

Abstract
Deregulation of mitochondrial function is a common feature in multiple aspects of aging. In addition to playing a role in aging-associated disease, decline in mitochondrial energy metabolism is likely to be important in the development of metabolic disease. Furthermore, altered mitochondrial function is a conserved feature in caloric restriction – a dietary intervention that delays aging in diverse species. The transcriptional co-activator PGC-1α is a critical regulator of mitochondrial energy metabolism and biogenesis. PGC-1α is uniquely poised as a potential target for correcting the effects of age on mitochondrial decline. We describe the cellular and tissue specific mechanisms of PGC-1α regulation and illustrate how these pathways may be involved in the aging process.
1. Metabolism and aging
There has been widespread public interest in aging research with particular attention given to aging-associated diseases and the factors that may contribute to disease onset. The fact that aging itself is the most significant risk factor for a range of diseases including, cancer, cardiovascular disease and diabetes [1], emphasizes the need for research on the biology of aging to better understand the underlying causes of aging-associated disease. The advent of transcriptional profiling has permitted the study of complex biological processes at the molecular level. The first studies of aging using this approach revealed that surprisingly few genes were altered by aging at the transcriptional level [2-4]. It is less surprising that the aging signature is distinct among different tissues [5]; however, microarray technology has provided clues to a few key cellular pathways that are universally regulated with aging. Foremost among these and pertinent to this review are genes involved in mitochondrial energy metabolism.
A comparison of genomic expression profiles from worms and flies reveals that there are common patterns of age related changes in gene expression [6]. Of particular note are the genes involved in mitochondrial energy metabolism whose expression declines with age. A similar approach, comparing mouse and human transcriptional data, reveals this same group of genes involved in the mitochondrial electron transport system (ETS) also declines with age in multiple tissues [7]. Mitochondrial function declines with age in skeletal muscle in humans [8], and increases in intramuscular triglycerides and lipid deposition due to mitochondrial insufficiency correlate with loss of insulin sensitivity, a prevalent phenotype of aging [9]. Dietary excess and the resulting obesity can lead to metabolic syndrome, the manifestation of multiple conditions previously associated with aging, including inflammation, hypertension and cardiovascular disease [10]. A consensus is emerging that describes adipose derived factors as key component in metabolic disorders linked to obesity [11, 12] and that alterations in mitochondrial metabolism may be important in the pathology of this disease [13]. The most robust and consistent regimen that opposes aging is caloric restriction (CR) [14, 15]. Alterations in mitochondrial energy metabolism are observed in multiple species on CR [16], including humans [17]. Taken together these findings suggest of a causal role of mitochondrial dysfunction in the aging process and a central role for mitochondrial adaptation in the mechanism of aging retardation by CR (Figure 1).
Figure 1. The role of mitochondrial metabolism in aging and disease.
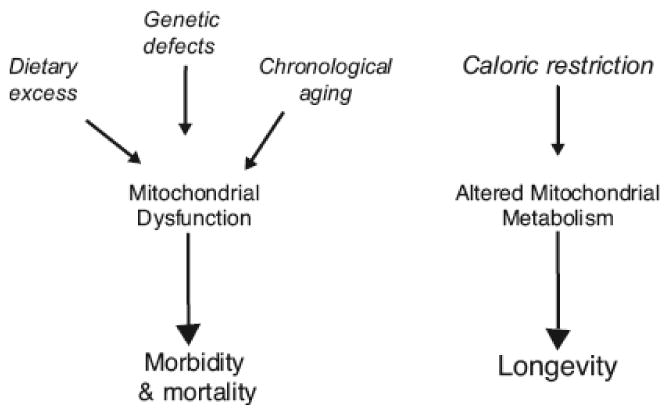
The loss of mitochondrial efficiency and energetic capacity is a unifying feature in the onset of morbidities due to dietary, genetic or aging influences. A causal role in the aging process is suggested and enabled through the complex interplay among distinct tissues in response to metabolic defects. In contrast, altered mitochondrial metabolism is a conserved feature of caloric restriction, the most robust intervention that delays aging in numerous species.
The PGC-1 family of transcriptional co-activators play a central role in regulation of mitochondrial biogenesis and function [18]. Three members of this family have been identified based on sequence similarity to the founding member PGC-1α (peroxisome proliferator activated receptor gamma coactivator 1 alpha). The genes encoding each are located on distinct chromosomes and show tissue specific patterns of expression. PGC-1β is larger than PGC-1α but shares functionally equivalent protein domain features and has many common downstream targets [19]. Both PGC-1α and PGC-1β are expressed preferentially in high oxidative capacity tissues [18]. PRC-1 is larger again with similar protein domain features but is not enriched in skeletal muscle and heart [20]. Although functionally similar, PGC-1α, PGC-1β and PRC-1 play distinct tissue specific roles in response to exercise [21], fasting [19, 22] and, in the case of PGC-1α and PGC-1β, the knockout mice show opposite phenotypes on high fat diets [23-26]. As PGC-1α is the best characterized of the three at this time and can be linked to aging through association with factors that influence longevity, it shall be the focus of this review.
2. Mitochondrial regulator PGC-1α
PGC-1α is a key regulator of mitochondrial biogenesis and respiration [27] that plays a critical role in the control of metabolism and energy homeostasis [28-30]. As the transcriptional co-activation of nuclear receptors involved in multiple aspects of metabolism, PGC-1α may play a pivotal role in organismal metabolic homeostasis [31]. PGC-1α is regulated at the protein level by alterations in cellular localization [32], protein stability [32-34] and post-translational modification (see below). It is regulated at the transcriptional level by CREB [35] and is involved in regulating its own transcription with YY1 [36]. Remarkably, PGC-1α is responsive to multiple diverse stimuli: i) changes in nutrient availability [37], ii) thermal fluctuation [27], iii) calcium/calmodulin [38, 39], iv) oxidative species [32, 40, 41], v) endocrine signaling through insulin [42] and T3 thyroid hormone [43, 44], vi) circadian clock [45] vii) cytokines [34], viii) energetic demand [46] and hypoxia [47, 48] (Figure 2). The importance of mitochondrial regulation and adaptive response in normal cellular function is implicit. The ability PGC-1α to respond to these disparate signals indicates that it is the principle medium through which external stimuli are communicated to the mitochondria.
Figure 2. Mitochondrial regulation through PGC-1α in response to diverse stimuli.
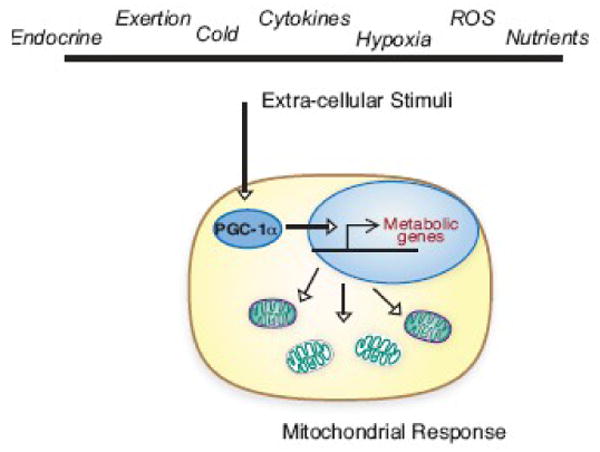
Mitochondrial metabolism can be regulated rapidly through changes in PGC-1α transcriptional activity. The wide range of stimuli that impact PGC-1α speaks to a central role for mitochondrial adaptation in the cellular response. Loss of mitochondrial plasticity and the ability to respond appropriately to changes in the cellular environment may contribute to metabolic derangements associated with the aging process.
3. Post translational modification of PGC-1α
PGC-1α localization, interaction with binding partners and transcriptional co-activation can be manipulated alone or in combination through protein modification. The multiplicity of regulatory points provides enormous versatility and a means to rapidly alter PGC-1α activity in response to a given stimulus. We focus on PGC-1α regulatory factors that have previously been associated with aging or CR and have been designated putative longevity factors in rodent and non-mammalian studies (Figure 3). There is a substantial volume of data on the mechanism of action of these key factors that reveal which elements of cell signaling they can influence, but the details of their actual role in normal physiology in wildtype animals remain less clear.
