

Abstract
Genomic translocations leading to the expression of chimeric transcripts characterize several hematologic, mesenchymal and epithelial malignancies. While several gene fusions have been linked to essential molecular events in hematologic malignancies, the identification and characterization of recurrent chimeric transcripts in epithelial cancers has been limited. However, the recent discovery of the recurrent gene fusions in prostate cancer has sparked a revitalization of the quest to identify novel rearrangements in epithelial malignancies. Here, the molecular mechanisms of gene fusions that drive several epithelial cancers and the recent technological advances that increase the speed and reliability of recurrent gene fusion discovery are explored.
Keywords: Translocation, Epithelial, Rearrangement, Gene Fusion, Chimera, MLL, ERG, ALK, HMGA2, COPA
Introduction
Throughout history, technological advances are often followed by discoveries that dramatically alter our perceptions of disease etiology. For example, after the term “chromosome” was introduced in the mid-1840’s, several German pathologists began using techniques to compare gross mitotic changes in tissue sections from different human malignancies.[1] Almost half of a century later, Theodore Boveri published a critical hypothesis that, “mammalian tumors might be initiated by mitotic abnormalities that resulted in a change in the number of chromosomes in the cell (aneuploidy),” based on the observation that sea urchin embryos would frequently engage in uncommon development following mitotic abnormality.[2] As time passed, breakthroughs arose that dramatically increased the quality and reproducibility of cytogenetic techniques such as the use of colchicine, which arrests cells in mitosis by inhibiting microtubule assembly. As a result of these observations, the general hypotheses regarding the evolution of human disease became increasingly complex; particular pathological conditions were associated with specific chromosomal abnormalities, such as Lejeune’s association of Down syndrome with an extra copy of chromosome 21.[3, 4]
Advances in technology once again spurred discovery when, in 1958, Rothfels and Siminovitch published a new cytogenetic, air-drying technique for flattening chromosomes.[5] The application of this technology later allowed Hungerford and Nowell to further characterize their initial observation that two patients with chronic myelogenous leukemia (CML) had a characteristic small chromosome.[6] Soon after the initial publication, Hungerford and Nowell were able to report on a series of seven patients, all of which harbored this minute chromosome.[7] This was coined the “Philadelphia chromosome” after the city in which the abnormal chromosome was discovered in accord with the Committee for the Standardization of Chromosomes.[8] The rearrangement leading to the Philadelphia chromosome was eventually characterized as a translocation between chromosomes 9 and 22 [9], resulting in the fusion of the breakpoint cluster region (BCR) gene on chromosome 22 with the v-abl Abelson murine leukemia viral oncogene homolog (ABL1) gene on chromosome 9.[10] Later in 1990, Lugo et al. demonstrated that the BCR-ABL1 fusion protein is an active tyrosine kinase, through immunoblotting cell lysates from Rat 1 transfected cells, revealing that cells transfected with either BCR-ABL1 or v-src, but not v-H-ras or v-myc, had a significant increase in total phosphotyrosine content.[11] Understanding the molecular mechanism of BCR-ABL1 led to the development of one of the first molecularly tailored therapies as the small molecule Imatinib was specifically selected for its ability to inhibit BCR-ABL1 kinase activity.[12, 13] The success of treating chronic myelogenous leukemia with a specific inhibitor of the BCR-ABL1 chimera led to a strong interest in the discovery of novel gene fusions in other cancer subtypes with the long term goal of designing disease specific therapeutics.
As techniques like the use of chromosome banding for karyotypic analysis were improved, the impact on discovery of novel gene fusions was immediately evident in leukemias and lymphomas. In fact, while BCR-ABL1 is perhaps the most famous gene fusion, the first molecularly characterized chimera was discovered by Zech et. al. through the use of karyotypic analysis and is actually involved in the pathogenesis of Burkett’s lymphoma and was identified. While this karyotypic analysis demonstrated absence of the distal region on the long arm of chromosome 8 and an extra band in the long arm chromosome 14 distal segment [14], the genes involved in the rearrangement remained elusive until 1982 when it was demonstrated that the translocation altered the c-MYC oncogene [15] and that the promoter and 5’ region of the immunoglobulin heavy chain (IGH) gene were rearranged such that the IGH promoter controls c-MYC expression.[16] Although this fusion does not lead to a chimeric protein, it was demonstrated that aberrant c-MYC expression through the IGH promoter is a necessary component of malignant transformation in Burkett’s lymphoma.[17]
As with lymphoma research, karyotypic analysis rapidly led to the identification of recurrent breakpoints that seemed to characterize subsets of myeloid leukemia. For example, in 1973, the acute myeloid leukemia 1 (AML1) gene was cloned from the breakpoint region of the first recurrent translocation described in leukemia, t(8;21).[18] In 1991, the AML1 gene was found to be fused to the eight-twenty one (ETO) gene on chromosome 21, which is also known as runt-related transcription factor 1 translocated to 1 (RUNX1T1).[19, 20] As the techniques of molecular biology improved, it became easier and easier to obtain the DNA sequence adjacent to chromosomal breakpoints. Since the original identification of AML1 in myeloid leukemia, over 10 genes have been described to participate in rearrangements with AML1.[21] In fact, advances in sequencing technology led to the realization that several genes are recurrently and promiscuously fused to multiple partners; the examples of which are ever increasing. In addition to AML1, the other notable example of a promiscuous fusion gene partner is the mixed lineage leukemia (MLL) gene, which is involved in over 40 different rearrangements (reviewed in [22]). In fact, because of the variety and difficulty of discussing all chromosomal aberrations in human malignancies, Mitelman et. al. maintain and frequently update an online database of rearrangements and chromosome aberrations from all malignant neoplasms.[23]
With the rapid development of current technologies like high-throughput sequencing, our perceptions as to the origins of disease have revealed a critical involvement of chromosomal aberrations, in particular, the role of translocations and gene fusions in malignant development. With a better understanding of the role of these chromosomal aberrations, therapies designed to inhibit the molecular function of chimeric proteins have recently been developed and, like Imatinib some have demonstrated a window of strong efficacy. Consequently, much hope has been generated by the potential for targeting existing and novel gene fusions that characterize specific cancer subtypes with rationally designed molecularly tailored therapies. Here, we review known genomic rearrangements in epithelial tumors that led to aberrant expression of chimeric transcripts and the emerging technologies that may lead to the identification of novel gene fusions.
Gene fusions in epithelial cancers
In order to highlight the number of genomic rearrangements leading to fusion genes that characterize epithelial cancers, we have surveyed some of the well-studied chimeras from several solid malignancies and describe the fusions in approximate chronological order. In the ensuing sections, we will analyze concepts from a global view of epithelial gene fusions with a few case studies of rearrangements from leukemia and endometrial stromal tumors. Gene fusions will be categorized into three different types: (1) those which alter the transcriptional regulation, (2) those which alter mRNA regulation and (3) those which alter protein activity. This will be followed by a discussion of the potential reasons why gene fusions have not been in the limelight of solid tumor pathogenesis and the developing technologies that are being used to find novel recurrent gene fusions in common epithelial tumors.
RET-NTRK1
The initial discovery of an epithelial gene fusion in the mid-1980’s comes directly from a novel screening technique used to identify transforming oncogenes. In this experimental approach, immortalized NIH3T3 cells were transfected with fragments of tumor cell genomic DNA, plated in soft agar. DNA is then isolated from cells and sequenced or sub-cloned to identify critical fragments. Using this approach, Martin-Zanca et. al. identified the RET-NTRK1 genomic translocation, providing some of the first insights into the possibility that recurrent genomic rearrangements were not specifically of hematologic phenomena.[24]
RET (rearranged during transfection) encodes a tyrosine kinase [25, 26] that was originally identified through transfection of DNA from a human T-cell lymphoma into NIH3T3 cells.[27] NTRK1 is a membrane-bound tyrosine kinase receptor that regulates neuronal cell growth, differentiation, and programmed cell death pathways.[28] Fusion of these two genes results in loss of the NTRK1 signal sequence giving rise to cytoplasmic localization and constitutive activation of the fusion protein.[29] Interestingly, although NTRK1 was the first identified RET fusion partner, RET has several other N-terminal fusion partners including H4 [30, 31], R1α [32], RFG5 [33] and ELE1.[34, 35] One possible explanation for the diversity of genomic rearrangements observed in PTC is that the underling pathology is simply dependent on deregulation of either the RET or NTRK1 tyrosine kinase domain. (reviewed in [36]) Consequently, the important determining event in PTC carcinogenesis may be constitutive activation of the mitogen-activated protein kinase (MAPK) signaling pathway, which can be caused by rearrangement of either the RET and/or NTRK1 gene. One reason for this hypothesis is that while the RET-NTRK1 rearrangement appears to be the predominant gene fusion responsible for childhood PTC, in adult-onset populations activating point mutations in the BRAF gene or, controversially, the RAS gene [37-43], also lead to constitutive activation of the MAPK pathway without RET and/or NTRK1 genomic rearrangement.[44]
In addition to differences in the age-related molecular onset of PTC, the proportion of cases with either a RET or NTRK1 rearrangement also appears to be based on the geographic area of origin, [45-47] possibly because thyroid cancer is established to be associated with exposure to ionizing radiation.[37, 48] Indeed, studies of patient populations exposed to either the Chernobyl nuclear power plant accident [49, 50] or the atomic bombings [51] have demonstrated that genomic rearrangements occur at a higher frequency than mutations following extreme exposure to radiation [37, 48], suggesting that under certain biological conditions exposure to high dose radiation may actually trigger specific DNA breaks leading to intentional genomic rearrangement. In fact, the fusion proteins that characterize PTC contain a number of different N-terminal partners fused the C-terminal tyrosine kinase domain of either RET or NTRK1 [52] that may depend on the environmental cues leading to genomic rearrangement.
CTNNB1-PLAG1
Within a year of publication of the RET-NTRK1 genomic rearrangement in PTC, another epithelial translocation was reported in pleomorphic adenoma (PA) [53], a slow-growing epithelial tumor that is responsible for more than 50% of salivary gland tumors [54], but less than 10% of tumors from the head and neck.[55] In contrast to RET-NTRK1 which was discovered by a screening technique, rearrangements in PA were first identified by karyotypic analysis of primary tumors. In fact, before any of the breakpoint genes were identified, PAs were already divided into four cytogenetic groups. (reviewed in [56]) Rearrangements of 8q12 account for about 40% of PAs with t(3;8)(p21;q12) comprising about half of rearrangements at this locus. Translocations of 12q14-15 account for about 8% of PAs with t(9;12)(p12-22;q13-15) or an ins(9;12)(p12-22;q13-15) responsible for these abnormalities.[57, 58] Tumors with non-recurrent clonal changes comprise about 20% of PAs, and tumors with apparently normal karyotypes account for the remaining cases.[56]
Almost 20 years after the initial karyotyping studies, Kas et. al. used a comprehensive breakpoint mapping approach, southern blot analysis and 5’ rapid amplification of cDNA ends (5’ RACE) to identify the genes involved in the most prevalent PA translocation, t(3;8)(p21;q12) as β-Catenin (CTNNB1) and PLAG1 (pleomorphic adenoma gene 1).[59] Specifically, the t(3;8)(p21;q12) rearrangement fuses the β-Catenin (CTNNB1) promoter and exon 1 to PLAG1 exon 2, resulting in a marked increase in PLAG1 expression (Figure 2). As such, because the gene fusion results in altered DNA level regulation of PLAG1 transcript, this gene fusion is characterized as type 1. Interestingly, the reciprocal translocation links the PLAG1 promoter and exon 1 to β-Catenin exon 2, reducing β-Catenin expression. As β-Catenin signals through several well characterized oncogenic pathways, (reviewed in [60]) the reduction in β-Catenin is curious. PLAG1, however, belongs to the PLAG family of proteins and encodes a zinc finger protein with two putative nuclear localization signals and can bind to either DNA or RNA. Forced expression of PLAG1 in NIH3T3 cells has demonstrated that this protein can induce the standard characteristics of neoplastic transformation including loss cell-cell contact inhibition, anchorage independent growth, and tumor formation in nude mice xenografts.[61] This suggests that the constitutive activity of the CTNNB1 promoter leads to sufficient PLAG1 expression for malignant transformation in PA.
Figure 2.
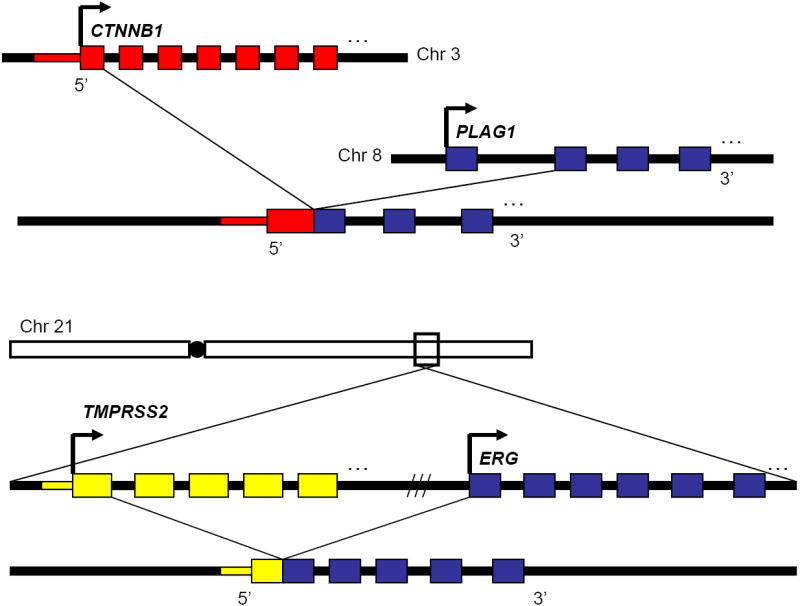
Genomic structure of gene fusions with altered transcriptional regulation. The CTNNB1-PLAG1 and TMPRSS2-ERG chimeras represent an important class of gene fusions in which the proto-oncogene remains largely intact, but the genomic rearrangement places a new promoter and 5’-UTR upstream of the main coding sequence, leading to aberrant expression of the proto-oncogene.
PRCC-TFE3
As cloning and molecular strategies improved in the early 1990’s, another recurrent gene fusion would soon be described in papillary renal cell carcinoma (PRCC), the second most common carcinoma of the renal tubules accounting for 15-20% of all renal cell carcinomas.[62-66] Karyotypic analysis as early as 1986 (de Jong et. al.) led to the identification of abnormalities in the Xp11.2 region characterized by a genomic rearrangement, t(X;1)(p11.2;q21.2).[62-66] Interestingly, before any of the genes surrounding the breakpoint were cloned a gene encoding TFE3, which was originally identified by their ability to bind to μE3 elements in the immunoglobin heavy chain intronic enhancer [67], was mapped to the Xp11.22 locus [68], and later shown to encode a member of the basic helix-loop-helix followed by a leucine zipper family (bHLHzip) of transcription factors. After the original genomic mapping, TFE3 was soon identified at the translocation breakpoint by southern blot analysis.[69] Subsequent 5’-RACE identified PRCC; a ubiquitously expressed gene that encodes a protein with a high proportion of prolines and glycines – including three P-X-X-P motifs that are known to interact with SH3 domains.[70, 71] Interestingly, the fusion event leading to the PRCC-TFE3 rearrangement also results in a reciprocal TFE3-PRCC gene fusion.[69, 72]
To elucidate the properties of these reciprocal gene fusions, Weterman et al. introduced wild type PRCC, wild type TFE3, PRCC-TFE3 and TFE3-PRCC expression vectors into COS cells and postulated that only the PRCC-TFE3 gene fusion was responsible for tumor formation based on its ability to activate a generalized report assay.[73] Thus, the PRCC-TFE3 genomic rearrangement is type 3 as the fusion protein gained a novel function through rearrangement. However, fusions of the PSF or NonO pre-mRNA splicing factors are also recurrently fused to TFE3, albeit at a much lower frequency than PRCC [69, 72, 74], suggesting that the TFE3 portion of the fusion is responsible for malignant transformation. Subsequent transcriptional activation assays demonstrated that of the PSF-TFE3, NonO-TFE3 and PRCC-TFE3 chimeras, only the PRCC-TFE3 fusion protein could activate the plasminogen activator inhibitor-1 (PAI-1) promoter, [75] suggesting that only this gene fusion retains transcriptional activity. However, recent co-immunoprecipitation experiments demonstrated that antibodies against the pre-mRNA splicing factors SC35, PRL1, and CDC5 were able to immunoprecipitate wild type PRCC, and an anti-SM antibody was able to immunoprecipitate the PRCC-TFE3 fusion protein.[75] This data suggests that the fusion protein functions may partially function through transcriptional pathways, it may also function by altering pre-mRNA splicing, but more conclusive experiments need to be conducted to demonstrate this phenotype.
