

Abstract
The photophysical and photochemical reactions, after light absorption by a photosynthetic pigment–protein complex, are among the fastest events in biology, taking place on timescales ranging from tens of femtoseconds to a few nanoseconds. The advent of ultrafast laser systems that produce pulses with femtosecond duration opened up a new area of research and enabled investigation of these photophysical and photochemical reactions in real time. Here, we provide a basic description of the ultrafast transient absorption technique, the laser and wavelength-conversion equipment, the transient absorption setup, and the collection of transient absorption data. Recent applications of ultrafast transient absorption spectroscopy on systems with increasing degree of complexity, from biomimetic light-harvesting systems to natural light-harvesting antennas, are presented. In particular, we will discuss, in this educational review, how a molecular understanding of the light-harvesting and photoprotective functions of carotenoids in photosynthesis is accomplished through the application of ultrafast transient absorption spectroscopy.
Keywords: Ultrafast spectroscopy, Photosynthesis, Light-harvesting antennas
Introduction
The process of photosynthesis relies upon the efficient absorption and conversion of the radiant energy from the Sun. Chlorophylls and carotenoids are the main players in the process. While the former are involved in light-harvesting and charge separation process, the latter also play vital photoprotective roles. Photosynthetic pigments are typically arranged in a highly organized fashion to constitute antennas and reaction centers, supramolecular devices where light harvesting and charge separation take place.
The very early steps in the photosynthetic process take place after the absorption of a photon by an antenna system, which harvests light and eventually delivers it to the reaction center (Van Grondelle et al. 1994). Despite the enormous variety of photosynthetic organisms, the primary events leading to photosynthetic energy storage are remarkably similar (Sundström 2008). In order to compete with internal conversion, intersystem crossing, and fluorescence, which inevitably lead to energy loss, the energy and electron transfer processes that fix the excited-state energy in photosynthesis must be extremely fast. In order to investigate these events, ultrafast techniques down to a sub-100 fs resolution must be used. In this way, energy migration within the system as well as the formation of new chemical species such as charge-separated states can be tracked in real time. This can be achieved by making use of ultrafast transient absorption spectroscopy. The basic principles of this technique, instrumentation, and some recent applications to photosynthetic systems that involve the light-harvesting and photoprotective functions of carotenoids are described in this educational review. For earlier reviews on ultrafast spectroscopy, see e.g., Jimenez and Fleming (1996), Groot and Van Grondelle (2008), and Zigmantas et al. (2008).
Ultrafast transient absorption spectroscopy
The principle of ultrafast transient absorption spectroscopy
The process of energy transfer in a photosynthetic membrane typically takes place on a time scale from less than 100 fs to hundreds of ps (Sundström et al. 1999; Van Amerongen and Van Grondelle 2001; Van Grondelle et al. 1994). The advent of ultrashort tunable laser systems in the early 1990s has opened up a new and extremely fascinating area of research. Nowadays, the high (sub 50 fs) time resolution has made it possible to investigate the very early events taking place within a light-harvesting antenna in real time (Sundström 2008). In transient absorption spectroscopy, a fraction of the molecules is promoted to an electronically excited state by means of an excitation (or pump) pulse. Depending on the type of experiment, this fraction typically ranges from 0.1% to tens of percents. A weak probe pulse (i.e., a pulse that has such a low intensity that multiphoton/multistep processes are avoided during probing) is sent through the sample with a delay τ with respect to the pump pulse (Fig. 1). A difference absorption spectrum is then calculated, i.e., the absorption spectrum of the excited sample minus the absorption spectrum of the sample in the ground state (ΔA). By changing the time delay τ between the pump and the probe and recording a ΔA spectrum at each time delay, a ΔA profile as a function of τ and wavelength λ, i.e., a ΔA(λ,τ) is obtained. ΔA(λ,τ) contains information on the dynamic processes that occur in the photosynthetic system under study, such as excited-state energy migration, electron and/or proton transfer processes, isomerization, and intersystem crossing. In order to extract this information, global analysis procedures may be applied (see below). One advantage of time-resolved absorption spectroscopy over time-resolved fluorescence is that with the former, the evolution of non-emissive states and dark states can be investigated. This is of particular importance in photosynthesis where carotenoid dark (non-emissive) states play a number of vital roles.
Fig. 1.

Left panel: Schematic depiction of the transient absorption spectroscopy principle. Right panel: Contributions to a ΔA spectrum: ground-state bleach (dashed line), stimulated emission (dotted line), excited-state absorption (solid line), sum of these contributions (gray line)
In general, a ΔA spectrum contains contributions from various processes:
The first contribution is by ground-state bleach. As a fraction of the molecules has been promoted to the excited state through the action of the pump pulse, the number of molecules in the ground state has been decreased. Hence, the ground-state absorption in the excited sample is less than that in the non-excited sample. Consequently, a negative signal in the ΔA spectrum is observed in the wavelength region of ground state absorption, as schematically indicated in Fig. 1 (dashed line).
The second contribution is by stimulated emission. For a two-level system, the Einstein coefficients for absorption from the ground to the excited state (A12) and stimulated emission from the excited to the ground state (A21) are identical. Thus, upon population of the excited state, stimulated emission to the ground state will occur when the probe pulse passes through the excited volume. Stimulated emission will occur only for optically allowed transitions and will have a spectral profile that (broadly speaking) follows the fluorescence spectrum of the excited chromophore, i.e., it is Stokes shifted with respect to the ground-state bleach. During the physical process of stimulated emission, a photon from the probe pulse induces emission of another photon from the excited molecule, which returns to the ground state. The photon produced by stimulated emission is emitted in the exact same direction as the probe photon, and hence both will be detected. Note that the intensity of the probe pulse is so weak that the excited-state population is not affected appreciably by this process. Stimulated emission results in an increase of light intensity on the detector, corresponding to a negative ΔA signal, as schematically indicated in Fig. 1 (dotted line). In many chromophores including bacteriochlorophyll (BChl), the Stokes shift may be so small that the stimulated emission band spectrally overlaps with ground-state bleach and merges into one band.
The third contribution is provided by excited-state absorption. Upon excitation with the pump beam, optically allowed transitions from the excited (populated) states of a chromophore to higher excited states may exist in certain wavelength regions, and absorption of the probe pulse at these wavelengths will occur. Consequently, a positive signal in the ΔA spectrum is observed in the wavelength region of excited-state absorption (Fig. 1, solid line). Again, the intensity of the probe pulse is so weak that the excited-state population is not affected appreciably by the excited-state absorption process.
A fourth possible contribution to the ΔA spectrum is given by product absorption. After excitation of the photosynthetic, or more generally photobiological or photochemical system, reactions may occur that result in a transient or a long-lived molecular state, such as triplet states, charge-separated states, and isomerized states. The absorption of such a (transient) product will appear as a positive signal in the ΔA spectrum. A ground-state bleach will be observed at the wavelengths where the chromophore on which the product state resides has a ground-state absorption. A well-known example of such a transient product state is the accessory bacteriochlorophyll (BChl) anion in the bacterial reaction center (RC), which acts as a transient intermediate in the electron transfer process from the primary donor P to the bacteriopheophytin (BPheo). The rise and decay of this species can be monitored through its specific product absorption at 1,020 nm (Arlt et al. 1993; Kennis et al. 1997a).
Pulse duration, time resolution, and spectral selectivity
Laser pulses as short as 5 fs are now available for transient absorption spectroscopy (see, e.g., Cerullo et al. (2002); and Nishimura et al. (2004)). A short pulse duration Δt implies a large spectral bandwidth Δv according to relation ΔtΔv = 0.44 for Gaussian-shaped pulses. This relation is known as the time–bandwidth product. For instance, a 10-fs pulse with a center wavelength of 800 nm has a spectral bandwidth of 4.4 × 1013 Hz at full-width at half maximum (FWHM), which corresponds to about 100 nm in this wavelength region. Thus, one has to make a trade-off between time resolution and spectral selectivity. Consider the example of the bacterial RC, which has the primary donor absorbing at 860 nm, the accessory BChls at 800 nm, and the BPheos at 760 nm. With a 10-fs pulse at 800 nm, one would simultaneously excite all the cofactors. In order to selectively excite one of the cofactor pairs to study its excited-state dynamics, spectral narrowing to ~30 nm is required, which implies a longer excitation pulse of ~30 fs (Streltsov et al. 1998; Vos et al. 1997). For the photosystem II (PSII) RC, where the energy gaps between the pigments are significantly smaller, the excitation bandwidth has to be narrowed even more to <10 nm for selective excitation, with corresponding pulse durations of ~100 fs (Durrant et al. 1992; Groot et al. 1997).
