

Abstract
Retinitis pigmentosa 1 (RP1) is a common inherited retinopathy with variable onset and severity. The RP1 gene encodes a photoreceptor-specific, microtubule-associated ciliary protein containing the doublecortin (DCX) domain. Here we show that another photoreceptor-specific Rp1-like protein (Rp1L1) in mice is also localized to the axoneme of outer segments (OSs) and connecting cilia in rod photoreceptors, overlapping with Rp1. Rp1L1−/− mice display scattered OS disorganization, reduced electroretinogram amplitudes, and progressive photoreceptor degeneration, less severe and slower than in Rp1−/− mice. In single rods of Rp1L1−/−, photosensitivity is reduced, similar to that of Rp1−/−. While individual heterozygotes are normal, double heterozygotes of Rp1 and Rp1L1 exhibit abnormal OS morphology and reduced single rod photosensitivity and dark currents. The electroretinogram amplitudes of double heterozygotes are more reduced than those of individual heterozygotes combined. In support, Rp1L1 interacts with Rp1 in transfected cells and in retina pull-down experiments. Interestingly, phototransduction kinetics are normal in single rods and whole retinas of individual or double Rp1 and Rp1L1 mutant mice. Together, Rp1 and Rp1L1 play essential and synergistic roles in affecting photosensitivity and OS morphogenesis of rod photoreceptors. Our findings suggest that mutations in RP1L1 could underlie retinopathy or modify RP1 disease expression in humans.
Introduction
Retinitis pigmentosa 1 (RP1) is an autosomal dominant RP (adRP) that causes characteristic night blindness and abnormal or extinguished electroretinogram (ERG) amplitudes and is associated with retinal atrophy, deposition of pigment, and attenuation of retinal vessels. Heterozygous carriers of RP1 mutations exhibit late onset of retinopathies, whereas homozygous carriers tend to have earlier onset and more severe symptoms (Haider et al., 2002; Daiger et al., 2006). RP1 locus mutations account for 5–10% of all adRPs; the RP1 mutant allele derived from the UCLA-RP01 pedigree is the third most frequent mutation causing adRP (Blanton et al., 1991; Bowne et al., 1999; Pierce et al., 1999; Sullivan et al., 1999; Jacobson et al., 2000). The onset and severity of RP1 disease symptoms can vary greatly, even within the same pedigree with the same mutation. Such variations suggest the influence of genetic modifiers or environmental factors (Daiger et al., 2006).
The RP1 gene encodes a 240 kDa retinal photoreceptor-specific protein (Pierce et al., 1999; Sullivan et al., 1999). The N terminus of RP1 (∼300 aa) shares significant homology with doublecortin (DCX), a known microtubule-associated protein that, when mutated, causes the double cortex syndrome in fetuses (des Portes et al., 1998; Sossey-Alaoui et al., 1998; Gleeson et al., 1999; Pilz et al., 1999). The outer segment (OS) axoneme of rod and cone photoreceptors localizes RP1 to the site of OS disc assembly (Liu et al., 2002, 2004). Two independent mouse lines with targeted disruptions of Rp1 have similar phenotypes of OS misalignment and dysplasia: one deletes the N-terminal conserved DCX tandem repeats (Gao et al., 2002), and the other introduces a truncation at the residue corresponding to the human UCLA-RP01 mutation, resulting in expression of only the DCX-containing N terminus (Q. Liu et al., 2003, 2004). Furthermore, Rp1 assembles and stabilizes microtubules in vitro and in vivo (Liu et al., 2004; Coquelle et al., 2006), providing evidence that Rp1 is a photoreceptor-specific microtubule-associated protein. A gene encoding an RP1-like protein 1 (RP1L1) has been identified through sequence analyses of human and mouse genomes (Bowne et al., 2003; Conte et al., 2003). RP1 and RP1L1 have similar DCX tandem repeats at their N termini followed by a 34 aa domain (RP1D) that is unique to them. The C termini have no significant homology to each other or to other proteins. The RP1 and RP1L1 genes have identical four-exon structures. Moreover, both are photoreceptor specific with identical temporal expression patterns during postnatal development. Although no mutations in RP1L1 have been identified, these striking similarities between RP1 and RP1L1 strongly suggest that they are colocalized and involved in similar functions in the photoreceptor.
Here we describe the creation and characterization of a knock-out mouse that lacks the Rp1L1 protein. Rp1L1 and Rp1 both localize to the OS axoneme, and their interactions are examined genetically and biochemically. We conclude that both are essential for OS morphogenesis and normal photosensitivity in rod photoreceptors. Our findings suggest that mutations in RP1L1 may cause autosomal recessive RP or modify RP1 disease expression.
Materials and Methods
Creation of Rp1L1−/− mice.
To generate the Rp1L1 knock-out mice, we used the recombineering approach developed by P. Liu et al. (2003) (see also Gao et al., 2004). Briefly, we first designed two sets of primers (AB and XY) to amplify two small fragments that flank the region from exon 2 to exon 4 in mouse Rp1L1 genomic DNA, and subcloned into the vector VP101. The VP101 vector was transformed into Escherichia coli cells (EL350), which contain the Rp1L1 BAC DNA, to retrieve the DNA fragment flanked by AB and XY into the VP101 vector (VP101-retrieve). Two sets of primers (CD and EF) were designed to amplify another two small fragments flanking the Rp1L1 genomic DNA starting from exon 2 to the middle of exon 4, and the PCR fragments were cloned into vector PL452 to flank the neomycin resistance cassette (PL452-mini target). The VP101-retrieve and PL452-mini target vectors were cotransformed into competent EL350 cells. AB2.2 embryonic stem cells (specialty medium) derived from the 129/SvEv strain were electroporated with NotI-linearized targeting vector. DNA from ES cells was digested by EcoRI and analyzed by Southern blotting.
Genotype.
PCR was performed using TaqDNA polymerase (Fisher Scientific). For Rp1L1−/−, PCR primers were designed as follows: upstream primer for wild-type allele, 5′-GTT GAG TGT TTG CCC AGT GTC-3′; upstream primer for knock-out allele, 5′-AAG CGC CTC CCC TAC CCG-3′; common downstream primer, 5′-GGC AAC CAA GCT CTC CTC TG-3′. The thermal parameters used in PCR were denaturation, 30 s at 94°C; annealing, 30 s at 54°C; extension, 30 s at 72°C. For the Rp1 and Rp1L1 cross, we used primers specific for the Rp1 targeted allele: 5′-CCT CTG CCC ATT GTT TGA GT-3′; 5′-CGT TGG CTA CCC GTG ATA TT-3′.
Light and transmission electron microscopic analysis of retinal sections.
All animal experiments were performed in accordance with National Institute of Health and institutional guidelines approved by the Animal Care and Use Committee. All mice were killed by cervical dislocation 8–12 h after the onset of the light phase (unless specified otherwise). The procedures for retinal histological analyses were described previously (Gao et al., 2002).
Constructs.
For mammalian expression vector of the N-terminal region (1–361) of Rp1L1 (accession number AAN86958), the oligonucleotides below were synthesized and used to amplify a fragment. The product was digested with EcoRI and SalI restriction enzymes, respectively, and subcloned into pcDNA3.1 mycHisA. Forward primer: 5′-ACC GAA TTC GCC ACC ATG AAC AGC ACC CCA GGA G-3′; reverse primer: 5′-CGC CCT CTA GAG GTT TTG GGG GGC TTC CTA TC-3′.
Purification of recombinant protein.
cDNA encoding mouse Rp1L1 (accession number AAN86958), which contains amino acid residues 1281–1531, was subcloned into pET-41 Ek/LIC bacterial expression vector (Novagen). The soluble fraction of the GST and His fusion protein was then affinity purified with a Ni-column (Qiagen) according to the manufacturer's protocol.
For the pull-down assay, cDNA encoding mouse Rp1 (accession number AF291754), which contains amino acid residues 27–365, was subcloned into pGEX-4T-1 bacterial expression vector (GE Healthcare). The soluble fraction of the GST fusion protein was then affinity-purified with glutathione-Sepharose 4B (GE Healthcare) according to the manufacturer's protocol.
Production of antibodies.
An antibody specific for Rp1L1 was obtained by immunizing rabbits with purified GST and His-fused Rp1L1 recombinant protein (described above) (Proteintech Group). Polyclonal antibody was then affinity-purified using the fusion protein coupled to NHS-activated Sepharose 4 (GE Healthcare). The antibody specificity was determined by immunoblotting and immunostain analysis as described below.
SDS-PAGE and immunoblot analysis.
To visualize the Rp1L1 band, 100 μg of retina whole-cell lysates were subjected to SDS-PAGE, followed by blotting onto a polyvinylidene difluoride (PVDF) membrane (Millipore). Primary antibodies used were rabbit anti-Rp1L1 antibody (0.5 μg/ml), mouse anti-FLAG M2 monoclonal antibody (F3165, 1:5000 dilution, Sigma), mouse anti-myc (9E10) antibody (M4439, 1:7000 dilution, Sigma), mouse anti-rhodopsin monoclonal antibody (MAB5316, 1:2000 dilution, Millipore Bioscience Research Reagents), rabbit anti-rod transducin α polyclonal antibody (sc-389, 1:5000 dilution, Santa Cruz), rabbit anti-PDE6α polyclonal antibody (ab5659, 1:1000 dilution, Abcam), goat anti-CNG1 polyclonal antibody (sc-13694, 1:1000 dilution, Santa Cruz), and mouse anti-GAPDH monoclonal antibody (ab8245, 1:5000 dilution, Abcam).
Immunocytochemistry.
