

Abstract
Closure of the active site phosphate gripper loop of orotidine 5′-monophosphate decarboxylase from Saccharomyces cerevisiae (ScOMPDC) over the bound substrate orotidine 5′-monophosphate (OMP) activates the bound substrate for decarboxylation by at least 104-fold [Amyes, T. L., Richard, J. P., and Tait, J. J. (2005) J. Am. Chem. Soc. 127, 15708-15709]. The 19 residue phosphate gripper loop of the mesophilic ScOMPDC is much larger than the 9 residue loop at the ortholog from the thermophile Methanothermobacter thermautotrophicus (MtOMPDC). This difference in loop size results in a small decrease in the total intrinsic phosphate binding energy of the phosphodianion group of OMP from 11.9 to 11.6 kcal/mol, along with a modest decrease in the extent of activation by phosphite dianion of decarboxylation of the truncated substrate 1-(β-D-erythrofuranosyl)orotic acid. The activation parameters ΔH‡ and ΔS‡ for kcat for decarboxylation of OMP are 3.6 kcal/mol and 10 cal/K/mol more positive, respectively, for MtOMPDC than for ScOMPDC. We suggest that these differences are related to the difference in size of the active site loops at the mesophilic ScOMPDC and the thermophilic MtOMPDC. The greater enthalpic transition state stabilization available from the more extensive loop-substrate interactions for the ScOMPDC-catalyzed reaction is largely balanced by a larger entropic requirement for immobilization of the larger loop at this enzyme.
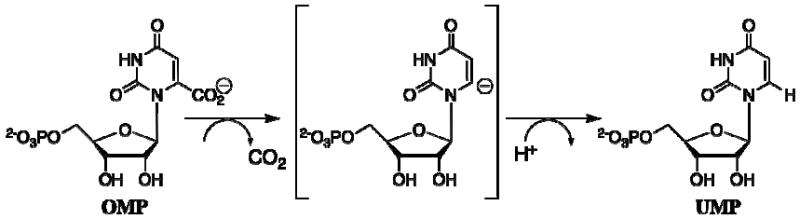
Orotidine 5′-monophosphate decarboxylase (OMPDC)1 is a remarkable enzyme because it employs no metal ions or other cofactors but yet effects an enormous ca. 30 kcal/mol stabilization of the transition state for the chemically very difficult decarboxylation of orotidine 5′-monophosphate (OMP) to give uridine 5′-monophosphate (UMP) (Scheme 1) (1-3). The enormous 1017-fold rate acceleration for decarboxylation of enzyme-bound OMP (kcat = 15 s-1 (4)) is a consequence of the exceedingly slow decarboxylation of OMP in water (t1/2 ≈ 78 million years (3)) through an unstable vinyl carbanion intermediate. This remarkable efficiency led to the proposal of several different mechanisms for the OMPDC-catalyzed reaction that avoid formation of an unstable carbanion (5-10). However, we recently showed that OMPDC meets its catalytic challenge head-on by stabilizing an enzyme-bound vinyl carbanion (Scheme 1) (11, 12).
Scheme 1.
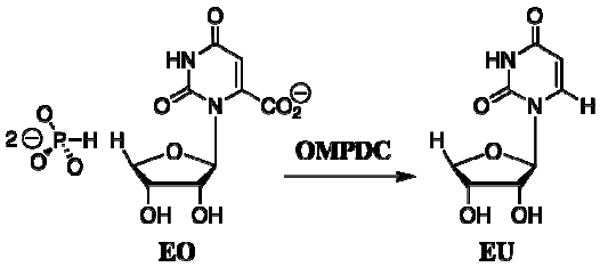
One of the principal differences between catalysis by enzymes and by small molecules is that enzymes have evolved unique mechanisms to utilize binding interactions with nonreacting portions of the substrate for transition state stabilization (13). We have shown that the binding of phosphite dianion to OMPDC from Saccharomyces cerevisiae (yeast, ScOMPDC) results in an 80,000-fold increase in the second-order rate constant kcat/Km for enzyme-catalyzed decarboxylation of the truncated substrate 1-(β-D-erythrofuranosyl)orotic acid (EO), which lacks a 5′-phosphodianion moiety, to give 1-(β-D-erythrofuranosyl)uridine (EU) (Scheme 2) (14). This shows that the nonreacting phosphodianion group of OMP does not function simply to anchor the substrate to the enzyme, but rather serves the more important role of activating the enzyme towards decarboxylation of bound OMP.
Scheme 2.
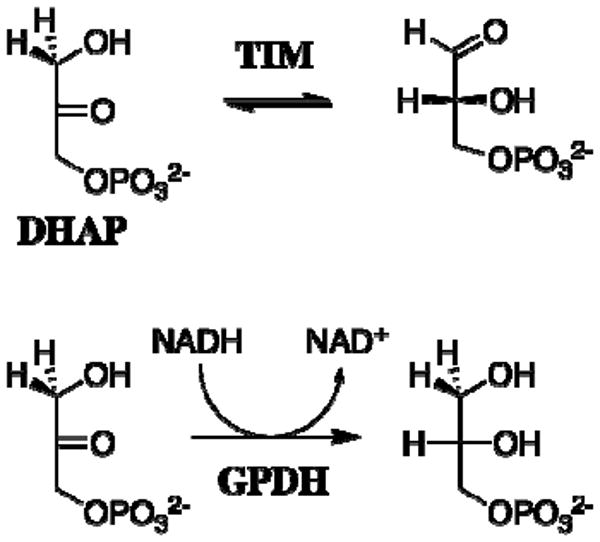
Similar experiments provided evidence that the “intrinsic phosphate binding energy” of the substrate phosphodianion group of dihydroxyacetone phosphate (DHAP) is utilized in stabilization of the transition state of the aldose-ketose isomerization reaction catalyzed by triosephosphate isomerase (TIM) (15, 16) and of the hydride transfer reaction catalyzed by glycerol 3-phosphate dehydrogenase (GPDH) (Scheme 3) (17).
Scheme 3.
OMPDC (10, 18-22), TIM (23-25) and GPDH (26, 27) each have a flexible “phosphate gripper” loop that is open at the free enzyme but closes over the phosphodianion group of bound substrate to sequester the substrate from bulk solvent. We have proposed that this phosphate-driven loop closure reduces the barrier for reaction at the loop-closed compared with the loop-open enzymes, and that it is the underlying origin of the 104 – 106-fold effect of the nonreacting substrate phosphodianion group on the chemical reactivity of enzyme-bound substrate (17).
The phosphate gripper loops of ScOMPDC and of OMPDC from Escherichia coli (EcOMPDC) both extend 19 residues, from the strictly conserved Pro-202 to Val-220 for ScOMPDC (21) (Figure 1), and from Pro-189 to Pro-207 for EcOMPDC (19, 20). By contrast, the corresponding loop of OMPDC from the thermophile Methanothermobacter thermautotrophicus (MtOMPDC) extends only 9 residues, from Pro-180 to Asp-188 (22) (Figure 1). These three enzymes show an otherwise high degree of structural homology (18) which then raises the question of whether there may be an underlying mechanistic imperative for the difference in the size of the flexible loops for enzymes from mesophiles and thermophiles. For example, if the size of this loop is related to catalytic efficiency, then the smaller loop for MtOMPDC might be expected to play a reduced role in promoting decarboxylation through utilization of the binding energy of the nonreacting substrate phosphodianion group of OMP or of a phosphite dianion activator.
