

Abstract
Background
One of the greatest impediments to cancer immunotherapy is the paucity of antigens that are tumor specific, sufficiently immunogenic, and shared among patients. Mucosa-restricted antigens that are expressed by tumor cells represent a novel class of vaccine targets that are characterized by immunologic privilege, which limits systemic tolerance to those antigens, and immunologic partitioning, which shields mucosae from systemic autoimmune responses. Here we defined the immunogenicity and antitumor efficacy of guanylyl cyclase C (GCC), a protein that is normally restricted to intestinal mucosa and universally expressed by metastatic colorectal cancer.
Methods
BALB/c mice (n = 197) were immunized with recombinant GCC-expressing viral vectors before (prophylactic) or after (therapeutic) a lethal challenge of GCC-expressing mouse colon cancer cells, and antitumor efficacy was monitored by quantifying metastasis and survival. Induction of autoimmunity was monitored by histopathology. Induction of GCC-specific B-cell and CD4+ and CD8+ T-cell responses were determined by enzyme-linked immunosorbent assay and ELISpot, respectively. Tolerance to GCC was quantified by comparing responses in GCC-deficient (n = 45) and wild-type (n = 69) C57BL/6 mice. Statistical tests were two-sided.
Results
Immunization with GCC-expressing viral vectors reduced the formation of metastases to liver (control vs GCC: mean = 30.4 vs 3.55 nodules, difference = 26.9 nodules, 95% confidence interval [CI] = 8.47 to 45.3 nodules; P = .008) and lung (control vs GCC: mean = 263 vs 55.7 nodules, difference = 207, 95% CI = 163 to 251; P < .001) and extended the median survival of mice with established lung metastases following therapeutic immunization (control vs GCC: 29 vs 38 days, P = .024), without autoimmunity. Antitumor efficacy reflected asymmetrical tolerance that was characterized by CD8+ T-cell, but not CD4+ T-cell or antibody, responses.
Conclusions
Immunologic partitioning together with immunologic privilege highlight the potential of mucosa-restricted antigens, particularly GCC, as therapeutic targets for metastatic cancer.
CONTEXT AND CAVEATS
Prior knowledge
Guanylyl cyclase C (GCC) is a protein that is expressed in the cells that line the normal intestine and by metastatic colorectal cancer cells.
Study design
Mouse models of colon cancer were used to test the immunotherapeutic efficacy of GCC.
Contributions
Mice that were immunized with GCC had fewer colon cancer metastases to the liver and lungs and survived longer than control-immunized mice. No autoimmunity was observed.
Implication
GCC is a potential therapeutic target for metastatic colon cancer.
Limitations
The study used cell lines and mouse models of cancer. It is unknown whether the same results would be observed in human cancer.
One of the greatest impediments to cancer immunotherapy is the paucity of antigens that are tumor-specific, sufficiently immunogenic, and shared among patients (1). In lieu of ideal targets, antitumor immune responses are generally directed to tissue-specific rather than tumor-specific proteins. Barriers to using self-antigens include the potential development of concomitant autoimmunity and tolerance, which limits immunotherapeutic efficacy (2). Attempts to circumvent these limitations have included the use of self-proteins that are expressed in immunologically privileged compartments, for example, cancer testis antigens (3). Their ectopic expression in tumors outside the restricted compartments provides opportunities for targeted immunologic responses that are essentially directed to tumor-specific antigens. The best characterized tumor-associated antigens derived from privileged compartments include cancer testis antigens (3), which represent more than 40 gene products, including MAGE and NY-ESO, whose expression is normally restricted to testis but that are anomalously expressed by many cancers. Although many cancer testis antigens exhibit characteristics that are suited to immunologic targeting in cancer, their use is restricted by heterogeneity of expression in tumors. With the exception of melanoma and lung cancers, individual cancer testis antigens are expressed in less than 20% of epithelial tumors, and colorectal tumors exhibit the lowest frequency of expression (4), rendering them suboptimal targets in that disease and limiting their clinical use.
An unexplored variation on this theme exploits the universal expression of mucosa-restricted antigens by mucosal tumors, in the context of the established asymmetry in immunologic cross talk between mucosal and systemic compartments (5). This asymmetry offers unique advantages that reflect the intersection of immunologic privilege and immunologic partitioning. Specifically, immunologic privilege may limit systemic tolerance to mucosal antigens and facilitate therapeutic antitumor responses, whereas immunologic partitioning may shield mucosae from systemic immune responses and limit autoimmunity (6–9).
Guanylyl cyclase C (GCC), the receptor for diarrheagenic bacterial heat-stable enterotoxins and the endogenous paracrine hormones guanylin and uroguanylin (10), is expressed in apical membranes of intestinal epithelial cells, restricting it to mucosal immune compartments (11–14). Moreover, GCC is universally expressed by primary and metastatic colorectal tumors (11–13,15,16). This pattern of expression suggests that GCC may qualify as an efficacious mucosa-restricted immunotherapeutic target in colorectal cancer, the second leading cause of cancer mortality in the United States and the fourth most common worldwide (17). Here, we define the immunogenicity and therapeutic utility of GCC in mouse models of metastatic colorectal cancer.
Materials and Methods
Guanylyl Cyclase Alignment
Mouse GCA (Genbank accession number NP_032753), GCB (NP_776149), GCC (NP_659504), GCE (NP_032218.2), GCF (NP_001007577), GCG (NP_001074545.1), and NPR3 (NP_001034270.1) were aligned using the CLUSTAL W algorithm (18).
Mice
C57BL/6 (n = 30) and BALB/c mice (n = 197) were obtained from the National Cancer Institute (NCI) Animal Production Program (Frederick, MD). GCC-deficient (GCC–/–) mice (19) were backcrossed with C57BL/6 mice for more than 10 generations to produce GCC–/– congenic C57BL/6 mice and wild-type (GCC+/+) littermates (20). Mouse protocols were approved by the Thomas Jefferson University Institutional Animal Care and Use Committee.
Plasmids
A truncated GCC1-430 construct (GCCECD) containing the extracellular ligand-binding domain and a C-terminus hexahistidine tag was generated by polymerase chain reaction (PCR) using GCC cDNA and subcloned to produce recombinant viruses. The membrane-bound recombinant GCC1-461 construct, GCCTM, was produced by PCR using the extracellular ligand-binding domain (residues 23–430) containing an N-terminus pentahistidine tag. The N-terminus signal peptide (residues 1–23) and C-terminus transmembrane domain (residues 431–461) containing a C-terminus hexahistidine tag were then added by PCR and subcloned into pMSCV2.2-Puro to generate the plasmid GCCTM-pMSCV2.2-Puro.
Recombinant Viruses
Replication-deficient human type 5 recombinant adenovirus (AV) was generated using the ViraPower Adenoviral Expression System and plasmid pAd/CMV/V5-DEST (Invitrogen, Carlsbad, CA). Adenovirus was purified using the Adeno-X Virus Purification Kit (Clontech, Mountain View, CA) and titered using the Adeno-X Rapid Titer Kit (Clontech). LacZ-AV was acquired from Clontech (LacZ-Adeno-X), expanded, purified, and titered in parallel with GCCECD-AV. GCCECD-rabies virus (RV) and control RV were generated using the BsiWI/Nhe restriction sites in the SPBN rabies virus backbone, and infectious virus was recovered as described previously (21). The control RV (RV-NP/SIIN) was described previously (22). GCCECD-vaccinia virus (VV) was generated using the pSC11 vaccinia plasmid (23). The control VV (VV-1686) was described previously (24). Mice were immunized with 1 × 108 infectious units (IFU) of adenovirus or 1 × 107 foci-forming units (FFU) of rabies virus by intramuscular injection of the anterior tibialis or with 1 × 107 plaque-forming units (PFU) of vaccinia virus by intraperitoneal injection (n = 322 AV, n = 10 AV and VV, n = 58 AV, RV, and VV).