HMGA2, evading let-7
While most of the gene fusions discovered until this point including PRCC-TFE3 were thought to define specific epithelial tumor types, a new gene fusion that was associated with several different tumor types, including pleomorphic adenoma (PA) (see above), lipoma, uterine leiomyoma and some myeloid malignancies [76], would refute the notion. In fact, the discovery of translocations involving 12q15 had been established by karyotypic analysis in multiple tumor types before the rearranged genes were actually identified and one of the genes involved in the t(9;12)(p12-22;q13-15) PA translocation was first identified in both mesenchymal tumors [77] and lipomas [78]. This first gene to be described was the 5’ gene fusion partner, HMGA2 (high mobility group AT-hook 2), belongs to the non-histone chromosomal high mobility group (HMG) protein family, which are small nuclear proteins (<30kDa) that undergo extensive post-translational modifications and contain nine amino acid segments that bind AT-rich DNA stretches in the minor groove (AT-hooks). (reviewed in [79]) Subsequent 3’ RACE of tumor samples revealed that HMGA2 has two different 3’ partners in PA, FHIT and NFIB, both of which contribute very little coding sequence to the resulting fusion gene. In fact, in one class of translocations, HMGA2 exon 3 is fused to FHIT exon 9 or 10, resulting in retention of the C-terminal 26 amino acids of FHIT [80], and in the other set, HMGA2 exon 3 or 4 fusion to NFIB exon 9 appends five amino acids (SWYLG) to the truncated HMGA2 protein.[81]
Surprisingly, transgenic mice overexpressing wild type HMGA2 were observed to have similar phenotypes to mice expressing the truncated protein HMGA2 protein found in the PA gene fusions.[82-84] To complicate this observation, in hereditary renal cell carcinoma, FHIT was previously demonstrated to be fused to the patched related gene TRC8 by t(3;8)(p14.2;q24.1) [85, 86] and the (SWYLG) amino acid motif found in the HMGA2-NFIB gene fusion were shown to be essential for NFIB function [81]. Recent research, however, has shed light onto the importance of these translocations to neoplastic transformation.
The discovery that small RNAs called microRNAs can negatively regulate gene expression through direct binding to a gene’s 3’-UTR has led to the hypothesis that certain microRNAs can function as tumor suppressors in cancer.[87] Bioinformatic analysis of the HMGA2 3’-UTR demonstrated that the mRNA contains seven conserved sites complementary to the let-7 microRNA [88]. (Depicted in figure 3) To show that the let-7 microRNA negatively influences HMGA2 expression, Mayr et. al. built a HMGA2 3’-UTR conjugated luciferase reporter and demonstrated that let-7 represses its expression.[89] As such, although the genomic rearrangements between HMGA2 and FHIT or NFIB yield fusion proteins, replacement of a Let-7 regulated 3’-UTR seems to be the critical event because it leads to HMGA2 over-expression, which is sufficient for neoplastic transformation. Thus, the HMGA2 genomic rearrangement represent the first of a novel class of gene fusions, type 2, in which fusion gene activity is enhanced by loss of mRNA level regulation (Figure 3).
Figure 3.
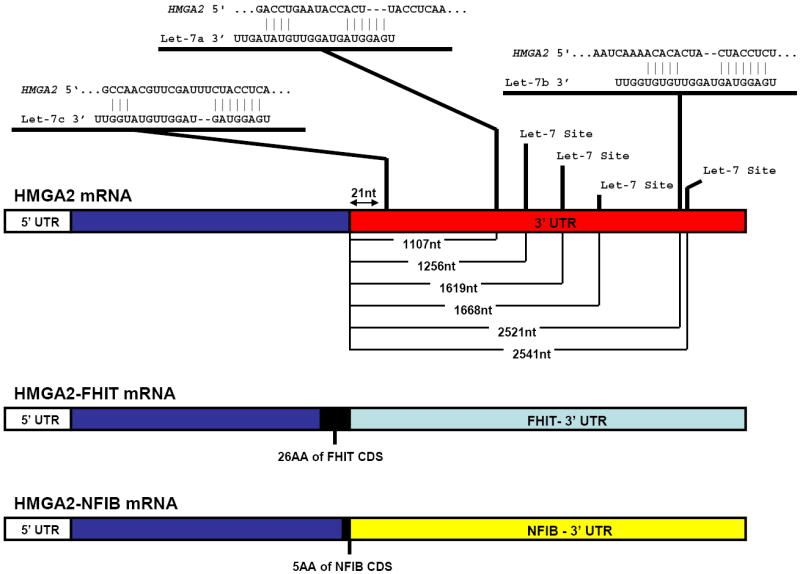
HMGA2 gene fusions elude the Let-7 family of microRNAs. The HMGA2 mRNA structure is shown along with putative Let-7 family binding sequences in the HMGA2 3’-UTR. Results were predicted by TargetScan [202] and three representative microRNAs are shown with there highest probability binding sites of the seven total predicted sites along the 3’UTR. Distance to each predicted binding site is annotated as nucleotides from the start of the 3’UTR. Below the wild type HMGA2 mRNA are the HMGA2-FHIT and HMGA2-NFIB mRNAs that result from these two gene fusions. TargetScan did not predict any microRNA binding sites in these genes. As such, the HMGA2 gene fusions represent a second class of gene fusions in which the recombination event allows the proto-oncogene mRNA to evade microRNA-mediated silencing.
Pax8-PPARγ
In 2000, Kroll et. al. employed fluorescence in situ hybridization (FISH), yeast artificial chromosome mapping and 3’ RACE to identify genes involved in a genomic rearrangement, t(2;3)(q13;p25) [90], that was originally identified by karyotype analysis of follicular thyroid carcinomas, a subset (10-20%) of all thyroid malignancies [91]. This translocation is thought to be specific to FTC as it has not been reported in other thyroid tumors or hyperplastic nodules [92] In the resulting gene fusion, the Pax8 (Paired box gene 8) gene is fused to PPARγ (Peroxisome proliferator-activated receptor-γ), a ubiquitously expressed transcription factor.[90] The Pax8 protein is involved in thyroid follicular cell development and regulation of thyroid-specific gene expression.[93] PPARγ plays a major role in a number of different diseases including obesity, atherosclerosis, diabetes as well as cancer. (reviewed in [94]). Because Pax8 is a thyroid specific transcription factor and because its DNA binding domain is fused to the c-terminal domains of PPARγ [90], the resulting protein chimera is thought to have constitutive re-distribution of PPARγ-directed transcription. In 2005, gene expression microarray profiling revealed that a distinct signature in follicular thyroid carcinomas harboring the Pax8-PPARγ gene fusion in which cell growth and chromatin remodeling pathways were over-represented and protein biosynthesis pathways were under-represented as compared to follicular thyroid carcinomas without the translocation [95], suggesting that PPARγ-transcription is indeed redefined by the gene fusion.
Interestingly, follicular thyroid carcinomas were originally thought to arise from disruption of distinct molecular pathways, either through the fusion of Pax8 to PPARγ, or through the acquisition of point mutations leading to the constitutive activation of the G-protein RAS. In fact, one study reported that 16/33 (49%) of follicular carcinomas had RAS mutations, 12/33 (36%) had Pax8-PPARγ rearrangement, only 1/33 (3%) had both, and 4/33 (12%) had neither.[96] However, in 2006, quantitative reverse transcription PCR analysis of follicular carcinoma clinical samples demonstrated loss of the tumor suppressor NORE1A in samples harboring the Pax8-PPARγ rearrangement, but not in other samples.[97] Because NORE1A binds to the GTP bound (activated) RAS protein and suppresses RAS activity, this discovery suggested that activation of the RAS pathway is a critical event in pathogenesis of thyroid carcinoma that is altered either directly by activating mutation, or indirectly by the Pax8-PPARγ rearrangement.
BRD-NUT
Soon after the discovery of the Pax8-PPARγ rearrangement, the translocation t(15;19)(q13;p13.1) was identified in a rare, highly aggressive carcinoma arising in the midline organs and upper respiratory tract of young people now termed nuclear protein in testis (NUT) midline carcinomas (NMC).[98-100] BRD4, which contains the chromosome 19 breakpoint, has two annotated transcripts encoding either short or long forms of the protein that both contain N-terminal bromodomains. The longer BRD4 transcript encodes a ubiquitously expressed 200kDa nuclear protein [101] with a c-terminal lysine rich region that is not found in the shorter transcript. The translocation resulting in fusion to the NUT gene (identified by southern blot analysis) only disrupts the longer BRD4 transcript resulting in loss of the lysine rich region in the fusion oncogene. Several studies of BRD4 in both murine and human cell line models have demonstrated a critical role in cell cycle progression and cell proliferation.[102, 103] In fact, Brd4 enhances cell growth by interacting with chromatin [104], replication factor C [102] and cyclinT1 and CDK1 that constitute core positive transcription elongation factor b (P-TEFb).[105] Likewise, chromatin immunoprecipitation assays demonstrated that Brd4 is required to recruit P-TEFb to active promoters, and that increased Brd4 leads to increased P-TEFb-dependent phosphorylation of RNA polymerase and enhanced transcription from promoters in vivo.[105]
More insight into the role of the BRD4-NUT fusion protein in NMC biology came from a screen for other NMC gene fusions. Because the BRD4-NUT translocation defines two-thirds of all NMCs, French et al. used a candidate gene approach to screen other NMC samples and discovered another recurrent translocation between BRD3 and NUT that defined large portion of the remaining NMC cases.[106] The BRD3-NUT fusion gene encodes a protein highly similar to that encoded by the BRD4-NUT transcript. It is composed of two tandem chromatin-binding bromodomains, an extra-terminal domain, a bipartite nuclear localization sequence, and a significant portion of NUT coding sequence. As such, the conserved protein structure gave insight into the mechanism by which the chimeric protein induces neoplastic properties.
Wild type NUT, which is normally only expressed in the testis [99], contains both nuclear localization and export signal sequences and is shuttled between the nucleus and cytoplasm via a leptomycin-sensitive pathway.[106] Importantly, however, the Brd3–NUT and Brd4–NUT proteins are retained in the nucleus, suggesting that interactions between the Brd3 or Brd4 bromodomains and chromatin are essential to the fusion protein.[106] (Figure 4) Further evidence for this hypothesis comes from an siRNA experiment in which knockdown of Brd-NUT fusion transcripts in NMC cell lines resulted in squamous differentiation and cell cycle arrest.[106] This suggested that the nuclear retention of NUT, not the loss of the Brd C-terminal domain, is responsible for promoting NMC carcinogenesis.[106] The realization that Brd-NUT gene fusions define a class of translocations that fuse bromodomains to the NUT protein suggests that oncogenic translocations will arise from multiple partners when critical domains are present in more than one gene.
Figure 4.
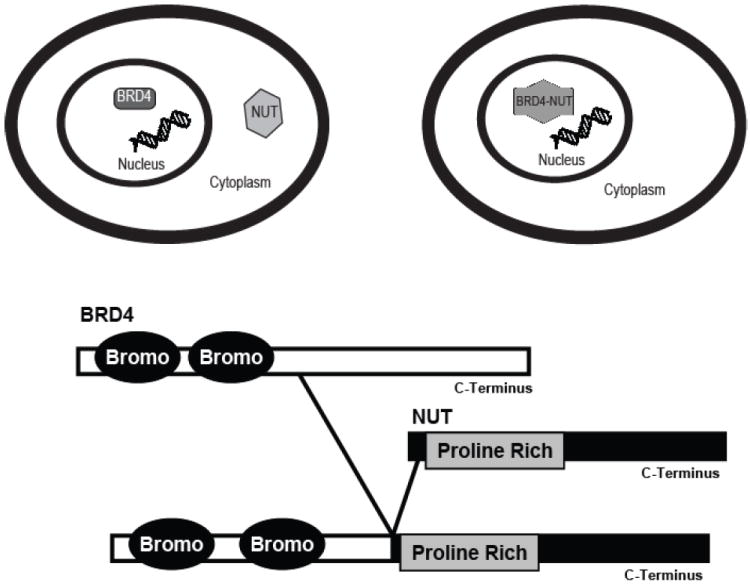
Nuclear retention of NUT. The BRD4-NUT gene fusion represents a third class of rearrangements in which the resulting protein gains activity to become a proto-oncogene. In this case, the two bromodomains of BRD4 are fused to NUT. Although NUT usually cycles between the nucleus and cytoplasm in a highly controlled manner, appendage of the BRD4 bromodomains to the majority of the NUT protein lead to nuclear retention of the protein and aberrant activity.
ETV6-NTRK3
The first major example of a recurrent epithelial rearrangement that appeared not only in multiple tumor types, but had also been reported in a large subset of hematologic malignancies was detected in several cases of secretory breast carcinoma, a rare subtype of infiltrating ductal carcinoma affecting both children and adults.[107] Tognon et al. detected the ETV6-NTRK3 fusion by comprehensive FISH analysis in 92% (12 of 13) secretory breast carcinoma cases.[108] ETV6 (also TEL) is an ETS family member that is involved in a large number of fusions to either a transcription factor like AML1 [109] or to a protein tyrosine kinase domain like that of ABL [110, 111], JAK2 [112-114], ARG [115, 116], PDGFRβ [117] or FGFR3 [118], each of which define a unique leukemia sub-type (reviewed in [119]) ETV6 contains a pointed oligomerization domain (PNT; also known as sterile alpha motif, SAM, or helix-loop-helix, HLH) and an ETS DNA binding domain, the expression of which is required for developmental processes such as hematopoeisis and yolk sac angiogenesis.[120] NTRK3 is a transmembrane neurotrophin-3 surface receptor that contains a c-terminal protein tyrosine kinase domain and plays a role in growth, development, and cell survival of neural cells in the central nervous system. (reviewed in [121]) The fusion of the N-terminal ETV6 pointed domain to the C-terminal tyrosine kinase domain of NTRK3 was first reported in congenital fibrosarcoma (CFS) [122], but has since been reported in multiple cell lineages including those that give rise to congenital mesoblastic nephroma (CMN), acute myelogenous leukemia, and secretory breast carcinoma [108]. (reviewed in [123])
Following the initial discovery, research focused on the transforming ability of the recombination product. By using retroviral gene delivery methods, the ETV6-NTRK3 fusion gene was shown to be sufficient to induce the non-tumorigenic murine breast cell lines Eph4 (epithelial) and Scg6 (myoepithelial) as well as NIH-3T3 fibroblasts to form tumors, glandular structures and to express epithelial antigens.[108] This discovery suggested that the fusion gene acts as a dominant oncogene in secretory breast cancer. ETV6-NTRK3 was also shown to inhibit TGF-β tumor suppressor activity in NIH3T3 cells [124], suggesting that it most likely regulates microRNA biogenesis indirectly, [125] but this has not yet been explored. Although it is known that adults have a less favorable prognosis than children and distant metastases are rare [126], local recurrences and nodal metastases have been observed [127] suggesting that the gene fusion leads to an invasion associated transcriptional program, but this also has not been explored. Despite this, it is known that constitutive activation of the fusion protein leads to activation of the Ras-mitogen-activated protein kinase (MAPK) pathway and the phosphoinositide-3-kinase (PI3K)-AKT pathway, the mechanism leading to activation of these pathways has remained elusive until recently, when the fusion protein was shown to associate with c-Src by immunoprecipitation from fusion-positive CFS and CMN human primary tumors.[128] More recently, however, a mouse knockin model was created by introducing the human NTRK3 cDNA into exon 6 of the mouse ETV6 locus, which induced a fully penetrant, multifocal breast cancer.[129] By using microarray analysis of unsorted and sorted tumors from this model, as well as NIH3T3 cells transduced with the fusion gene, the authors showed that ETV6-NTRK3 enriches for WNT target genes through activation of the AP1 complex.[129] The requirement for AP1 activity in ETV6-NTRK3-mediated transformation was confirmed by showing that the co-expression of a dominant negative component of AP1 complex, c-JUN TAM67, with the gene fusion blocked tumorigenic properties both in vitro and in vivo.[129] The ETV6-NTRK3 gene fusion represents one of the last gene fusions to be discovered by traditional biological techniques.