For immunostaining of retina sections, either the eye cup or retina was taken out and fixed with 4% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4, overnight. Primary antibodies used were rabbit anti-Rp1L1 polyclonal antibody (20 μg/ml), chicken anti-Rp1 polyclonal antibody (1:100 dilution, kindly donated by Eric Pierce), mouse acetylated α-tubulin monoclonal antibody (T6793, 1:200 dilution, Sigma), mouse γ-tubulin monoclonal antibody (T6557, 1:200 dilution, Sigma), mouse GFAP monoclonal antibody (G3893, 1:100 dilution, Sigma), and peanut agglutinin (PNA, RL1072, 1:10 dilution, Vector Laboratories). The immunofluorescence was visualized with Alexa Fluor 488 goat anti-mouse IgG (H+L), Alexa Fluor 488 goat anti-rabbit IgG (H+L), Alexa Fluor 546 goat anti-mouse IgG (H+L), or Alexa Fluor 546 goat anti-chicken IgG (H+L) (Invitrogen). Fluorescence images were analyzed with a Zeiss Axiophot2 microscope using a LSM510 Meta confocal laser scanning image system (Carl Zeiss).
Preparation of photoreceptor OSs.
Individual photoreceptor OSs were isolated with a slight modification of established techniques (Hong et al., 2001; Yang et al., 2002). In brief, fresh retinas from C57BL/6 mice, Rp1L1−/− mice, or Rp1L1+/+ mice 21 d postnatally (P21) were collected in a microcentrifuge tube containing 0.5 ml of PBS consisting of 0.01 m phosphate, 0.138 m NaCl, and 0.0027 m KCl, pH 7.4. The tube was shaken vigorously for a few seconds and allowed to settle. Next, 10 μl of the buffer containing intact OSs was transferred to a poly-l-lysine-treated glass coverslip and then subjected to immunostaining described above.
TUNEL assay.
Cell death was detected by TUNEL assay using the ApopTag Peroxidase In Situ Apoptosis Detection Kit (Millipore Bioscience Research Reagents) according to the manufacturer's protocol.
ERG measurements and fundus photography.
Mouse eyes were dilated with phenylephrine hydrochloride 2.5% and tropicamide 2%, dark-adapted 4.5 h to overnight and prepared for recording under a dim red light while the dilation was reinforced with hyoscine. Mice were anesthetized with intraperitoneal injection of 0.008 ml/g of a mixture of ketamine (20%), xylazine (0.5%), and sodium chloride solution (79.5%). The eye was numbed with Alcaine, and Refresh Celluvisc was added to the contact-lens electrode to protect the eye. Signals were amplified (×10,000) and filtered (eight-pole Butterworth 60 Hz notch filter) to remove line noise before averaging (n = 20–200) by computer. A Ganzfeld dome and the incorporated Grass photostimulator, similar to systems used in human testing, was used to produce flashes comparable to those of the International Society for the Clinical Electrophysiology of Vision standard (Marmor and Fishman, 1989). Rod a-waves were elicited by a high-intensity Xenon flash (Novatron). Cone b-wave responses were obtained in the presence of a rod-saturating background (3.2 log Td). Fundus photographs and fluorescein angiographs for Rp1L1+/+ and Rp1L1−/− mice were taken at 6 and 12 months of age using a Kowa RC-2 handheld fundus camera. The eyes were dilated with phenylephrine hydrochloride 2.5% 1 h before photography.
Single-cell recordings.
Mice were dark-adapted overnight before experiments. Animals were killed in dim red illumination by CO2 asphyxiation followed by cervical dislocation. The eye was removed, and the retina was isolated and finely chopped under infrared illumination. Small pieces of the retina were placed in the experimental chamber on the stage of an inverted microscope and perfused with bicarbonate-buffered solution (112.5 mm NaCl, 3.6 mm KCl, 2.4 mm MgCl2, 1.2 mm CaCl2, 10 mm HEPES, pH 7.4, 20 mm NaHCO3, 3 mm sodium succinate, 0.5 mm sodium glutamate, 0.02 mm EDTA, and 10 mm glucose). The solution was bubbled with 95% O2/5% CO2 and warmed to 36–38°C in a flow heater before it entered the experimental chamber. Membrane current was recorded with a suction electrode from a rod OS projecting from a piece of retina. The recording electrode was filled with 140 mm NaCl, 3.6 mm KCl, 2.4 mm MgCl2, 1.2 mm CaCl2, 3 mm HEPES, pH 7.4, 0.02 mm EDTA, and 10 mm glucose. Flashes (20 ms) were delivered from a calibrated light source via computer-controlled shutters. Light intensity and wavelength were changed by using calibrated neutral-density and interference filters. The current was amplified, low-pass filtered at 30 Hz, digitized at 1 kHz, and stored on a computer for subsequent analysis. Dark current in individual rods was measured as the amplitude of a saturating light response. Sensitivity was estimated from the intensity–response curve for each cell as the light intensity required to produce a half-saturating response. Single-photon response was estimated as the ratio of variance and mean of a family of 30 responses to identical dim flashes. Time to peak was measured as the time between the midpoint of the flash and the peak of the response. Integration time was calculated as the time integral of the dim-flash response divided by its peak amplitude.
Tissue cultures and transfection.
Human embryonic kidney cells (293T) and COS7 cells were maintained in DMEM (Invitrogen) supplemented with 10% (v/v) heat-inactivated FBS at 37°C in a humidified atmosphere containing 5% CO2. The cells were seeded at a density of 1.2 × 106 cells on each 100 mm dish the day before transfection. Transfection was performed using Fugene6 according to manufacturer's protocol.
In vitro coimmunoprecipitation assay.
COS7 cells (2 × 106 cells per 100 mm dish) were transfected with pcDNA3.1mycHis-Rp1L1 (1–361) or p3XFLAG-Rp1. One day after transfection, cells were lysed in TNE buffer (50 mm Tris-HCl, pH 7.5, 1% NP-40, 5 mm EDTA, and 145 mm NaCl) containing a protease inhibitor cocktail (Sigma). The insoluble fraction was excluded by centrifugation at 18,000 × g for 30 min, and the supernatants were used for coimmunoprecipitation by incubation with anti-FLAG M2 resin (Sigma) or protein G Sepharose 4 Fast Flow (GE Healthcare) as a negative control for 1 h at 4°C. The immunoprecipitates were washed three times with TNE buffer and eluted with Laemmli sample buffer.
In vitro and in vivo pull-down assay.
Purified GST-Rp1 (residues 27–365) fusion protein and the Myc/His-fused Rp1L1-transfected 293T cell lysate were used in vitro pull-down assays. For the in vivo experiment, mouse retinal homogenates were prepared in TNE buffer (50 mm Tris-HCl, pH 7.5, 1% NP-40, 5 mm EDTA, and 145 mm NaCl) containing a protease inhibitor cocktail (Sigma), using a glass/Teflon homogenizer (20 passes). The insoluble fraction was excluded by centrifugation at 18,000 × g for 30 min. To inhibit polymerization of microtubules during the experiment, the supernatants were mixed with 5 μg/ml (17 μm) nocodazole and then incubated with 10 μg of GST-RP1 (residues 27–365) or GST (negative control) coupled to glutathione-Sepharose 4B (GE Healthcare) at 4°C for 6 h. The GST beads were washed three times with TNE buffer.
Results
Generation of Rp1L1−/− mice
To study in vivo the function of Rp1L1 and to provide negative controls for Rp1L1 localization studies, we created an Rp1L1 knock-out mouse by deleting exons 2 and 3 and the first part of exon 4, which encodes the conserved DCX tandem repeat and the 34 aa RP1D domain of Rp1L1 (Fig. 1A, B). Homozygous mutant mice were viable and displayed no detectable behavioral abnormalities; their retinas contained no Rp1L1 mRNA, as shown by RT-PCR analysis of the deleted region of Rp1L1 (Fig. 1C).
Figure 1.

Generation of Rp1L1 mutant mice. A, Targeted disruption of Rp1L1. Solid rectangles represent exons 2 through 4. The targeting vector contains a Neo-selectable marker and deletes exons 2 and 3 and the N-terminal portion of exon 4 in the Rp1L1 locus by homologous recombination. B, Genomic Southern blot analysis of Rp1L1 mutant mice. After EcoRI digestion of genomic DNA, an external probe as indicated detected a 9.2 kb band in the wild-type allele and a 7.6 kb band in the targeted allele. C, RT-PCR analysis of retinas of Rp1L1 mutant mice. Exons 3 and 4 were amplified and subjected to 1% agarose gel. No products from the targeted allele were detected (top). β-Actin was amplified as a control. D, Western blot analysis of retinal homogenates from Rp1L1 mutant mice using an affinity-purified Rp1L1 antibody. Each lane contains 100 μg of homogenates of the retinas of one mouse at P30. A 200 kDa band seen in Rp1L1+/+ mice is absent in Rp1L1−/− mice. E, Immunofluorescent staining on frozen sections (projected images) of retinas from wild-type and Rp1L1−/− mice at P30 using an affinity-purified C-terminal Rp1L1 antibody (green) and TOPRO3 (blue). RPE, Retinal pigment epithelium. Scale bar, 10 μm.
We developed a polyclonal antibody against the middle segment of Rp1L1 that is specific to Rp1L1 and is undeleted in Rp1L1−/− mice. The antibody's efficacy was tested by Western blot analysis using bacterially expressed recombinant fusion protein GST-Rp1L1 (1281–1531). The anti-Rp1L1 antibody specifically recognized GST-Rp1L1 but it did not recognize GST alone (data not shown). In Western blot analysis using retinal lysate, the antibody labeled multiple bands clustered around ∼200 kDa, consistent with the predicted size of Rp1L1 (Bowne et al., 2003) (Fig. 1D). These bands were absent in the knock-out lysate. Several faint bands below 130 kDa were also detected in retinal lysates; however, they were present in both genotypes, indicating that these bands were labeled nonspecifically and were not degraded products or isoforms of Rp1L1. Together, these findings showed that the knock-out mice were Rp1L1-null. We performed immunostaining of Rp1L1 using this polyclonal antibody on retinal sections of both genotypes (Fig. 1E). The Rp1L1 antibody labeled a subcellular region in the vicinity of connecting cilia (CCs) between the inner segment (IS) and OS in wild-type retinas, and this signal was completely absent in knock-out retinas. Furthermore, no signals were detected in other regions of the wild-type retinas that were absent in the knock-out retinas (data not shown).