Cell Lines
C57BL/6-derived MC38 colorectal cancer cells were provided by Jeffrey Schlom (NCI, National Institutes of Health, Bethesda, MD). BALB/c-derived CT26 colorectal cancer cells were from the American Type Culture Collection (Manassas, VA). Both cell lines lack endogenous GCC expression, as determined by radiolabeled ligand binding and quantitative reverse transcriptase–PCR. Stable CT26-GCCTM cell lines were generated by transducing CT26 cells with retrovirus produced from 293T cells that were transiently transfected with pCL-Ampho (Imgenex, San Diego, CA) and GCCTM-pMSCV2.2-Puro, followed by antibiotic selection. GCCTM expression was quantified by fluorescence activated cell sorting (FACS) analysis after staining for the extracellular pentahistidine on GCCTM using mouse monoclonal anti–pentahistidine-Alexa 488 antibody (Qiagen, Valencia, CA) and by binding of radiolabeled heat-stable enterotoxin (ST) to purified membranes (25).
GCCECD Protein Purification
Hexahistidine-tagged GCCECD (GCCECD-6xHis) was purified from supernatants of 293A cells that had been transduced with GCCECD-AV using Ni-NTA agarose beads (Qiagen). A baculovirus expression system was used to produce a hexahistidine-tagged signal peptide-deficient GCC extracellular domain protein (GCC23–430) using the transfer vector pVL1393 and the Sapphire Baculovirus DNA Kit (Orbigen, San Diego, CA). Baculovirus-produced GCCECD-6xHis was purified using Ni-NTA agarose beads (Qiagen). 293A-expressed GCCECD was used in enzyme-linked immunosorbent assays (ELISAs), and baculovirus-expressed GCCECD was used in CD4+ T-cell assays.
Antigen-Specific IgG Antibody Detection by Enzyme-Linked Immunosorbent Assay
BALB/c mice were naive (n = 4) or immunized with control AV (n = 4) or GCCECD-AV (n = 4), and GCC+/+ (+/+) and GCC–/– (−/−) C57BL/6 mice were immunized with control AV (n = 4+/+ and n = 4−/−), GCCECD-AV (n = 4+/+ and n = 4−/−), or an irrelevant GST-fusion protein as a negative control (n = 3+/+ and n = 3–/–). Mice were killed by CO2 asphyxiation, and serum was collected 10–14 days after immunization. Immunosorbent plates (Nunc, Rochester, NY) were coated with purified GCCECD-6xHis at 10 μg/mL or with irrelevant adenoviral particles at 1 × 107 IFU/mL to detect GCCECD-specific or AV-specific responses, respectively. Coated plates were incubated with serum or with mouse anti–pentahistidine IgG (Qiagen) as a positive control. Specific antibodies were detected with HRP-conjugated goat anti–mouse immunoglobulin (Jackson Laboratories, Bar Harbor, ME) and ABTS substrate (Pierce, Rockford, IL). Data represent the mean absorbance at 405 nm of individual mice.
Antigen-Specific T-Cell Response Detection by IFN[gamma] ELISpot
BALB/c mice were immunized with control AV (n = 16) or GCCECD-AV (n = 16), and GCC+/+ (+/+) and GCC–/– (−/−) C57BL/6 mice were immunized with control AV (n = 24+/+ and n = 12−/−) or GCCECD-AV (n = 32+/+ and n = 20−/−). Mice were killed by CO2 asphyxiation, and spleens were collected 10 days after immunization. Mice were assayed by pooling spleens from two mice per experiment and assaying in triplicate. Data are representative of 2–6 independent experiments, as indicated in figure legends. Multiscreen filtration plates (Millipore, Billerica, MA) were coated with anti–mouse interferon gamma (IFNγ)–capture antibody (BD Pharmingen, San Jose, CA). To assay CD8+ T-cell responses, splenocytes or CD8+ T cells that had been separated from splenocytes by magnetic activated cell sorting (MACS) (Miltenyi Biotec, Bergisch Gladbach, Germany) were plated at 10 000–250 000 effector cells per well and incubated with stimulator cells (MC38 for C57BL/6 mice or CT26 for BALB/c mice). To measure GCCECD or β-galactosidase–specific responses, stimulator cells were transduced with replication-deficient GCCECD- or LacZ-adenovirus, respectively, at a multiplicity of infection of 500 (MC38) or 100 (CT26) and treated with 1000 U/mL recombinant mouse IFNγ (EMD Biosciences, San Diego, CA) for 48 hours to increase MHC expression. To assay CD4+ T-cell responses, MACS-sorted CD4+ T cells (Miltenyi Biotec) were mixed with naive splenocytes as antigen presenting cells and incubated on antibody-coated plates with GCCECD-6xHis (0–100 μg/mL) or irrelevant purified adenovirus particles (0–1 × 108 IFU/mL) to measure GCCECD-specific or AV-specific responses, respectively. After 24–28 hours of stimulation, cells were removed by washing, and spots were developed with biotinylated anti-IFNγ detection antibody (BD Pharmingen) and alkaline phosphatase-conjugated streptavidin (Pierce), followed by Nitro-Blue Tetrazolium Chloride and 5-Bromo-4-Chloro-3’-Indolyphosphate p-Toluidine Salt substrate (Pierce). Spot numbers were quantified using computer-assisted video imaging analysis (ImmunoSpot v3, Cellular Technology, Shaker Heights, OH). Data are reported as spots (IFNγ-producing cells) per 106 cells or per well.
GCC Immunization Against Subcutaneous Colon Tumors
BALB/c mice received a prophylactic immunization with AV (n = 5 control and GCCECD), with AV and VV at 28-day intervals (n = 5 control and GCCECD), or with AV, RV, and VV at 28-day intervals (n = 7–8 control and GCCECD). Subcutaneous tumors were then established on the flanks of mice with 1 × 105 CT26-GCCTM cells 1 week after the final immunization, and tumor growth was quantified twice a week for 45 days by measuring three orthogonal diameters and calculating volumes using: 4/3π × r1 × r2 × r3, in which (r) is half the diameter. For survival analysis, mice were killed by CO2 asphyxiation when tumors achieved a volume of 1200 mm3, a surrogate endpoint used in compliance with institutional animal care guidelines. Twenty-one days was selected as the maximum for the growth curves because most control mice reached the survival endpoint around day 25.
GCC Immunization Against Colon Cancer Metastases to Liver
BALB/c mice were immunized with control AV (n = 11) or GCCECD-AV (n = 12). Liver metastases were established by injecting 1 × 105 CT26-GCCTM cells into the exteriorized spleens of mice 7 days after immunization (26). Mice were killed by CO2 asphyxiation, and metastases were counted in excised livers 21 days after tumor challenge.
GCC Immunization Against Colon Cancer Metastases to Lung
To examine prophylaxis, BALB/c mice were immunized with control AV (n = 21) or GCCECD-AV (n = 31), and lung metastases were established by injecting 5 × 105 CT26-GCCTM cells into the tail veins of mice 7 days after immunization. Some mice (n = 14) were imaged by positron emission tomography (PET) and micro computer tomography (CT) 14 days later, then killed by CO2 asphyxiation, and metastases were counted (27). Other mice (n = 36) were monitored for survival daily for 45 days. In studies of therapeutic immunization, lung metastases were established in BALB/c mice (n = 39) by tail vein injection with 1 × 105 CT26-GCCTM cells. Mice were then immunized with control (n = 22) or GCCECD (n = 17) AV on day 3, followed by sequential boosting with RV and VV every 12–26 days such that most mice received all three immunizations before dying from metastasis growth. Survival was monitored and recorded daily for 55 days.
