMS1 is the old and divergent haplogroup lineages. After taking into account levels of population structure, the estimated TMRCA and average pairwise divergence among haplogroups are not unusual (table 1), suggesting these characteristics occur with appreciable frequency in highly structured regions of the genome (see also Cornejo and Escalante 2006; Garrigan and Hammer 2006). We note, however, that our analyses have primarily focused on elucidating the recent evolutionary history of ALMS1, which shows compelling evidence for recent directional selection acting on preexisting variation in Eurasian populations; additional studies will be necessary to better delimit the contribution of additional models, such as balancing selection to the long-term evolutionary history of ALMS1. Indeed, ALMS1 possesses higher levels of LD in the YRI relative to the genome-at-large (data not shown), a finding that is surprising given its ancient TMRCA, suggesting some form of nonneutral evolution, such as balancing or frequency dependent selection, in Africa.
We estimate the selection coefficient, s, for ALMS1 to be approximately 0.01–0.05, which is commensurate with magnitudes of selection observed for genes underlying lactase persistence (LCT, s = 0.01–0.05; Bersaglieri et al. 2004; Enattah et al. 2007; Tishkoff et al. 2007) and resistance to malaria (G6PD, s = 0.02–0.05; Tishkoff et al. 2001). Thus, the estimated strength of selection for ALMS1 is among the strongest identified in humans, which begs the question as to the historical selective pressure acting on ALMS1 genetic variation. Although it is clear from Alström Syndrome patients that ALMS1 mutations can influence a spectrum of phenotypes, including obesity, type 2 diabetes, and metabolic disorders, the phenotypic consequences of nonsyndromic variation are unknown.
To explore the role of ALMS1 in metabolic phenotypes further, we reanalyzed the results from a number of genomewide association studies for type 2 diabetes (t Hart et al. 2003; Patel et al. 2006; Wellcome Trust Case Control Consortium 2007; Saxena et al. 2007), none of which implicate the ALMS1 region. However, 18 metabolic traits were measured in Saxena et al. (2007) that were not extensively discussed in the original publication. We obtained association data from this study to test the hypothesis that ALMS1 genetic variation is associated with insulin or glucose-related phenotypes, given the observed clinical manifestations of individuals with Alström Syndrome. Interestingly, in nondiabetic controls ALMS1 SNPs show nominal levels of association to five insulin and glucose related phenotypes (supplementary table S3, Supplementary Material online). The strongest association was observed between rs7598660 and 2-h insulin levels (P = 1.38 × 10−4; supplementary fig. S3, Supplementary Material online), which ranked as the 43rd most significant association among the approximately 380,000 genotyped SNPs. Although these results should be interpreted with caution because of the modest statistical evidence supporting them, which do not attain genomewide significance, they suggest that nonsyndromic variation in ALMS1 may contribute to interindividual variation in the same metabolic phenotypes that are perturbed in Alström Syndrome patients. Additional studies will ultimately be necessary to more clearly define the functional and phenotypic consequences of ALMS1 genetic variation, which in turn will inform inferences about the historical selective pressures acting on this genomic region.
A closer inspection of the genomewide association results for ALMS1 also provides insight into how past adaptive evolution may influence present day distribution and susceptibility to disease. Specifically, the strongest association between ALMS1 genetic variation and metabolic phenotypes was observed between rs7598660 and 2-h insulin levels (supplementary fig. S3, Supplementary Material online). The ancestral allele of rs7598660 is associated with higher 2-h insulin levels (i.e., greater insulin resistance), whereas the derived allele is associated with lower 2-h insulin levels (i.e., less insulin resistance; supplementary fig. S3, Supplementary Material online). As the derived allele is only present on a subset of Haplogroup D2 chromosomes (supplementary table S5, Supplementary Material online) it is unlikely that the rs7598660 polymorphism (or linked variation) was the direct target of selection, but rather increased in frequency in non-African populations by hitchhiking. Therefore, geographically varying selective pressures on ALMS1 resulted in large allele frequency differences of a putative polymorphism (rs7598660 or linked variant) that influences insulin resistance, which is tangential to the primary selective force. Interestingly, the frequency of the rs7598660 derived allele among the HapMap YRI, CEU, and ASN samples is 0.008, 0.669, and 0.367, respectively, which is consistent with a higher prevalence of insulin resistance in African-Americans relative to European-Americans (Haffner et al. 1996; Reiner et al. 2007). Thus, models that attempt to place human disease into an evolutionary context (Di Rienzo and Hudson 2005; Biswas and Akey 2006) may also need to account for indirect selective effects, where susceptibility alleles are not causally related to historical selective pressures but merely go along for the ride on a selected haplotype.