Subcellular localization of Rp1L1 in photoreceptors
Because ∼97% of photoreceptors in mouse retinas are rods, we investigated whether Rp1L1 was present in cones as well. By double staining Rp1L1 with PNA [a cone-specific marker that labels the plasma membrane of the OS and IS (Rattner et al., 2001)], we analyzed dissociated OS preparations (Hong et al., 2001; Yang et al., 2002; Liu et al., 2004) and frozen sections from wild-type mouse retinas (Fig. 2A, top). In >200 PNA-positive dissociated cones, we found none with Rp1L1 staining (Fig. 2A, bottom; data not shown). Furthermore, we used confocal microscopy to analyze retinal sections of >200 PNA-positive cones in serial Z-sections 0.8 μm apart and found no Rp1L1 staining within the PNA-labeled plasma peripheral boundaries of the cones (Fig. 2A, top). The possibility still exists that it is more difficult to stain Rp1L1 in cones than in rods. However, given that we did not observe any Rp1L1 staining in a large number of cones using two distinct methods that both clearly showed Rp1L1 expression in rods, we believe that Rp1L1 is not expressed in cones.
Figure 2.

Cellular and subcellular distribution of Rp1L1 in mouse retinas. A, Cone receptors were visualized with rhodamine-conjugated PNA. A series of 0.8-μm-thick optical sections oriented close to the plane of the retina at the level of the connecting cilium. Top, Rp1L1 immunoreactivity (green) was not seen in a small zone within the cone sheath at the level of the IS–OS junction. Scale bar, 5 μm. Bottom, Rp1L1 proteins (green) and cone photoreceptors (red) were immunostained in dissociated OSs of mouse retinas at P21. Scale bar, 2.5 μm. B, Dissociated OSs from mice at P21 were labeled with anti-Rp1L1 antibody (green), anti-rhodopsin antibody (red, top), anti-γ-tubulin antibody (red, middle), and anti-Rp1 antibody (red, bottom). Rhodopsin staining indicates OSs. γ-Tubulin staining indicates the position of the basal bodies (white arrowheads). Rp1 staining indicates OS axoneme. Scale bar, 0.1 μm. C, Frozen sections of retinas from mice at P30 were stained with antibodies to Rp1L1 (green) and acetylated α-tubulin (red), and viewed by confocal microscopy (projected images). Boxed areas are merged and enlarged in the top; acetylated α-tubulin staining indicates axoneme. Scale bar (top, 10 μm; bottom, 1 μm) applies in all panels in C.
To determine the subcellular localization of Rp1L1 in rods, we double stained Rp1L1 and rhodopsin, Rp1, or γ-tubulin in dissociated OS preparations (Fig. 2B). Rhodopsin localizes to OS discs, Rp1 localizes to the axoneme of the basal OS (Zhao et al., 2003; Liu et al., 2004), and γ-tubulin localizes to the basal bodies of the CCs (Hong et al., 2001). We found that Rp1L1 localized to the axoneme of both the OS and CCs but not to the axoneme of the IS or on the basal bodies (Figs. 1E, 2B). Recent photoreceptor sensory cilium complex proteome revealed Rp1L1 expression in the OS but not in the IS, consistent with our observations (Liu et al., 2007). Furthermore, Rp1L1 coexisted with Rp1 only in the OS axoneme but not in the CCs; Rp1L1 and Rp1 appeared localized along the entire length of OS axoneme but were more concentrated at the basal third OS, consistent with previous findings (Fig. 2B, bottom) (Zhao et al., 2003; Liu et al., 2004). To determine whether Rp1L1 was colocalized with microtubules in the axoneme, we stained Rp1L1 in retinal sections with α-acetylated tubulin, which is a marker of polymerized microtubules, including those of the axoneme (Fig. 2C). All Rp1L1 signals were colocalized with axoneme-positive signals. Only a small amount of α-acetylated tubulin-positive axoneme did not appear to include Rp1L1. These were probably the axonemes of cones, which lack Rp1L1 (Fig. 2C, long arrowhead). These findings suggest that both Rp1L1 and Rp1 are expressed in the rod photoreceptor axoneme.
Outer nuclear layer and OS abnormalities in Rp1L1−/− retinas
We next examined Rp1L1−/− retinal photoreceptor phenotypes. We first examined the morphology of Rp1L1−/− and wild-type retinas at 1, 6, and 12 months using plastic sections. The retinas of Rp1L1−/− mice appeared overall nearly normal at these ages, with only a few minor abnormalities. For example, the outer nuclear layer (ONL) of the retinas of Rp1L1−/− mice at all ages was significantly thinner than that of wild-type retinas (Fig. 3A, B). At 12 months, the number of photoreceptors in Rp1L1 knock-out retinas was ∼60% of that in wild-type littermate retinas (Fig. 3B). The rate of ONL reduction appeared constant over a period of 12 months, indicating that at any given time, similar numbers of photoreceptor cells were dying, resulting in a progressive degeneration at a rate significantly slower than that of Rp1−/− retinas (Gao et al., 2002). Retina sections of Rp1L1−/− mice also revealed consistent but unique alterations in OS morphology (Fig. 3A, C). The OS was shorter at 1, 6, and 12 months. The average percentages of obviously abnormal OS observed in Rp1L1−/− retinas at ultrastructural levels increased from 15.1 ± 0.9% at 1 month (p < 0.05 compared with wild-type), to 25.7 ± 6.8% at 6 months (p < 0.05 compared with wild-type), to 19.9 ± 10.8% at 12 months (p < 0.05 compared with wild-type) in Rp1L1−/− retinas, relative to those observed in wild-type controls (3.1 ± 1.1% at 1 month, 6.5 ± 2.0% at 6 months, and 2.1 ± 1.8% at 12 months; due to preparation artifacts). Sporadically, the OS appeared to form vacuoles, and discs were disarrayed or swirling. Within the vacuoles, some dark spots appeared (Fig. 3C, long arrow). The content of these vacuoles in the OS is unknown, but they appeared similar to those in Purkinje cell degeneration (pcd−/−) retinal photoreceptors as early as P13. Moreover, abnormal OSs were often flanked by neighboring OS with a normal appearance (Fig. 3C). Swirling OS discs sometimes formed sharp boundaries in the basal or middle portion of the OS. The membranes appeared to take a different orientation than the normal OS discs and formed discrete segments along the entire OS length.
Figure 3.

Photoreceptor abnormalities in Rp1L1−/− retinas. A, Light micrographs of epoxy-embedded sections of central retinas of Rp1L1 mutant mice at ages 1 (1M), 6 (6M), and 12 (12M) months. Scale bar, 5 μm. The OS and ONL are significantly thinner in retinas of 6-month-old Rp1L1−/− mice than in retinas of wild-type littermates. B, Average numbers of photoreceptor rows in the ONL of central retinas located 300–400 μm from the optic nerve at ages 1, 6, and 12 months as described before (Gao et al., 2002). Group mean values and SDs (bars) are shown. Progressive degeneration of photoreceptors (manifested as the numbers of photoreceptor rows) in Rp1L1−/− mice is much milder than in Rp1−/− mice (Gao et al., 2002). C, Ultrastructural (transmission electron) micrographs of OSs at ages 1, 6, and 12 months. Scale bar, 2 μm. Note the appearance of vacuoles in several OSs (1M) and abnormal discs in isolated OSs flanked by normal-appearing OSs (1, 6, and 12M). Interestingly, the abnormal discs that are in the middle or basal segment of the OS and are bordered by normal-appearing discs in the same OS (6 and 12M). D, GFAP induction in retinal Müller glia in Rp1L1−/− retinas at 3 months (3M). Rp1L1+/+ (left) and Rp1L1−/− (right) retinas were stained with monoclonal anti-GFAP antibodies and fluorescein-conjugated secondary antibody. Scale bar, 10 μm.
Photoreceptor degeneration in Rp1L1−/− mice
To investigate photoreceptor cell death in Rp1L1−/− retinas, we performed in situ labeling of DNA fragmentation using the TUNEL method. At 3 months, TUNEL-positive photoreceptor cells were detected in Rp1L1−/− mice (2.6 ± 1.7 cells per retinal section) but not in their wild-type littermates. There were far fewer TUNEL-positive photoreceptors in Rp1L1−/− mice at 3 months than in Rp1−/− mice (26.8 ± 12.0 cells per retinal section) (Gao et al., 2002). Additionally, we examined activation of Müller glia by immunostaining with the glial fibrillary acidic protein (GFAP) antibody in retinas of Rp1L1−/− mice. Significant GFAP staining was clearly observed in Rp1L1−/− retinas in the ONL, inner nuclear layer, and ganglion cell layer at 3 months (Fig. 3D). The GFAP staining was much weaker at 1 month than at 3 months; no GFAP signal was observed in Rp1L1−/− retinas at 6 months except in the inner limiting membrane region (data not shown). These labeled fibers were probably from Müller cells reactive to photoreceptor cell degeneration, similar to those observed in Rp1−/− and RP GTPase regulator (Rpgr)−/− retinas (Hong et al., 2000; Gao et al., 2002). These observations were consistent with our measurements of ONL thickness in Rp1L1−/− mice and indicated that the lack of Rp1L1 caused photoreceptor cell death. Staining with the cone marker PNA was normal in Rp1L1−/− mice at 6 months (data not shown), showing that, as in Rp1−/− mice, cones did not degenerate in Rp1L1−/− mice at least until 6 months of age (Gao et al., 2002). These findings were consistent with our observation that Rp1L1 is absent from cones.