TMPRSS2-ETS
In 2005, advances in bioinformatics led to the discovery of rearrangements on chromosome 21 between TMPRSS2 (transmembrane protease, serine 2) and ERG (v-ets erythroblastosis virus E26 oncogene homolog (avian)) resulting in the TMPRSS2-ERG gene fusion. Thus far, genomic rearrangements leading to an ERG gene fusion have been reported in approximately 50% of clinically localized prostate cancers published. (reviewed in [130]) TMPRSS2 is a prostate-specific, androgen-regulated gene [131-133] that has two annotated transcription variants, both of which are involved in the fusion with ERG; the annotated TMPRSS2 in about 50% of the gene fusions, an alternative TMPRSS2 variant in 10% of gene fusions, and both variants in slightly more than 40% of analyzed gene fusions.[134] ERG belongs to the ETS family of transcription factors and has two transcription variants that differ only slightly in the 5’-UTR (deleted in the gene fusion) and in the usage of an in-frame exon, the role of which remains undefined. The most common TMPRSS2-ERG gene fusion variants involve TMPRSS2 exon 1 or 2 fused to ERG exon 2, 3, 4, or 5 [134-143] and less frequently rearrangements of TMPRSS2 exon 4 or 5 fused to ERG exon 4 or 5.[141] In line with the combinatorial complexity of TMPRSS2-ERG rearrangements, different fusions have correlated with slightly different phenotypic outcomes. For example, TMPRSS2 exon 2 fused with ERG exon 4 is associated with aggressive disease, while others have been associated with seminal vesicle invasion and poor outcome.[143]
Like TMPRSS2, the TMPRSS2-ERG gene fusion is androgen regulated in an androgen responsive cell line (VCAP) carrying the rearrangement [135], but not in an androgen insensitive cell line harboring the fusion (NCI-H660).[144] We have shown that VCaP cells and benign prostate cells forced to overexpress ERG drive components of the plasminogen activation pathway to mediate cellular invasion using transwell migration assays.[145] We have also reported that primary or immortalized benign prostate epithelial cells overexpressing ERG have a transcriptional program with high levels of several invasion-associated genes, but did not display phenotypic increases in cellular proliferation or anchorage-independent growth.[145] Despite this, one group recently identified c-MYC as a downstream target of ERG and demonstrated that ERG knockdown in TMPRSS2-ERG expressing CaP cells resulted in loss of cell growth in vitro and loss of tumorgenicity in vivo, with only 22% (2/9) mice developing detectable tumors at day 42 in siRNA treated cells as compared to 100% (5/5) in the control group.[146] Interestingly, transgenic mice expressing an androgen-regulated ERG fusion gene develop mouse prostatic intraepithelial neoplasia (PIN), a precursor lesion of prostate cancer, not prostate cancer. Taken together with our in vitro data, these results suggest that, without secondary molecular lesions such as loss of the tumor suppressors PTEN or NKX3-1, the TMPRSS2-ERG gene fusion may not be sufficient for transformation.[145, 147, 148]
Although ERG clearly participates in the majority of ETS family gene fusions in prostate cancer, other ETS family members including ETV1 [135], ETV4 [149, 150] and ETV5 [151] also contribute to gene fusions in prostate cancer, albeit at a much lower frequency. In contrast to TMPRSS2, which is the only known 5’ partner to ERG, the other ETS family members may have a variety of 5’ partners including those with androgen-responsive promoters (TMPRSS2, SLC45A3, KLK2, HERV-K_22q11.23 and CANT1), one with an androgen-insensitive promoter, but constitutively active promoter (HNRPA2B1), and one with an androgen-repressed promoter (C15orf21).[135, 149, 151-153] As in the case of ERG, forced expression of ETV1 under the control of a CMV promoter did not enhance cell proliferation in benign prostate epithelial cell lines and did not lead to anchorage-independent colony formation in soft agar, but did lead to the enrichment of genes associated with invasion.[145] Consequently, knockdown of ETV1 in LNCAP cells prevented transwell invasion through matrigel.[145, 154]
EML4-ALK
Recently, Soda et. al. reported retroviral-mediated transformation screen, in which they created a cDNA expression library from a surgically resected lung adenocarcinoma.[155] Following transformation of NIH3T3 cells, cDNAs were recovered from cells by PCR amplification and sequenced. One of these sequenced transcripts contained a fusion between EML4 (echinoderm microtubule-associated protein-like 4) and ALK (anaplastic lymphoma kinase) that was later confirmed as an inversion of chromosome 2p in 6.7% (5 of 75) NSCLC patients.[155] Wild type EML4 is a member of the EMAP family of proteins and the amino-terminus (amino acids 1-249) were previously demonstrated to be essential for microtubule formation in HeLa cells.[156] ALK encodes a tyrosine kinase and a MAM domain (a domain frequently found on the extracellular side of the membrane on many receptors). Despite the apparent low frequency EML4-ALK gene fusions in NSCLC, the transforming ability of EML4-ALK gene fusion variant 1, 2, and 3b, but not a kinase inactive mutant (K589M) has been demonstrated by engrafting NIH-3T3 cells infected with retroviral expression vectors and showing that tumors arise in 8/8 mice from all groups except for the kinase dead mutant.[157]
To corroborate the low frequency EML4-ALK rearrangements in NSCLC, careful PCR-based analysis was completed on NSCLC cases to identify novel in-frame EML4-ALK gene fusions that led to the identification of two novel fusion isoforms called variant 3a and 3b.[157] Even more recently, analysis of a cohort of 253 lung adenocarcinoma patient samples identified two new EML4-ALK fusions in which either exon 14 or exon 2 of EML4 was fused to Exon 20 of ALK (variants 4 and 5, respectively), however, only 4.35% of patients were found to express any of the 5 known EML4-ALK genomic rearrangements.[158] A similarly low rate of the ELM4-ALK fusion was reported in a study of 104 lung cancer surgical specimens with only one fusion positive case [159] and, in a study of different lung cancers, the fusion was identified in 3.4% (5 of 149) adenocarcinomas, but not in 48 squamous cell carcinomas, 3 large-cell neuroendocrine carcinomas, or 21 small-cell carcinomas.[160] However, this is to be expected, given the small sample size from non-adenocarcinomas. The ALK gene has previously been identified as the 3’ fusion partner of NPM- [161], TPM3- [162], CLTC- [163], ATIC- [164-166] and TFG- [167]. In light of this observation, RT-PCR analysis was used to screen all known hematologic ALK fusion partners in a cohort of 77 NSCLC samples, however, no redundant fusion partners were identified and only 2.6% (2 of 77) NSCLC cases harbored the EML4-ALK fusion.[168] To supplement the existing RT-PCR data in the literature, our group developed a break-apart FISH assay to analyze ELM4-ALK fusion as well as the amplification of each gene. We reported the fusion occurred in less than 3% of NSCLC cases analyzed, and that, in most cases harboring the lesion, not all cells exhibited the fusion. We also found that EML4 and/or ALK amplification occurred, indicating that other mechanisms of genomic rearrangement leading to amplification may arise.[169]
SLC34A2-ROS
In 2007, a survey of phosphotyrosine signaling in lung cancer not only led to the re-identification of the EML4-ALK fusion, but also the discovery of a novel translocation between chromosomes 4p15 and 6q22, in which the transmembrane domain containing N-terminal region of the solute carrier family 32, member 2 (SLC34A2) is fused to an N-terminal transmembrane domain of the c-ros oncogenes 1 (ROS), respectively, in the lung cell line HCC78.[170] SLC34A2 is encoded from a single transcription variant and ROS, which is a type I integral membrane bound tyrosine kinase is a known oncogene that is highly expressed in several tumor cell lines, and also encoded from a single transcript. Interestingly, while the authors did not identify SLC34A2 rearrangements with ROS in patient samples, a gene fusion between CD74, located at 5q32, and ROS was observed, in which the tandem transmembrane domain structure was again observed.[170] This suggests not only that ROS is another promiscuous gene fusion partner, but the tandem transmembrane structure is one mechanism leading to constitutive activation of the tyrosine kinase. Indeed, forced expression of the SLC34A2-ROS chimera demonstrated constitutive kinase activity in the cellular membrane fraction.[170]
SLC45A3-ELK4
With the recent advent of next generation sequencing technology (described below), our group has recently identified another recurrent gene fusion in prostate cancer.[171] Using this technology we identified the fusion of SLC45A3 to ELK4, an ETS family member. Here exon 4 of SLC45A3 is fused to exon 1 of ELK4. Interestingly, this novel gene fusion was identified from the RNA of a cell line harboring a known gene fusion involving another ETS family member gene, ETV1. Likewise this novel gene fusion involves SLC45A3, which is known to fuse with ETV1 in other prostate cancer cases. Unlike other gene fusions described to this point, SLC45A3-ELK4 seems to result from polymerase read-through and intergenic splicing rather than genomic rearrangement as no detectable alterations were detected on the DNA level by fluorescence in situ hybridization (FISH), array comparative hybridization (aCGH) or high-density single nucleotide polymorphism (SNP) arrays.[171] RNA level gene fusions were recently identified in endometrial stromal tumors and are discussed below.
Lessons from MLL translocations
While the list of epithelial derived gene fusions continues to expand, it is important to highlight unique mechanisms of oncogene formation through specific genomic rearrangements from the hematological malignancies. Translocations altering the mixed-lineage leukemia (MLL) gene on 11q23 frequently lead to fusions with over 40 different genes on different chromosomes with MLL-AF4 and MLL-AF9 among the most frequent chimeras. (reviewed in [172], [173]) Interestingly, different MLL fusions are highly associated with either acute myeloid leukemia (AML) or acute lymphoid leukemia (ALL, depending on the fusion partner.[174] MLL is the mammalian homologue of a Drosophila gene called trithorax, which was shown to play a critical role in axial morphogenesis and patterning during embryogenesis through the regulation of HOX genes (HOM-C in Drosophila).[175, 176] Multiple studies have suggested that deregulation of HOX gene expression contributes to leukemogenesis.[177] Additionally, retroviral transduction of a MLL fusion gene construct was able to transform wild type, but not the Hoxa9-deficient, bone marrow cells providing direct evidence that specific HOX gene expression may be required for leukemogenesis.[178] Because MLL chimeras often lose large fragments and different domains from either the N- or C-terminal regions, the seemingly critical role of MLL-associated HOX gene expression to leukemogenesis led to the question of whether the molecular mechanisms by which wild type MLL regulates gene expression are mutually exclusive from those employed by MLL chimeras.[179]
As the molecular mechanisms of MLL target gene regulation continue to unravel, several studies have shed light on the fact that molecular function between wild type and fusion gene settings may be unique, though the outcome of gene activity is ultimately similar. Wild type MLL encodes a multi-domain protein with three AT hooks used for binding AT-rich DNA sequences and a histone methyltransferase domain [180] and assembles into supercomplexes containing several different chromatin remodeling enzymes on target DNA motifs like those found in HOX genes.[181] Chimeric MLL proteins, on the other hand, appear to utilize different mechanisms to modulate HOX gene expression and initiate leukemogenesis. For example, fusion of coiled-coil domains from GAS7 or AF1p to MLL endow the chimeric protein with the ability to dimerize on the target gene promoters and have been suggested to stimulate transcription through the inappropriate recruitment of members of the MLL supercomplex.[182] This suggested that preventing dimerization of the coiled-coil domains with targeted small molecules could inhibit MLL activity in this subset of MLL fusions. In contrast, some MLL fusions lead to constitutive nuclear retention while maintaining similar binding patterns as the dimerizable MLL chimeras on the HoxA9 locus.[183] In the absence of a partner gene, MLL can acquire an in-frame partial tandem duplication (PTD) of exons 5 through 11 (occurring in approximately 4%–7% of AML cases) that causes overexpression of HoxA7, HoxA9, and HoxA10 in spleen, BM, and blood in a knockin mouse model.[184] As such, altering downstream HOX gene expression appears to be one critical role of MLL gene fusions and rearrangements.
Given that wild type and chimeric MLL proteins appear to accomplish at least one similar molecular function (HOX gene regulation), the question of how epithelial gene fusions will function in comparison to their wild type counterparts remains intriguing. For example, we have very little understanding of the normal molecular mechanisms utilized by ERG and ETV1 to control gene expression (prostate cancer gene fusions, discussed above), let alone the critical co-factors required for transcriptional regulation. Although we may expect the molecular mechanisms of ERG and ETV1 mediated gene regulation to be the same in the wild type and fusion settings (because the encoded proteins are nearly identical), this remains to be proven. Perhaps the ability to design rational drug targets against specific fusion proteins without obvious molecular susceptibilities (like the tyrosine kinase activity of BCR-ABL) will depend as much on our understanding of each fusion protein’s function and critical co-factors as on their downstream targets.
