

Abstract
The oncogene v-akt was isolated from a retrovirus that induced naturally occurring thymic lymphomas in AKR mice. We hypothesized that constitutive activation of Akt2 could serve as a first hit for the clonal expansion of malignant T-cells by promoting cell survival and genomic instability, leading to chromosome alterations. Furthermore, genes that cooperate with Akt2 to promote malignant transformation may reside at translocation/inversion junctions found in spontaneous thymic lymphomas from transgenic mice expressing constitutively active Akt2 specifically in T cells. Cytogenetic analysis revealed that thymic tumors from multiple founder lines exhibited either of two recurrent chromosomal rearrangements, inv(6)(A2B1) or t(14;15)(C2;D1). Fluorescence in situ hybridization, array-CGH, and PCR analysis was used to delineate the inv(6) and t(14;15) breakpoints. Both rearrangements involved T-cell receptor loci. The inv(6) results in robust up regulation of the homeobox/transcription factor gene Dlx5 due to its relocation near the Tcrb enhancer. The t(14;15) places the Tcra enhancer in the vicinity of the Myc proto-oncogene, resulting in up regulated Myc expression. These findings suggest that activation of the Akt pathway can act as the initial hit to promote cell survival and genomic instability, while the acquisition of T-cell-specific overexpression of Dlx5 or Myc leads to lymphomagenesis.
INTRODUCTION
The first evidence incriminating Akt in oncogenesis was provided by studies of acute transforming viruses. A retrovirus, isolated from an AKR mouse T-cell lymphoma (Staal et al., 1977), was found to harbor transduced sequences of cellular origin (Staal, 1987). The predicted oncoprotein contained viral Gag sequences fused to a cellularly-derived protein related to protein kinase C. The oncogenic potential of v-akt was found to arise from the creation of a myristylation signal at the amino-terminus, with consequent constitutive kinase activity (Ahmed et al., 1993).
Akt includes a family of related kinases, designated Akt1, Akt2 and Akt3 (Bellacosa et al., 2005). The Akt kinases are downstream targets of receptor tyrosine kinases that signal through phosphatidylinositol 3-kinase (PI3K) (Brazil et al., 2004; Bellacosa et al., 2005; Manning and Cantley, 2007). Processes regulated by Akt include cell proliferation and survival, genomic instability, cell size and response to nutrient availability, intermediary metabolism, angiogenesis and tissue invasion, each considered cancer hallmarks (Bellacosa et al., 2005).
The development of transgenic mouse models in which Akt is selectively overexpressed in various tissues has permitted investigators to genetically define its role in vivo. To achieve constitutive activation of Akt, myristylated forms of the Akt kinases (MyrAkt) have been used. Such mutants bypass the need for activation of phosphoinositides 3,4,5-trisphosphate (PIP3) and PIP2 generated by PI3K, and thus cannot be inhibited by Pten, a tumor suppressor that acts as a negative regulator of the PI3K pathway (Cantley and Neel, 1999; Di Cristofano and Pandolfi, 2000). Transgenic mice in which the Lck promoter is used to drive expression of MyrAkt (Lck-MyrAkt mice) in early thymocyte development are unique among Akt transgenic models in that they develop tumors spontaneously. The Lck promoter directs initial expression in T cells just prior to rearrangement of the T-cell receptor (TCR) loci (Perlmutter et al., 1993). Transgenic founder lines expressing high levels of MyrAkt1 developed aggressive thymic lymphomas within 10-20 weeks (Malstrom et al., 2001; Rathmell et al., 2003). T lymphocytes from these mice show increased cell size and proliferation, as well as resistance to apoptosis. Similarly, a preliminary study of mice expressing MyrAkt2, driven by the Lck promoter, revealed that at least 70% of animals developed lymphomas (Mende et al., 2001). The latency for tumor development and the fact that not all T lymphocytes from Lck-MyrAkt mice undergo transformation suggest that additional genetic and/or epigenetic events are required for full transformation.
Recurrent, clonal chromosomal translocations and inversions are common features of lymphoid tumors in humans and mice (Tycko and Sklar, 1990; Rabbitts, 1994; Davila et al., 2001; Rabbitts, 2001). Characterization of such rearrangements has led to the identification of genes critical to the origin of these tumors (Rabbitts, 1997; Greaves, 2004). In lymphoid malignancies, such rearrangements classically involve juxtaposition of a proto-oncogene, often encoding a transcription factor or signaling molecule (Look, 1997), with an immunoglobulin or TCR gene. The fact that immunoglobulin or TCR genes are so frequently involved suggests that mechanisms mediating normal V(D)J recombination play a role in generating these tumor-specific chromosome alterations, consequently resulting in gene fusion or gene activation (Sanchez-Garcia, 1997). In the T-cell lineage, cells not displaying functional TCR molecules are normally eliminated by apoptosis in the thymus (Petrie et al., 1995). Positioning of regulatory elements with new genes at the recombination junction may result in altered gene expression. Thus, in T- or B-lymphoid progenitor cells, proto-oncogenes that are silent or expressed at low levels in progenitor cells are activated when translocated under the control of potent enhancer elements within the regulatory region of a TCR or immunoglobulin gene (Look, 1997). In T-cell malignancies, activation of transcription factor genes, including the HOX11 homeobox gene, MYC, LMO1/2, TAL1/2 and LYL1, via juxtaposition to TCR genes are common (Look, 1997). Another example is the TCL1 locus, which is activated in mature T-cell leukemias by translocations and inversions that juxtapose it to regulatory elements of TCR genes (Pekarsky et al., 2001). Interestingly, functional analysis of TCL1 has revealed its involvement in an Akt prosurvival pathway by binding to Akt kinase and enhancing its enzymatic activity.
