

Abstract
Background
The oomycete Aphanomyces astaci is regarded as the causative agent of crayfish plague and represents an evident hazard for European crayfish species. Native crayfish populations infected with this pathogen suffer up to 100% mortality. The existence of multiple transmission paths necessitates the development of a reliable, robust and efficient test to detect the pathogen. Currently, A. astaci is diagnosed by a PCR-based assay that suffers from cross-reactivity to other species. We developed an alternative closed-tube assay for A. astaci, which achieves robustness through simultaneous amplification of multiple functionally constrained genes.
Results
Two novel constitutively expressed members of the glycosyl hydrolase (GH18) gene family of chitinases were isolated from the A. astaci strain Gb04. The primary amino acid sequence of these chitinase genes, termed CHI2 and CHI3, is composed of an N-terminal signal peptide directing the post-translational transport of the protein into the extracellular space, the catalytic GH18 domain, a proline-, serine-, and threonine-rich domain and a C-terminal cysteine-rich putative chitin-binding site. The A. astaci mycelium grown in a pepton-glucose medium showed significant temporal changes in steady-state CHI2 and CHI3 mRNA amounts indicating functional constraint. Their different temporal occurrence with maxima at 48 and 24 hours of incubation for CHI2 and CHI3, respectively, is in accordance with the multifunctionality of GH18 family members. To identify A. astaci-specific primer target sites in these novel genes, we determined the partial sequence homologs in the related oomycetes A. frigidophilus, A. invadans, A. helicoides, A. laevis, A. repetans, Achlya racemosa, Leptolegnia caudata, and Saprolegnia parasitica, as well as in the relevant fungi Fusarium solani and Trichosporon cutaneum. An A. astaci-specific primer pair targeting the novel genes CHI2 and CHI3 as well as CHI1 - a third GH18 family member - was multiplexed with primers targeting the 5.8S rRNA used as an endogenous control. A species was typed unambiguously as A. astaci if two peaks were concomitantly detected by melting curve analysis (MCA). For sensitive detection of the pathogen, but also for quantification of agent levels in susceptible crayfish and carrier crayfish, a TaqMan-probe based real-time PCR (qPCR) assay was developed. It targets the same chitinase genes and allows quantification down to 25 target sequences.
Conclusion
The simultaneous qualitative detection of multiple sequences by qPCR/MCA represents a promising approach to detect species with elevated levels of genetic variation and/or limited available sequence information. The homogenous closed-tube format, reduced detection time, higher specificity, and the considerably reduced chance of false negative detection achieved by targeting multiple genes (CHI1, CHI2, CHI3, and the endogenous control) at least two of which are subject to high functional constraint, are the major advantages of this multiplex assay compared to other diagnostic methods.
Sensitive quantification achieved with TaqMan qPCR facilitates to monitor infection status and pathogen distribution in different tissues and can help prevent disease transmission.
Background
Oomycetes are a group of filamentous, unicellular heterokonts. They are fungus-like in their growth form, adsorptive and parasitic lifestyles and formation of spores, but are relatively closely related to photosynthetic algae such as brown algae and diatoms [1]. Among oomycetes, also known as water molds, there are economically important pathogens that comprise severe pests, like Phytophthora infestans [2,3] causing potato late blight, A. euteiches causing seedling blight or legumes root rot [4], A. astaci [5], - the causative agent of crayfish plague, and several fish pathogens from the genera Aphanomyces [6], Achlya and Saprolegnia [7]. There is also at least one species with zoonotic potential, namely Pythium insidiosum - the etiologic agent of the human disease pythiosis insidiosii, which can be life-threatening [8]. The oomycetes A. astaci and Phytophthora cinnamomi are listed among the world's 100 worst invasive species (Global Invasive Species Database: http://www.issg.org/database, alphabetical list as of November 2008).
The crayfish plague, representing the most severe disease among Asian, Australian, and European crayfish species, is caused by the oomycete A. astaci (Saprolegniales, Oomycetes). Crayfish plague-associated die-offs in Austrian waters were first reported in 1879 [9] and in the 1920s [10], and continue sporadically into the present. An estimated 80% of all native Austrian crayfish populations disappeared in the 20th century (Pöckl, personal communication). A high percentage of these die-offs are associated with crayfish plague, which represents one of the major threats to the recovery of populations of native crayfish species in Central Europe [11]. For example, Astacus astacus, formerly a very abundant species in Europe, is now considered threatened by the International Union for Conservation of Nature and Natural Resources (IUCN) [12]. In many countries this economically valuable crayfish is on the Red List and its current harvest is probably less than 10% of the harvest rate before introduction of the crayfish-plague pathogen [13,14]. A. astaci was introduced from North America, where various species harbour the pathogen without showing clinical signs of infection. Crayfish-plague outbreaks among such populations often occur only under stress conditions. The introduction of resistant North American species like the signal crayfish (Pacifastacus leniusculus), the red-swamp crayfish (Procambarus clarkii) and the spiny-cheek crayfish (Orconectes limosus) http://www.issg.org/database has established a permanent reservoir for the pest in Europe. The transmission of the pathogen occurs via crayfish cadavers, crayfish-feeding fish [15], fish scales [16] and all kinds of equipment, which have been in contact with contaminated water [10]. The adaptive life style, high fecundity, and resistance to the pathogen make introduced crayfish species a potent bioinvador and the most dangerous vector for pathogen transmission.
Biflagellated secondary zoospores, measuring 8 × 12 μm, represent the infective unit of A. astaci. They target host tissue by various mechanisms including chemotaxis [17,18] on soft parts of the crayfish integument, especially at the joints, the bottom side of the abdomen and even near the eyestalks [19] as well as fresh wounds [20]. Once zoospores reach the upper lipoprotein-layer of the crayfish cuticle, they discard their flagellae, and develop a penetration peg, that weakens the lipid layer enzymatically [21]. Soon after the germ tube has penetrated the cuticle by mechanical force, the developing hyphae begin to secrete chitinases and proteases [22]. In this phase different chitinases [18] jointly degrade chitin polymers in order to release nutrients and facilitate further growth mainly parallel to the chitin fibrils of the endocuticula [23]. Based on their substrate affinity these enzymes can be classified into three groups: (i) endochitinases, which randomly cleave glycosidic linkages, generating free ends and long chitooligosaccharides that are processed by (ii) exochitinases (chitobiosidases), which release diacetylchitobiose (chitobiose) and (iii) N-acetylglucosaminidases (chitobiases), which hydrolyse chitobiose or release N-acetylglucosamine monomer from chitin chains [24,25].
High-level production of extracellular chitinase in the absence of substrate is one of the most prominent features of the specialised crayfish-parasite A. astaci [26,18]. The GH18 family-chitinase Chi1 was the first chitinase described for A. astaci [18]. Here we selected two additional members of this gene family as targets for an A. astaci-specific diagnostic assay. GH18 chitinases can be divided into three clusters, two of which (A and B) differentiated before the appearance of the eukaryotic lineage [27]. For example, fungal GH18 families comprise between one and twenty genes represented by members of all three clusters [28]. We demonstrate the temporally regulated expression of two novel members of the A. astaci-GH18 family. This functional constraint was regarded as a basic criterion for the development of a closed-tube diagnostic method for qualitative and quantitative detection of A. astaci. In conclusion, simultaneously targeting multiple chitinase sequences including the novel, functionally constrained chitinase sequences, facilitates a robust analysis of clinical samples with a maximum reduced chance of false-negative detection.
Results
Strain identification
Two putative A. astaci strains were recovered from healthy signal crayfish in two small streams in the Austrian province of Burgenland (Gb04 - Ganaubach and Z12 - Zöbernbach). A third strain (GKS07) was isolated from the subabdominal cuticle of a moribund noble crayfish specimen collected during an acute crayfish-plague outbreak in the lake „Gleinkersee” (Austrian province: Upper Austria) in March 2007 (Table 1). ITS-sequence data and constitutive chitinase secretion specific for A. astaci (Additional file 1) confirm the assumed species assignment for all three strains. The strain Gb04 was used to identify two new chitinase genes, test for their functional constraint and finally to develop the diagnostic assay for A. astaci.
The Aphanomyces strain LK29 was isolated from a healthy signal crayfish (Pacifastacus leniusculus). Physiological and genetic evidence showed that the strain does not fit into any previously identified group of A. astaci. It exhibited properties like repeated zoospore emergence and lack of sexual reproduction commonly associated with parasitic species. In contrast to A. astaci, the strain LK29 does not express chitinase constitutively during growth or sporulation. Phylogenetic analysis of ITS sequences (Additional file 1A) demonstrated clustering within the A. laevis-repetans clade [29]. In addition, a Blastn search with the 28SrDNA sequence of LK29 (GenBank:GQ152606, this work) showed close homology to A. laevis (99%, GenBank:AF320584), but clear difference (97% identity) to the A. astaci strains Hö, FDL, GB04 and Z12 (AF320583, AF320582, GQ374534, GQ374535, respectively). Until their taxonomic status is fully elucidated the new isolate was assigned to A. repetans. This species is not capable of killing crayfish following standardised experimental infection and is characterised by a high growth rate, and germination in response to nutrients [30].
Sequence determination of the novel A. astaci genes CHI2 and CHI3
Fungal species contain one to twenty GH18 chitinase family genes [28]. In order to develop a robust diagnostic assay for A. astaci, we asked whether the chitinolytic system of the pathogen would contain multiple genes of this ancient gene family widely expressed in archea, prokaryotes and eukaryotes [31].