Figure 3. PGC-1α is regulated through post-translational modification.
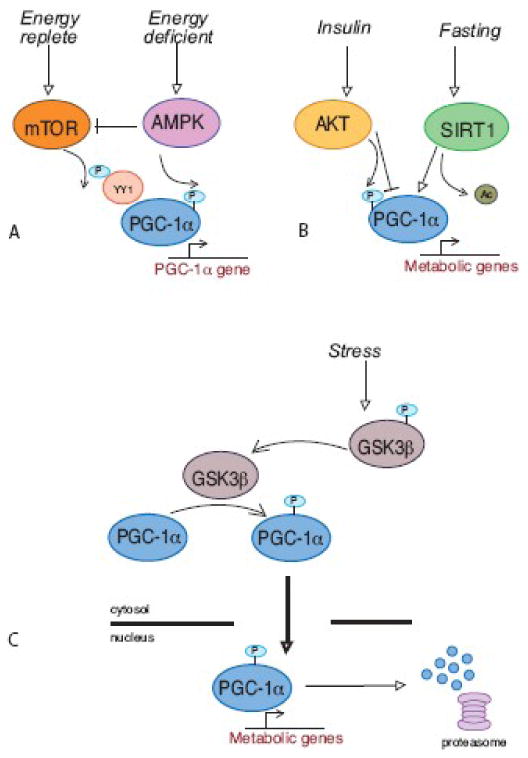
These highly simplified illustrations are intended to convey conceptually how the metabolic impact of any given stimulus can be tailored through alterations in PGC-1α localization, binding partners and gene target specificity. A. Mitochondrial adaptation in response to changes in nutrient availability is achieved through a balance of factors that regulate PGC-1α levels and activity. It is likely that PGC-1α function is not equivalent in each response due to simultaneous regulation of PGC-1α binding partners. B. Tissue specificity plays an important role in the response of PGC-1α to the fed or fasted state. The appropriate response to fasting requires increased glucose output from liver and reduced glucose utilization in skeletal muscle, both of which involve PGC-1α. The use of the same regulator in each tissue ensures a coordinated response. C. PGC-1α localization and stability is regulated in response to stress. Transient activation of mitochondrial metabolism is an early event in the stress response. PGC-1α accumulates in the nucleus and is degraded following transcriptional activation. A strategy of altering PGC-1α protein turnover provides a sensitive and rapid mechanism for regulation of mitochondrial metabolism.
3.1 AMPK
AMP-activated protein kinase (AMPK) is involved in the adaptive response to energy deficit. Activation of AMPK induces PGC-1α, resulting in increase mitochondrial biogenesis and increased activity of mitochondrial enzymes in skeletal muscle in response to exercise [49, 50]. A comparison of young (3 mo.) and old (28 mo.) rats reveals that this metabolic response to AMPK activation becomes blunted with age [51]. AMPK is involved in PGC-1α autoregulation; direct phosphorylation by AMPK promotes PGC-1α dependent induction at the PGC-1α promoter [52]. PGC-1α is also induced in response to cold in skeletal muscle and a feed forward activation of AMPK occurs [53]. In addition to activating PGC-1α, AMPK also inhibits mTOR [54], a nutrient activated factor that regulates PGC-1α gene expression [36]. This dual arrangement allows for a PGC-1α response to energy deficit and to increased nutrient availability (Figure 3A).
3.2 mTOR
Target of rapamycin (TOR) is a nutrient sensing kinase that promotes cell growth in conditions of nutrient abundance [55]. Mitochondrial oxygen consumption and oxidative capacity is positively influenced by mTOR in mammalian cells [56]. Inhibition of TOR extends lifespan in yeast, worms and flies but it is not clear if simply reducing mTOR signaling will achieve a similar effect in mammalian systems [16]. Mice treated with rapamycin develop symptoms of diabetes [36] and CR increases mTOR signaling in adipose tissue [57]. In mammalian cells, PGC-1α gene transcription is regulated by mTOR through interaction of PGC-1α with transcription factor YY1 and mTOR1 complex I [36]. The ability of cells to respond to changes in nutritional status through TOR is highly conserved and the role of this nutrient senstive kinase in regulating mitochondrial function is likely to be important in the aging process [58].
3.3 SIRT1
Activity of PGC-1α is regulated through inhibitory acetylation by GCN5 [59] and stimulatory deacetlyation by SIRT1 [60-62]. SIRT1 is a member of the Sirtuin family that has been implicated in longevity in yeast, worms and flies [63]. PGC-1α is regulated by cellular localization and in response to oxidative stress, accumulates in the nucleus in a SIRT1 dependent manner [32]. Deacetylation of PGC-1α is required to sequester it in the nucleus and this cellular redistribution is prevented by nicotinamide, a potent SIRT1 inhibitor [64]. Sirtuins have been implicated in the mechanism of lifespan extension by CR [65] and SIRT1 is regulated by CR in mice in a tissue specific manner [66].
3.4 AKT
The serine threonine kinase AKT is a key component of the insulin signaling pathway whose downstream targets include regulators of metabolism, the stress response and apoptosis [67]. AKT is a negative regulator of PGC-1α (Figure 3B). AKT phosphorylates and inhibits PGC-1α in response to insulin in liver [68] and activation of AKT causes a reduction in PGC-1α levels in heart [69]. AKT positively regulates mTOR (activator of PGC-1α) by direct inhibition of the inhibitory complex TSC1/2 and by inhibition of the mTOR inhibitors AMPK (activator of PGC-1α) and GSK3b (regulator of PGC-1α turnover) [67, 70]. Clearly there remains much to be discovered about how these signaling molecules coordinately respond to cell stimuli and, importantly, to aging.
3.5 GSK3β
The nutrient sensitive kinase GSK3β (Glycogen synthase kinase 3 beta) targets PGC-1α for nuclear proteasomal degradation [32], regulating PGC-1α turnover and thereby mitochondrial function (Figure 3C). A role for GSK3β in CR is suggested as inhibitory of GSK3β phosphorylation is detected along with increased PGC-1α protein levels in adipose tissue [32]. Regulation of PGC-1α turnover by GSK3β is also a component of the oxidative stress response in cultured cells and in vivo. GSK3β is downstream of the insulin and the Wnt signaling pathways both of which have been negatively associated with longevity [71, 72]. GSK3β has been linked to the pathogenesis of Alzheimer's disease [73], although it's role in PGC-1α regulation has not been explored in this context.
4. Regulation of PGC-1α activity
Increasing evidence suggests that modifications can result in distinct effects on PGC-1α activity in a manner that is a) stimulus dependent and b) tissue dependent. For example, the mitogen activated kinase p38 has been reported to increase PGC-1α activity upon stress in cardiac myocytes [74], in response to cytokines in myotubes [34] and is required for activation of PGC-1α through calcium calmodulin signaling pathway [75] or in response to trophic factors at the neuromuscular junction [76]. Phosphorylation of PGC-1α by p38 prevents interaction with p160 repressor [77, 78] and increases protein stability [34]. In contrast, a p38-dependent reduction in transcript levels of PGC-1α is observed in myotubes treated with the saturated fatty acid palmitate [79]. The activation of PGC-1α is not likely to be an isolated event in normal physiology. The interplay between PGC-1α and other factors downstream of a given stimulus is expected to be complex. It would appear that factors like PGC-1α are regulated as part of a coordinate response that is influenced by combinations of stimuli.