Deletion of Pten results in activation of the PI3K/Akt pathway and may promote genomic instability (Shen et al., 2007). Recently, Pten deletion in mouse hematopoietic stem cells was shown to lead to a myeloproliferative disorder, followed by acute T-lymphoblastic leukemia (TALL); transformation to the T-ALL phase was associated with acquisition of a recurring translocation involving chromosomes 14 and 15, t(14;15) (Guo et al., 2008). The rearrangement resulted in overexpression of Myc due to juxtaposition with Tcra regulatory elements. In this murine model, Pten inactivation serves as the first hit, which activates the PI3K/Akt pathway to promote cell survival and genomic instability, leading to additional alterations that may confer a proliferative advantage and subsequent clonal expansion. We hypothesized that constitutive activation of a downstream target of PI3K signaling, Akt2, could lead to genomic instability and clonal expansion of malignant T cells with recurring chromosome rearrangements. We speculated that genes that cooperate with Akt2 to promote malignant transformation might be identified at translocation/inversion junctions in T-cell lymphomas arising in Lck-Myr-Akt2 mice.
Here, we report consistent chromosome abnormalities in thymic lymphomas from Lck-MyrAkt2 mice. Independently-derived transgenic founder lines each developed tumors with recurring chromosome anomalies, including an inversion of chromosome 6, inv(6)(A2B1), that places the Tcrb enhancer near the Dlx5 gene, as well as a translocation t(14;15)(C2;D1) that juxtaposes the Tcra enhancer in chromosome 14 with the Myc locus in chromosome 15. The respective rearrangements result in up regulation of the transcription factors Dlx5 and Myc, respectively. The consistent involvement of these rearrangements in thymic tumors from various Lck-MyrAkt2 founder lines suggest that Dlx5 and Myc play a critical role in oncogenic transformation by cooperating with activated Akt2 to promote development of T-cell lymphomas.
MATERIAL AND METHODS
Transgenic Mice
Transgenic founder lines were generated using a previously described vector (Mende et al., 2001), which employs the Lck promoter to drive expression of MyrAkt2 specifically in early stages of thymocyte development. Mice were genotyped by PCR using primers for HA and Akt2, as reported (Tan et al., 2008).
Cell Lines and Reagents
Thymocytes were isolated and cultured from thymic lymphomas as described (Tan et al., 2008). Antibodies raised against Akt2, phospho-Akt, mTOR, phospho-mTOR and Myc were from Cell Signaling Technologies (Danvers, MA), and anti-β-actin antibodies were from Santa Cruz or Sigma (St Louis, MO).
Karyotypic Analysis and FISH
Preparation of metaphase spreads and G-banding were performed using standard procedures. Chromosome identification and karyotype designations were in accordance with U. Washington guidelines located at: http://www.pathology.washington.edu/research/cytopages/idiograms/mouse/.
BAC clones for FISH were purchased from Children's Hospital Oakland Research Institute. BAC DNA was extracted and labeled as described (Tan et al., 2008). Smaller FISH probes were labeled by nick translation using DNA polymerase I/Dnase I (Invitrogen, Carlsbad, CA). Hybridization of probes to metaphases and detection of FISH signals were performed using standard protocols.
Cloning and Sequence Analysis
A PCR-based strategy was used to clone genomic breakpoints of the inv(6), using primers near breakpoints in Tcrb and Dss1 (Tan et al., 2008). For the t(14;15), we used primers for exon 1 of Pvt1 (ACTTAGCATTCCCAGAGCC) and constant region of Tcra (GGTCTTCAGATCCAGGAGA). PCR products were cloned using TOPO-TA (Invitrogen).
Array-CGH Analysis
DNA copy analysis was performed according to Agilent's Oligonucleotide Array-based CGH protocol for Genomic DNA Analysis, Version 4.0. Genomic DNA (0.5 to 3 μg) from thymic tumors was digested with AluI and RsaI. Digested DNA was labeled using Agilent's Genomic DNA Labeling Kit PLUS. Test and reference DNA samples were labeled with either cyanine 5- or cyanine 3-dUTP, according to the manufacturer's recommendations. Labeled DNA products were purified using Microcon YM-30 filtration devices (Millipore, Billerica, MA). DNA yield and level of dye incorporation were measured using a ND-1000 Spectrophotometer. Appropriate cyanine 5- and cyanine 3-labeled DNA sample pairs were combined and mixed with mouse Cot-1 DNA, Agilent 10X Blocking Agent, and Agilent 2X Hybridization Buffer. Labeled target solution was hybridized to Agilent's 244K Mouse Genome CGH Microarray (G4415A) using SureHyb chambers. After hybridization, microarrays were washed and dried according to procedures described in Agilent's protocol. Microarray slides were scanned immediately using an Agilent microarray scanner. Data for individual features on microarray were extracted from the scan image, using Agilent's Feature Extraction Software. Output files were imported into Agilent's CGH Analytics for DNA copy number analysis.