In summary, we have shown that the evolutionary history of ALMS1 is considerably more complex than might have been expected based on its identification as an outlier locus in genomewide scans for selection, involving the interaction of demographic history, geographically restricted selection, and selection from standing variation. An emerging question in the evolution of natural populations is to what extent selection acts on new or preexisting mutations (Orr and Betancourt 2001; Hermisson and Pennings 2005; Przeworski et al. 2005; Barrett and Schluter 2008). This issue has important implications for the evolutionary trajectory of populations (Hermisson and Pennings 2005) and more practically on the types of signatures to pursue in the search for selected loci (Przeworski et al. 2005). A number of examples in humans have been described that are consistent with selection from standing variation such as FY (Hamblin et al. 2002), LCT (Tishkoff et al. 2007; Enattah et al. 2008), and NAT2 (Magalon et al. 2008). Thus, we suspect that when additional candidate selection genes are examined with more scrutiny, selection from standing variation will be found to be a common mechanism of adaptation, driven by the rapid dispersal of humans into new environments during the last 60 ky.
Supplementary Materials
Supplementary tables S1–S5 and supplementary figures S1 and S2 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We thank members of the Akey Lab and Willie Swanson for helpful discussions and comments on the manuscript. This work was supported by a research grant (1R01GM076036-01A1) from the NIH and a Sloan Fellowship in Computational Biology to J.M.A. and by an NHGRI Interdisciplinary Training in Genomic Sciences grant (HG00035) to L.B.S.
References
- Akey JM, Eberle MA, Rieder MJ, Carlson CS, Shriver MD, Nickerson DA, Kruglyak L. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol. 2004;2:e286. doi: 10.1371/journal.pbio.0020286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsov T, Silva DG, O'Bryan MK, et al. (13 co-authors) Fat aussie—a new Alstrom syndrome mouse showing a critical role for ALMS1 in obesity, diabetes, and spermatogenesis. Mol Endocrinol. 2006;20:1610–1622. doi: 10.1210/me.2005-0494. [DOI] [PubMed] [Google Scholar]
- Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genom Hum Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- Bahlo M, Griffiths RC. Inference from gene trees in a subdivided population. Theor Popul Biol. 2000;57:79–95. doi: 10.1006/tpbi.1999.1447. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Barrett RD, Schluter D. Adaptation from standing genetic variation. Trends Ecol Evol. 2008;23:38–44. doi: 10.1016/j.tree.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Beaumont MA, Zhang W, Balding DJ. Approximate Bayesian computation in population genetics. Genetics. 2002;162:2025–2035. doi: 10.1093/genetics/162.4.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersaglieri T, Sabeti PC, Patterson N, Vanderploeg T, Schaffner SF, Drake JA, Rhodes M, Reich DE, Hirschhorn JN. Genetic signatures of strong recent positive selection at the lactase gene. Am J Hum Genet. 2004;74:1111–1120. doi: 10.1086/421051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhangale TR, Stephens M, Nickerson DA. Automating resequencing-based detection of insertion–deletion polymorphisms. Nat Genet. 2006;38:1457–1462. doi: 10.1038/ng1925. [DOI] [PubMed] [Google Scholar]