Tissue specificity of the role of PGC-1α is a further consideration in assessing the impact of PGC-1α manipulation. The metabolic shifts that occur in response to fasting highlight the non-equivalence of PGC-1α function in distinct tissues. In liver PGC-1α is upregulated by fasting and induces a shift in metabolism toward increased glucose output [80]. The liver fasting response involves activation of PGC-1α by SIRT1 dependent deacytylation [61]. Fasting also causes SIRT1 dependent activation of PGC-1α in skeletal muscle but the consequence is the induction of mitochondrial fatty acid oxidation [62]. The role of PGC-1α in insulin resistance is also tissue specific. In the pancreas, PGC-1α is elevated in mouse models of obesity and overexpression of PGC-1α in wildtype mice reproduces the defect in glucose regulatored insulin secretion [81]. PGC-1α regulates expression of UCP2 (uncoupling protein 2), a negative regulator of insulin secretion [43]. Interestingly, SIRT1 represses UCP2 expression in pancreas [82] indicating that SIRT1 inhibits rather than activates PGC-1α in this tissue. In cultured islets, PGC-1α expression is increased by elevated fatty acids and decreased by elevated glucose [83], presumably impacting the requisite changes in UCP2 to reduce or increase insulin secretion. PGC-1α levels are decreased and expression of PGC-1α target genes is coordinately down-regulated in skeletal muscle in human diabetics [84, 85]. Skeletal muscle specific knockout of PGC-1α in mice causes reduced insulin tolerance and there are overt defects in pancreatic islet morphology indicting that metabolic deficiency in muscle impacts pancreatic function [86]. Heterozygotes display increased levels of pro-inflammatory cytokines IL-6 and TNFα in skeletal muscle, and the inverse relationship between PGC-1α levels and expression of IL-6 and TNFα is also observed in glucose intolerant humans [86]. In contrast to skeletal muscle, PGC-1α is elevated in liver in mouse models of diabetes and regulates the expression of TRB3, an inhibitor of insulin-dependent AKT activation [87, 88]. Metabolic derangements in the diabetic heart are consistent with chronic activation of PPARα/PGC-1α [89, 90], a phenotype reminiscent of that observed with cardiac specific overexpression of PGC-1α [91].
The pleiotropic phenotypes of the knock out mice point to an integrative role for PGC-1α across tissues that is particularly important in the adaptive response [24, 25]. The loss of PGC-1α was characterized in two independent studies involving animals ∼3-4 months or younger. Not surprisingly, the thermogenic response and the response to exercise are defective in knock outs and both mouse models display abnormalities in the central nervous system. Unexpectedly, loss of PGC-1α affords protection against diet-induced insulin resistance [24] and obesity [25]. It is possible that a compensatory mechanism regulating this response is in place. The total lack of PGC-1α necessitates that functions normally provided by PGC-1α be supplied by alternate means.
5. Tissue specific aging and the influence of PGC-1α
The effect of age is often most overt in post-mitotic tissues. Although few studies have directly measured changes in PGC-1α function and activity with age, there are an abundance studies that provide indirect evidence in support of a role for PGC-1α in aging and in anti-aging.
5.1. Skeletal muscle
Aging causes a gradual loss of muscle mass that is characterized by fiber shrinkage and fiber loss [92]. With age the girth of limb muscles shrink considerably and contain significantly more fat and connective tissue. The fibers that make up skeletal muscle are heterogenous and differ mechanically and metabolically [93]. Type II glycolytic fibers are most vulnerable to the effects of age in contrast to the more resilient Type I oxidative fibers.
In skeletal muscle, PGC-1α expression is activated by CaMKIV resulting in increased and augmented mitochondrial content and activity [27, 94]. Activation of PGC-1α triggers an autoregulatory mechanism where PGC-1α coactivates muscle specific transcription factor MEF2 to drive expression from its own promoter [38]. PGC1-α is involved in fiber type switching, shifting fiber type distribution toward Type I oxidative fibers [39]. There is a rapid induction of PGC-1α in response to exercise in rats [95] and may be regulated through β-adrenergic receptor signaling [96]. PGC-1α increases are fiber type specific [46] and may contribute to the changes in fiber type distribution as a result of endurance training. PGC-1α overexpression ameliorates defects in β-oxidation caused by high lipid load in myocytes [97]. Similarly, overexpression of PPARδ in skeletal muscle in mice induces a switch to Type I fiber and not only improves running endurance, but also protects against obesity [98].
Despite its many beneficial effects, exercise is not thought to slow the primary causes of aging and has no impact on maximum lifespan in rodents [99]. Although the role of PGC-1α in primary muscle aging is not clear, the decline in mitochondrial function is well documented. Activation of PGC-1α prevents the onset of myopathy and extends lifespan in a mouse model of mitochondrial myopathy [100], suggesting that increase in PGC-1α can partially compensate for pervasive mitochondrial defects. Another mouse model of myopathy involves tissue-specific deletion of the COX10 gene through the use of the Cre-LoxP system. In this mouse model, Cre recombination occurs over time in the muscle nuclei, leading to a partial and segmental COX deficiency [101]. This pattern is more similar to that observed in disorders associated with mtDNA deletions, and also in natural aging. The induction of mitochondrial biogenesis through transgenic expression of PGC-1α in skeletal muscle, or by administration of bezafibrate, a PPAR agonist that induces PGC-1α in multiple tissues, led to enhanced OXPHOS capacity, delayed onset of myopathy and markedly prolonged lifespan [100].
The contribution of defects in skeletal muscle metabolism to aging-associated insulin resistance implicates diminished PGC-1α activity in onset of morbidity. Mitochondrial dysfunction has been implicated in Type 2 diabetes and may be a contributing factor to reduced insulin sensitivity and defects in pancreatic insulin secretion: both critical factors in the pathogenesis of the disease [102]. Mitochondrial activity is reduced in skeletal muscle from insulin resistant individuals and fiber type composition is altered as reflected in a reduced proportion of Type I oxidative fibers [103]. Transcriptional analysis reveals that expression of PGC-1α target genes is coordinately down-regulated in human diabetics [84, 85] and PGC-1α genetic polymorphisms have been linked to Type II Diabetes [104, 105]. PGC-1α levels are reduced in genetic and dietary induced rodent obesity models [79]. Treatments that successfully treat diabetes also increase PGC-1α expression [106] and muscular oxidative capacity [107].
5.2. Heart
The major source of energy in the adult heart is derived from fatty acid oxidation [108]. In old age, a shift away from lipid metabolism toward carbohydrate metabolism is suggested by changes in gene transcript abundance. Genes involved in lipid transport, lipolysis and fatty acid oxidation are down-regulated and genes involved in carbohydrate metabolism are induced [4]. These metabolic alterations are also observed in pathological hypertrophy, where loss of PPARα/PGC-1α activity is associated with mitochondrial insufficiency in fatty acid oxidation [109]. CR opposes the age-induced metabolic shift in heart and furthermore, induces the expression of genes involved in mitochondrial electron transport system. [4]. Deregulation of mitochondrial energy metabolism is a shared phenotype of aging and heart disease and implementation of CR anti-aging regimen targets this same process.
PGC-1α is required for maintenance of cardiac mitochondrial function and is regulated by MEF2 and HDAC5 [110]. Normal growth in response to development or exercise is associated with increased PGC-1α activity [111]; however, inappropriate increase in PGC-1α activity through overexpression causes uncontrolled mitochondrial proliferation that results in cardiomyopathy [91]. Indeed, diabetic cardiac myopathy is associated with derangements in myocardial energy metabolism and chronic activation of fatty acid metabolism [90]. In contrast, reduced activity of PGC-1α is associated with pathological forms of cardiac hypertrophy [109]. Deregulation of PGC-1α has clear adverse effects in heart, consistent with a critical role for maintenance of cardiac mitochondrial function in heart health.