As indicated by the two cross-reacting bands detected in western-blot analysis with antibodies raised against the catalytic GH18 domain, A. astaci contains more than one chitinase-like protein (Figure 2). Therefore, we attempted to identify homologous genes using PCR amplification with consensus primers targeting the amino acid motifs DSWND and AGSW (Figure 3). For various A. astaci strains representing all four genotype groups described (type A: L1, Sv, Ra; B: Hö, Yx, Ti; C: Kv; D: Pc; [32]) and the Austrian strain Gb04 isolated in this work (Figure 1), partial GH18 domain sequences were amplified and subsequently sequenced. Analysis revealed a mixture of sequences derived from two new chitinase genes (CHI2 and CHI3, see below), as concluded by retrospective evaluation. Only synonymous substitutions were found in these genes (data not shown). Starting from the consensus sequence obtained for the "core" of CHI2 and CHI3 mRNAs, their complete mRNA sequences were identified by Rapid Amplification of cDNA Ends (RACE)-PCR and submitted to the GenBank (accessions FJ439177 and FJ386997, respectively).
Figure 2.
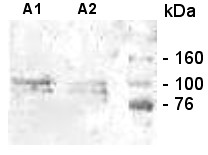
Western-blot analysis of chitinfree PG1-supernatant of a ten-day old A. astaci (strain Hö) broth culture. Two bands of about 100 kDa and slightly below this size were detected by antibodies A1 and A2 raised against epitopes in the catalytic domain of the first A. astaci GH18 chitinase family member Chi1.
Figure 3.

Domains completeley homologous in the novel chitinases Chi2 and Chi3 as well as in the first A. astaci chitinase (Chi1, GenBank:AJ416354, [18]) were selected as primer target sites in the diagnostic assays for A. astaci. In blue: primer target sites. Note that only the homologous part of Chi1 is shown. The chitinase-like protein Clp mRNA (GenBank:FJ439176) was amplified from cDNA, but failed to amplify from genomic DNA for unknown reasons (data not shown). Chi1 peptide sequences selected to generate antibodies for Western blot analysis are underlined. Highly conserved motifs in the GH18 domain (grey boxes) were selected as primer target sites to identify the homologous genes of related oomycetes and relevant fungi (see text). Dots indicate missing sequence homology. The triangle marks the signal peptide cleavage site in Chi2 and Chi3. The catalytic-site residues D154, D156 and E158 putatively required for catalytic activity [27] are indicated by vertical arrows. Residues given as red or black letters represent mismatches and conservative changes, respectively. The conserved cysteines in the CB site 2 are highlighted in bold.
Figure 1.

Biological material used in this work.
Genomic DNA amplified with gene specific primers designed near the start and stop codons of CHI2 and CHI3, yielded fragments of 1810 bp and 1617 bp for CHI2 and CHI3, respectively. Subsequent sequence analysis performed with a primer-walking strategy (data not shown) confirmed the absence of the consensus sequence for exon-intron junctions (5'-GTRNGT...YAG-3' [33]) and identity of cDNA and genomic sequences (GenBank:DQ974157 and FJ457089 for genomic sequences of CHI2 and CHI3).
Characterization of cDNA and deduced amino acid sequences of CHI2 and CHI3
Without the poly(A) tail, the cDNA sequences of CHI2 and CHI3 measure 1807 and 1591-bp in length and exhibit a relatively high guanine and cytosine base content of 59.9% and 60.3%, respectively, a typical feature of oomycete genes [34]. CHI2 and CHI3 code for open reading frames of 596 and 522 amino acids (Figure 3) with molecular masses of 64.0 kDa and 56.7 kDa and isoelectric points of pH 6.14 and 6.63 predicted for the mature secreted enzymes Chi2 and Chi3 (see below), respectively. The mRNAs possess an identical 42-bp 5' untranslated region (UTR) carrying the major part of the oomycete consensus sequence for the start site of transcription (TATTCAATTTGCCAT, [33]). The 3' UTRs of CHI2 and CHI3 contain the polyadenylation signal WAUAAC (W = A or T) [35] (Additional file 2). In both genes the translation start codon is part of the eukaryotic consensus ACCATGA [33]. The enzymes are predicted to be cleaved by signal peptidase between positions A20 and A21 producing a hydrophobic signal peptide of 20 amino acids (Figure 3).
Overall, the deduced amino acid sequences of CHI2 and CHI3 are highly homologous with an identity of up to 79.0% (overlapping residues 1 to 596 and 1 to 522, respectively). The proline-, serine-, and threonine-rich domain [36] of Chi2 contains extra residues resulting in an extended amino acid sequence of the whole protein compared to Chi3 (Figure 3). This domain also represents the most heterologous part of the enzymes regarding primary sequence. Chi2 and Chi3 possess an oomycete-type catalytic GH18 domain (A21 to G400/403, Figure 4). It contains a conserved chitin-binding (CB) site [37] (CB site 1 in Figure 3), and an active site consensus [LIVMFY] - [DN] - G - [LIVMF] - [DN] - [LIVMF] - [DN] - x - E (Prosite no. PS01095) being variant at one position (Additional file 3). The catalytic-site residues D154, D156 and E158 are putatively required for catalytic activity [27]. A second putative, highly homologous CB site was identified in the C-terminal part of the chitinases (CB site 2 in Figure 4). It contains four cysteines, instead of the five residues found in a diatom chitinase (GenBank:EED92972) or six in most insect chitinases [38].
Figure 4.
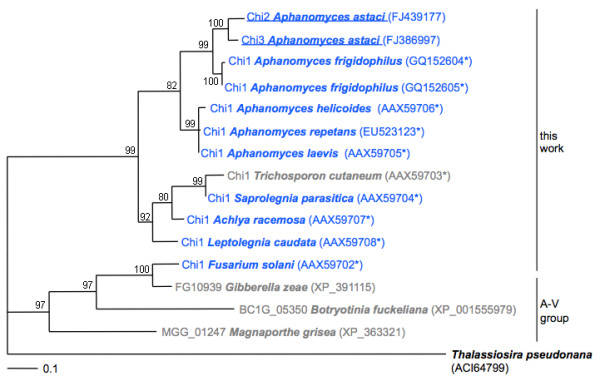
The A. astaci chitinases Chi2 and Chi3 possess an oomycete type-GH18 catalytic domain. Maximum likelihood phylogenetic analysis was performed with TreePuzzle using the diatom Thalassiosira pseudonana as an outgroup. Oomycete and fungal sequences are given in blue and grey, respectively. GenBank accession numbers of partial or complete amino acid GH18 domain sequences are indicated in parentheses. The scale bar represents 0.1 substitutions per site. The numbers at the nodes are quartet puzzling values indicating the frequencies of occurrence for 1,000 replicate trees and can be interpreted in much the same way as bootstrap values. The group A-V - one of six separate fungal groups classified [27,28] - showing the closest homology to the sequences identified in this work, is represented by two members. An asterisk denotes partial sequences.
Chi2 and Chi3 sequences contain sites for amidation, N-myristoylation or O-linked glycosylation (Additional file 4). The latter type of glycosylation predicted for the C-terminal protein parts occurs often at serine and threonine residues that would otherwise be phosphorylated; one illustration of the complex interplay among eukaryotic post-translational modification systems [39]. N-glycosylation at N165/165 (site: NDS) and N296/298 (site: NFT) was predicted for Chi2/Chi3, respectively. These posttranslational modifications may account for the discrepant masses deduced from primary protein sequences and calculated on the basis of the electrophoretic mobility (Figure 2). Putative sites for C-linked glycosylation (C-mannosylation, [39]) were not found. The tripeptide 'RGD' mediating cell adhesion (R81 to D83) was predicted for Chi2. Potential sites for phosphorylation at serine, threonine and tyrosine residues are listed in the Additional file 4.
Temporal mRNA expression analysis for CHI2 and CHI3
Next, we verified that target genes selected for the DNA-based diagnostic crayfish-plague assay are subject to functional constraint. This could be assumed if temporal expression of target genes significantly changes during physiological conditions relevant to the infection in vivo.
The CHI2 and CHI3 mRNA copy numbers expressed in the A. astaci mycelium, grown in chitin-free culture were quantified over three days at intervals of twelve hours using one-step qRT-PCR. A partial sequence of the nuclear gene NDUFV1 encoding the mitochondrial protein NADH dehydrogenase (ubiquinone) flavoprotein 1, which is part of mitochondrial respiratory chain complex I, was identified in this work (data not shown, GenBank:EU500726). We used this sequence as target for an endogenous positive control qRT-PCR assay reporting deviations in extraction, reverse transcription and PCR amplification including mRNA integrity, quality, and quantity. Overall, levels of NDUFV1 mRNA changed only slightly across the time points studied (< 2.5-fold), including, however, expression changes which were near or below the level of significance (p = 0.05) but not matching the temporal expression patterns of the chitinases. In detail, the dynamic growth of the mycelium during the first hours in drop culture (12 to 24 hours, [18]) was reflected by the higher NDUFV1 expression found after 12 and 24 hours of culture (P = 0.03 and 0.07, respectively). Mycelium growth reached its plateau after 72 hours of incubation. The decreasing energy requirement and the beginning of autolytic processes at this stage are reflected by a lower NDUFV1-transcript copy number (P = 0.05 for expressions at 72 and 24 hours).
The chitinase genes CHI2 and CHI3 were both constitutively expressed in mycelium grown in a medium lacking the substrate chitin. However, different mRNA amounts and temporal expression patterns, including the time point at which the maximum level was reached, were observed (Figure 5). Most prominent was the significant maximum in the CHI2 mRNA level reached after 48 hours (P = 0.013).
Figure 5.