- Biswas S, Akey JM. Genomic insights into positive selection. Trends Genet. 2006;22:437–446. doi: 10.1016/j.tig.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Collin GB, Cyr E, Bronson R, et al. (11 co-authors) Alms1-disrupted mice recapitulate human Alstrom syndrome. Hum Mol Genet. 2005;14:2323–2333. doi: 10.1093/hmg/ddi235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium IH. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium WTCC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornejo OE, Escalante AA. The origin and age of Plasmodium vivax. Trends Parasitol. 2006;22:558–563. doi: 10.1016/j.pt.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rienzo A, Hudson RR. An evolutionary framework for common diseases: the ancestral-susceptibility model. Trends Genet. 2005;21:596–601. doi: 10.1016/j.tig.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Edmonds CA, Lillie AS, Cavalli-Sforza LL. Mutations arising in the wave front of an expanding population. Proc Natl Acad Sci USA. 2004;101:975–979. doi: 10.1073/pnas.0308064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enattah NS, Jensen TG, Nielsen M, et al. (22 co-authors) Independent introduction of two lactase-persistence alleles into human populations reflects different history of adaptation to milk culture. Am J Hum Genet. 2008;82:57–72. doi: 10.1016/j.ajhg.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enattah NS, Trudeau A, Pimenoff V, et al. (27 co-authors) Evidence of still-ongoing convergence evolution of the lactase persistence T-13910 alleles in humans. Am J Hum Genet. 2007;81:615–625. doi: 10.1086/520705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Fay JC, Wu CI. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP-Phylogeny Inference Package. Cladistics. 1989;5:164–166. [Google Scholar]
- Felsenstein J. 2005 PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. [Google Scholar]
- Frazer KA, Ballinger DG, Cox DR, et al. (233 co-authors) A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrigan D, Hammer MF. Reconstructing human origins in the genomic era. Nat Rev Genet. 2006;7:669–680. doi: 10.1038/nrg1941. [DOI] [PubMed] [Google Scholar]
- Gillespie JH. Population genetics: a concise guide. Baltimore (MD): The Johns Hopkins University Press; 1998. [Google Scholar]
- Goebel T, Waters MR, O'Rourke DH. The late Pleistocene dispersal of modern humans in the Americas. Science. 2008;319:1497–1502. doi: 10.1126/science.1153569. [DOI] [PubMed] [Google Scholar]
- Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Gordon D, Desmarais C, Green P. Automated finishing with autofinish. Genome Res. 2001;11:614–625. doi: 10.1101/gr.171401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner SM, D'Agostino R, Saad MF, et al. (11 co-authors) Increased insulin resistance and insulin secretion in nondiabetic African-Americans and Hispanics compared with non-Hispanic whites. The Insulin Resistance Atherosclerosis Study. Diabetes. 1996;45:742–748. doi: 10.2337/diab.45.6.742. [DOI] [PubMed] [Google Scholar]
- Hallatschek O, Nelson DR. Gene surfing in expanding populations. Theor Popul Biol. 2008;73:158–170. doi: 10.1016/j.tpb.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Hamblin MT, Thompson EE, Di Rienzo A. Complex signatures of natural selection at the Duffy blood group locus. Am J Hum Genet. 2002;70:369–383. doi: 10.1086/338628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearn T, Spalluto C, Phillips VJ, Renforth GL, Copin N, Hanley NA, Wilson DI. Subcellular localization of ALMS1 supports involvement of centrosome and basal body dysfunction in the pathogenesis of obesity, insulin resistance, and type 2 diabetes. Diabetes. 2005;54:1581–1587. doi: 10.2337/diabetes.54.5.1581. [DOI] [PubMed] [Google Scholar]
- Hermisson J, Pennings PS. Soft sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics. 2005;169:2335–2352. doi: 10.1534/genetics.104.036947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F, Otto E. Cilia and centrosomes: a unifying pathogenic concept for cystic kidney disease? Nat Rev Genet. 2005;6:928–940. doi: 10.1038/nrg1727. [DOI] [PubMed] [Google Scholar]
- Hill WG. Linkage disequilibrium in finite populations. Theor Appl Genet. 1968;38:226–231. doi: 10.1007/BF01245622. [DOI] [PubMed] [Google Scholar]