On very fast timescales, transient absorption signals have contributions from processes additional to those described in the previous section. These non-resonant contributions are often lumped together under the terms “coherent artifact” and “cross-phase modulation.” As transient absorption signals result from light–matter interaction through the third-order non-linear susceptibility χ(3) (Mukamel 1995), non-sequential light interactions that do not represent population dynamics of electronic states will contribute to the signals. Such undesired signals can be ignored by excluding the initial phases of the femtosecond dynamics from the data interpretation and analysis. On the other hand, they may be explicitly included in the analysis by considering their physical origin. In such a case, assumptions need to be made about the lineshapes and dephasing times of the chromophore in question (Novoderezhkin et al. 2004). Cross-phase modulation effects are due to a change in the index of refraction of solvent and cuvette induced by the pump beam and give rise to oscillatory patterns around zero delay (Kovalenko et al. 1999). These artifacts can in principle be subtracted from the data by recording an experiment in a cuvette with the solvent.
Equipment: amplified Ti:sapphire laser systems and optical parametric amplifiers
Generally speaking, two types of ultrafast transient absorption spectroscopy setups are widely used today for photosynthesis research, distinguished by the repetition rate and pulse energies at which they operate: the first type involves systems with a repetition rate of 1–5 kHz with a relatively high pulse energy. The second type involves systems with a repetition rate in the range 40–250 kHz with a relatively low pulse energy. In addition, the direct or cavity-dumped output from a Ti:sapphire oscillator has frequently been employed for transient absorption spectroscopy, but will not be discussed here (Arnett et al. 1999; Kennis et al. 1997b; Nagarajan et al. 1996; Streltsov et al. 1998; Vulto et al. 1999).
The first type of spectroscopy typically provides the experimenter with excitation energies of 5–100 nJ, which when focused on 150–200 μm diameter (the regular focusing conditions in our laboratory) typically results in 2–20% of the molecules being promoted to the excited state. This value is only approximate, since the accurate estimate of the excitation density depends on several factors, namely, the exact size of the focus, the concentration of the chromophores, and their extinction coefficient. The relatively high excitation densities achieved with these systems make them suitable to study complexes with a relatively small number of connected pigments such as pigments in solution (Billsten et al. 2002; Cong et al. 2008; De Weerd et al. 2003; Niedzwiedzki et al. 2007; Polivka et al. 1999), isolated reaction centers (De Weerd et al. 2002; Holzwarth et al. 2006a, 2006b; Wang et al. 2007), isolated light-harvesting antenna complexes (Croce et al. 2001; Gradinaru et al. 2000, 2001; Ilagan et al. 2006; Krueger et al. 2001; Papagiannakis et al. 2002, 2003; Polívka et al. 2002; Polivka and Sundström 2004; Zigmantas et al. 2002), artificial antenna systems (Berera et al. 2006, 2007; Kodis et al. 2004; Pan et al. 2002), and photoreceptor proteins that bind only a single chromophore (Kennis and Groot 2007; Wilson et al. 2008). With appropriate detection schemes that involve multichannel detection on a shot-to-shot basis, signal detection sensitivities of ~10−5 units of absorbance over a broad wavelength range can be achieved, implying that molecular species with a small extinction coefficient or that accumulate in very low (transient) concentrations can be detected (Berera et al. 2006; Wilson et al. 2008). A drawback of a 1–5-kHz system is that with its relatively high excitation densities, multiple excited states may appear in a single multichromophoric complex, resulting in singlet–singlet annihilation processes among (B)Chls (Van Grondelle 1985).
With the laser systems that operate at 40–250 kHz, a lower pulse energy can be used for excitation with respect to the kHz systems owing to their higher repetition rate, which allows more laser shots to be averaged per unit time. Typically, pulse energies of 0.5–10 nJ are used, roughly corresponding to excited-state populations of <1–10%. Under the right circumstances, detection sensitivities of ~10−6 units of absorbance can be achieved. Accordingly, this kind of system has been used to study exciton migration in large systems with many connected pigments such as chloroplasts and light-harvesting complex (LHC) II aggregates (Holt et al. 2005; Ma et al. 2003; Ruban et al. 2007). In addition, it has been used to examine exciton migration in isolated LH complexes under annihilation-free conditions (Monshouwer et al. 1998; Novoderezhkin et al. 2004; Palacios et al. 2006; Papagiannakis et al. 2002). Drawbacks of this type of systems involve the shorter time between pulses (4–20 μs), which may lead to the build-up of relatively long-lived species such as triplet or charge-separated states. In addition, multichannel detection on a shot-to-shot basis has been limited to 14 channels at such high repetition rates (Ruban et al. 2007), although significant strides are currently being made in our laboratory to resolve this limitation.
Figure 2 shows a scheme of an ultrafast transient absorption setup, as it exists today in the Biophysics Laboratory of the Laser Center at the Vrije Universiteit (LCVU) in Amsterdam, The Netherlands. A broadband oscillator (Coherent Vitesse) generates pulses of ~30 fs duration with a wavelength of 800 nm, a bandwidth of ~35 nm at a repetition rate of 80 MHz. The pulses from the oscillator are too weak to perform any meaningful spectroscopy and therefore have to be amplified. Femtosecond pulse amplification is not a trivial matter because at high energies, the peak power in a femtosecond pulse becomes so high that amplification and pulse-switching media such as crystals and Pockels cells easily get damaged. A Pockels cell is an electro-optical device containing a crystal, such as potassium dihydrogenphosphate (KH2PO4), capable of switching the polarization of light when an electrical potential difference is applied to it. In this way, the amount of stimulated emission from the laser cavity can be controlled. For this reason, femtosecond pulse amplification is carried out through the chirped-pulse amplification principle: the pulse from the oscillator (hereafter, referred to as “seed pulse”) is first stretched to ~200 ps by a stretcher, which temporally delays the “blue” wavelengths within the pulse bandwidth of ~35 nm with respect to the “red” wavelengths by means of a grating pair. Then, the seed pulse is coupled into a regenerative amplifier (Coherent Legend-UltraShort Pulse (USP)). There, the seed pulse travels through a Pockels cell which sets its polarization in such a way that it becomes trapped within the amplifier’s cavity. On traveling back and forth in the cavity, it passes through a Ti:sapphire crystal that is pumped at 1-kHz repetition rate by a diode-pumped Nd:YLF pump laser at 527 nm (Coherent Evolution, 30 W). At each passage through the crystal, the trapped seed pulse is amplified until saturation is reached. Then, the Pockels cell switches the polarization of the amplified pulse which results in its ejection from the amplifier. The amplified pulse is compressed to ~45 fs by temporally synchronizing the “blue” and “red” wavelengths within the pulse bandwidth, essentially the reverse of the “stretching” procedure. At this point, the output from the laser system is a 40-fs pulse at an energy of 2.5 mJ, a center wavelength of 800 nm, a bandwidth of 30 nm, and a repetition rate of 1 kHz.
Fig. 2.