PET/Micro CT Metastasis Quantification
Mice were immunized with control AV (n = 5) or GCCECD-AV (n = 9) 7 days before being injected by tail vein with 5 × 105 GCCTM cells. Fourteen days later, mice received 0.45 mCi 18F-fluorodeoxyglucose by tail vein, and PET images were collected 2 hours later on a Mosaic scanner (Philips Medical Systems, Andover, MA). CT images were acquired on a microCAT II (Imtek, Inc, Knoxville, TN). For quantification of 18F-fluorodeoxyglucose uptake and, consequently, tumor burden, a volumetric region of interest was defined in the lungs. Using calibration parameters that were derived from a cylinder of known size and activity, absolute quantification values of percent of injected dose per g of tissue were calculated. Average lung values were normalized to uptake in liver within each mouse (lung minus liver).
Detection of Serum Antinuclear Antibodies
Antinuclear antibodies were quantified in serum using an anti-ANA ELISA Kit (Alpha Diagnostics, San Antonio, TX) in C57BL/6 mice 10 days after immunization with LacZ-AV (n = 6) or GCCECD-AV (n = 6). Serum from 8-week-old male MRL/MpJ-Faslpr/J mice (Jackson Laboratories) (n = 3) served as positive controls, and serum from naïve mice (n = 2) served as negative controls. Antinuclear IgG was quantified using a standard curve and reported as micrograms of IgG per milliliter.
Autoimmune Pathology
Chemistry profiles were commercially determined (Charles River Laboratories, Wilmington, MA) on fresh serum from previously immunized GCC+/+ (n = 5 control AV and n = 6 GCCECD-AV) and GCC–/– (n = 6 control AV and n = 5 GCCECD-AV) mice. Tissues from immunized BALB/c mice (n = 3 for each immunization) were formalin-fixed, paraffin-embedded, stained by hematoxylin and eosin, blindly labeled, and analyzed by a pathologist (R. Birbe).
Statistical Analyses
Cohort sizes were selected to provide 80% power to detect twofold differences between groups. Survival analysis used Mantel–Haenszel log-rank test (GraphPad Prism Software, San Diego, CA). Two-way analysis of variance (GraphPad Prism) was used for subcutaneous tumor growth studies, antigen-specific antibody assays, and T-cell assays. Student t test or Welch t test (for tests with unequal intergroup variances) were used for all other statistical analyses. All statistical tests were two-sided. P values less than .05 were considered statistically significant.
Results
Vaccine Strategy
The seven members of the particulate guanylyl cyclase family have a canonical structure with highly homologous cytoplasmic catalytic and regulatory domains and extracellular domains possessing minimal sequence homology, reflecting the diversity of their cognate peptide ligands (14) (Figure 1, A). Conservation of cytoplasmic domains among isoforms (Figure 1, B) and the broad tissue distribution of guanylyl cyclases A, B, and G identified the extracellular ligand-binding domain of GCC as the only unique segment of the molecule, therefore focusing vaccine generation on only the GCC extracellular ligand-binding domain. Because none of the established mouse cell lines express endogenous GCC, CT26 mouse colon cancer cells were transduced with a retrovirus containing a recombinant mouse GCC (Figure 1, C), which was truncated beyond the transmembrane domain and lacked intracellular domains (GCCTM; Figure 1, B), at expression levels within the range of normal murine enterocytes and human colon cancer cells (Figure 1, D). Viral vaccine vectors were constructed to express a secreted form of GCC that was truncated beyond the ligand-binding domain and lacked transmembrane and intracellular domains (GCCECD; Figure 1, B). We used constructs containing the nonhomologous (unique) extracellular domain because the full-length molecule has homology with the cytoplasmic domains of other guanylyl cyclases.
Figure 1.

Vaccine design. A) Protein alignment of membrane-bound mouse guanylyl cyclases. Shaded boxes represent amino acid homology. Extracellular ligand-binding domains are nearly devoid of homology across the family. In contrast, there is a high degree of homology within the intracellular domains of the different isoforms. For a full-size version of the alignment, see Supplementary Figure 1 (available online). B) GCC constructs used in these studies: GCCTM lacks cytosolic domains but includes the transmembrane domain and traffics to the cell surface; GCCECD lacks cytosolic and transmembrane domains and is secreted. GCCTM was used in tumor models, and GCCECD was used in viral vaccine vectors. C–D) Cell surface expression of GCCTM in transduced CT26 mouse colon cancer cells, as assessed using anti–pentahistidine antibody and FACS analysis (C) and binding of 125I-ST to membranes prepared from CT26-GCCTM cells (D). Membranes from C57BL/6 mouse small intestine and colon, which endogenously express GCC, and T84 human colon cancer cells were used as positive controls. Binding data indicate means and upper 95% confidence intervals of triplicate measurements, representing two independent experiments.
Prophylactic GCC Immunotherapy Against Subcutaneous Colon Tumors
BALB/c mice received a prophylactic immunization comprising an escalating heterologous prime-boost regimen of replication-deficient recombinant adenovirus, recombinant attenuated rabies virus, and recombinant vaccinia virus, with 28 days between each immunization. Seven days after the last immunization, mice were subcutaneously injected with 1 × 105 CT26 mouse colon carcinoma cells expressing GCCTM (CT26-GCCTM cells), and tumor volume was monitored for 21 days. A single immunization with GCCECD-AV did not alter the growth of CT26-GCCTM cells compared with a control AV immunization (Figure 2, A). However, a prophylactic heterologous prime-boost strategy using GCCECD-AV followed by GCCECD-VV reduced tumor growth (Figure 2, B; mean day 21 tumor volume: control vs GCCECD, 449 vs 124 mm3, difference = 325 mm3, 95% CI = 202 to 448 mm3; P < .001). Moreover, GCCECD-AV followed sequentially by GCCECD-RV and GCCECD-VV reduced tumor growth (Figure 2, C and D; mean day 21 tumor volume: control vs GCCECD, 526 vs 29 mm3, difference = 487 mm3, 95% CI = 337 to 656 mm3, P < .001). Further, both the AV-VV and AV-RV-VV heterologous prime-boost regimens extended survival of GCCECD-immunized mice compared with control immunization (Figure 2, E; median survival in control AV-VV vs GCCECD AV-VV, 28 vs 33 days, P = .018, and Figure 2, F; control AV-RV-VV vs GCCECD AV-RV-VV, 24 vs 42 days, P < .001).
Figure 2.

Prophylactic guanylyl cyclase C (GCC)–specific immunity against subcutaneous colon tumor progression. BALB/c mice were prophylactically immunized with recombinant viruses expressing the extracellular domain of GCC (GCCECD) or control viruses using an escalating heterologous prime-boost strategy and were then injected on day 0 with 1 × 105 CT26-GCCTM cells. A) Adenovirus (AV). B) AV followed by vaccinia virus (VV). C) AV followed sequentially by rabies virus (RV) and VV. Data in (A–C) are means of n = 5–8 mice per immunization, and error bars indicate 95% confidence intervals (***P < .001, two-way analysis of variance of tumor growth in control- vs GCCECD-immunized mice). D) Images of control and GCCECD AV-RV-VV–immunized mice on day 24 with tumors in control mice outlined for clarity. E) Survival analysis of mice from (B) (P = .018, two-sided Mantel–Haenszel log-rank test) in which a tumor volume greater than 1200 mm3 was used as a surrogate endpoint for death. F) Survival analysis of mice from (C) (P < .001, two-sided Mantel–Haenszel log-rank test), in which a tumor volume greater than 1200 mm3 was used as a surrogate endpoint for death. Solid lines in (E) and (F) indicate percent survival; dashed lines indicate 95% confidence intervals.