Further insight into the effect of mitochondrial dysfunction in heart is revealed in mutant and knockout studies. Activation of PPARα and γ reduces blood pressure and cardiac hypertrophy in spontaneously hypertensive heart failure rats [112]. Furthermore, cardiac specific knockout of PPARα causes deficiencies in mitochondrial β-oxidation, leading to eventual myocardial damage and fibrosis [113]. Mitochondrial regulation may also be important in the adaptive response, communicating metabolic status to the nucleus. PPARα ligand dependent transcriptional activity and co-activation by PGC-1α is activated in heart by stress including ischemia, and hypoxia [74] suggesting that PGC-1α is part of a proactive response to cardiac tissue damage. Progressive myocardial disease due to cardiac specific knockout of Tfam, causes a shift in metabolism away from fatty acid metabolism toward glucose concomitant with progression of cardiac mitochondrial dysfunction [114]. Deregulation of mitochondrial energy metabolism is a central feature of cardiac disease indicating that the balance of PGC-1α function is of utmost importance in cardiac biology.
5.3. Adipose
Increasingly evident that adipose tissue, far from being an inert storage depot for unused fat, acts as an endocrine organ and is involved in organismal metabolic homeostasis [115, 116]. Adipogenic signaling influences energy balance and inflammation [117] and adipose derived factors are likely to be key components in metabolic disorders linked to obesity [11, 12]. Aging is associated with adverse alterations in body fat distribution resulting in a higher ratio of visceral to subcutaneous fat, and is associated with deregulated adipose function [118-120].
CR has a dramatic effect on adipose tissue adiposity, morphology and metabolism and the expression of pro-inflammatory genes is reduced [121, 122]. PGC-1α protein levels are increased in adipose from CR animals, and gene targets of PGC-1α are increased. CR induces inhibitory phosphoryation of GSK3β, the regulator of PGC-1α stability, and an increase in levels of PGC-1α activator, SIRT1, in adipose tissue [32]. The metabolic shifts caused by in CR are consistent with the activation of the PGC-1α regulated pathways, including the increased expression of the β3 adrenergic receptor (β3AR) and UCP3. In white adipose tissue, activation of the βARs leads to mobilization of fat stores and regulates the release of several adipokines [123].
The increase in PGC-1α may be critical in the activation of adipose tissue by CR. Overexpression of PGC-1α increases the expression of ETS components and FAO enzymes in human adipocytes and transcription profiling indicates a metabolic activation of the fat cells [124]. In contrast, expression of PGC-1α is reduced in adipose tissue from insulin-resistant [125] and morbidly obese [126] individuals. Fat accumulation correlates with systemic oxidative stress humans, and in cultured cells oxidative stress causes deregulated secretion of adipokines [127]. Adipogenic capacity is important in preventing lipid accumulation outside of fat depots and declines with age. CR partially attenuates the age effect and is associated with increased levels of PPARγ and adiponectin [128]. Increased adiponectin stimulates mitochondrial metabolism in skeletal muscle through AMPK [129] providing a link between adipose tissue function and skeletal muscle metabolism presumably through PGC-1α.
A high fat diet causes down-regulation of genes involved in oxidative phosphorylation in skeletal muscle from humans and levels of PGC-1α transcript are modestly reduced within days [130]. A similar decease in PGC-1α is observed following infusion of a triglyceride emulsion to increase serum levels of free fatty acids [131]. PGC-1α levels are dramatically reduced in skeletal muscle in genetic and sustained diet induced mouse models of obesity [79]. A separate study demonstrated increases in PGC-1α upon introduction of a high fat diet in rats [132]. A key difference between these two studies was the source of the high fat in the diet, where PGC-1α was increased, fat was provided in the form of flax seed and olive oil and where PGC-1α was decreased milk fat was the primary fat source. These data support the concept that PGC-1α activity may be sensitive to aging-associated changes in serum triglycerides.
5.4. Nervous system
Abnormalities in mitochondrial function are associated with neurodegenerative disorders including Parkinson's disease [133], Alzheimer's disease [134] and Huntington's disease [135]. The coincidence of mitochondrial dysfunction in these distinct neurodegenerative disorders [136] suggests that mitochondrial efficiency is important in maintaining neural function and plasticity [137]. Progressive neurodegeneration is observed in mice with mosaic respiratory chain deficiency in the cerebral cortex, not only in the affected neurons but also in neighboring cells affected in trans [138]. In wildtype mice brain mitochondrial function declines with age [139] and although increased oxidative stress is observed, it is unclear whether increased oxidative damage is causal or symptomatic in aging-associated behavioral deficits. Absence of functional peroxisomes in oligodendrocytes causes axonal degeneration and neuroinflammation [140], lending support for a metabolic influence on maintenance of neural function.
Defects in mitochondrial energy metabolism have been implicated in the pathogenesis of Huntington's disease [141]. Levels of PGC-1α are reduced in brain from Huntington's disease patients due to repression of PGC-1α gene expression by mutant huntingtin. Expression of PGC-1α partially reverses the toxic effects and provides neuroprotection in the HD mutant mouse. In a spontaneous mutant of the Wallerian axonal degeneration mouse model with delayed axonal degeneration, improved outcome has been linked to SIRT1 [142], a known activator of PGC-1α [60, 61]. In the peripheral nervous system, PGC-1α has been shown to regulate gene expression at the neuromuscular junction and influences expression of acetylcholine receptors in muscle fibers [76]. In addition to regulating mitochondrial energetics, PGC-1α induces the expression of many ROS-detoxifying enzymes, including GPx1 and SOD2. Increase in PGC-1α levels protects neural cells in culture from oxidative-stressor mediated cell death [40].
6. Pharmacological anti-aging strategies: resveratrol
Targeting PGC-1α activity may be a fruitful approach to delay the aging process. Differences in tissue specific roles of PGC-1α and the cross-talk between factors that regulate PGC-1α pose a challenge for design of an agent can actually produce the desired effect. One promising candidate is resveratrol, a plant polyphenol commonly found in red wine that extends lifespan in yeast, worms and flies [143, 144]. Resveratrol treatment activates SIRT1, PGC-1α and AMPK [29, 145-148] and inhibits AKT [149] (Figure 4). The effect of resveratrol on downstream effectors is dose dependent. High doses of resveratrol feeding reduce the adverse effects of feeding a high-fat diet to mice [150]. Mice receiving resveratrol in diets (22 or 186 mg/Kg) have increased survival and reduced liver damage. Because mice on the high (50%) fat diet have a dramatically reduced average lifespan, the effects of resveratrol likely represents prevention of fat-induced pathology. Several parameters in resveratrol-treated mice are consistent with metabolic activation, including increased mitochondria number, activation of AMPK and PGC-1α deacetylation [145, 150]. Since many of the beneficial effects of resveratrol are similar to those that would be expected from a calorie-restricted diet [151], these studies support the concept that activation of PGC-1α may be one of the central mechanisms of action of CR. Feeding of a much lower dose of resveratrol (4.9 mg/Kg) also results in health benefits, including prevention of age-related cardiac dysfunction [152]. Gene expression profiling showed significant overlap between CR-treated and resveratrol-treated mice. Measurement of levels of transcriptional targets of PGC-1α, including PDK4, NRF1, UCP3 and Tfam revealed that PDK4 was induced in CR muscle, heart and brain, and strong induction of UCP3 was observed in CR heart and muscle. Surprisingly, the only PGC-1α target induced by low dose resveratrol was PDK4 in skeletal muscle. These findings suggest that although CR clearly induces PGC-1α or its targets in multiple tissues, resveratrol is not likely to induce PGC-1α activity at low doses [152].
Figure 4. Activation of PGC-1α by resveratrol.