Difficulty in identifying epithelial cancers gene fusions
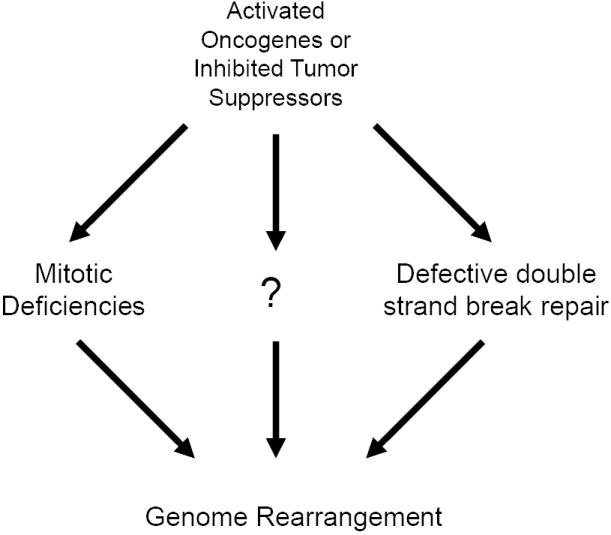
With the discovery of the TMPRSS2-ERG gene fusion in prostate cancer, we look back on the history of cancer biology and wonder why gene fusions have not been identified in some of the most well studied epithelial cancers? Part of the problem was methodological, as the chromosome quality in epithelial neoplasms is very poor when compared to hematologic neoplasms. However, cytogenetic techniques have improved dramatically since the discovery of the “minute” chromosome in 1960.[6] In fact, in the 1960s, chromosome patterns in epithelial tumors were already being described as abnormal [185] and it was often thought that the degree of cytogenetic changes corresponded proportionally with clinical progression [186], making the identification of individual and recurrent translocations difficult. In fact, the idea that the induction of genomic instability is a critical and intended step in the malignant progression of solid tumors has gained considerable momentum.[187, 188] Recently, it was demonstrated that overexpression of Separase, a protein that is over-expressed in a subset of breast cancers, leads to can induce chromosome instability and aneuploidy in the mutant p53 mouse mammary epithelial cell line FSK3.[189] Likewise, deregulation of Mad2, which regulates separase activity, has been shown to promote chromosomal instability, induce aneuploidy and lead tumorigenesis.[190] Interestingly, once Mad2-induced neoplastic transformation has occurred, Sotillo et. al. demonstrated that expression of Mad2 is no longer required for tumor progression suggesting that the induction of chromosomal instability could be a transient event in oncogenesis.[190] In fact, it is possible that specific gene fusions induce genomic instability through deregulation of normal mitotic events like separase or Mad2 activity or through novel mechanisms yet to be described. If induction of chromosomal instability was a mechanism of oncogenesis employed by a specific gene fusion, then induction of other secondary “carrier” chromosomal rearrangements would simply serve to mask the identification of the recurrent genetic rearrangement. Such a progression pattern in epithelial tumors could explain the complex heterogeneity often observed in such malignancies. In contrast, leukemias, lymphomas and mesenchymal tumors are almost 95% clonal.[191] As such, the complexity and shear number of genomic rearrangements in epithelial malignancies has led to difficulty in defining primary aberrations in these neoplasms. This difficulty eventually led to the incorrect notion that genomic rearrangements leading to gene fusions were simply less common in epithelial tumors.
Mitelman Hypothesis
In order to address this notion that fusion genes are almost exclusively a hematologic phenomena, Mitelman et al. completed a comprehensive study of all known cytogenetically abnormal neoplasms reported in the literature.[192] Importantly, data published by the group supported the counter-hypothesis that, in every tumor type, the numbers of recurrent balanced chromosome abnormalities, gene fusions and balanced rearrangements are a function of the total number of analyzed cases.[192] In this study, 271 gene fusions and 59 potential gene fusions (only one gene identified at the breakpoint) were catalogued, of which 275 unique genes were involved in the rearrangements.[192] This indicated that a substantial number of genes were present in more than one chimeric transcript (e.g., MLL, ETV6 and RET as described above). In classifying each gene fusion by the class to which each member of the chimera belonged, the group demonstrated that the proportion of fusions belonging to each class was approximately equal in both hematologic and solid tumor malignancies, with the transcription factor class accounting for 38-44% and tyrosine kinase class tabulating 5-7%.[192] This study suggested that the occurrence of gene fusions is a general molecular event that has no fundamental tissue-specific differences. However, gene rearrangements must at least encourage function in specific genetic backgrounds such as the TMPRSS2-ERG fusion, which requires active androgen signaling, and thus encourages prostate specificity.
Tissue-specific gene fusions
The idea that genomic rearrangements are tissue specific is an emerging concept in the field of gene fusion biology. For example, TMPRSS2 is a strongly androgen regulated and prostate specific gene that is fused to the ETS family members ERG and ETV1 in prostate cancer.[135] While other ETS family members form fusion genes that give rise to other malignancies, chimeras between androgen regulated genes and ETS genes have only been observed in prostate cancer.[130] Likewise, the ALK tyrosine kinase is frequently fused to multiple partners in hematopoietic (myelogenous leukemia), mesenchymal (congenital fibrosarcoma) and epithelial (secretory breast carcinoma) malignancies, but no redundant fusion partners have been identified across tissue types.[159] Retention of the TFE3 DNA binding domain in follicular thyroid carcinoma is another example of this, as TFE3 is a thyroid-specific transcription factor.[93] Importantly, little is understood about the molecular mechanisms leading to gene rearrangement and the underlying reasons that particular chimeras are formed recurrently. The idea that tissue specific rearrangements occur by fusing highly transcribed genes holds promise and would at least partially explain the apparent tissue specificity observed in the formation of chimeric transcripts even between genes that are fused in multiple cancer types.
The idea that gene fusions are tissue specific could have profound implications on the discovery of novel gene fusions. Clearly, however, gene fusions do not always confer tissue specificity. HMGA2 has a 3’-UTR that is negatively regulated by the Let7 microRNA and simply replaces its 3’-UTR through rearrangement with another gene (described above), therefore representing a gene fusion that most likely retains functionality in multiple tissue types. As such, while this concept may have its largest impact on underlying molecular mechanisms of newly discovered gene fusions, it will probably not alter the rate gene fusion discovery.
Discovery of novel gene fusions
Although the rate recurrent chromosomal rearrangement discovery in epithelial tumors has been modest, the recent discovery of gene fusions in prostate cancer has led to a renewed interest in gene fusions identification in other epithelial cancer subtypes. Perhaps the best explanation for the sudden increase in the characterization of recurrent gene fusions is the advent of novel technologies. For example, the use of existing gene expression data in the discovery of novel gene fusions was limited until the emergence of cancer outlier profile analysis (COPA), which ranks genes by normalizing expression values based on median absolute deviation of gene expression to accentuate outlier profiles (reviewed in [130]). When COPA was applied to gene-expression datasets in the Oncomine database [193-196], the analysis was able to identify several hallmark cancer related genes and led to the discovery of the ERG and ETV1 outlier profiles in prostate cancer.[135] Subsequent exon-walking quantitative PCR was used to demonstrate loss of the 5’ exons in both ERG and ETV1, giving rise to the notion that a gene fusion event was responsible for the outlier expression of these genes in prostate cancer. Finally, 5’-RNA ligase-mediated rapid amplification of cDNA ends (5’-RACE) was used to identify the 5’ untranslated region of TMPRSS2, a prostate-specific, androgen-regulated, transmembrane serine protease gene [131, 132, 197]. Fusion specific PCR and fluorescence in situ hybridization (FISH) were used to confirm the genomic rearrangement.
In contrast to using COPA and exon-walking quantitative PCR to identify fusion gene candidates, several labs are now employing next generation sequence methods wherein DNA or mRNA can be fragmented, sequenced and mapped to the genome in a matter of weeks to identify gene fusions. Various commercial platforms have been developed with the intent of sequencing as much of the genome or transcriptome as possible and are classified based on the length of the templates each platform sequences. Long read technologies, like 454, can sequence long templates (>1kb) whereas short read technologies, like SOLEXA and SOLID, are currently capable of sequencing 35-50 nucleotide templates. At first glance, long read technologies may appear to have the advantage of making genome (or transcriptome) re-assembly much simpler than short read technologies. However, a major advantage of short read technologies is the depth of coverage, or number of times a segment of the genome is sequenced, which is currently much higher for short read than long read technologies. As such, the choice of technology is still dependent on the scientific question.
If our question is to identify the best method for novel fusion gene discovery, we assume that sequencing the transcriptome space will be much efficient than sequencing cancer genomes. In theory, the discovery of gene fusions by long read technology will require sequencing across the actual gene fusion boundary of the chimeric transcript. In contrast, short read technologies may be able to identify gene fusions by two different methods. The first and most straight forward method is the identification of sufficient short reads that do not map directly to the transcriptome, but correspond to the gene fusion boundary; and these short reads should identify both contributing genes with high probability. Second, because transcripts are thought to be sequenced with a uniform distribution across the length of the transcript, except for at the extreme 5’ and 3’ ends, exon expression for each transcript can be analyzed. Genes involved in rearrangements, leading to chimeric transcripts, would be expected to lack any exon expression on one of the transcript ends. However, this method will need to be carefully developed, as mapping of short reads to duplicated sequences (or sequences that appear more than one time in the genome) remains challenging.
To test whether short or long read technology was better for the discovery of recurrent gene fusions, we recently sought to “re-discovered” the known gene fusions BCR-ABL1 and TMPRSS2-ERG by sequencing the RNA transcriptome of either the leukemia cell line K562 or the prostate cell line VCAP, respectively, with both short and long read platforms.[171] Initially both technologies were able to identify the known gene fusion from the sample, but were also able to identify several other candidate gene fusions. For example, the Illumina short read platform nominated 428 candidates from the VCAP cell line.[171] However, most of these candidates were likely to result from either trans-splicing [198], co-transcription of adjacent genes followed by intergenic splicing [199], or as a consequence of the sample preparation protocol. In order to reduce the list of potential candidate genes, we intersected the results of the two platforms to yield a much more condensed list. Indeed by integrating the short read and long read platforms rather than constraining the analysis to either short or long read technology, we were able to significantly reduce the percent of false positive gene fusions discovered.[171]
In the future, an even newer adaptation of next generation sequencing will likely replace the current reliance on both short and long read technologies for fusion gene discovery. Paired end sequencing is a method in which short read technology is used to sequence nucleotides from both the 5’ and 3’ ends of 200-300 nucleotide fragments of the genome (or transcriptome). By sequencing both ends of a fragmented RNA, paired end sequencing enhances not only the reliability of mapping and assembly, but also maintains significant sequencing depth. In a manner similar to our recent integration of short and long read platforms, the use of paired end sequencing technology for gene fusion discovery should first be examined by comparing the ability of matched mate-pairs to identify known gene fusions from control samples. With paired end sequencing, a single sample preparation and individual sequencing run will hopefully provide sufficient coverage for gene fusion discovery and these improvements as well as other advancements in modern sequencing technologies will likewise lead to a dramatic improvement in our ability to identify novel, pathogenic gene fusions.
Lessons from the JAZF1-JJAZ1 chimera
Advances in sequencing technology will most likely lead to a rapid increase in the number of characterized gene fusions over the next few years. However, a much more pertinent question may address the reasons for chromosomal rearrangements leading to gene fusions. Could fusion transcripts be a part of normal cell biology? It is also plausible that tissue specific fusions could impart growth advantages that allow a cell to survive traumatic stress. Nonetheless, while the underlying molecular mechanisms triggering genomic rearrangement are still unclear; we surmise that once a genomic rearrangement occurs, cells harboring favorable gene fusions will be selected over time.
Insight into the development of genomic rearrangements may come from fundamental observations made following the study of endometrial stromal (EMS) tumors. In 2001, a recurrent translocation t(7;17)(p15;q21) was demonstrated to occur in EMS tumors that led to expression of the chimeric JAZF1/JJAZ1 mRNA transcript.[200] Although the mechanism leading to this rearrangement remains unknown, a recent study demonstrated that trans-splicing of RNAs in normal human endometrial stromal cells can lead to the chimeric JAZF1/JJAZ1 RNA and protein independent of chromosomal rearrangement.[201] This observation suggests that certain gene fusions may be generated by trans-splicing of RNAs, which then lead to chromosomal rearrangement due to their pro-neoplastic nature. Interestingly, the group also demonstrated that the RNA trans-splicing event leading to the JAZF1/JJAZ1 chimera was inhibited at high concentrations of either estrogen or progesterone, further suggesting that certain RNA fusions may occur in a hormone-dependent manner. The question of whether or not other specific gene fusions arise due to abnormal exposure to specific hormones has not been studied.
Conclusions
A limited number of epithelial gene fusions have been described and the quest for novel recurrent gene fusions, like the discovery of TMPRSS2-ERG gene fusions in prostate cancer, may provide major advances in cancer research in the near future. Here, we have demonstrated that gene fusions lead to over-expression or constitutive activation of oncogenes by a variety of unique mechanisms including fusion of housekeeping or tissue-specific gene promoters to oncogenes, as in the case of TMPRSS2 gene promoter and 5’-UTR to ERG or, as in the case of HMGA2, through evasion of a microRNA by replacement of an oncogene’s 3’-UTR. Despite the multitude of mechanisms used by chimeric transcripts to drive malignancy, several important lessons can be taken from characterized epithelial gene fusions, studies of MLL translocations, as well as the very recent discovery of JAZF1-JJAZ1 RNA fusions, which precede genomic rearrangement in specific cell types.
As in the case of Imatinib and BCR-ABL1, perhaps the one of the best methods for interfering with the development of specific malignancies will be through inhibition of well-characterized, pathogenic fusion genes with rationally designed molecularly tailored therapies. In the future, the use of both COPA and high throughput massively parallel sequencing will greatly increase the speed and reliability of fusion gene discovery on both the genomic and transcriptomic levels. We expect many more gene fusions to be reported over the next several years in various tumor types, many of which will hopefully serve as rational drug targets.
Figure 1.
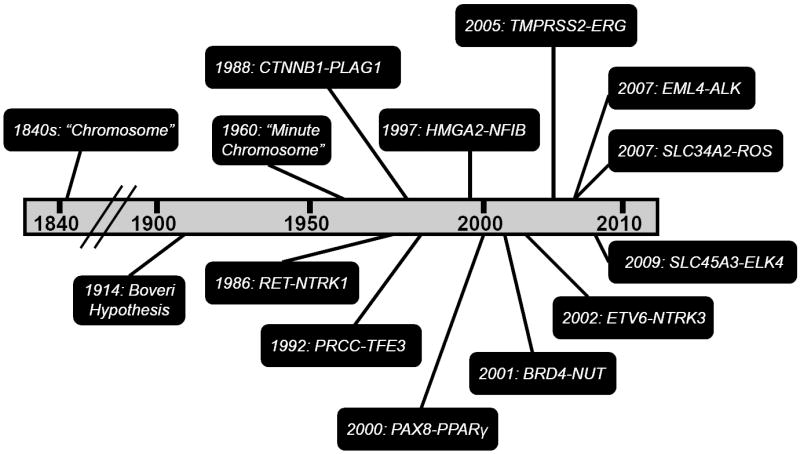
Chronology of gene fusion discoveries in epithelial cancers.
Figure 5.
Difficulty in discovering gene fusions. One possibility is that a critical function of oncogenes in epithelial cancers is to alter genomic structure and it has been suggested that such changes could lead to cancer progression. However, if such a model were true, it would give a reason for the genomic heterogeneity observed in epithelial cancers that has allowed recurrent gene fusions to go unnoticed in solid tumors.
Table 1.