Prophylactic and Therapeutic GCC Immunotherapy Against Parenchymal Colon Cancer Metastases
More than 50% of patients with colorectal cancer die of metastatic disease, primarily in liver and lung (5). To mimic human disease in parenchymal metastasis models, mice were administered 1 × 105 CT26-GCCTM cells by intrasplenic injection to establish liver metastases 7 days after immunization with GCCECD-expressing or control AV. Three weeks later, livers were collected and tumor burden was quantified by counting nodules and measuring liver wet weight, a marker of metastatic disease. In contrast to efficacy against subcutaneous tumor growth, immunization with GCCECD-AV alone reduced the formation of liver nodules by approximately 90% (Figure 3, A and C; control AV vs GCCECD-AV: 30.4 vs 3.55 nodules, difference = 26.9 nodules, 95% CI = 8.47 to 45.3 nodules, P = .008) and liver wet weight by 25% (Figure 3, B; control AV vs GCCECD-AV: 1.86 vs 1.44 g, difference = 0.42 g, 95% CI = −0.018 to 0.948 g, P = .088). In addition, immunization with GCCECD-AV 7 days before intravenous seeding of lung metastases with 5 × 105 CT26-GCCTM cells reduced the formation of lung nodules by approximately 80% (Figure 4, B and D; control AV vs GCCECD-AV: 263 vs 56 nodules, difference = 207 nodules, 95% CI = 163 to 251 nodules, P < .001) and tumor burden by approximately 80%, as quantified by 18F-fluorodeoxyglucose PET (Figure 4, A and C; relative uptake in control AV vs GCCECD-AV: 1.27 vs 0.25, difference = 1.02, 95% CI = 0.71 to 1.32, P < .001). Furthermore, GCCECD-AV immunization extended survival of mice bearing lung metastases by approximately 50% (Figure 4, E; median survival in control AV vs GCCECD-AV, 23 vs 35 days, P < .001). Moreover, beyond prophylaxis, GCC-directed immunization was efficacious in treating established parenchymal tumor metastases because sequential immunization with GCCECD-AV, -RV, and -VV in mice carrying lung metastases extended median survival (Figure 4, F; control AV-RV-VV vs GCCECD AV-RV-VV, 29 vs 38 days, P = .024), with an immunotherapeutic efficacy comparable to self-antigens that have advanced from mouse models to human trials (28,29).
Figure 3.
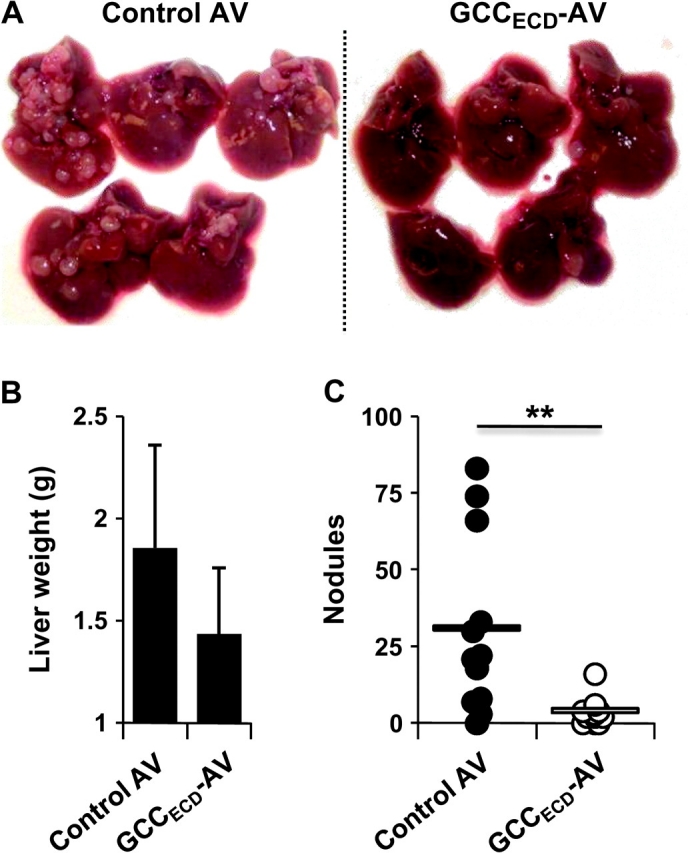
Prophylactic guanylyl cyclase C (GCC)–specific immunity against colon cancer metastases in liver. BALB/c mice were immunized with control adenovirus (AV) or GCCECD-AV and challenged 1 week later by intrasplenic injection (on day 0) with 1 × 105 CT26-GCCTM mouse colon cancer cells. Mice were killed on day 21, and livers were collected. A) Livers of control- and GCCECD-immunized mice. B) Liver weights of control- and GCCECD-immunized mice. Data shown are means of n = 5 mice per immunization, and error bars indicate upper 95% confidence intervals (P = .088, two-sided Student t test). C) Numbers of liver nodules (n = 11–12 mice per immunization). Bars indicate means (**P = .008, two-sided Welch t test).
Figure 4.

Guanylyl cyclase C (GCC)–specific immunotherapy of colon cancer metastases in lung. BALB/c mice were immunized with control adenovirus (AV) or GCCECD-AV (day 7) and then challenged with 5 × 105 CT26-GCCTM cells by tail vein injection on day 0. A) On day 14, metastases were visualized by positron emission tomography/micro computer tomography (PET/microCT). Lungs are outlined for clarity. B) Lungs surgically removed following PET/microCT analysis were stained with India ink to visualize lung nodules. C) PET/microCT–quantified tumor burden, determined by absolute quantification of the percent of injected dose per gram of tissue (% ID/g) in a defined region of interest. Average lung values were normalized to uptake in liver within each mouse (lung minus liver). Bars indicate means of n = 5–9 mice per immunization (***P < .001, two-sided Welch t test). D) Numbers of lung nodules as determined by visual inspection of lungs in (C). Bar indicates mean of n = 6–10 mice per immunization (***P < .001, two-sided Welch t test). E) Survival analysis of prophylactic GCCECD immunization. F) Survival analysis of therapeutic GCCECD immunization. Mice were challenged with 1 × 105 CT26-GCCTM cells by tail vein injection. Beginning 3 days after tumor challenge, mice were immunized sequentially with control or GCCECD AV, rabies virus (RV), and vaccinia virus (VV), and survival was monitored (P = .024, two-sided Mantel–Haenszel log-rank test). Solid lines in (E) and (F) indicate percent survival, and dashed lines indicate 95% confidence intervals.
CD8+ T Cells, but Not CD4+ T Cells or Antibodies, as Mediators of GCC-Targeted Immunity
Protection against metastatic tumor cells by immunization with recombinant viruses presumably reflects mucosal compartmentalization of GCC expression, resulting in incomplete systemic tolerance and selected immune cell responses to that antigen. Immunization of BALB/c mice with GCCECD-AV or LacZ-AV elicited AV-specific, but not GCC-specific, IgG responses (Figure 5, A and B). Similarly, AV-specific, but not GCC-specific, Th1 CD4+ T-cell responses were induced in immunized mice (Figure 5, C and D). In contrast, antigen-specific CD8+ T-cell responses directed against β-galactosidase (LacZ), a foreign antigen, or GCC were nearly equivalent (Figure 5, E and F; mean specific spots per well at 250 000 effector cells: control vs GCCECD, 49.3 vs 33.0, difference = 16.3, 95% CI = 2.41 to 30.3, P = .086). Inhibition of parenchymal metastases that are associated with enhanced survival in the context of CD8+ T-cell responses, but not CD4+ T or B cell responses, induced by GCC immunization suggests that antitumor immunity is CD8+ T cell mediated and underscores the central role of CD8+ T lymphocytes as key antitumor effectors of the adaptive immune system.
Figure 5.