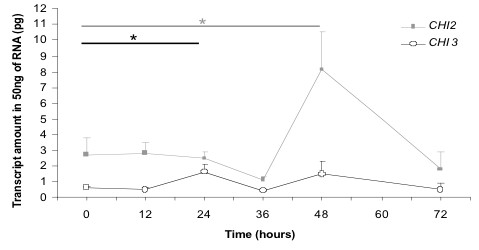
Significant changes of temporal expression of CHI2 and CHI3 mRNAs. The transcript abundance changes during 72 hours of growth in chitinless, liquid PG-1 medium. The significant differences in temporal expression indicate functional constraint and are in accordance with the plurifunctionality of GH18 family members, respectively. Error bars (only the positive error bar is shown) represent the standard errors of the mean obtained from three independent time-course experiments. The asterisk designates significance at p < 0.05.
Analogous to data obtained for CHI1 [18], we demonstrated, exemplarily for CHI3, that neither the amplitude of expression nor its pattern was influenced by substrate addition (0.6% colloidal chitin instead of glucose, data not shown).
Assay development for qualitative and quantitative detection of A. astaci in clinical samples based on chitinase gene sequences
Compared to other crayfish-afflicting oomycetes, permanent chitinase expression and activity represents a unique feature of A. astaci [18,40]. Due to the assumed functional constraints demonstrated by the significant alterations of temporal gene expression (Figure 5), its chitinolytic system was chosen as a target for the development of a diagnostic test.
qPCR/MCA
A BLASTp search with the deduced amino acid sequence of CHI1 as query identified two conserved motifs within the GH18 chitinase domain (83-DSWND and 229-MTYDLAGSW, Figure 3). The nucleotide sequences of these motifs were used as target sites for the design of degenerated PCR primers. Using these primers we were able to amplify and sequence the homologous sequences of nine strains from eight oomycete species and two fungi which are known to live on or in proximity of crayfish (species and GenBank accessions in Figure 6a). On the basis of these sequences, we designed a diagnostic primer pair producing a 93 bp-amplicon from each of the three related chitinase genes (CHI1: [18], CHI2 and CHI3: this work, Figure 6a). Its melting temperature of 86.7°C in MCA was regarded as criterion for identification of an A. astaci strain. For assay robustness the chitinase primer pair was multiplexed with primers targeting the 5.8S rRNA gene as an endogenous control (Additional file 5) and yielding a peak in MCA at 81.5 to 83.5°C depending from the species investigated.
Figure 6.

Qualitative and quantitative detection of the oomycete A. astaci. A: Diagnostic qPCR/MCA primers (blue arrows) target A. astaci-specific sites in the homologous chitinase genes CHI1, CHI2 and CHI3, but not homologous sequences of related oomycetes and fungi. Parentheses contain GenBank accession numbers. Dots and letters represent identical and substituted nucleotides compared to the A. astaci sequence, respectively. B: Qualititative detection of A. astaci by qPCR/MCA. The left and right peaks are derived from amplification of the endogenous control, and the chitinase genes CHI2 &CHI3, respectively. Red plot: A. astaci, blue plot: A. frigidophilus. C: Quantitative detection of A. astaci by TaqMan qPCR. The standard curve of the assay demonstrates quantification down to 25 copies.
The qPCR/MCA assay was tested for specificity against the oomycetes A. frigidophilus, A. invadans, A. laevis, A. helicoides, A. irregularis, and Leptolegnia caudata. Only the endogenous control was recorded, but not the A. astaci-specific chitinase peak.
qPCR/MCA-based detection of A. astaci was used to elucidate several spontanous crayfish mortalities in Austrian waterbodies. In detail, A. astaci was identified as causative agent of acute crayfish-plague outbreaks among noble crayfish inhabiting a small unnamed pond-system (Hartberg, district Hartberg, province Styria), in the noble crayfish-pond Bäckerteich (Velden am Wörthersee, district Villach-Land, province Carinthia), in the brook Hahntrattenbach near St. Andrä (district Wolfsberg, province Carinthia) known for its large stone crayfish population and in a noble crayfish population of the lake Gleinkersee (Roßleithen, district Kirchdorf an der Krems, province Upper Austria). A. astaci was also detected by MCA in necrobiopsy pools each derived from up to five euthanised signal crayfish specimens collected at the streams Ganaubach, Zöbernbach, Strem, Tauchenbach and Güns (province Burgenland). Clinical samples tested positive by MCA were subjected to pathogen isolation. In case of isolation failure the qPCR/MCA amplicon was sequenced.
TaqMan qPCR
For sensitive detection of the pathogen, but also for quantification of agent levels in susceptible crayfish and carrier crayfish, a TaqMan-probe-based qPCR assay was developed. TaqMan qPCR uses the same primers as qPCR/MCA except the additional nucleotide at the very 5' end of the reverse primer compared to qPCR/MCA. Using amplicon standards with known copy numbers spiked into genomic crayfish DNA, a quantitative detection limit of 25 target sequences was determined (Figure 6c). No amplication, i.e. CT > 50, was obtained for A. frigidophilus, A. invadans, A. leaevis and A. irregularis, In the case of the oomycete species A. helicoides and Leptolegnia caudata a cross-amplification signal corresponding to 28 and 44 copies was detected, respectively.
Discussion
Qualitative detection of two or multiple target sequences by MCA has been reported before. Single-tube SNP genotyping [41], sex determination [42], identification of methylation in promoter sequences [43] or the simultaneous detection of multiple pathogens [44,45] are exemplarily mentioned.
In this work we have used MCA of multiplex qPCR [46] for rapid species identification of the crayfish-plague pathogen A. astaci in a closed-tube format. The diagnostic assay for qualitative detection is highly discriminative, robust, inexpensive, and reliable.
High discrimination was aimed at since new Aphanomyces ITS sequences, probably representing new Aphanomyces spp. and including sequences closely related to A. astaci were reported [47,48]. Current molecular techniques for A. astaci detection based on ITS sequences suffer from a lack of specificity ([47,48], Additional file 6), or are laborious and time-consuming due to agarose electrophoresis and subsequent amplicon sequence analysis [11]. To facilitate unambiguous species identification, we considered the unique feature of constitutive chitinase gene expression of A. astaci, not found in closely related Aphanomyces species [18,26]. In a search for additional GH18 family members the novel chitinase genes CHI2 and CHI3 were identified in this work. The genes differ in their 3' UTRs including variant putative polyadenylation signals. Their temporal mRNA expressions change differently during mycelium growth in chitin-free medium. The deduced extracellular protein sequences are different in proline-, serine-, and threonine-rich domain size, and either possess or lack a putative cell attachement site. This speaks in favour of a joint action during the infection process. Therefore, we regarded CHI2 and CHI3 as different members of the GH18 gene family rather than allelic sequences. Altogether, three genes (CHI1, CHI2 and CHI3) encoding constitutively expressed GH18 chitinases in the absence of chitin were identified as unique characteristics of A. astaci and selected as targets for species-specific detection.
Assay robustness, characterised by a low risk of false negatives related to genotypic variation of pathogenic strains, was another issue for assay design. This was especially important since A. astaci belongs to the group of asexual organisms, for which a low level of genetic variation turns out to be the exception rather than the rule [49]. We argued that targeting one or even several functionally constrained sequences would restrict the genotypic variations allowed. The novel chitinase genes CHI2 and CHI3 being functionally constrained as concluded from their significant changes in temporal mRNA expression during growth (Figure 5) were regarded to be appropriate candidates to achieve this aim. Together with the first member of the GH18 gene family of A. astaci (CHI1: [18]) they served as targets in the diagnostic assays based on qPCR/MCA or TaqMan qPCR. In the qPCR/MCA-based assay for qualitative detection, a further level of robustness was achieved by multiplexing with a primer pair targeting the 5.8S rRNA gene as an endogenous control. This DNA sequence is naturally present at multiple copies [50] and harbours two completely homologous primer target sites in each experimental oomycete species (Figure 6a). The simultaneous amplification of this 5.8S rRNA sequence controling for the DNA extraction and amplification steps reduces the chance of false negative detection due to insufficient sample quality. The chitinase gene targets and the endogenous control can be considered to be present at comparable copy numbers [50,28]. Therefore, if non-limited primer concentrations are applied like here, the simultaneous amplification of more than one target in a single PCR, i.e. multiplexing, leads to competition between multiple targets for a finite number of reagents. Representing a welcomed side effect, this further enhances assay discrimination (see above). Co-amplification of an endogenous control adds another level to assay robustness and represents an improvement compared to the ITS1-based TaqMan minor-groove binder qPCR assay for A. astaci-detection reported recently [51]. Coextraction of an homologous (competitive) internal positive control (IPC) with the clinical samples and coamplification in the qPCR or qPCR/MCA assays with the same primers used for the target DNA ensures accurate control of the entire molecular assay and represents the state of the art for internal controls. It was shown that the addition of an IPC at levels resulting in 100 copies per PCR did not affect the amplification of the target sequence [52,53]. A competitive IPC compatible with the qPCR/MCA and TaqMan qPCR assays developed in this work is presented as Additional file 7.