Chromosomal rearrangements in epithelial cancers.
| Malignancy | Gene Fusion | Chromosome Rearrangement | Method of Discovery | Study | Ref |
|---|---|---|---|---|---|
| Follicular thyroid carcinoma | PAX8-PPARγ | t(2;3)(q13;p25) | Primary tumor karyotypic analysis/FISH/3’ RACE | Kroll et. al. | 90 |
| Midline carcinoma | BRD3-NUT | t(9;15)(q34;q14) | Canidate gene FISH Screen | French et. al. | 106 |
| BRD4-NUT | t(15;19)(q14;p13) | Primary tumor karyotypic analysis/FISH/southern blot | French et. al. | 98 | |
| Non-small cell lung cancer | EML4-ALK | inv(2p) | Transformation assay/direct sequencing | Soda et. al. | 155 |
| TFG-ALK | t(2;3)(p23;q12) | Tyrosine Kinase Activity Screen/5’ RACE | Rikova et. al. | 170 | |
| SLC34A2-ROS | t(4;6)(p15;q22) | ||||
| Papillary renal cell carcinoma | PRCC-TFE3 | t(X;1)(p11;q23) | Primary tumor karyotypic analysis/southern blot/5’ RACE | Sidhar et. al. | 69 |
| Papillary thyroid carcinoma | RET-NTRK1 | t(1;10)(q21;q11) | Transformation assay/direct sequencing | Martin-Zanca et. al. | 24 |
| Pleomorphic adenoma | CTTNB1-PLAG1 | t(3;8)(p21;q12) | Primary tumor karyotypic analysis/Breakpoint mapping/southern blot/5’ Race | Kas et. al. | 59 |
| HMGA2-FHIT | t(3;12)(p14;q15) | Primary tumor karyotypic analysis/3’ RACE | Geurts et. al. | 80 | |
| HMGA2-NFIB | t(9;12)(q24;q15) | Primary tumor karyotypic analysis/3’ RACE | Geurts et. al. | 81 | |
| Prostate cancer | TMPRSS2-ERG | del(21)(q22) | COPA/Exon walking/5’ RACE | Tomlins et. al. | 135 |
| TMPRSS2-ETV1 | t(7;21)(p21;q22) | ||||
| TMPRSS2-ETV4 | t(17;21)(q21;q22) | Tomlins et. al. | 149 | ||
| TMPRSS2-ETV5 | t(3;21)(p28;q22) | Helgeson et. al. | 151 | ||
| SLC45A3-ELK4 | del(1)(q32) | Intergrated high throughput sequencing | Maher et. al. | 171 | |
| DDX5-ETV4 | t(17)(q24;q21) | Canidate gene FISH Screen/5’ RACE | Han et. al. | 150 | |
| Secretory breast carcinoma | ETV6-NTRK3 | t(12;15)(q13;q25) | Primary tumor karyotypic analysis/FISH | Tognon et. al. | 108 |
Acknowledgments
We thank Jill Granger for critically reading the manuscript. A.M.C. is supported by a Burroughs Welcome Foundation Award in Clinical Translational Research. This work was supported in part by the National Institutes of Health Prostate SPORE P50CA69568 to A.M.C., and the Early Detection Research Network (UO1 UO1 CA113913 to A.M.C.) and the Department of Defense (PC051081 to A.M.C., BC083217 to J.C.B.).
References
- 1.Von Hansemann D. Ueber asymmetrische zelltheilung in epithelhresbsen und deren biologische bedeutung. Virchows Arch A Pathol Anat. 1890;119:299–326. [Google Scholar]
- 2.Boveri T. Zur frage der entstehung maligner tumoren. Jena, Germany: 1914. [Google Scholar]
- 3.Lejeune J, Gautier M, Turpin R. Study of somatic chromosomes from 9 mongoloid children. Comptes rendus hebdomadaires des seances de l’Academie des sciences. 1959;248:1721–1722. [PubMed] [Google Scholar]
- 4.Lejeune J, Turpin R, Gautier M. Mongolism; a chromosomal disease (trisomy) Bulletin de l’Academie nationale de medecine. 1959;143:256–265. [PubMed] [Google Scholar]
- 5.Rothfels KH, Siminovitch L. An air-drying technique for flattening chromosomes in mammalian oells grown in vitro. Stain technology. 1958;33:73–77. doi: 10.3109/10520295809111827. [DOI] [PubMed] [Google Scholar]
- 6.Nowell P, Hungerford D. A minute chromosome in human chronic granulocytic leukemia. Science (New York, N Y) 1960;132:1497. doi: 10.1126/science.144.3623.1229. Abstract. [DOI] [PubMed] [Google Scholar]
- 7.Nowell PC, Hungerford DA. Chromosome studies in human leukemia. II. Chronic granulocytic leukemia. Journal of the National Cancer Institute. 1961;27:1013–1035. [PubMed] [Google Scholar]
- 8.Tough IM, Court Brown WM, Baikie AG, Buckton KE, Harnden DG, Jacobs PA, King MJ, Mc BJ. Cytogenetic studies in chronic myeloid leukaemia and acute leukaemia associated with monogolism. Lancet. 1961;1:411–417. doi: 10.1016/s0140-6736(61)90001-0. [DOI] [PubMed] [Google Scholar]
- 9.Rowley JD. Ph1-positive leukaemia, including chronic myelogenous leukaemia. Clinics in haematology. 1980;9:55–86. [PubMed] [Google Scholar]
- 10.Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984;36:93–99. doi: 10.1016/0092-8674(84)90077-1. [DOI] [PubMed] [Google Scholar]
- 11.Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science (New York, N Y) 1990;247:1079–1082. doi: 10.1126/science.2408149. [DOI] [PubMed] [Google Scholar]
- 12.Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, Capdeville R, Talpaz M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. The New England journal of medicine. 2001;344:1038–1042. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 13.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. The New England journal of medicine. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 14.Zech L, Haglund U, Nilsson K, Klein G. Characteristic chromosomal abnormalities in biopsies and lymphoid-cell lines from patients with Burkitt and non-Burkitt lymphomas. International journal of cancer. 1976;17:47–56. doi: 10.1002/ijc.2910170108. [DOI] [PubMed] [Google Scholar]
- 15.Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:7824–7827. doi: 10.1073/pnas.79.24.7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.ar-Rushdi A, Nishikura K, Erikson J, Watt R, Rovera G, Croce CM. Differential expression of the translocated and the untranslocated c-myc oncogene in Burkitt lymphoma. Science (New York, N Y) 1983;222:390–393. doi: 10.1126/science.6414084. [DOI] [PubMed] [Google Scholar]
- 17.Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- 18.Rowley JD. Identificaton of a translocation with quinacrine fluorescence in a patient with acute leukemia. Annales de genetique. 1973;16:109–112. [PubMed] [Google Scholar]
- 19.Gao J, Erickson P, Gardiner K, Le Beau MM, Diaz MO, Patterson D, Rowley JD, Drabkin HA. Isolation of a yeast artificial chromosome spanning the 8;21 translocation breakpoint t(8;21)(q22;q22.3) in acute myelogenous leukemia. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:4882–4886. doi: 10.1073/pnas.88.11.4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nucifora G, Rowley JD. AML1 and the 8;21 and 3;21 translocations in acute and chronic myeloid leukemia. Blood. 1995;86:1–14. [PubMed] [Google Scholar]
- 21.Roulston D, Espinosa R, 3rd, Nucifora G, Larson RA, Le Beau MM, Rowley JD. CBFA2(AML1) translocations with novel partner chromosomes in myeloid leukemias: association with prior therapy. Blood. 1998;92:2879–2885. [PubMed] [Google Scholar]
- 22.Meyer C, Schneider B, Jakob S, Strehl S, Attarbaschi A, Schnittger S, Schoch C, Jansen MW, van Dongen JJ, den Boer ML, Pieters R, Ennas MG, Angelucci E, Koehl U, Greil J, Griesinger F, Zur Stadt U, Eckert C, Szczepanski T, Niggli FK, Schafer BW, Kempski H, Brady HJ, Zuna J, Trka J, Nigro LL, Biondi A, Delabesse E, Macintyre E, Stanulla M, Schrappe M, Haas OA, Burmeister T, Dingermann T, Klingebiel T, Marschalek R. The MLL recombinome of acute leukemias. Leukemia. 2006;20:777–784. doi: 10.1038/sj.leu.2404150. [DOI] [PubMed] [Google Scholar]
- 23.Mitelman F, Johansson B, Mertens F. Mitelman Database of Chromosome Aberrations in Cancer. 2008 http://cgap.nci.nih.gov/Chromosomes/Mitelman.
- 24.Martin-Zanca D, Hughes SH, Barbacid M. A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature. 1986;319:743–748. doi: 10.1038/319743a0. [DOI] [PubMed] [Google Scholar]
- 25.Ishizaka Y, Itoh F, Tahira T, Ikeda I, Sugimura T, Tucker J, Fertitta A, Carrano AV, Nagao M. Human ret proto-oncogene mapped to chromosome 10q11.2. Oncogene. 1989;4:1519–1521. [PubMed] [Google Scholar]
- 26.Ishizaka Y, Tahira T, Ochiai M, Ikeda I, Sugimura T, Nagao M. Molecular cloning and characterization of human ret-II oncogene. Oncogene research. 1988;3:193–197. [PubMed] [Google Scholar]
- 27.Takahashi M, Ritz J, Cooper GM. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell. 1985;42:581–588. doi: 10.1016/0092-8674(85)90115-1. [DOI] [PubMed] [Google Scholar]
- 28.Kaplan DR, Miller FD. Signal transduction by the neurotrophin receptors. Current opinion in cell biology. 1997;9:213–221. doi: 10.1016/s0955-0674(97)80065-8. [DOI] [PubMed] [Google Scholar]
- 29.Greco A, Fusetti L, Miranda C, Villa R, Zanotti S, Pagliardini S, Pierotti MA. Role of the TFG N-terminus and coiled-coil domain in the transforming activity of the thyroid TRK-T3 oncogene. Oncogene. 1998;16:809–816. doi: 10.1038/sj.onc.1201596. [DOI] [PubMed] [Google Scholar]
- 30.Grieco M, Santoro M, Berlingieri MT, Melillo RM, Donghi R, Bongarzone I, Pierotti MA, Della Porta G, Fusco A, Vecchio G. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell. 1990;60:557–563. doi: 10.1016/0092-8674(90)90659-3. [DOI] [PubMed] [Google Scholar]
- 31.Pierotti MA, Santoro M, Jenkins RB, Sozzi G, Bongarzone I, Grieco M, Monzini N, Miozzo M, Herrmann MA, Fusco A, et al. Characterization of an inversion on the long arm of chromosome 10 juxtaposing D10S170 and RET and creating the oncogenic sequence RET/PTC. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:1616–1620. doi: 10.1073/pnas.89.5.1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sozzi G, Bongarzone I, Miozzo M, Borrello MG, Blutti MG, Pilotti S, Della Porta G, Pierotti MA. A t(10;17) translocation creates the RET/PTC2 chimeric transforming sequence in papillary thyroid carcinoma. Genes, chromosomes & cancer. 1994;9:244–250. doi: 10.1002/gcc.2870090404. [DOI] [PubMed] [Google Scholar]
- 33.Klugbauer S, Demidchik EP, Lengfelder E, Rabes HM. Detection of a novel type of RET rearrangement (PTC5) in thyroid carcinomas after Chernobyl and analysis of the involved RET-fused gene RFG5. Cancer research. 1998;58:198–203. [PubMed] [Google Scholar]
- 34.Fugazzola L, Pierotti MA, Vigano E, Pacini F, Vorontsova TV, Bongarzone I. Molecular and biochemical analysis of RET/PTC4, a novel oncogenic rearrangement between RET and ELE1 genes, in a post-Chernobyl papillary thyroid cancer. Oncogene. 1996;13:1093–1097. [PubMed] [Google Scholar]
- 35.Santoro M, Dathan NA, Berlingieri MT, Bongarzone I, Paulin C, Grieco M, Pierotti MA, Vecchio G, Fusco A. Molecular characterization of RET/PTC3; a novel rearranged version of the RETproto-oncogene in a human thyroid papillary carcinoma. Oncogene. 1994;9:509–516. [PubMed] [Google Scholar]
- 36.Alberti L, Carniti C, Miranda C, Roccato E, Pierotti MA. RET and NTRK1 proto-oncogenes in human diseases. Journal of cellular physiology. 2003;195:168–186. doi: 10.1002/jcp.10252. [DOI] [PubMed] [Google Scholar]
- 37.Hamatani K, Eguchi H, Ito R, Mukai M, Takahashi K, Taga M, Imai K, Cologne J, Soda M, Arihiro K, Fujihara M, Abe K, Hayashi T, Nakashima M, Sekine I, Yasui W, Hayashi Y, Nakachi K. RET/PTC rearrangements preferentially occurred in papillary thyroid cancer among atomic bomb survivors exposed to high radiation dose. Cancer research. 2008;68:7176–7182. doi: 10.1158/0008-5472.CAN-08-0293. [DOI] [PubMed] [Google Scholar]
- 38.Fukushima T, Suzuki S, Mashiko M, Ohtake T, Endo Y, Takebayashi Y, Sekikawa K, Hagiwara K, Takenoshita S. BRAF mutations in papillary carcinomas of the thyroid. Oncogene. 2003;22:6455–6457. doi: 10.1038/sj.onc.1206739. [DOI] [PubMed] [Google Scholar]
- 39.Cohen Y, Xing M, Mambo E, Guo Z, Wu G, Trink B, Beller U, Westra WH, Ladenson PW, Sidransky D. BRAF mutation in papillary thyroid carcinoma. Journal of the National Cancer Institute. 2003;95:625–627. doi: 10.1093/jnci/95.8.625. [DOI] [PubMed] [Google Scholar]
- 40.Nikiforova MN, Kimura ET, Gandhi M, Biddinger PW, Knauf JA, Basolo F, Zhu Z, Giannini R, Salvatore G, Fusco A, Santoro M, Fagin JA, Nikiforov YE. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. The Journal of clinical endocrinology and metabolism. 2003;88:5399–5404. doi: 10.1210/jc.2003-030838. [DOI] [PubMed] [Google Scholar]
- 41.Namba H, Nakashima M, Hayashi T, Hayashida N, Maeda S, Rogounovitch TI, Ohtsuru A, Saenko VA, Kanematsu T, Yamashita S. Clinical implication of hot spot BRAF mutation, V599E, in papillary thyroid cancers. The Journal of clinical endocrinology and metabolism. 2003;88:4393–4397. doi: 10.1210/jc.2003-030305. [DOI] [PubMed] [Google Scholar]
- 42.Frattini M, Ferrario C, Bressan P, Balestra D, De Cecco L, Mondellini P, Bongarzone I, Collini P, Gariboldi M, Pilotti S, Pierotti MA, Greco A. Alternative mutations of BRAF, RET and NTRK1 are associated with similar but distinct gene expression patterns in papillary thyroid cancer. Oncogene. 2004;23:7436–7440. doi: 10.1038/sj.onc.1207980. [DOI] [PubMed] [Google Scholar]
- 43.Esapa CT, Johnson SJ, Kendall-Taylor P, Lennard TW, Harris PE. Prevalence of Ras mutations in thyroid neoplasia. Clinical endocrinology. 1999;50:529–535. doi: 10.1046/j.1365-2265.1999.00704.x. [DOI] [PubMed] [Google Scholar]
- 44.Bongarzone I, Fugazzola L, Vigneri P, Mariani L, Mondellini P, Pacini F, Basolo F, Pinchera A, Pilotti S, Pierotti MA. Age-related activation of the tyrosine kinase receptor protooncogenes RET and NTRK1 in papillary thyroid carcinoma. The Journal of clinical endocrinology and metabolism. 1996;81:2006–2009. doi: 10.1210/jcem.81.5.8626874. [DOI] [PubMed] [Google Scholar]
- 45.Tallini G, Asa SL. RET oncogene activation in papillary thyroid carcinoma. Advances in anatomic pathology. 2001;8:345–354. doi: 10.1097/00125480-200111000-00005. [DOI] [PubMed] [Google Scholar]
- 46.Chua EL, Wu WM, Tran KT, McCarthy SW, Lauer CS, Dubourdieu D, Packham N, O’Brien CJ, Turtle JR, Dong Q. Prevalence and distribution of ret/ptc 1, 2, and 3 in papillary thyroid carcinoma in New Caledonia and Australia. The Journal of clinical endocrinology and metabolism. 2000;85:2733–2739. doi: 10.1210/jcem.85.8.6722. [DOI] [PubMed] [Google Scholar]