Adaptive systemic cellular responses to guanylyl cyclase C (GCC) immunization. BALB/c mice were naive or immunized with LacZ-adenovirus (AV) or GCCECD-AV, and sera and splenocytes were collected 10–14 days later for quantification of antibody and T-cell responses. A) Enzyme-linked immunosorbent assay (ELISA) analysis of AV-specific IgG antibody responses in immunized BALB/c mice. Bars indicate mean absorbance at 405 nm of reciprocal serum dilutions of 200, 400, 800, and 1600 of n = 4 mice per group, and error bars indicate upper 95% confidence intervals (CIs). B) ELISA analysis of GCCECD-specific IgG antibody responses in immunized BALB/c mice. Bars indicate means at reciprocal serum dilutions of 25, 50, 100, and 200 of n = 4 mice per group, and error bars indicate upper 95% CIs. Anti–hexahistidine mouse IgG (αHis) (concentrations of 200, 100, 50, and 25 ng/mL αHis IgG) served as a positive control for immunodetection of the plated hexahistidine-tagged GCCECD. C) AV-specific CD4+ T-cell responses in immunized BALB/c mice as measured by interferon gamma (IFNγ) ELISpot. Data indicate pooled analysis of n = 2 mice per group, and error bars indicate 95% CIs of triplicate measurements. Two independent experiments were performed with similar results. D) GCCECD-specific CD4+ T-cell responses in immunized BALB/c mice as measured by IFNγ ELISpot. Data indicate pooled analysis of n = 2 mice per group, and error bars indicate 95% CIs of triplicate measurements. Two independent experiments were performed with similar results. E) β-galactosidase–specific CD8+ T-cell responses in LacZ-AV–immunized BALB/c mice as measured by IFNγ ELISpot. Data indicate pooled analysis of n = 2 mice per group, and error bars indicate 95% CIs of triplicate measurements. Results are representative of those obtained in four independent experiments (***P < .001, two-way analysis of variance [ANOVA]). F) GCCECD-specific CD8+ T-cell responses in GCCECD-AV–immunized BALB/c mice as measured by IFNγ ELISpot. Data indicate pooled analysis of n = 2 mice per group, and error bars indicate 95% CIs of triplicate measurements and are representative of four independent experiments (***P < .001, two-way ANOVA). Symbols indicate means in (C–F).
Lineage-Specific Systemic Tolerance to GCC
CD8+ T-cell responses, but not CD4+ T or B cell responses, to GCC suggest that mucosa-restricted expression induces selective systemic tolerance reflecting structural characteristics of GCC influencing antigenic competence and/or compartmentalization (30,31). Tolerance was explored by comparing responses of wild-type (GCC+/+) C57BL/6 mice to those in GCC–/– mice (19). GCC+/+ and GCC–/– mice that were immunized with GCCECD-AV or LacZ-AV produced identical adenovirus-specific antibody responses (Figure 6, A), Th1 CD4+ T-cell responses (Figure 6, C), and β-galactosidase-specific CD8+ T-cell responses (Figure 6, G), suggesting that GCC deficiency does not broadly alter antigen-specific immune responses. However, GCC–/– mice but not GCC+/+ mice produced antibodies (Figure 6, B) and Th1 CD4+ T cells (Figure 6, D) that were directed against GCCECD. In contrast, GCC+/+ C57BL/6 mice, like GCC+/+ BALB/c mice (see Figure 5, E and F), produced CD8+ T-cell responses that specifically recognized β-galactosidase or GCCECD following immunization with LacZ-AV or GCCECD-AV, respectively (Figure 6, E and F). However, although CD8+ T-cell responses to β-galactosidase were comparable in GCC–/– mice following immunization with LacZ-AV (Figure 6, G), responses to GCC were attenuated in GCC+/+ mice compared with GCC–/– mice following immunization with GCCECD-AV (Figure 6, H). Robust GCC-directed antibody responses and CD4+ T-cell responses in GCC–/– mice following immunization with GCCECD-AV underscore the antigenic competence of GCC to induce responses in all arms of the adaptive immune system. In contrast, incomplete tolerance to GCC in wild-type BALB/c and C57BL/6 mice reflects mucosal partitioning and restricted cross-compartmental antigen availability (30,31).
Figure 6.

Differential systemic immune cell tolerance to guanylyl cyclase C (GCC). A) Enzyme-linked immunosorbent assay (ELISA) analysis of adenovirus (AV)-specific IgG antibody responses in GCC+/+ (+/+) or GCC–/– (−/−) C57BL/6 mice 10–14 days after immunization with AV or a GST-fusion protein (negative control). Data represent means (symbols) of n = 8 AV-immunized mice per genotype or pooled samples of n = 3 control-immunized mice per genotype at reciprocal serum dilutions from 100 to 800. Error bars indicate 95% confidence intervals (CIs). B) ELISA analysis of GCCECD-specific antibody responses in +/+ or −/− C57BL/6 mice 14 days after immunization with GCCECD-AV or LacZ-AV. Responses from individual GCCECD-immunized mice are shown, and those from controls are presented as means (n = 3 per group, P < .001, two-way analysis of variance [ANOVA] for GCCECD-AV−/− vs all other groups). Data are representative of three independent experiments. C) AV-specific CD4+ T-cell responses in GCCECD-AV–immunized +/+ or −/− C57BL/6 mice as measured by interferon gamma (IFNγ) ELISpot. D) GCCECD-specific CD4+ T-cell responses in LacZ-AV– or GCCECD-AV–immunized +/+ and −/− C57BL/6 mice as measured by IFNγ ELISpot. Data in (C) and (D) indicate pooled analysis of n = 2 mice per group, and error bars indicate 95% CIs of triplicate measurements and are representative of four independent experiments (***P < .001, two-way ANOVA for GCCECD-AV−/− vs all other groups). E) β-galactosidase–specific CD8+ T-cell responses in GCC+/+ C57BL/6 mice following LacZ-AV immunization as measured by IFNγ ELISpot. F) GCCECD-specific CD8+ T-cell responses in GCC+/+ C57BL/6 mice following GCCECD-AV immunization and measured by IFNγ ELISpot. Data in (E) and (F) indicate pooled analysis of n = 2 mice per group, and error bars indicate 95% CIs of triplicate measurements and are representative of six independent experiments (***P < .001, two-way ANOVA). G) β-galactosidase–specific CD8+ T-cell responses in +/+ and −/− C57BL/6 mice following LacZ-AV immunization measured by IFNγ ELISpot. Data indicate pooled analysis of n = 2 mice per group, and error bars indicate 95% CIs of two independent experiments. H) GCCECD-specific CD8+ T-cell responses in +/+ and −/− C57BL/6 mice following GCCECD-AV immunization measured by IFNγ ELISpot. Data indicate pooled analysis of n = 2 mice per group, and error bars indicate 95% CIs of two independent experiments (*P = .019, two-way ANOVA).
Antitumor Immunity Without Collateral Autoimmune Disease
The established restriction of immunologic cross talk between systemic and mucosal compartments in conjunction with systemic tolerance in CD4+ T-cell responses to mucosa-restricted antigens identified herein should limit autoimmune disease in response to GCC-based immunization. Mice that were serially immunized with three viruses encoding GCCECD and exhibiting maximum antitumor responses (see Figure 2, C, D, and F) were healthy, with no signs or symptoms of inflammatory bowel disease, including weight loss, failure to thrive, altered bowel habits, or rectal bleeding. Similarly, GCC+/+ and GCC–/– mice that were immunized with GCCECD-AV exhibited normal organ and metabolic function, as quantified by serum chemistries (Table 1). Further, autoimmune-mediated tissue damage, which was quantified by serum antinuclear antibodies, was absent in GCC+/+ mice that were immunized with GCCECD-AV (Figure 7, A). Moreover, the gastrointestinal tract, lungs, liver, and kidneys of mice that were serially immunized with three viruses encoding GCCECD exhibiting maximum antitumor responses were free of damage and immune cell infiltration (Figure 7, B; Supplementary Table 1, available online). In that context, GCC is a cancer mucosa antigen that yields efficacious immunotherapeutic antitumor responses without autoimmune tissue damage, underscoring the potential for mucosa-restricted proteins as vaccine targets for derivative metastatic tumors.