ERG and fundus analyses of Rp1L1−/− mice
ERG recordings were made for a comparison of retinal function in wild-type and Rp1L1−/− mice (Fig. 4A–C). Maximum rod (scotopic) a-wave responses to high-intensity flashes (which are proportional to the total number of functioning photoreceptor cells) at 6 and 12 months were reduced to 70% and 54% compared with age-matched wild-type mice (p < 0.01, unpaired Student's t test) (Fig. 4A), consistent with ONL length reductions at equivalent ages (70% and 60%) (Fig. 3B). Such a close correlation (correlation coefficiency of 1.00) between ERG rod a-wave amplitudes and ONL reductions in Rp1L1−/− retinas at 6 and 12 months further suggests that only a small number of photoreceptor cells degenerated at any time and that the remaining photoreceptors were still functional. The maximum rod (scotopic) b-waves in Rp1L1−/− mice at 12 months of age were significantly reduced to 79% of those of wild-type controls (p < 0.05, unpaired Student's t test) (Fig. 4B). The rod ERG b-wave represents the responses of secondary retinal neurons (i.e., bipolar and Müller cells). As in many other retinal rod mutants (Gao et al., 2002; Q. Liu et al., 2003), the fact that it was much less affected than the rod a-wave is consistent with the notion that rod dysfunctions are the primary cause of abnormalities in Rp1L1−/− retinas. The cone (photopic) b-waves were reduced significantly only at 12 months to 74% of the wild-type level (p < 0.01, unpaired Student's t test) (Fig. 4C). The fact that the cone responses were reduced at a slower rate than the rod responses is consistent with the normal appearance of cones in Rp1L1−/− mice at 6 months, similar to the effect seen in many other rod mutants (Gao et al., 2002; Q. Liu et al., 2003). The kinetics of ERG waves appeared normal in Rp1L1−/− mice at all ages examined (data not shown). Fundus images at 6 and 12 months revealed subretinal deposits, atrophy, and thinner arteries in Rp1L1−/− mice (data not shown), consistent with the histologic findings from their retinas.
Figure 4.
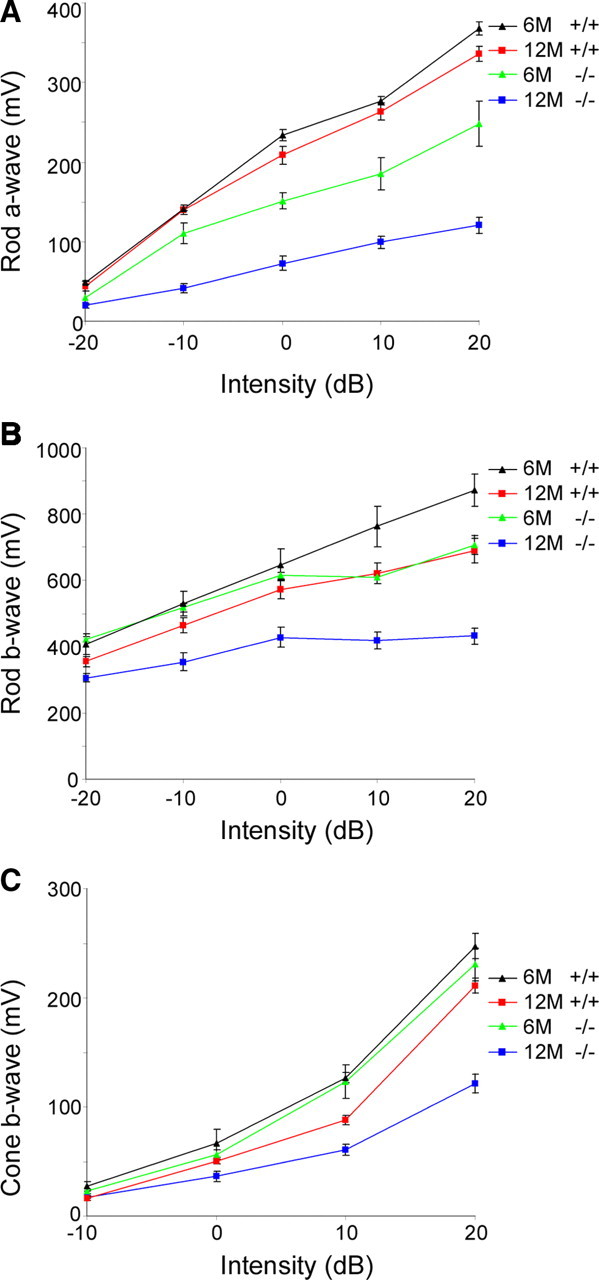
Dark-adapted ERG. A, Amplitudes (in millivolts) of scotopic a-waves with increasing light intensities. B, Amplitudes (in millivolts) of scotopic b-waves with increasing light intensities. C, Amplitudes (in millivolts) of photopic b-waves with increasing light intensities. Light intensity is in decibels; a series of light intensities were used from −20 dB to +20 dB (i.e., −2 to 2 log cd · s · m−2) in steps of 1 log cd · s · m−2. ERGs were obtained from 16 mice between the ages of 6 months (6M; black, wild-type; green, Rp1L1−/−) and 12 months (12M; red, wild-type; blue, Rp1L1−/−): 5 mice were Rp1L1−/− and 5 mice were Rp1L1+/+ at 12 months; 3 were Rp1L1−/− and 3 were Rp1L1+/+ at 6 months. The rod phototransduction model was fit to the leading edges of a-waves to high-intensity stimuli. Error bars give SEM.
Genetic interactions between Rp1 and Rp1L1
Because Rp1 and Rp1L1 exhibited identical temporal and spatial mRNA expression patterns in rod photoreceptors (Bowne et al., 2003) and protein distributions in the OS axoneme, we next examined potential genetic interactions between Rp1 and Rp1L1. Transmission electron microscopic studies of OS from Rp1+/−:Rp1L1+/− mice at 6 months of age revealed OS abnormalities in double heterozygous retinas similar to those in Rp1L1−/− retinas, but not in Rp1+/− and Rp1L1+/− retinas (Fig. 5A). The average percentage of obviously abnormal OSs, which includes vacuoles, disarrayed or swirling discs, observed in Rp1+/−:Rp1L1+/− retinas was 16.0 ± 2.3% at 6 months (p < 0.05 compared with Rp1+/+:Rp1L1+/−), much higher than those observed in individual heterozygous retinas (2.8% in Rp1+/−:Rp1L1+/+, 2.3 ± 2.2% in Rp1+/+:Rp1L1+/−) but lower than that in Rp1L1−/− retinas (25.7 ± 6.8%). The two-way ANOVA showed a significant effect of Rp1-Rp1L1 interaction (p = 0.0067). The ONL length appeared to be reduced at 6 months in double heterozygotes, although not significantly (Fig. 5B). These findings indicate that Rp1 and Rp1L1 play essential roles in disc organization in a synergistic manner.
Figure 5.
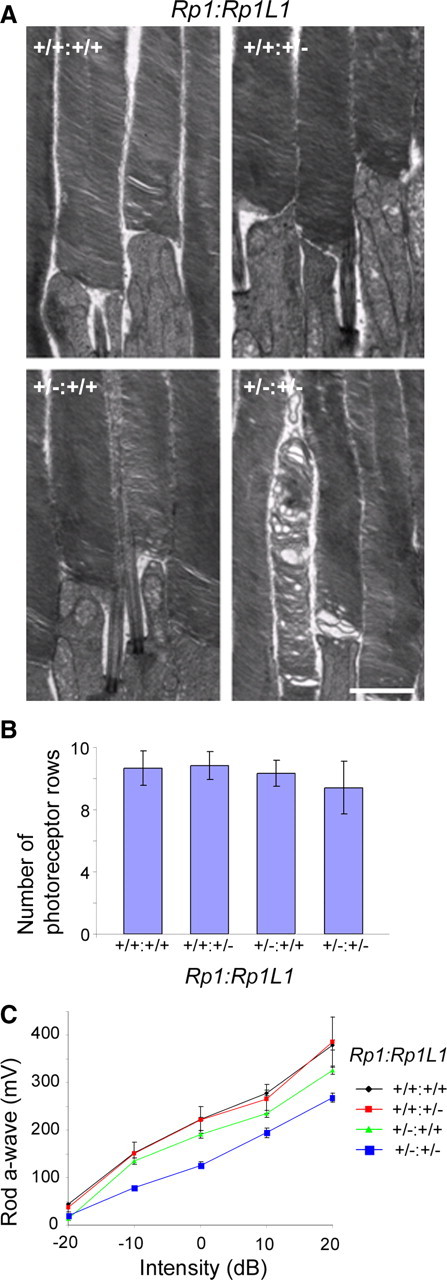
Genetic interactions of Rp1L1 and Rp1. A, Transmission electron microscopy of OSs of retinas of a double heterozygous mouse at 6 months and its wild-type littermate. Note the appearance of abnormal discs only in the double heterozygote. Scale bar, 2 μm. B, Average numbers of photoreceptor rows in ONL of central retinas at 6 months. Group mean values, SDs (bars), and numbers of mice examined in A and B in each genotype and age are shown. C, Amplitudes (in millivolts) of scotopic a-waves with increasing light intensities. A series of light intensities were used from −20 dB to +20 dB (i.e., −2 to 2 log cd · s · m−2) in steps of 1 log cd · s · m−2. ERG amplitudes were recorded from 20 mice at 6 months: 5 Rp1+/− (green), 3 Rp1L1+/− (red), 7 Rp1+/−:Rp1L1+/− (blue), and 5 wild-type (black) mice. Error bars give SEM.
The abnormality of rods from double heterozygotes was confirmed by ERG recordings of their dark-adapted flash responses (Fig. 5C). The maximum rod a-wave responses in Rp1+/− mice at 6 months were reduced and consistent with previous findings (223.47 ± 7.33 mV vs 190.59 ± 17.63 mV at 0 dB) (Gao et al., 2002; Q. Liu et al., 2003), whereas those in Rp1L1+/− mice were similar to those of wild-type mice (p > 0.05, unpaired Student's t test) and the average reduction was 15% (at 0 dB intensity). To confirm the synergistic interaction between them, we have added together the effects of each heterozygote and compared with the effect of the double heterozygote. The a-wave amplitudes of double heterozygotes were similar to those in Rp1L1−/− mice (150.98 ± 17.14 mV vs 125.92 ± 22.38 mV at 0 dB intensity p > 0.05, unpaired Student's t test) but significantly more severe than additive amplitudes of individual heterozygotes (p = 0.018, unpaired Student's t test).
Photoresponses of individual rods
To gain insight into photoreceptor degeneration in RP1 diseases and to confirm the genetic interactions between Rp1 and Rp1L1, we recorded photoresponses from individual rods from Rp1−/−, Rp1L1−/−, Rp1+/−:Rp1L1+/−, Rp1+/−, and wild-type retinas at P21–P30 using suction electrodes. At this age, rods in these mutant retinas were already mature but displayed fairly mild alterations.