Western Blotting
Cells were incubated in lysis buffer on ice for 15 min. Protein quantitation was determined by the Bradford method. Washed pellets were resuspended in 2X reducing buffer and loaded onto Novex SDS-PAGE gels (Invitrogen). For immunoblotting, samples (50 μg) were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes. Immunoblots were incubated with primary antibodies at 4°C overnight, followed by incubation with secondary antibody conjugated with horseradish peroxidase for 60 min. at room temperature.
RESULTS
Lck-MyrAkt2 Mice Develop Aggressive Thymic Lymphomas
Six Lck-MyrAkt2 transgenic founder lines were generated, three of which were followed to evaluate tumor incidence and survival. In these three founder lines, 95%-100% of animals developed thymic lymphomas, with median survivals of 75-140 days (Fig. 1). Immunoblot analysis demonstrated increased levels of total Akt2 and activation of the Akt pathway in thymic lymphomas relative to normal thymocytes from wild-type mice (Fig. 6).
Figure 1.
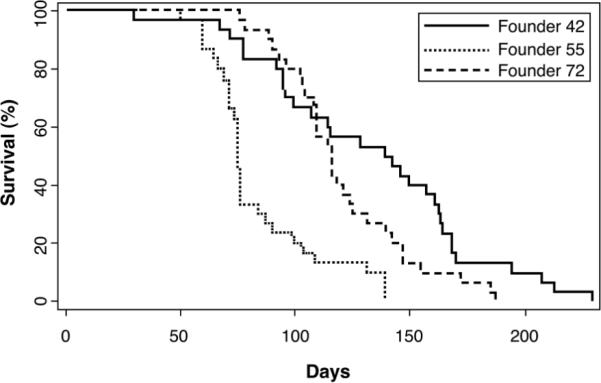
Survival curves of three Lck-Akt2 founder lines (30 animals/curve). Median survivals ranged from 75-140 days.
Figure 6.
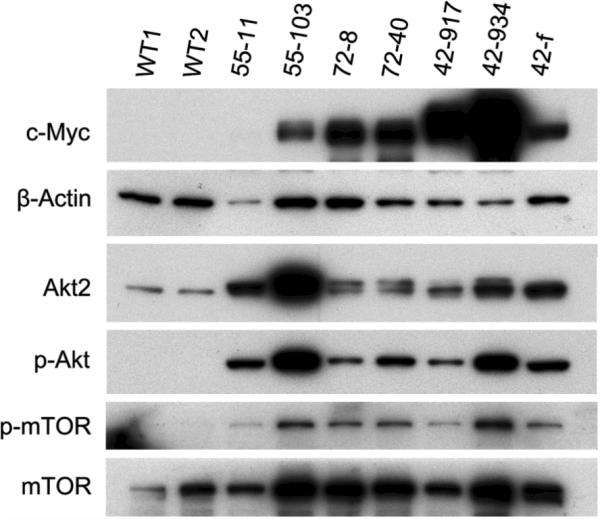
Western blot analysis demonstrating overexpression of Myc in thymic lymphomas compared to thymocytes from wild-type littermates (WT1, WT2). Figure also shows increased expression of phosphorylated Akt, mTor, and p70S6K in tumors, indicating activation of Akt/mTor pathway.
Thymic Lymphomas from Lck-MyrAkt2 Mice Develop Recurrent Chromosome Alterations
Karyotypic analysis was performed on two or more tumors from each of six transgenic founder lines, and findings are summarized in Table 1. Complete karyotypic details are shown in Supplemental Table 1. In founder line 42, all 15 tumors examined cytogenetically had an inv(6)(A2B1) (Fig. 2a), whereas non-malignant tissues from the same mice had normal karyotypes (Tan et al., 2008). In founder lines 72 and 54, a t(14;15)(C2;D1) (Fig. 2b) was observed in all seven lymphomas karyotyped. Tumors from founder lines 55, 62 and 79 had either an inv(6), a t(14;15), or other cytogenetic alterations.
TABLE 1.
Clonal chromosome alterations in thymic lymphomas from Lck-MyrAkt2 transgenic mice.
| Founder line | No. tumors karyotyped | Recurrent rearrangements | Other recurrent alterations |
|---|---|---|---|
| 42 | 15 | inv(6)(A2B1) [15]* | +14 [6], +15 [12], +16 [3] |
| 54 | 2 | t(14;15)(C2;D1) [2] | +10 [1], +14 [1], +der(14)t(14;15) [1], +15 [1], +der(15)t(14;15) [2] |
| 55 | 7 | inv(6)(A2B1) [1] t(14;15)(C2;D1) [4] | +10 [3], +14 [1], +der(14)t(14;15) [1], +15 [2], +der(15)t(14;15) [4], +16 [2] |
| 62 | 5 | t(14;15)(C2;D1) [2] | +10 [1], +14 [2], +der(14)t(14;15) [1], +15 [3], +der(15)t(14;15) [2], +16 [2] |
| 72 | 5 | t(14;15)(C2;D1) [5] | +10 [3], +14 [1], +der(14)t(14;15) [2], +der(15)t(14;15) [5] |
| 79 | 3 | inv(6)(A2B1) [1] t(14;15)(C2;D1) [1] | +10 [1], +14 [1], +15 [2], +der(15)t(14;15) [1] |
Numbers in brackets indicate number of tumors showing given chromosomal alteration.