Schematic representation of an experimental ultrafast transient absorption setup
In order to perform transient absorption spectroscopy with a Ti:sapphire laser alone, one is restricted to a wavelength region for the excitation pulse around 800 nm, allowing only the study of some BChl a-containing systems (Arnett et al. 1999; Kennis et al. 1997b; Nagarajan et al. 1996; Novoderezhkin et al. 1999; Streltsov et al. 1998; Vulto et al. 1999). In order to shift the wavelength to other parts of the visible and near-IR spectra, optical parametric amplifiers (OPAs) or optical parametric generators (OPGs) are typically used. In an OPA, non-linear birefringent crystals such as beta barium borate (BBO) are pumped by the direct output of the amplified laser system at 800 nm or frequency-doubled pulses at 400 nm. The pump is temporally and spatially overlapped with a white-light continuum in the crystal, and depending on the angle between the laser beam and the symmetry axis of the crystal, two particular wavelengths of the white-light continuum called “signal” and “idler” are amplified through the second-order non-linear polarizability of the crystal, of which the signal has the shortest wavelength and is routinely selected for further use. Since pump, signal, and idler beams have different polarizations, the group velocity of pump, signal, and idler beams can be made equal by varying the angle between the laser beam and the symmetry axis of the birefringent crystal. This allows energy from the pump beam to be converted to the signal and idler beams over a large propagation length up to millimeters. This is the so-called phase-matching condition. Conservation of energy requires that the sum of the frequencies of signal and idler add up to the frequency of the pump beam. Thus, 800-nm-pumped OPAs operate in the near-InfraRed (IR) (1,100–1,600 nm for the signal) while 400-nm-pumped OPAs operate in the visible (475–750 nm for the signal) spectrum. Using the output of an OPA as a basis, essentially all wavelengths from the UltraViolet (UV) to mid-IR can be generated at relatively high pulse energies by using non-linear mixing processes such as frequency-doubling, sum-frequency generation, and difference-frequency generation in suitable non-linear crystals. Obviously, visible and near-IR light are the most useful wavelengths for the study of photosynthetic systems. In addition, mid-IR wavelengths are very useful for probing molecular vibrations of chlorophylls and carotenoids (Groot et al. 2005, 2007). The pulse duration out of the OPA roughly corresponds to that of the amplified Ti:sapphire laser system. The pulse energy from our regenerative laser amplifier of 2.5 mJ allows simultaneous pumping of several OPAs. The latter option is important for experiments that require multiple pump pulses, such as pump–dump or pump–repump experiments (Kennis et al. 2004; Larsen et al. 2003; Papagiannakis et al. 2004).
The transient absorption setup
In order to vary the time delay between the excitation and probe pulses, the excitation pulse generated by the OPA is sent through an optical delay line, which consists of a retroreflector mounted on a high-precision motorized computer-controlled translation stage. The translation stage employed in our experiments has an accuracy and reproducibility of 0.1 μm, which corresponds to a timing accuracy of 0.5 fs. The delay line can be moved over 80 cm, implying that time delays up to 5 ns can be generated between excitation and probe beams. The excitation beam is focused in the sample to a diameter of 130–200 μm and blocked after the sample. In most cases, the polarization of the pump beam is set at the magic angle (54.7°) with respect to that of the probe to eliminate polarization and photoselection effects (Lakowicz 2006).
For the detection of the pump-induced absorbance changes, a part of the amplified 800-nm light is focused on a sapphire or calcium fluoride plate (though other materials such as quartz, MgF2, water, and ethylene glycol can also be used) to generate a white-light continuum. In the absence of special precautions, the white-light continuum may range from ~400 to ~1,100 nm (depending on the material) and be used as a broadband probe; its intensity is so weak that it does not transfer an appreciable population from the ground to the excited state (or vice versa). It is focused on the sample to a diameter slightly smaller than the pump, spatially overlapped with the pump, collimated, and sent into a spectrograph. There, it is spectrally dispersed and projected on a silicon diode array that consists of tens to hundreds of elements. The diode array is read out by a computer on a shot-to-shot basis, in effect measuring an absorption spectrum with each shot.
Under some experimental conditions, detection with a diode array is not possible or appropriate. For instance, for many experiments in the near-IR and the UV, other detector types need to be employed that, in combination with the white-light continuum intensities at those wavelengths, lack the sensitivity required for array detection. In these cases, single wavelength detection is often employed. In the mid-IR (~3–10 μm), mercury cadmium telluride (MCT) arrays that consist of 32 or 64 elements are available (Groot et al. 2007). Another detection method in the visible spectrum employs a charge-coupled device (CCD) detector. Frequently, a reference beam is used to account for shot-to-shot intensity fluctuations in the white-light continuum. In such a case, the white-light continuum beam is split in two beams, the probe and the reference. The probe is overlapped with the pump beam in the sample, while the reference beam is led past the sample (or through the sample past the excited volume). The probe and reference beams are then projected on separate diode arrays. During data collection, the probe beam is divided by the reference beam, which may lead to improved signal to noise because the intensity fluctuations of the white-light continuum are eliminated.
By the nature of the white-light generation process, the white light is “chirped” on generation, i.e., the “blue” wavelengths are generated later in time than the “red” wavelengths. The exact temporal properties depend on the specific generation conditions. Hence, the white-light continuum has an “intrinsic” group-velocity dispersion. When traveling through optically dense materials such as lenses and cuvettes, the group velocity dispersion in the white light readily increases to picoseconds. This effect can be minimized by using parabolic mirrors for collimation and focusing of the white-light beam between its point of generation and the sample. The group velocity dispersion may be accounted for in the data analysis and described by a polynomial function. Alternatively, the white-light continuum can be compressed by means of a grating pair or prism pair in such a way that the “red” and “blue” wavelengths in the probe beam coincide in time.
The instrument response function of this particular transient absorption apparatus, which can be measured by frequency mixing in a non-linear crystal placed at the sample spot or by the transient birefringence in CS2 or water, can usually be modeled with a Gaussian with a FWHM of 120 fs. If required, the white-light continuum can be compressed down to ~10 fs by means of a grating pair or prism pair; in such a case, the instrument response function is generally limited by the duration of the pump pulse.
For measurements at room temperature, the sample is placed in a 1–2-mm quartz cuvette which is either connected to a flow system or mounted on a shaker to prevent exposure of the same excited volume to multiple laser shots and to prevent sample degradation.
Collection of transient absorption spectra
A transient absorption experiment proceeds as follows: the time delay between excitation and probe beams is fixed. Before reaching the sample, the excitation beam (that delivers a pulse every 1 ms) passes through a mechanical chopper that is synchronized to the amplifier in such a way that every other excitation pulse is blocked. Thus, alternately the sample is being excited and not excited. Consequently, the white-light continuum that is incident on the detector diode array alternately corresponds to a “pumped” and “unpumped” sample, and the detector alternately measures the intensity of the probe beam of a “pumped” and “unpumped” sample, I(λ)pumped and I(λ)unpumped. I(λ)pumped and I(λ)unpumped are stored in separate buffers (while keeping the time delay between pump and probe fixed), and a number of shots that is sufficient for an acceptable signal-to-noise ratio is measured, usually 103–104. With the shot-to-shot detection capability of the multichannel detection system, particular spectra that deviate from the average (“outliers”) can in real time be rejected during data collection, significantly improving signal-to-noise ratio. A second white-light beam (the reference beam) not overlapping with the pump pulse can also be used to further increase the signal-to-noise ratio. From the averaged values of I(λ)pumped and I(λ)unpumped, an absorbance difference spectrum ΔA(λ) is constructed according to
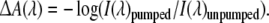 |
Then, the delay line is moved to another time delay between pump and probe, and the above procedure is repeated. In total, absorbance difference spectra at approximately 100–200 time points between 0 fs and ~5 ns are collected, along with absorbance difference spectra before time zero to determine the baseline. In addition, many spectra are collected around the time that pump and probe pulse overlap in time (“zero delay”) to enable accurate recording of the instrument response function. This whole procedure is repeated several times to test reproducibility, sample stability, and long-term fluctuations of the laser system. In this way, an entire dataset ΔA(λ,τ) is collected.
Anisotropy experiments in transient absorption spectroscopy
In photosynthetic antennae and reaction centers, the pigments are bound in a well-defined way. Energy and electron transfer processes and pathways can be specifically assessed through the use of polarized excitation and probe beams. The time-dependent anisotropy is defined as
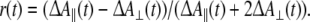 |
With ΔA∥(t), the time-dependent absorbance difference signal with pump and probe beams is polarized parallel, and with ΔA⊥(t), the time-dependent absorbance difference signal with pump and probe beams is polarized perpendicular. In light-harvesting antennae, the decay of r(t) indicates the elementary timescales of exciton migration, be it through incoherent hopping or exciton relaxation (Kennis et al. 1997b; Nagarajan et al. 1996; Novoderezhkin et al. 1998; Savikhin et al. 1994, 1998, 1999; Vulto et al. 1999; Vulto et al. 1997). Energy transfer or exciton relaxation processes often occur among (pools of) Chls that have their absorption maxima at similar wavelengths. Consequently, these processes are associated with small spectral shifts of the ΔA spectra and are therefore difficult to observe under magic angle detection conditions. Through time-resolved anisotropy experiments, the timescales of such fast exciton migration events can accurately be determined.