Another level of diagnostic uncertainty in the assay developed for A. astaci detection [51] is added by the use of a synthetic amplicon mimicking one of the closest relatives, A. frigidophilus. This approach supposes the intragenomic homogeneity of the ITS regions which has already been rebutted in many organisms [54,55]. The addition of a minor-groove binder to a TaqMan probe in the assay reported by Vralstad et al. allows to use shorter probes. However, probe cost increases by about 2.5-fold compared to our conventional TaqMan qPCR designed for quantitative detection. It also elevates the chance of detection failure when varying genotypes are present. Generally, the avoidance of false negatives represents a major challenge in molecular diagnostics. Particularly, in TaqMan qPCR assays the possibility of false-negative testing poses a substantial problem because mutations within the probe-binding site can prevent annealing of the probe and subsequent detection [56,57]. For example, TaqMan qPCR failed to detect any target with more than two mutations at the probe-binding site in contrast to a dye-based assay [56]. The dilemma of false-negative detection due to probe-binding site variation can be overcome, for example, by combining a DNA probe with a fluorescent, double-stranded DNA-binding dye for specific nucleic acid quantification by probe-based qPCR and MCA [58]. In this case the dye would report a detection failure if the probe-binding site of a clinical specimen is mutated. However, "compensation" for mutations in the probe-binding site is no longer an issue if only two instead of three regions of conserved sequence are required for assay design as in the dye-based qPCR/MCA developed in this work. If very limited prior target sequence information exists from a population of interest like in our case, a dye-based detection approach represents a favourable strategy for species confirmation.
A welcomed side effect of dye- instead of probe-mediated monitoring is the cost reduction for screening clinical samples.
Last but not least, the reliability of the diagnostic assay was proven on a set of relevant related pathogens and during an acute crayfish-plague outbreak in the small, noble crayfish (Astacus astacus) population inhabiting the lake "Gleinkersee" located at an altitude of about 800 meters above sea level at the foothills of the Austrian Alps. In addition to qPCR/MCA typing (not shown), the presence of the pathogen A. astaci was independently confirmed by ITS-sequence analysis and testing for constitutive chitinase activity (A. astaci-strain GKS07 in Additional file 1). Finally, the A. astaci strain GKS07 was isolated on PG-1 agar from an infected noble crayfish. Numerous crayfish individuals were found to be affected but were still alive during the outbreak of late March 2007. At that time the ice of the lake Gleinkersee was melting and the physiological activities of both pathogen and victim would have been expected to be at a minimum. These circumstances strongly indicated the acuteness of the outbreak. The suspicion of a deliberate introduction of the pathogen could not be confirmed by an inquiry led by the local criminal investigation department. Fish stocking performed in autumn 2006 may be the most likely source of disease transmission.
Sensitive quantitative detection of the crayfish-plague pathogen is currently of increasing importance for screening natural non-native crayfish populations or for certifying a pathogen-free status of hatchery fish before introduction into natural habitats or aquaculture facilities. Samples of fish transport water including sediments can be filtered via membrane filters [59] and subsequently screened by TaqMan qPCR (see Results and Additional file 8). This circumvents pathogen transmission via transport water, fish faeces, mucus and scales.
Conclusion
The identification of two new chitinase genes showing specific patterns of constitutive temporal expression in the absence of substrate has facilitated the development of a discriminative, robust and reliable method for qualitative and quantitative detection for A. astaci.
Methods
Biological material
Isolates of Oomycetes and related fungi used to validate the molecular assays were either obtained from The Centraalbureau voor Schimmelcultures (CBS) Fungal Biodiversity Centre (Utrecht, The Netherlands), the German Collection of Microorganisms and Cell Cultures (DSMZ) (Braunschweig, Germany), the American Type Culture Collection (ATCC) or cultured from lesioned tissue by standard methods [60,61]. The A. astaci-types 1 to 4 were purchased from Lage Cerenius (Uppsala University, Uppsala, Sweden). Javier Diéguez-Uribeondo (Real Jardín Botánico CSIC, Madrid, Spain) provided the A. frigidophilus isolate SAP472 [29]. A DNA aliquot of A. frigidophilus NJM 9665 [6,62] and A. invadans WIC [6] was obtained from Mark W. Vandersea (Center for Coastal Fisheries and Habitat Research, National Ocean Service, National Oceanic Atmospheric Administration, Beaufort, North Carolina, USA).
The Austrian A. astaci strains Gb04, Z12, and the A. repetans strain Lk29 were isolated from dissected melanised spots found in the integument of signal crayfish [19]. The A. astaci strain GKS07 was grown out of a moribund noble crayfish collected during an acute crayfish-plague outbreak. Melanised necrobiopsies were incubated in peptone-glucose (PG1) medium (3 g/l glucose, 6 g/l peptone, 0.37 g/l KCl, 0.17 g/l MgCl2·6H2O, 0.15 g/l CaCl2·2H2O, 20 mg/l FeCl3·6H2O, 44 mg/l Na2EDTA, 13 mM sodium phosphate buffer (pH 6.3); [63]) for three days at 18°C [19] in a humidified chamber and subcultured every two weeks on PG1 agar medium. The same growth and subculturing conditions were applied to the strains obtained from the culture collections.
Fungal contamination of oomycete culture encountered when culturing the A. astaci strain Z12 and the A. repetans strain LK29 were overcome as follows. A piece of agar culture was incubated for one day at 20°C in autoclaved pond water (pH 6.5 to 7) collected at the central biotop of the University campus. This depletion of nutrients induced the sporulation of the oomycete [64]. Under an inverted microscope the swimm spores were aspired into a 100 μL Gilson pipette and re-cultured on PG1 agar medium.
A fungus isolated from horse food was assigned to Aspergillus sp. based on morphological evaluation and added to the strain collection of the Institute of Bacteriology, Mycology and Hygiene (University of Veterinary Medicine, Vienna).
An overview on the biological material used in this work is presented in Table 1.
Species assignment of Austrian Aphanomyces strains
ITS sequences of nuclear rDNA were analysed to allow species assignation of the Austrian A. astaci strains GB04, GKS07, and Z12 as well as of the A. repetans strain LK29 (Table 1, Additional file 1). For this purpose DNA was extracted from 25 mg drop culture mycelium using the DNeasy Tissue Kit (Qiagen, Hilden, Germany). A DNA fragment of about 1,000 bp was amplified and sequenced using the universal primers V9D (5'-TTACGTCCCTGCCCTTTGTA) [65] and LSU266 (5'-GCATTCCCAAACAACTCGACTC, [66]). Sequences obtained were compared with reference homologs of Aphanomyces [29] retrieved from GenBank. For sequence alignment the CodonCode Aligner software (version 3.0.1; CodonCode, Dedham, USA) was used. Molecular phylogenetic relationships were reconstructed using default settings in a program package for quartet-based maximum-likelihood analysis (TREE-PUZZLE, version 5.2 [67]) and TreeView for graphical illustration [68].
Additional evidence for species assignation was obtained from sequence analysis of the large subunit ribosomal RNA gene using the primers nuLSU-5' (5'-CGCTGATTTTTCCAAGCCC) and nuLSU-3' (5'-GAGATAGGGAGGAAGCCATGG) for amplification and sequencing.
Thus far A. astaci represents the only species within the genus Aphanomyces known to produce significant amounts of chitinase in chitin-free medium [18]. This unique feature was additionally used for species assignment. In detail, chitinase activity accumulated in broth culture supernatant was measured in a reaction volume of 100 μL containing 5 mM sodium-phosphate buffer (pH 7), 180 μM 4-methylumbelliferyl-β-D-N,N',N''-triacetylchitotrioside (4-MU-chitotrioside; Sigma-Aldrich, Vienna, Austria) as substrate, and 75 μl of supernatant [18]. Following incubation at room temperature for 10 min, the fluorescence intensity was evaluated under UV light.
DNA isolation from mycelium of oomycetes
The mycelium was transferred to a 2 ml-extraction tube containing 0.7 g Precellys® ceramic beads of 1.4 mm diameter (Peqlab Biotechnology, Erlangen, Germany) and 180 μl buffer ATL, the lysis buffer of the DNeasy® Blood & Tissue Kit (Qiagen, Hilden, Germany). Samples were homogenised twice for 15 s at 5000 rpm using the MagNA Lyser (Roche). Further isolation was performed according to the protocol "Purification of Total DNA from Animal Tissues (Spin-Column Protocol)" provided by the manufacturer.
De novo sequencing of partial GH domain using degenerate PCR primers
Partial GH18 domains of chitinases from various A. astaci strains representing all four genotype groups described (A: L1, Sv, Ra; B: Hö, Yx, Ti; C: Kv; D: Pc; [32]), the Austrian strain Gb04 isolated in this work and six related oomycete species (A. laevis, A. helicoides, A. repetans, A. irregularis, Saprolegnia parasitica, Achlya racemosa, Leptolegnia caudata (Table 1) were amplified using the primers SEQ685F (5'-CCGGAGACTCGTGGAACGAC) and SEQ1159R (5'-TTGCTCCAGCTGCCCGC). Primers targeting the amino acid motifs DSWND and AGSW, respectively, amplified an approximately 475-bp product by qPCR. The 20-μL reaction consisted of 0.4 × EvaGreen™ dye (Biotium, Hayward, USA), 4 mM MgCl2, 200 μM of each dNTP, 375 nM of each primer, 2 μl template DNA, 1 U GoTaq® DNA polymerase - a proprietary formulation of Taq DNA polymerase (Promega, Madison, USA), and 1 × Colorless GoTaq® Flexi Reaction Buffer (Promega) not containing magnesium. Amplification was performed in the Rotor-Gene 6000 (Corbett Life Science, Sydney, Australia) using denaturation for 4 min at 94°C, amplification for 35 cycles (1 min at 94°C, 1 min at 63°C and 1 min at 72°C), and final elongation of 7 min at 72°C followed by MCA.
Amplicons from Fusarium solani and Trichosporon cutaneum, representing fungi, were obtained with the degenerate primer SEQuni-F (5'-CGCCGGAGAYTCTTGGAAYGA, Y = C or T) in combination with the primer SEQuni-R (5'-CCAGCATAGTCGTAGGCCAT) targeting the amino acid motifs xxDSWND and MTDYAG, respectively.
