

Abstract
The metabolism of tamoxifen is being redefined in the light of several important pharmacological observations. Recent studies have identified 4-hydroxy N-desmethyl tamoxifen (endoxifen) as an important metabolite of tamoxifen necessary for antitumor actions. The metabolite is formed through the enzymatic product of CYP2D6 which also interacts with specific selective serotonin reuptake inhibitors (SSRIs) used to prevent the hot flashes observed in up to 45% of patients taking tamoxifen. Additionally, the finding that enzyme variants of CYP2D6 do not promote the metabolism of tamoxifen to endoxifen means that significant numbers of women might not receive optimal benefit from tamoxifen treatment. Clearly these are particularly important issues not only for breast cancer treatment but also for selecting premenopausal women, at high risk for breast cancer, as candidates for chemoprevention using tamoxifen.
Keywords: selective serotonin reuptake inhibitors, raloxifene, selective estrogen receptor modulators, ospemifene, arzoxifene
Introduction
The aim of the body’s biotransformation mechanisms is to prevent potentially toxic xenobiotic substances that include drugs, from damaging the body. That being the case, an orally active medicine must overcome numerous challenges to reach a target organ and produce the appropriate pharmacological effect at a receptor system. There is not one but several stages of biotransformation of a lipophilic drug such as tamoxifen that are designed to enhance the hydrophilic nature of the chemical so it can be rapidly eliminated. The stages of biotransformation are called phases I, II and III.
Phase I metabolism enhances the water solubility of a lipophilic chemical by hydroxylating an aromatic compound to become a phenol or hydrolyzing an esterified compound. These reactions are conducted by the family of cytochrome P450 enzymes referred to as CYP’s. Phase II metabolism further increases the water solubility of the Phase I product by attaching highly water soluble entities. In the case of selective estrogen receptor modulators (SERMs) sugars (glucuronic acid) and salts (sulfates) are the most important conjugation products. In contrast, the phase III system are efflux pump molecules (also known as p-glycoproteins and multi-drug resistance transports protein) that exclude unmetabolized drugs from the epithelial cells of the intestinal tract immediately upon absorption.
In general terms, the ingested SERM must survive “first pass” metabolism from the intestine to the liver to have any chance of reaching target organs around the body. The general principles are illustrated in Figure 1 where the SERM is biotransformed by CYPs in the intestinal wall and Phase II metabolism occurs via intestinal bacteria. A fraction of the administered dose is then absorbed into the hepatic portal vein and further biotransformed by phase I CYPs and/or glucuronidated or sulfated in phase II metabolism in the liver. By way of example, only 2% of the administered raloxifene survives and is bioavailable for systemic distribution[1].
Figure 1.

The stylized representation of the absorption of two selective estrogen receptor modulators (SERMS) tamoxifen (TAM) or raloxifene (RAL) into the circulation as bioactive molecules. The polyphenolic SERM raloxifene must transverse phase II and phase III obstacles in the gut and the liver to get into the general circulation. This results in very little of the ingested drug being bioavailable at target sites. In contrast, tamoxifen is extremely lipophilic and 98% protein bound to serum albumin. This extends the duration of action of tamoxifen because phase II metabolism to phenolic compounds is retarded.
Tamoxifen, the first SERM
The nonsteroidal antiestrogen tamoxifen (ICI 46,474 Nolvadex®) is a pioneering medicine[2] used to treat all stages of breast cancer in more than 120 countries throughout the world. The compound ICI 46,474 was discovered in the Fertility Control Program at Imperial Chemical Industries (ICI Pharmaceuticals Division, now AstraZeneca) in Alderley Park, Cheshire, England in the early 1960’s [3–5]. The drug was found to be an extremely potent postcoital contraceptive in the rat [4, 5]. Unfortunately, ICI 46,474 did not exhibit antifertility properties in women, in fact, quite the opposite, it induced ovulation [6, 7]. As a result, the medicine was, at one time, marketed in the United Kingdom for the induction of ovulation in subfertile women with a functional hypothalamo-pituitary-ovarian axis.
There is a known link between estrogen and the initiation and growth of some breast cancers[8] so the nonsteroidal antiestrogen ICI 46,474 was tested as a potential treatment for advanced breast cancer in postmenopausal women. The antiestrogen produced response rates of 25–35% in unselected patients comparable to diethylstilbestrol and high dose androgen therapy, the standard endocrine therapies at the time [9, 10]. However, fewer side effects were noted with tamoxifen [9, 10]. As a result, the drug was approved as a palliative option for the hormonal treatment of breast cancer in the UK in 1973. There the story may have ended had not tamoxifen been reinvented as the first targeted therapy for breast cancer[2].
The seminal observations by Elwood Jensen that estrogen action is mediated by the estrogen receptor (ER)[11, 12] in its target tissues (uterus, vagina pituitary and breast tumors) opened the door to targeting tamoxifen to select patients with the ER in their metastatic tumor[13, 14]. However, a strategic plan was developing to use tamoxifen in a broader range of patient populations. Laboratory studies conducted in the 1970’s showed that tamoxifen blocked estrogen binding to the ER [15–17], should be used as a long-term adjuvant therapy to suppress tumor recurrence [18–20] and the drug also had potential as a chemopreventive agent [21, 22].
Clinical studies subsequently confirmed that long-term adjuvant tamoxifen therapy, targeted to the patients with ER positive breast cancers, significantly decreased the death rate from the disease [23] and contributes to the current decline in death from breast cancer nationally [24]. Overall, the strategy of targeted long-term “antiestrogenic” [25] treatment for breast cancer has presaged the current fashion of targeting anticancer agents to other organ sites in the body.
Despite the fact that aromatase inhibitors show superiority over tamoxifen as adjuvant therapy in postmenopausal women[26–29], several issues have surfaced that have retained tamoxifen as a useful therapeutic agent worldwide. The medicine is extremely cheap compared to aromatase inhibitors so tamoxifen remains an essential anticancer agent in undeveloped countries or in countries with under-funded managed healthcare systems. Furthermore, tamoxifen is the only appropriate antiestrogenic therapy for premenopausal women whether they are being treated for breast cancer or whether chemoprevention is being considered[30]. For these reasons, new knowledge that can enhance the appropriate use of an established drug is of value to improve healthcare.
There are current initiatives to translate emerging knowledge on genetic variations in drug metabolism to target patient populations.[31] It is reasoned that by applying pharmacogenomic tests to specific patient populations, there will be fewer surprises with side effects, drug interactions, and a higher probability of increasing therapeutic effectiveness in the treatment or prevention of disease. The promise of practical progress is exemplified in this article using tamoxifen as the model drug.
Tamoxifen is a prodrug and can be metabolically activated to 4-hydroxytamoxifen[32–34] or alternatively can be metabolically routed via N-desmethyltamoxifen to 4-hydroxy-N-desmethyltamoxifen [35, 36] (Figure 2). The hydroxy metabolites of tamoxifen have a high binding affinity for the ER[32, 37]. The finding that the enzyme produced by CYP2D6 activates tamoxifen to hydroxylated metabolites 4-hydroxytamoxifen and endoxifen[38] has implications for cancer therapeutics. Women with enzyme variants that cannot make endoxifen may not have as successful an outcome with tamoxifen therapy. Alternatively, women who have a normal enzyme may make high levels of the potent antiestrogen endoxifen and experience hot flashes. As a result, these women may take selective serotonin reuptake inhibitors (SSRIs) to ameliorate hot flashes but there are potential pharmacological consequences to this strategy. Some of the SSRIs are metabolitically altered by the CYP2D6 enzyme product[39]. It is therefore possible to envision a drug interaction whereby SSRIs block the metabolic activation of tamoxifen.
Figure 2.
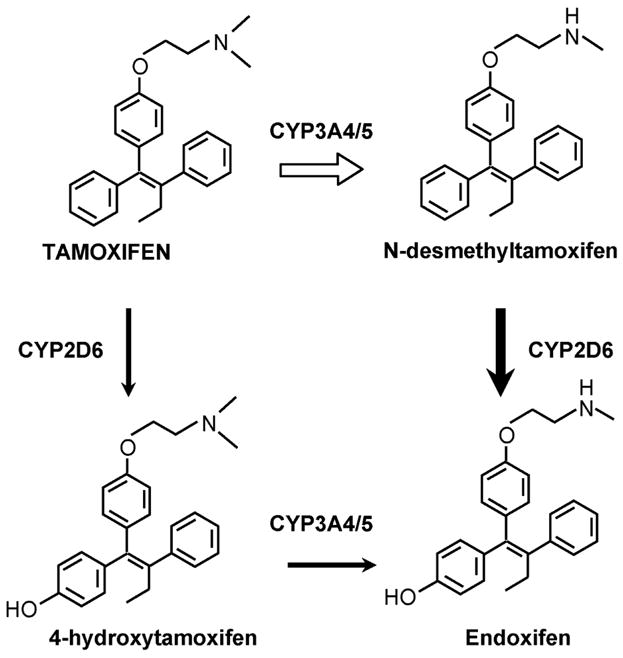
The metabolic activation of tamoxifen to phenolic metabolites that have a high binding activity for the human estrogen receptor. Both 4-hydroxytamoxifen and endoxifen are potent antiestrogens in vitro.
This article will describe the scientific twists and turns that tamoxifen and its metabolites have taken over the past 30 years. The story is naturally dependent on the fashions in therapeutic research at the time. What seems obvious to us as a successful research strategy today, with millions of women taking tamoxifen, was not so 30 years ago at the beginning when the clinical community and pharmaceutical industry did not see “antihormones” as a priority at all for drug development[25]. In 1972, tamoxifen was declared an orphan drug with no prospects[2].
Basic mechanisms of tamoxifen metabolism
The original survey of the putative metabolites of tamoxifen was conducted in the laboratories of ICI Pharmaceuticals Division and published in 1973 [40]. A number of hydroxylated metabolites were noted (Figure 3) following the administration of 14C labeled tamoxifen to various species (rat, mouse, monkey, and dog). The major route of excretion of radioactivity was in the feces. The rat and dog were used to show that up to 53% of the radioactivity derived from tamoxifen was excreted via the bile and up to 69% of this was reabsorbed via a enterohepatic recirculation until eventual elimination occurs[40]. The hydroxylated metabolites are excreted as glucuronides. However, no information about their biological activity was available until the finding that 4-hydroxytamoxifen had a binding affinity for the ER equivalent to 17β estradiol [32]. Similarly, 3,4 dihydroxytamoxifen (Figure 3) bound to the human ER but interestingly enough, 3,4 dihydroxytamoxifen was not significantly estrogen-like in the rodent uterus despite being antiestrogenic [32].