Table 1.
Serum chemistries of AV-immunized GCC+/+ and GCC–/– mice*
| Control AV | GCCECD AV | |||||
| Analyte | GCC+/+ (n = 5) | GCC–/– (n = 6) | GCC+/+ (n = 6) | GCC–/– (n = 5) | Normal range | System |
| Total protein, g/dL | 4.9 | 4.8 | 4.9 | 4.8 | 4.4–6.2 | Liver |
| Glucose, mg/dL | 206 | 209 | 197 | 190 | 186–265 | Pancreas |
| Creatinine, mg/dL | 0.2 | 0.1 | 0.1 | 0.1 | 0.2–0.7 | Kidney |
| Albumin, g/dL | 2.7 | 2.7 | 2.9 | 2.7 | 2.6–4.6 | Liver |
| Na, meq/L | 136 | 135 | 138 | 139 | 147–167 | Electrolytes |
| Phosphorous, mg/dL | 7 | 6 | 7 | 7 | 6–13 | Kidney |
| Alanine transaminase, U/L | 41 | 28 | 35 | 25 | 24–140 | Liver, heart, skeletal muscle |
| Cholesterol, mg/dL | 73 | 80 | 69 | 72 | 63–174 | Liver |
| Total bilirubin, mg/dL | 0.3 | 0.2 | 0.3 | 0.2 | 0.0–0.9 | Heme catabolism, cholestasis |
| Aspartate transaminase, U/L | 135 | 99 | 103 | 71 | 69–191 | Liver, heart, skeletal muscle |
| Triglycerides, mg/dL | 58 | 57 | 53 | 67 | 71–164 | Lipids |
| Cl, meq/L | 99 | 97 | 100 | 100 | 104–120 | Electrolytes |
| Blood urea nitrogen, mg/dL | 19 | 19 | 24 | 19 | 19–34 | Kidney |
*AV = adenovirus; GCC = guanylyl cyclase C. Values indicate the mean for each group. No statistically significant differences were seen between groups.
Figure 7.

Absence of autoimmunity upon systemic guanylyl cyclase C (GCCECD) immunization. A) Serum antinuclear antibody (ANA) quantification from naive, LacZ-adenovirus (AV)–, or GCCECD-AV–immunized C57BL/6 or naive MRL/MpJ-Faslpr/J mice (positive control). Data represent mean serum ANA concentrations and upper 95% confidence intervals. B) Histopathologic analysis of tissues collected from BALB/c mice immunized with control or GCCECD-AV or control or GCCECD-ARV (sequential AV, rabies, and vaccinia virus from Figure 2, C–F). Images are representative sections from n = 3 mice per immunization group. Bar = 200 μm.
Discussion
Here we have identified GCC as the first in a newly defined category of self-antigens, cancer mucosa antigens, that are expressed normally in the immunologically privileged mucosa and by tumors (5). Viral vector immunization with GCC elicited systemic CD8+ T-cell responses in both strains of mice tested. Moreover, these responses effectively opposed tumor growth in multiple prophylaxis models of GCC-expressing metastatic colorectal cancer and in a model of immunotherapy of established parenchymal metastases. Comparison of responses in GCC+/+ and GCC–/– mice revealed CD4+ T-cell and antibody responses in GCC–/–, but not in two strains of GCC+/+, mice, suggesting that tolerance to cancer mucosa antigens may be universally characterized by absent CD4+ T-cell and antibody responses but only partially attenuated CD8+ T-cell responses, making them attractive targets for tumor immunotherapy. Histological examination of tissues and measurement of serum markers of autoimmunity and tissue damage in mice with effective concomitant antitumor immunity revealed an absence of adverse effects induced by GCC immunotherapy.
In contrast to currently available cancer testis antigens, the universal expression of mucosa-restricted antigens by derivative tumors offers a unique solution to the application of self-antigens from immunologically privileged sites to tumor immunotherapy. This approach leverages the established immunologic partitioning of systemic and mucosal compartments (6–9). Asymmetry in signaling across compartments, wherein systemic immune responses rarely extend to mucosae, limits the risk of autoimmunity following systemic immunization. Conversely, expression that is restricted to mucosae limits antigen access to the systemic compartment and opposes tolerance to mucosal antigens and, therefore, may overcome the inherent limitations of immunotherapy directed to self-antigens. However, systemic tolerance to mucosa-restricted antigens, which can limit antitumor efficacy, has been only incompletely defined. Here, immunization revealed lineage-specific T-cell tolerance in the systemic compartment, which contained CD8+ T-cell responses, but not CD4+ T-cell responses, to GCC. Tolerance reflects thymic and/or peripheral mechanisms rather than antigenicity, because GCC–/– mice responded to GCC in all arms of the adaptive immune system, whereas in two strains of GCC+/+ mice GCC elicited only CD8+ T-cell responses. Incomplete central tolerance to GCC presumably reflects anatomical, functional, and immunologic compartmentalization wherein sequestration of mucosal antigens provides insufficient antigen for complete systemic tolerance (30,31).
In that context, common molecular mechanisms mediating incomplete systemic tolerance to mucosal antigens have not yet emerged. Indeed, tolerance to the gastric-specific H+/K+-ATPase is mediated peripherally (32) and that to a mucosa-selective carcinoembryonic antigen transgene is mediated by the thymus (33). Here, incomplete tolerance may reflect CD4+ T-cell–independent induction of antigen-specific CD8+ T-cell responses (34). Alternatively, virus-specific CD4+ T cells may provide sufficient support to develop efficacious GCC-specific CD8+ T-cell responses (35). Incomplete tolerance may be mediated, in part, by AIRE-regulated peripheral antigen expression within thymic epithelial cells, resulting in T-cell deletion (36,37). In addition, intestinal antigens may be acquired by mucosal dendritic cells and transported to mesenteric lymph nodes, where they induce regulatory CD4+CD25+ T cells, deletion/anergy of naive T cells, or other mechanisms that diminish immune responsiveness to GCC (38,39). Although mechanisms that mediate incomplete systemic tolerance remain to be defined, these advantageous immune cell responses underscore the potential of cancer mucosa antigens as immunotherapeutic targets to prevent tumor metastases in the absence of collateral autoimmune tissue damage.
GCC immunization produced effective antitumor immune responses likely mediated by CD8+ T cells in the systemic compartment in the absence of mucosal autoimmune disease, reflecting compartment-restricted lymphocyte recirculation imprinted by tissue-specific activation (6,7,40–43). T cells that are activated in mesenteric lymph nodes home to lymphoid structures that are associated with the gut wall, including the lamina propria and Peyer’s patches, and those that are activated systemically home primarily to the spleen and to peripheral lymph nodes and associated tissues (41–44). Compartmentalized recirculation reciprocally increases the efficiency of regional immune responses while decreasing tissue antigen cross-reactivity (41). In that context, the functional independence of compartments is reflected by the paucity of mucosal responses to systemic immunization (6–9). Here, this functional independence, wherein systemic immune responses rarely extend to the mucosal compartment, has been exploited to generate therapeutic responses to metastatic cancer without collateral autoimmune disease by using a self-antigen whose expression is normally restricted to mucosa but is universally expressed in metastatic colorectal tumors.