First, we analyzed the flash sensitivity of individual rods in these mice. The flash sensitivity is defined as the peak amplitude of the linear response divided by the strength of the flash and commonly measured as the half-saturating flash strength. We found that the flash sensitivity was significantly decreased in Rp1−/−, Rp1L1−/−, and Rp1+/−:Rp1L1+/− rods (∼50%, p < 0.01) (Fig. 6, Table 1), whereas that in Rp1+/− rods was normal. In contrast, the disruption of rootletin, another ciliary rootlet protein, apparently resulted in shortening, disorganization, and loss of the OS at 18 months; however, single-photoreceptor recordings showed normal response kinetics and flash sensitivity (Yang et al., 2005). Therefore, the reduced flash sensitivity in Rp1−/−, Rp1L1−/−, and Rp1+/−:Rp1L1+/− rods may depend on OS abnormalities that are unique to the functions of Rp1 and Rp1L1.
Figure 6.
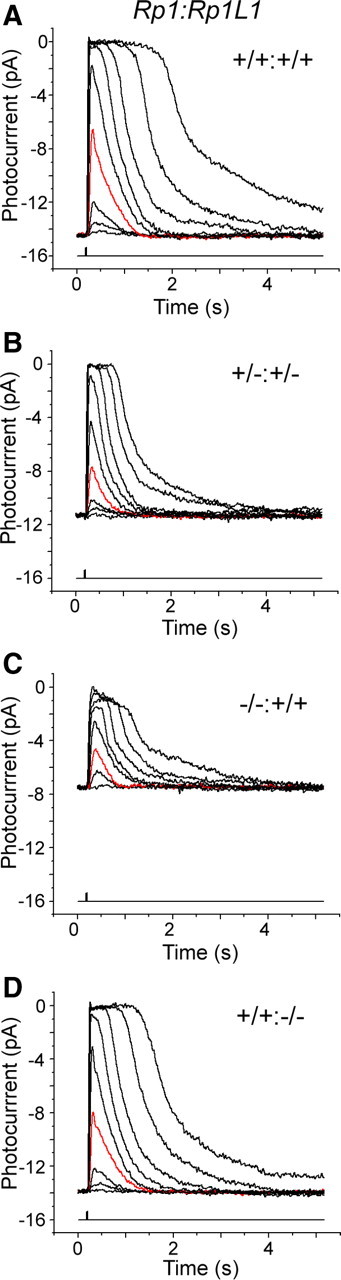
Photoresponses of rod receptors from wild-type, Rp1+/−:Rp1L1+/−, Rp1−/−, and Rp1L1−/− mice. Representative families of responses from control (A), Rp1+/−:Rp1L1+/− (B), Rp1−/− (C), and Rp1L1−/− (D) rods to flashes delivering 1.3, 3.7, 10.7, 40, 118, 359, 1054, 5111, and 15,024 photons · μm−2. Each trace is the average of 4–30 responses. The red trace in each figure represents the photoresponse to a flash delivering 40 photons · μm−2, which is close to the half-saturating intensity for wild-type rods.
Table 1.
Flash response parameters of Rp1+/−, Rp1−/−, Rp1L1−/−, and Rp1+/−:Rp1L1+/− mice and wild-type controls
| Genotype (Rp1:Rp1L1) | Sensitivity i0 (photons · μm−2) | Maximal response rmax (pA) | Single-photon response a (pA) | Dim-flash-response kinetics | |
|---|---|---|---|---|---|
| tp (ms) | ti (ms) | ||||
| +/+:+/+ | 41 ± 1 (58) | 11.9 ± 0.4 (58) | 0.45 ± 0.03 (45) | 179 ± 5 (57) | 395 ± 14 (57) |
| +/−:+/− | 60 ± 4 (21) | 9.3 ± 0.8 (21) | 0.46 ± 0.05 (15) | 170 ± 8 (21) | 385 ± 22 (21) |
| −/−:+/+ | 52 ± 5 (19) | 6.6 ± 0.7 (19) | 0.73 ± 0.12 (16) | 215 ± 9 (18) | 377 ± 38 (18) |
| +/+:−/− | 57 ± 4 (37) | 13.3 ± 0.8 (37) | 0.44 ± 0.03 (31) | 167 ± 5 (36) | 351 ± 14 (36) |
| +/−:+/+ | 45 ± 2 (26) | 11.8 ± 0.3(26) | 0.37 ± 0.04 (24) | 182 ± 4 (26) | 408 ± 17 (26) |
Values are mean ± SEM with the number of cells studied in parentheses; rmax is the saturating photoresponse amplitude; i0 (inversely proportional to sensitivity) is the intensity required to produce a half-saturating response, which was measured from the intensity–response curve for each rod; a is the amplitude of the single photon response; tp is the time to peak of the dim flash response; and ti is the integration time of the dim flash response. Bold indicates a significant difference (p < 0.01) between Rp mutants and wild-type control analyzed by a two-population t test of variance.
To investigate whether rhodopsin function was intact in each rod, we estimated the amplitude of the single-photon response in mutant rods as the ratio of variance/mean of a series of photoresponses to identical dim flashes. We found that single-photon response amplitude was normal in Rp1L1−/− and double heterozygous rods but elevated (1.6 times) in the Rp1−/− rods (Table 1) (p < 0.01). These findings indicate that phototransduction cascades are normal in Rp1L1−/− and double heterozygous rods. For the increased single-photon responses in Rp1−/− rods, we noticed that flashes producing substantial responses occasionally failed to generate any response (data not shown), which resulted in increased response variance. This higher than normal response variance would result in overestimation of the single-photon response in these rods, as estimated from very dim flashes. Therefore, we speculate that some of the flashes were absorbed by the normal discs of the Rp1−/− rods and generated normal responses, while other flashes were absorbed by the disorganized discs and failed to produce responses. Alternatively, it is possible that most OS discs in Rp1−/− rods were misaligned and therefore remained immature, similar to the nascent normal discs at the base of the OS that are known to have higher single-photon responses (Schnapf, 1983).
We also used saturating flash responses to measure the dark current flowing through the cyclic nucleotide-gated (CNG) channels of rod OS. Using bright flashes that completely block this current and produce saturating photoresponses, we found that the amplitudes of the saturating responses were normal in individual Rp1L1−/− and Rp1+/− rods (Fig. 6, Table 1), despite reductions in ERG a-wave amplitudes in their mice (Fig. 4) (Gao et al., 2002; Q. Liu et al., 2003). In contrast, the amplitudes of saturating responses were significantly decreased in Rp1+/−:Rp1L1+/− and Rp1−/− rods (22 ± 7% and 45 ± 6%, p < 0.01) (Fig. 6, Table 1), supporting synergistic genetic interactions between Rp1 and Rp1L1. At least two factors can contribute to dark current: OS length and number of CNG channels (Mendez et al., 2001). Because the OS length was not significantly reduced at 6 months in double heterozygous rods (Fig. 5B), the reduction of the dark current is probably not directly related to the OS abnormalities. The OS of Rp1−/− rods was significantly shorter than that of wild-type rods (Liu et al., 2004). Therefore, this reduced dark current of Rp1−/− rods was most likely the result of the shorter OS.
To further examine the roles of Rp1 and Rp1L1 in phototransduction, we investigated how their deletions affect the kinetics of photoresponses of individual rods. First, we measured the time to peak of dim flash responses, which reflects the rate of activation of the phototransduction cascade. We found that the time to peak in Rp1L1−/− and double heterozygotes was indistinguishable from that of control rods, but the time to peak in Rp1−/− rods was 1.2 times longer than that of control rods (Table 1) (p < 0.01). Consistent with these observations from single-cell recordings, the kinetics of a-wave ERG in both Rp1L1−/− and double heterozygous mice appeared normal (see above), whereas that of another independent Rp1-mutant allele (Rp1myc/myc) appeared slower than that in controls at 30 ms (Q. Liu et al., 2003). This slower activation of Rp1−/− rods was most likely the result of deficiencies in the interactions between phototransduction cascade proteins (i.e., downstream of transducins) caused by the severe OS disc disorganization in these mice.
Finally, we examined the integration time of the normalized dim flash response, which is a measure of the overall kinetics of the response and is dominated by the shutoff of the phototransduction cascade. No significant differences were observed between the four mutant rods and the wild-type rods in this parameter (Table 1), indicating that phototransduction shutoff was not affected by the deletion of Rp1 or Rp1L1. A key step in phototransduction shutoff is the inactivation of rhodopsin by arrestin (Xu et al., 1997), a molecule whose distribution in the OS is controlled by light (McGinnis et al., 2002; Strissel et al., 2006). The normal shutoff of rhodopsin, as indicated from the unaffected integration time of the mutant rods, suggests lack of significant alterations in arrestin distribution under light-to-dark conditions (Fig. 6, Table 1). Furthermore, based on our measurements of integration time and sensitivity from the single-cell recording experiments, it is most likely that the Rp1 and Rp1L1 deletions have not affected the normal OS/IS movement of arrestin and transducin (Fig. 6, Table 1). However, to directly establish that, measurements of kinetics of transducin and arrestin translocation will be needed (Sokolov et al., 2002, 2004; Sampath et al., 2005; Strissel et al., 2006).
Biochemical interactions between Rp1 and Rp1L1
Both Rp1 and Rp1L1 mRNAs are photoreceptor specific with identical temporal expression patterns during postnatal development (Bowne et al., 2003). In addition, both Rp1L1 and Rp1 proteins were localized along the length of OS axoneme in rods (Fig. 2C). One possible mechanism for the synergistic action is that the proteins function in a physical complex to regulate OS disc organization. To seek evidence of biochemical interactions between Rp1 and Rp1L1 proteins, we cotransfected full-length FLAG-tagged Rp1 and Myc/His-tagged Rp1L1 (1–361) into COS7 cells and examined their association by coimmunoprecipitation (Fig. 7A). ∼12% of FLAG-Rp1 was coimmunoprecipitated when Myc/His-tagged Rp1L1 was immunoprecipitated from the cotransfected cell lysates (Fig. 7A, left). The reciprocal immunoprecipitation confirmed the association (Fig. 7A, right). It would be desirable to repeat this experiment with full-length Rp1L1 protein. Unfortunately, we could not access the potential interaction between full-length Rp1 and Rp1L1 in vitro due to technical difficulties.