Figure 3.
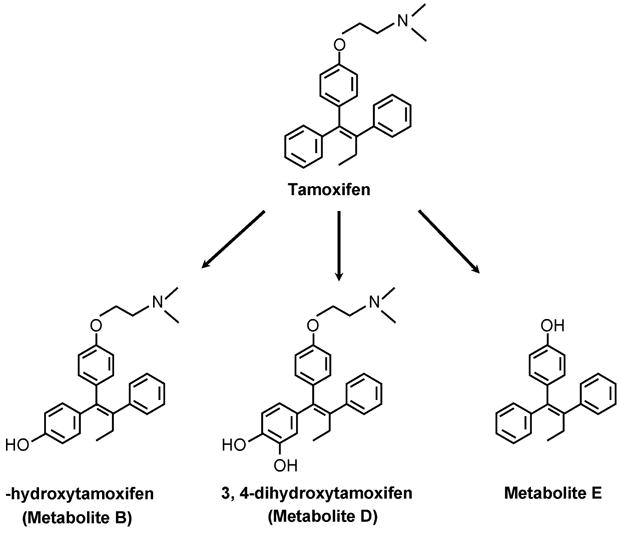
The original hydroxylated metabolites of tamoxifen noted in animals by Fromson et al.[40]
Additional studies on the metabolism of tamoxifen in four women [41] identified 4- hydroxytamoxifen as the primary metabolite using a thin layer chromatographic technique to identify 14C labeled metabolites. This assumption, coupled with the potent antiestrogenic actions of 4-hydroxytamoxifen [32] and the conclusion that it was an advantage, but not a requirement for tamoxifen to be metabolically activated [33, 42] seemed to confirm the idea that 4-hydroxytamoxifen was the active metabolite that bound in rat estrogen target tissues to block estrogen action[34]. However, the original analytical methods used to identify 4-hydroxytamoxifen as the major metabolite in humans were flawed[43] and subsequent studies identified N-desmethyltamoxifen (Figure 4) as the major metabolite circulating in human serum[44]. The metabolite was found to be further demethylated to N-desdimethyltamoxifen (Metabolite Z)[45] and then deaminated to Metabolite Y, a glycol derivative of tamoxifen[46, 47]. The metabolites (Figure 4) that are not hydroxylated at the 4 position of tamoxifen (equivalent to the 3 phenolic hydroxyl of estradiol) are all weak antiestrogens that would each contribute to the overall antitumor actions of tamoxifen at the ER based on their relative binding affinities for the ER and their actual concentrations locally.
Figure 4.

The serial metabolic demethylation and deamination of the antiestrogenic side chain of tamoxifen. Each of the metabolites is a weak antiestrogen with poor binding affinity for the estrogen receptor.
At the end of the 1980’s the identification of another metabolite tamoxifen 4-hydroxy N-desmethyltamoxifen in animals[48] and man [35, 36] was anticipated but viewed as obvious and uninteresting. The one exception that was of interest was Metabolite E (Figure 3) identified in the dog [40]. This phenolic metabolite without the dimethylaminoethyl side chain is a full estrogen[47, 49]. The dimethylaminoethoxy side chain of tamoxifen is necessary for antiestrogenic action[49].
It is not a simple task to study the actions of metabolites in vivo. Problems of pharmacokinetics, absorption and subsequent metabolism all conspire to confuse the interpretation of data. Studies in vitro using cell systems of estrogen target tissues were defined and refined in the early 1980’s to create an understanding of the actual structure function relationships of tamoxifen metabolites. Systems were developed to study the regulation of the prolactin gene in primary cultures of immature rat pituitary gland cells[42, 50] or cell replication in ER positive breast cancer cells[51–54]. Overall, these models were used to describe the importance of a phenolic hydroxyl to tether a triphenylethylenes appropriately in the ligand binding domain of the ER and to establish the appropriate positioning of an “antiestrogenic” side chain in the “antiestrogen region” of the ER[50] to modulate gene activation and growth[42, 50, 55–58]. These structure function studies, that created hypothetical models of the ligand-ER/complex, were rapidly advanced with the first reports of the x-ray crystallography of the estrogen, 4-hydroxytamoxifen[59] or raloxifene ER[60] complexes. The ligand-receptor protein interaction was subsequently interrogated by examining the interaction of the specific amino acid, asp 351 with the antiestrogenic side chain of the ligand[61]. A mutation was found as the dominant ER species in a tamoxifen-stimulated breast tumor grown in athymic mice[61, 62]. The structure function relationships studies, that modulated estrogen action at a transforming growth factor alpha gene target, demonstrated that the ligand shape would ultimately program the shape of the ER complex in a target tissue [30, 63–65]. This concept is at the heart of metabolite pharmacology and is required to switch on and switch off target sites around the body. The other piece of the mechanism of SERMs puzzle that was eventually solved was the need for another player to partner with the ER complex. Coactivators[66] can enhance the estrogen-like effects of compounds at a target site[67]. However, in the early 1990’s, the molecular and clinical use of this knowledge with the development and application of SERMs was in the future[68].
The urgent focus of translational research in the early 1990’s was to discover why tamoxifen was a complete carcinogen in rat liver[69, 70] and to determine whether there was a link between metabolism and the development of endometrial cancer noted in very small but significant numbers of postmenopausal women taking adjuvant tamoxifen[71, 72].
All interest in the metabolism of tamoxifen focused on the production of DNA adducts[73] that were responsible for rat liver carcinogenesis and, at the time, believed to be potentially responsible for carcinogenesis in humans[74]. Although many candidates were described[75–78], the metabolite found to be responsible for the initiation of rat liver carcinogenesis is α-hydroxytamoxifen[79–83] (Figure 5) Alpha-hydroxytamoxifen has been resolved into R- (+) and S- (−) enantiomers. Metabolism by rat liver microsomes gave equal amounts of the two forms, but in hepatocytes the R form gave 8x the level of DNA adducts as the S form. As both had the same chemical reactivity towards DNA, Osborne and coworkers[84] suggested that the R form was a better sulfotransferase substrate. This enzyme is believed to catalyze DNA adduct formation. Subsequently, Osborne and coworkers[85] conducted studies with alpha-hydroxy-N-desmethyltamoxifen; the R-(+) gave 10x the level of adducts in rat hepatocytes as the S-(−).
Figure 5.

The putative metabolite of tamoxifen, α hydroxytamoxifen that produces DNA adducts through covalent binding to deoxyguanosine.
There were reasonable concerns that the hepatocarcinogenicity of tamoxifen in rats would eventually translate to humans but fortunately this is now known to be untrue[86]. The demonstration of carcinogenesis in the rat liver appears to be related to poor DNA repair mechanisms in the inbred strains of rats. In contrast, it appears that the absence of liver carcinogenesis in women exposed to tamoxifen [87] is believed to result from the sophisticated mechanisms of DNA repair inherent in humans cells..
It is clear from this background about the early development of tamoxifen and the fact that tamoxifen was considered to be such a safe drug in comparison to other cytotoxic agents used in therapy during the 1970’s and 1980’s, that there was little enthusiasm for in-depth studies of tamoxifen metabolism. However, this perspective was to change in the 1990’s with the widespread use of tamoxifen as the gold standard for the treatment and prevention of breast cancer. Questions needed to be addressed: 1) what happens to tamoxifen in patients? and 2) can improvements be made to the molecule?
Clinical pharmacology
A number of analytical techniques are available to evaluate blood levels of tamoxifen and its metabolites once the drug is absorbed. The early method of thin layer chromatography, and the current method of high performance liquid chromatography (HPLC) both depend on the conversion of the triphenylethylenes to fluorescent phenanthrenes for their detection (Figure 6). The original description of the reaction [88] was successfully adapted [89] to identify tamoxifen, N-desmethytamoxifen and 4-hydroxytamoxifen in plasma samples.
Figure 6.

The UV activation of a triphenylethylenes to a florescent phenanthrene. This basic reaction is exploited in the detection of serum tamoxifen levels.
Subsequent improvements were made [90] but the method significantly underestimated phenolic metabolites (4-hydroxytamoxifen) and had no internal standardization. In contrast, a method of post column fluorescence activation [91] or preliminary purification from interfering substance using a Sep-Pack C18 cartridge (Waters Association, Milford MA) [92] with internal standardization considerably improved accuracy. The detection of tamoxifen metabolites in serum was further improved by Lien and coworkers [93] and recently by Lee and coworkers[94] who adapted the methods [95, 96] developed to perform “on line” extraction and post column cyclization. Using this methodology the limits of detection for 4-hydroxy tamoxifen and endoxifen are 0.5 and 0.25 ng/ml respectively [97]. Since there was such initial controversy about the identification of metabolites in patient serum, it is perhaps important to describe the validation of 4-hydroxy-desmethyltamoxifen as a metabolite of tamoxifen in patients. Tamoxifen metabolites were investigated in a 57 year old female patient receiving tamoxifen treatment[35]. Two major chromatographic peaks were identified in bile following treatment with β-glucuronidase. On major peak co-elevated with 4-hydroxytamoxifen but the second peak was proven to be 4-hydroxy-N-desmethyltamoxifen using a) co-elution with an authentic standard on reversed-phase chromatography and formation of fluorescent derivative by cyclization; b), the detection of a molecular ion (M+l)+ of 374 m/2 as determined by liquid chromatography-mass spectrometry; and c) a fragmatogram identical to that of the authentic standard, obtained by mass spectrometry. Subsequent refinement of the technology improved detection for identification of 4-hydroxy-N-desmethyltamoxifen in human serum, tissues[36] and rat tissues[93].
Studies confirm that tamoxifen is 98% bound to serum albumin which ultimate creates a long biological half life (plasma half life 7 days)[93]. A single oral dose of 10 mg tamoxifen (half the daily dose) produces peak serum levels of 20–30 ng of tamoxifen/ml within 3–6 hours but it must be stressed that patient variation is very large [98]. Nevertheless, continuous therapy with either 10mg bid [98] or 20 mg bid [99] produces steady state levels within 4 weeks. Blood levels of tamoxifen can average around 150 ng/ml for 10 mg tamoxifen bid and 300 ng/ml for 20 mg tamoxifen bid. A strategy of using loading doses [98, 100] to elevate blood levels rapidly has not produced any therapeutic benefit.