Agarose gel electrophoresis was used to the determine amplicon size. The MSB® Spin PCRapace Kit (Invitek, Berlin, Germany) was used for amplicon purification in case of a single band showing the expected length. Multiple bands were excised from the gel and purified with the Xact DNA Cleanup kit (genXpress, Wiener Neudorf, Austria).
The BigDye® Terminator sequencing chemistry (Applied Biosystems, Foster City, USA) was used for sequence analysis of amplicons performed at VBC Genomics Bioscience Research GmbH (Vienna, Austria).
Identification and phylogenetic analysis of GH18 domains
The GH18 domain in the amino acid sequences of CHI2 and CHI3 were identified using the Reversed Position Specific Blast (rpsblast) search modus and the conserved domain database [69]. Domain sequences were aligned to GH18 domain sequences of related species with the ClustalW alignment program implemented in the graphical multiple sequence alignment editor SeaView version 4 [70]. Quartet-based maximum likelihood analysis for aligned amino acid sequences was performed using TreePuzzle with default settings [67]. The graphical display of the phylogram was generated as described above.
Western blot analysis of A. astaci culture supernatant
The peptides DEFKTLPWKAE and LYEDPNHPPGAKY were selected from the deduced amino acid sequence of the A. astaci gene CHI1 (GenBank:AJ416354). Conjugates of these peptides with bovine serum albumin (BSA) were obtained from PSL GmbH (Heidelberg, Germany). Coupling to BSA was achieved via the SH group of a cysteine residue introduced at the C terminus of the peptide to be synthesised. Conjugates were used for the production of polyclonal rabbit serum antibodies served as primary antibodies. Peroxidase-labelled goat anti-rabbit IgG antibodies (K&P Laboratories, Gaithersburg, USA) were used as secondary antibodies.
Western-immunoblot analysis was performed as follows. The A. astaci strain Hö was grown in broth culure. The culture supernatant was boiled for 5 min in a buffer consisting of 25 mM Tris-HCl (pH 6.8), 2.2% sodium dodecyl sulfate (SDS), 15% glycerol and 0.001% bromophenol blue. Insoluble debris was removed by centrifugation. Proteins were resolved by SDS-polyacrylamide gel electrophoresis on a 12% polyacrylamide Tris-glycine gel and electroblotted onto a polyvinylidene difluoride (PVDF) membrane (Bio-Rad Laboratories, Hercules, USA) using a tank blot system (Bio-Rad). The Opti-4CN™ substrate detection kit (Bio-Rad) was used for colorimetric detection of secondary antibodies conjugated to horseradish peroxidase.
Determination of complete cDNA- and genomic-DNA sequences for CHI2 and CHI3
Mycelium derived from the A. astaci-strain Gb04 was grown in liquid PG1 medium for three days and transferred to fresh medium for another 24 h. Total RNA was isolated from mycelium using the Plant and Fungi Protocol provided with the RNeasy Plant Mini Kit (Qiagen). Treatment with DNase I (Promega, Mannheim, Germany) was performed at 37°C for 40 min according to the supplier's instructions. The complete cDNA sequences of CHI2 and CHI3 were generated by RACE-PCR using the 5'/3' RACE Kit (Roche Applied Science, Vienna, Austria).
To amplify genomic sequences corresponding to the cDNAs determined, we designed primers in the region of the start and stop codons of CHI2 and CHI3. The common forward primer (Chi5'f: 5'-AGCAAACTGCAACAAGCATG) targeting a region immediately upstream of the start codon of putative CHI2 and CHI3 genomic sequences, was combined with a gene-specific reverse primer binding adjacent to the stop codon (Chi2.3'r: 5'-GGGCACCAGATGAACGACGC or Chi3.3'r: 5'-ACTAACATACACAACGAATGCGC for CHI2 and CHI3, respectively). The matching fragment size between cDNA and respective DNA sequences shown by agarose gel electrophoresis, and the identity of genomic and cDNA sequences identified by a primer-walking strategy (data not shown), were considered as experimental demonstration for the absence of intronic sequences within CHI2 and CHI3 genes.
In silico analysis of amino acid sequences deduced from CHI2 and CHI3
Multiple matching subsegments in two protein sequences were identified with the LALIGN program http://www.ch.embnet.org/software/LALIGN_form.html implementing the algorithm of Huang & Miller [71].
The theoretical isoelectric points for the protein sequences were calculated using the Protein Isoelectric Point menu within the Sequence Manipulation Suite [72].
The presence and location of signal peptide cleavage sites in the amino acid sequences of CHI2 and CHI3 were predicted with the SignalP 3.0 Server http://www.cbs.dtu.dk/services/SignalP;[73]). Protein phosphorylation at serine, threonine or tyrosine residues was predicted with the NetPhos 2.0 Server [74]. Putative sites for amidation, N-myristoylation and cell attachment were identified by a protein pattern search against the Prosite database http://www.expasy.org/prosite/; [75]). O-, N-, and C-glycosylated sites were predicted with EnsembleGly - a web server for prediction of O-, N-, and C-linked glycosylation sites with ensemble learning [39].
Transcript quantification by real-time reverse transcription PCR (qRT-PCR)
Propagules of the strain Gb04 were grown in PG1 medium for three days, washed in fresh medium for 2 min and transferred to another portion of fresh medium (time point 0). Twelve, 24, 36, 48 or 72 hours later the mycelium was shortly washed with distilled water, quick-frozen in liquid nitrogen and stored at -80°C. RNA was isolated from three independent samples grown per time point.
For quantification of transcript mass expressed from the chitinase genes CHI2 and CHI3 as well as the endogenous positive control NDUFV1, sense strand transcript standards were generated by in vitro transcription from a PCR product template tailed with the T7 phage promoter sequence. In more detail, for template construction a minimum sequence of 19 bases (5'-TAATACGACTCACTATAGG) required for efficient transcription was selected out of the 23 nt T7 phage promoter sequence and added to the 5' end of the respective PCR primer. In vitro transcription was performed with the RNAMaxx™ High Yield Transcription Kit (Stratagene, Amsterdam, The Netherlands) according to the manufacturer's instructions. Transcription was terminated by adding 1 μl DNase I (10 units/μl RQ1 RNase-Free DNase, Promega) and incubation at 37°C for 40 minutes. The amount of the in vitro transcript was determined by UV-absorbance measurement performed at 260 nm on a GeneQuantII RNA/DNA Calculator (Pharmacia Biotech, Cambridge, UK). Ten-fold serial dilutions were used as absolute concentration standards.
The 10-μl one-step qRT-PCR contained 125 nM of each primer (5'-CCATCACGAACCCCCTTGAG and 5'-GGGCACCAGATGAACGACG for CHI2, 5'-GTGGCCCCATCACGAACC and 5'-ACTAACATACACAACGAATGCGC for CHI3, 5'-TCGGCTGTCGCACTTCTACA and 5'-ATCCACCCCGTTCCTTCG for NDUV1), 75 nM TaqMan probe (Hexachloro-6-carboxyfluorescein (HEX)-5'-CTGCGGCCAATGTACCCCTTGCC black-hole quencher 1 (BHQ1) and 6-carboxyfluorescein (FAM)-5'-TTGTTGCCCTTGCACTGGTCGCC-BHQ1 for NDUV1 and CHI2/CHI3, respectively), 0.1 μl of the QuantiTect RT Mix, 5 μl of the 2 × QuantiTect Probe PCR Master Mix (Qiagen) and 50 ng total RNA or 1 μL in vitro transcript. In minus RT controls the QuantiTect RT Mix was replaced by water. Reverse transcription of one-step RT-PCR was conducted at 50°C for 30 min followed by a 15 min-activation of the HotStartTaq DNA polymerase at 95°C and amplification for 35 cycles (94°C for 20 s, 60°C for 1 min).
Qualitative detection of A. astaci using qPCR/MCA
The 20-μl duplex qPCR/MCA contained 2 μl 10 × PCR buffer B (Solis BioDyne, Tartu, Estonia), 200 nM of forward and reverse chitinase gene(s) primers (5'-TCAAGCAAAAGCAAAAGGCT and 5'-CCGTGCTCGCGATGGA), 125 nM of forward and reverse 5.8S rRNA primers (5'-ATACAACTTTCAACAGTGGATGTCT and 5'-ATTCTGCAATTCGCATTACG, Figure 6a), 200 μM of each dNTP (Fermentas, St. Leon-Rot, Germany), 0.4 × EvaGreen™ (Biotium), 3.0 mM MgCl2, 1 U Taq DNA polymerase chemically modified for "hot start" (Hot FirePol®; Solis BioDyne, Tartu, Estonia) and 10 ng DNA template or water in the case of the no-template control. QPCR/MCA was performed on the StepOnePlus™ Real-Time PCR System (Applied Biosystems) run under the StepOne™ software version 2.0. Polymerase activation (95°C for 15 min) was followed by amplification for 35 cycles (95°C for 15 s, 59°C for 15 s and 72°C for 10 s). After an initial denaturation step at 95°C for 15 s, amplicon melting was recorded during a gradual increase of the temperature from 60°C to 95°C.
Oligonucleotides (Sigma-Aldrich, Steinheim, Germany) were designed with Primer Express Software Version 2.0 (Applied Biosystems). The difference between amplicon melting temperatures was calculated using the Nearest Neighbor mode implemented in the online oligonucleotide properties calculator OligoCalc [76].
