

SUMMARY
Members of the Sirtuin (SIRT) family of NAD+-dependent deacetylases promote longevity in multiple organisms. Deficiency of mammalian SIRT6 leads to shortened lifespan and an aging-like phenotype in mice, but the underlying molecular mechanisms are unclear. Here we show that SIRT6 functions at chromatin to attenuate NF-κB signaling. SIRT6 interacts with the NF-κB RELA subunit and deacetylates histone H3 lysine 9 (H3K9) at NF-κB target gene promoters. In SIRT6-deficient cells, hyperacetylation of H3K9 at these target promoters is associated with increased RELA promoter occupancy, and enhanced NF-κB-dependent modulation of gene expression, apoptosis and cellular senescence. Computational genomics analyses revealed increased activity of NF-κB-driven gene expression programs in multiple Sirt6-deficient tissues in vivo. Moreover, haploinsufficiency of RelA rescues the early lethality and degenerative syndrome of Sirt6-deficient mice. We propose that SIRT6 attenuates NF-κB signaling via H3K9 deacetylation at chromatin, and hyperactive NF-κB signaling may contribute to premature and normal aging.
INTRODUCTION
The Sirtuin (SIRT) family of nicotinamide adenine dinucleotide (NAD+)-dependent protein deacetylases is implicated in lifespan regulation in yeast, worms, and flies (Haigis and Guarente, 2006). In mammalian genomes, there are seven SIRT genes (SIRT1-SIRT7). Inactivation of the Sirt6 gene in mice leads to dramatically shortened lifespan and a premature aging-like phenotype (Mostoslavsky et al., 2006). Specifically, Sirt6-deficient animals develop normally for several weeks after birth, but succumb to an acute multi-organ degenerative syndrome that is uniformly lethal by one month of age. This phenotype includes bone mineral density defects reminiscent of osteoporosis, spinal curvature abnormalities, loss of subcutaneous fat, lymphocyte attrition, and severe metabolic defects with precipitous drops in serum glucose and IGF-1 levels (Mostoslavsky et al., 2006). In addition, Sirt6-deficient mouse cells are hypersensitive to certain forms of DNA damage and show increased genomic instability (Mostoslavsky et al., 2006). However, relatively little was understood regarding the fundamental molecular activity of SIRT6, and the cellular pathways in which it functions. Recently, we discovered that SIRT6 is a histone H3-lysine 9 (H3K9) deacetylase, and identified a role for this SIRT6 activity at telomeric chromatin, where it prevents telomere dysfunction in human cells (Michishita et al., 2008). However, telomere dysfunction is not observed in Sirt6-deficient mouse cells (Michishita et al., 2008), and the molecular pathways that contribute to the phenotypes of Sirt6-/- mice remain unclear. It is also unknown whether SIRT6 deacetylates H3K9 at genomic loci beyond telomeres, or whether it regulates other chromatin-templated processes. Thus, much remains to be discovered regarding the molecular basis of SIRT6 action.
Another emerging modulator of aging-related pathways in mammals is the NF-κB family of transcription factors, which controls the activity of genes involved in apoptosis, cell senescence, inflammation, and immunity (reviewed by (Adler et al., 2008; Hayden and Ghosh, 2008). NF-κB target gene activities increase with age in many mammalian tissues and in stem cells (Adler et al., 2007; Chambers et al., 2007; Helenius et al., 1996; Korhonen et al., 1997). Moreover, blockade of NF-κB in the skin of aged mice can reverse the global gene expression program and tissue characteristics to that of younger animals (Adler et al., 2007). NF-κB is also implicated in age-dependent induction of cellular senescence in epithelial and hematopoietic progenitor cells (Adler et al., 2007; Chambers et al., 2007). Therefore, NF-κB appears to be an important regulator of aging-related cellular processes.
NF-κB activity is controlled by multiple layers of regulation (reviewed by (Hayden and Ghosh, 2008). There are five NF-κB family members, each of which contains a Rel-homology DNA binding domain. These NF-κB proteins typically bind to their target DNA sequences as dimers. In unstimulated cells, NF-κB is sequestered in the cytoplasm by inhibitory IκB proteins. Upon stimulation by diverse cell stresses (such as cytotoxic cytokine TNF-α, oxidative stress, DNA damage, or infection), IκBs are degraded, allowing NF-κB to translocate into the nucleus and activate target genes. NF-κB induces the transcription of IκBα and other negative regulators of the pathway (Hayden and Ghosh, 2008), which contribute to signal inactivation. Thus, in individual cells, NF-κB signaling is characterized by dynamic patterns of periodic NF-κB nuclear localization and target gene activation interspersed with nuclear exit and gene deactivation. Chromatin regulation likely plays an important role in modulating NF-κB target gene expression patterns. For example, certain NF-κB target genes are primed while others are prevented from reactivation following a pioneering round of NF-κB activity, and these effects are associated with distinct chromatin modifications at the target genes (Foster et al., 2007). Additional factors that control chromatin dynamics in NF-κB regulation remain to be explored.