Data analysis
In time-resolved spectroscopic experiments, the very large amounts of data collected can be analyzed by global and target analysis techniques (Van Stokkum et al. 2004). A typical time-resolved experiment ΔA(λ,τ) in fact consists of a collection of thousands of data points, i.e., tens to hundreds wavelengths times one to two hundred data points. In order to extract valuable information, one could simply take slices of the data; for instance, one could take one wavelength and look at its evolution in time (a so-called kinetic trace), or one could plot the signal at different wavelengths for a given time point (a ΔA spectrum). This is normally the first stage of the data analysis where the experimentalist has a glimpse of an expected (or unexpected) process. The next step in the data analysis is to apply the so-called global analysis techniques, in an attempt to distill the overwhelming amount of data into a relatively small number of components and spectra. In the most basic model, the femtosecond transient absorption data are globally analyzed using a kinetic model consisting of sequentially interconverting evolution-associated difference spectra (EADS), i.e., 1→2→3→··· in which the arrows indicate successive monoexponential decays of increasing time constants, which can be regarded as the lifetime of each EADS. The first EADS correspond to the time-zero difference spectrum. This procedure enables a clear visualization of the evolution of the (excited) states of the system. Based on the insight obtained from this model and from the raw data, one can then take a further step in the analysis and apply a so-called target kinetic scheme. The EADS that follow from the sequential analysis are generally made up from a mixture of various molecular species. In general, the EADS may well reflect mixtures of molecular states. In order to disentangle the contributions from these molecular species and obtain the spectrum signature of the “pure” excited- and product state intermediates (the so-called species-associated difference spectra, SADS), a specific kinetic model must be applied in a so-called target analysis procedure. In this way, the energy and electron transfer mechanisms can be assessed in terms of a number of discrete reaction intermediates. A comprehensive review of global and target analysis techniques has been published (Van Stokkum et al. 2004). In the next section, we illustrate a few examples of time-resolved experiments and data analysis. We will start with the description of elementary energy transfer processes in artificial systems followed by more complex examples in natural light-harvesting compounds.
Example 1: the light-harvesting function of carotenoids
Carotenoids play an important role in light-harvesting antennae, not only in photoprotection but also by harvesting blue and green light and transferring the excited-state energy to nearby (B)Chls (Frank et al. 1999; Polivka and Sundström 2004; Ritz et al. 2000). Carotenoids have a complicated excited-state manifold: they have a strongly allowed transition from the ground state (which has Ag− symmetry in ideal polyenes) to a state with Bu+ symmetry called S2. This transition is responsible for their strong absorption of blue-green light. Below the S2 state lies the optically forbidden S1 state that has Ag− symmetry, along with a number of additional optically forbidden states, the physical nature of which remains unclear (Polivka and Sundström 2004).
Ultrafast spectroscopy has proven to be a valuable tool to map out the energy transfer pathways from carotenoid to (B)Chl and understand these processes at the molecular level. In particular, simple artificial photosynthetic light-harvesting systems have given important insights into the physical mechanisms that underlie the various energy transfer and relaxation processes (Berera et al. 2007; Kodis et al. 2004; Marino-Ochoa et al. 2002). Figure 3a shows a minimal artificial light-harvesting mimic suitable for the study of the light-harvesting role of carotenoids. The model system, referred to as dyad 1, is made up of two moieties: a carotenoid with nine conjugated double bonds in its π-electron system and a phthalocyanine (Pc) molecule. The Pc molecule has a maximal absorption at 680 nm (called the Q band), and it acts as a Chl a mimic. The carotenoid to Pc energy transfer efficiency is very high in this particular dyad, ~90% (Berera et al. 2007).
Fig. 3.

a Molecular structure of a carotenophthalocyanine light-harvesting dyad 1. b Evolution-associated difference spectra (EADS) that result from a global analysis on transient absorption experiments on dyad 1. The excitation wavelength was 475 nm. c Kinetic traces at 560 nm (upper panel) and 680 nm (lower panel). d Kinetic scheme that describes the excited-state processes in dyad 1 upon carotenoid excitation. Solid lines denote energy transfer, dotted denote internal conversion, dashed denotes intersystem crossing processes. Source: Berera et al. (2007)
In order to map out the energy transfer pathways from carotenoid to Pc, the carotenoid moiety of dyad 1 was excited at 475 nm for ultrafast time-resolved spectroscopy (Berera et al. 2007). Figure 3b shows the results of a global analysis of the time-resolved data. Figure 3c shows kinetic traces at selected wavelengths for dyad 1. Six time constants were needed for a satisfactory fit of the data. The first EADS (Fig. 3b, dotted line) is formed instantaneously at time zero and represents population of the optically allowed S2 state of the carotenoid. It presents a region of negative signal below 570 nm originating from the carotenoid ground-state bleach and from stimulated emission (SE). In addition, the Pc Q region around 680 nm shows a band shift-like signal. The latter is due the response of the Pc molecule to the charge redistribution on the nearby carotenoid upon excitation to the S2 state.
The first EADS evolve in 40 fs into the second EADS (Fig. 3b, dashed line), which is characterized by a strong bleach/SE signal at 680 nm. This corresponds to a population of the Pc excited state (the Q state) indicating that the carotenoid S2 state is active in transferring energy to Pc. The dip at 610 nm originates from a vibronic band of the Pc Q state. In addition, excited-state absorption is observed in the 480–600 nm region, which can be assigned to the optically forbidden S1 state and the so-called S* state (Gradinaru et al. 2001). This observation indicates that internal conversion from the carotenoid S2 state to the lower-lying states has taken place in competition with energy transfer to Pc. The S1 excited-state absorption has a maximum around 560 nm while that of the S* state is around 525 nm.
The evolution to the third EADS (Fig. 3b, dash-dotted line) takes place in 500 fs. It corresponds to a decrease of excited state absorption (ESA) at the red wing of the S1 absorption, which may be assigned to vibrational cooling of the S1 state (Polivka and Sundström 2004). Moreover, an increase of the Pc Q bleach at 680 nm is observed which is likely to originate from the energy transfer from the S1 and possibly the S* state to Pc. Note that the third EADS overlap with the fourth EADS (Fig. 3b, solid line) in the Pc Q region and is not visible.
The fourth EADS (Fig. 3b, solid line) appear after 900 fs and has a lifetime of 7.8 ps. The signal at 525 nm, where the main contribution to the spectrum is given by S*, has decreased, whereas the signal in the 540–620 nm region, where the absorption is mainly due to S1, has slightly increased, indicating the decay of S* in about 0.9 ps, partly by internal conversion to S1.
The evolution to the fifth EADS (Fig. 3b, dash–dot–dot line) takes place in 8 ps. At this stage, the carotenoid ESA has decayed, and the fifth EADS correspond very well to that of the excited Pc Q state with a flat ESA in the 450–600 nm region. Around 680 nm, the bleach increases with respect to the previous EADS, which implies that the carotenoid S1 state has transferred energy to Pc.
The final EADS (Fig. 3b, short-dotted line) is formed in 2 ns and represents the component that does not decay on the time scale of the experiment. It features the typical carotenoid triplet ESA in the 475–550 nm region as well as a bleach/band shift-like signal in the Pc Q region. Thus, the carotenoid triplet state rises directly upon decay of the singlet excited state of Pc. This observation implies that triplet–triplet energy transfer from Pc to the carotenoid occurs much faster than the inter system crossing (ISC) process in Pc, which effectively occurs in 2 ns.
Figure 3c shows the kinetic trace recorded at 680 nm (lower panel) and at 560 nm (upper panel), corresponding to the maximum of the Pc Q absorption and the maximum of carotenoid S1 excited state absorption. At 680 nm, the ultrafast rise of the bleach corresponding to the carotenoid S2 → Pc energy transfer (40 fs) is followed by two slower rise corresponding to hot S1 and/or S* → Pc (500–900 fs) and S1 → Pc energy transfer (8 ps). At 560 nm, the carotenoid S1 signal decays in 8 ps and matches the 8 ps rise of the Pc bleach. The energy transfer pathways in dyad 1 are summarized with the kinetic scheme in Fig. 3d. Note that this scheme is simplified; a full account of the kinetic modeling of energy transfer pathways in dyad 1 along with the SADS of the involved molecular species is given in Berera et al. (2007). The carotenoid to Pc energy transfer dynamics in dyad 1 is reminiscent of several natural light-harvesting antennas where high energy transfer efficiency from carotenoids to chlorophylls is obtained; this occurs by transfer of energy to Chl from multiple excited states of the carotenoid (Holt et al. 2004; Kennis et al. 2001; Papagiannakis et al. 2002; Polivka and Sundström 2004; Ritz et al. 2000; Walla et al. 2000, 2002; Wehling and Walla 2005; Zhang et al. 2000; Zigmantas et al. 2002).