Figure 2.

Recurrent chromosome abnormalities seen in thymic lymphomas from Lck-MyrAkt2 mice. (A) Left, Partial karyotype from tumor exhibiting inv(6)(A2B1). Bracket indicates inverted segment of inv(6) (arrow). Right, Idiograms with bracket indicating inverted segment in inv(6) and arrows denoting band locations of breakpoints beside normal homolog (N6). (B) Upper, Partial karyotype of tumor with t(14;15)(C2;D1) and extra copy of der(15). Arrows indicate abnormal chromosomes; brackets denote translocated segments of der(14) and der(15). Lower, Idiograms with brackets indicating translocated segments and arrows identifying band locations of breakpoints beside normal chromosomes 14 and 15, N14 and N15, respectively.
Altogether, 31 of 37 (84%) tumors examined karyotypically had either an inv(6) or a t(14;15). Of the remaining six tumors, two showed trisomy 16 only, two had both trisomy 10 and trisomy 15 only, and two had different complex karyotypes with multiple numerical and structural chromosome changes. Altogether, trisomy of chromosome 14 and/or 15 was observed in 21 of 37 tumors. Trisomy 14 and trisomy 15 have also been reported to be common changes in other studies of T-cell malignancies (Gaudet et al., 2003).
All 14 tumors with t(14;15) had one, or occasionally two, extra copies of the derivative chromosome 15, der(15)t(14;15), but only five of these tumors showed an extra copy of the der(14)t(14;15).
The positions of the inv(6) and t(14;15) breakpoints suggested the involvement of TCR loci, which undergo RAG-mediated recombination during T-cell development in the thymus at a time when the Lck transcriptional control region driving the MyrAkt2 transgene is active (Perlmutter et al., 1993; Mende et al., 2001).
Analysis of the inv(6)
To map the inv(6) breakpoints, fluorescence in situ hybridization (FISH) was performed on tumor metaphases using bacterial artificial chromosome (BAC) probes corresponding to known map locations. The distal breakpoint was mapped to Tcrb in band 6B1, specifically near the enhancer, and the proximal breakpoint was localized to a 100-kb region encompassing the Dss1 gene in band 6A2 (Tan et al., 2008). To more precisely map the proximal breakpoint, long range PCR-generated probes corresponding to 3' and 5' genomic sequences in Dss1 were next used as FISH probes; these experiments revealed that the proximal inv(6) breakpoint resides between exons 1 and 3 of Dss1 (Fig. 3).
Figure 3.

Analysis of inv(6). (A) Left, Schematic map of Dss1 gene showing corresponding genomic location of DNA probes used for FISH. Right, FISH analysis of inv(6) using PCR probes. (B) DNA sequences from 5 inv(6)-positive thymic lymphomas indicating proximal breakpoint junction of inv(6). Top and bottom sequences correspond to genomic DNA in Tcrb and Dss1, respectively, of normal chromosome 6. Nucleotides located at junctions (red box) between Dss1 and Tcrb are variable. Horizontal lines between Tcrb and Dss1 segments indicate joining without any intervening sequence between nucleotides connected by line.
The proximal inv(6) breakpoint junction was cloned, and sequence analysis revealed the presence of recombination signal sequences at the breakpoints in both Tcrb and Dss1, suggesting RAG-mediated V(D)J recombination. The proximal breakpoint sequences from five inv(6)-positive thymic lymphomas are shown in Fig. 3b.
Northern and RT-PCR analysis demonstrated that the inv(6) lymphomas exhibit a Dss1 exon1-Tcrb fusion transcript, but immunoblot analysis did not detect a Dss1-Tcrb fusion protein (Tan et al., 2008). However, Dlx5, which is placed within 300 kb of the Tcrb enhancer was consistently and robustly up regulated in inv(6) lymphomas compared to that observed in normal thymocytes or lymphomas without the inv(6) (Tan et al., 2008).
Analysis of t(14;15)
FISH analysis of metaphases from tumors with the t(14;15), using a BAC probe encompassing the Tcra enhancer, showed signals on both copies of the der(15)t(14;15) as well as on the normal chromosome 14 (Fig. 4, Left). FISH mapping of the chromosome 15 breakpoint, using a BAC encompassing the entire Myc and Pvt1 loci, showed signals on the native site of the normal chromosome 15 as well as a split signal hybridizing to both the der(14)t(14;15) and both copies of the der(15)t(14;15)(Fig. 4, Middle). Co-hybridization of probes encompassing Tcra (green signals) or Myc(red signals) is shown in Fig. 4(Right).
Figure 4.