Figure 1.
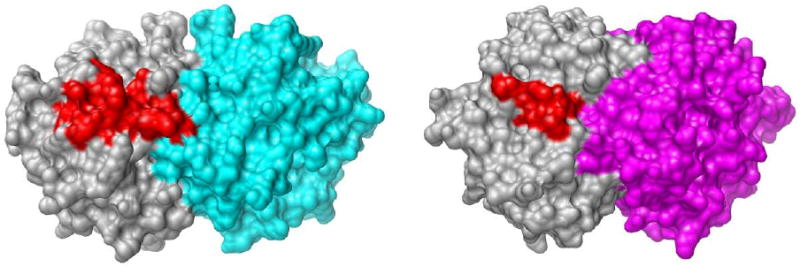
Comparison of the closed active site loops of the dimeric OMPDCs from S. cerevisiae (19 residues, left structure, PDB accession code 3GDL (46)) and M thermautotrophicus (9 residues, right structure, PDB accession code 3G1A (46)) liganded with 6-azauridine 5′-monophosphate. The active site loops are shown in red and the remainder of the monomer is shown in grey. The second monomer is shown in cyan (ScOMPDC) or magenta (MtOMPDC). The loop for ScOMPDC extends from Pro-202 to Val-220 and the loop for MtOMPDC extends from Pro-180 to Asp-188. The images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081).
We report here the results of experiments that were designed to compare the intrinsic phosphate binding energy and the thermodynamic activation parameters for OMPDCs from mesophilic and thermophilic organisms. The thermophilic enzyme with the shorter phosphate gripper loop, MtOMPDC, shows an intrinsic phosphate binding energy of the phosphodianion group of OMP and phosphite activation of decarboxylation of the truncated substrate EO that are similar to those observed for the mesophilic ScOMPDC. By contrast there is a significant difference in the activation parameters ΔH‡ and ΔS‡ for the decarboxylation of enzyme-bound OMP such that MtOMPDC pays a substantially smaller entropic price, but a larger enthalpic price, for conversion of the Michaelis complex to the transition state for decarboxylation than does ScOMPDC.
Experimental
Materials
Orotidine 5′-monophosphate trisodium salt (99%) was purchased from Sigma or was prepared by chemical or enzymatic methods from uridine 5′-monophosphate using modifications of literature procedures (28-30). 1-(β-D-Erythrofuranosyl)orotic acid (EO) and 1-(β-D-erythrofuranosyl)uridine (EU) were available from an earlier study (14). Sodium phosphite (dibasic, pentahydrate), 3-(N-morpholino)propanesulfonic acid (MOPS, ≥ 99.5%) and ammonium acetate (≥ 99%) were purchased from Fluka. Water was from a Milli-Q Academic purification system. All other chemicals were reagent grade or better and were used without further purification.
Preparation of OMPDCs
The C155S mutant OMPDC from Saccharomyces cerevisiae (ScOMPDC) was prepared as described previously (31, 32). This mutant is more stable than, but kinetically and structurally essentially identical with, wildtype yeast OMPDC (33). Wildtype OMPDC from Methanothermobacter thermautotrophicus (MtOMPDC) was prepared as described previously (32). The gene for wildtype OMPDC from Escherichia coli (EcOMPDC) was cloned from Escherichia coli K12 genomic DNA by Dr. Wen Shan Yew and the protein was expressed in E. coli BL21 (DE3) using a modified pET-15b plasmid with a His10-tag, as described previously for OMPDCs with a His6-tag (32). The cells were grown at 37 °C, induced with IPTG (0.5 mM) when the cell density reached an OD600 of 0.6, and harvested after 18 h. Purification was carried out as described previously for ScOMPDC and MtOMPDC (32) except that no Q-Sepharose column was used. The protein was dialyzed at 5 °C against the storage buffer (20 mM Tris-HCl, pH 7.0, 100 mM NaCl and 20% glycerol), concentrated to ca. 25 mg/mL via ultrafiltration, flash frozen in liquid nitrogen as 25 μL pellets and stored at -80 °C.
Preparation of Solutions
Solution pH was determined at 25 °C using an Orion Model 720A pH meter equipped with a Radiometer pHC4006-9 combination electrode that was standardized at pH 7.00 and 10.00 at 25 °C. Stock solutions of OMP were prepared in water and the OMP concentration was determined from the absorbance in 0.1 M HCl at 267 nm using ε = 9430 M-1 cm-1 (34). Stock solutions of 1-(β-D-erythrofuranosyl)orotic acid (EO) were prepared and neutralized to pH ≈ 6 as described in earlier work (14, 31) and the EO concentration was determined from the absorbance in 0.1 M HCl at 267 nm using ε = 9570 M-1 cm-1 reported for orotidine (34).
The stock solution of phosphite (100 mM, 80% free base, I = 0.28) was prepared by addition of a measured amount of 1 M HCl to the sodium salt to give the desired acid/base ratio. MOPS buffers were prepared by addition of measured amounts of 1 M HCl and solid NaCl to give the desired acid/base ratio and ionic strength.
Samples of OMPDCs that had been stored at -80 °C were defrosted and dialyzed at 4 °C against 10 mM MOPS (50% free base) at pH 7.1 containing 100 mM NaCl, unless noted otherwise.
Determination of Values of kcat and kcat/Km for Turnover of OMP by OMPDCs at Various Temperatures
All assays were carried out in 10 mM MOPS (50% free base) at pH 7.1 and I = 0.105 (NaCl) using a Cary 3E spectrophotometer equipped with a temperature-controlled Peltier block multicell changer. The temperature dependence of the difference between the extinction coefficients of OMP and UMP at 279 nm, Δε (M-1 cm-1), was determined using the following procedure: First the spectrophotometer was zeroed at 279 nm using a solution of 10 mM MOPS at pH 7.1 and I = 0.105 (NaCl) at 25 °C. A small aliquot of OMP was added to give a final concentration of 100 μM OMP and the absorbance of the resulting solution was determined at several temperatures between 10 and 75 °C. The temperature was then returned to 25 °C and 1 μL of a solution of ScOMPDC was added to give a final enzyme concentration of 21 nM. The ensuing decarboxylation of OMP was allowed to proceed to completion to give a quantitative yield of UMP and the final absorbance of the solution was again determined at several temperatures between 10 and 75 °C. The observed absorbance changes were used to calculate the following values of Δε (M-1 cm-1): 10 °C, 2290; 25 °C, 2400 (4); 35 °C, 2490; 45 °C, 2560; 55 °C, 2610; 65 °C, 2660; 75 °C, 2720. The following values of Δε at other temperatures were obtained by extrapolation or interpolation using the two linear regions of the biphasic linear correlation between Δε (M-1 cm-1) and T (°C): 5 °C, 2250; 12 °C, 2300; 15 °C, 2330; 17 °C, 2340.