Example 2: carotenoids in non-photochemical quenching in photosystem II and artificial systems
When exposed to high light illumination, oxygenic photosynthetic organisms protect themselves by switching to a protective mode where the excess energy in photosystem II (PSII) is dissipated as heat through a mechanism known as non-photochemical quenching (NPQ) (Demmig-Adams et al. 2006; Horton et al. 1996; Müller et al. 2001). The mechanism of energy dissipation in the PSII antenna has long remained elusive but over the last years, significant progress has been made in resolving its molecular basis. In particular, the involvement of carotenoids in the quenching of Chl singlet excited states has clearly been demonstrated. Yet, controversy persists on whether the quenching process(es) involve energy or electron transfer processes among Chls and carotenoids, and which particular Chl and carotenoid pigments constitute the quenching site (Ahn et al. 2008; Berera et al. 2006; Holt et al. 2005; Ma et al. 2003; Ruban et al. 2007).
On the basis of energetic considerations, it was speculated that the low-lying optically forbidden S1 state of carotenoids could act as an energy sink to Chl singlet excited states (Frank et al. 1994). In order to address this issue, ultrafast transient absorption spectroscopy was applied on the same artificial light-harvesting dyad as discussed previously, but with extended conjugated π-electron system of the carotenoid moiety with 10 or 11 C=C double bonds, implying lower excited-state energies (Fig. 4a). Strikingly, the Pc lifetime is reduced from its natural lifetime of 3 ns to 15–300 ps, depending on the length of the carotenoid’s conjugated π-electron system (Fig. 4b) and the solvent polarity. Furthermore, Berera et al. (2006) have demonstrated that the carotenoid S1 excited state acts as the acceptor of excited-state energy from the covalently linked Pc, as schematically shown in Fig. 4c, thereby providing an efficient channel for energy dissipation.
Fig. 4.

a Molecular structure of a carotenophthalocyanine light-harvesting dyad 1, 2, and 3. The carotenoids of dyad 1, 2 and 3 contain 9, 10 and 11 conjugated C=C double bonds, respectively. b Upper panel: kinetic traces at 680 nm of dyad 1, 2, and 3 and a model Pc in tetrahydrofuran (THF). Lower panel: kinetic traces of dyad 3 dissolved in acetone detected at 480 nm (solid line) and 576 nm (dashed line). Excitation wavelength for b and d was 680 nm. c Kinetic scheme that describes the excited-state decay processes In dyad 2 and 3 upon Pc excitation. Solid line denotes energy transfer, dotted line denotes internal conversion process. d Evolution-associated difference spectra (EADS) that result from a global analysis on transient absorption experiments on dyad 3 dissolved in acetone. Source: Berera et al. (2006)
A crucial aspect of Pc and Chl excited-state quenching by the carotenoid S1 state is the notion that such processes occur through a so-called inverted kinetic scheme, i.e., the quenching state S1 is slowly populated by rate constant kslow (in 15–300 ps) and quickly depopulated with rate constant kfast (in ~5 ps). The latter time constant is inherent to the photophysics of the carotenoid S1 state, i.e., internal conversion to the ground state occurs on this timescale through efficient vibronic coupling between the ground and S1 states (Chynwat and Frank 1995). In such an inverted kinetic scheme, the donor (Pc) decays with a single rate constant kslow. The acceptor (carotenoid S1) will rise with rate constant kfast and decay in parallel with the donor with rate constant kslow, and reach a maximum transient concentration that remains low, and with sufficiently separated rate constants, it is approximately equal to kslow/kfast. Thus, in the specific case of the artificial light-harvesting dyads, the carotenoid S1 signal is expected to rise with a rate constant that corresponds to the internal conversion rate of S1 to the ground state and to have a low amplitude throughout the Pc excited-state lifetime. This is exactly what is observed experimentally: Figure 4d shows the EADS for dyad 3, whose carotenoid has 11 conjugated double bonds, dissolved in the very polar solvent acetone. Under these circumstances, the Pc is strongly quenched to a main lifetime of 15 ps and the quenching state is expected to accumulate to a readily observable transient concentration of approximately a third of the Pc population at time zero. The Pc moiety of dyad 3 was excited at 680 nm. Four components are needed to obtain a satisfactory fit of the data, with lifetimes of 4.9, 15, and 89 ps and a non-decaying component. A closer examination of the EADS reveals the nature of the quenching process: the first component (Fig. 4d, solid line), appearing at time zero, shows bleach of the Pc Q state in the 680 nm region—an almost flat excited state absorption region that represents the excited Pc molecule. The first EADS evolve in 4.9 ps to the second EADS (Fig. 4d, dashed line), characterized by an increase of the amplitude in the 530–600 nm region and a decrease below 530 nm. The Pc bleach at 680 nm remains the same. This change indicates that another species is populated in 4.9 ps. In fact, the positive signal in the 530–600 nm region is due to the carotenoid S1 ESA, while the region below 530 nm corresponds to the carotenoid ground-state bleach. Thus, the second EADS is a superposition of Pc singlet excited state and a contribution from the carotenoid S1 state. The second EADS evolve to the third EADS (Fig. 4d, dotted line) in 15.6 ps. The third EADS is characterized by an overall decrease of the Pc and carotenoid S1 signal with respect to the second EADS, indicating that these molecular species have decayed together. The third EADS has a lifetime of 89 ps and represents a fraction of dyad 3 that decays more slowly, presumably as a result of conformational heterogeneity (Berera et al. 2006). A target analysis that fully accounts for the spectral evolution in terms of distinct SADS for the Pc and carotenoid S1 excited states is given in Berera et al. (2006).
The inverted kinetics of the carotenoid S1 state are illustrated in the lower panel of Fig. 4b, where kinetic traces at 480 and 576 nm are shown upon excitation of Pc at 680 nm. The 576 nm trace represents the carotenoid S1 excited state absorption region and shows a rise with a time constant of 4.9 ps that mainly decays in 15 ps. Thus, population of the carotenoid S1 state rises in 4.9 ps and then decays in parallel with excited Pc. Likewise, the 480 nm trace first gets a positive amplitude that originates from Pc ESA. Then, the signal apparently decays in 4.9 ps. The latter is interpreted as a growing in of the carotenoid ground-state bleach that results from a population of the carotenoid S1 state. Thus, the 480 and 576 nm traces show the rise in 4.9 ps and decay in 15 ps of the quenching state, i.e., the carotenoid S1 state.
Time-resolved experiments in LH complexes of oxygenic photosynthesis have revealed kinetic patterns similar to those observed for the artificial dyads, in particular the major light harvesting antenna of plants, LHCII (Ruban et al. 2007) and the aggregated IsiA antenna complexes from cyanobacteria (Berera et al. 2009). Figure 5 shows selected kinetic traces for LHCII in the unquenched, trimeric state (panel a) and in a quenched aggregated state (panel b), following a 100 fs, 10 nJ laser pulse at 675 nm. In the quenched state, the trace at 537 nm not only represents the carotenoid S1 ESA, but it also has a positive amplitude coming from Chl ESA. It clearly shows a slower decay in the first ~10 ps compared to the decay of the Chl Qy state at 679 nm. The opposite trend is seen at 489 nm (carotenoid ground state absorption region), where the trace shows a faster decay in the first ~10 ps. If only Chl signals were to contribute to the kinetics, one would expect homogeneous decay. Thus, in analogy with the dyad case (vide supra), the observed ΔA signals show that concomitantly with the decay of the Chl excited state, a carotenoid excited state is populated. Application of a target analysis with a kinetic model that incorporates quenching and singlet–singlet annihilation (Fig. 5, panel c) revealed the SADS of the quenching state, which correspond to the carotenoid S1 state. On the basis of the wavelength of its maximum ground-state bleach, Ruban et al. (2007) concluded that Lutein 1 likely acts as a quencher of Chl excited states in this isolated system.