Sensitive detection and quantification of A. astaci using TaqMan qPCR
Duplicate TaqMan qPCR was carried out in a total volume of 20 μl containing 2 μl 10 × PCR buffer A2 (Solis BioDyne), 0.2 mM of each dNTP, 4 mM MgCl2, 300 nM of each primer (Chi3-324f20 and AaChi-Tmr), 150 nM TaqMan probe (AaChi-FAM), 1 U HOT FIREPol DNA polymerase (Solis BioDyne), 20 ng template DNA or water in the case of the no-template control.
Reactions were amplified in the StepOnePlus™ Real-Time PCR System (Applied Biosystems) under the StepOne™ software version 2.0 using thermal cycling conditions of 15 min at 95°C, followed by 50 cycles of 15 s at 95°C and 1 min at 64°C. A standard curve was generated by plotting the logarithm of the standards copy numbers versus measured CT values.
Isolation of spike-in DNA for use in serial dilutions
A crayfish sample extracted from the abdomen of Cherax quadricarinatus (Australian red-claw crayfish) was transferred to a 2 ml-extraction tube containing 0.7 g Precellys® ceramic beads of 1.4 mm diameter (Peqlab Biotechnology, Erlangen, Germany) and 180 μl buffer ATL, the lysis buffer of the DNeasy® Blood & Tissue Kit (Qiagen). The MagNA Lyser (Roche) was used for three mechanical lysis cycles consisting of 30 s at 6,500 rpm followed by 60 s on a cooling block held at 4°C. Further isolation was performed according to the protocol "Purification of Total DNA from Animal Tissues (Spin-Column Protocol)" provided by the manufacturer. DNA concentration was determined spectrophotometrically using the Hellma® TrayCell (Hellma, Müllheim/Baden, Germany) on the Eppendorf BioPhotometer 6131.
Generation of copy standards
A DNA template stock consisting of CHI1, CHI2 and CHI3 sequences was generated as follows. Genomic DNA from chitinase sequences were amplified with the primers Chi3-324f20 (5'-TCAAGCAAAAGCAAAAGGCT) and AaChi-Tmr (5'-TCCGTGCTCGCGATGGA). Amplification was evaluated by the signal generated from the TaqMan® probe AaChi-FAM (5'-FAM-TCAACGTCCACCCGCCAATGG-BHQ-1). Amplification was performed in a total volume of 20 μl containing 2 μl 10 × PCR buffer A2 (Solis BioDyne), 0.2 mM of each dNTP, 4 mM MgCl2, 250 nM of each primer, 150 nM TaqMan probe, 1 U HOT FIREPol® DNA polymerase (Solis BioDyne) and 20 ng DNA or water in the case of the no-template control. DNA denaturation and enzyme activation were performed for 15 min at 95°C. DNA was amplified over 50 cycles consisting of 95°C for 15 s, 60°C for 1 min. QPCR was run on StepOnePlus™ Real-Time PCR System (Applied Biosystems) under the StepOne™ software version 2.0.
PCR fragments were purified with the MSB® Spin PCRapace Kit (Invitek, Berlin, Germany).
The copy number of the target template was determined spectrophotometrically using the Hellma® TrayCell (Hellma, Müllheim/Baden, Germany) on the Eppendorf BioPhotometer 6131. Serial dilutions of the target sequence (108 to 102, 50, 25 and 12.5 copies per 2 μl) prepared in 10 ng/μl C. quadricarinatus DNA were used to determine the amplification efficiency and the quantitative detection limit.
Statistical analysis of expression changes
A univariate one-way analysis of variance (ANOVA) with Scheffè's post-hoc test was used to evaluate the significance of changes in temporal mRNA expression. The dependent variable was the log-transformed mRNA amount. The time was considered a fixed effect. A value of p < 0.05 calculated by the Scheffè's post-hoc test was regarded as significant. The normality assumption was tested using the Kolmogoroff-Smirnow test.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
GH, RB-R and RS conceived and designed the experiments. RB-R performed Western blot analysis, GH, RT and RS provided the diagnostic assays. GH performed all other experiments. RS supervised the experimental work and the interpretation of data and planned the manuscript. EL provided funding. GH and RS wrote the paper. All authors analysed the data, commented on and approved the manuscript.
Supplementary Material
Species identification of Austrian A. astaci strains Gb04, Z12, and GKS07 based on phylogenetic analysis and constitutive chitinase activity in substrate-free medium. ITS sequence and chitinase expression in chitin-free medium are criteria to classify a strain as A. astaci
Sequences of 3' untranslated regions (UTRs) of CHI2 and CHI3 mRNAs. Alignment shows differences between 3' UTRs of CHI2 and CHI3 mRNAs
Amino-acid substitutions in the GH18 catalytic site of oomycete species. Table lists amino-acid substitutions in the GH18 catalytic site of oomycete species
O-linked glycosylation and phosphorylation predicted for Chi2 and Chi3. Predicted O-linked glycosylations and phosporylations at serine and threonine residues for Chi2 and Chi3 are listed in a table
Alignment of primer target sites for the 5.8S rRNA gene used as endogenous control in qPCR/MCA. Primers target conserved sites in the 5.8S rRNA gene of various oomycete species
A conventional PCR assay for detection of A. astaci that may fail to discriminate between closely related species. Alignment of primer sites for a conventional PCR assay reported for detection of A. astaci
Design of a homologous IPC for use in the qPCR/MCA or qPCR assays. The IPC monitored by a characteristic melting temperature or by an alternatively labeled hydrolysis probe in the qPCR/MCA or qPCR assays, respectively, helps to prevent false-negative detection due to insufficient extraction and/or amplification.
TaqMan qPCR assay design for sensitive detection and quantification of A. astaci. Primers, but also TaqMan probe facilitate discrimination between A. astaci and various related or relevant oomycete species.
Contributor Information
Gerald Hochwimmer, Email: Gerald.Hochwimmer@fischfarm.co.at.
Reinhard Tober, Email: Reinhard.Tober@vetmeduni.ac.at.
Renè Bibars-Reiter, Email: Bibars-Reiter@gmx.at.
Elisabeth Licek, Email: Elisabeth.Licek@vetmeduni.ac.at.
Ralf Steinborn, Email: Ralf.Steinborn@vetmeduni.ac.at.
Acknowledgements
We are very grateful to Javier Diéguez-Uribeondo and Mark W. Vandersea for providing samples of closely related Aphanomyces strains. We thank Christian Holler, Gerhard Woschitz, Stefan Magg, Rudolf Lengauer, Hannes Hager and Reinhard Pekny for providing crayfish samples. Georg Mair, Joachim Spergser, Gunther Vogl, Klaus Kotschy, Claus Vogl, Renate Rosengarten, Fritz Pittner and Michael Hess are acknowledged for support. We are indebted to Steve Weiss for comments on the manuscript. This project was supported by the Austrian Federal Ministry of Agriculture, Forestry, Environment and Water Management (grant no. 1362 to EL).
Role of the Sponsor
The funding organisation had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript.