FISH mapping of breakpoints in tumor with t(14;15)(C2;D1). Left, Hybridization with BAC probe encompassing Tcra enhancer, showing signals on normal chromosome 14 (N 14) as well as on two copies of der(15) (Ab 15). In abnormal chromosome 15, Tcra enhancer is placed near Myc locus. Middle, Hybridization of BAC clone encompassing both Myc and Pvt1 to normal chromosome 15; in addition split signals are found on both der(14) (Ab 14) and der(15) (Ab 15). Right, Co-hybridization of BAC clones encompassing Tcra enhancer (green signals) and Myc (red signals) in lymphoma with t(14;15). Note adjacent Tcra and Myc signals on der(15) (Ab 15). N 14, normal 14; N 15, normal 15.
Although balanced translocations cannot be detected by array-CGH analysis, the presence of an extra copy of der(15)t(14;15) provided a unique opportunity to identify the precise translocation breakpoints by this technology. Array-CGH analysis of a tumor with the t(14;15) and extra copy of the der(15)t(14;15) revealed gains of both distal chromosome 14 and proximal chromosome 15; and closer examination revealed that the gain in chromosome 14 starts from the proximal part of the Tcra locus, whereas the gain in chromosome 15 begins in the Pvt1 locus and results in increased Myc gene copy number (Fig. 5A). Notably, chromosome 14 also exhibited a prominent ~1-Mb deletion encompassing most of the Tcra locus. The fact that the deletion appears to be homozygous suggests that in addition to aberrant V(D)J rearrangement involving the structurally rearranged chromosome 14, a normal V(D)J rearrangement occurred on the other homolog.
Figure 5.

Array-CGH and cDNA sequence analysis of thymic tumors with t(14;15). (A) Array-CGH profiles of thymic tumor with t(14;15) and extra copy of the der(15), as in Fig. 2b. Presence of second copy of der(15) results in duplication of distal portion of chromosome 14 and proximal chromosome 15, permitting precise demarcation of chromosome breakpoints at 14C2 and 15D1. High-resolution views of chromosome 14 and 15 breakpoint regions reveal that increased DNA copy number begins at distal part of Tcra locus in chromosome 14 (left) and at Pvt1 locus in chromosome 15 (right), resulting in increased Myc gene copy number. Figure also shows prominent loss of most of Tcra locus near breakpoint in chromosome 14 (see text). (B) cDNA sequences from two t(14;15)-positive tumors indicating translocation breakpoint junction. Top and bottom sequences correspond to cDNA of constant region of Tcra and exon 1 of Pvt1, respectively, of normal chromosomes 14 and 15.
To more precisely define the breakpoints of the t(14;15), RT-PCR analysis, using various primers corresponding to coding sequences from Myc-Pvt1 and Tcra loci, was performed on cDNA from 10 lymphomas with this translocation. A 740-bp PCR product was detected using primers from exon 1 of Pvt1 and the constant region of Tcra. Sequence analysis of two tumors confirmed the presence of Pvt1 exon 1 sequences fused in frame to the constant region of Tcra (Fig. 5B).
As predicted based on the transposition of Tcra and Myc due to the t(14;15), immunoblot analysis demonstrated that expression of Myc was up regulated in lymphomas from founder line 72, which consistently show this chromosome rearrangement. In addition to up regulation of Dlx5 in connection with the inv(6), Myc was also overexpressed in lymphomas from founder line 42, which consistently exhibited this inversion (Fig. 6). Tumor 55-103, from founder line 55 mouse, had a t(14;15) and up regulation of Myc. In contrast, the only karyotypic abnormality seen in lymphoma 55-11 was +16, and this tumor did not show abundant expression of Myc.
DISCUSSION
In this report, we provide cytogenetic and molecular genetic evidence for stepwise events leading to the development of thymic lymphoma in a transgenic mouse model. We show that constitutive activation of Akt2, followed by recurrent inv(6) or t(14;15) that result in up regulation of the transcription factors Dlx5 or Myc, respectively, are responsible for malignant transformation of thymic cells. AKT2 is frequently activated in human cancer, including ovarian and pancreatic carcinomas (Yuan et al., 2000; Altomare et al., 2003). While there is a voluminous literature documenting the role of MYC in various cancers (Meyer and Penn, 2008), an understanding of the involvement of DLX5 in human cancer is only now beginning to emerge. To date, up regulation of DLX5 has been reported in a subset of human T-cell lymphomas (Tan et al., 2008) as well as many lung cancers (Kato et al., 2008).
Tumors from Lck-MyrAkt2 mice consistently showed elevated levels of active Akt (Fig. 6) due to expression of the MyrAkt2 transgene. Among its pleiotropic effects, activated Akt is a well-established survival factor (Bellacosa et al., 2005). Although T lymphocytes with aberrant TCR rearrangements should normally undergo apoptosis (Petrie et al., 1995), the concurrent expression of activated Akt is known to influence thymocyte selection and promote T-cell survival (Na et al., 2003). Early evidence supporting the idea that subversion of cell death pathways might lead to malignant transformation comes from the discovery of the BCL2 protooncogene at the site of the t(14;18) in human follicular lymphomas (Tsujimoto et al., 1984). While overexpression of the anti-apoptotic protein BCL2 does not appear to affect cell proliferation, it does promote accumulation of cells that otherwise would die; and some of these cells may acquire additional mutations that result in full malignant conversion (Look, 1997).