Fig. 5.

Selected kinetic traces for unquenched LHCII trimers (a) and quenched LHCII aggregates (b) at 677 nm (top), 489 nm (middle) and 537 nm (bottom), following a 100 fs, 10 nJ laser pulse at 675 nm. The vertical axis shows the measured change in absorption, the horizontal axis is linear up to 1 ps and logarithmic thereafter. The long short-dashed line represents the 1 ps phase due to chlorophyll excited state relaxation, the dotted line the excited state decay of chlorophyll, the dashed line the absorption changes due to the quencher Q, and the dash-dotted line the build-up of the triplet state. The kinetic model is shown in (c) and the corresponding species-associated difference spectra (SADS) in (d). Source: Ruban et al. (2007)
In conclusion, carotenoids can accept energy from a neighboring tetrapyrrole thereby acting as strong quenchers (Berera et al. 2006, 2009; Ruban et al. 2007). The carotenoid S1 state acts as a quencher and effective energy dissipator since its lifetime is 100–1,000 times shorter compared to the lifetime of the Pc or Chl excited state. By making use of ultrafast spectroscopy, we have been able to follow the process of energy dissipation in real time and to determine the underlying physical mechanism. In particular, it is important to note that the quenching phenomena in the artificial dyads, PSII, and IsiA antenna systems occur through inverted kinetic schemes where the lifetime of the quencher is inherently shorter lived than the Chl excited state. This results in low transient concentrations of the quenching states, which requires transient absorption data at high-signal to noise. Further, application of target analysis techniques utilizing specific kinetic models is required to extract the spectroscopic signature of the quenching states and to identify the molecular mechanism of non-photochemical quenching.
Acknowledgments
J.T.M.K. and R.B. were supported by the Earth and Life Sciences council of the Netherlands Foundation for Scientific Research (NWO-ALW) through a VIDI and a Rubicon grant, respectively. The authors thank Cosimo Bonetti for providing Fig. 2. This manuscript was edited by Govindjee.
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- (B)Chl
(Bacterio)Chlorophyll
- BPheo
Bacteriopheophtin
- EADS
Evolution-associated difference spectra
- ESA
Excited-state absorption
- FWHM
Full-width at half maximum
- LHC
Light-harvesting complex
- PSII
Photosystem II
- RC
Reaction center
- SADS
Species-associated difference spectra
- SE
Stimulated emission
References
- Ahn TK, Avenson TJ, Ballottari M, Cheng YC, Niyogi KK, Bassi R, Fleming GR (2008) Architecture of a charge-transfer state regulating light harvesting in a plant antenna protein. Science 320:794–797 [DOI] [PubMed]
- Arlt T, Schmidt S, Kaiser W, Lauterwasser C, Meyer M, Scheer H, Zinth W (1993) The accessory bacteriochlorophyll—a real electron carrier in primary photosynthesis. Proc Natl Acad Sci USA 90:11757–11761 [DOI] [PMC free article] [PubMed]
- Arnett DC, Moser CC, Dutton PL, Scherer NF (1999) The first events in photosynthesis: electronic coupling and energy transfer dynamics in the photosynthetic reaction center from Rhodobacter sphaeroides. J Phys Chem B 103:2014–2032 [DOI]
- Berera R, Herrero C, Van Stokkum IHM, Vengris M, Kodis G, Palacios RE, Van Amerongen H, Van Grondelle R, Gust D, Moore TA, Moore AL, Kennis JTM (2006) A simple artificial light-harvesting dyad as a model for excess energy dissipation in oxygenic photosynthesis. Proc Natl Acad Sci USA 103:5343–5348 [DOI] [PMC free article] [PubMed]
- Berera R, Van Stokkum IHM, Kodis G, Keirstead AE, Pillai S, Herrero C, Palacios RE, Vengris M, Van Grondelle R, Gust D, Moore TA, Moore AL, Kennis JTM (2007) Energy transfer, excited-state deactivation, and exciplex formation in artificial caroteno-phthalocyanine light-harvesting antennas. J Phys Chem B 111:6868–6877 [DOI] [PubMed]
- Berera R, Van Stokkum IHM, D’Haene S, Kennis JTM, Van Grondelle R, Dekker JP (2009) A mechanism of energy dissipation in cyanobacteria. Biophys J 96:2261–2267 [DOI] [PMC free article] [PubMed]
- Billsten HH, Zigmantas D, Sundström V, Polivka T (2002) Dynamics of vibrational relaxation in the S1 state of carotenoids having 11 conjugated C=C bonds. Chem Phys Lett 355:465–470 [DOI]
- Cerullo G, Polli D, Lanzani G, De Silvestri S, Hashimoto H, Cogdell RJ (2002) Photosynthetic light harvesting by carotenoids: detection of an intermediate excited state. Science 298:2395–2398 [DOI] [PubMed]
- Chynwat V, Frank HA (1995) The application of the energy-gap law to the S1 energies and dynamics of carotenoids. Chem Phys 194:237–244 [DOI]
- Cong H, Niedzwiedzki DM, Gibson GN, Frank HA (2008) Ultrafast time-resolved spectroscopy of xanthophylls at low temperature. J Phys Chem B 112:3558–3567 [DOI] [PubMed]
- Croce R, Muller MG, Bassi R, Holzwarth AR (2001) Carotenoid-to-chlorophyll energy transfer in recombinant major light-harvesting complex (LHCII) of higher plants. I. Femtosecond transient absorption measurements. Biophys J 80:901–915 [DOI] [PMC free article] [PubMed]
- De Weerd FL, Van Stokkum IHM, Van Amerongen H, Dekker JP, Van Grondelle R (2002) Pathways for energy transfer in the core light-harvesting complexes CP43 and CP47 of Photosystem II. Biophys J 82:1586–1597 [DOI] [PMC free article] [PubMed]
- De Weerd FL, Dekker JP, Van Grondelle R (2003) Dynamics of beta-carotene-to-chlorophyll singlet energy transfer in the core of photosystem II. J Phys Chem B 107:6214–6220 [DOI]
- Demmig-Adams B, Adams W Jr, Mattoo A (eds) (2006) Photoprotection, photoinhibition, gene regulation, and environment. In: Govindjee (Series ed) Advances in photosynthesis and respiration, vol 21. Springer, Dordrecht
- Durrant JR, Hastings G, Joseph DM, Barber J, Porter G, Klug DR (1992) Subpicosecond equilibration of excitation-energy in isolated photosystem-II reaction centers. Proc Natl Acad Sci USA 89:11632–11636 [DOI] [PMC free article] [PubMed]
- Frank HA, Cua A, Chynwat V, Young A, Gosztola D, Wasielewski MR (1994) Photophysics of the carotenoids associated with the xanthophyll cycle in photosynthesis. Photosynth Res 41:389–395 [DOI] [PubMed]
- Frank HA, Britton G, Cogdell RJ (eds) (1999) The photochemistry of carotenoids. In: Govindjee (Series ed) Advances in photosynthesis and respiration, vol 9. Springer, Dordrecht
- Gradinaru CC, Van Stokkum IHM, Pascal AA, Van Grondelle R, Van Amerongen H (2000) Identifying the pathways of energy transfer between carotenoids and chlorophylls in LHCII and CP29. A multicolor, femtosecond pump-probe study. J Phys Chem B 104:9330–9342 [DOI]
- Gradinaru CC, Kennis JTM, Papagiannakis E, Van Stokkum IHM, Cogdell RJ, Fleming GR, Niederman RA, Van Grondelle R (2001) An unusual pathway of excitation energy deactivation in carotenoids: singlet-to-triplet conversion on an ultrafast timescale in a photosynthetic antenna. Proc Natl Acad Sci USA 98:2364–2369 [DOI] [PMC free article] [PubMed]