For the determination of values of kcat the reaction mixtures (1 mL total) containing buffer and OMP ([OMP]o = 50 - 250 μM, ≫ 10Km] were equilibrated at the temperature of interest and the reaction was initiated by the addition of 1 - 3 μL of a stock solution of OMPDC using a cuvette admixer without removal of the cuvette from the temperature-controlled block. The initial velocity of decarboxylation of OMP under these conditions, Vmax (M s-1), was determined within 1 min by monitoring the decrease in absorbance at 279 nm using the appropriate value of Δε (M-1 cm-1) for the temperature of interest. The observed values of Vmax were generally shown to be proportional to enzyme concentration in the following ranges: ScOMPDC, 11 - 42 nM; MtOMPDC, 76 - 150 nM; EcOMPDC, 30 - 230 nM. At some temperatures the use of lower concentrations of OMPDC resulted in reduced activity, presumably as a result of dissociation of the active dimer to give the inactive monomeric form (4). These conditions were: 38 nM MtOMPDC at 45 or 55 °C, kcat was reduced by 20%; 30 nM EcOMPDC at 10 °C, kcat was reduced by 30% (the data were therefore obtained using 60 nM enzyme); 30 nM EcOMPDC at 5 °C, kcat was reduced by 50% (the data were therefore obtained using 90 – 230 nM enzyme); 15 nM EcOMPDC at 25 °C, kcat was reduced by 20%. In order to verify that the temperature variation did not result in a loss of enzyme activity in the timeframe of the assays which were conducted over a period of ≤ 1 min (see above), periodic controls were conducted in which the stock solution of OMPDC was incubated in a water bath at the assay temperature of interest for 1 – 5 min prior to its use in the assay at this temperature. It was found that this preincubation did not significantly affect the observed activity at the temperature of interest.
Values of kcat (s-1) were calculated from the values of Vmax (M s-1) using eq 1. In all cases the concentration of ScOMPDC in the stock solution was calculated from the values of Vmax (M s-1) determined in side-by-side standard assays at 25 °C using eq 1 with kcat = 15 s-1 (4). For determination of the values of kcat at 25 °C for MtOMPDC (4.7 s-1) and EcOMPDC (13 s-1) the concentration of OMPDC was determined from the absorbance of the protein at 280 nm in 10 mM MOPS (50% free base) at pH 7.1 containing 100 mM NaCl and the extinction coefficients of 6100 M-1 cm-1 (MtOMPDC) and 10100 M-1 cm-1 (EcOMPDC) that were calculated using the ProtParam tool available on the ExPASy server (35, 36). For determination of the values of kcat at other temperatures the concentrations of MtOMPDC and EcOMPDC were determined from the values of Vmax (M s-1) determined in side-by-side standard assays at 25 °C using eq 1 with kcat = 4.7 s-1 and 13 s-1, respectively.
| (1) |
Values of kcat/Km for turnover of OMP by ScOMPDC and MtOMPDC at various temperatures were determined in experiments in which the complete disappearance of a relatively low initial concentration OMP was monitored at 279 nm. Reactions (1 mL total, [OMP]o = 4 – 40 μM) were initiated by the addition of OMPDC as described above to give a final concentration of 20 – 70 nM ScOMPDC or 50 – 420 nM MtOMPDC. The observed rate constants kobsd (s-1) for the first-order decay of OMP in the final stages of the reactions where [OMP]t ≤ 0.3 – 0.5Km were obtained from the fits of the absorbance vs time data to a single exponential. Experiments were conducted at several enzyme concentrations in the indicated ranges to ensure that the observed first-order decay represents enzyme-catalyzed reaction of OMP rather than a loss of enzyme activity with time, and the values of kobsd were shown to be proportional to [E]. Values of kcat/Km (M-1 s-1) were then calculated using the relationship kcat/Km = kobsd/[E] and values of Km for OMP were obtained by combining the experimental values of kcat (s-1) and kcat/Km (M-1 s-1) (see Table 1). In all cases the concentration of OMPDC was determined from the values of Vmax (M s-1) determined in side-by-side standard assays at 25 °C using eq 1 with the appropriate value of kcat. This analysis is possible because the initial concentration of OMP in these experiments ([OMP]o ≤ 40 μM) was chosen (see below) such that the amount of the UMP product generated (≤ 40 μM) is insufficient to result in significant inhibition of the OMPDC of interest. The initial concentration of OMP ([OMP]o) was chosen based on the following: For ScOMPDC a value of Ki = 400 μM has been reported for binding of UMP to ScOMPDC at 25 °C under our experimental conditions (4), and there was no significant change in the value of Km determined in our experiments at 45 °C for [OMP]o = 11 – 33 μM. For MtOMPDC at 25 °C experiments conducted at [OMP]o = 200 – 400 μM yielded apparent values of Km that are consistent with values of Ki for binding of UMP to MtOMPDC of ca. 100 μM at 25 °C and ca. 200 μM at 55 °C. There was no significant change in the value of Km determined in our experiments when the initial concentration of OMP was varied in the range 6 – 30 μM at 5 °C, 4 – 12 μM at 25 °C, 12 – 22 μM at 35 °C, or 18 – 39 μM at 55 °C.
Table 1.
Kinetic parameters and intrinsic phosphate binding energies for decarboxylation of OMP and EO catalyzed by ScOMPDC and MtOMPDC and for the phosphite-activated reactions of EO at pH 7.0 and 25 °C.
| kcat/Km(M-1 s-1) | IPBE b | (kcat/Km)E·HPi/Kdc | Phosphite Activation d | ||
|---|---|---|---|---|---|
| OMP a | EO | M-2s-1 | |||
| ScOMPDC | 1.1 × 107 | 2.1 × 10-2 e | 11.9 kcal/mol | 11,700 e | 5.6 × 105 M-1 |
| MtOMPDC | 3.1 × 106 | 8.7 × 10-3 | 11.6 kcal/mol | 2,500 | 2.9 × 105 M-1 |
Data at pH 7.1 from Table 2.
Intrinsic phosphate binding energy calculated from the ratio of the second-order rate constants for the reactions of OMP and EO.
Third-order rate constant for phosphite-activated decarboxylation of EO catalyzed by OMPDC, calculated as the slope of the plot of (kcat/Km)obsd against [HPO32-] at low concentrations of phosphite (Figure 2).
Ratio of the third-order rate constant for the phosphite-activated decarboxylation of EO and the second-order rate constant for the unactivated decarboxylation of EO.
Data at pH 7.1 from Ref. 14.
Turnover of EO by MtOMPDC in the Absence and Presence of Phosphite Dianion
The decarboxylation of the truncated substrate 1-(β-D-erythrofuranosyl)orotic acid (EO, 5 mM) in 50 mM MOPS (45% free base) at pH 7.0, 25 °C and I = 0.14 (NaCl) catalyzed by MtOMPDC (350 μM) was followed in a discontinuous assay in which the initial velocity of formation of the product 1-(β-D-erythrofuranosyl)uridine (EU) was monitored by HPLC. For these experiments MtOMPDC was dialyzed at 4 °C against 100 mM MOPS (45% free base) at pH 7.0 and I = 0.28 (NaCl). The reaction in a total volume of 200 μL was initiated by the addition of EO to a solution of the enzyme in buffer that was equilibrated at 25 °C. The reaction was followed for ca. 7 h, during which time there was 8% reaction of EO. At various times an aliquot (20 μL) was withdrawn and quenched to pH 3.8 by the addition of 180 μL of ice-cold 5 mM formic acid. The enzyme was removed by ultrafiltration using an Amicon Microcon filtration device (10K MWCO) and the filtrate (150 μL) was analyzed by HPLC using a using a Waters Atlantis dC18 3 μm column (3.9 × 150 mm) with an isocratic flow of 1 mL/min 10 mM NH4OAc (pH 4.2) and peak detection at 262 nm. Under these conditions the unreacted EO eluted close to the void volume and the product EU eluted at ca. 7 min. The concentration of the product EU in the reaction mixture at time t, [EU]t, was obtained from the HPLC peak area by interpolation of a standard curve that was constructed using authentic EU. The concentration of MtOMPDC in the reaction mixture was determined by periodic standard assay (see above) and it was shown that there was no significant decrease in enzyme activity during the reaction. The initial velocity of the reaction, vi (M s-1), was determined as the slope of the linear plot of [EU]t against time. The second-order rate constant (kcat/Km)o (M-1 s-1) for MtOMPDC-catalyzed decarboxylation of EO was calculated using the relationship (kcat/Km)o = vi/[E] [S]o.