- 47.Learoyd DL, Messina M, Zedenius J, Guinea AI, Delbridge LW, Robinson BG. RET/PTC and RET tyrosine kinase expression in adult papillary thyroid carcinomas. The Journal of clinical endocrinology and metabolism. 1998;83:3631–3635. doi: 10.1210/jcem.83.10.5152. [DOI] [PubMed] [Google Scholar]
- 48.Nakachi K, Hayashi T, Hamatani K, Eguchi H, Kusunoki Y. Sixty years of follow-up of Hiroshima and Nagasaki survivors: current progress in molecular epidemiology studies. Mutation research. 2008;659:109–117. doi: 10.1016/j.mrrev.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 49.Kazakov VS, Demidchik EP, Astakhova LN. Thyroid cancer after Chernobyl. Nature. 1992;359:21. doi: 10.1038/359021a0. [DOI] [PubMed] [Google Scholar]
- 50.Astakhova LN, Anspaugh LR, Beebe GW, Bouville A, Drozdovitch VV, Garber V, Gavrilin YI, Khrouch VT, Kuvshinnikov AV, Kuzmenkov YN, Minenko VP, Moschik KV, Nalivko AS, Robbins J, Shemiakina EV, Shinkarev S, Tochitskaya SI, Waclawiw MA. Chernobyl-related thyroid cancer in children of Belarus: a case-control study. Radiation research. 1998;150:349–356. [PubMed] [Google Scholar]
- 51.Imaizumi M, Usa T, Tominaga T, Neriishi K, Akahoshi M, Nakashima E, Ashizawa K, Hida A, Soda M, Fujiwara S, Yamada M, Ejima E, Yokoyama N, Okubo M, Sugino K, Suzuki G, Maeda R, Nagataki S, Eguchi K. Radiation dose-response relationships for thyroid nodules and autoimmune thyroid diseases in Hiroshima and Nagasaki atomic bomb survivors 55-58 years after radiation exposure. Jama. 2006;295:1011–1022. doi: 10.1001/jama.295.9.1011. [DOI] [PubMed] [Google Scholar]
- 52.Pierotti MA, Bongarzone I, Borello MG, Greco A, Pilotti S, Sozzi G. Cytogenetics and molecular genetics of carcinomas arising from thyroid epithelial follicular cells. Genes, chromosomes & cancer. 1996;16:1–14. doi: 10.1002/(SICI)1098-2264(199605)16:1<1::AID-GCC1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 53.Bullerdiek J, Raabe G, Boschen C, Bartnitzke S. Translocation (3;8;8)(p22 or p23;p23;q12) in a case of pleomorphic adenoma: similarity to a primary cytogenetic abnormality detected in an endometrial adenocarcinoma. Cancer genetics and cytogenetics. 1987;27:177–180. doi: 10.1016/0165-4608(87)90273-1. [DOI] [PubMed] [Google Scholar]
- 54.Waldron C. Mixed tumor (Pleomorphic adenoma and myoepithelioma) In: Gnepp GEPAD, editor. Surgical Pathology of the salivary glands. W.B Saunders Co.; Philadelphia: 1991. pp. 165–184. [Google Scholar]
- 55.Shah JP, Ihde JK. Salivary gland tumors. Current problems in surgery. 1990;27:775–883. doi: 10.1016/0011-3840(90)90032-z. [DOI] [PubMed] [Google Scholar]
- 56.Stenman G. Fusion oncogenes and tumor type specificity--insights from salivary gland tumors. Seminars in cancer biology. 2005;15:224–235. doi: 10.1016/j.semcancer.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 57.Bullerdiek J, Haubrich J, Meyer K, Bartnitzke S. Translocation t(11;19)(q21;p13.1) as the sole chromosome abnormality in a cystadenolymphoma (Warthin’s tumor) of the parotid gland. Cancer genetics and cytogenetics. 1988;35:129–132. doi: 10.1016/0165-4608(88)90131-8. [DOI] [PubMed] [Google Scholar]
- 58.Bullerdiek J, Wobst G, Meyer-Bolte K, Chilla R, Haubrich J, Thode B, Bartnitzke S. Cytogenetic subtyping of 220 salivary gland pleomorphic adenomas: correlation to occurrence, histological subtype, and in vitro cellular behavior. Cancer genetics and cytogenetics. 1993;65:27–31. doi: 10.1016/0165-4608(93)90054-p. [DOI] [PubMed] [Google Scholar]
- 59.Kas K, Voz ML, Roijer E, Astrom AK, Meyen E, Stenman G, Van de Ven WJ. Promoter swapping between the genes for a novel zinc finger protein and beta-catenin in pleiomorphic adenomas with t(3;8)(p21;q12) translocations. Nature genetics. 1997;15:170–174. doi: 10.1038/ng0297-170. [DOI] [PubMed] [Google Scholar]
- 60.Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science (New York, N Y) 2004;303:1483–1487. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hensen K, Van Valckenborgh IC, Kas K, Van de Ven WJ, Voz ML. The tumorigenic diversity of the three PLAG family members is associated with different DNA binding capacities. Cancer research. 2002;62:1510–1517. [PubMed] [Google Scholar]
- 62.de Jong B, Molenaar IM, Leeuw JA, Idenberg VJ, Oosterhuis JW. Cytogenetics of a renal adenocarcinoma in a 2-year-old child. Cancer genetics and cytogenetics. 1986;21:165–169. doi: 10.1016/0165-4608(86)90042-7. [DOI] [PubMed] [Google Scholar]
- 63.Meloni AM, Dobbs RM, Pontes JE, Sandberg AA. Translocation (X;1) in papillary renal cell carcinoma. A new cytogenetic subtype. Cancer genetics and cytogenetics. 1993;65:1–6. doi: 10.1016/0165-4608(93)90050-v. [DOI] [PubMed] [Google Scholar]
- 64.Meloni AM, Sandberg AA, Pontes JE, Dobbs RM., Jr Translocation (X;1)(p11.2;q21). A subtype of renal adenocarcinomas. Cancer genetics and cytogenetics. 1992;63:100–101. doi: 10.1016/0165-4608(92)90388-o. [DOI] [PubMed] [Google Scholar]
- 65.Shipley JM, Birdsall S, Clark J, Crew J, Gill S, Linehan M, Gnarra J, Fisher S, Craig IW, Cooper CS. Mapping the X chromosome breakpoint in two papillary renal cell carcinoma cell lines with a t(X;1)(p11.2;q21.2) and the first report of a female case. Cytogenetics and cell genetics. 1995;71:280–284. doi: 10.1159/000134127. [DOI] [PubMed] [Google Scholar]
- 66.Tonk V, Wilson KS, Timmons CF, Schneider NR, Tomlinson GE. Renal cell carcinoma with translocation (X;1). Further evidence for a cytogenetically defined subtype. Cancer genetics and cytogenetics. 1995;81:72–75. doi: 10.1016/s0165-4608(94)00195-2. [DOI] [PubMed] [Google Scholar]
- 67.Beckmann H, Su LK, Kadesch T. TFE3: a helix-loop-helix protein that activates transcription through the immunoglobulin enhancer muE3 motif. Genes & development. 1990;4:167–179. doi: 10.1101/gad.4.2.167. [DOI] [PubMed] [Google Scholar]
- 68.Henthorn PS, Stewart CC, Kadesch T, Puck JM. The gene encoding human TFE3, a transcription factor that binds the immunoglobulin heavy-chain enhancer, maps to Xp11.22. Genomics. 1991;11:374–378. doi: 10.1016/0888-7543(91)90145-5. [DOI] [PubMed] [Google Scholar]
- 69.Sidhar SK, Clark J, Gill S, Hamoudi R, Crew AJ, Gwilliam R, Ross M, Linehan WM, Birdsall S, Shipley J, Cooper CS. The t(X;1)(p11.2;q21.2) translocation in papillary renal cell carcinoma fuses a novel gene PRCC to the TFE3 transcription factor gene. Human molecular genetics. 1996;5:1333–1338. doi: 10.1093/hmg/5.9.1333. [DOI] [PubMed] [Google Scholar]
- 70.Sudol M. From Src Homology domains to other signaling modules: proposal of the ‘protein recognition code’. Oncogene. 1998;17:1469–1474. doi: 10.1038/sj.onc.1202182. [DOI] [PubMed] [Google Scholar]
- 71.Ren R, Mayer BJ, Cicchetti P, Baltimore D. Identification of a ten-amino acid proline-rich SH3 binding site. Science (New York, N Y) 1993;259:1157–1161. doi: 10.1126/science.8438166. [DOI] [PubMed] [Google Scholar]
- 72.Weterman MA, Wilbrink M, Geurts van Kessel A. Fusion of the transcription factor TFE3 gene to a novel gene, PRCC, in t(X;1)(p11;q21)-positive papillary renal cell carcinomas. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:15294–15298. doi: 10.1073/pnas.93.26.15294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weterman MJ, van Groningen JJ, Jansen A, van Kessel AG. Nuclear localization and transactivating capacities of the papillary renal cell carcinoma-associated TFE3 and PRCC (fusion) proteins. Oncogene. 2000;19:69–74. doi: 10.1038/sj.onc.1203255. [DOI] [PubMed] [Google Scholar]
- 74.Clark J, Lu YJ, Sidhar SK, Parker C, Gill S, Smedley D, Hamoudi R, Linehan WM, Shipley J, Cooper CS. Fusion of splicing factor genes PSF and NonO (p54nrb) to the TFE3 gene in papillary renal cell carcinoma. Oncogene. 1997;15:2233–2239. doi: 10.1038/sj.onc.1201394. [DOI] [PubMed] [Google Scholar]
- 75.Skalsky YM, Ajuh PM, Parker C, Lamond AI, Goodwin G, Cooper CS. PRCC, the commonest TFE3 fusion partner in papillary renal carcinoma is associated with pre-mRNA splicing factors. Oncogene. 2001;20:178–187. doi: 10.1038/sj.onc.1204056. [DOI] [PubMed] [Google Scholar]
- 76.Odero MD, Grand FH, Iqbal S, Ross F, Roman JP, Vizmanos JL, Andrieux J, Lai JL, Calasanz MJ, Cross NC. Disruption and aberrant expression of HMGA2 as a consequence of diverse chromosomal translocations in myeloid malignancies. Leukemia. 2005;19:245–252. doi: 10.1038/sj.leu.2403605. [DOI] [PubMed] [Google Scholar]
- 77.Schoenmakers EF, Wanschura S, Mols R, Bullerdiek J, Van den Berghe H, Van de Ven WJ. Recurrent rearrangements in the high mobility group protein gene, HMGI-C, in benign mesenchymal tumours. Nature genetics. 1995;10:436–444. doi: 10.1038/ng0895-436. [DOI] [PubMed] [Google Scholar]
- 78.Ashar HR, Fejzo MS, Tkachenko A, Zhou X, Fletcher JA, Weremowicz S, Morton CC, Chada K. Disruption of the architectural factor HMGI-C: DNA-binding AT hook motifs fused in lipomas to distinct transcriptional regulatory domains. Cell. 1995;82:57–65. doi: 10.1016/0092-8674(95)90052-7. [DOI] [PubMed] [Google Scholar]
- 79.Agresti A, Bianchi ME. HMGB proteins and gene expression. Current opinion in genetics & development. 2003;13:170–178. doi: 10.1016/s0959-437x(03)00023-6. [DOI] [PubMed] [Google Scholar]
- 80.Geurts JM, Schoenmakers EF, Roijer E, Stenman G, Van de Ven WJ. Expression of reciprocal hybrid transcripts of HMGIC and FHIT in a pleomorphic adenoma of the parotid gland. Cancer research. 1997;57:13–17. [PubMed] [Google Scholar]
- 81.Geurts JM, Schoenmakers EF, Roijer E, Astrom AK, Stenman G, van de Ven WJ. Identification of NFIB as recurrent translocation partner gene of HMGIC in pleomorphic adenomas. Oncogene. 1998;16:865–872. doi: 10.1038/sj.onc.1201609. [DOI] [PubMed] [Google Scholar]
- 82.Fedele M, Battista S, Kenyon L, Baldassarre G, Fidanza V, Klein-Szanto AJ, Parlow AF, Visone R, Pierantoni GM, Outwater E, Santoro M, Croce CM, Fusco A. Overexpression of the HMGA2 gene in transgenic mice leads to the onset of pituitary adenomas. Oncogene. 2002;21:3190–3198. doi: 10.1038/sj.onc.1205428. [DOI] [PubMed] [Google Scholar]
- 83.Battista S, Fidanza V, Fedele M, Klein-Szanto AJ, Outwater E, Brunner H, Santoro M, Croce CM, Fusco A. The expression of a truncated HMGI-C gene induces gigantism associated with lipomatosis. Cancer research. 1999;59:4793–4797. [PubMed] [Google Scholar]
- 84.Baldassarre G, Fedele M, Battista S, Vecchione A, Klein-Szanto AJ, Santoro M, Waldmann TA, Azimi N, Croce CM, Fusco A. Onset of natural killer cell lymphomas in transgenic mice carrying a truncated HMGI-C gene by the chronic stimulation of the IL-2 and IL-15 pathway. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:7970–7975. doi: 10.1073/pnas.141224998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ohta M, Inoue H, Cotticelli MG, Kastury K, Baffa R, Palazzo J, Siprashvili Z, Mori M, McCue P, Druck T, Croce CM, Huebner K. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell. 1996;84:587–597. doi: 10.1016/s0092-8674(00)81034-x. [DOI] [PubMed] [Google Scholar]
- 86.Gemmill RM, West JD, Boldog F, Tanaka N, Robinson LJ, Smith DI, Li F, Drabkin HA. The hereditary renal cell carcinoma 3;8 translocation fuses FHIT to a patched-related gene. TRC8, Proceedings of the National Academy of Sciences of the United States of America. 1998;95:9572–9577. doi: 10.1073/pnas.95.16.9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 88.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 89.Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science (New York, N Y) 2007;315:1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kroll TG, Sarraf P, Pecciarini L, Chen CJ, Mueller E, Spiegelman BM, Fletcher JA. PAX8-PPARgamma1 fusion oncogene in human thyroid carcinoma. Science (New York, N Y) 2000;289:1357–1360. doi: 10.1126/science.289.5483.1357. corrected. [DOI] [PubMed] [Google Scholar]
- 91.Gimm O. Thyroid cancer. Cancer letters. 2001;163:143–156. doi: 10.1016/s0304-3835(00)00697-2. [DOI] [PubMed] [Google Scholar]
- 92.Nikiforova MN, Nikiforov YE. Molecular genetics of thyroid cancer: implications for diagnosis, treatment and prognosis. Expert review of molecular diagnostics. 2008;8:83–95. doi: 10.1586/14737159.8.1.83. [DOI] [PubMed] [Google Scholar]
- 93.Puppin C, Presta I, D’Elia AV, Tell G, Arturi F, Russo D, Filetti S, Damante G. Functional interaction among thyroid-specific transcription factors: Pax8 regulates the activity of Hex promoter. Molecular and cellular endocrinology. 2004;214:117–125. doi: 10.1016/j.mce.2003.10.061. [DOI] [PubMed] [Google Scholar]
- 94.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annual review of biochemistry. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 95.Lui WO, Foukakis T, Liden J, Thoppe SR, Dwight T, Hoog A, Zedenius J, Wallin G, Reimers M, Larsson C. Expression profiling reveals a distinct transcription signature in follicular thyroid carcinomas with a PAX8-PPAR(gamma) fusion oncogene. Oncogene. 2005;24:1467–1476. doi: 10.1038/sj.onc.1208135. [DOI] [PubMed] [Google Scholar]
- 96.Nikiforova MN, Lynch RA, Biddinger PW, Alexander EK, Dorn GW, 2nd, Tallini G, Kroll TG, Nikiforov YE. RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors: evidence for distinct molecular pathways in thyroid follicular carcinoma. The Journal of clinical endocrinology and metabolism. 2003;88:2318–2326. doi: 10.1210/jc.2002-021907. [DOI] [PubMed] [Google Scholar]
- 97.Foukakis T, Au AY, Wallin G, Geli J, Forsberg L, Clifton-Bligh R, Robinson BG, Lui WO, Zedenius J, Larsson C. The Ras effector NORE1A is suppressed in follicular thyroid carcinomas with a PAX8-PPARgamma fusion. The Journal of clinical endocrinology and metabolism. 2006;91:1143–1149. doi: 10.1210/jc.2005-1372. [DOI] [PubMed] [Google Scholar]