In the scope of current clinical practice and management of patients with colorectal cancer, vaccination using cancer mucosa antigens generally, and GCC specifically, should have the greatest impact on survival in patients who are at risk for developing metastatic disease. Patients who are ostensibly free of regional metastases at the time of diagnosis and staging (tumor, node and metastases [TNM] stages 1, 2) are at substantial risk for developing metastatic disease, reflecting the presence of occult micrometastases (17). In this clinically heterogeneous population, some of whom have occult residual disease, adjuvant immunotherapy could reduce recurrences and extend disease-free survival, reflecting maximum therapeutic efficacy in the context of minimal metastatic tumor burden. Similarly, cancer mucosa antigen vaccines could impact fluoropyrimidine-based adjuvant therapeutic regimens, which are the mainstay for patients with TNM stage 3 disease with regional lymph node metastases (17). Further, GCC-based vaccines might improve clinical outcomes in patients with esophageal and gastric cancer, reflecting the role of intestinal metaplasia and the associated novel ectopic expression of that antigen in those malignancies (45). Moreover, the efficacy of adjuvant immunotherapy in gastrointestinal malignancies might benefit from polyvalent vaccines that incorporate cancer mucosa antigens other than GCC, for example, Cdx2 and sucrase-isomaltase, which also are intestinally restricted and highly expressed in mucosa-derived malignancies.
These observations establish a framework for exploiting immunologic compartmentalization beyond the gastrointestinal tract to achieve antimetastatic therapy in tumors that originate from other mucosae, including oral, respiratory, mammary, and urogenital, for the treatment of cancers of the head and neck, lung, breast, and vagina and bladder, respectively. This potential for cancer mucosa antigens as immunotherapeutic targets highlights the gap in understanding mechanisms underlying systemic and peripheral tolerance and the segregation of adaptive immune responses across mucosae. Those considerations notwithstanding, the overarching principles of immunologic compartmentalization apply outside the gastrointestinal tract wherein systemic immune responses do not extend to extraintestinal mucosal surfaces (8,46). The established principles of immune partitioning in the context of the present results with GCC underscore the importance of defining the generalizability of cancer mucosa antigens as targets for immunotherapy of mucosa-derived tumors.
Limitations in this study relate to cell lines and mice as preclinical models of human malignancy. Cell lines may not accurately reflect the spectrum of antigen presentation or immunogenicity exhibited by human tumors. Also, the CT26 mouse tumor cell line used herein was genetically engineered to express GCC, albeit at levels comparable with those endogenously expressed in normal mouse intestinal cells and human tumor cells. Further, subcutaneous and parenchymal tumor metastasis models in mice may incompletely reflect the pathophysiology and immunology of metastatic disease in patients. Moreover, although observational data suggest the compartmentalization of mucosal and systemic immunity, its relevance and underlying mechanistic contribution to antitumor immunotherapy in humans are unknown. In addition, the immunogenicity of GCC in humans is not yet known. Finally, the generalizability of cancer mucosa antigens across different vaccine targets and mucosae for immunotherapy in animals and humans remains to be defined.
Funding
National Institutes of Health (CA75123, CA95026 to S.A.W.); Targeted Diagnostic and Therapeutics Inc. (to S.A.W); Measey Foundation Fellowship (to A.E.S.). S. A. Waldman is the Samuel M. V. Hamilton Endowed Professor.
Supplementary Material
Footnotes
J. L. Rothstein holds stock in Amgen, Inc., and is currently doing research supported by Amgen. This research was performed before his employment at Amgen. S. A. Waldman is a paid consultant for Merck Research Laboratories and is Chair of the Scientific Advisor Board for Targeted Diagnostics and Therapeutics, Inc. (uncompensated), which sponsors research and has a license to commercialize diagnostic and therapeutic products related to guanylyl cyclase C. The sponsors of the study had no role in the study design, data collection and analysis, interpretation of the results, the preparation of the manuscript, or the decision to submit the manuscript for publication.
References
- 1.Dalerba P, Maccalli C, Casati C, Castelli C, Parmiani G. Immunology and immunotherapy of colorectal cancer. Crit Rev Oncol Hematol. 2003;46(1):33–57. doi: 10.1016/s1040-8428(02)00159-2. [DOI] [PubMed] [Google Scholar]
- 2.Blattman JN, Greenberg PD. Cancer immunotherapy: a treatment for the masses. Science. 2004;305(5681):200–205. doi: 10.1126/science.1100369. [DOI] [PubMed] [Google Scholar]
- 3.Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev. 2002;188:22–32. doi: 10.1034/j.1600-065x.2002.18803.x. [DOI] [PubMed] [Google Scholar]
- 4.Alves PM, Levy N, Bouzourene H, et al. Molecular and immunological evaluation of the expression of cancer/testis gene products in human colorectal cancer. Cancer Immunol Immunother. 2007;56(6):839–847. doi: 10.1007/s00262-006-0228-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snook A, Stafford B, Eisenlohr L, Rothstein J, Waldman S. Mucosally-restricted antigens as novel immunological targets for anti-tumor therapy. Biomarkers Med. 2007;1(1):187–202. doi: 10.2217/17520363.1.1.187. [DOI] [PubMed] [Google Scholar]
- 6.Belyakov IM, Derby MA, Ahlers JD, et al. Mucosal immunization with HIV-1 peptide vaccine induces mucosal and systemic cytotoxic T lymphocytes and protective immunity in mice against intrarectal recombinant HIV-vaccinia challenge. Proc Natl Acad Sci USA. 1998;95(4):1709–1714. doi: 10.1073/pnas.95.4.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belyakov IM, Ahlers JD, Brandwein BY, et al. The importance of local mucosal HIV-specific CD8+ cytotoxic T lymphocytes for resistance to mucosal viral transmission in mice and enhancement of resistance by local administration of IL-12. J Clin Invest. 1998;102(12):2072–2081. doi: 10.1172/JCI5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eo SK, Gierynska M, Kamar AA, Rouse BT. Prime-boost immunization with DNA vaccine: mucosal route of administration changes the rules. J Immunol. 2001;166(9):5473–5479. doi: 10.4049/jimmunol.166.9.5473. [DOI] [PubMed] [Google Scholar]
- 9.Stevceva L, Abimiku AG, Franchini G. Targeting the mucosa: genetically engineered vaccines and mucosal immune responses. Genes Immun. 2000;1(5):308–315. doi: 10.1038/sj.gene.6363680. [DOI] [PubMed] [Google Scholar]
- 10.Schulz S, Green CK, Yuen PS, Garbers DL. Guanylyl cyclase is a heat-stable enterotoxin receptor. Cell. 1990;63(5):941–948. doi: 10.1016/0092-8674(90)90497-3. [DOI] [PubMed] [Google Scholar]
- 11.Carrithers SL, Barber MT, Biswas S, et al. Guanylyl cyclase C is a selective marker for metastatic colorectal tumors in human extraintestinal tissues. Proc Natl Acad Sci USA. 1996;93(25):14827–14832. doi: 10.1073/pnas.93.25.14827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolfe HR, Mendizabal M, Lleong E, et al. In vivo imaging of human colon cancer xenografts in immunodeficient mice using a guanylyl cyclase C-specific ligand. J Nucl Med. 2002;43(3):392–399. [PubMed] [Google Scholar]
- 13.Frick GS, Pitari GM, Weinberg DS, Hyslop T, Schulz S, Waldman SA. Guanylyl cyclase C: a molecular marker for staging and postoperative surveillance of patients with colorectal cancer. Expert Rev Mol Diagn. 2005;5(5):701–713. doi: 10.1586/14737159.5.5.701. [DOI] [PubMed] [Google Scholar]