The decarboxylation of EO (0.12 mM) in the presence of 2 – 36 mM phosphite dianion and 5 mM MOPS at pH 7.0, 25 °C and I = 0.14 (NaCl) catalyzed by MtOMPDC (22 – 38 μM) was monitored spectrophotometrically at 283 nm. Reactions (1 mL total) were initiated by the addition of 50 μL of a solution of MtOMPDC in 100 mM MOPS (pH 7.0, I = 0.28, NaCl) and were monitored for up to 6 h. The concentration of MtOMPDC in the reaction mixture was determined by standard assay and it was shown that there was no significant decrease in enzyme activity during the reaction. These reactions obeyed excellent first-order kinetics with stable endpoints and values of kobsd (s-1) for the reaction of EO were obtained from the fits of the absorbance vs time data to a single exponential. The apparent second-order rate constants (kcat/Km)obsd (M-1 s-1) for MtOMPDC-catalyzed decarboxylation of EO at the various concentrations of phosphite dianion were calculated using the relationship (kcat/Km)obsd = kobsd/[E].
Results
The second-order rate constant for decarboxylation of the truncated substrate 1-(β-D-erythrofuranosyl)orotic acid (EO) lacking a 5′-phosphodianion group catalyzed by OMPDC from Methanothermobacter thermautotrophicus (MtOMPDC) at pH 7.0, 25 °C and I = 0.14 (NaCl) was determined as (kcat/Km)o = 8.7 × 10-3 M-1 s-1 (Table 1).
Figure 2 shows the dependence of the apparent second-order rate constant (kcat/Km)obsd (Table 1) for decarboxylation of the truncated substrate EO catalyzed by MtOMPDC on the concentration of added phosphite dianion at pH 7.0, 25 °C and I = 0.14 (NaCl). There is no evidence for saturation of MtOMPDC by phosphite dianion and the data for [HPO32-] < 30 mM show a good fit to eq 2 that was derived for the mechanism shown in Scheme 4 with [HPO32-] ≪ Kd. The slope of the correlation gives the third-order rate constant for the phosphite-activated reaction as (kcat/Km)E·HPi/Kd = 2500 M-2 s-1 (Table 1). However, there are small (ca. 30%) positive deviations of the data at [HPO32-] = 36 mM from the linear correlation established for [HPO32-] < 30 mM (Figure 2). A control experiment at [HPO32-] = 18 mM, [Cl-] = 12 mM and [NO3-] = 63 mM (I = 0.14) gave a value of (kcat/Km)obsd = 6.3 M-1 s-1, which is 7-fold lower than the corresponding value of 46 M-1 s-1 determined at [HPO32-] = 18 mM and [Cl-] = 75 mM (I = 0.14). This suggests that there are specific salt effects on the activity of MtOMPDC associated with the replacement of chloride with other anions.
Figure 2.
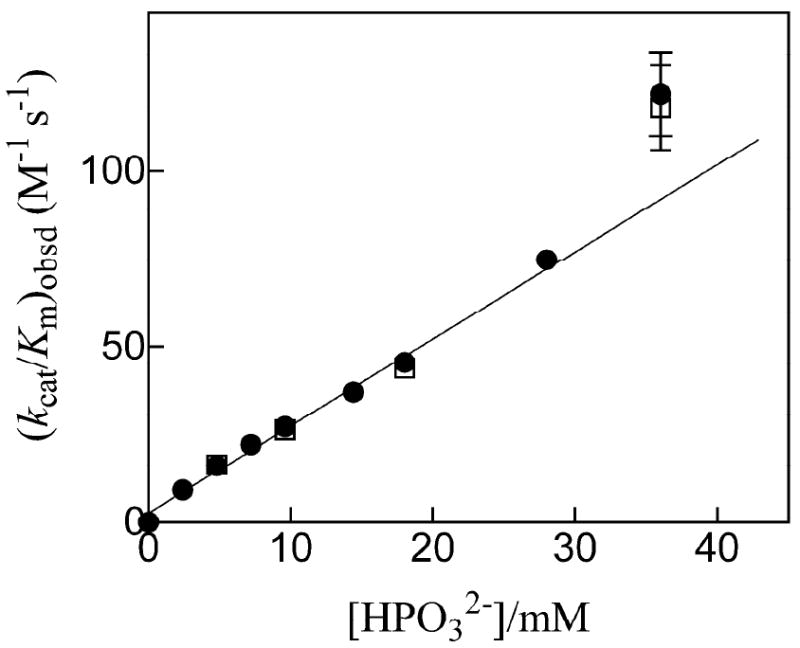
Dependence of (kcat/Km)obsd for the decarboxylation of EO catalyzed by MtOMPDC on the concentration of added phosphite dianion at pH 7.0, 25 °C and I = 0.14 (NaCl). The closed and open symbols represent data obtained in two independent experiments that were reproducible to within 5%. The slope of the correlation gives the third-order rate constant (kcat/Km)/Kd = 2500 M-1 s-1 for the phosphite-activated reaction. The data at [HPO32-] = 36 mM include error bars that indicate the estimated maximum range of experimental error (± 10%) and they were not included in the correlation (see text).
Scheme 4.
| (2) |
Table 2 reports the kinetic parameters kcat, kcat/Km and Km for decarboxylation of OMP catalyzed by ScOMPDC, MtOMPDC and EcOMPDC at various temperatures between 5 °C and 55 °C. Care was taken to work at concentrations of OMPDC where the enzyme exists mainly in the active dimeric form (4). It was routinely shown that the kinetic parameters are independent of the concentration of both OMPDC and the initial concentration of OMP used for their determination. Control experiments were conducted to ensure that there that the temperature variation did not result in a loss of enzyme activity in the timeframe of the assays.
Table 2.