Overall, the results from the metabolic studies with tamoxifen during the 1970’s and 80’s did not help clinicians to use tamoxifen more effectively. The structures of metabolites were in fact used as leads to create new molecules for clinical development.
Metabolic Mimicry
The demonstration [32] that the class of compounds referred to as nonsteroidal antiestrogens were metabolically activated to compounds with high binding affinity for the ER created additional opportunities for the medicinal chemists within the pharmaceutical industry to develop new agents. This was particularly true once the nonsteroidal antiestrogens were recognized to be SERMs [101–103] and had applications not only for the treatment and prevention of breast cancer but also as potential agents to treat osteoporosis and coronary heart disease[104, 105]. The reader is referred to other recent review articles to obtain further details of new medicines under investigation [104, 105] but some current examples are worthy of note and will be mentioned briefly. Compounds of interest that have their structural origins as metabolites from nonsteroidal antiestrogens are summarized in Figure 7. Raloxifene is an agent that originally was destined to be a drug to treat breast cancer but it failed in that application [106]. It appears that the pharmacokinetics and bioavailability of raloxifene are a challenge. Only about 2% of administered raloxifene is bioavailable [1] but despite this, the drug is known to have a long biological half life of 27 hr. The reason for this disparity is that raloxifene is a polyphenolic drug that can be glucuronidated and sulfated by bacteria in the gut so the drug cannot be absorbed[107, 108]. This phase II metabolism in turn controls enterohepatic recirculation and ultimately impairs the drug from reaching and interacting with receptors in the target. This concern has been addressed with the development of the long-acting raloxifene derivative arzoxifene that is known to be superior to raloxifene as a chemopreventive in rat mammary carcinogenesis. [109]. One of the phenolic groups (Figure 7) is methylated to provide protection from Phase II metabolism. Nevertheless, arzoxifene has not performed well as a treatment for breast cancer [110, 111]; higher doses are less effective than lower doses. These data imply that effective absorption is impaired by phase III metabolism. That being said, the results of trials evaluating the effects of arzoxifene as a drug to treat osteoporosis, using lower doses, are eagerly awaited. Perhaps arzoxifene will be a better breast cancer preventive than a treatment.
Figure 7.

The formulae of SERMs that have been developed based on the knowledge of the metabolic activation of tamoxifen (and nafoxidine, see text) as well as the metabolism of the antiestrogen side chain of tamoxifen to a glycol.
Unfortunately, the bioavailability of phenolic drugs is also dependent on Phase II metabolism to inactive conjugates in the target tissue. 4-Hydroxytamoxifen,[32] is only sulfated by three of seven sulfotransferase isoforms whereas raloxifene is sulfated by all seven [112]. Maybe local phase II metabolism plays a role in neutralizing the antiestrogen action of raloxifene in the breast. Falany and coworkers [112] further report that SULT1E1, that sulfates raloxifene in the endometrium, is only expressed in the secretory phase. In contrast, 4-hydroxytamoxifen is sulfated at all stages of the uterine cycle.
Lasofoxifene is a diaryltetrahydronaphthalene derivative referred to as CP336156 [113] that has been reported to have high binding affinity for ER and have potent activity in preserving bone density in the rat[114, 115]. The structure of CP336156 is reminiscent of the putative antiestrogenic metabolite of nafoxidine[116] that failed to become a breast cancer drug because of unacceptable side effects[117]. There are two disasterometiric salts of the chemical shown in Figure 7. CP336156 is the l enantiomer that has 20 times the binding affinity for the ER as the d enantiomer. Studies demonstrate that the l enantiomer had twice the bioavailbility of the d enantiomer. The authors [113] ascribed the difference to enantioselective glucuronidation of the d isomer. An evaluation of CP336156 in the prevention and treatment of rat mammary tumors induced by N-nitroso-N-methylurea shows activity similar to that of tamoxifen[118].
Ospemifene or deaminohydroxytoremifene is related to metabolite Y formed by the deamination of tamoxifen[47]. Metabolite Y has a very low binding affinity for the ER[47, 119] and has weak antiestrogenic properties compared with tamoxifen. Ospermifene is a known metabolite of toremifene (4 chlorotoremifene) but unlike tamoxifen, there is little carcinogenic potential in animals[120]. It is possible that the large chlorine atom on the 4 position of toremifene and ospermifene reduces α hydroxylation to the ultimate carcinogen related to α hydroxy tamoxifen (Figure 6) D eaminohydroxytoremifene has very weak estrogenic and antiestrogenic properties in vivo[121] but demonstrates SERM activity in bone and lowers cholesterol. The compound is proposed to be used as a preventative for osteoporosis. Preliminary clinical data in healthy men and postmenopausal women demonstrate pharmacokinetics suitable for daily dosing between 25 and 200 mg[122]. Interestingly enough, unlike raloxifene, ospermifene has a strong estrogen-like action in the vagina but neither ospermifene nor raloxifene affect endometrial histology[123, 124]. Overall, the goal of developing a bone specific agent is reasonable, but the key to commercial success will be the prospective demonstration of the prevention of breast and endometrial cancer as beneficial side effects. This remains a possibility based on prevention studies completed in the laboratory[125, 126].
Tamoxifen Metabolism Today
A comprehensive evaluation of the sequential biotransformation of tamoxifen has been completed by Desta and colleagues[38]. They used human liver microsomes and experiments with specifically expressed human cytochrome P450’s to identify the prominent enzymes involved in Phase I metabolism. Their results are summarized in Figure 2 with the relevant CYP genes indicated for the metabolic transformations. The authors make a strong case that N-desmethyltamoxifen, the principal metabolite of tamoxifen that accumulates in the body, is converted to endoxifen by the enzymatic product of CYP2D6. The CYP2D6 product is also important to produce the potent primary metabolite 4-hydroxytamoxifen but the metabolite can also be formed by the enzymatic products: CYP2B6, CYP2C9, CY2C19 and CYP3A4.
The CYP2D6 phenotype is defined as the metabolic ratio (MR) by dividing the concentration of an unchanged probe drug, known to be metabolized by the CYP2D6 gene product, by the concentration of the relevant metabolite at a specific time. These measurements have resulted in the division of the CYP2D6 phenotype in four metabolic classes; poor metabolizers (PM), intermediate metabolizers (IM), extensive metabolizers (EM) and ultrarapid metabolizes (UM). Over 80 different single nucleotide polymorphisms have been identified but there are inconsistencies in the precise definitions of the ascribing a genotype to a phenotype[127, 128]. Bradford[128] and Raimondo and coworkers[129] have described the frequency of common alleles for CYP2D6. Pertinent to the current discussion of tamoxifen metabolism, the CYP2D6*4 allele[130] is estimated to have a frequency of 12–23% in Causasians, 1.2–7% in black Africans and 0–2.8% in Asians[127, 128]. A lower estimate of (<10%) of the PM phenotype is presented by Bernard and coworkers[131].
The molecular pharmacology of endoxifen has recently been reported [37, 132, 133]. Endoxifen and 4-hydroxytamoxifen were equally potent at inhibiting estrogen stimulated growth of ER positive breast cancer cells MCF-7, T47D and BT474. Both metabolites are significantly superior in vitro to tamoxifen the parent drug. Additionally, the estrogen-responsive genes pS2 and progesterone receptor were both blocked to an equivalent degree by endoxifen and 4-hydroxytamoxifen[132, 133]. Lim and co-workers[133] have extended the comparison of endoxifen and 4-hydroxytamoxifen in MCF-7 cells by comparing and contrasting global gene regulation using the Affymetrix U133A Gene Chip Array. There were 4062 total genes that were either up or down regulated by estradiol whereas, in the presence of estradiol, 4-hydroxytamoxifen or endoxifen affected 2444 and 2390 genes respectively. Overall, the authors[133] demonstrated good correlation between RTPCR and select genes from the microarray and concluded that the global effects of endoxifen and 4-hydroxytamoxifen were similar.
Stearns and coworkers[97] and Jin and coworkers[134] have confirmed and significantly extended Lien’s original identificatiaon of endoxifen and observation[35, 36] that there are usually higher circulating levels of endoxifen than 4-hydroxytamoxifen in patients receiving adjuvant tamoxifen therapy. However, Flockhart’s group[97] have advanced the pharmacogenomics and drug interactins surrounding tamoxifen therapy that should be a consideration in the antihormonal treatment of breast cancer.
The ubiquitous use of tamoxifen for the treatment of node negative women[135] during the 1990’s, the use of tamoxifen plus radiotherapy following lumpectomy for the treatment of ductal carcinoma in situ (DCIS)[136] as well as the option to use tamoxifen for chemoprevention in high risk pre and postmenopausal women[137] enhanced awareness of the menopausal side effects experienced by women when taking tamoxifen. Up to 45% of women with hot flashes grade them as severe[137] therefore there have been efforts to improve quality of life. Treatments with the SSRIs are popular [97, 138, 139] (Figure 8). The SSRIs are twice as effective as the “placebo” effect at reducing menopausal symptoms in randomized clinical trials[138–140], so there is naturally an increased usage of SSRIs with long-term tamoxifen treatment to maintain compliance. Unfortunately, the metabolism of tamoxifen to hydroxylated metabolites[141–143] and the metabolism of SSRIs[39, 144–147] both occur via the CYP2D6 gene product. Indeed Stearns and coworkers[97] showed that the SSRI inhibitor paroxetine reduced the levels of endoxifen during adjuvant tamoxifen therapy and endoxifen levels decrease by 64% in women with wild type CYP2D6 enzyme. Patients were examined who were taking venlafaxine, sertraline, and paroxetine and compared with those women who were homozygotes for the CYP2D6 *4/*4 inactive genotype. Patients with the wild type gene who took the most potent inhibitor paroxetine had serum levels of endoxifen equivalent to the patients with the aberrant CYP2D6 gene. In fact, the clinical data were consistent with the inhibition constants for the inhibition of CYP2D6 by paroxetine (potent), fluoxetine, sertraline, citalopram (intermediate) and venlafaxine (weak) which are 0.05, 0.17, 1.5, 7 and 33μmol/L respectively.
Figure 8.