- 14.Lucas KA, Pitari GM, Kazerounian S, et al. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000;52(3):375–414. [PubMed] [Google Scholar]
- 15.Birbe R, Palazzo JP, Walters R, Weinberg D, Schulz S, Waldman SA. Guanylyl cyclase C is a marker of intestinal metaplasia, dysplasia, and adenocarcinoma of the gastrointestinal tract. Hum Pathol. 2005;36(2):170–179. doi: 10.1016/j.humpath.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 16.Schulz S, Hyslop T, Haaf J, et al. A validated quantitative assay to detect occult micrometastases by reverse transcriptase-polymerase chain reaction of guanylyl cyclase C in patients with colorectal cancer. Clin Cancer Res. 2006;12(15):4545–4552. doi: 10.1158/1078-0432.CCR-06-0865. [DOI] [PubMed] [Google Scholar]
- 17.Jemal A, Murray T, Ward E, et al. Cancer statistics, 2005. CA Cancer J Clin. 2005;55(1):10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 18.Thompson JD, Higgins DG, Gibson TJ, clustal W. improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schulz S, Lopez MJ, Kuhn M, Garbers DL. Disruption of the guanylyl cyclase-C gene leads to a paradoxical phenotype of viable but heat-stable enterotoxin-resistant mice. J Clin Invest. 1997;100(6):1590–1595. doi: 10.1172/JCI119683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li P, Schulz S, Bombonati A, et al. Guanylyl cyclase C suppresses intestinal tumorigenesis by restricting proliferation and maintaining genomic integrity. Gastroenterology. 2007;133(2):599–607. doi: 10.1053/j.gastro.2007.05.052. [DOI] [PubMed] [Google Scholar]
- 21.McGettigan JP, Pomerantz RJ, Siler CA, et al. Second-generation rabies virus-based vaccine vectors expressing human immunodeficiency virus type 1 gag have greatly reduced pathogenicity but are highly immunogenic. J Virol. 2003;77(1):237–244. doi: 10.1128/JVI.77.1.237-244.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plesa G, McKenna PM, Schnell MJ, Eisenlohr LC. Immunogenicity of cytopathic and noncytopathic viral vectors. J Virol. 2006;80(13):6259–6266. doi: 10.1128/JVI.00084-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zook MB, Howard MT, Sinnathamby G, Atkins JF, Eisenlohr LC. Epitopes derived by incidental translational frameshifting give rise to a protective CTL response. J Immunol. 2006;176(11):6928–6934. doi: 10.4049/jimmunol.176.11.6928. [DOI] [PubMed] [Google Scholar]
- 24.Golovina TN, Morrison SE, Eisenlohr LC. The impact of misfolding versus targeted degradation on the efficiency of the MHC class I-restricted antigen processing. J Immunol. 2005;174(5):2763–2769. doi: 10.4049/jimmunol.174.5.2763. [DOI] [PubMed] [Google Scholar]
- 25.Carrithers SL, Parkinson SJ, Goldstein SD, Park PK, Urbanski RW, Waldman SA. Escherichia coli heat-stable enterotoxin receptors. A novel marker for colorectal tumors. Dis Colon Rectum. 1996;39(2):171–181. doi: 10.1007/BF02068072. [DOI] [PubMed] [Google Scholar]
- 26.Wai PY, Mi Z, Guo H, et al. Osteopontin silencing by small interfering RNA suppresses in vitro and in vivo CT26 murine colon adenocarcinoma metastasis. Carcinogenesis. 2005;26(4):741–751. doi: 10.1093/carcin/bgi027. [DOI] [PubMed] [Google Scholar]
- 27.Mule JJ, Shu S, Schwarz SL, Rosenberg SA. Adoptive immunotherapy of established pulmonary metastases with LAK cells and recombinant interleukin-2. Science. 1984;225(4669):1487–1489. doi: 10.1126/science.6332379. [DOI] [PubMed] [Google Scholar]
- 28.Aarts WM, Schlom J, Hodge JW. Vector-based vaccine/cytokine combination therapy to enhance induction of immune responses to a self-antigen and antitumor activity. Cancer Res. 2002;62(20):5770–5777. [PubMed] [Google Scholar]
- 29.Hodge JW, Grosenbach DW, Aarts WM, Poole DJ, Schlom J. Vaccine therapy of established tumors in the absence of autoimmunity. Clin Cancer Res. 2003;9(5):1837–1849. [PubMed] [Google Scholar]
- 30.Ando K, Guidotti LG, Cerny A, Ishikawa T, Chisari FV. CTL access to tissue antigen is restricted in vivo. J Immunol. 1994;153(2):482–488. [PubMed] [Google Scholar]
- 31.Kurts C, Miller JF, Subramaniam RM, Carbone FR, Heath WR. Major histocompatibility complex class I-restricted cross-presentation is biased towards high dose antigens and those released during cellular destruction. J Exp Med. 1998;188(2):409–414. doi: 10.1084/jem.188.2.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allen S, Read S, DiPaolo R, et al. Promiscuous thymic expression of an autoantigen gene does not result in negative selection of pathogenic T cells. J Immunol. 2005;175(9):5759–5764. doi: 10.4049/jimmunol.175.9.5759. [DOI] [PubMed] [Google Scholar]
- 33.Bos R, van Duikeren S, van Hall T, et al. Expression of a natural tumor antigen by thymic epithelial cells impairs the tumor-protective CD4+ T-cell repertoire. Cancer Res. 2005;65(14):6443–6449. doi: 10.1158/0008-5472.CAN-05-0666. [DOI] [PubMed] [Google Scholar]
- 34.Mozdzanowska K, Maiese K, Gerhard W. Th cell-deficient mice control influenza virus infection more effectively than Th- and B cell-deficient mice: evidence for a Th-independent contribution by B cells to virus clearance. J Immunol. 2000;164(5):2635–2643. doi: 10.4049/jimmunol.164.5.2635. [DOI] [PubMed] [Google Scholar]
- 35.Qiu F, Cui Z. CD4+ T helper cell response is required for memory in CD8+ T lymphocytes induced by a poly(I:C)-adjuvanted MHC I-restricted peptide epitope. J Immunother (1997) 2007;30(2):180–189. doi: 10.1097/01.cji.0000211330.61019.6f. [DOI] [PubMed] [Google Scholar]
- 36.Anderson MS, Venanzi ES, Klein L, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298(5597):1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 37.Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC. Aire regulates negative selection of organ-specific T cells. Nat Immunol. 2003;4(4):350–354. doi: 10.1038/ni906. [DOI] [PubMed] [Google Scholar]
- 38.Niess JH, Brand S, Gu X, et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307(5707):254–258. doi: 10.1126/science.1102901. [DOI] [PubMed] [Google Scholar]
- 39.Rescigno M, Urbano M, Valzasina B, et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001;2(4):361–367. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 40.Mayer L. Mucosal immunity and gastrointestinal antigen processing. J Pediatr Gastroenterol Nutr. 2000:30. doi: 10.1097/00005176-200001001-00002. suppl:S4–S12. [DOI] [PubMed] [Google Scholar]
- 41.Kunkel EJ, Butcher EC. Chemokines and the tissue-specific migration of lymphocytes. Immunity. 2002;16(1):1–4. doi: 10.1016/s1074-7613(01)00261-8. [DOI] [PubMed] [Google Scholar]
- 42.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21(4):527–538. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 43.Mora JR, Bono MR, Manjunath N, et al. Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature. 2003;424:88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- 44.Campbell DJ, Butcher EC. Rapid acquisition of tissue-specific homing phenotypes by CD4+ T cells activated in cutaneous or mucosal lymphoid tissues. J Exp Med. 2002;195(1):135–141. doi: 10.1084/jem.20011502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Debruyne PR, Witek M, Gong L, et al. Bile acids induce ectopic expression of intestinal guanylyl cyclase C through nuclear factor-kappaB and Cdx2 in human esophageal cells. Gastroenterology. 2006;130(4):1191–1206. doi: 10.1053/j.gastro.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 46.Tan GS, McKenna PM, Koser ML, et al. Strong cellular and humoral anti-HIV Env immune responses induced by a heterologous rhabdoviral prime-boost approach. Virology. 2005;331(1):82–93. doi: 10.1016/j.virol.2004.10.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.