Akt kinases are frequently activated in primary human lymphomas and play a key role in lymphoma cell survival (Fillmore et al., 2005). Among members of the Akt family, AKT2 may have particular importance in mediating PI3K-dependent oncogenic effects (Arboleda et al., 2003). Our studies demonstrate that Akt2 activation predisposes to the development of thymic lymphomas in a mouse model. However, the invariable presence of clonal chromosome alterations in tumors from Lck-MyrAkt2 mice indicates that besides Akt2 activation, additional genetic changes are necessary for malignant transformation.
Illegitimate V(D)J recombination can place early thymocytes at risk for malignant transformation when DNA breaks are rejoined and cause the kind of chromosome rearrangements commonly seen in lymphomas (Look, 1997). The two recurrent chromosome rearrangements identified here, inv(6) and t(14;15), each involve somatic juxtaposition of a TCR locus and a transcription factor gene, either Dlx5 or Myc, respectively.
In T-cell malignancies, tumor-specific translocations and inversions classically juxtapose regulatory elements of a TCR gene with a proto-oncogene often encoding a transcription factor such as MYC or the homeobox gene HOX11 (Look, 1997). Thus, proto-oncogenes that are normally silent in T-lymphoid progenitor cells become activated when translocated under the control of these potent enhancer elements. The t(14;15) places Myc next to Tcra enhancer elements, leading to altered expression of Myc. This same t(14;15) was recently reported in the acute T-ALL phase of a Pten-null leukemic stem cell model in which this chromosomal rearrangement resulted in overexpression of Myc due to juxtaposition with Tcra regulatory elements (Guo et al., 2008). Moreover, a similar translocation involving chromosomes 8 and 14 is known to be associated with a subset of T-ALL patients (Erikson et al., 1986).
The Pvt1 locus, first identified in 1984 (Webb et al., 1984), resides ~40 kb distal to Myc on murine chromosome 15, and is a common site of reciprocal translocations to immunoglobulin loci. In Burkitt's lymphoma, immunoglobulin kappa or lambda light chain genes can be fused to PVT1, resulting in chimeric transcripts containing the first exon of PVT1 and the constant region of kappa or lambda (Cory et al., 1985; Shtivelman and Bishop, 1990). The Pvt1 locus is also a common site of retroviral integration in murine leukemia virus-induced T-cell lymphomas in mice (Graham et al., 1985), and Pvt1 is also disrupted as a result of a t(6;15) or a t(12;15) in mouse plasmacytomas (Huppi et al., 1990). Although Pvt1 encodes several alternative transcripts, there is no known protein product in normal mouse tissue or mouse plasmacytomas (Huppi et al., 1990), and the Burkitt's lymphoma translocations that reposition immunoglobulin sequences near the MYC locus result in overexpression of MYC (Erikson et al., 1983). The t(14;15) seen in the T lymphoblasts of the Pten-null leukemic stem cell model (Guo et al., 2008) was examined by FISH mapping, which revealed that the translocation breakpoints resided in the constant region/enhancer of Tcra and in a 680-kb fragment between the genes Pvt1 and Tsg101-ps. In our model, FISH analysis of the t(14;15) revealed that the breakpoint in chromosome 15 resides within a 30-kb region containing Pvt1, which was confirmed by array-CGH analysis; moreover, sequence analysis of the chimeric transcript showed a fusion between exon 1 of Pvt1 and the constant region of Tcra.
The inv(6) reported here juxtaposes the Dlx5 homeobox gene with Tcrb enhancer elements, resulting in up-regulated expression of this transcription factor. Although not extensively studied in lymphoid progenitor cells, deregulation of homeobox genes is increasingly recognized as contributing to hematological malignancies. Evidence in support of an oncogenic role of DLX5 comes from our earlier experiments demonstrating that overexpression of Dlx5 in mammalian cells leads to increased cell proliferation and colony formation, and that knockdown of Dlx5 in inv(6)-positive thymic lymphoma cells can result in decreased cell proliferation (Tan et al., 2008). More recently, DLX5 has been reported to be overexpressed in most human lung cancers (Kato et al., 2008). Overexpression of DLX5 was found to correlate with tumor size and poorer prognosis of non-small cell lung cancer patients and also was shown to be an independent prognostic factor. Moreover, as in our studies of inv(6)-positive murine tumor cells, treatment of human lung cancer cells with siRNAs against DLX5 effectively knocked down its expression and suppressed cell proliferation.
Collectively, data presented here as well as findings in the Pten-null leukemic stem cell model (Guo et al., 2008) suggest that activation of the PI3K/Akt pathway acts as the first hit to promote cell survival and genomic instability, while the acquisition of T-cell-specific overexpression of Myc, either directly via a t(14;15) or indirectly through an inv(6), leads to aggressive T-cell malignancies.