The structures of selective serotonin reuptake inhibitors (SSRIs) that have low intermediate or high affinity for the CYP2D6 enzyme system. High affinity binders for CYP2D6 block the metabolic activation of tamoxifen to endoxifen (Figure 2).
The CYP2D6 gene product that is fully functional (wild type) is classified as the CYP2D6*1. A large number of alleles are associated with no enzyme activity or reduced activity. Conversely, high metabolizers can have multiple copies of the CYP2D6 allele[31]. A recent study by Borges[148] continues to expand our understanding of the detrimental effect of CYP2D6 variants plus concomitant administration of SSRIs on endoxifen levels. But, it is the clinical correlations with tumor responses and side effects that are starting to provide clues about the importance of pharmacogenomics for tamoxifen to be optimally effective as a breast cancer drug.
Clinical Correlations
The significance of genotyping on clinical outcomes of a tamoxifen trial have been addressed using paraffin-embedded tumor blocks from a North Central Center Treatment Group (NCCTG) trial NCCTG 89-30-52[149]. The postmenopausal women with ER positive tumors received 5 years of adjuvant tamoxifen therapy. The tumor blocks were used to determine CY2D6 (*4 and *6) and CYP3A5 (*3) and 17 buccal swabs were used to test the veracity of the tumor genotyping. The concordance rate for the buccal swabs was 100%. Overall, the CYP3A5*3 variant was not associated with any adverse clinical outcomes but the women with the CYP2D6*4/*4 genotype had a higher risk of disease relapse but a lower incidence of side effects such as hot flashes.[149] The implication is that tamoxifen must be converted to endoxifen, a more potent antiestrogen.
In a follow up study[150] using the same database established for trial NCCTG 89-30-52, patient records were screened to determine the extent of SSRI prescribing. The goal was to establish the combined effect of genotyping and SSRI inhibition of the CYP2D6 enzyme. Overall, the authors[150] concluded that a mutated CYP2D6 gene or the inadvertent use of SSRIs that inhibit the CYP2D6 enzyme product are independent predictors of breast cancer outcomes for postmenopausal women with breast cancer taking tamoxifen. In a recent complimentary study, Mortimer and coworkers[151] demonstrated that hot flashes were a strong predictor of positive outcomes for adjuvant tamoxifen treatment.
Although all of the current emphasis has been on the biological effects of tamoxifen in patients with the CYPD6*4 variant, studies of CYP3A5* 1 AND *3 1A1 *1 and 2 and UGT2B15 * and *2 have been undertaken and compared with carriers of CYP2D6*4. In contrast to the studies of Goetz and colleagues[149], patients who carry the SULT1A1*1, CYP2D7*4 and CYP3A5*3 alleles, and would be predicted to give rise to lower concentrations of metabolites with high affinity for the ER, might actually benefit from tamoxifen[152–155]. No differences were noted between genotypes CYP2D6, SULT1A1 or UGT 2B15 and tamoxifen treatment but Wegman and coworkers[155] claim that genetic variants of CYP3A5 may predict response to tamoxifen. Clearly, reasons for the different conclusions need to be advanced. The hypothesis that variants of metabolizing enzymes can affect patient outcomes for the treatment of breast cancer must now be addressed in large populations and with prospective studies.
Conclusions
Overall, the study of tamoxifen metabolism has provided important clues which guided medicinal chemists to synthesize and develop new medicines. The study of metabolites has also provided valuable insight into the mechanism of action of SERMs at their target the ER. However, it is the recent research on the value of genotyping CYPs in breast cancer patients to improve response rates to tamoxifen therapy that is showing important promise. Genotyping patients for CYP2D6 appears to be valuable to exclude the suboptimal use of tamoxifen in select individuals. Additionally, and perhaps more importantly, an effect of SSRIs on the blood levels of endoxifen has raised the possibility that the cheap and effective veteran tamoxifen could be targeted further to select populations of women to improve response rates. Avoiding SSRIs with a high affinity for CYP2D6 gene product could improve tamoxifen’s efficacy. Since tamoxifen is still the antihormonal treatment of choice for premenopausal patients and the only choice for breast cancer risk reduction in premenopausal women, then genotyping from buccal swabs appears to be a cheap and effective way of ensuring that tamoxifen is used to treat the appropriate woman.
It is necessary, however, to close on a note of caution. Very few patients have been studied to create definitive guidelines. That being said, the task of proving the value of these tantalizing clues and hypotheses is the responsibility of clinicians to organize prospective clinical trials or at least there must be investment in the further analysis of archival material from randomized trials. The value of committing resources to establish hypothesis as fact is clear. An important cheap medicine should potentially be given only to women who will benefit from it. Indeed, it may be the role of CYP2D6 in tamoxifen metabolism that is creating the small but significant advantage of aromatase inhibitors vs. tamoxifen in postmenopausal women.[26, 27]. Again, this can be tested as the tumor blocks and patient records could be reviewed to determine genotyping and whether SSRIs were used. It would be remarkable to discover that the pharmacology of tamoxifen is undermining activity rather than the current view that aromatase inhibitors were better medicines because they have, unlike the SERMs, no estrogen-like actions at the level of the tumor.
Acknowledgments
Dr. Jordan is supported by the Department of Defense Breast Program under award number BC050277 Center of Excellence (Views and opinions of, and endorsements by the author(s) do not reflect those of the US Army or the Department of Defense), SPORE in Breast Cancer CA 89018, R01 GM067156, FCCC Core Grant NIH P30 CA006927, the Avon Foundation and the Weg Fund of Fox Chase Cancer Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Snyder KR, Sparano N, Malinowski JM. Raloxifene hydrochloride. Am J Health Syst Pharm. 2000;57(18):1669–1675. quiz 1676–1678. [PubMed] [Google Scholar]
- 2.Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nature Reviews Drug Discovery. 2003;2:205–213. doi: 10.1038/nrd1031. [DOI] [PubMed] [Google Scholar]
- 3.Harper MJ, Walpole AL. Contrasting endocrine activities of cis and trans isomers in a series of substituted triphenylethylenes. Nature. 1966;212(57):87. doi: 10.1038/212087a0. [DOI] [PubMed] [Google Scholar]
- 4.Harper MJ, Walpole AL. Mode of action of I.C.I. 46,474 in preventing implantation in rats. J Endocrinol. 1967;37(1):83–92. doi: 10.1677/joe.0.0370083. [DOI] [PubMed] [Google Scholar]
- 5.Harper MJ, Walpole AL. A new derivative of triphenylethylene: effect on implantation and mode of action in rats. J Reprod Fertil. 1967;13:101–119. doi: 10.1530/jrf.0.0130101. [DOI] [PubMed] [Google Scholar]
- 6.Klopper A, Hall M. New synthetic agent for the induction of ovulation. Preliminary trial in women Br Med J. 1971;1:152–154. doi: 10.1136/bmj.1.5741.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williamson JG, Ellis JP. The induction of ovulation by tamoxifen. J Obstet Gynaec Br Commonw. 1973;80:844–847. doi: 10.1111/j.1471-0528.1973.tb11230.x. [DOI] [PubMed] [Google Scholar]
- 8.Kennedy BJ. Hormone therapy for advanced breast cancer. Cancer. 1965;18:1551–1557. doi: 10.1002/1097-0142(196512)18:12<1551::aid-cncr2820181206>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 9.Cole MP, Jones CT, Todd ID. A new anti-oestrogenic agent in late breast cancer. An early clinical appraisal of ICI 46474. Br J Cancer. 1971;25:270–275. doi: 10.1038/bjc.1971.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ward HW. Anti-oestrogen therapy for breast cancer: a trial of tamoxifen at two dose levels. Br Med J. 1973;1(844):13–4. doi: 10.1136/bmj.1.5844.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jensen EV, Jacobson HI. Basic guides to the mechanism of estrogen action. Recent Progr Hormone Res. 1962;18:387–414. [Google Scholar]
- 12.Jensen EV, Block GE, Smith S, Kyser K, DeSombre ER. Estrogen receptors and breast cancer response to adrenalectomy. Natl Cancer Inst Monogr. 1971;34:55–70. [PubMed] [Google Scholar]
- 13.Kiang DT, Kennedy BJ. Tamoxifen (antiestrogen) therapy in advanced breast cancer. Ann Intern Med. 1977;87(6):687–90. doi: 10.7326/0003-4819-87-6-687. [DOI] [PubMed] [Google Scholar]
- 14.Jensen EV, Jordan VC. The estrogen receptor: a model for molecular medicine. The Dorothy P. Landon AACR Prize for Translational Research. Clin Cancer Res. 2003;9:1980–1989. [PubMed] [Google Scholar]