Figure 7.

Biochemical interaction of Rp1L1 and Rp1. A, C-terminal mycHis-tagged Rp1L1 (1–361) (Rp1L1-mycHis) and N-terminal FLAG-tagged full-length Rp1 (FLAG-Rp1) were coexpressed in COS7 cells. Lysates of the transfected cells were immunoprecipitated (IP) with anti-myc antibody or resin alone (left). Conversely, lysates were immunoprecipitated with anti-FLAG antibody or resin alone (right). The immunoprecipitates were electrophoresed and immunoblotted with anti-myc (9E10) (top) or anti-FLAG M2 (bottom) monoclonal antibodies. The strength of the interactions can be assessed by comparing the amount of protein immunoprecipitated in the test lanes to the amount in the 10% of input for immunoprecipitation run the gel in parallel. B, The GST-Rp1 fusion protein was incubated with retinal lysate followed by pull-down with glutathione-Sepharose 4B. Bound proteins were analyzed by immunoblotting using anti-Rp1L1 antibody. C, The GST-Rp1 fusion protein was incubated with 1 mg of Rp1L1-mycHis-transfected 293T cell lysate (as a source for Rp1L1) followed by pull-down with glutathione-Sepharose 4B. Bound proteins were analyzed by immunoblotting. The first lane shows a negative control in which a GST protein, instead of the GST-Rp1 fusion protein, was incubated with Rp1L1-mycHis-transfected 293T cell lysate. D, Localization of Rp1L1 (green) in dissociated rods of Rp1−/−:Rp1L1+/− retinas at P21. Red signal shows Rp1 distribution. The DIC image is superimposed (right). Note that the truncated-Rp1 protein lacking DCX domains is mislocalized into the CCs in Rp1−/− rods, consistent with previous study reporting the mislocalization of the residual Rp1 protein in Rp1−/− rods (Liu et al., 2004). Scale bar, 0.2 μm.
The potential interaction between Rp1 and Rp1L1 was next examined by in vitro and in vivo GST pull-down assays using bacterial-expressed GST-Rp1 N-terminal fusion protein. The Rp1 recombinant protein coprecipitated Rp1L1 from Rp1L1-transfected cells (Fig. 7C) as well as multiple bands around ∼200 kDa that was detected by anti-Rp1L1 antibody from retinal lysates (Fig. 7B). This form of multiple bands in Figure 7B is specific for Rp1L1 as shown in Figure 1D. Notably, when normalized to GST-Rp1, which was bound to the glutathione-Sepharose beads (Fig. 7B, bottom), the relative amounts of the precipitants appeared similar regardless of whether nocodazole (a microtubule disrupting reagent) was present (Fig. 7B, top). The complementary in vivo immunoprecipitation assay using ciliary protein would require antibodies for Rp1 and Rp1L1. Sufficient quantities of these antibodies could not be obtained for this experiment. Nevertheless, these results further support the idea that their synergistic interactions can be mediated by their physical association in rod OS axoneme, although it remains possible that such interactions are indirect or transient through other proteins in the complex in vivo.
No mislocalization of Rp1L1 was observed in Rp1−/−:Rp1L1+/− rod photoreceptors (Fig. 7D). Conversely, Rp1 is most likely localized correctly in Rp1L1−/− rod photoreceptors; otherwise, Rp1L1−/− retinas would display severe phenotypes as in Rp1−/− retinas (Gao et al., 2002). Therefore, Rp1 and Rp1L1 are not dependent on the expression of one another, although they coexisted.
Protein levels of rhodopsin and other members of the phototransduction cascade
To determine whether the decrease in rhodopsin concentration reduced photosensitivity, we measured rod opsin, rod transducin α, phosphodiesterase (PDE) 6α, and the CNG1 channel by immunoblotting analysis. The immunoblotting analysis of whole-retinal lysates from Rp1−/−, Rp1L1−/−, and wild-type controls did not indicate significant differences in their expression levels, consistent with our single cell recording analysis (Fig. 8). No mislocalization of rhodopsin was observed in Rp1L1−/− retinas (data not shown). Therefore, the density of the disk is probably normal in Rp1L1−/− retinas.
Figure 8.

Expression of other phototransduction proteins, rod opsin (A), rod transducin α (B), PDE6α (C), and CNG1 channel (D), in Rp1+/−, Rp1−/−, Rp1L1−/−, and wild-type mice that had been exposed to ambient light for ∼8 h. The retinas from Rp1L1−/− and wild-type mice at P46–P47 were analyzed by SDS electrophoresis and probed with antibodies as shown (left). Cerebellum whole-cell lysates were used as a negative control. Because of the severe degeneration of photoreceptors, the whole-retina lysates from Rp1+/−, Rp1−/−, and wild-type mice were analyzed at P21 (right). An anti-GAPDH antibody was used for the detection of the loading control. Note slight reductions of rod opsin, rod transducin α, and PDE6α protein in Rp1−/− retinal lysates, attributable to the shortened OS in Rp1−/− rods, as described by Liu et al. (2004). WB, Western blot.
Discussion
We have elucidated the subcellular localization and in vivo function of Rp1L1, a paralog of the RP1 disease gene, and their relations. Our studies also have implications for the function of other DCX-containing microtubule-associated proteins and ciliary proteins.
Similarities and differences between Rp1 and Rp1L1
It is striking that Rp1 and Rp1L1 share significant homology in their N-terminal sequences and have identical photoreceptor-specific postnatal expression patterns, similar axoneme localization, and similar OS functions in rod photoreceptors. Although RP1 is present in cones (Liu et al., 2002), human RP1 patients do not lose their cone-mediated color vision as long as visual acuities are better than 20/20 (Jacobson et al., 2000). Rp1−/− mice do not display cone degeneration or reduction in cone ERG until 10 months of age (Gao et al., 2002), suggesting that RP1 does not play a critical role in cones. Rp1L1 is absent in cones (Fig. 2A), and Rp1L1−/− retinas do not exhibit significant cone loss at least until 6 months of age (data not shown), as expected. It is already clear that Rp1L1 cannot fully complement the lack of Rp1 function. Therefore, this differential distribution may reflect the functional disparity between the two proteins.
A small percentage of photoreceptors in Rp1L1−/− retinas were abnormal and randomly scattered at any time we examined them over the period of 1 year, whereas all rod photoreceptors in Rp1−/− retinas were abnormal starting at P7–P10. In Tubby−/− retinas, although rhodopsin is mislocalized in every photoreceptor cell, arrestin is mislocalized in only a small number of photoreceptor cells that are randomly scattered among normal-appearing photoreceptor cells (Kong et al., 2006) in a manner similar to the scattered rod loss in Rp1L1−/− retinas (Fig. 3C) and scattered rhodopsin mislocalization in Rp1−/− retinas (Gao et al., 2002).
The photoreceptor degeneration in Rp1L1−/− retinas is much slower than that in Rp1−/− retinas. At 12 months, only a single row of photoreceptors (mostly cones) remains, and no normal OS ever forms in Rp1−/− retinas (Gao et al., 2002), whereas nearly 60% of photoreceptors remain in Rp1L1−/− retinas. Abnormal OS discs often form in discrete boundaries along the length of the OS, flanked by normal-appearing discs in Rp1L1−/− retinas. Possibly an unidentified OS protein is mislocalized or absent in such a segment along the OS. Alternatively, fluctuations in light or oxygen levels might trigger the abnormal formation of these disc segments (Pierce et al., 1999). However, because OS discs are renewed every 10 d in mice and our light–dark cycle was 12 h on and 12 h off, we would expect ∼10 such segments to coexist in a single OS if light were the trigger. Higher oxygen consumption during exercise is another possible trigger of abnormal OS discs, consistent with the fact that RP1 was originally identified as oxygen-regulated protein (Pierce et al., 1999). It would be interesting to raise Rp1L1−/− mice in darkness or in steady light to directly test this hypothesis.
Functions of Rp1 and Rp1L1
Photosensitivity was reduced in Rp1−/− and Rp1L1−/− rods. This can be explained either by altered expression levels or assembly of phototransduction proteins or by the morphologic defects of the rod OS. Conversely, single photoresponses were normal in either homozygote (Fig. 6, Table 1), indicating that the phototransduction cascade (the gain and kinetics of any phototransduction reaction downstream of rhodopsin) functions normally. The total amounts of rod opsin, transducins, PDEs, and CNG channels were also normal in rods of either homozygote, indicating no defects in the assembly, total amounts, and ratios of these phototransduction molecules (Fig. 8). Therefore, we believe that Rp1 and Rp1L1 are required for normal OS morphogenesis. Swirling OS discs existed only in a small percentage of Rp1L1−/− rods (Fig. 3C). The variations of single-cell photoresponse in Rp1L1−/− rods were greater than those in wild-type controls, possibly reflecting the small percentage of abnormal rods in Rp1L1−/− retinas.
The steady-state localization of arrestin and transducin in dark-adapted mutant retinas of Rp1 or Rp1L1 appeared normal (Fig. 6). The OS/IS movement also appeared normal. These results further support the notion that deletion of Rp1 or Rp1L1 affects primarily rod morphology, but not the molecular mechanisms that deliver phototransduction proteins to the OS.