References
- Lamour KH, Win J, Kamoun S. Oomycete genomics: new insights and future directions. FEMS Microbiol Lett. 2007;274(1):1–8. doi: 10.1111/j.1574-6968.2007.00786.x. [DOI] [PubMed] [Google Scholar]
- Judelson HS. The genetics and biology of Phytophthora infestans: Modern approaches to a historical challenge. Fung Genet Biol. 1997;22(2):65–76. doi: 10.1006/fgbi.1997.1006. [DOI] [PubMed] [Google Scholar]
- Tyler BM. Genetics and genomics of the oomycete host interface. Trends Genet. 2001;17(11):611–614. doi: 10.1016/S0168-9525(01)02517-3. [DOI] [PubMed] [Google Scholar]
- Gaulin E, Madoui MA, Bottin A, Jacquet C, Mathe C, Couloux A, Wincker P, Dumas B. Transcriptome of Aphanomyces euteiches: New Oomycete putative pathogenicity factors and metabolic pathways. PLoS One. 2008;3(3) doi: 10.1371/journal.pone.0001723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerenius L, Söderhäll K, Persson M, Ajaxon R. The crayfish plague fungus Aphanomyces astaci - diagnosis, isolation and pathobiology. Freshw Crayfish. 1988;7:131–144. [Google Scholar]
- Vandersea MW, Litaker RW, Yonnish B, Sosa E, Landsberg JH, Pullinger C, Moon-Butzin P, Green J, Morris JA, Kator H, Noga EJ, Tester PA. Molecular assays for detecting Aphanomyces invadans in ulcerative mycotic fish lesions. Appl Environ Microbiol. 2006;72(2):1551–1557. doi: 10.1128/AEM.72.2.1551-1557.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerenius L, Söderhäll K. In: Advances in Zoosporic Fungi. Dayal R, editor. New Delhi: M D Publications Pvd Ltd; 1996. Saprolegniaceae: zoospore formation, virulence and pathogenesis in animal hosts; pp. 97–116. [Google Scholar]
- Mendoza L, Hernandez F, Ajello L. Life cycle of the human and animal oomycete pathogen Pythium insidiosum. J Clin Microbiol. 1993;31(11):2967–2973. doi: 10.1128/jcm.31.11.2967-2973.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schikora F. Die Krebspest. Fischerei-Zeitung. 1906;9:529. [Google Scholar]
- Alderman DJ. Geographical spread of bacterial and fungal diseases of crustaceans. Rev Sci Tech. 1996;15(2):603–632. doi: 10.20506/rst.15.2.943. [DOI] [PubMed] [Google Scholar]
- Kozubíková E, Petrusek A, Duris Z, Martín MP, Diéguez-Uribeondo J, Oidtmann B. The old menace is back: Recent crayfish plague outbreaks in the Czech Republic. Aquaculture. 2008;274(2-4):208–217. doi: 10.1016/j.aquaculture.2007.11.015. [DOI] [Google Scholar]
- Baillie J, Groombridge B. 1996 IUCN Red List of Threatened Animals. Gland, Switzerland: The World Conservation Union (IUCN), Species Survival Commission (SSC); 1996. [Google Scholar]
- Skurdal J, Taugbol T, Tuusti J. Crayfish in Europe an Alien Species. How to Make the Best of a Bad Situation? Rotterdam, Netherlands: A. A. Balkema; 1999. Crayfish introductions in the Nordic and Baltic countries; pp. 193–219. [Google Scholar]
- Westman K, Pursiainen M, Westman P. Status of crayfish stocks, fisheries, diseases and culture in Europe. Finnish Game and Fisheries Research Institute, Report No. 3, Helsinki, Finland; 1990. [Google Scholar]
- Oidtmann B, Bausewein S, Holzle L, Hoffmann R, Wittenbrink M. Identification of the crayfish plague fungus Aphanomyces astaci by polymerase chain reaction and restriction enzyme analysis. Vet Microbiol. 2002;85(2):183–194. doi: 10.1016/S0378-1135(01)00505-3. [DOI] [PubMed] [Google Scholar]
- Hall L, Unestam T. The effect of fungicides on survival of the crayfish plague fungus, Aphanomyces astaci, Oomycetes, growing on fish scales. Mycopathologia. 1980;72(3):131–134. doi: 10.1007/BF00572654. [DOI] [PubMed] [Google Scholar]
- Cerenius L, Söderhäll K. Chemotaxis in Aphanomyces astaci, an Arthropod-Parasitic Fungus. J Invertebr Pathol. 1984;43(2):278–281. doi: 10.1016/0022-2011(84)90150-2. [DOI] [Google Scholar]
- Andersson MG, Cerenius L. Analysis of chitinase expression in the crayfish plague fungus Aphanomyces astaci. Dis Aquat Organ. 2002;51(2):139–147. doi: 10.3354/dao051139. [DOI] [PubMed] [Google Scholar]
- Oidtmann B, Schmid I, Rogers D, Hoffmann RW. An improved isolation method for the cultivation of crayfish plague fungus, Aphanomyces astaci. Freshw Crayfish. 1999;12:303–312. [Google Scholar]
- Nyhlén L, Unestam T. Wound reactions and Aphanomyces astaci growth in crayfish cuticle. J Invertebr Pathol. 1980;36(2):187–197. doi: 10.1016/0022-2011(80)90023-3. [DOI] [Google Scholar]
- Svensson E. Interactions between a parasitic fungus, Aphanomyces astaci, Oomycetes, and its crayfish host: I. Motility, encystment, attachment, and germination of the zoospore. Acta Univ Ups. 1978;457:1–18. [Google Scholar]
- Söderhäll K, Unestam T. Properties of extracellular enzymes from Aphanomyces astaci and their relevance in the penetration process of crayfish cuticle. Physiol Plant. 1975;35:140–146. doi: 10.1111/j.1399-3054.1975.tb03883.x. [DOI] [Google Scholar]
- Nyhlen L, Unestam T. Ultrastructure of the crayfish integument by the fungal parasite, Aphanomyces astaci, Oomycetes. J Invertebr Pathol. 1975;26(3):353–366. doi: 10.1016/0022-2011(75)90233-5. [DOI] [Google Scholar]
- Chernin L, Ismailov Z, Haran S, Chet I. Chitinolytic Enterobacter agglomerans antagonistic to fungal plant pathogens. Appl Environ Microbiol. 1995;61(5):1720–1726. doi: 10.1128/aem.61.5.1720-1726.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tews I, Vincentelli R, Vorgias CE. N-Acetylglucosaminidase (chitobiase) from Serratia marcescens: gene sequence, and protein production and purification in Escherichia coli. Gene. 1996;170(1):63–67. doi: 10.1016/0378-1119(95)00848-9. [DOI] [PubMed] [Google Scholar]
- Ballesteros I, Martín MP, Diéguez-Uribeondo J. First isolation of Aphanomyces frigidophilus (Saprolegniales) in Europe. Mycotaxon. 2006;95:335–340. [Google Scholar]
- Karlsson M, Stenlid J. Comparative evolutionary histories of the fungal chitinase gene family reveal non-random size expansions and contractions due to adaptive natural selection. Evol Bioinform. 2008;4:47–60. doi: 10.4137/ebo.s604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl V, Huemer B, Seiboth B, Kubicek CP. A complete survey of Trichoderma chitinases reveals three distinct subgroups of family 18 chitinases. FEBS J. 2005;272(22):5923–5939. doi: 10.1111/j.1742-4658.2005.04994.x. [DOI] [PubMed] [Google Scholar]
- Dieguez-Uribeondo J, Garcia MA, Cerenius L, Kozubikova E, Ballesteros I, Windels C, Weiland J, Kator H, Soderhall K, Martin MP. Phylogenetic relationships among plant and animal parasites, and saprotrophs in Aphanomyces (Oomycetes) Fungal Genet Biol. 2009;46(5):365–376. doi: 10.1016/j.fgb.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Royo F, Andersson G, Bangyeekhun E, Muzquiz JL, Soderhall K, Cerenius L. Physiological and genetic characterisation of some new Aphanomyces strains isolated from freshwater crayfish. Vet Microbiol. 2004;104(1-2):103–112. doi: 10.1016/j.vetmic.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Funkhouser JD, Aronson NN Jr. Chitinase family GH18: evolutionary insights from the genomic history of a diverse protein family. BMC Evol Biol. 2007;7:96. doi: 10.1186/1471-2148-7-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T, Cerenius L, Söderhäll K. Analysis of genetic diversity in the crayfish plague fungus, Aphanomyces astaci, by random amplification of polymorphic DNA. Aquaculture. 1994;126(1-2):1–10. doi: 10.1016/0044-8486(94)90243-7. [DOI] [Google Scholar]
- Kamoun S. Molecular genetics of pathogenic oomycetes. Eukaryot Cell. 2003;2(2):191–199. doi: 10.1128/EC.2.2.191-199.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torto-Alalibo T, Tian M, Gajendran K, Waugh ME, van West P, Kamoun S. Expressed sequence tags from the oomycete fish pathogen Saprolegnia parasitica reveal putative virulence factors. BMC Microbiol. 2005;5:46. doi: 10.1186/1471-2180-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Win J, Kanneganti TD, Torto-Alalibo T, Kamoun S. Computational and comparative analyses of 150 full-length cDNA sequences from the oomycete plant pathogen Phytophthora infestans. Fungal Genet Biol. 2006;43(1):20–33. doi: 10.1016/j.fgb.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Shen Z, Jacobs-Lorena M. Characterization of a novel gut-specific chitinase gene from the human malaria vector Anopheles gambiae. J Biol Chem. 1997;272(14):28895–28900. doi: 10.1074/jbc.272.46.28895. [DOI] [PubMed] [Google Scholar]
- Liu ZH, Yang Q, Hu S, Zhang JD, Ma J. Cloning and characterization of a novel chitinase gene (chi46) from Chaetomium globosum and identification of its biological activity. Appl Microbiol Biotechnol. 2008;80(2):241–252. doi: 10.1007/s00253-008-1543-x. [DOI] [PubMed] [Google Scholar]
- Kramer KJ, Muthukrishnan S. Insect chitinases: molecular biology and potential use as biopesticides. Insect Biochem Mol Biol. 1997;27(11):887–900. doi: 10.1016/S0965-1748(97)00078-7. [DOI] [PubMed] [Google Scholar]
- Caragea C, Sinapov J, Silvescu A, Dobbs D, Honavar V. Glycosylation site prediction using ensembles of Support Vector Machine classifiers. BMC Bioinformatics. 2007;8:438. doi: 10.1186/1471-2105-8-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unestam T. Chitinolytic, cellulolytic, and pectinolytic activity in vitro of some parasitic and saprophytic oomycetes. Physiol Plant. 1966;19(1):15–30. doi: 10.1111/j.1399-3054.1966.tb09070.x. [DOI] [Google Scholar]
- Wang J, Chuang K, Ahluwalia M, Patel S, Umblas N, Mirel D, Higuchi R, Germer S. High-throughput SNP genotyping by single-tube PCR with Tm-shift primers. Biotechniques. 2005;39(6):885–893. doi: 10.2144/000112028. [DOI] [PubMed] [Google Scholar]