Temperature dependence of the kinetic parameters for decarboxylation of OMP catalyzed by ScOMPDC, MtOMPDC and EcOMPDC at pH 7.1 and I = 0.1 (NaCl).a
| ScOMPDC | MtOMPDC | EcOMPDC | |||||
|---|---|---|---|---|---|---|---|
| Temp °C |
kcatb s-1 |
kcat/Kmc M-1 s-1 |
Kmd μM |
kcatb s-1 |
kcat/Kmc M-1 s-1 |
Kmd μM |
kcatb s-1 |
| 5 | 3.1 ± 0.5 | 2.7 ± 0.2 × 106 | 1.1 ± 0.2 | 0.64 ± 0.03 | 6.0 ± 2.2 × 105 | 1.1 ± 0.4 | 2.5 ± 0.1 |
| 10 | 4.6 ± 0.6 | 3.2 ± 0.2 × 106 | 1.4 ± 0.2 | 3.7 ± 0.1 | |||
| 12 | 5.1 ± 0.5 | ||||||
| 15 | 6.9 ± 0.9 | 5.6 ± 0.4 × 106 | 1.2 ± 0.2 | 2.0 ± 0.3 | 1.4 ± 0.2 × 106 | 1.5 ± 0.3 | 6.4 ± 0.1 |
| 17 | 7.8 ± 0.8 | ||||||
| 25 | 15 ± 1 | 1.1 ± 0.1 × 107 | 1.4 ± 0.2 | 4.7 ± 0.3 | 3.1 ± 0.3 × 106 | 1.5 ± 0.2 | 13 ± 1 |
| 35 | 27 ± 1 | 1.7 ± 0.4 × 107 | 1.6 ± 0.4 | 11 ± 1 | 4.2 ± 0.9 × 106 | 2.8 ± 0.6 | 23 ± 1 |
| 45 | 45 ± 4 | 1.4 ± 0.2 × 107 | 3.3 ± 0.6 | 24 ± 2 | 5.0 ± 1.3 × 106 | 4.8 ± 1.4 | 35 ± 1 |
| 50 | 59 ± 6 | 7.5 ± 0.3 × 106 | 7.9 ± 0.9 | ||||
| 55 | 44 ± 2 | 6.1 ± 0.9 × 106 | 7.3 ± 1.1 | ||||
The quoted errors in kcat and kcat/Km are standard errors that were obtained from multiple experiments to determine these parameters.
Determined from the initial velocity of decarboxylation of OMP when [OMP]o ≥ 20Km. The observed values of kcat were independent of enzyme concentration in the range 11 – 42 nM for ScOMPDC, 76 - 152 nM for MtOMPDC and 30 – 230 nM for EcOMPDC (see text).
Determined from the first-order rate constant for decarboxylation of OMP ([OMP]o ≤ 40 μM) when [OMP]t = 0.3 – 0.5Km (see text).
Calculated from the values of kcat and kcat/Km. The standard error was obtained by propagation of the standard errors in kcat and kcat/Km.
Discussion
A comparison of the kinetic parameters for decarboxylation of the whole substrate OMP and the truncated substrate EO (Table 1) shows that the binding interactions between the enzyme and the 5′-phosphodianion group of OMP are responsible for the 5.2 × 108 and 3.6 × 108-fold increases, respectively, in the second-order rate constant for decarboxylation of the truncated substrate EO catalyzed by ScOMPDC and MtOMPDC. These rate increases correspond to similar intrinsic phosphate binding energies of 11.9 and 11.6 kcal/mol, respectively (14). These results are consistent with a somewhat larger transition state stabilization from interactions of the phosphodianion group of the enzyme-bound ligand with the long flexible phosphate gripper loop of the mesophilic ScOMPDC (19 residues) compared with the shorter flexible loop of the thermophilic MtOMPDC (9 residues) (Figure 1).
We have reported intrinsic phosphate binding energies of 12.2 and ≥ 11.0 kcal/mol for the deprotonation of R-glyceraldehyde 3-phosphate catalyzed by TIM (16) and the reduction of DHAP by NADH catalyzed by GPDH (17), respectively, each of which has a phosphate gripper loop that results in optimal interaction of the substrate phosphodianion group with the protein catalyst (23-27). The similarity in the intrinsic phosphate binding energies for three enzymes that catalyze a variety of chemical reactions suggests that 12 kcal/mol serves as the effective upper limit on the intrinsic phosphate binding energy obtained after billions of years of evolutionary pressure to select for optimal protein catalysts of these different reactions. We suggest that phosphate gripper loops are essential to obtaining this large intrinsic phosphate binding energy, because they provide the protein with the means to make a maximal number of contacts with the substrate, which binds initially at an open active site cleft and then becomes buried in the protein interior upon loop closure (13, 37).
We reported earlier that the binding of phosphite dianion to ScOMPDC strongly activates the enzyme toward decarboxylation of the truncated substrate EO lacking a 5′-phosphodianion group (14). Figure 2 shows that phosphite dianion is also a potent activator of the decarboxylation of EO catalyzed by MtOMPDC. The ratio of the third-order rate constant (kcat/Km)E·HPi/Kd (Scheme 4) for the phosphite-activated decarboxylation of EO and the second-order rate constant (kcat/Km)o for the unactivated decarboxylation of EO provides a good measure of the extent of activation by sub-saturating concentrations of phosphite dianion. The values of the ratios of {(kcat/Km)E·HPi/Kd}/(kcat/Km)o = 5.6 × 105 M-1 and 2.9 × 105 M-1 for the reactions catalyzed by ScOMPDC and MtOMPDC (Table 1) shows that the larger flexible phosphate gripper loop at the enzyme from yeast results in slightly larger advantage from phosphite activation. We conclude that the large reduction in loop size for the thermophilic compared with mesophilic enzyme does not have a severe effect on either phophodianion activation of the whole OMP, or phosphite dianion activation of the truncated substrate EO.
Temperature Dependence of kcat for OMPDCs from Various Organisms
Figure 3 shows Eyring plots of kcat for decarboxylation of OMP catalyzed by ScOMPDC, MtOMPDC and EcOMPDC over the temperature range 5 – 55 °C (278 – 328 K) according to eq 3. Table 3 gives the derived values of ΔH‡ and ΔS‡ that were calculated from the slopes (-ΔH‡/R) and intercepts (ΔS‡/R) of these correlations, respectively. The activation parameters for ScOMPDC and EcOMPDC are very similar so that there is almost no change in the relative values of kcat for these enzymes with changing temperature. However, the ratio of the values of kcat for ScOMPDC and MtOMPDC decreases from 4.8 at 5 °C to 1.9 at 45 °C, which reflects the larger value of ΔH‡ for MtOMPDC (14.8 kcal/mol, Table 3) than for ScOMPDC (11.2 kcal/mol). These results show that, compared with ScOMPDC, MtOMPDC makes better use of the greater thermal energy that is available at the higher temperature maximum for this thermophilic enzyme.
Figure 3.
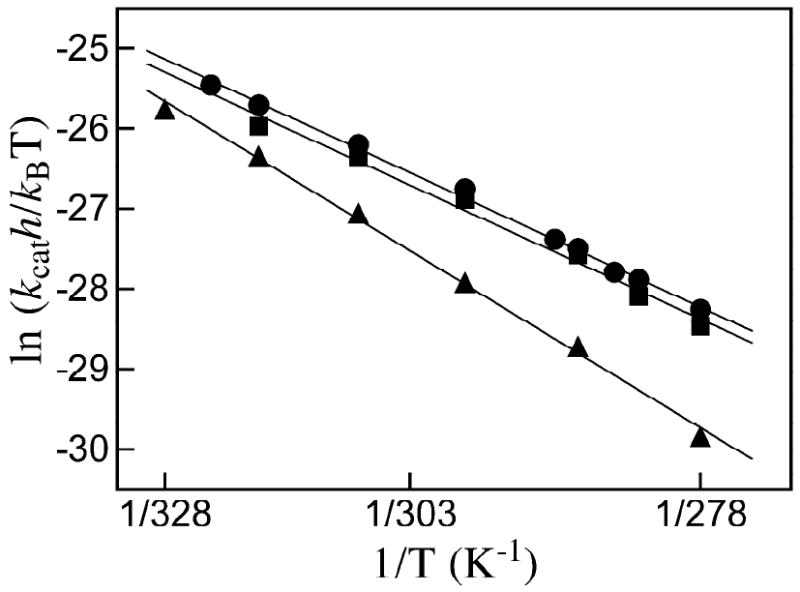
The temperature dependence of kcat (s-1) for the decarboxylation of saturating OMP catalyzed by OMPDCs from various organisms plotted according to the Eyring equation (eq 3). Key: ●, ScOMPDC; ■, EcOMPDC; ▲, MtOMPDC.