- 98.French CA, Miyoshi I, Aster JC, Kubonishi I, Kroll TG, Dal Cin P, Vargas SO, Perez-Atayde AR, Fletcher JA. BRD4 bromodomain gene rearrangement in aggressive carcinoma with translocation t(15;19) The American journal of pathology. 2001;159:1987–1992. doi: 10.1016/S0002-9440(10)63049-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.French CA, Miyoshi I, Kubonishi I, Grier HE, Perez-Atayde AR, Fletcher JA. BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer research. 2003;63:304–307. [PubMed] [Google Scholar]
- 100.French CA. Molecular pathology of NUT midline carcinomas. Journal of clinical pathology. 2008 doi: 10.1136/jcp.2007.052902. [DOI] [PubMed] [Google Scholar]
- 101.Dey A, Ellenberg J, Farina A, Coleman AE, Maruyama T, Sciortino S, Lippincott-Schwartz J, Ozato K. A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G(2)-to-M transition. Molecular and cellular biology. 2000;20:6537–6549. doi: 10.1128/mcb.20.17.6537-6549.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Maruyama T, Farina A, Dey A, Cheong J, Bermudez VP, Tamura T, Sciortino S, Shuman J, Hurwitz J, Ozato K. A Mammalian bromodomain protein, brd4, interacts with replication factor C and inhibits progression to S phase. Molecular and cellular biology. 2002;22:6509–6520. doi: 10.1128/MCB.22.18.6509-6520.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Farina A, Hattori M, Qin J, Nakatani Y, Minato N, Ozato K. Bromodomain protein Brd4 binds to GTPase-activating SPA-1, modulating its activity and subcellular localization. Molecular and cellular biology. 2004;24:9059–9069. doi: 10.1128/MCB.24.20.9059-9069.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8758–8763. doi: 10.1073/pnas.1433065100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Molecular cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 106.French CA, Ramirez CL, Kolmakova J, Hickman TT, Cameron MJ, Thyne ME, Kutok JL, Toretsky JA, Tadavarthy AK, Kees UR, Fletcher JA, Aster JC. BRD-NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene. 2008;27:2237–2242. doi: 10.1038/sj.onc.1210852. [DOI] [PubMed] [Google Scholar]
- 107.Oberman HA. Secretory carcinoma of the breast in adults. The American journal of surgical pathology. 1980;4:465–470. doi: 10.1097/00000478-198010000-00006. [DOI] [PubMed] [Google Scholar]
- 108.Tognon C, Knezevich SR, Huntsman D, Roskelley CD, Melnyk N, Mathers JA, Becker L, Carneiro F, MacPherson N, Horsman D, Poremba C, Sorensen PH. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer cell. 2002;2:367–376. doi: 10.1016/s1535-6108(02)00180-0. [DOI] [PubMed] [Google Scholar]
- 109.Golub TR, Barker GF, Bohlander SK, Hiebert SW, Ward DC, Bray-Ward P, Morgan E, Raimondi SC, Rowley JD, Gilliland DG. Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:4917–4921. doi: 10.1073/pnas.92.11.4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Papadopoulos P, Ridge SA, Boucher CA, Stocking C, Wiedemann LM. The novel activation of ABL by fusion to an ets-related gene, TEL. Cancer research. 1995;55:34–38. [PubMed] [Google Scholar]
- 111.Golub TR, Goga A, Barker GF, Afar DE, McLaughlin J, Bohlander SK, Rowley JD, Witte ON, Gilliland DG. Oligomerization of the ABL tyrosine kinase by the Ets protein TEL in human leukemia. Molecular and cellular biology. 1996;16:4107–4116. doi: 10.1128/mcb.16.8.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffe M, Berthou C, Lessard M, Berger R, Ghysdael J, Bernard OA. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science (New York, N Y) 1997;278:1309–1312. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- 113.Peeters P, Raynaud SD, Cools J, Wlodarska I, Grosgeorge J, Philip P, Monpoux F, Van Rompaey L, Baens M, Van den Berghe H, Marynen P. Fusion of TEL, the ETS-variant gene 6 (ETV6), to the receptor-associated kinase JAK2 as a result of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. Blood. 1997;90:2535–2540. [PubMed] [Google Scholar]
- 114.Schwaller J, Frantsve J, Aster J, Williams IR, Tomasson MH, Ross TS, Peeters P, Van Rompaey L, Van Etten RA, Ilaria R, Jr, Marynen P, Gilliland DG. Transformation of hematopoietic cell lines to growth-factor independence and induction of a fatal myelo- and lymphoproliferative disease in mice by retrovirally transduced TEL/JAK2 fusion genes. The EMBO journal. 1998;17:5321–5333. doi: 10.1093/emboj/17.18.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cazzaniga G, Tosi S, Aloisi A, Giudici G, Daniotti M, Pioltelli P, Kearney L, Biondi A. The tyrosine kinase abl-related gene ARG is fused to ETV6 in an AML-M4Eo patient with a t(1;12)(q25;p13): molecular cloning of both reciprocal transcripts. Blood. 1999;94:4370–4373. [PubMed] [Google Scholar]
- 116.Iijima Y, Ito T, Oikawa T, Eguchi M, Eguchi-Ishimae M, Kamada N, Kishi K, Asano S, Sakaki Y, Sato Y. A new ETV6/TEL partner gene, ARG (ABL-related gene or ABL2), identified in an AML-M3 cell line with a t(1;12)(q25;p13) translocation. Blood. 2000;95:2126–2131. [PubMed] [Google Scholar]
- 117.Golub TR, Barker GF, Lovett M, Gilliland DG. Fusion of PDGF receptor beta to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell. 1994;77:307–316. doi: 10.1016/0092-8674(94)90322-0. [DOI] [PubMed] [Google Scholar]
- 118.Yagasaki F, Wakao D, Yokoyama Y, Uchida Y, Murohashi I, Kayano H, Taniwaki M, Matsuda A, Bessho M. Fusion of ETV6 to fibroblast growth factor receptor 3 in peripheral T-cell lymphoma with a t(4;12)(p16;p13) chromosomal translocation. Cancer research. 2001;61:8371–8374. [PubMed] [Google Scholar]
- 119.Bohlander SK. ETV6: a versatile player in leukemogenesis. Seminars in cancer biology. 2005;15:162–174. doi: 10.1016/j.semcancer.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 120.Wang LC, Swat W, Fujiwara Y, Davidson L, Visvader J, Kuo F, Alt FW, Gilliland DG, Golub TR, Orkin SH. The TEL/ETV6 gene is required specifically for hematopoiesis in the bone marrow. Genes & development. 1998;12:2392–2402. doi: 10.1101/gad.12.15.2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Current opinion in neurobiology. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 122.Knezevich SR, McFadden DE, Tao W, Lim JF, Sorensen PH. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nature genetics. 1998;18:184–187. doi: 10.1038/ng0298-184. [DOI] [PubMed] [Google Scholar]
- 123.Lannon CL, Sorensen PH. ETV6-NTRK3: a chimeric protein tyrosine kinase with transformation activity in multiple cell lineages. Seminars in cancer biology. 2005;15:215–223. doi: 10.1016/j.semcancer.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 124.Jin W, Kim BC, Tognon C, Lee HJ, Patel S, Lannon CL, Maris JM, Triche TJ, Sorensen PH, Kim SJ. The ETV6-NTRK3 chimeric tyrosine kinase suppresses TGF-beta signaling by inactivating the TGF-beta type II receptor. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:16239–16244. doi: 10.1073/pnas.0503137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008 doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Herz H, Cooke B, Goldstein D. Metastatic secretory breast cancer. Non-responsiveness to chemotherapy: case report and review of the literature. Ann Oncol. 2000;11:1343–1347. doi: 10.1023/a:1008387800525. [DOI] [PubMed] [Google Scholar]
- 127.de Bree E, Askoxylakis J, Giannikaki E, Chroniaris N, Sanidas E, Tsiftsis DD. Secretory carcinoma of the male breast. Annals of surgical oncology. 2002;9:663–667. doi: 10.1007/BF02574482. [DOI] [PubMed] [Google Scholar]
- 128.Jin W, Yun C, Hobbie A, Martin MJ, Sorensen PH, Kim SJ. Cellular transformation and activation of the phosphoinositide-3-kinase-Akt cascade by the ETV6-NTRK3 chimeric tyrosine kinase requires c-Src. Cancer research. 2007;67:3192–3200. doi: 10.1158/0008-5472.CAN-06-3526. [DOI] [PubMed] [Google Scholar]
- 129.Li Z, Tognon CE, Godinho FJ, Yasaitis L, Hock H, Herschkowitz JI, Lannon CL, Cho E, Kim SJ, Bronson RT, Perou CM, Sorensen PH, Orkin SH. ETV6-NTRK3 fusion oncogene initiates breast cancer from committed mammary progenitors via activation of AP1 complex. Cancer cell. 2007;12:542–558. doi: 10.1016/j.ccr.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kumar-Sinha C, Tomlins SA, Chinnaiyan AM. Recurrent gene fusions in prostate cancer. Nature reviews. 2008;8:497–511. doi: 10.1038/nrc2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lin B, Ferguson C, White JT, Wang S, Vessella R, True LD, Hood L, Nelson PS. Prostate-localized and androgen-regulated expression of the membrane-bound serine protease TMPRSS2. Cancer research. 1999;59:4180–4184. [PubMed] [Google Scholar]
- 132.Afar DE, Vivanco I, Hubert RS, Kuo J, Chen E, Saffran DC, Raitano AB, Jakobovits A. Catalytic cleavage of the androgen-regulated TMPRSS2 protease results in its secretion by prostate and prostate cancer epithelia. Cancer research. 2001;61:1686–1692. [PubMed] [Google Scholar]
- 133.Vaarala MH, Porvari K, Kyllonen A, Lukkarinen O, Vihko P. The TMPRSS2 gene encoding transmembrane serine protease is overexpressed in a majority of prostate cancer patients: detection of mutated TMPRSS2 form in a case of aggressive disease. International journal of cancer. 2001;94:705–710. doi: 10.1002/ijc.1526. [DOI] [PubMed] [Google Scholar]
- 134.Lapointe J, Kim YH, Miller MA, Li C, Kaygusuz G, van de Rijn M, Huntsman DG, Brooks JD, Pollack JR. A variant TMPRSS2 isoform and ERG fusion product in prostate cancer with implications for molecular diagnosis. Mod Pathol. 2007;20:467–473. doi: 10.1038/modpathol.3800759. [DOI] [PubMed] [Google Scholar]
- 135.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montie JE, Shah RB, Pienta KJ, Rubin MA, Chinnaiyan AM. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science (New York, N Y) 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 136.Cerveira N, Ribeiro FR, Peixoto A, Costa V, Henrique R, Jeronimo C, Teixeira MR. TMPRSS2-ERG gene fusion causing ERG overexpression precedes chromosome copy number changes in prostate carcinomas and paired HGPIN lesions. Neoplasia (New York, N Y) 2006;8:826–832. doi: 10.1593/neo.06427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Clark J, Merson S, Jhavar S, Flohr P, Edwards S, Foster CS, Eeles R, Martin FL, Phillips DH, Crundwell M, Christmas T, Thompson A, Fisher C, Kovacs G, Cooper CS. Diversity of TMPRSS2-ERG fusion transcripts in the human prostate. Oncogene. 2007;26:2667–2673. doi: 10.1038/sj.onc.1210070. [DOI] [PubMed] [Google Scholar]
- 138.Hermans KG, van Marion R, van Dekken H, Jenster G, van Weerden WM, Trapman J. TMPRSS2:ERG fusion by translocation or interstitial deletion is highly relevant in androgen-dependent prostate cancer, but is bypassed in late-stage androgen receptor-negative prostate cancer. Cancer research. 2006;66:10658–10663. doi: 10.1158/0008-5472.CAN-06-1871. [DOI] [PubMed] [Google Scholar]
- 139.Iljin K, Wolf M, Edgren H, Gupta S, Kilpinen S, Skotheim RI, Peltola M, Smit F, Verhaegh G, Schalken J, Nees M, Kallioniemi O. TMPRSS2 fusions with oncogenic ETS factors in prostate cancer involve unbalanced genomic rearrangements and are associated with HDAC1 and epigenetic reprogramming. Cancer research. 2006;66:10242–10246. doi: 10.1158/0008-5472.CAN-06-1986. [DOI] [PubMed] [Google Scholar]
- 140.Rajput AB, Miller MA, De Luca A, Boyd N, Leung S, Hurtado-Coll A, Fazli L, Jones EC, Palmer JB, Gleave ME, Cox ME, Huntsman DG. Frequency of the TMPRSS2:ERG gene fusion is increased in moderate to poorly differentiated prostate cancers. Journal of clinical pathology. 2007;60:1238–1243. doi: 10.1136/jcp.2006.043810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Soller MJ, Isaksson M, Elfving P, Soller W, Lundgren R, Panagopoulos I. Confirmation of the high frequency of the TMPRSS2/ERG fusion gene in prostate cancer. Genes, chromosomes & cancer. 2006;45:717–719. doi: 10.1002/gcc.20329. [DOI] [PubMed] [Google Scholar]
- 142.Yoshimoto M, Joshua AM, Chilton-Macneill S, Bayani J, Selvarajah S, Evans AJ, Zielenska M, Squire JA. Three-color FISH analysis of TMPRSS2/ERG fusions in prostate cancer indicates that genomic microdeletion of chromosome 21 is associated with rearrangement. Neoplasia (New York, N Y) 2006;8:465–469. doi: 10.1593/neo.06283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang J, Cai Y, Ren C, Ittmann M. Expression of variant TMPRSS2/ERG fusion messenger RNAs is associated with aggressive prostate cancer. Cancer research. 2006;66:8347–8351. doi: 10.1158/0008-5472.CAN-06-1966. [DOI] [PubMed] [Google Scholar]
- 144.Mertz KD, Setlur SR, Dhanasekaran SM, Demichelis F, Perner S, Tomlins S, Tchinda J, Laxman B, Vessella RL, Beroukhim R, Lee C, Chinnaiyan AM, Rubin MA. Molecular characterization of TMPRSS2-ERG gene fusion in the NCI-H660 prostate cancer cell line: a new perspective for an old model. Neoplasia (New York, N Y) 2007;9:200–206. doi: 10.1593/neo.07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, Helgeson BE, Cao Q, Prensner JR, Rubin MA, Shah RB, Mehra R, Chinnaiyan AM. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia (New York, N Y) 2008;10:177–188. doi: 10.1593/neo.07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sun C, Dobi A, Mohamed A, Li H, Thangapazham RL, Furusato B, Shaheduzzaman S, Tan SH, Vaidyanathan G, Whitman E, Hawksworth DJ, Chen Y, Nau M, Patel V, Vahey M, Gutkind JS, Sreenath T, Petrovics G, Sesterhenn IA, McLeod DG, Srivastava S. TMPRSS2-ERG fusion, a common genomic alteration in prostate cancer activates C-MYC and abrogates prostate epithelial differentiation. Oncogene. 2008;27:5348–5353. doi: 10.1038/onc.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, Menon A, Jing X, Cao Q, Han B, Yu J, Wang L, Montie JE, Rubin MA, Pienta KJ, Roulston D, Shah RB, Varambally S, Mehra R, Chinnaiyan AM. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–599. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 148.Shaffer DR, Pandolfi PP. Breaking the rules of cancer. Nature medicine. 2006;12:14–15. doi: 10.1038/nm0106-14. [DOI] [PubMed] [Google Scholar]