- 15.Jordan VC, Koerner S. Tamoxifen (ICI 46,474) and the human carcinoma 8S oestrogen receptor. Eur J Cancer. 1975;11:205–206. doi: 10.1016/0014-2964(75)90119-x. [DOI] [PubMed] [Google Scholar]
- 16.Jordan VC, Dowse LJ. Tamoxifen as an anti-tumour agent: effect on oestrogen binding. J Endocrinol. 1976;68(02):297–303. doi: 10.1677/joe.0.0680297. [DOI] [PubMed] [Google Scholar]
- 17.Jordan VC, Jaspan T. Tamoxifen as an antitumour agent: oestrogen binding as a predictive test for tumour response. J Endocrinol. 1976;68:453–460. doi: 10.1677/joe.0.0680453. [DOI] [PubMed] [Google Scholar]
- 18.Jordan VC. Use of the DMBA-induced rat mammary carcinoma system for the evaluation of tamoxifen as a potential adjuvant therapy. Reviews on Endocrine-Related Cancer. 1978;(October Supplement):49–55. [Google Scholar]
- 19.Jordan VC, Dix CJ, Allen KE. The effectiveness of long term tamoxifen treatment in a laboratory model for adjuvant hormone therapy of breast cancer. Adjuvant Therapy of Cancer. 1979;2:19–26. [Google Scholar]
- 20.Jordan VC, Allen KE. Evaluation of the antitumour activity of the non-steroidal antioestrogen monohydroxytamoxifen in the DMBA-induced rat mammary carcinoma model. Eur J Cancer. 1980;16:239–251. doi: 10.1016/0014-2964(80)90156-5. [DOI] [PubMed] [Google Scholar]
- 21.Jordan VC. Antitumour activity of the antioestrogen ICI 46,474 (tamoxifen) in the dimethylbenzanthracene (DMBA)-induced rat mammary carcinoma model. J Steroid Biochem. 1974;5:354. [Google Scholar]
- 22.Jordan VC. Effect of tamoxifen (ICI 46,474) on initiation and growth of DMBA- induced rat mammary carcinoma. Eur J Cancer. 1976;12:419–424. doi: 10.1016/0014-2964(76)90030-x. [DOI] [PubMed] [Google Scholar]
- 23.EBCTCG. Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet. 1998;354:1451–1467. [PubMed] [Google Scholar]
- 24.EBCTCG. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 25.Jordan VC, Brodie AMH. Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer. Steroids. 2007;72:7–25. doi: 10.1016/j.steroids.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Howell A, Cuzick J, Baum M, Buzdar A, Dowsett M, Forbes JF, et al. Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years’ adjuvant treatment for breast cancer. Lancet. 2005;365:60–62. doi: 10.1016/S0140-6736(04)17666-6. [DOI] [PubMed] [Google Scholar]
- 27.Thurlimann B, Keshaviah A, Coates AS, Mouridsen H, Mauriac L, Forbes JF, et al. A comparison of letrozole and tamoxifen in postmenopausal women with early breast cancer. N Engl J Med. 2005;353:2747–57. doi: 10.1056/NEJMoa052258. [DOI] [PubMed] [Google Scholar]
- 28.Goss PE, Ingle JN, Marino S, Robert NJ, Muss HB, Piccart MJ, et al. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N Engl J Med. 2003;349:1–10. doi: 10.1056/NEJMoa032312. [DOI] [PubMed] [Google Scholar]
- 29.Coombes RC, Hall E, Gibson LJ, Paridaens R, Jassem J, Delozier T, et al. A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer. N Engl J Med. 2004;350(11):1081–1092. doi: 10.1056/NEJMoa040331. [DOI] [PubMed] [Google Scholar]
- 30.Jordan VC. Chemoprevention of breast cancer with selective oestrogen-receptor modulators. Nature Reviews Cancer. 2007;7:46–53. doi: 10.1038/nrc2048. [DOI] [PubMed] [Google Scholar]
- 31.Andersson T, Flockhart DA, Goldstein DB, Huang SM, Kroetz DL, Milos PM, et al. Drug-metabolizing enzymes: evidence for clinical utility of pharmacogenomic tests. Clin Pharmacol Ther. 2005;78:559–581. doi: 10.1016/j.clpt.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 32.Jordan VC, Collins MM, Rowsby L, Prestwich G. A monohydroxylated metabolite of tamoxifen with potent antioestrogenic activity. J Endocrinol. 1977;75(2):305–316. doi: 10.1677/joe.0.0750305. [DOI] [PubMed] [Google Scholar]
- 33.Allen KE, Clark ER, Jordan VC. Evidence for the metabolic activation of non-steroidal antioestrogens: a study of structure-activity relationships. Br J Pharmacol. 1980;71(1):83–91. doi: 10.1111/j.1476-5381.1980.tb10912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Borgna JL, Rochefort H. Hydroxylated metabolites of tamoxifen are formed in vivo and bound to estrogen receptor in target tissues. J Biol Chem. 1981;256(2):859–68. [PubMed] [Google Scholar]
- 35.Lien EA, Solheim E, Kvinnsland S, Ueland PM. Identification of 4-hydroxy-N-desmethyltamoxifen as a metabolite of tamoxifen in human bile. Cancer Res. 1988;48(8):2304–8. [PubMed] [Google Scholar]
- 36.Lien EA, Solheim E, Lea OA, Lundgren S, Kvinnsland S, Ueland PM. Distribution of 4-hydroxy-N-desmethyltamoxifen and other tamoxifen metabolites in human biological fluids during tamoxifen treatment. Cancer Res. 1989;49(8):2175–83. [PubMed] [Google Scholar]
- 37.Johnson MD, Zuo H, Lee KH, Trebley JP, Rae JM, Weatherman RV, et al. Pharmacological characterization of 4-hydroxy-N-desmethyl tamoxifen, a novel active metabolite of tamoxifen. Breast Cancer Res Treat. 2004;85(2):151–159. doi: 10.1023/B:BREA.0000025406.31193.e8. [DOI] [PubMed] [Google Scholar]
- 38.Desta Z, Ward BA, Soukhova NV, Flockhart DA. Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: prominent roles for CYP3A and CYP2D6. J Pharmacol Exp Ther. 2004;310(3):1062–75. doi: 10.1124/jpet.104.065607. [DOI] [PubMed] [Google Scholar]
- 39.Crewe HK, Lennard MS, Tucker GT, Woods FR, Haddock RE. The effect of selective serotonin re-uptake inhibitors on cytochrome P4502D6 (CYP2D6) activity in human liver microsomes. Br J Clin Pharmacol. 1992;34(3):262–5. doi: 10.1111/j.1365-2125.1992.tb04134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fromson JM, Pearson S, Bramah S. The metabolism of tamoxifen (I.C.I. 46,474). I. In laboratory animals. Xenobiotica. 1973;3(11):693–709. doi: 10.3109/00498257309151594. [DOI] [PubMed] [Google Scholar]
- 41.Fromson JM, Pearson S, Bramah S. The metabolism of tamoxifen (ICI 46,474) Part II in female patients. Xenobiotica. 1973;3:711–713. doi: 10.3109/00498257309151595. [DOI] [PubMed] [Google Scholar]
- 42.Lieberman ME, Jordan VC, Fritsch M, Santos MA, Gorski J. Direct and reversible inhibition of estradiol-stimulated prolactin synthesis by antiestrogens in vitro. J Biol Chem. 1983;258(8):4734–40. [PubMed] [Google Scholar]
- 43.Adam HK, Gay MA, Moore RH. Measurement of tamoxifen in serum by thin layer densitometry. Journal of Endocrinology. 1980;84:35–41. doi: 10.1677/joe.0.0840035. [DOI] [PubMed] [Google Scholar]
- 44.Adam HK, Douglas EJ, Kemp JV. The metabolism of tamoxifen in humans. Biochem Pharmacol. 1979;27:145–147. doi: 10.1016/0006-2952(79)90283-1. [DOI] [PubMed] [Google Scholar]
- 45.Kemp JV, Adam HK, Wakeling AE, Slater R. Identification and biological activity of tamoxifen metabolites in human serum. Biochem Pharmacol. 1983;32:2045–2052. doi: 10.1016/0006-2952(83)90425-2. [DOI] [PubMed] [Google Scholar]
- 46.Bain RR, Jordan VC. Identification of a new metabolite of tamoxifen in patient serum during breast cancer therapy. Biochem Pharmacol. 1983;32(2):373–5. doi: 10.1016/0006-2952(83)90571-3. [DOI] [PubMed] [Google Scholar]
- 47.Jordan VC, Bain RR, Brown RR, Brown RR, Gosden B, Santos MA. Determination and pharmacology of a new hydroxylated metabolite of tamoxifen observed in patient during therapy for advanced breast cancer. Cancer Res. 1983;43:1446–1450. [PubMed] [Google Scholar]
- 48.Robinson SP, Langan-Fahey SM, Jordan VC. Implications of tamoxifen metabolism in the athymic mouse for the study of antitumor effects upon human breast cancer xenografts. Eur J Cancer Clin Oncol. 1989;25(12):1769–76. doi: 10.1016/0277-5379(89)90347-7. [DOI] [PubMed] [Google Scholar]
- 49.Jordan VC, Gosden B. Importance of the alkylamino-ethoxy side chain for the estrogenic and antiestrogenic actions of tamoxifen and trioxifene in the immature rat uterus. Mol Cell Endocrinol. 1982;27:291–306. doi: 10.1016/0303-7207(82)90095-8. [DOI] [PubMed] [Google Scholar]
- 50.Lieberman ME, Gorski J, Jordan VC. An estrogen receptor model to describe the regulation of prolactin synthesis by antiestrogens in vitro. J Biol Chem. 1983;258(8):4741–5. [PubMed] [Google Scholar]
- 51.Katzenellenbogen JA, Carlson KE, Katzenellenbogen BS. Facile geometric isomerization of phenolic nonsteroidal estrogens and antiestrogens: limitations to the interpretation of experiments characterizing the activity of individual isomers. J Steroid Biochem. 1985;22:589–96. doi: 10.1016/0022-4731(85)90210-9. [DOI] [PubMed] [Google Scholar]
- 52.Katzenellenbogen BS, Norman MJ, Eckert RL, Peltz SW, Mangel WF. Bioactivities, estrogen receptor interactions, and plasminogen activator-inducing activities of tamoxifen and hydroxy-tamoxifen isomers in MCF-7 human breast cancer cells. Cancer Res. 1984;44(1):112–9. [PubMed] [Google Scholar]