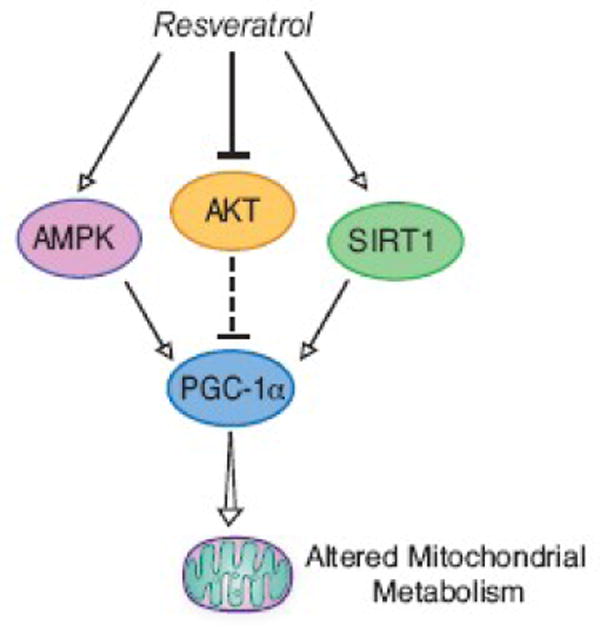
Increased PGC-1α activity is accomplished through regulation of a combination of factors involved in modulating PGC-1α. The positive effect of resveratrol on SIRT1 and AMPK, activators of PGC-1α, is augmented by the negative effect on AKT inhibitor of PGC-1α. Resveratrol treatment has been shown to improve parameters of aging at low doses in mice (see text) although its impact on aging in primates is not known.
An alternate approach to induce PGC-1α in tissues may be through the use of fibrates. In a mouse model of myopathy, the induction of mitochondrial biogenesis through transgenic expression of PGC-1α in skeletal muscle, or by administration of bezafibrate, a PPAR agonist that induces PGC-1α in skeletal muscle and heart [153], led to enhanced OXPHOS capacity, delayed onset of myopathy and markedly prolonged lifespan [100]. If PGC-1α overexpression can markedly improve such overt phenotypes, it seems likely that its modulation may be beneficial in the context of age-related mitochondrial dysfunction.
Conclusion
The quest for new and effective means to delay aging and the onset of aging-associated disease has never looked better. Mechanistic insights in the aging process are being gleaned at an overwhelming rate through studies in cells in culture and in short-lived non-mammalian and mammalian species. The impact of mitochondrial dysfunction is pervasive and as described here likely extends to multiple aspects of normal physiology of aging. We propose that PGC-1α is a very good candidate target to correct or compensate for age-induced changes in mitochondrial function. Extension of these extremely promising mechanistic studies to primates should reveal factors that may be unique to aging in a long-lived species.
Acknowledgments
This work was supported by grants from NIH/NIA (AG11915), and NIH/NCRR/CTSA (UL1RR025011). Thanks to Dhanu Shanmuganayagam for critical reading of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Masoro EJ. Physiology of aging. Int J Sport Nutr Exerc Metab. 2001;11(Suppl):S218–222. doi: 10.1123/ijsnem.11.s1.s218. [DOI] [PubMed] [Google Scholar]
- 2.Lee CK, Klopp RG, Weindruch R, Prolla TA. Gene expression profile of aging and its retardation by caloric restriction. Science. 1999;285:1390–1393. doi: 10.1126/science.285.5432.1390. [DOI] [PubMed] [Google Scholar]
- 3.Lee CK, Weindruch R, Prolla TA. Gene-expression profile of the ageing brain in mice. Nat Genet. 2000;25:294–297. doi: 10.1038/77046. [DOI] [PubMed] [Google Scholar]
- 4.Lee CK, Allison DB, Brand J, Weindruch R, Prolla TA. Transcriptional profiles associated with aging and middle age-onset caloric restriction in mouse hearts. Proc Natl Acad Sci U S A. 2002;99:14988–14993. doi: 10.1073/pnas.232308999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park SK, Prolla TA. Lessons learned from gene expression profile studies of aging and caloric restriction. Ageing Res Rev. 2005;4:55–65. doi: 10.1016/j.arr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 6.McCarroll SA, Murphy CT, Zou S, et al. Comparing genomic expression patterns across species identifies shared transcriptional profile in aging. Nat Genet. 2004;36:197–204. doi: 10.1038/ng1291. [DOI] [PubMed] [Google Scholar]
- 7.Zahn JM, Sonu R, Vogel H, et al. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet. 2006;2:e115. doi: 10.1371/journal.pgen.0020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Short KR, Bigelow ML, Kahl J, et al. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005;102:5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petersen KF, Befroy D, Dufour S, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–1428. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 11.Lago F, Dieguez C, Gomez-Reino J, Gualillo O. The emerging role of adipokines as mediators of inflammation and immune responses. Cytokine Growth Factor Rev. 2007;18:313–325. doi: 10.1016/j.cytogfr.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 12.Wisse BE. The inflammatory syndrome: the role of adipose tissue cytokines in metabolic disorders linked to obesity. J Am Soc Nephrol. 2004;15:2792–2800. doi: 10.1097/01.ASN.0000141966.69934.21. [DOI] [PubMed] [Google Scholar]
- 13.Guarente L. Sirtuins as potential targets for metabolic syndrome. Nature. 2006;444:868–874. doi: 10.1038/nature05486. [DOI] [PubMed] [Google Scholar]
- 14.Masoro EJ. Caloric restriction and aging: an update. Exp Gerontol. 2000;35:299–305. doi: 10.1016/s0531-5565(00)00084-x. [DOI] [PubMed] [Google Scholar]
- 15.Weindruch R, Naylor PH, Goldstein AL, Walford RL. Influences of aging and dietary restriction on serum thymosin alpha 1 levels in mice. J Gerontol. 1988;43:B40–42. doi: 10.1093/geronj/43.2.b40. [DOI] [PubMed] [Google Scholar]
- 16.Anderson RM, Weindruch R. Metabolic reprogramming in dietary restriction. Interdiscip Top Gerontol. 2007;35:18–38. doi: 10.1159/000096554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Civitarese AE, Carling S, Heilbronn LK, et al. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007;4:e76. doi: 10.1371/journal.pmed.0040076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- 19.Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta), a novel PGC-1-related transcription coactivator associated with host cell factor. J Biol Chem. 2002;277:1645–1648. doi: 10.1074/jbc.C100631200. [DOI] [PubMed] [Google Scholar]
- 20.Andersson U, Scarpulla RC. Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol Cell Biol. 2001;21:3738–3749. doi: 10.1128/MCB.21.11.3738-3749.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mortensen OH, Plomgaard P, Fischer CP, Hansen AK, Pilegaard H, Pedersen BK. PGC-1beta is downregulated by training in human skeletal muscle: no effect of training twice every second day vs. once daily on expression of the PGC-1 family. J Appl Physiol. 2007;103:1536–1542. doi: 10.1152/japplphysiol.00575.2007. [DOI] [PubMed] [Google Scholar]
- 22.Lin J, Tarr PT, Yang R, et al. PGC-1beta in the regulation of hepatic glucose and energy metabolism. J Biol Chem. 2003;278:30843–30848. doi: 10.1074/jbc.M303643200. [DOI] [PubMed] [Google Scholar]
- 23.Lelliott CJ, Medina-Gomez G, Petrovic N, et al. Ablation of PGC-1beta results in defective mitochondrial activity, thermogenesis, hepatic function, and cardiac performance. PLoS Biol. 2006;4:e369. doi: 10.1371/journal.pbio.0040369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leone TC, Lehman JJ, Finck BN, et al. PGC-1alpha deficiency causes multisystem energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin J, Wu PH, Tarr PT, et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 26.Sonoda J, Mehl IR, Chong LW, Nofsinger RR, Evans RM. PGC-1beta controls mitochondrial metabolism to modulate circadian activity, adaptive thermogenesis, and hepatic steatosis. Proc Natl Acad Sci U S A. 2007;104:5223–5228. doi: 10.1073/pnas.0611623104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Z, Puigserver P, Andersson U, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 28.Corton JC, Brown-Borg HM. Peroxisome proliferator-activated receptor gamma coactivator 1 in caloric restriction and other models of longevity. J Gerontol A Biol Sci Med Sci. 2005;60:1494–1509. doi: 10.1093/gerona/60.12.1494. [DOI] [PubMed] [Google Scholar]