Recently, we have found that treatment of Lck-MyrAkt2 mice with an Akt inhibitor significantly inhibits tumor progression (D. Altomare et al., in preparation). Should inhibitors of DLX5 or MYC-Max prove to be clinically feasible, combination therapies with an Akt pathway inhibitor could have therapeutic synergy in certain human malignancies.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Binaifer Balsara for technical assistance with early FISH studies, Dr. Dietmar Kappes for expert scientific advice, Dr. Philip Tsichlis for providing one of the transgenic founder lines, and Dr. Jinfei Xu and Lili Zhang for technical assistance with the animal colony. The following Fox Chase Cancer Center shared facilities were used: Laboratory Animal, Genomics, Transgenic Mouse, Biochemistry & Biotechnology, Cell Culture, and Histopathology.
Supported by: NCI; Grant numbers: CA77429 and CA06927; an appropriation from the Commonwealth of Pennsylvania; and the Pennsylvania Department of Health. The Department of Health specifically disclaims responsibility for any analyses, interpretations or conclusions.
REFERENCES
- Ahmed NN, Franke TF, Bellacosa A, Datta K, Gonzalez-Portal ME, Taguchi T, Testa JR, Tsichlis PN. The proteins encoded by c-akt and v-akt differ in post-translational modification, subcellular localization and oncogenic potential. Oncogene. 1993;8:1957–1963. [PubMed] [Google Scholar]
- Altomare DA, Tanno S, De Rienzo A, Klein-Szanto A, Skele KL, Hoffman JP, Testa JR. Frequent activation of AKT2 kinase in human pancreatic carcinomas. J Cell Biochem. 2003;87:470–476. doi: 10.1002/jcb.10287. [DOI] [PubMed] [Google Scholar]
- Arboleda MJ, Lyons JF, Kabbinavar FF, Bray MR, Snow BE, Ayala R, Danino M, Karlan BY, Slamon DJ. Overexpression of AKT2/protein kinase Bbeta leads to up-regulation of beta1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer Res. 2003;63:196–206. [PubMed] [Google Scholar]
- Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci. 2004;29:233–242. doi: 10.1016/j.tibs.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory S, Graham M, E. W, Corcoran L, Adams JM. Variant (6;15) translocations in murine plasmacytomas involve a chromosome 15 locus at least 72 kb from the c-myc oncogene. EMBO J. 1985;4:675–681. doi: 10.1002/j.1460-2075.1985.tb03682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila M, Foster S, Kelsoe G, Yang K. A role for secondary V(D)J recombination in oncogenic chromosomal translocations? Adv Cancer Res. 2001;81:61–92. doi: 10.1016/s0065-230x(01)81002-2. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, Pandolfi PP. The multiple roles of PTEN in tumor suppression. Cell. 2000;100:387–390. doi: 10.1016/s0092-8674(00)80674-1. [DOI] [PubMed] [Google Scholar]
- Erikson J, ar-Rushdi A, Drwinga HL, Nowell PC, Croce CM. Transcriptional activation of the c-myc oncogene in Burkitt lymphoma. Proc Natl Acad Sci USA. 1983;80:820–824. doi: 10.1073/pnas.80.3.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erikson J, Finger L, Sun L, ar-Rushdi A, Nishikura K, Minowada J, Finan J, Emanuel BS, Nowell PC, Croce CM. Deregulation of c-myc by translocation of the alpha-locus of the T-cell receptor in T-cell leukemias. Science. 1986;232:884–886. doi: 10.1126/science.3486470. [DOI] [PubMed] [Google Scholar]
- Fillmore GC, Wang Q, Carey MJ, Kim CH, Elenitoba-Johnson KS, Lim MS. Expression of Akt (protein kinase B) and its isoforms in malignant lymphomas. Leuk Lymphoma. 2005;46:1765–1773. doi: 10.1080/10428190500159944. [DOI] [PubMed] [Google Scholar]
- Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489–492. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- Graham M, Adams JM, Cory S. Murine T lymphomas with retroviral inserts in the chromosomal 15 locus for plasmacytoma variant translocations. Nature. 1985;314:740–743. doi: 10.1038/314740a0. [DOI] [PubMed] [Google Scholar]
- Greaves MF. Biological models for leukaemia and lymphoma. IARC Sci Publ. 2004;157:351–372. [PubMed] [Google Scholar]
- Guo W, Lasky JL, Chang CJ, Mosessian S, Lewis X, Xiao Y, Yeh JE, Chen J, Iruela-Arispe ML, Varella-Garcia M, Wu H. Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature. 2008;453:529–533. doi: 10.1038/nature06933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppi K, Siwarski D, Skurla R, Klinman D, Mushinski JF. Pvt-1 transcripts are found in normal tissues and are altered by reciprocal(6;15) translocations in mouse plasmacytomas. Proc Natl Acad Sci USA. 1990;7:6964–6968. doi: 10.1073/pnas.87.18.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Sato N, Takano A, Miyamoto M, Nishimura H, Tsuchiya E, Kondo S, Nakamura Y, Daigo Y. Activation of placenta-specific transcription factor distal-less homeobox 5 predicts clinical outcome in primary lung cancer patients. Clin Cancer Res. 2008;14:2363–2370. doi: 10.1158/1078-0432.CCR-07-1523. [DOI] [PubMed] [Google Scholar]