- Hinds DA, Stuve LL, Nilsen GB, Halperin E, Eskin E, Ballinger DG, Frazer KA, Cox DR. Whole-genome patterns of common DNA variation in three human populations. Science. 2005;307:1072–1079. doi: 10.1126/science.1105436. [DOI] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Karafet T, Zegura SL, Vuturo-Brady J, et al. (14 co-authors) Y chromosome markers and Trans-Bering Strait dispersals. Am J Phys Anthropol. 1997;102:301–314. doi: 10.1002/(SICI)1096-8644(199703)102:3<301::AID-AJPA1>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Kelley JL, Madeoy J, Calhoun JC, Swanson W, Akey JM. Genomic signatures of positive selection in humans and the limits of outlier approaches. Genome Res. 2006;16:980–989. doi: 10.1101/gr.5157306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley JL, Swanson WJ. Positive selection in the human genome: from genome scans to biological significance. Annu Rev Genom Hum Genet. 2008;9:143–160. doi: 10.1146/annurev.genom.9.081307.164411. [DOI] [PubMed] [Google Scholar]
- Kelly JK. A test of neutrality based on interlocus associations. Genetics. 1997;146:1197–1206. doi: 10.1093/genetics/146.3.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura R, Fujimoto A, Tokunaga K, Ohashi J. A practical genome scan for population-specific strong selective sweeps that have reached fixation. PLoS ONE. 2007;2:e286. doi: 10.1371/journal.pone.0000286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfstein S, Currat M, Excoffier L. The fate of mutations surfing on the wave of a range expansion. Mol Biol Evol. 2006;23:482–490. doi: 10.1093/molbev/msj057. [DOI] [PubMed] [Google Scholar]
- Kreitman M, Di Rienzo A. Balancing claims for balancing selection. Trends Genet. 2004;20:300–304. doi: 10.1016/j.tig.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Leitch CC, Zaghloul NA, Davis EE, et al. (14 co-authors) Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet–Biedl syndrome. Nat Genet. 2008;40:443–448. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- Li G, Vega R, Nelms K, Gekakis N, Goodnow C, McNamara P, Wu H, Hong NA, Glynne R. A role for Alstrom syndrome protein, alms1, in kidney ciliogenesis and cellular quiescence. PLoS Genet. 2007;3:e8. doi: 10.1371/journal.pgen.0030008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JZ, Absher DM, Tang H, et al. (11 co-authors) Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- Magalon H, Patin E, Austerlitz F, Hegay T, Aldashev A, Quintana-Murci L, Heyer E. Population genetic diversity of the NAT2 gene supports a role of acetylation in human adaptation to farming in Central Asia. Eur J Hum Genet. 2008;16:243–251. doi: 10.1038/sj.ejhg.5201963. [DOI] [PubMed] [Google Scholar]
- Maynard-Smith JM, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res. 1974;23:23–35. [PubMed] [Google Scholar]
- McPeek MS, Strahs A. Assessment of linkage disequilibrium by the decay of haplotype sharing, with application to fine-scale genetic mapping. Am J Hum Genet. 1999;65:858–875. doi: 10.1086/302537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekel-Bobrov N, Gilbert SL, Evans PD, Vallender EJ, Anderson JR, Hudson RR, Tishkoff SA, Lahn BT. Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens. Science. 2005;309:1720–1722. doi: 10.1126/science.1116815. [DOI] [PubMed] [Google Scholar]
- Mulligan CJ, Hunley K, Cole S, Long JC. Population genetics, history, and health patterns in native americans. Annu Rev Genom Hum Genet. 2004;5:295–315. doi: 10.1146/annurev.genom.5.061903.175920. [DOI] [PubMed] [Google Scholar]
- Orr HA, Betancourt AJ. Haldane's sieve and adaptation from the standing genetic variation. Genetics. 2001;157:875–884. doi: 10.1093/genetics/157.2.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Minton JA, Weedon MN, Frayling TM, Ricketts C, Hitman GA, McCarthy MI, Hattersley AT, Walker M, Barrett TG. Common variations in the ALMS1 gene do not contribute to susceptibility to type 2 diabetes in a large white UK population. Diabetologia. 2006;49:1209–1213. doi: 10.1007/s00125-006-0227-2. [DOI] [PubMed] [Google Scholar]