- 53.Berthois Y, Katzenellenbogen JA, Katzenellenbogen BS. Phenol red in tissue culture media is a weak estrogen: implications concerning the study of estrogen-responsive cells in culture. Proc Natl Acad Sci U S A. 1986;83(8):2496–500. doi: 10.1073/pnas.83.8.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murphy CS, Meisner LF, Wu SQ, Jordan VC. Short- and long-term estrogen deprivation of T47D human breast cancer cells in culture. Eur J Cancer Clin Oncol. 1989;25(12):1777–88. doi: 10.1016/0277-5379(89)90348-9. [DOI] [PubMed] [Google Scholar]
- 55.Jordan VC, Lieberman ME, Cormier E, Koch R, Bagley JR, Ruenitz PC. Structural requirements for the pharmacological activity of nonsteroidal antiestrogens in vitro. Mol Pharmacol. 1984;26(2):272–8. [PubMed] [Google Scholar]
- 56.Jordan VC, Koch R, Mittal S, Schneider MR. Oestrogenic and antioestrogenic actions in a series of triphenylbut-1-enes: modulation of prolactin synthesis in vitro. Br J Pharmacol. 1986;87:217–23. doi: 10.1111/j.1476-5381.1986.tb10174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murphy CS, Langan-Fahey SM, McCague R, Jordan VC. Structure-function relationships of hydroxylated metabolites of tamoxifen that control the proliferation of estrogen-responsive T47D breast cancer cells in vitro. Mol Pharmacol. 1990;38(5):737–43. [PubMed] [Google Scholar]
- 58.Murphy CS, Parker CJ, McCague R, Jordan VC. Structure-activity relationships of nonisomerizable derivatives of tamoxifen: importance of hydroxyl group and side chain positioning for biological activity. Mol Pharmacol. 1991;39(3):421–8. [PubMed] [Google Scholar]
- 59.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/co-activator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 60.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389(6652):753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 61.Wolf DM, Jordan VC. The estrogen receptor from a tamoxifen stimulated MCF-7 tumor variant contains a point mutation in the ligand binding domain. Breast Cancer Res Treat. 1994;31(1):129–38. doi: 10.1007/BF00689683. [DOI] [PubMed] [Google Scholar]
- 62.Wolf DM, Jordan VC. Characterization of tamoxifen stimulated MCF-7 tumor variants grown in athymic mice. Breast Cancer Res Treat. 1994;31(1):117–27. doi: 10.1007/BF00689682. [DOI] [PubMed] [Google Scholar]
- 63.MacGregor-Shafer JI, Liu H, Bentrem D, Zapf J, Jordan VC. Allosteric silencing of activating function 1 in the 4-hydroxytamoxifen estrogen receptor complex by substituting glycine for aspartate at amino acid 351. Cancer Res. 2000;60:5097–5105. [PubMed] [Google Scholar]
- 64.Levenson AS, Jordan VC. The key to the antiestrogenic mechanism of raloxifene is amino acid 351 (aspartate) in the estrogen receptor. Cancer Res. 1998;58(9):1872–5. [PubMed] [Google Scholar]
- 65.Liu H, Lee E-S, De Los Reyes A, Zapf JW, Jordan VC. Silencing and Reactivation of the Selective Estrogen Receptor Modulator (SERM)-ER alpha Complex. Cancer Res. 2001;61:3632–3639. [PubMed] [Google Scholar]
- 66.Onate SA, Tsai SY, Tsai MJ, O’Malley BW. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science. 1995;270(5240):1354–1357. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- 67.Jordan VC, O’Malley BM. Selective estrogen receptor modulators and antihormonal resistance in breast cancer. J Clin Oncol. doi: 10.1200/JCO.2007.11.3886. (in press) [DOI] [PubMed] [Google Scholar]
- 68.Jordan VC, Gapstur S, Morrow M. Selective estrogen receptor modulation and reduction in risk of breast cancer, osteoporosis, and coronary heart disease. J Natl Cancer Inst. 2001;93(19):1449–57. doi: 10.1093/jnci/93.19.1449. [DOI] [PubMed] [Google Scholar]
- 69.Greaves P, Goonetilleke R, Nunn G, Topham J, Orton T. Two-year carcinogenicity study of tamoxifen in Alderley Park Wistar-derived rats. Cancer Res. 1993;53(17):3919–24. [PubMed] [Google Scholar]
- 70.Hard GC, Iatropoulos MJ, Jordan K, Radi L, Kaltenberg OP, Imondi AR, et al. Major difference in the hepatocarcinogenicity and DNA adduct forming ability between toremifene and tamoxifen in female Crl:CD(BR) rats. Cancer Res. 1993;53(19):4534–41. [PubMed] [Google Scholar]
- 71.Fornander T, Rutqvist LE, Cedermark B, Glas U, Mattsson A, Silfversward C, et al. Adjuvant tamoxifen in early breast cancer: occurrence of new primary cancers. Lancet. 1989;1(8630):117–120. doi: 10.1016/s0140-6736(89)91141-0. [DOI] [PubMed] [Google Scholar]
- 72.Fisher B, Costantino JP, Redmond CK, Fisher ER, Wickerham DL, Cronin WM. Endometrial cancer in tamoxifen-treated breast cancer patients: findings from the National Surgical Adjuvant Breast and Bowel Project (NSABP) B-14 [see comments] J Natl Cancer Inst. 1994;86(7):527–37. doi: 10.1093/jnci/86.7.527. [DOI] [PubMed] [Google Scholar]
- 73.Han XL, Liehr JG. Induction of covalent DNA adducts in rodents by tamoxifen. Cancer Res. 1992;52:1360–3. [PubMed] [Google Scholar]
- 74.Rutqvist LE, Johansson H, Signomklao T, Johansson U, Fornander T, Wilking N. Adjuvant tamoxifen therapy for early stage breast cancer and second primary malignancies. Journal of the National Cancer Institute. 1995;87:645–651. doi: 10.1093/jnci/87.9.645. [DOI] [PubMed] [Google Scholar]
- 75.Styles JA, Davies A, Lim CK, De Matteis F, Stanley LA, White IN, et al. Genotoxicity of tamoxifen, tamoxifen epoxide and toremifene in human lymphoblastoid cells containing human cytochrome P450s. Carcinogenesis. 1994;15(1):5–9. doi: 10.1093/carcin/15.1.5. [DOI] [PubMed] [Google Scholar]
- 76.Lim CK, Zhi-Xin Y, Lamb JH, Whihte INH, DeMatteis F, Smith LL. A comparative study of tamoxifen metabolism in female rat, mouse and human liver microsomes. Carcinogenesis. 1994;15:589–593. doi: 10.1093/carcin/15.4.589. [DOI] [PubMed] [Google Scholar]
- 77.Moorthy B, Sriram P, Pathak DN, Bodell WJ, Randerath K. Tamoxifen metabolic activation: comparison of DNA adducts formed by microsomal and chemical activation of tamoxifen and 4-hydroxytamoxifen with DNA adducts formed in vivo. Cancer Res. 1996;56(1):53–7. [PubMed] [Google Scholar]
- 78.Pongracz K, Pathak DN, Nakamura T, Burlingame AL, Bodell WJ. Activation of the tamoxifen derivative metabolite E to form DNA adducts: comparison with the adducts formed by microsomal activation of tamoxifen. Cancer Res. 1995;55(14):3012–5. [PubMed] [Google Scholar]
- 79.Potter GA, McCague R, Jarman M. A mechanistic hypothesis for DNA adduct formation by tamoxifen following hepatic oxidative metabolism. Carcinogenesis. 1994;15(3):439–42. doi: 10.1093/carcin/15.3.439. [DOI] [PubMed] [Google Scholar]
- 80.Phillips DH, Carmichael PL, Hewer A, Cole KJ, Poon GK. alpha-Hydroxytamoxifen, a metabolite of tamoxifen with exceptionally high DNA-binding activity in rat hepatocytes. Cancer Res. 1994;54(21):5518–22. [PubMed] [Google Scholar]
- 81.Phillips DH, Potter GA, Horton MN, Hewer A, Crofton-Sleigh C, Jarman M, et al. Reduced genotoxicity of [D5-ethyl]-tamoxifen implicates alpha- hydroxylation of the ethyl group as a major pathway of tamoxifen activation to a liver carcinogen. Carcinogenesis. 1994;15(8):1487–92. doi: 10.1093/carcin/15.8.1487. [DOI] [PubMed] [Google Scholar]
- 82.Osborne MR, Hewer A, Hardcastle IR, Carmichael PL, Phillips DH. Identification of the major tamoxifen-deoxyguanosine adduct formed in the liver DNA of rats treated with tamoxifen. Cancer Res. 1996;56(1):66–71. [PubMed] [Google Scholar]
- 83.Phillips DH, Carmichael PL, Hewer A, Cole KJ, Hardcastle IR, Poon GK, et al. Activation of tamoxifen and its metabolite alpha-hydroxytamoxifen to DNA-binding products: comparisons between human, rat and mouse hepatocytes. Carcinogenesis. 1996;17(1):89–94. doi: 10.1093/carcin/17.1.89. [DOI] [PubMed] [Google Scholar]
- 84.Osborne MR, Hewer A, Phillips DH. Resolution of alpha-hydroxytamoxifen; R-isomer forms more DNA adducts in rat liver cells. Chem Res Toxicol. 2001;14(7):888–93. doi: 10.1021/tx010027b. [DOI] [PubMed] [Google Scholar]
- 85.Osborne MR, Hewer A, Phillips DH. Stereoselective metabolic activation of alpha-hydroxy-N-desmethyltamoxifen: the R-isomer forms more DNA adducts in rat liver cells. Chem Res Toxicol. 2004;17(5):697–701. doi: 10.1021/tx049957w. [DOI] [PubMed] [Google Scholar]
- 86.Phillips DH. Understanding the genotoxicity of tamoxifen? Carcinogenesis. 2001;22(6):839–849. doi: 10.1093/carcin/22.6.839. [DOI] [PubMed] [Google Scholar]
- 87.Jordan VC. What if tamoxifen (ICI 46,474) had been found to produce rat liver tumors in 1973? A personal perspective. Ann Oncol. 1995;6(1):29–34. doi: 10.1093/oxfordjournals.annonc.a059035. [DOI] [PubMed] [Google Scholar]
- 88.Mallory FB, Wood CS, Gordon JT. Photochemistry of stilbenes III. Some aspects of photocylisation to phenanthrenes. Journal of American Chem Society. 1986;86:3094–3102. [Google Scholar]
- 89.Mendenhall lW, Kobayashi H, Shih FML, Sternson LA, Higuchi T, Fabian C. Clinical analysis of tamoxifen, an antineoplastic agent, in plasma. Clin Chem. 1978;24:1518–1524. [PubMed] [Google Scholar]
- 90.Golander Y, Sternson LA. Paired-ion chromatographis analysis of tamoixfen and two major metabolites in plasma. J Cromatogr. 1980;181:41–49. doi: 10.1016/s0378-4347(00)81267-0. [DOI] [PubMed] [Google Scholar]