- Look AT. Oncogenic transcription factors in the human acute leukemias. Science. 1997;278:1059–1064. doi: 10.1126/science.278.5340.1059. [DOI] [PubMed] [Google Scholar]
- Malstrom S, Tili E, Kappes D, Ceci JD, Tsichlis PN. Tumor induction by an Lck-MyrAkt transgene is delayed by mechanisms controlling the size of the thymus. Proc Natl Acad Sci USA. 2001;98:14967–14972. doi: 10.1073/pnas.231467698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mende I, Malstrom S, Tsichlis PN, Vogt PK, Aoki M. Oncogenic transformation induced by membrane-targeted Akt2 and Akt3. Oncogene. 2001;20:4419–4423. doi: 10.1038/sj.onc.1204486. [DOI] [PubMed] [Google Scholar]
- Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8:976–990. doi: 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- Na SY, Patra A, Scheuring Y, Marx A, Tolaini M, Kioussis D, Hemmings B, Hunig T, Bommhardt U. Constitutively active protein kinase B enhances Lck and Erk activities and influences thymocyte selection and activation. J Immunol. 2003;171:1285–1296. doi: 10.4049/jimmunol.171.3.1285. [DOI] [PubMed] [Google Scholar]
- Pekarsky Y, Hallas C, Croce CM. Molecular basis of mature T-cell leukemia. JAMA. 2001;286:2308–2314. doi: 10.1001/jama.286.18.2308. [DOI] [PubMed] [Google Scholar]
- Perlmutter RM, Levin SD, Appleby MW, Anderson SJ, Alberola-Ila J. Regulation of lymphocyte function by protein phosphorylation. Ann Rev Immunol. 1993;11:451–499. doi: 10.1146/annurev.iy.11.040193.002315. [DOI] [PubMed] [Google Scholar]
- Petrie HT, Livak F, Burtrum D, Mazel S. T cell receptor gene recombination patterns and mechanisms: cell death, rescue, and T cell production. J Exp Med. 1995;182:121–127. doi: 10.1084/jem.182.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabbitts TH. Chromosomal translocations in human cancer. Nature. 1994;372:143–149. doi: 10.1038/372143a0. [DOI] [PubMed] [Google Scholar]
- Rabbitts TH. Chromosomal breakpoints hit the spot. Nat Med. 1997;3:496–497. doi: 10.1038/nm0597-496. [DOI] [PubMed] [Google Scholar]
- Rabbitts TH. Chromosomal translocation master genes, mouse models and experimental therapeutics. Oncogene. 2001;20:5763–5777. doi: 10.1038/sj.onc.1204597. [DOI] [PubMed] [Google Scholar]
- Rathmell JC, Elstrom RL, Cinalli RM, Thompson CB. Activated Akt promotes increased resting T cell size, CD28-independent T cell growth, and development of autoimmunity and lymphoma. Eur J Immunol. 2003;33:2223–2232. doi: 10.1002/eji.200324048. [DOI] [PubMed] [Google Scholar]
- Sanchez-Garcia I. Consequences of chromosomal abnormalities in tumor development. Annu Rev Genet. 1997;31:429–453. doi: 10.1146/annurev.genet.31.1.429. [DOI] [PubMed] [Google Scholar]
- Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:25–28. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- Shtivelman E, Bishop JM. Effects of translocations on transcription from PVT. Mol Cell Biol. 1990;10:1835–1839. doi: 10.1128/mcb.10.4.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci USA. 1987;84:5034–5037. doi: 10.1073/pnas.84.14.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal SP, Hartley JW, Rowe WP. Isolation of transforming murine leukemia viruses from mice with a high incidence of spontaneous lymphoma. Proc Natl Acad Sci USA. 1977;74:3065–3067. doi: 10.1073/pnas.74.7.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Timakhov RA, Rao M, Altomare DA, Xu J, Liu Z, Gao Q, Jhanwar SC, Di Cristofano A, Wiest DL, Knepper JE, Testa JR. A novel recurrent chromosomal inversion implicates the homeobox gene Dlx5 in T-cell lymphomas from Lck-Akt2 transgenic mice. Cancer Res. 2008;68:1296–1302. doi: 10.1158/0008-5472.CAN-07-3218. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984;226:1097–1099. doi: 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
- Tycko B, Sklar J. Chromosomal translocations in lymphoid neoplasia: a reappraisal of the recombinase model. Cancer Cells. 1990;2:1–8. [PubMed] [Google Scholar]
- Webb E, Adams JM, Cory S. Variant (6; 15) translocation in a murine plasmacytoma occurs near an immunoglobulin kappa gene but far from the myc oncogene. Nature. 1984;312:777–779. doi: 10.1038/312777a0. [DOI] [PubMed] [Google Scholar]
- Yuan ZQ, Sun M, Feldman RI, Wang G, Ma X, Jiang C, Coppola D, Nicosia SV, Cheng JQ. Frequent activation of AKT2 and induction of apoptosis by inhibition of phosphoinositide-3-OH kinase/Akt pathway in human ovarian cancer. Oncogene. 2000;19:2324–2330. doi: 10.1038/sj.onc.1203598. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.