Table 3.
Activation parameters for kcat for decarboxylation of OMP catalyzed by OMPDCs from S. cerevisiae, E. Coli and M. thermautotrophicus.a
| (3) |
The entropy of activation for MtOMPDC (-6.0 cal K-1 mol-1, Table 3) is more positive than for ScOMPDC (-16 cal K-1 mol-1), so that there is a larger unfavorable enthalpic and smaller unfavorable entropic change for conversion of the ground state Michaelis complex to the transition state for MtOMPDC-catalyzed decarboxylation than for the ScOMPDC-catalyzed reaction. We suggest that the difference in the size of the active site loops at MtOMPDC and ScOMPDC results in the observed difference in the activation parameters for these enzymes. The greater enthalpic transition state stabilization may reflect the more extensive loop-substrate interactions for the ScOMPDC-catalyzed reaction, and these are suggested to be balanced by a greater entropic requirement for immobilization of the larger loop at the ES complex and transition state for the yeast enzyme. A similar difference in the enthalpy and entropy of activation has been observed for the mesophilic lactate dehydrogenase from Deinococcus radiourans and the corresponding thermophilic enzyme from Thermus thermophilus where the role of loops in catalysis is less well defined (13).
For many enzymes it has been shown that increases in protein thermostability are often the result of enhanced networks of stabilizing ionic interactions between the side chains of amino acid residues (38). These added ionic interactions have the effect of restricting protein conformational changes, so that, at the same temperature, enzymes from thermophiles are often more rigid than their mesophilic counterparts (39, 40). For example, a study of neutron scattering by the thermophilic malate dehydrogenase from Methanococcus jannachii and a mesophilic analog, lactate dehydrogenase from Orytolagus cunniliculus (rabbit muscle), shows that the root mean square fluctuations (1.5 Å) are similar for these enzymes at the temperature of their optimal activity (41). The similar conformational flexibility observed for enzymes at their temperature optimum suggests that the restriction of protein motions below a threshold level might cripple catalytic activity (40, 42, 43). Similarly, loop movement is required both to trap OMPDC-bound substrate (loop closure) and to release the product (loop opening), but yet the loop must remain fixed as the loop-closed enzyme-substrate complex is converted to product.
We suggest that efficient substrate binding and catalysis requires both flexibility of the loop to allow for efficient transport of the ligand to and from OMPDC, but also rigidity at the closed ES and ES‡ complexes. Truncation of the large flexible phosphate gripper loop for the mesophilic enzyme on moving to the thermophilic MtOMPDC might maintain this balance by reducing high-temperature motion of the loop when it is closed over the substrate. This would have the additional effect of reducing the stabilizing interactions between the loop and the substrate phosphodianion group, thereby leading to an increase in ΔH‡ and a decrease in the kinetic parameters for the reaction catalyzed by the thermophilic enzyme at 25 °C (Figure 3). However, the increase in ΔH‡ also sharpens the temperature dependence for the thermophilic enzyme-catalyzed reaction and helps to ensure that the thermophilic and mesophilic enzymes have similar kinetic parameters at their respective operating temperatures.
Temperature Effects on kcat/Km for OMPDCs from Various Organisms
Figure 4 shows the effect of changing temperature on kcat/Km for decarboxylation of OMP catalyzed by ScOMPDC and MtOMPDC. For both enzymes roughly linear correlations of ln (kcat/Km) with 1/T are observed at low temperature (< 35 °C, 308 K) where the value of Km is essentially constant and the overall change in kcat/Km is due to the increase in kcat with increasing T (Table 2). For ScOMPDC there is a sharp downward break in the correlation at 35 °C (308 K) where kcat/Km = 1.7 × 107 M-1 s-1 reaches a maximum (Table 2). This downward break reflects the sharp increases in the Michaelis constant Km with increasing temperature, which outweigh the increases in kcat. These observations are consistent with a change in rate-determining step for kcat/Km for ScOMPDC from rate-determining decarboxylation (kcat, k-1 > kcat, eq 4) for reactions at low temperature, to rate-determining substrate binding (k1, k-1 < kcat, eq 4) for reactions at higher temperatures (Scheme 5) (44, 45). The fractional dependence of kcat/Km on solvent viscosity (32) and the experimental rate constant ratio (k-1/kcat) ≈ 4 (4) determined for ScOMPDC-catalyzed decarboxylation of OMP at 25 °C both show that substrate binding (k1) is already partly rate-determining at this temperature. Therefore, if the temperature dependence of k-1 for breakdown of the E·OMP complex is small, a change in rate-determining step with increasing temperature should be observed (Scheme 5).
Figure 4.
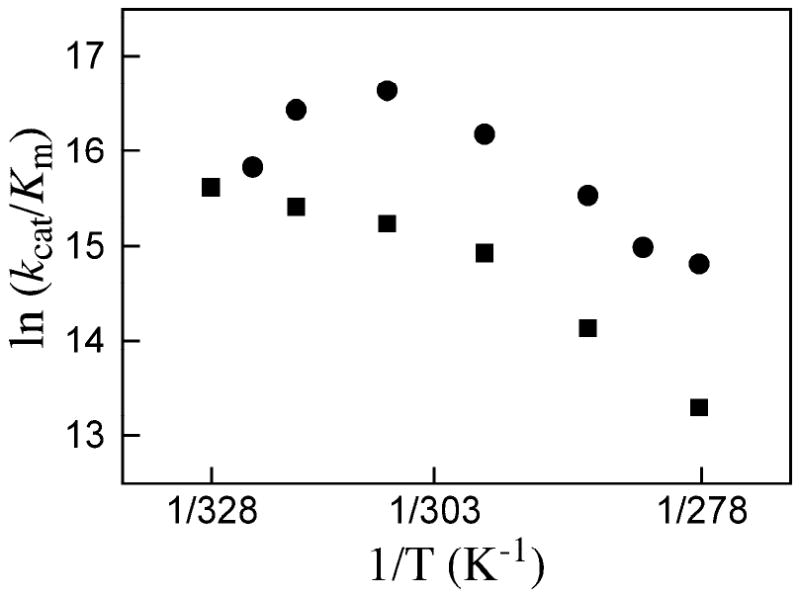
Correlations of ln (kcat/Km) with 1/T for the decarboxylation of sub-saturating OMP catalyzed by ScOMPDC (●) and MtOMPDC (■).
Scheme 5.
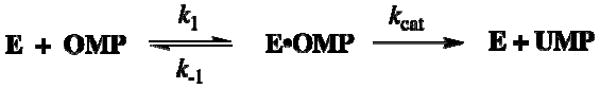
| (4) |
For MtOMPDC the downward break in the correlation in Figure 4 at 25 °C (298 K) where kcat/Km = 3.1 × 106 M-1 s-1 is probably due to a similar change in rate-determining step for the reaction of OMP. The difference in the limiting values of kcat/Km = 6.1 × 106 M-1 s-1 (T = 55 °C, Table 2) and 1.7 × 107 M-1 s-1 (T = 25 °C, Table 2) for the reactions catalyzed by MtOMPDC and ScOMPDC, respectively, suggests that simple diffusion of substrate to the enzyme active sites cannot be rate determining for both reactions. We suggest that the barrier to loop-reorganization to “trap” the substrate at the enzyme active site contributes to the activation barriers for encounter-complex formation, and that this former barrier is larger for the MtOMPDC-catalyzed reaction.