- 91.Brown RR, Bain R, Jordan VC. Determination of tamoxifen and metabolites in human serum by high-performance liquid chromatography with post-column fluorescence activation. J Chromatogr. 1983;272(2):351–8. doi: 10.1016/s0378-4347(00)86138-1. [DOI] [PubMed] [Google Scholar]
- 92.Camaggi CM, Strocchi E, Canova N. High performance liquid chromatographic analysis or tamoixfen and major metabolites in human plasma. J Chromatogr. 1983;275:436–442. doi: 10.1016/s0378-4347(00)84393-5. [DOI] [PubMed] [Google Scholar]
- 93.Lien EA, Solheim E, Ueland PM. Distribution of tamoxifen and its metabolites in rat and human tissues during steady-state treatment. Cancer Res. 1991;51:4837–4844. [PubMed] [Google Scholar]
- 94.Lee KH, Ward BA, Desta Z, Flockhart DA, Jones DR. Quantification of tamoxifen and three metabolites in plasma by high-performance liquid chromatography with fluorescence detection: application to a clinical trial. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;791:245–253. doi: 10.1016/s1570-0232(03)00218-6. [DOI] [PubMed] [Google Scholar]
- 95.Fried KM, Wainer IW. Direct determination of tamoxifen and its four major metabolites in plasma using coupled column high-performance liquid chromatography. J Chromatogr B Biomed Appl. 1994;655(2):261–8. doi: 10.1016/0378-4347(94)00054-9. [DOI] [PubMed] [Google Scholar]
- 96.Kikuta C, Schmid R. Specific high-performance liquid chromatographic analysis of tamoxifen and its major metabolites by “on-line” extraction and post-column photochemical reaction. J Pharm Biomed Anal. 1989;7(3):329–37. doi: 10.1016/0731-7085(89)80100-1. [DOI] [PubMed] [Google Scholar]
- 97.Stearns V, Johnson MD, Rae JM, Morocho A, Novielli A, Bhargava P, et al. Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J Natl Cancer Inst. 2003;95(23):1758–1764. doi: 10.1093/jnci/djg108. [DOI] [PubMed] [Google Scholar]
- 98.Fabian C, Sternson L, Barnett M. Clinical pharmacology of tamoxifen in patients with breast cancer: comparison of traditional and loading dose schedules. Cancer Treat Rep. 1980;64(6–7):765–73. [PubMed] [Google Scholar]
- 99.Patterson JS, Settatree RS, Adam HK, Kemp JV. Serum concentrations of tamoxifen and major metabolite during long-term nolvadex therapy, correlated with clinical response. Eur J Cancer. 1980;(Suppl 1):89–92. [PubMed] [Google Scholar]
- 100.Wilkinson PM, Ribiero GG, Adama HK, Kemp JV, Patterson JS. Tamoxifen (Nolvadex) therapy - rationale for loading dose followed by maintenance dose for patients with metastatic breast cancer. Cancer Chemother Pharmacol. 1982;10:33–35. doi: 10.1007/BF00257234. [DOI] [PubMed] [Google Scholar]
- 101.Jordan VC. Chemosuppression of breast cancer with tamoxifen: laboratory evidence and future clinical investigations. Cancer Invest. 1988;6(5):589–595. doi: 10.3109/07357908809082124. [DOI] [PubMed] [Google Scholar]
- 102.Lerner LJ, Jordan VC. The development of antiestrogens for the treatment of breast cancer: Eighth Cain Memorial Award Lecture. Cancer Res. 1990;50:4177–4189. [PubMed] [Google Scholar]
- 103.Jordan VC. Selective estrogen receptor modulation: a personal perspective. Cancer Res. 2001;61:5683–5687. [PubMed] [Google Scholar]
- 104.Jordan VC. Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. 2. Clinical considerations and new agents. J Med Chem. 2003;46(7):1081–1111. doi: 10.1021/jm020450x. [DOI] [PubMed] [Google Scholar]
- 105.Ariazi EA, Ariazi JL, Cordera F, Jordan VC. Estrogen receptors as therapeutic targets in breast cancer. Current Topics in Medicinal Chemistry. 2006;6:195–216. [PubMed] [Google Scholar]
- 106.Buzdar A, Marcus C, Holmes F, Hug V, Hortobagyi G. Phase II evaluation of Ly156758 in metastatic breast cancer. Oncology. 1988;45(5):344–5. doi: 10.1159/000226637. [DOI] [PubMed] [Google Scholar]
- 107.Kemp DC, Fan PW, Stevens JC. Characterization of raloxifene glucuronidation in vitro: contribution of intestinal metabolism to presystemic clearance. Drug Metab Dispos. 2002;30:694–700. doi: 10.1124/dmd.30.6.694. [DOI] [PubMed] [Google Scholar]
- 108.Jeong EJ, Liu Y, Lin H, Hu M. Species- and disposition model-dependent metabolism of raloxifene in gut and liver: role of UGT1A10. Drug Metab Dispos. 2005;33:785–94. doi: 10.1124/dmd.104.001883. [DOI] [PubMed] [Google Scholar]
- 109.Suh N, Lamph WW, Glasebrook AL, Grese TA, Palkowitz AD, Williams CR, et al. Prevention and treatment of experimental breast cancer with the combination of a new selective estrogen receptor modulator, arzoxifene, and a new rexinoid, LG 100268. Clin Cancer Res. 2002;8(10):3270–5. [PubMed] [Google Scholar]
- 110.Baselga J, Llombart-Cussac A, Bellet M, Guillem-Porta V, Enas N, Krejcy K, et al. Randomized, double-blind multicenter trial comparing two doses of arzoxifene ( LY353381) in hormone-sensitive advanced or metastatic breast cancer patients. Annals of Oncology. 2003;14:1383–1390. doi: 10.1093/annonc/mdg368. [DOI] [PubMed] [Google Scholar]
- 111.Buzdar A, O’Shaughnessy JA, Booser DJ, Pippen JE, Jr, Jones SE, Munster PN, et al. Phase II, randomized double-blind study of two dose levels of arzoxifene in patients with locally advanced or metastatic breast cancer. Journal of Clinical Oncology. 2003;21:1007–1014. doi: 10.1200/JCO.2003.06.108. [DOI] [PubMed] [Google Scholar]
- 112.Falany JL, Pilloff DE, Leyh TS, Falany CN. Sulfation of raloxifene and 4-hydroxytamoxifen by human cytosolic sulfotransferases. Drug Metab Dispos. 2006;34:361–368. doi: 10.1124/dmd.105.006551. [DOI] [PubMed] [Google Scholar]
- 113.Rosati RL, Da Silva Jardine P, Cameron KO, Thompson DD, Ke HZ, Toler SM, et al. Discovery and preclinical pharmacology of a novel, potent, nonsteroidal estrogen receptor agonist/antagonist, CP-336156, a diaryltetrahydronaphthalene. J Med Chem. 1998;41(16):2928–2931. doi: 10.1021/jm980048b. [DOI] [PubMed] [Google Scholar]
- 114.Ke HZ, Paralkar VM, Grasser WA, Crawford DT, Qi H, Simmons HA, et al. Effects of CP-336,156, a new, nonsteroidal estrogen agonist/antagonist, on bone, serum cholesterol, uterus and body composition in rat models. Endocrinology. 1998;139(4):2068–76. doi: 10.1210/endo.139.4.5902. [DOI] [PubMed] [Google Scholar]
- 115.Ke HZ, Qi H, Crawford DT, Chidsey-Frink KL, Simmons HA, Thompson DD. Lasofoxifene (CP-336,156), a selective estrogen receptor modulator, prevents bone loss induced by aging and orchidectomy in the adult rat. Endocrinology. 2000;141(4):1338–44. doi: 10.1210/endo.141.4.7408. [DOI] [PubMed] [Google Scholar]
- 116.Tatee T, Carlson KE, Katzenellenbogen JA, Robertson DW, Katzenellenbogen BS. Antiestrogens and antiestrogen metabolites: preparation of tritium-labeled (+/−)-cis-3-[p-(1,2,3,4-tetrahydro-6-methoxy-2-phenyl-1-naphthyl)phenoxyl]-1,2-propanediol (U-23469) and characterization and synthesis of a biologically important metabolite. J Med Chem. 1979;22(12):1509–17. doi: 10.1021/jm00198a015. [DOI] [PubMed] [Google Scholar]
- 117.Legha SS, Slavik M, Carter SK. Nafoxidine--an antiestrogen for the treatment of breast cancer. Cancer. 1976;38(4):1535–41. doi: 10.1002/1097-0142(197610)38:4<1535::aid-cncr2820380415>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 118.Cohen LA, Pittman B, Wang CX, Aliaga C, Yu L, Moyer JD. Lasofoxifene, a novel selective estrogen receptor modulator with chemopreventive and therapeutic activity in the N-nitroso-N-methylurea-induced rat mammary tumor model. Cancer Res. 2001;61(24):8683–8688. [PubMed] [Google Scholar]
- 119.Robertson DW, Katzenellenbogen JA, Hayes JR, Katzenellenbogen BS. Antiestrogen basicity-activity relationships: a comparison of the estrogen receptor binding and antiuterotrophic potencies of several analogues of (Z)-1,2 dipheny1-1-[4-[2-(dimethylamino)ethoxy]phenyl]-1-butene (tamoxifen Nolvadex) having altered basicity. J Med Chem. 1982;25:167–171. doi: 10.1021/jm00344a015. [DOI] [PubMed] [Google Scholar]
- 120.Hellmann-Blumberg U, Taras TL, Wurz GT, DeGregorio MW. Genotoxic effects of the novel mixed antiestrogen FC-1271a in comparison to tamoxifen and toremifene. Breast Cancer Res Treat. 2000;60(1):63–70. doi: 10.1023/a:1006311214152. [DOI] [PubMed] [Google Scholar]
- 121.Qu Q, Zheng H, Dahllund J, Laine A, Cockcroft N, Peng Z, et al. Selective estrogenic effects of a novel triphenylethylene compound, FC1271a, on bone, cholesterol level, and reproductive tissues in intact and ovariectomized rats. Endocrinology. 2000;141(2):809–20. doi: 10.1210/endo.141.2.7342. [DOI] [PubMed] [Google Scholar]
- 122.DeGregorio MW, Wurz GT, Taras TL, Erkkola RU, Halonen KH, Huupponen RK. Pharmacokinetics of (deaminohydroxy)toremifene in humans: a new, selective estrogen-receptor modulator. Eur J Clin Pharmacol. 2000;56(6–7):469–75. doi: 10.1007/s002280000176. [DOI] [PubMed] [Google Scholar]
- 123.Rutanen EM, Heikkinen J, Halonen K, Komi J, Lammintausta R, Ylikorkala O. Effects of ospemifene, a novel SERM, on hormones, genital tract, climacteric symptoms, and quality of life in postmenopausal women: a double-blind, randomized trial. Menopause. 2003;10(5):433–9. doi: 10.1097/01.GME.0000063609.62485.27. [DOI] [PubMed] [Google Scholar]