- 29.Feige JN, Auwerx J. Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol. 2007;17:292–301. doi: 10.1016/j.tcb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 30.Puigserver P, Rhee J, Donovan J, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 31.Handschin C, Spiegelman BM. The role of exercise and PGC1alpha in inflammation and chronic disease. Nature. 2008;454:463–469. doi: 10.1038/nature07206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson RM, Barger JL, Edwards MG, et al. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell. 2008;7:101–111. doi: 10.1111/j.1474-9726.2007.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sano M, Tokudome S, Shimizu N, et al. Intramolecular control of protein stability, subnuclear compartmentalization, and coactivator function of peroxisome proliferator-activated receptor gamma coactivator 1alpha. J Biol Chem. 2007;282:25970–25980. doi: 10.1074/jbc.M703634200. [DOI] [PubMed] [Google Scholar]
- 34.Puigserver P, Rhee J, Lin J, et al. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol Cell. 2001;8:971–982. doi: 10.1016/s1097-2765(01)00390-2. [DOI] [PubMed] [Google Scholar]
- 35.Wu Z, Huang X, Feng Y, et al. Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1alpha transcription and mitochondrial biogenesis in muscle cells. Proc Natl Acad Sci U S A. 2006;103:14379–14384. doi: 10.1073/pnas.0606714103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- 37.Ichida M, Nemoto S, Finkel T. Identification of a specific molecular repressor of the peroxisome proliferator-activated receptor gamma Coactivator-1 alpha (PGC-1alpha) J Biol Chem. 2002;277:50991–50995. doi: 10.1074/jbc.M210262200. [DOI] [PubMed] [Google Scholar]
- 38.Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM. An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1alpha expression in muscle. Proc Natl Acad Sci U S A. 2003;100:7111–7116. doi: 10.1073/pnas.1232352100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin J, Wu H, Tarr PT, et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 40.St-Pierre J, Drori S, Uldry M, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 41.Valle I, Alvarez-Barrientos A, Arza E, Lamas S, Monsalve M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res. 2005;66:562–573. doi: 10.1016/j.cardiores.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 42.Southgate RJ, Bruce CR, Carey AL, et al. PGC-1alpha gene expression is down-regulated by Akt- mediated phosphorylation and nuclear exclusion of FoxO1 in insulin-stimulated skeletal muscle. FASEB J. 2005;19:2072–2074. doi: 10.1096/fj.05-3993fje. [DOI] [PubMed] [Google Scholar]
- 43.Oberkofler H, Klein K, Felder TK, Krempler F, Patsch W. Role of peroxisome proliferator-activated receptor-gamma coactivator-1alpha in the transcriptional regulation of the human uncoupling protein 2 gene in INS-1E cells. Endocrinology. 2006;147:966–976. doi: 10.1210/en.2005-0817. [DOI] [PubMed] [Google Scholar]
- 44.Wulf A, Harneit A, Kroger M, Kebenko M, Wetzel MG, Weitzel JM. T3-mediated expression of PGC-1alpha via a far upstream located thyroid hormone response element. Mol Cell Endocrinol. 2008;287:90–95. doi: 10.1016/j.mce.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 45.Liu C, Li S, Liu T, Borjigin J, Lin JD. Transcriptional coactivator PGC-1alpha integrates the mammalian clock and energy metabolism. Nature. 2007;447:477–481. doi: 10.1038/nature05767. [DOI] [PubMed] [Google Scholar]
- 46.Russell AP, Feilchenfeldt J, Schreiber S, et al. Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-gamma coactivator-1 and peroxisome proliferator-activated receptor-alpha in skeletal muscle. Diabetes. 2003;52:2874–2881. doi: 10.2337/diabetes.52.12.2874. [DOI] [PubMed] [Google Scholar]
- 47.Arany Z, Foo SY, Ma Y, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- 48.O'Hagan KA, Cocchiglia S, Zhdanov AV, et al. PGC-1alpha is coupled to HIF-1alpha-dependent gene expression by increasing mitochondrial oxygen consumption in skeletal muscle cells. Proc Natl Acad Sci U S A. 2009;106:2188–2193. doi: 10.1073/pnas.0808801106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Putman CT, Kiricsi M, Pearcey J, et al. AMPK activation increases uncoupling protein-3 expression and mitochondrial enzyme activities in rat muscle without fibre type transitions. J Physiol. 2003;551:169–178. doi: 10.1113/jphysiol.2003.040691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zong H, Ren JM, Young LH, et al. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99:15983–15987. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reznick RM, Zong H, Li J, et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007;5:151–156. doi: 10.1016/j.cmet.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oliveira RL, Ueno M, de Souza CT, et al. Cold-induced PGC-1alpha expression modulates muscle glucose uptake through an insulin receptor/Akt-independent, AMPK-dependent pathway. Am J Physiol Endocrinol Metab. 2004;287:E686–695. doi: 10.1152/ajpendo.00103.2004. [DOI] [PubMed] [Google Scholar]
- 54.Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 56.Schieke SM, Phillips D, McCoy JP, et al. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J Biol Chem. 2006;281:27643–27652. doi: 10.1074/jbc.M603536200. [DOI] [PubMed] [Google Scholar]
- 57.Linford NJ, Beyer RP, Gollahon K, et al. Transcriptional response to aging and caloric restriction in heart and adipose tissue. Aging Cell. 2007;6:673–688. doi: 10.1111/j.1474-9726.2007.00319.x. [DOI] [PubMed] [Google Scholar]
- 58.Schieke SM, Finkel T. Mitochondrial signaling, TOR, and life span. Biol Chem. 2006;387:1357–1361. doi: 10.1515/BC.2006.170. [DOI] [PubMed] [Google Scholar]
- 59.Lerin C, Rodgers JT, Kalume DE, Kim SH, Pandey A, Puigserver P. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab. 2006;3:429–438. doi: 10.1016/j.cmet.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 60.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 61.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 62.Gerhart-Hines Z, Rodgers JT, Bare O, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Longo VD, Kennedy BK. Sirtuins in aging and age-related disease. Cell. 2006;126:257–268. doi: 10.1016/j.cell.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 64.Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 65.Guarente L. Mitochondria--a nexus for aging, calorie restriction, and sirtuins? Cell. 2008;132:171–176. doi: 10.1016/j.cell.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen D, Bruno J, Easlon E, et al. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008;22:1753–1757. doi: 10.1101/gad.1650608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell. 2007;12:487–502. doi: 10.1016/j.devcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 68.Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature. 2007;447:1012–1016. doi: 10.1038/nature05861. [DOI] [PubMed] [Google Scholar]
- 69.Cook SA, Matsui T, Li L, Rosenzweig A. Transcriptional effects of chronic Akt activation in the heart. J Biol Chem. 2002;277:22528–22533. doi: 10.1074/jbc.M201462200. [DOI] [PubMed] [Google Scholar]
- 70.Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem. 2005;280:32081–32089. doi: 10.1074/jbc.M502876200. [DOI] [PubMed] [Google Scholar]
- 71.Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 72.Liu H, Fergusson MM, Castilho RM, et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317:803–806. doi: 10.1126/science.1143578. [DOI] [PubMed] [Google Scholar]
- 73.Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J Neurochem. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barger PM, Browning AC, Garner AN, Kelly DP. p38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor alpha: a potential role in the cardiac metabolic stress response. J Biol Chem. 2001;276:44495–44501. doi: 10.1074/jbc.M105945200. [DOI] [PubMed] [Google Scholar]
- 75.Wright DC, Geiger PC, Han DH, Jones TE, Holloszy JO. Calcium induces increases in peroxisome proliferator-activated receptor gamma coactivator-1alpha and mitochondrial biogenesis by a pathway leading to p38 mitogen-activated protein kinase activation. J Biol Chem. 2007;282:18793–18799. doi: 10.1074/jbc.M611252200. [DOI] [PubMed] [Google Scholar]