Synergistic interactions between Rp1 and Rp1L1
Several lines of evidence show that Rp1 and Rp1L1 interact in the rod OS axoneme. First, they are both localized to the axoneme of the rod OS. Second, OS discs of double heterozygotes are abnormal, similar to those of Rp1L1−/− but different from those of Rp1−/− retinas; the ONL length of double heterozygotes appeared reduced, although insignificantly, at 6 months. No morphologic OS abnormalities have ever been identified in Rp1+/− or Rp1L1+/− retinas at any ages despite extensive efforts (Gao et al., 2002; Q. Liu et al., 2003; this report). Third, ERG a-wave amplitudes of double heterozygotes are significantly reduced at 6 months, similar to those of Rp1L1−/− mice, but significantly more than the combined 15% reduction in Rp1+/− mice and 0% reduction in Rp1L1+/− mice (Gao et al., 2002; Q. Liu et al., 2003) (Fig. 5C). Fourth, at the single-cell level, photosensitivity in Rp1−/−, Rp1L1−/−, and Rp1+/−:Rp1L1+/− rods were significantly decreased but not in Rp1+/− rods (Fig. 6, Table 1). Because the ERG a-wave amplitude, the single-cell photosensitivity, for Rp1L1+/− mice was normal, these results indicate that the photosensitivity for double heterozygous rods was reduced in a synergistic manner, caused by the abnormal OS discs. Finally, dark current in single rods of double heterozygotes is reduced whereas those of individual heterozygotes are normal (Table 1). Expression of Rp1L1 overlaps with that of Rp1 in the OS axoneme, whereas only Rp1L1 alone exists in the CCs (Fig. 2B). Although the overlap between Rp1 and Rp1L1 distribution in rod OS is partial and biochemical interactions between Rp1 and Rp1L1 are possibly transient, such interactions could have significant functions for rod photoreceptors, as evident in our genetic interaction results. DCX and doublecortin-like kinase (DCLK) are collectively involved in neuronal migration and axon growth in embryonic cortex (Koizumi et al., 2006; Deuel et al., 2006). DCLK coimmunoprecipitated DCX from P0 whole-brain lysate and the transfected cells (Koizumi et al., 2006). It would be attractive to test whether the function in pairs is conserved among DCX-domain proteins. How Rp1 and Rp1L1 play synergistic roles in the OS morphogenesis remains to be further investigated.
Implications for the RP1 disease and other retinopathy
Nearly all human RP1 patients are heterozygous carriers (Jacobson et al., 2000; Daiger et al., 2006). It remains unclear why 15% reduction of ERG amplitudes in Rp1+/− mice (Gao et al., 2002; Q. Liu et al., 2003) (Fig. 5C) occurs despite normal morphology, normal protein levels of members of the phototransduction cascade (Fig. 8), and normal sensitivity in Rp1+/− rods (Table 1).
No mutations in RP1L1 were found in ∼60 patients with adRP of unknown causes (Bowne et al., 2003). Given the mild and recessive nature of Rp1L1−/− mouse retinal phenotypes, mutations in RP1L1 may cause age-related recessive RP in humans. Alternatively, given the genetic interactions between Rp1 and Rp1L1, mutations or polymorphisms in RP1L1 could modify the phenotypic expression of RP1 in humans (Jacobson et al., 2000; Daiger et al., 2006).
Implication for other DCX-containing and ciliary proteins
Among a total of 11 DCX-containing proteins found in the mouse and human genomes (Coquelle et al., 2006), only RP1 and RP1L1 are photoreceptor specific and coexist in the axoneme of the rod OS. Because the DCX domain is a microtubule-binding domain, Rp1L1 and Rp1 can assemble and stabilize axonemal microtubules (Liu et al., 2004; Coquelle et al., 2006).
Because the RP1D domain, a 34 aa segment following the DCX domains, is conserved only in Rp1 and Rp1L1 but is absent in other DCX-containing proteins, a mutant Rp1 protein with both DCX repeats and RP1D is targeted to the axoneme in photoreceptors in vivo, whereas another truncated Rp1 protein without these domains is not (Liu et al., 2004; Coquelle et al., 2006). It is therefore likely that the DCX tandem repeats and the RP1D domain are necessary and sufficient for OS axoneme localization.
The mouse photoreceptor sensory cilium complex, which comprises OS axoneme and its cytoskeleton, has been estimated to include ∼2000 distinct proteins (Liu et al., 2007). Mutations in genes encoding ciliary proteins result in progressive photoreceptor degeneration [i.e., Rpgr−/−, Rpgrip−/−, BBS, IFT88 (Pazour et al., 2002; Pazour and Rosenbaum, 2002; Hong et al., 2003, 2005; Zhao et al., 2003; Kim et al., 2004; Nishimura et al., 2004; Pazour, 2004; Yen et al., 2006)]. In addition, many existing mutants display photoreceptor phenotypes similar to those of Rp1−/− and Rp1L1−/− retinas [Rds+/− or Rds−/−, pcd−/−, Rom1−/− (Blanks et al., 1982; Pazour et al., 2002; Pazour and Rosenbaum, 2002; Hong et al., 2003, 2005; Kim et al., 2004; Nishimura et al., 2004; Pazour, 2004)]. It is therefore attractive to hypothesize that RP1 and RP1L1 work together with some of these ciliary or OS proteins in photoreceptors. Further investigation of identifying RP1- and RP1L1-interacting proteins in photoreceptors is warranted.
Footnotes
This work is supported in part by National Institutes of Health (NIH) Grants EY12950, CA21765, and CA023944 and American Lebanese Syrian Associated Charities (to J.Z.); NIH Grants EY14428, EY14448, and EY018660, Foundation Fighting Blindness, the Macular Vision Research Foundation, and Research to Prevent Blindness (to K.Z.); a Career Development Award from Research to Prevent Blindness (to V.J.K.); and a Fight For Sight Postdoctoral Fellowship from Fight For Sight, Inc., a fellowship from Association for Research in Vision and Ophthalmology/Japan National Society for the Prevention of Blindness, and a St. Jude Children's Research Hospital Academic Programs Special Fellowship (to T.Y.). We thank Sharon Frase, Andrew Romeo, Clare F. Cook, Rebakkha Robbins, Sana Mujahid, and Jason Porter for technical assistance, Dr. Eric Pierce for chicken anti-Rp1 antibody, Dr. Ching-Kang Jason Chen for rabbit S-antigen, and Drs. Kim Howes, Tiansen Li, and Wei Cao for critical comments.
References
- Blanks JC, Mullen RJ, LaVail MM. Retinal degeneration in the pcd cerebellar mutant mouse. II. Electron microscopic analysis. J Comp Neurol. 1982;212:231–246. doi: 10.1002/cne.902120303. [DOI] [PubMed] [Google Scholar]
- Blanton SH, Heckenlively JR, Cottingham AW, Friedman J, Sadler LA, Wagner M, Friedman LH, Daiger SP. Linkage mapping of autosomal dominant retinitis pigmentosa (RP1) to the pericentric region of human chromosome 8. Genomics. 1991;11:857–869. doi: 10.1016/0888-7543(91)90008-3. [DOI] [PubMed] [Google Scholar]
- Bowne SJ, Daiger SP, Hims MM, Sohocki MM, Malone KA, McKie AB, Heckenlively JR, Birch DG, Inglehearn CF, Bhattacharya SS, Bird A, Sullivan LS. Mutations in the RP1 gene causing autosomal dominant retinitis pigmentosa. Hum Mol Genet. 1999;8:2121–2128. doi: 10.1093/hmg/8.11.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowne SJ, Daiger SP, Malone KA, Heckenlively JR, Kennan A, Humphries P, Hughbanks-Wheaton D, Birch DG, Liu Q, Pierce EA, Zuo J, Huang Q, Donovan DD, Sullivan LS. Characterization of RP1L1, a highly polymorphic paralog of the retinitis pigmentosa 1 (RP1) gene. Mol Vis. 2003;9:129–137. [PMC free article] [PubMed] [Google Scholar]
- Conte I, Lestingi M, den Hollander A, Alfano G, Ziviello C, Pugliese M, Circolo D, Caccioppoli C, Ciccodicola A, Banfi S. Identification and characterisation of the retinitis pigmentosa 1-like1 gene (RP1L1): a novel candidate for retinal degenerations. Eur J Hum Genet. 2003;11:155–162. doi: 10.1038/sj.ejhg.5200942. [DOI] [PubMed] [Google Scholar]
- Coquelle FM, Levy T, Bergmann S, Wolf SG, Bar-El D, Sapir T, Brody Y, Orr I, Barkai N, Eichele G, Reiner O. Common and divergent roles for members of the mouse DCX superfamily. Cell Cycle. 2006;5:976–983. doi: 10.4161/cc.5.9.2715. [DOI] [PubMed] [Google Scholar]
- Daiger SP, Shankar SP, Schindler AB, Sullivan LS, Bowne SJ, King TM, Daw EW, Stone EM, Heckenlively JR. Genetic factors modifying clinical expression of autosomal dominant RP. Adv Exp Med Biol. 2006;572:3–8. doi: 10.1007/0-387-32442-9_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, Carrié A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, Catala M, Kahn A, Beldjord C, Chelly J. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51–61. doi: 10.1016/s0092-8674(00)80898-3. [DOI] [PubMed] [Google Scholar]
- Deuel TAS, Liu JS, Corbo JC, Yoo SY, Rorke-Adams LB, Walsh CA. Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron. 2006;49:41–53. doi: 10.1016/j.neuron.2005.10.038. [DOI] [PubMed] [Google Scholar]
- Gao J, Cheon K, Nusinowitz S, Liu Q, Bei D, Atkins K, Azimi A, Daiger SP, Farber DB, Heckenlively JR, Pierce EA, Sullivan LS, Zuo J. Progressive photoreceptor degeneration, outer segment dysplasia, and rhodopsin mislocalization in mice with targeted disruption of the retinitis pigmentosa-1 (Rp1) gene. Proc Natl Acad Sci U S A. 2002;99:5698–5703. doi: 10.1073/pnas.042122399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Wu X, Zuo J. Targeting hearing genes in mice. Brain Res Mol Brain Res. 2004;132:192–207. doi: 10.1016/j.molbrainres.2004.06.035. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257–271. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- Haider NB, Ikeda A, Naggert JK, Nishina PM. Genetic modifiers of vision and hearing. Hum Mol Genet. 2002;11:1195–1206. doi: 10.1093/hmg/11.10.1195. [DOI] [PubMed] [Google Scholar]