- Groot ML, Van Grondelle R (2008) Femtosecond time-resolved infrared spectroscopy. In: Aartsma TJ, Matysik J (eds) Biophysical techniques in photosynthesis, volume II. Advances in photosynthesis and respiration, vol 28. Springer, Dordrecht, pp 191–200
- Groot ML, Van Mourik F, Eijckelhoff C, Van Stokkum IHM, Dekker JP, Van Grondelle R (1997) Charge separation in the reaction center of photosystem II studied as a function of temperature. Proc Natl Acad Sci USA 94:4389–4394 [DOI] [PMC free article] [PubMed]
- Groot ML, Pawlowicz NP, Van Wilderen L, Breton J, Van Stokkum IHM, Van Grondelle R (2005) Initial electron donor and acceptor in isolated photosystem II reaction centers identified with femtosecond mid-IR spectroscopy. Proc Natl Acad Sci USA 102:13087–13092 [DOI] [PMC free article] [PubMed]
- Groot ML, Van Wilderen L, Di Donato M (2007) Time-resolved methods in biophysics. 5. Femtosecond time-resolved and dispersed infrared spectroscopy on proteins. Photochem Photobiol Sci 6:501–507 [DOI] [PubMed]
- Holt NE, Kennis JTM, Fleming GR (2004) Femtosecond fluorescence upconversion studies of light harvesting by beta-carotene in oxygenic photosynthetic core proteins. J Phys Chem B 108:19029–19035 [DOI]
- Holt NE, Zigmantas D, Valkunas L, Li XP, Niyogi KK, Fleming GR (2005) Carotenoid cation formation and the regulation of photosynthetic light harvesting. Science 307:433–436 [DOI] [PubMed]
- Holzwarth AR, Muller MG, Niklas J, Lubitz W (2006a) Ultrafast transient absorption studies on Photosystem I reaction centers from Chlamydomonas reinhardtii. 2: mutations near the P700 reaction center chlorophylls provide new insight into the nature of the primary electron donor. Biophys J 90:552–565 [DOI] [PMC free article] [PubMed]
- Holzwarth AR, Muller MG, Reus M, Nowaczyk M, Sander J, Rogner M (2006b) Kinetics and mechanism of electron transfer in intact photosystem II and in the isolated reaction center: pheophytin is the primary electron acceptor. Proc Natl Acad Sci USA 103:6895–6900 [DOI] [PMC free article] [PubMed]
- Horton P, Ruban AV, Walters RG (1996) Regulation of light harvesting in green plants. Annu Rev Plant Physiol Plant Mol Biol 47:655–684 [DOI] [PubMed]
- Ilagan RP, Koscielecki JF, Hiller RG, Sharples FP, Gibson GN, Birge RR, Frank HA (2006) Femtosecond time-resolved absorption spectroscopy of main-form and high-salt peridinin-chlorophyll a-proteins at low temperatures. Biochemistry 45:14052–14063 [DOI] [PubMed]
- Jimenez R, Fleming GR (1996) Ultrafast spectroscopy of photosynthetic systems. In: Amesz J, Hoff AJ (eds) Biophysical techniques in photosynthesis. Advances in photosynthesis and respiration (Series ed. Govindjee), vol 3. Springer, Dordrecht, pp 63–73
- Kennis JTM, Groot ML (2007) Ultrafast spectroscopy of biological photoreceptors. Curr Opin Struct Biol 17:623–630 [DOI] [PubMed]
- Kennis JTM, Shkuropatov AY, Van Stokkum IHM, Gast P, Hoff AJ, Shuvalov VA, Aartsma TJ (1997a) Formation of a long-lived P(+)B(A)(−)state in plant pheophytin-exchanged reaction centers of Rhodobacter sphaeroides R26 at low temperature. Biochemistry 36:16231–16238 [DOI] [PubMed]
- Kennis JTM, Streltsov AM, Vulto SIE, Aartsma TJ, Nozawa T, Amesz J (1997b) Femtosecond dynamics in isolated LH2 complexes of various species of purple bacteria. J Phys Chem B 101:7827–7834 [DOI]
- Kennis JTM, Gobets B, Van Stokkum IHM, Dekker JP, Van Grondelle R, Fleming GR (2001) Light harvesting by chlorophylls and carotenoids in the photosystem I core complex of Synechococcus elongatus: a fluorescence upconversion study. J Phys Chem B 105:4485–4494 [DOI]
- Kennis JTM, Larsen DS, Van Stokkum NHM, Vengris M, Van Thor JJ, Van Grondelle R (2004) Uncovering the hidden ground state of green fluorescent protein. Proc Natl Acad Sci USA 101:17988–17993 [DOI] [PMC free article] [PubMed]
- Kodis G, Herrero C, Palacios R, Marino-Ochoa E, Gould S, De la Garza L, Van Grondelle R, Gust D, Moore TA, Moore AL, Kennis JTM (2004) Light harvesting and photoprotective functions of carotenoids in compact artificial photosynthetic antenna designs. J Phys Chem B 108:414–425 [DOI]
- Kovalenko SA, Dobryakov AL, Ruthmann J, Ernsting NP (1999) Femtosecond spectroscopy of condensed phases with chirped supercontinuum probing. Phys Rev A 59:2369–2384 [DOI]
- Krueger BP, Lampoura SS, Van Stokkum IHM, Papagiannakis E, Salverda JM, Gradinaru CC, Rutkauskas D, Hiller RG, Van Grondelle R (2001) Energy transfer in the peridinin chlorophyll-a protein of Amphidinium carterae studied by polarized transient absorption and target analysis. Biophys J 80:2843–2855 [DOI] [PMC free article] [PubMed]
- Lakowicz JR (2006) Principles of fluorescence spectroscopy, 3rd edn. Springer, Berlin
- Larsen DS, Papagiannakis E, Van Stokkum IHM, Vengris M, Kennis JTM, Van Grondelle R (2003) Excited state dynamics of beta-carotene explored with dispersed multi-pulse transient absorption. Chem Phys Lett 381:733–742 [DOI]
- Ma YZ, Holt NE, Li XP, Niyogi KK, Fleming GR (2003) Evidence for direct carotenoid involvement in the regulation of photosynthetic light harvesting. Proc Natl Acad Sci USA 100:4377–4382 [DOI] [PMC free article] [PubMed]
- Marino-Ochoa E, Palacios R, Kodis G, Macpherson AN, Gillbro T, Gust D, Moore TA, Moore AL (2002) High-efficiency energy transfer from carotenoids to a phthalocyanine in an artificial photosynthetic antenna. Photochem Photobiol 76:116–121 [DOI] [PubMed]
- Monshouwer R, Baltuska A, Van Mourik F, Van Grondelle R (1998) Time-resolved absorption difference spectroscopy of the LH-1 antenna of Rhodopseudomonas viridis. Am Chem Soc:4360–4371
- Mukamel S (1995) Principles of nonlinear optical spectroscopy. Oxford University Press, New York
- Müller P, Li XP, Niyogi KK (2001) Non-photochemical quenching. A response to excess light energy. Plant Physiol 125:1558–1566 [DOI] [PMC free article] [PubMed]
- Nagarajan V, Alden RG, Williams JC, Parson WW (1996) Ultrafast exciton relaxation in the B850 antenna complex of Rhodobacter sphaeroides. Proc Natl Acad Sci USA 93:13774–13779 [DOI] [PMC free article] [PubMed]
- Niedzwiedzki D, Koscielecki JF, Cong H, Sullivan JO, Gibson GN, Birge RR, Frank HA (2007) Ultrafast dynamics and excited state spectra of open-chain carotenoids at room and low temperatures. J Phys Chem B 111:5984–5998 [DOI] [PubMed]
- Nishimura K, Rondonuwu FS, Fujii R, Akahane J, Koyama Y, Kobayashi T (2004) Sequential singlet internal conversion of 1B(u)(+)→3A(g)(−)→1B(u)(−)→2A(g)(−)→(1 A(g)(−) ground) in all-trans-spirilloxanthin revealed by two-dimensional sub-5-fs spectroscopy. Chem Phys Lett 392:68–73 [DOI]
- Novoderezhkin VI, Taisova AS, Fetisova Z, Blankenship RE, Savikhin S, Buck DR, Struve WS (1998) Energy transfers in the B808-866 antenna from the green bacterium Chloroflexus aurantiacus. Biophys J 74:2069–2075 [DOI] [PMC free article] [PubMed]
- Novoderezhkin V, Monshouwer R, Van Grondelle R (1999) Exciton (de)localization in the LH2 antenna of Rhodobacter sphaeroides as revealed by relative difference absorption measurements of the LH2 antenna and the B820 subunit. J Phys Chem B 103:10540–10548 [DOI]