- 124.Komi J, Lankinen KS, Harkonen P, DeGregorio MW, Voipio S, Kivinen S, et al. Effects of ospemifene and raloxifene on hormonal status, lipids, genital tract, and tolerability in postmenopausal women. Menopause. 2005;12(2):202–9. doi: 10.1097/00042192-200512020-00015. [DOI] [PubMed] [Google Scholar]
- 125.Namba R, Young LJ, Maglione JE, McGoldrick ET, Liu S, Wurz GT, et al. Selective estrogen receptor modulators inhibit growth and progression of premalignant lesions in a mouse model of ductal carcinoma in situ. Breast Cancer Res. 2005;7(6):R881–9. doi: 10.1186/bcr1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wurz GT, Read KC, Marchisano-Karpman C, Gregg JP, Beckett LA, Yu Q, et al. Ospemifene inhibits the growth of dimethylbenzanthracene-induced mammary tumors in Sencar mice. J Steroid Biochem Mol Biol. 2005;97(3):230–40. doi: 10.1016/j.jsbmb.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 127.Beverage JN, Sissung TM, Sion AM, Danesi R, Figg WD. CYP2D6 polymorphisms and the impact on tamoxifen therapy. Journal of Pharmaceutical Sciences. 2007 doi: 10.1002/jps.20892. Published online in Wiley InterScience ( www.interscience.wiley.com) [DOI] [PubMed]
- 128.Bradford LD. CYP2D6 allele frequency in European Caucasians, Asians, Africans and their descendants. Pharmacogenomics. 2002;3(2):229–43. doi: 10.1517/14622416.3.2.229. [DOI] [PubMed] [Google Scholar]
- 129.Raimundo S, Toscano C, Klein K, Fischer J, Griese EU, Eichelbaum M, et al. A novel intronic mutation, 2988G>A, with high predictivity for impaired function of cytochrome P450 2D6 in white subjects. Clin Pharmacol Ther. 2004;76(2):128–38. doi: 10.1016/j.clpt.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 130.Hanioka N, Kimura S, Meyer UA, Gonzalez FJ. The human CYP2D locus associated with a common genetic defect in drug oxidation: a G1934----A base change in intron 3 of a mutant CYP2D6 allele results in an aberrant 3′ splice recognition site. Am J Hum Genet. 1990;47(6):994–1001. [PMC free article] [PubMed] [Google Scholar]
- 131.Bernard S, Neville KA, Nguyen AT, Flockhart DA. Interethnic differences in genetic polymorphisms of CYP2D6 in the U.S. population: clinical implications. Oncologist. 2006;11(2):126–35. doi: 10.1634/theoncologist.11-2-126. [DOI] [PubMed] [Google Scholar]
- 132.Lim YC, Desta Z, Flockhart DA, Skaar TC. Endoxifen (4-hydroxy-N-desmethyl-tamoxifen) has anti-estrogenic effects in breast cancer cells with potency similar to 4-hydroxy-tamoxifen. Cancer Chemother Pharmacol. 2005;55:471–478. doi: 10.1007/s00280-004-0926-7. [DOI] [PubMed] [Google Scholar]
- 133.Lim YC, Li L, Desta Z, Zhao Q, Rae JM, Flockhart DA, et al. Endoxifen, a secondary metabolite of tamoxifen, and 4-OH-tamoxifen induce similar changes in global gene expression patterns in MCF-7 breast cancer cells. J Pharmacol Exp Ther. 2006;318(2):503–512. doi: 10.1124/jpet.105.100511. [DOI] [PubMed] [Google Scholar]
- 134.Jin Y, Desta Z, Stearns V, Ward B, Ho H, Lee KH, Skaar T, Storniolo AM, Li L, Araba A, Blanchard R, Nguyen A, Ullmer L, Hayden J, Lemler S, Weinshilboum RM, Rae JM, Hayes DF, Flockhart DA. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. JNCI. 2005;97:30–39. doi: 10.1093/jnci/dji005. [DOI] [PubMed] [Google Scholar]
- 135.Fisher B, Costantino J, Redmond C, Poisson R, Bowman D, Couture J, et al. A randomized clinical trial evaluating tamoxifen in the treatment of patients with node-negative breast cancer who have estrogen-receptor- positive tumors. N Engl J Med. 1989;320(8):479–84. doi: 10.1056/NEJM198902233200802. [DOI] [PubMed] [Google Scholar]
- 136.Fisher B, Dignam J, Wolmark N, Wickerham DL, Fisher ER, Mamounas E, et al. Tamoxifen in treatment of intraductal breast cancer: National Surgical Adjuvant Breast and Bowel Project B-24 randomised controlled trial. Lancet. 1999;353(9169):1993–2000. doi: 10.1016/S0140-6736(99)05036-9. [DOI] [PubMed] [Google Scholar]
- 137.Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst. 1998;90:1371–1388. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 138.Loprinzi CL, Kugler JW, Sloan JA, Mailliard JA, LaVasseur BI, Barton DL, et al. Venlafaxine in management of hot flashes in survivors of breast cancer: a randomised controlled trial. Lancet. 2000;356(9247):2059–63. doi: 10.1016/S0140-6736(00)03403-6. [DOI] [PubMed] [Google Scholar]
- 139.Loprinzi CL, Sloan JA, Perez EA, Quella SK, Stella PJ, Mailliard JA, et al. Phase III evaluation of fluoxetine for treatment of hot flashes. J Clin Oncol. 2002;20(6):1578–83. doi: 10.1200/JCO.2002.20.6.1578. [DOI] [PubMed] [Google Scholar]
- 140.Stearns V, Beebe KL, Iyengar M, Dube E. Paroxetine controlled release in the treatment of menopausal hot flashes: a randomized controlled trial. JAMA. 2003;289(21):2827–34. doi: 10.1001/jama.289.21.2827. [DOI] [PubMed] [Google Scholar]
- 141.Dehal SS, Kupfer D. CYP2D6 catalyzes tamoxifen 4-hydroxylation in human liver. Cancer Res. 1997;57(16):3402–6. [PubMed] [Google Scholar]
- 142.Crewe HK, Ellis SW, Lennard MS, Tucker GT. Variable contribution of cytochromes P450 2D6, 2C9 and 3A4 to the 4-hydroxylation of tamoxifen by human liver microsomes. Biochem Pharmacol. 1997;53(2):171–8. doi: 10.1016/s0006-2952(96)00650-8. [DOI] [PubMed] [Google Scholar]
- 143.Coller JK, Krebsfaenger N, Klein K, Endrizzi K, Wolbold R, Lang T, et al. The influence of CYP2B6, CYP2C9 and CYP2D6 genotypes on the formation of the potent antioestrogen Z-4-hydroxy-tamoxifen in human liver. Br J Clin Pharmacol. 2002;54(2):157–67. doi: 10.1046/j.1365-2125.2002.01614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lessard E, Yessine MA, Hamelin BA, Gauvin C, Labbe L, O’Hara G, et al. Diphenhydramine alters the disposition of venlafaxine through inhibition of CYP2D6 activity in humans. J Clin Psychopharmacol. 2001;21(2):175–84. doi: 10.1097/00004714-200104000-00009. [DOI] [PubMed] [Google Scholar]
- 145.Albers LJ, Reist C, Vu RL, Fujimoto K, Ozdemir V, Helmeste D, et al. Effect of venlafaxine on imipramine metabolism. Psychiatry Res. 2000;96(3):235–43. doi: 10.1016/s0165-1781(00)00213-4. [DOI] [PubMed] [Google Scholar]
- 146.Yoon YR, Cha IJ, Shon JH, Kim KA, Cha YN, Jang IJ, et al. Relationship of paroxetine disposition to metoprolol metabolic ratio and CYP2D6*10 genotype of Korean subjects. Clin Pharmacol Ther. 2000;67(5):567–76. doi: 10.1067/mcp.2000.106128. [DOI] [PubMed] [Google Scholar]
- 147.Jeppesen U, Gram LF, Vistisen K, Loft S, Poulsen HE, Brosen K. Dose-dependent inhibition of CYP1A2, CYP2C19 and CYP2D6 by citalopram, fluoxetine, fluvoxamine and paroxetine. Eur J Clin Pharmacol. 1996;51(1):73–8. doi: 10.1007/s002280050163. [DOI] [PubMed] [Google Scholar]
- 148.Borges S, Desta Z, Li L, Skaar TC, Ward BA, Nguyen A, et al. Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: implication for optimization of breast cancer treatment. Clin Pharmacol Ther. 2006;80(1):61–74. doi: 10.1016/j.clpt.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 149.Goetz MP, Rae JM, Suman VJ, Safgren SL, Ames MM, Visscher DW, et al. Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J Clin Oncol. 2005;23(36):9312–9318. doi: 10.1200/JCO.2005.03.3266. [DOI] [PubMed] [Google Scholar]
- 150.Goetz MP, Knox SK, Suman VJ, Rae JM, Safgren SL, Ames MM, et al. The impact of cytochrome P450 2D6 metabolism in women receiving adjuvant tamoxifen. Breast Cancer Res Treat. 2007;101(1):113–21. doi: 10.1007/s10549-006-9428-0. [DOI] [PubMed] [Google Scholar]
- 151.Mortimer JE, Flatt SW, Parker BA, Gold EB, Wasserman L, Natarajan L, et al. Tamoxifen, hot flashes and recurrence in breast cancer. Breast Cancer Res Treat. 2007 doi: 10.1007/s10549-007-9612-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nowell S, Sweeney C, Winters M, Stone A, Lang NP, Hutchins LF, et al. Association between sulfotransferase 1A1 genotype and survival of breast cancer patients receiving tamoxifen therapy. J Natl Cancer Inst. 2002;94(21):1635–40. doi: 10.1093/jnci/94.21.1635. [DOI] [PubMed] [Google Scholar]
- 153.Nowell SA, Ahn J, Rae JM, Scheys JO, Trovato A, Sweeney C, et al. Association of genetic variation in tamoxifen-metabolizing enzymes with overall survival and recurrence of disease in breast cancer patients. Breast Cancer Res Treat. 2005;91(3):249–58. doi: 10.1007/s10549-004-7751-x. [DOI] [PubMed] [Google Scholar]
- 154.Wegman PP, Wingren S. CYP2D6 variants and the prediction of tamoxifen response in randomized patients: author response. Breast Cancer Res. 2005;7(6):E7. doi: 10.1186/bcr1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wegman P, Elingarami S, Carstensen J, Stal O, Nordenskjold B, Wingren S. Genetic variants of CYP3A5, CYP2D6, SULT1A1, UGT2B15 and tamoxifen response in postmenopausal patients with breast cancer. Breast Cancer Res. 2007;9(1):R7. doi: 10.1186/bcr1640. [DOI] [PMC free article] [PubMed] [Google Scholar]