- Hong DH, Pawlyk BS, Shang J, Sandberg MA, Berson EL, Li T. A retinitis pigmentosa GTPase regulator (RPGR)-deficient mouse model for X-linked retinitis pigmentosa (RP3) Proc Natl Acad Sci U S A. 2000;97:3649–3654. doi: 10.1073/pnas.060037497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong DH, Yue G, Adamian M, Li T. Retinitis pigmentosa GTPase regulator (RPGRr)-interacting protein is stably associated with the photoreceptor ciliary axoneme and anchors RPGR to the connecting cilium. J Biol Chem. 2001;276:12091–12099. doi: 10.1074/jbc.M009351200. [DOI] [PubMed] [Google Scholar]
- Hong DH, Pawlyk B, Sokolov M, Strissel KJ, Yang J, Tulloch B, Wright AF, Arshavsky VY, Li T. RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Invest Ophthalmol Vis Sci. 2003;44:2413–2421. doi: 10.1167/iovs.02-1206. [DOI] [PubMed] [Google Scholar]
- Hong DH, Pawlyk BS, Adamian M, Sandberg MA, Li T. A single, abbreviated RPGR-ORF15 variant reconstitutes RPGR function in vivo. Invest Ophthalmol Vis Sci. 2005;46:435–441. doi: 10.1167/iovs.04-1065. [DOI] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Iannaccone A, Weleber RG, Fishman GA, Maguire AM, Affatigato LM, Bennett J, Pierce EA, Danciger M, Farber DB, Stone EM. Disease expression of RP1 mutations causing autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2000;41:1898–1908. [PubMed] [Google Scholar]
- Kim JC, Badano JL, Sibold S, Esmail MA, Hill J, Hoskins BE, Leitch CC, Venner K, Ansley SJ, Ross AJ, Leroux MR, Katsanis N, Beales PL. The Bardet-Biedl protein BBS4 targets cargo to the pericentriolar region and is required for microtubule anchoring and cell cycle progression. Nat Genet. 2004;36:462–470. doi: 10.1038/ng1352. [DOI] [PubMed] [Google Scholar]
- Koizumi H, Tanaka T, Gleeson JG. Doublecortin-like kinase functions with doublecortin to mediate fiber tract decussation and neuronal migration. Neuron. 2006;49:55–66. doi: 10.1016/j.neuron.2005.10.040. [DOI] [PubMed] [Google Scholar]
- Kong L, Li F, Soleman CE, Li S, Elias RV, Zhou X, Lewis DA, McGinnis JF, Cao W. Bright cyclic light accelerates photoreceptor cell degeneration in tubby mice. Neurobiol Dis. 2006;21:468–477. doi: 10.1016/j.nbd.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Zhou J, Daiger SP, Farber DB, Heckenlively JR, Smith JE, Sullivan LS, Zuo J, Milam AH, Pierce EA. Identification and subcellular localization of the RP1 protein in human and mouse photoreceptors. Invest Ophthalmol Vis Sci. 2002;43:22–32. [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Lyubarsky A, Skalet JH, Pugh EN, Jr, Pierce EA. RP1 is required for the correct stacking of outer segment discs. Invest Ophthalmol Vis Sci. 2003;44:4171–4183. doi: 10.1167/iovs.03-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Zuo J, Pierce EA. The retinitis pigmentosa 1 protein is a photoreceptor microtubule-associated protein. J Neurosci. 2004;24:6427–6436. doi: 10.1523/JNEUROSCI.1335-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Tan G, Levenkova N, Li T, Pugh EN, Jr, Rux JJ, Speicher DW, Pierce EA. The proteome of the mouse photoreceptor sensory cilium complex. Mol Cell Proteomics. 2007;6:1299–1317. doi: 10.1074/mcp.M700054-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmor MF, Fishman GA. At last. A standard electroretinography protocol. Arch Ophthalmol. 1989;107:813–814. doi: 10.1001/archopht.1989.01070010835022. [DOI] [PubMed] [Google Scholar]
- McGinnis JF, Matsumoto B, Whelan JP, Cao W. Cytoskeleton participation in subcellular trafficking of signal transduction proteins in rod photoreceptor cells. J Neurosci Res. 2002;67:290–297. doi: 10.1002/jnr.10120. [DOI] [PubMed] [Google Scholar]
- Mendez A, Burns ME, Sokal I, Dizhoor AM, Baehr W, Palczewski K, Baylor DA, Chen J. Role of guanylate cyclase-activating proteins (GCAPs) in setting the flash sensitivity of rod photoreceptors. Proc Natl Acad Sci U S A. 2001;98:9948–9953. doi: 10.1073/pnas.171308998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura DY, Fath M, Mullins RF, Searby C, Andrews M, Davis R, Andorf JL, Mykytyn K, Swiderski RE, Yang B, Carmi R, Stone EM, Sheffield VC. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci U S A. 2004;101:16588–16593. doi: 10.1073/pnas.0405496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ. Intraflagellar transport and cilia-dependent renal disease: the ciliary hypothesis of polycystic kidney disease. J Am Soc Nephrol. 2004;15:2528–2536. doi: 10.1097/01.ASN.0000141055.57643.E0. [DOI] [PubMed] [Google Scholar]
- Pazour GJ, Rosenbaum JL. Intraflagellar transport and cilia-dependent diseases. Trends Cell Biol. 2002;12:551–555. doi: 10.1016/s0962-8924(02)02410-8. [DOI] [PubMed] [Google Scholar]
- Pazour GJ, Baker SA, Deane JA, Cole DG, Dickert BL, Rosenbaum JL, Witman GB, Besharse JC. The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J Cell Biol. 2002;157:103–113. doi: 10.1083/jcb.200107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce EA, Quinn T, Meehan T, McGee TL, Berson EL, Dryja TP. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat Genet. 1999;22:248–254. doi: 10.1038/10305. [DOI] [PubMed] [Google Scholar]
- Pilz DT, Kuc J, Matsumoto N, Bodurtha J, Bernadi B, Tassinari CA, Dobyns WB, Ledbetter DH. Subcortical band heterotopia in rare affected males can be caused by missense mutations in DCX (XLIS) or LIS1. Hum Mol Genet. 1999;8:1757–1760. doi: 10.1093/hmg/8.9.1757. [DOI] [PubMed] [Google Scholar]
- Rattner A, Smallwood PM, Williams J, Cooke C, Savchenko A, Lyubarsky A, Pugh EN, Nathans J. A photoreceptor-specific cadherin is essential for the structural integrity of the outer segment and for photoreceptor survival. Neuron. 2001;32:775–786. doi: 10.1016/s0896-6273(01)00531-1. [DOI] [PubMed] [Google Scholar]
- Sampath AP, Strissel KJ, Elias R, Arshavsky VY, McGinnis JF, Chen J, Kawamura S, Rieke F, Hurley JB. Recoverin improves rod-mediated vision by enhancing signal transmission in the mouse retina. Neuron. 2005;46:413–420. doi: 10.1016/j.neuron.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Schnapf JL. Dependence of the single photon response on longitudinal position of absorption in toad rod outer segments. J Physiol. 1983;343:147–159. doi: 10.1113/jphysiol.1983.sp014886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov M, Lyubarsky AL, Strissel KJ, Savchenko AB, Govardovskii VI, Pugh EN, Jr, Arshavsky VY. Massive light-driven translocation of transducin between the two major compartments of rod cells: a novel mechanism of light adaptation. Neuron. 2002;34:95–106. doi: 10.1016/s0896-6273(02)00636-0. [DOI] [PubMed] [Google Scholar]
- Sokolov M, Strissel KJ, Leskov IB, Michaud NA, Govardovskii VI, Arshavsky VY. Phosducin facilitates light-driven transducin translocation in rod photoreceptors. Evidence from the phosducin knockout mouse. J Biol Chem. 2004;279:19149–19156. doi: 10.1074/jbc.M311058200. [DOI] [PubMed] [Google Scholar]
- Sossey-Alaoui K, Hartung AJ, Guerrini R, Manchester DK, Posar A, Puche-Mira A, Andermann E, Dobyns WB, Srivastava AK. Human doublecortin (DCX) and the homologous gene in mouse encode a putative Ca2+-dependent signaling protein which is mutated in human X-linked neuronal migration defects. Hum Mol Genet. 1998;7:1327–1332. doi: 10.1093/hmg/7.8.1327. [DOI] [PubMed] [Google Scholar]
- Strissel KJ, Sokolov M, Trieu LH, Arshavsky VY. Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin. J Neurosci. 2006;26:1146–1153. doi: 10.1523/JNEUROSCI.4289-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LS, Heckenlively JR, Bowne SJ, Zuo J, Hide WA, Gal A, Denton M, Inglehearn CF, Blanton SH, Daiger SP. Mutations in a novel retina-specific gene cause autosomal dominant retinitis pigmentosa. Nat Genet. 1999;22:255–259. doi: 10.1038/10314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Dodd RL, Makino CL, Simon MI, Baylor DA, Chen J. Prolonged photoresponses in transgenic mouse rods lacking arrestin. Nature. 1997;389:505–509. doi: 10.1038/39068. [DOI] [PubMed] [Google Scholar]
- Yang J, Liu X, Yue G, Adamian M, Bulgakov O, Li T. Rootletin, a novel coiled-coil protein, is a structural component of the ciliary rootlet. J Cell Biol. 2002;159:431–440. doi: 10.1083/jcb.200207153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Gao J, Adamian M, Wen XH, Pawlyk B, Zhang L, Sanderson MJ, Zuo J, Makino CL, Li T. The ciliary rootlet maintains long-term stability of sensory cilia. Mol Cell Biol. 2005;25:4129–4137. doi: 10.1128/MCB.25.10.4129-4137.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen HJ, Tayeh MK, Mullins RF, Stone EM, Sheffield VC, Slusarski DC. Bardet-Biedl syndrome genes are important in retrograde intracellular trafficking and Kupffer's vesicle cilia function. Hum Mol Genet. 2006;15:667–677. doi: 10.1093/hmg/ddi468. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Hong DH, Pawlyk B, Yue G, Adamian M, Grynberg M, Godzik A, Li T. The retinitis pigmentosa GTPase regulator (RPGR)-interacting protein: subserving RPGR function and participating in disk morphogenesis. Proc Natl Acad Sci U S A. 2003;100:3965–3970. doi: 10.1073/pnas.0637349100. [DOI] [PMC free article] [PubMed] [Google Scholar]