- Przeworski M, Coop G, Wall JD. The signature of positive selection on standing genetic variation. Evolution. 2005;59:2312–2323. [PubMed] [Google Scholar]
- Reiner AP, Carlson CS, Ziv E, Iribarren C, Jaquish CE, Nickerson DA. Genetic ancestry, population sub-structure, and cardiovascular disease-related traits among African-American participants in the CARDIA Study. Hum Genet. 2007;121:565–575. doi: 10.1007/s00439-007-0350-2. [DOI] [PubMed] [Google Scholar]
- Sabeti PC, Reich DE, Higgins JM, et al. (17 co-authors) Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- Saxena R, Voight BF, Lyssenko V, et al. (66 co-authors) Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- Schaffner SF, Foo C, Gabriel S, Reich D, Daly MJ, Altshuler D. Calibrating a coalescent simulation of human genome sequence variation. Genome Res. 2005;15:1576–1583. doi: 10.1101/gr.3709305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet. 2005;76:449–462. doi: 10.1086/428594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- t Hart LM, Maassen JA, Dekker JM, Heine RJ. Lack of association between gene variants in the ALMS1 gene and Type 2 diabetes mellitus. Diabetologia. 2003;46:1023–1024. doi: 10.1007/s00125-003-1138-0. [DOI] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K, Thornton KR, Stoneking M. A new approach for using genome scans to detect recent positive selection in the human gnome. PLoS Biol. 2007;5:e171. doi: 10.1371/journal.pbio.0050171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templeton A. Out of Africa again and again. Nature. 2002;416:45–51. doi: 10.1038/416045a. [DOI] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PD, Kejariwal A, Guo N, Mi H, Campbell MJ, Muruganujan A, Lazareva-Ulitsky B. Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res. 2006;34:W645–W650. doi: 10.1093/nar/gkl229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson R, Pritchard JK, Shen P, Oefner PJ, Feldman MW. Recent common ancestry of human Y chromosomes: evidence from DNA sequence data. Proc Natl Acad Sci USA. 2000;97:7360–7365. doi: 10.1073/pnas.97.13.7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton KR, Jensen JD. Controlling the false-positive rate in multilocus genome scans for selection. Genetics. 2007;175:737–750. doi: 10.1534/genetics.106.064642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Reed FA, Ranciaro A, et al. (19 co-authors) Convergent adaptation of human lactase persistence in Africa and Europe. Nat Genet. 2007;39:31–40. doi: 10.1038/ng1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Varkonyi R, Cahinhinan N, et al. (17 co-authors) Haplotype diversity and linkage disequilibrium at human G6PD: recent origin of alleles that confer malarial resistance. Science. 2001;293:455–462. doi: 10.1126/science.1061573. [DOI] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen XPritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volodko NV, Starikovskaya EB, Mazunin IO, Eltsov NP, Naidenko PV, Wallace DG, Sukernik RI. Mitochondrial genome diversity in arctic Siberians, with particular reference to the evolutionary history of Beringia and Pleistocenic peopling of the Americas. Am J Hum Genet. 2008;82:1084–1100. doi: 10.1016/j.ajhg.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Kodama G, Baldi P, Moyzis RK. Global landscape of recent inferred Darwinian selection for Homo sapiens. Proc Natl Acad Sci USA. 2006;103:135–140. doi: 10.1073/pnas.0509691102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir BS. Genetic data analysis II. Sunderland: Sinauer Associates, Inc Publishers; 1996. [Google Scholar]
- Zhang C, Bailey DK, Awad T, et al. (12 co-authors) A whole genome long-range haplotype (WGLRH) test for detecting imprints of positive selection in human populations. Bioinformatics. 2006;22:2122–2128. doi: 10.1093/bioinformatics/btl365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.