- 76.Handschin C, Kobayashi YM, Chin S, Seale P, Campbell KP, Spiegelman BM. PGC-1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev. 2007;21:770–783. doi: 10.1101/gad.1525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fan M, Rhee J, St-Pierre J, et al. Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1alpha: modulation by p38 MAPK. Genes Dev. 2004;18:278–289. doi: 10.1101/gad.1152204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Knutti D, Kressler D, Kralli A. Regulation of the transcriptional coactivator PGC-1 via MAPK-sensitive interaction with a repressor. Proc Natl Acad Sci U S A. 2001;98:9713–9718. doi: 10.1073/pnas.171184698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Crunkhorn S, Dearie F, Mantzoros C, et al. Peroxisome proliferator activator receptor gamma coactivator-1 expression is reduced in obesity: potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. J Biol Chem. 2007;282:15439–15450. doi: 10.1074/jbc.M611214200. [DOI] [PubMed] [Google Scholar]
- 80.Yoon JC, Puigserver P, Chen G, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 81.Yoon JC, Xu G, Deeney JT, et al. Suppression of beta cell energy metabolism and insulin release by PGC-1alpha. Dev Cell. 2003;5:73–83. doi: 10.1016/s1534-5807(03)00170-9. [DOI] [PubMed] [Google Scholar]
- 82.Bordone L, Motta MC, Picard F, et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 2006;4:e31. doi: 10.1371/journal.pbio.0040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang P, Liu C, Zhang C, et al. Free fatty acids increase PGC-1alpha expression in isolated rat islets. FEBS Lett. 2005;579:1446–1452. doi: 10.1016/j.febslet.2005.01.046. [DOI] [PubMed] [Google Scholar]
- 84.Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 85.Patti ME, Butte AJ, Crunkhorn S, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Handschin C, Choi CS, Chin S, et al. Abnormal glucose homeostasis in skeletal muscle-specific PGC-1alpha knockout mice reveals skeletal muscle-pancreatic beta cell crosstalk. J Clin Invest. 2007;117:3463–3474. doi: 10.1172/JCI31785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300:1574–1577. doi: 10.1126/science.1079817. [DOI] [PubMed] [Google Scholar]
- 88.Koo SH, Satoh H, Herzig S, et al. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat Med. 2004;10:530–534. doi: 10.1038/nm1044. [DOI] [PubMed] [Google Scholar]
- 89.Finck BN, Han X, Courtois M, et al. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci U S A. 2003;100:1226–1231. doi: 10.1073/pnas.0336724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Finck BN, Kelly DP. Peroxisome proliferator-activated receptor alpha (PPARalpha) signaling in the gene regulatory control of energy metabolism in the normal and diseased heart. J Mol Cell Cardiol. 2002;34:1249–1257. doi: 10.1006/jmcc.2002.2061. [DOI] [PubMed] [Google Scholar]
- 91.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lexell J. Human aging, muscle mass, and fiber type composition. J Gerontol A Biol Sci Med Sci. 1995;50(Spec No):11–16. doi: 10.1093/gerona/50a.special_issue.11. [DOI] [PubMed] [Google Scholar]
- 93.Bassel-Duby R, Olson EN. Signaling pathways in skeletal muscle remodeling. Annu Rev Biochem. 2006;75:19–37. doi: 10.1146/annurev.biochem.75.103004.142622. [DOI] [PubMed] [Google Scholar]
- 94.Wu H, Kanatous SB, Thurmond FA, et al. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science. 2002;296:349–352. doi: 10.1126/science.1071163. [DOI] [PubMed] [Google Scholar]
- 95.Baar K, Wende AR, Jones TE, et al. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. Faseb J. 2002;16:1879–1886. doi: 10.1096/fj.02-0367com. [DOI] [PubMed] [Google Scholar]
- 96.Miura S, Kawanaka K, Kai Y, et al. An increase in murine skeletal muscle peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) mRNA in response to exercise is mediated by beta-adrenergic receptor activation. Endocrinology. 2007;148:3441–3448. doi: 10.1210/en.2006-1646. [DOI] [PubMed] [Google Scholar]
- 97.Koves TR, Li P, An J, et al. Peroxisome proliferator-activated receptor-gamma co-activator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J Biol Chem. 2005;280:33588–33598. doi: 10.1074/jbc.M507621200. [DOI] [PubMed] [Google Scholar]
- 98.Wang YX, Zhang CL, Yu RT, et al. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004;2:e294. doi: 10.1371/journal.pbio.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Holloszy JO. Exercise and food restriction in rats. J Nutr. 1992;122:774–777. doi: 10.1093/jn/122.suppl_3.774. [DOI] [PubMed] [Google Scholar]
- 100.Wenz T, Diaz F, Spiegelman BM, Moraes CT. Activation of the PPAR/PGC-1alpha pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab. 2008;8:249–256. doi: 10.1016/j.cmet.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 101.Diaz F, Thomas CK, Garcia S, Hernandez D, Moraes CT. Mice lacking COX10 in skeletal muscle recapitulate the phenotype of progressive mitochondrial myopathies associated with cytochrome c oxidase deficiency. Hum Mol Genet. 2005;14:2737–2748. doi: 10.1093/hmg/ddi307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 103.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ek J, Andersen G, Urhammer SA, et al. Mutation analysis of peroxisome proliferator-activated receptor-gamma coactivator-1 (PGC-1) and relationships of identified amino acid polymorphisms to Type II diabetes mellitus. Diabetologia. 2001;44:2220–2226. doi: 10.1007/s001250100032. [DOI] [PubMed] [Google Scholar]
- 105.Hara K, Tobe K, Okada T, et al. A genetic variation in the PGC-1 gene could confer insulin resistance and susceptibility to Type II diabetes. Diabetologia. 2002;45:740–743. doi: 10.1007/s00125-002-0803-z. [DOI] [PubMed] [Google Scholar]
- 106.Al-Khalili L, Forsgren M, Kannisto K, Zierath JR, Lonnqvist F, Krook A. Enhanced insulin-stimulated glycogen synthesis in response to insulin, metformin or rosiglitazone is associated with increased mRNA expression of GLUT4 and peroxisomal proliferator activator receptor gamma co-activator 1. Diabetologia. 2005;48:1173–1179. doi: 10.1007/s00125-005-1741-3. [DOI] [PubMed] [Google Scholar]
- 107.Mensink M, Hesselink MK, Russell AP, Schaart G, Sels JP, Schrauwen P. Improved skeletal muscle oxidative enzyme activity and restoration of PGC-1 alpha and PPAR beta/delta gene expression upon rosiglitazone treatment in obese patients with type 2 diabetes mellitus. Int J Obes (Lond) 2007;31:1302–1310. doi: 10.1038/sj.ijo.0803567. [DOI] [PubMed] [Google Scholar]
- 108.Huss JM, Kelly DP. Nuclear receptor signaling and cardiac energetics. Circ Res. 2004;95:568–578. doi: 10.1161/01.RES.0000141774.29937.e3. [DOI] [PubMed] [Google Scholar]
- 109.Lehman JJ, Kelly DP. Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clin Exp Pharmacol Physiol. 2002;29:339–345. doi: 10.1046/j.1440-1681.2002.03655.x. [DOI] [PubMed] [Google Scholar]
- 110.Czubryt MP, McAnally J, Fishman GI, Olson EN. Regulation of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1 alpha) and mitochondrial function by MEF2 and HDAC5. Proc Natl Acad Sci U S A. 2003;100:1711–1716. doi: 10.1073/pnas.0337639100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.O'Neill BT, Kim J, Wende AR, et al. A conserved role for phosphatidylinositol 3-kinase but not Akt signaling in mitochondrial adaptations that accompany physiological cardiac hypertrophy. Cell Metab. 2007;6:294–306. doi: 10.1016/j.cmet.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Alibin CP, Kopilas MA, Anderson HD. Suppression of cardiac myocyte hypertrophy by conjugated linoleic acid: role of peroxisome proliferator-activated receptors alpha and gamma. J Biol Chem. 2008;283:10707–10715. doi: 10.1074/jbc.M800035200. [DOI] [PubMed] [Google Scholar]