- Novoderezhkin VI, Palacios MA, Van Amerongen H, Van Grondelle R (2004) Energy-transfer dynamics in the LHCII complex of higher plants: modified redfield approach. J Phys Chem B 108:10363–10375 [DOI] [PubMed]
- Palacios MA, Standfuss J, Vengris M, Van Oort BF, Van Stokkum IHM, Kuhlbrandt W, Van Amerongen H, Van Grondelle R (2006) A comparison of the three isoforms of the light-harvesting complex II using transient absorption and time-resolved fluorescence measurements. Photosynth Res 88:269–285 [DOI] [PubMed]
- Pan J, Benko G, Xu YH, Pascher T, Sun LC, Sundström V, Polivka T (2002) Photoinduced electron transfer between a carotenoid and TiO2 nanoparticle. J Am Chem Soc 124:13949–13957 [DOI] [PubMed]
- Papagiannakis E, Kennis JTM, Van Stokkum IHM, Cogdell RJ, Van Grondelle R (2002) An alternative carotenoid-to-bacteriochlorophyll energy transfer pathway in photosynthetic light harvesting. Proc Natl Acad Sci USA 99:6017–6022 [DOI] [PMC free article] [PubMed]
- Papagiannakis E, Das SK, Gall A, Van Stokkum IHM, Robert B, Van Grondelle R, Frank HA, Kennis JTM (2003) Light harvesting by carotenoids incorporated into the B850 light-harvesting complex from Rhodobacter sphaeroides R-26.1: excited-state relaxation, ultrafast triplet formation, and energy transfer to bacteriochlorophyll. J Phys Chem B 107:5642–5649 [DOI]
- Papagiannakis E, Larsen DS, Van Stokkum IHM, Vengris M, Hiller R, Van Grondelle R (2004) Resolving the excited state equilibrium of peridinin in solution. Biochemistry 43:15303–15309 [DOI] [PubMed]
- Polivka T, Sundström V (2004) Ultrafast dynamics of carotenoid excited states—from solution to natural and artificial systems. Chem Rev 104:2021–2071 [DOI] [PubMed]
- Polivka T, Herek JL, Zigmantas D, Akerlund HE, Sundström V (1999) Direct observation of the (forbidden) S-1 state in carotenoids. Proc Natl Acad Sci USA 96:4914–4917 [DOI] [PMC free article] [PubMed]
- Polívka T, Zigmantas D, Sundström V, Formaggio E, Cinque G, Bassi R (2002) Carotenoid S1 state in a recombinant light-harvesting complex of photosystem II. Biochemistry 41:439–450 [DOI] [PubMed]
- Ritz T, Damjanovic A, Schulten K, Zhang JP, Koyama Y (2000) Efficient light harvesting through carotenoids. Photosynth Res 66:125–144 [DOI] [PubMed]
- Ruban AV, Berera R, Ilioaia C, Van Stokkum IHM, Kennis JTM, Pascal AA, Van Amerongen H, Robert B, Horton P, Van Grondelle R (2007) Identification of a mechanism of photoprotective energy dissipation in higher plants. Nature 450:575–579 [DOI] [PubMed]
- Savikhin S, Vanamerongen H, Kwa SLS, Van Grondelle R, Struve WR (1994) Low-temperature energy-transfer in LHC-II trimers from the Chl-a/b light-harvesting antenna of photosystem-II. Biophys J 66:1597–1603 [DOI] [PMC free article] [PubMed]
- Savikhin S, Buck DR, Struve WS (1998) Toward level-to-level energy transfers in photosynthesis: the Fenna-Matthews-Olson protein. J Phys Chem B 102:5556–5565 [DOI]
- Savikhin S, Xu W, Soukoulis V, Chitnis PR, Struve WS (1999) Ultrafast primary processes in photosystem I of the cyanobacterium Synechocystis sp. PCC 6803. Biophys J 76:3278–3288 [DOI] [PMC free article] [PubMed]
- Streltsov AM, Vulto SIE, Shkuropatov AY, Hoff AJ, Aartsma TJ, Shuvalov VA (1998) B-A and B-B absorbance perturbations induced by coherent nuclear motions in reaction centers from Rhodobacter sphaeroides upon 30-fs excitation of the primary donor. J Phys Chem B 102:7293–7298 [DOI]
- Sundström V (2008) Femtobiology. Annu Rev Phys Chem 59:53–77 [DOI] [PubMed]
- Sundström V, Pullerits T, Van Grondelle R (1999) Photosynthetic light-harvesting: reconciling dynamics and structure of purple bacterial LH2 reveals function of photosynthetic unit. J Phys Chem B 103:2327–2346 [DOI]
- Van Amerongen H, Van Grondelle R (2001) Understanding the energy transfer function of LHCII, the major light-harvesting complex of green plants. J Phys Chem B 105:604–617 [DOI]
- Van Grondelle R (1985) Excitation energy transfer, trapping and annihilation in photosynthetic systems. Biochim Biophys Acta 811:147–195
- Van Grondelle R, Dekker JP, Gillbro T, Sundström V (1994) Energy-transfer and trapping in photosynthesis. Biochim Biophys Acta 1187:1–65 [DOI]
- Van Stokkum IHM, Larsen DS, Van Grondelle R (2004) Global and target analysis of time-resolved spectra. Biochim Biophys Acta 1657:82–104 [DOI] [PubMed]
- Vos MH, Breton J, Martin JL (1997) Electronic energy transfer within the hexamer cofactor system of bacterial reaction centers. J Phys Chem B 101:9820–9832 [DOI]
- Vulto SIE, Streltsov AM, Aartsma TJ (1997) Excited state energy relaxation in the FMO complexes of the green bacterium Prosthecochloris aestuarii at low temperatures. J Phys Chem B 101:4845–4850 [DOI]
- Vulto SIE, Kennis JTM, Streltsov AM, Amesz J, Aartsma TJ (1999) Energy relaxation within the B850 absorption band of the isolated light-harvesting complex LH2 from Rhodopseudomonas acidophila at low temperature. J Phys Chem B 103:878–883 [DOI]
- Walla PJ, Linden PA, Hsu CP, Scholes GD, Fleming GR (2000) Femtosecond dynamics of the forbidden carotenoid S-1 state in light-harvesting complexes of purple bacteria observed after two-photon excitation. Proc Natl Acad Sci USA 97:10808–10813 [DOI] [PMC free article] [PubMed]
- Walla PJ, Linden PA, Ohta K, Fleming GR (2002) Excited-state kinetics of the carotenoid S-1 state in LHC II and two-photon excitation spectra of lutein and beta-carotene in solution: efficient car S-1→Chl electronic energy transfer via hot S-1 states? J Phys Chem A 106:1909–1916 [DOI]
- Wang HY, Lin S, Allen JP, Williams JC, Blankert S, Laser C, Woodbury NW (2007) Protein dynamics control the kinetics of initial electron transfer in photosynthesis. Science 316:747–750 [DOI] [PubMed]
- Wehling A, Walla PJ (2005) Time-resolved two-photon spectroscopy of photosystem I determines hidden carotenoid dark-state dynamics. J Phys Chem B 109:24510–24516 [DOI] [PubMed]
- Wilson A, Punginelli C, Gall A, Bonettit C, Alexandre M, Routaboul JM, Kerfeld CA, Van Grondelle R, Robert B, Kennis JTM, Kirilovsky D (2008) A photoactive carotenoid protein acting as light intensity sensor. Proc Natl Acad Sci USA 105:12075–12080 [DOI] [PMC free article] [PubMed]
- Zhang JP, Fujii R, Qian P, Inaba T, Mizoguchi T, Koyama Y, Onaka K, Watanabe Y, Nagae H (2000) Mechanism of the carotenoid-to-bacteriochlorophyll energy transfer via the S1 state in the LH2 complexes from purple bacteria. J Phys Chem B 104:3683–3691 [DOI]
- Zigmantas D, Hiller RG, Sundström V, Polivka T (2002) Carotenoid to chlorophyll energy transfer in the peridinin-chlorophyll-a-protein complex involves an intramolecular charge transfer state. Proc Natl Acad Sci USA 99:16760–16765 [DOI] [PMC free article] [PubMed]
- Zigmantas D, Read EL, Fleming GR (2008) Non-linear femtosecond optical spectroscopy in photosynthesis. In: Aartsma TJ, Matysik J (ed) Biophysical techniques in photosynthesis, volume II. Advances in photosynthesis and respiration, vol 26. Springer, Dordrecht, pp 201–222