Conclusions and Speculation
A large positive ΔH‡ allows thermophilic enzymes to make efficient use of the large thermal heat energy available from their environment by maximizing the increase in reactivity as the temperature is increased. A small negative ΔS‡ will minimize unfavorable temperature effects associated with an increase in “order” on proceeding from the enzyme-substrate complex to the transition state. We suggest that these thermodynamic constraints favor the evolution of truncated flexible active site loops at enzymes from thermophilic organisms. The results reported here support the conclusion that the relatively short flexible phosphate gripper loop at OMPDC from the thermophile M. thermautotrophicus provides a smaller enthalpic transition state stabilization, but that this enthalpic stabilization carries a lower entropic price, compared with the transition state stabilization provided by the larger loops at OMPDCs from the mesophiles S. cerevisiae and E. coli. This tentative conclusion is not meant to minimize the possible relevance and contribution of other protein motions to the activation barriers for enzyme-catalyzed reactions.
Footnotes
Abbreviations: OMP, orotidine 5′-monophosphate; UMP, uridine 5′-monophosphate; OMPDC, orotidine 5′-monophosphate decarboxylase; ScOMPDC, orotidine 5′-monophosphate decarboxylase from Saccharomyces cerevisiae (yeast); EcOMPDC, orotidine 5′-monophosphate decarboxylase from Escherichia coli; MtOMPDC, orotidine 5′-monophosphate decarboxylase from Methanothermobacter thermautotrophicus; EO, 1-(β-D-erythrofuranosyl)orotic acid; EU, 1-(β-D-erythrofuranosyl)uridine; DHAP, dihydroxyacetone phosphate; TIM, triosephosphate isomerase; GPDH, glycerol 3-phosphate dehydrogenase; MOPS, 3-(N-morpholino)propanesulfonic acid; IPTG, isopropyl β-D-thiogalactopyranoside; NADH, nicotinamide adenine dinucleotide, reduced form.
This work was supported by Grants GM39754 to JPR and GM65155 to JAG from the National Institutes of Health.
References
- 1.Callahan BP, Miller BG. OMP decarboxylase - An enigma persists. Bioorg Chem. 2007;35:465–469. doi: 10.1016/j.bioorg.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 2.Miller BG, Wolfenden R. Catalytic proficiency: the unusual case of OMP decarboxylase. Annu Rev Biochem. 2002;71:847–885. doi: 10.1146/annurev.biochem.71.110601.135446. [DOI] [PubMed] [Google Scholar]
- 3.Radzicka A, Wolfenden R. A proficient enzyme. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- 4.Porter DJT, Short SA. Yeast Orotidine-5′-Phosphate Decarboxylase: Steady-State and Pre-Steady-State Analysis of the Kinetic Mechanism of Substrate Decarboxylation. Biochemistry. 2000;39:11788–11800. doi: 10.1021/bi001199v. [DOI] [PubMed] [Google Scholar]
- 5.Stanton CL, Kuo IF, Mundy CJ, Laino T, Houk KN. QM/MM metadynamics study of the direct decarboxylation mechanism for orotidine-5′-monophosphate decarboxylase using two different QM regions: acceleration too small to explain rate of enzyme catalysis. J Phys Chem B. 2007;111:12573–12581. doi: 10.1021/jp074858n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silverman R, Groziak M. Model chemistry for a covalent mechanism of action of orotidine 5′-phosphate decarboxylase. J Am Chem Soc. 1982;104:6434–6439. [Google Scholar]
- 7.Shostak K, Jones ME. Orotidylate decarboxylase: insights into the catalytic mechanism from substrate specificity studies. Biochemistry. 1992;31:12155–12161. doi: 10.1021/bi00163a026. [DOI] [PubMed] [Google Scholar]
- 8.Lee JK, Houk KN. A proficient enzyme revisited: the predicted mechanism for orotidine monophosphate decarboxylase. Science. 1997;276:942–945. doi: 10.1126/science.276.5314.942. [DOI] [PubMed] [Google Scholar]
- 9.Lee TS, Chong LT, Chodera JD, Kollman PA. An alternative explanation for the catalytic proficiency of orotidine 5′-phosphate decarboxylase. J Am Chem Soc. 2001;123:12837–12848. doi: 10.1021/ja011096f. [DOI] [PubMed] [Google Scholar]
- 10.Appleby TC, Kinsland C, Begley TP, Ealick SE. The crystal structure and mechanism of orotidine 5′-monophosphate decarboxylase. Proc Natl Acad Sci U S A. 2000;97:2005–2010. doi: 10.1073/pnas.259441296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toth K, Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. Product Deuterium Isotope Effect for Orotidine 5′-Monophosphate Decarboxylase: Evidence for the Existence of a Short-Lived Carbanion Intermediate. J Am Chem Soc. 2007;129:12946–12947. doi: 10.1021/ja076222f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. Formation and Stability of a Vinyl Carbanion at the Active Site of Orotidine 5′-Monophosphate Decarboxylase: pKa of the C-6 Proton of Enzyme-Bound UMP. J Am Chem Soc. 2008;130:1574–1575. doi: 10.1021/ja710384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jencks WP. Binding energy, specificity, and enzymic catalysis: the Circe effect. Adv Enzymol Relat Areas Mol Biol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 14.Amyes TL, Richard JP, Tait JJ. Activation of orotidine 5′-monophosphate decarboxylase by phosphite dianion: The whole substrate is the sum of two parts. J Am Chem Soc. 2005;127:15708–15709. doi: 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- 15.Amyes TL, Richard JP. Enzymatic catalysis of proton transfer at carbon: activation of triosephosphate isomerase by phosphite dianion. Biochemistry. 2007;46:5841–5854. doi: 10.1021/bi700409b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amyes TL, O'Donoghue AC, Richard JP. Contribution of phosphate intrinsic binding energy to the enzymatic rate acceleration for triosephosphate isomerase. J Am Chem Soc. 2001;123:11325–11326. doi: 10.1021/ja016754a. [DOI] [PubMed] [Google Scholar]
- 17.Tsang WY, Amyes TL, Richard JP. A Substrate in Pieces: Allosteric Activation of Glycerol 3-Phosphate Dehydrogenase (NAD+) by Phosphite Dianion. Biochemistry. 2008;47:4575–4582. doi: 10.1021/bi8001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Begley TP, Ealick SE. Enzymatic reactions involving novel mechanisms of carbanion stabilization. Curr Opin Chem Biol. 2004;8:508–515. doi: 10.1016/j.cbpa.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 19.Harris P, Poulsen JCN, Jensen KF, Larsen S. Structural basis for the catalytic mechanism of a proficient enzyme: orotidine 5′-monophosphate decarboxylase. Biochemistry. 2000;39:4217–4224. doi: 10.1021/bi992952r. [DOI] [PubMed] [Google Scholar]