- Chang HW, Cheng CA, Gu DL, Chang CC, Su SH, Wen CH, Chou YC, Chou TC, Yao CT, Tsai CL, Cheng CC. High-throughput avian molecular sexing by SYBR green-based real-time PCR combined with melting curve analysis. BMC Biotechnol. 2008;8:12. doi: 10.1186/1472-6750-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorente A, Mueller W, Urdangarin E, Lazcoz P, von Deimling A, Castresana JS. Detection of methylation in promoter sequences by melting curve analysis-based semiquantitative real time PCR. BMC Cancer. 2008;8:61. doi: 10.1186/1471-2407-8-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvapandiyan A, Stabler K, Ansari NA, Kerby S, Riemenschneider J, Salotra P, Duncan R, Nakhasi HL. A novel semiquantitative fluorescence-based multiplex polymerase chain reaction assay for rapid simultaneous detection of bacterial and parasitic pathogens from blood. J Mol Diagn. 2005;7(2):268–275. doi: 10.1016/S1525-1578(10)60554-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibellini D, Gardini F, Vitone F, Schiavone P, Furlini G, Re MC. Simultaneous detection of HCV and HIV-1 by SYBR Green real time multiplex RT-PCR technique in plasma samples. Mol Cell Probes. 2006;20(3-4):223–229. doi: 10.1016/j.mcp.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Guion CE, Ochoa TJ, Walker CM, Barletta F, Cleary TG. Detection of diarrheagenic Escherichia coli by use of melting-curve analysis and real-time multiplex PCR. J Clin Microbiol. 2008;46(5):1752–1757. doi: 10.1128/JCM.02341-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros I, Martín MP, Cerenius L, Söderhäll K, Tellería MT, Diéguez-Uribeondo J. Lack of specificity of the molecular diagnostic method for identification of Aphanomyces astaci. Bull Fr Pêche Piscic. 2007;385:17–24. doi: 10.1051/kmae:2007002. [DOI] [Google Scholar]
- Kozubíková E, Filipova L, Kozák P, Duris Z, Martín MP, Diéguez-Uribeondo J, Oidtmann B, Petrusek A. Prevalence of the crayfish plague pathogen Aphanomyces astaci in invasive American crayfishes in the Czech Republic. Conserv Biol. 2009. in press . [DOI] [PubMed]
- Van Doninck K, Schon I, Martens K, Backeljau T. Clonal diversity in the ancient asexual ostracod Darwinula stevensoni assessed by RAPD-PCR. Heredity. 2004;93(2):154–160. doi: 10.1038/sj.hdy.6800486. [DOI] [PubMed] [Google Scholar]
- Drouin G, de Sa MM. The concerted evolution of 5S ribosomal genes linked to the repeat units of other multigene families. Mol Biol Evol. 1995;12(3):481–493. doi: 10.1093/oxfordjournals.molbev.a040223. [DOI] [PubMed] [Google Scholar]
- Vralstad T, Knutsen AK, Tengs T, Holst-Jensen A. A quantitative TaqMan MGB real-time polymerase chain reaction based assay for detection of the causative agent of crayfish plague Aphanomyces astaci. Vet Microbiol. 2009;137(1-2):146–155. doi: 10.1016/j.vetmic.2008.12.022. [DOI] [PubMed] [Google Scholar]
- Betsou F, Beaumont K, Sueur JM, Orfila J. Construction and evaluation of internal control DNA for PCR amplification of Chlamydia trachomatis DNA from urine samples. J Clin Microbiol. 2003;41(3):1274–1276. doi: 10.1128/JCM.41.3.1274-1276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory JB, Litaker RW, Noble RT. Rapid one-step quantitative reverse transcriptase PCR assay with competitive internal positive control for detection of enteroviruses in environmental samples. Appl Environ Microbiol. 2006;72(6):3960–3967. doi: 10.1128/AEM.02291-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bart A, Heijden HM van der, Greve S, Speijer D, Landman WJ, van Gool T. Intragenomic variation in the internal transcribed spacer 1 region of Dientamoeba fragilis as a molecular epidemiological marker. J Clin Microbiol. 2008;46(10):3270–3275. doi: 10.1128/JCM.00680-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worheide G, Nichols SA, Goldberg J. Intragenomic variation of the rDNA internal transcribed spacers in sponges (phylum Porifera): implications for phylogenetic studies. Mol Phylogenet Evol. 2004;33(3):816–830. doi: 10.1016/j.ympev.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Papin JF, Vahrson W, Dittmer DP. SYBR Green-based real-time quantitative PCR assay for detection of West Nile virus circumvents false-negative results due to strain variability. J Clin Microbiol. 2004;42(4):1511–1518. doi: 10.1128/JCM.42.4.1511-1518.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson TP, Werno AM, Beynon KA, Murdoch DR. Failure to genotype herpes simplex virus by real-time PCR assay and melting curve analysis due to sequence variation within probe binding sites. J Clin Microbiol. 2003;41(5):2135–2137. doi: 10.1128/JCM.41.5.2135-2137.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind K, Ståhlberg A, Zoric N, Kubista M. Combining sequence-specific probes and DNA binding dyes in real-time PCR for specific nucleic acid quantification and melting curve analysis. Biotechniques. 2006;40(3):315–319. doi: 10.2144/000112101. [DOI] [PubMed] [Google Scholar]
- Reischer GH, Kasper DC, Steinborn R, Mach RL, Farnleitner AH. Quantitative PCR method for sensitive detection of ruminant fecal pollution in freshwater and evaluation of this method in alpine karstic regions. Appl Environ Microbiol. 2006;72(8):5610–5614. doi: 10.1128/AEM.00364-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazer VS, Vogelbein WK, Densmore CL, May EB, Lilley JH, Zwerner DE. Aphanomyces as a cause of ulcerative skin lesions of menhaden from chesapeake bay tributaries. J Aquat Anim Health. 1999;11(4):340–349. doi: 10.1577/1548-8667(1999)011<0340:AAACOU>2.0.CO;2. [DOI] [Google Scholar]
- Willoughby LG, Roberts RJ. Improved methodology for isolation of the Aphanomyces fungal pathogen of epizootic ulcerative syndrome (EUS) in Asian fish. J Fish Dis. 1994;17(5):541–543. doi: 10.1111/j.1365-2761.1994.tb00250.x. [DOI] [Google Scholar]
- Kitancharoen N, Hatai K. Some biochemical characteristics of fungi isolated from salmonid eggs. Mycoscience. 1998;39(3):249–255. doi: 10.1007/BF02464005. [DOI] [Google Scholar]
- Lilley JH, Hart D, Panyawachira V, Kanchanakhan S, Chinabut S, Söderhäll K, Cerenius L. Molecular characterization of the fish-pathogenic fungus Aphanomyces invadans. J Fish Dis. 2003;26(5):263–275. doi: 10.1046/j.1365-2761.2003.00448.x. [DOI] [PubMed] [Google Scholar]
- Beakes GW. In: The Growing Fungus. Gow NAR, Gadd GM, editor. London: Chapman & Hall; 1995. Sporulation of lower fungi. [Google Scholar]
- de Hoog GS, Gerrits van den Ende AH. Molecular diagnostics of clinical strains of filamentous Basidiomycetes. Mycoses. 1998;41(5-6):183–189. doi: 10.1111/j.1439-0507.1998.tb00321.x. [DOI] [PubMed] [Google Scholar]
- Masclaux F, Gueho E, de Hoog GS, Christen R. Phylogenetic relationships of human-pathogenic Cladosporium (Xylohypha) species inferred from partial LS rRNA sequences. J Med Vet Mycol. 1995;33(5):327–338. doi: 10.1080/02681219580000651. [DOI] [PubMed] [Google Scholar]
- Schmidt HA, Strimmer K, Vingron M, von Haeseler A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18(3):502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- Page RDM. TREEVIEW: An application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996;12(4):357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson JB, Derbyshire MK, DeWeese-Scott C, Gonzales NR, Gwadz M, Hao L, He S, Hurwitz DI, Jackson JD, Ke Z, Krylov D, Lanczycki CJ, Liebert CA, Liu C, Lu F, Lu S, Marchler GH, Mullokandov M, Song JS, Thanki N, Yamashita RA, Yin JJ, Zhang D, Bryant SH. CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res. 2007. pp. D237–240. [DOI] [PMC free article] [PubMed]
- Galtier N, Gouy M, Gautier C. SEA VIEW and PHYLO_WIN Two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci. 1996;12(6):543–548. doi: 10.1093/bioinformatics/12.6.543. [DOI] [PubMed] [Google Scholar]
- Huang X, Miller W. A time-efficient, linear-space local similarity algorithm. Adv Appl Math. 1991;12(3):337–357. doi: 10.1016/0196-8858(91)90017-D. [DOI] [Google Scholar]
- Stothard P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques. 2000;28(6):1102–1104. doi: 10.2144/00286ir01. [DOI] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340(4):783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294(5):1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- Hulo N, Bairoch A, Bulliard V, Cerutti L, Cuche BA, de Castro E, Lachaize C, Langendijk-Genevaux PS, Sigrist CJ. The 20 years of PROSITE. Nucleic Acids Res. 2008. pp. D245–249. [DOI] [PMC free article] [PubMed]
- Kibbe WA. OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Res. 2007. pp. W43–46. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Species identification of Austrian A. astaci strains Gb04, Z12, and GKS07 based on phylogenetic analysis and constitutive chitinase activity in substrate-free medium. ITS sequence and chitinase expression in chitin-free medium are criteria to classify a strain as A. astaci
Sequences of 3' untranslated regions (UTRs) of CHI2 and CHI3 mRNAs. Alignment shows differences between 3' UTRs of CHI2 and CHI3 mRNAs
Amino-acid substitutions in the GH18 catalytic site of oomycete species. Table lists amino-acid substitutions in the GH18 catalytic site of oomycete species
O-linked glycosylation and phosphorylation predicted for Chi2 and Chi3. Predicted O-linked glycosylations and phosporylations at serine and threonine residues for Chi2 and Chi3 are listed in a table
Alignment of primer target sites for the 5.8S rRNA gene used as endogenous control in qPCR/MCA. Primers target conserved sites in the 5.8S rRNA gene of various oomycete species
A conventional PCR assay for detection of A. astaci that may fail to discriminate between closely related species. Alignment of primer sites for a conventional PCR assay reported for detection of A. astaci
Design of a homologous IPC for use in the qPCR/MCA or qPCR assays. The IPC monitored by a characteristic melting temperature or by an alternatively labeled hydrolysis probe in the qPCR/MCA or qPCR assays, respectively, helps to prevent false-negative detection due to insufficient extraction and/or amplification.
TaqMan qPCR assay design for sensitive detection and quantification of A. astaci. Primers, but also TaqMan probe facilitate discrimination between A. astaci and various related or relevant oomycete species.