- 149.Tomlins SA, Mehra R, Rhodes DR, Smith LR, Roulston D, Helgeson BE, Cao X, Wei JT, Rubin MA, Shah RB, Chinnaiyan AM. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer research. 2006;66:3396–3400. doi: 10.1158/0008-5472.CAN-06-0168. [DOI] [PubMed] [Google Scholar]
- 150.Han B, Mehra R, Dhanasekaran SM, Yu J, Menon A, Lonigro RJ, Wang X, Gong Y, Wang L, Shankar S, Laxman B, Shah RB, Varambally S, Palanisamy N, Tomlins SA, Kumar-Sinha C, Chinnaiyan AM. A fluorescence in situ hybridization screen for E26 transformation-specific aberrations: identification of DDX5-ETV4 fusion protein in prostate cancer. Cancer research. 2008;68:7629–7637. doi: 10.1158/0008-5472.CAN-08-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Helgeson BE, Tomlins SA, Shah N, Laxman B, Cao Q, Prensner JR, Cao X, Singla N, Montie JE, Varambally S, Mehra R, Chinnaiyan AM. Characterization of TMPRSS2:ETV5 and SLC45A3:ETV5 gene fusions in prostate cancer. Cancer research. 2008;68:73–80. doi: 10.1158/0008-5472.CAN-07-5352. [DOI] [PubMed] [Google Scholar]
- 152.Hermans KG, Bressers AA, van der Korput HA, Dits NF, Jenster G, Trapman J. Two unique novel prostate-specific and androgen-regulated fusion partners of ETV4 in prostate cancer. Cancer research. 2008;68:3094–3098. doi: 10.1158/0008-5472.CAN-08-0198. [DOI] [PubMed] [Google Scholar]
- 153.Mehra R, Tomlins SA, Yu J, Cao X, Wang L, Menon A, Rubin MA, Pienta KJ, Shah RB, Chinnaiyan AM. Characterization of TMPRSS2-ETS gene aberrations in androgen-independent metastatic prostate cancer. Cancer research. 2008;68:3584–3590. doi: 10.1158/0008-5472.CAN-07-6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Cai C, Hsieh CL, Omwancha J, Zheng Z, Chen SY, Baert JL, Shemshedini L. ETV1 is a novel androgen receptor-regulated gene that mediates prostate cancer cell invasion. Molecular endocrinology (Baltimore, Md) 2007;21:1835–1846. doi: 10.1210/me.2006-0480. [DOI] [PubMed] [Google Scholar]
- 155.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, Aburatani H, Niki T, Sohara Y, Sugiyama Y, Mano H. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 156.Pollmann M, Parwaresch R, Adam-Klages S, Kruse ML, Buck F, Heidebrecht HJ. Human EML4, a novel member of the EMAP family, is essential for microtubule formation. Experimental cell research. 2006;312:3241–3251. doi: 10.1016/j.yexcr.2006.06.035. [DOI] [PubMed] [Google Scholar]
- 157.Choi YL, Takeuchi K, Soda M, Inamura K, Togashi Y, Hatano S, Enomoto M, Hamada T, Haruta H, Watanabe H, Kurashina K, Hatanaka H, Ueno T, Takada S, Yamashita Y, Sugiyama Y, Ishikawa Y, Mano H. Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer research. 2008;68:4971–4976. doi: 10.1158/0008-5472.CAN-07-6158. [DOI] [PubMed] [Google Scholar]
- 158.Takeuchi K, Choi YL, Soda M, Inamura K, Togashi Y, Hatano S, Enomoto M, Takada S, Yamashita Y, Satoh Y, Okumura S, Nakagawa K, Ishikawa Y, Mano H. Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clin Cancer Res. 2008;14:6618–6624. doi: 10.1158/1078-0432.CCR-08-1018. [DOI] [PubMed] [Google Scholar]
- 159.Fukuyoshi Y, Inoue H, Kita Y, Utsunomiya T, Ishida T, Mori M. EML4-ALK fusion transcript is not found in gastrointestinal and breast cancers. British journal of cancer. 2008;98:1536–1539. doi: 10.1038/sj.bjc.6604341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Inamura K, Takeuchi K, Togashi Y, Nomura K, Ninomiya H, Okui M, Satoh Y, Okumura S, Nakagawa K, Soda M, Choi YL, Niki T, Mano H, Ishikawa Y. EML4-ALK fusion is linked to histological characteristics in a subset of lung cancers. J Thorac Oncol. 2008;3:13–17. doi: 10.1097/JTO.0b013e31815e8b60. [DOI] [PubMed] [Google Scholar]
- 161.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science (New York, N Y) 1994;263:1281–1284. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 162.Lamant L, Dastugue N, Pulford K, Delsol G, Mariame B. A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood. 1999;93:3088–3095. [PubMed] [Google Scholar]
- 163.Touriol C, Greenland C, Lamant L, Pulford K, Bernard F, Rousset T, Mason DY, Delsol G. Further demonstration of the diversity of chromosomal changes involving 2p23 in ALK-positive lymphoma: 2 cases expressing ALK kinase fused to CLTCL (clathrin chain polypeptide-like) Blood. 2000;95:3204–3207. [PubMed] [Google Scholar]
- 164.Colleoni GW, Bridge JA, Garicochea B, Liu J, Filippa DA, Ladanyi M. ATIC-ALK: A novel variant ALK gene fusion in anaplastic large cell lymphoma resulting from the recurrent cryptic chromosomal inversion, inv(2)(p23q35) The American journal of pathology. 2000;156:781–789. doi: 10.1016/S0002-9440(10)64945-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ma Z, Cools J, Marynen P, Cui X, Siebert R, Gesk S, Schlegelberger B, Peeters B, De Wolf-Peeters C, Wlodarska I, Morris SW. Inv(2)(p23q35) in anaplastic large-cell lymphoma induces constitutive anaplastic lymphoma kinase (ALK) tyrosine kinase activation by fusion to ATIC, an enzyme involved in purine nucleotide biosynthesis. Blood. 2000;95:2144–2149. [PubMed] [Google Scholar]
- 166.Trinei M, Lanfrancone L, Campo E, Pulford K, Mason DY, Pelicci PG, Falini B. A new variant anaplastic lymphoma kinase (ALK)-fusion protein (ATIC-ALK) in a case of ALK-positive anaplastic large cell lymphoma. Cancer research. 2000;60:793–798. [PubMed] [Google Scholar]
- 167.Hernandez L, Pinyol M, Hernandez S, Bea S, Pulford K, Rosenwald A, Lamant L, Falini B, Ott G, Mason DY, Delsol G, Campo E. TRK-fused gene (TFG) is a new partner of ALK in anaplastic large cell lymphoma producing two structurally different TFG-ALK translocations. Blood. 1999;94:3265–3268. [PubMed] [Google Scholar]
- 168.Shinmura K, Kageyama S, Tao H, Bunai T, Suzuki M, Kamo T, Takamochi K, Suzuki K, Tanahashi M, Niwa H, Ogawa H, Sugimura H. EML4-ALK fusion transcripts, but no NPM-, TPM3-, CLTC-, ATIC-, or TFG-ALK fusion transcripts, in non-small cell lung carcinomas. Lung cancer (Amsterdam, Netherlands) 2008 doi: 10.1016/j.lungcan.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 169.Perner S, Wagner PL, Demichelis F, Mehra R, Lafargue CJ, Moss BJ, Arbogast S, Soltermann A, Weder W, Giordano TJ, Beer DG, Rickman DS, Chinnaiyan AM, Moch H, Rubin MA. EML4-ALK fusion lung cancer: a rare acquired event. Neoplasia (New York, N Y) 2008;10:298–302. doi: 10.1593/neo.07878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, Hu Y, Tan Z, Stokes M, Sullivan L, Mitchell J, Wetzel R, Macneill J, Ren JM, Yuan J, Bakalarski CE, Villen J, Kornhauser JM, Smith B, Li D, Zhou X, Gygi SP, Gu TL, Polakiewicz RD, Rush J, Comb MJ. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 171.Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X, Sam L, Barrette T, Palanisamy N, Chinnaiyan AM. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009 doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ayton PM, Cleary ML. Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene. 2001;20:5695–5707. doi: 10.1038/sj.onc.1204639. [DOI] [PubMed] [Google Scholar]
- 173.Collins EC, Rabbitts TH. The promiscuous MLL gene links chromosomal translocations to cellular differentiation and tumour tropism. Trends in molecular medicine. 2002;8:436–442. doi: 10.1016/s1471-4914(02)02397-3. [DOI] [PubMed] [Google Scholar]
- 174.Daser A, Rabbitts TH. Extending the repertoire of the mixed-lineage leukemia gene MLL in leukemogenesis. Genes & development. 2004;18:965–974. doi: 10.1101/gad.1195504. [DOI] [PubMed] [Google Scholar]
- 175.Schumacher A, Magnuson T. Murine Polycomb- and trithorax-group genes regulate homeotic pathways and beyond. Trends Genet. 1997;13:167–170. [PubMed] [Google Scholar]
- 176.Maconochie M, Nonchev S, Morrison A, Krumlauf R. Paralogous Hox genes: function and regulation, Annual review of genetics. 1996;30:529–556. doi: 10.1146/annurev.genet.30.1.529. [DOI] [PubMed] [Google Scholar]
- 177.Look AT. Oncogenic transcription factors in the human acute leukemias. Science (New York, N Y) 1997;278:1059–1064. doi: 10.1126/science.278.5340.1059. [DOI] [PubMed] [Google Scholar]
- 178.Ayton PM, Cleary ML. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes & development. 2003;17:2298–2307. doi: 10.1101/gad.1111603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hsu K, Look AT. Turning on a dimer: new insights into MLL chimeras. Cancer cell. 2003;4:81–83. doi: 10.1016/s1535-6108(03)00192-2. [DOI] [PubMed] [Google Scholar]
- 180.Zeleznik-Le NJ, Harden AM, Rowley JD. 11q23 translocations split the “AT-hook” cruciform DNA-binding region and the transcriptional repression domain from the activation domain of the mixed-lineage leukemia (MLL) gene. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:10610–10614. doi: 10.1073/pnas.91.22.10610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, Dubois G, Mazo A, Croce CM, Canaani E. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Molecular cell. 2002;10:1119–1128. doi: 10.1016/s1097-2765(02)00740-2. [DOI] [PubMed] [Google Scholar]
- 182.So CW, Lin M, Ayton PM, Chen EH, Cleary ML. Dimerization contributes to oncogenic activation of MLL chimeras in acute leukemias. Cancer cell. 2003;4:99–110. doi: 10.1016/s1535-6108(03)00188-0. [DOI] [PubMed] [Google Scholar]
- 183.Milne TA, Martin ME, Brock HW, Slany RK, Hess JL. Leukemogenic MLL fusion proteins bind across a broad region of the Hox a9 locus, promoting transcription and multiple histone modifications. Cancer research. 2005;65:11367–11374. doi: 10.1158/0008-5472.CAN-05-1041. [DOI] [PubMed] [Google Scholar]
- 184.Dorrance AM, Liu S, Yuan W, Becknell B, Arnoczky KJ, Guimond M, Strout MP, Feng L, Nakamura T, Yu L, Rush LJ, Weinstein M, Leone G, Wu L, Ferketich A, Whitman SP, Marcucci G, Caligiuri MA. Mll partial tandem duplication induces aberrant Hox expression in vivo via specific epigenetic alterations. The Journal of clinical investigation. 2006;116:2707–2716. doi: 10.1172/JCI25546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sandberg AA. The chromosomes and causation of human cancer and leukemia. Cancer research. 1966;26:2064–2081. [PubMed] [Google Scholar]
- 186.DeGrouchy J. Chromosomes in neoplastic tissues. In: Neel JCJ, editor. Proceedings of the Third International Congress of Human Genetics. Johns Hopkins University Press; Chicago: 1967. [Google Scholar]
- 187.Aguilera A, Gomez-Gonzalez B. Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet. 2008;9:204–217. doi: 10.1038/nrg2268. [DOI] [PubMed] [Google Scholar]
- 188.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science (New York, N Y) 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 189.Zhang N, Ge G, Meyer R, Sethi S, Basu D, Pradhan S, Zhao YJ, Li XN, Cai WW, El-Naggar AK, Baladandayuthapani V, Kittrell FS, Rao PH, Medina D, Pati D. Overexpression of Separase induces aneuploidy and mammary tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13033–13038. doi: 10.1073/pnas.0801610105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sotillo R, Hernando E, Diaz-Rodriguez E, Teruya-Feldstein J, Cordon-Cardo C, Lowe SW, Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Gorunova L, Hoglund M, Andren-Sandberg A, Dawiskiba S, Jin Y, Mitelman F, Johansson B. Cytogenetic analysis of pancreatic carcinomas: intratumor heterogeneity and nonrandom pattern of chromosome aberrations. Genes, chromosomes & cancer. 1998;23:81–99. doi: 10.1002/(sici)1098-2264(199810)23:2<81::aid-gcc1>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 192.Mitelman F, Johansson B, Mertens F. Fusion genes and rearranged genes as a linear function of chromosome aberrations in cancer. Nature genetics. 2004;36:331–334. doi: 10.1038/ng1335. [DOI] [PubMed] [Google Scholar]
- 193.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia (New York, N Y) 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Barrette TR, Ghosh D, Chinnaiyan AM. Mining for regulatory programs in the cancer transcriptome. Nature genetics. 2005;37:579–583. doi: 10.1038/ng1578. [DOI] [PubMed] [Google Scholar]
- 195.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P, Varambally S, Ghosh D, Chinnaiyan AM. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia (New York, N Y) 2007;9:166–180. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Rhodes DR, Kalyana-Sundaram S, Tomlins SA, Mahavisno V, Kasper N, Varambally R, Barrette TR, Ghosh D, Varambally S, Chinnaiyan AM. Molecular concepts analysis links tumors, pathways, mechanisms, and drugs. Neoplasia (New York, N Y) 2007;9:443–454. doi: 10.1593/neo.07292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Jacquinet E, Rao NV, Rao GV, Zhengming W, Albertine KH, Hoidal JR. Cloning and characterization of the cDNA and gene for human epitheliasin. European journal of biochemistry / FEBS. 2001;268:2687–2699. doi: 10.1046/j.1432-1327.2001.02165.x. [DOI] [PubMed] [Google Scholar]
- 198.Takahara T, Tasic B, Maniatis T, Akanuma H, Yanagisawa S. Delay in synthesis of the 3’ splice site promotes trans-splicing of the preceding 5’ splice site. Molecular cell. 2005;18:245–251. doi: 10.1016/j.molcel.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 199.Communi D, Suarez-Huerta N, Dussossoy D, Savi P, Boeynaems JM. Cotranscription and intergenic splicing of human P2Y11 and SSF1 genes. The Journal of biological chemistry. 2001;276:16561–16566. doi: 10.1074/jbc.M009609200. [DOI] [PubMed] [Google Scholar]
- 200.Koontz JI, Soreng AL, Nucci M, Kuo FC, Pauwels P, van Den Berghe H, Dal Cin P, Fletcher JA, Sklar J. Frequent fusion of the JAZF1 and JJAZ1 genes in endometrial stromal tumors. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6348–6353. doi: 10.1073/pnas.101132598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Li H, Wang J, Mor G, Sklar J. A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science (New York, N Y) 2008;321:1357–1361. doi: 10.1126/science.1156725. [DOI] [PubMed] [Google Scholar]
- 202.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]