- 20.Harris P, Poulsen JC, Jensen KF, Larsen S. Substrate binding induces domain movements in orotidine 5′-monophosphate decarboxylase. J Mol Biol. 2002;318:1019–1029. doi: 10.1016/S0022-2836(02)00200-0. [DOI] [PubMed] [Google Scholar]
- 21.Miller BG, Hassell AM, Wolfenden R, Milburn MV, Short SA. Anatomy of a proficient enzyme: the structure of orotidine 5′-monophosphate decarboxylase in the presence and absence of a potential transition state analog. Proc Natl Acad Sci U S A. 2000;97:2011–2016. doi: 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu N, Mo Y, Gao J, Pai EF. Electrostatic stress in catalysis: structure and mechanism of the enzyme orotidine monophosphate decarboxylase. Proc Natl Acad Sci U S A. 2000;97:2017–2022. doi: 10.1073/pnas.050417797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sampson NS, Knowles JR. Segmental movement: definition of the structural requirements for loop closure in catalysis by triosephosphate isomerase. Biochemistry. 1992;31:8482–8487. doi: 10.1021/bi00151a014. [DOI] [PubMed] [Google Scholar]
- 24.Sampson NS, Knowles JR. Segmental motion in catalysis: investigation of a hydrogen bond critical for loop closure in the reaction of triosephosphate isomerase. Biochemistry. 1992;31:8488–8494. doi: 10.1021/bi00151a015. [DOI] [PubMed] [Google Scholar]
- 25.Pompliano DL, Peyman A, Knowles JR. Stabilization of a reaction intermediate as a catalytic device: definition of the functional role of the flexible loop in triosephosphate isomerase. Biochemistry. 1990;29:3186–3194. doi: 10.1021/bi00465a005. [DOI] [PubMed] [Google Scholar]
- 26.Ou X, Ji C, Han X, Zhao X, Li X, Mao Y, Wong LL, Bartlam M, Rao Z. Crystal structures of human glycerol 3-phosphate dehydrogenase 1 (GPD1) J Mol Biol. 2006;357:858–869. doi: 10.1016/j.jmb.2005.12.074. [DOI] [PubMed] [Google Scholar]
- 27.Suresh S, Turley S, Opperdoes FR, Michels PAM, Hol WGH. A potential target enzyme for trypanocidal drugs revealed by the crystal structure of NAD-dependent glycerol-3-phosphate dehydrogenase from Leishmania mexicana. Structure. 2000;8:541–552. doi: 10.1016/s0969-2126(00)00135-0. [DOI] [PubMed] [Google Scholar]
- 28.Smiley JA, Paneth P, O'Leary MH, Bell JB, Jones ME. Investigation of the enzymic mechanism of yeast orotidine-5′-monophosphate decarboxylase using carbon-13 kinetic isotope effects. Biochemistry. 1991;30:6216–6223. doi: 10.1021/bi00239a020. [DOI] [PubMed] [Google Scholar]
- 29.Miller BG, Butterfoss GL, Short SA, Wolfenden R. Role of Enzyme-Ribofuranosyl Contacts in the Ground State and Transition State for Orotidine 5′-Phosphate Decarboxylase: A Role for Substrate Destabilization? Biochemistry. 2001;40:6227–6232. doi: 10.1021/bi0028993. [DOI] [PubMed] [Google Scholar]
- 30.Van Vleet JL, Reinhardt LA, Miller BG, Sievers A, Cleland WW. Carbon isotope effect study on orotidine 5′-monophosphate decarboxylase: support for an anionic intermediate. Biochemistry. 2008;47:798–803. doi: 10.1021/bi701664n. [DOI] [PubMed] [Google Scholar]
- 31.Barnett SA, Amyes TL, Wood BM, Gerlt JA, Richard JP. Dissecting the Total Transition State Stabilization Provided by Amino Acid Side Chains at Orotidine 5′-Monophosphate Decarboxylase: A Two-Part Substrate Approach. Biochemistry. 2008;47:7785–7787. doi: 10.1021/bi800939k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wood BM, Chan KK, Amyes TL, Richard JP, Gerlt JA. Mechanism of the Orotidine 5′-Monophosphate Decarboxylase-Catalyzed Reaction: Effect of Solvent Viscosity on Kinetic Constants. Biochemistry. 2009;48:5510–5517. doi: 10.1021/bi9006226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sievers A, Wolfenden R. The effective molarity of the substrate phosphoryl group in the transition state for yeast OMP decarboxylase. Bioorg Chem. 2005;33:45–52. doi: 10.1016/j.bioorg.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 34.Moffatt JG. The synthesis of orotidine 5′-phosphate. J Am Chem Soc. 1963;85:1118–1123. [Google Scholar]
- 35.Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. In: Proteomics Protocols Handbook. Walker JM, editor. Humana Press Inc.; Totowa, NJ: 2005. pp. 571–607. [Google Scholar]
- 37.Herschlag D. The role of induced fit and conformational changes of enzymes in specificity and catalysis. Bioorg Chem. 1988;16:62–96. [Google Scholar]
- 38.Vielle C, Zeikus GJ. Hyperthermophilic Enzymes: Sources, Uses, and Molecular Mechanisms for Thermostability. Microbiol Mol Biol Rev. 2001;65:1–43. doi: 10.1128/MMBR.65.1.1-43.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wrba A, Schweiger A, Schultes V, Jaenicke R, Zavodszky P. Extremely thermostable D-glyceraldehyde-3-phosphate dehydrogenase from the eubacterium Thermotoga maritima. Biochemistry. 1990;29:7584–7592. doi: 10.1021/bi00485a007. [DOI] [PubMed] [Google Scholar]
- 40.Závodszky P, Kardos J, Svingor, Petsko GA. Adjustment of conformational flexibility is a key event in the thermal adaptation of proteins. Proc Natl Acad Sci U S A. 1998;95:7406–7411. doi: 10.1073/pnas.95.13.7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tehei M. Neutron Scattering Reveals the Dynamic Basis of Protein Adaptation to Extreme Temperature. J Biol Chem. 2005;280:40974–40979. doi: 10.1074/jbc.M508417200. [DOI] [PubMed] [Google Scholar]
- 42.Liang Z, Lee T, Resing KA, Ahn NG, Klinman J. Thermal-activated protein mobility and its correlation with catalysis in thermophilic alcohol dehydrogenase. Proc Natl Acad Sci U S A. 2004;101:9556–9561. doi: 10.1073/pnas.0403337101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang ZX, Tsigos I, Bouriotis V, Klinman JP. Impact of Protein Flexibility on Hydride-Transfer Parameters in Thermophilic and Psychrophilic Alcohol Dehydrogenases. J Am Chem Soc. 2004;126:9500–9501. doi: 10.1021/ja047087z. [DOI] [PubMed] [Google Scholar]
- 44.Brouwer A, Kirsch J. Investigation of diffusion-limited rates of chymotrypsin reactions by viscosity variation. Biochemistry. 1982;21:1302–1307. doi: 10.1021/bi00535a030. [DOI] [PubMed] [Google Scholar]
- 45.Lim W, Raines R, Knowles J. Triosephosphate isomerase catalysis is diffusion controlled. Appendix: Analysis of triose phosphate equilibria in aqueous solution by phosphorus-31 NMR. Biochemistry. 1988;27:1165–1167. doi: 10.1021/bi00404a013. [DOI] [PubMed] [Google Scholar]
- 46.Chan KK, Wood BM, Fedorov AA, Fedorov EV, Imker HJ, Amyes TL, Richard JP, Almo SC, Gerlt JA. Mechanism of the Orotidine 5′-Monophosphate Decarboxylase-Catalyzed Reaction: Evidence for Substrate Destabilization. Biochemistry. 2009;48:5518–5531. doi: 10.1021/bi900623r. [DOI] [PMC free article] [PubMed] [Google Scholar]