- 113.Watanabe K, Fujii H, Takahashi T, et al. Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor alpha associated with age-dependent cardiac toxicity. J Biol Chem. 2000;275:22293–22299. doi: 10.1074/jbc.M000248200. [DOI] [PubMed] [Google Scholar]
- 114.Hansson A, Hance N, Dufour E, et al. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc Natl Acad Sci U S A. 2004;101:3136–3141. doi: 10.1073/pnas.0308710100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89:2548–2556. doi: 10.1210/jc.2004-0395. [DOI] [PubMed] [Google Scholar]
- 116.Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. 2006;444:847–853. doi: 10.1038/nature05483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6:772–783. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 118.Clement K, Langin D. Regulation of inflammation-related genes in human adipose tissue. J Intern Med. 2007;262:422–430. doi: 10.1111/j.1365-2796.2007.01851.x. [DOI] [PubMed] [Google Scholar]
- 119.Das M, Gabriely I, Barzilai N. Caloric restriction, body fat and ageing in experimental models. Obes Rev. 2004;5:13–19. doi: 10.1111/j.1467-789x.2004.00115.x. [DOI] [PubMed] [Google Scholar]
- 120.Kirkland JL, Tchkonia T, Pirtskhalava T, Han J, Karagiannides I. Adipogenesis and aging: does aging make fat go MAD? Exp Gerontol. 2002;37:757–767. doi: 10.1016/s0531-5565(02)00014-1. [DOI] [PubMed] [Google Scholar]
- 121.Higami Y, Barger JL, Page GP, et al. Energy restriction lowers the expression of genes linked to inflammation, the cytoskeleton, the extracellular matrix, and angiogenesis in mouse adipose tissue. J Nutr. 2006;136:343–352. doi: 10.1093/jn/136.2.343. [DOI] [PubMed] [Google Scholar]
- 122.Higami Y, Pugh TD, Page GP, Allison DB, Prolla TA, Weindruch R. Adipose tissue energy metabolism: altered gene expression profile of mice subjected to long-term caloric restriction. Faseb J. 2004;18:415–417. doi: 10.1096/fj.03-0678fje. [DOI] [PubMed] [Google Scholar]
- 123.Collins S, Cao W, Robidoux J. Learning new tricks from old dogs: beta-adrenergic receptors teach new lessons on firing up adipose tissue metabolism. Mol Endocrinol. 2004;18:2123–2131. doi: 10.1210/me.2004-0193. [DOI] [PubMed] [Google Scholar]
- 124.Tiraby C, Tavernier G, Lefort C, et al. Acquirement of brown fat cell features by human white adipocytes. J Biol Chem. 2003;278:33370–33376. doi: 10.1074/jbc.M305235200. [DOI] [PubMed] [Google Scholar]
- 125.Hammarstedt A, Jansson PA, Wesslau C, Yang X, Smith U. Reduced expression of PGC-1 and insulin-signaling molecules in adipose tissue is associated with insulin resistance. Biochem Biophys Res Commun. 2003;301:578–582. doi: 10.1016/s0006-291x(03)00014-7. [DOI] [PubMed] [Google Scholar]
- 126.Semple RK, Crowley VC, Sewter CP, et al. Expression of the thermogenic nuclear hormone receptor coactivator PGC-1alpha is reduced in the adipose tissue of morbidly obese subjects. Int J Obes Relat Metab Disord. 2003 doi: 10.1038/sj.ijo.0802482. [DOI] [PubMed] [Google Scholar]
- 127.Furukawa S, Fujita T, Shimabukuro M, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhu M, Lee GD, Ding L, et al. Adipogenic signaling in rat white adipose tissue: modulation by aging and calorie restriction. Exp Gerontol. 2007;42:733–744. doi: 10.1016/j.exger.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Civitarese AE, Ukropcova B, Carling S, et al. Role of adiponectin in human skeletal muscle bioenergetics. Cell Metab. 2006;4:75–87. doi: 10.1016/j.cmet.2006.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sparks LM, Xie H, Koza RA, et al. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes. 2005;54:1926–1933. doi: 10.2337/diabetes.54.7.1926. [DOI] [PubMed] [Google Scholar]
- 131.Richardson DK, Kashyap S, Bajaj M, et al. Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J Biol Chem. 2005;280:10290–10297. doi: 10.1074/jbc.M408985200. [DOI] [PubMed] [Google Scholar]
- 132.Hancock CR, Han DH, Chen M, et al. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc Natl Acad Sci U S A. 2008;105:7815–7820. doi: 10.1073/pnas.0802057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Banerjee R, Starkov AA, Beal MF, Thomas B. Mitochondrial dysfunction in the limelight of Parkinson's disease pathogenesis. Biochim Biophys Acta. 2008 doi: 10.1016/j.bbadis.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rhein V, Eckert A. Effects of Alzheimer's amyloid-beta and tau protein on mitochondrial function -- role of glucose metabolism and insulin signalling. Arch Physiol Biochem. 2007;113:131–141. doi: 10.1080/13813450701572288. [DOI] [PubMed] [Google Scholar]
- 135.Browne SE. Mitochondria and Huntington's disease pathogenesis: insight from genetic and chemical models. Ann N Y Acad Sci. 2008;1147:358–382. doi: 10.1196/annals.1427.018. [DOI] [PubMed] [Google Scholar]
- 136.Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. 2005;58:495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- 137.Mattson MP. Mitochondrial regulation of neuronal plasticity. Neurochem Res. 2007;32:707–715. doi: 10.1007/s11064-006-9170-3. [DOI] [PubMed] [Google Scholar]
- 138.Dufour E, Terzioglu M, Sterky FH, et al. Age-associated mosaic respiratory chain deficiency causes trans-neuronal degeneration. Hum Mol Genet. 2008;17:1418–1426. doi: 10.1093/hmg/ddn030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Navarro A, Sanchez Del Pino MJ, Gomez C, Peralta JL, Boveris A. Behavioral dysfunction, brain oxidative stress, and impaired mitochondrial electron transfer in aging mice. Am J Physiol Regul Integr Comp Physiol. 2002;282:R985–992. doi: 10.1152/ajpregu.00537.2001. [DOI] [PubMed] [Google Scholar]
- 140.Kassmann CM, Lappe-Siefke C, Baes M, et al. Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes. Nat Genet. 2007;39:969–976. doi: 10.1038/ng2070. [DOI] [PubMed] [Google Scholar]
- 141.Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 142.Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- 143.Howitz KT, Bitterman KJ, Cohen HY, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 144.Wood JG, Rogina B, Lavu S, et al. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- 145.Lagouge M, Argmann C, Gerhart-Hines Z, et al. Resveratrol Improves Mitochondrial Function and Protects against Metabolic Disease by Activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 146.Ulrich S, Loitsch SM, Rau O, et al. Peroxisome proliferator-activated receptor gamma as a molecular target of resveratrol-induced modulation of polyamine metabolism. Cancer Res. 2006;66:7348–7354. doi: 10.1158/0008-5472.CAN-05-2777. [DOI] [PubMed] [Google Scholar]
- 147.Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 148.Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci U S A. 2007;104:7217–7222. doi: 10.1073/pnas.0610068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chan AY, Dolinsky VW, Soltys CL, et al. Resveratrol inhibits cardiac hypertrophy via AMP-activated protein kinase and Akt. J Biol Chem. 2008;283:24194–24201. doi: 10.1074/jbc.M802869200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Baur JA, Pearson KJ, Price NL, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pearson KJ, Baur JA, Lewis KN, et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008;8:157–168. doi: 10.1016/j.cmet.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Barger JL, Kayo T, Vann JM, et al. A low dose of dietary resveratrol partially mimics caloric restriction and retards aging parameters in mice. PLoS ONE. 2008;3:e2264. doi: 10.1371/journal.pone.0002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hondares E, Pineda-Torra I, Iglesias R, Staels B, Villarroya F, Giralt M. PPARdelta, but not PPARalpha, activates PGC-1alpha gene transcription in muscle. Biochem Biophys Res Commun. 2007;354:1021–1027. doi: 10.1016/j.bbrc.2007.01.092. [DOI] [PubMed] [Google Scholar]