Given the intriguing links of both SIRT6 and NF-κB to aging-associated processes, we hypothesized that these two pathways may functionally interact. Here we report that SIRT6 binds to the NF-κB subunit RELA and attenuates NF-κB signaling by modifying chromatin at NF-κB target genes. Genetic and genomic studies further reveal that SIRT6-mediated control of NF-κB prevents aging-associated, hyperactive NF-κB dependent gene expression, and inhibition of NF-κB can rescue the early lethality of Sirt6 knockout mice.
RESULTS
SIRT6 Interacts Physically with NF-κB Subunit RELA
To probe a potential connection between SIRT6 and NF-κB, we asked whether SIRT6 physically interacts with the NF-κB subunit RELA. FLAG-tagged SIRT6 or several other mammalian SIRT proteins were expressed in 293T cells and immunoprecipitated with anti-FLAG antibodies. Western analysis of the immunoprecipitates (IPs) revealed that RELA associates with FLAG-SIRT6 (Figure 1A). Other FLAG-tagged SIRTs did not associate significantly with RELA under these conditions. The interaction between FLAG-SIRT6 and RELA is not due to DNA bridging, because it is resistant to ethidium bromide (Figure S1). Western analysis of endogenous SIRT6 IP’s also revealed a specific interaction with RELA (Figure 1B), but not other NF-κB family members (Figure S1), and conversely, SIRT6 was detected in IPs of endogenous RELA (Figure S1). We note that on long exposures, the previously reported interaction between RELA and SIRT1 (Yeung et al., 2004) was also observed, but much more weakly than the RELA-SIRT6 interaction (data not shown). Notably, the SIRT6-RELA interaction was augmented by TNF-α stimulation (Figure 1C), suggesting that TNFα-dependent translocation of RELA to the nucleus facilitates its interaction with SIRT6, a predominantly nuclear protein. In vitro pull-down assays using purified GST-fusion proteins revealed a specific interaction between GST-SIRT6 and in vitro translated (IVT) RELA (Figure 1D), and between a GST-RELA protein and IVT SIRT6 (Figure 1E), suggesting that the SIRT6-RELA interaction is direct. Together, the data indicate that SIRT6 interacts physically with RELA in vitro and in cells.
Figure 1. SIRT6 Interacts with RELA.
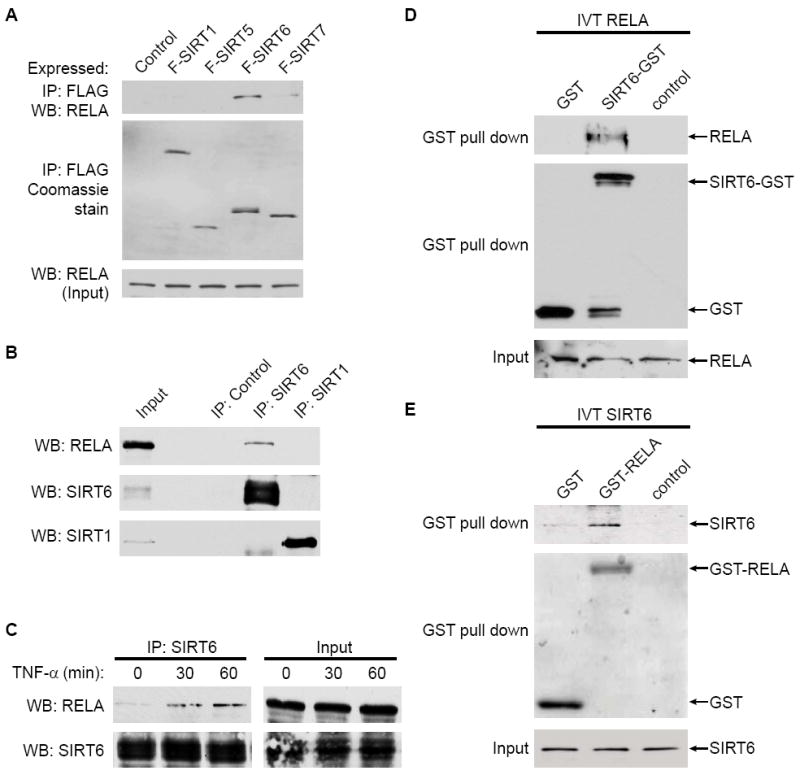
(A) Co-IP of RELA with FLAG-tagged SIRT6 protein. Western blot (WB) analysis reveals RELA protein in immunoprecipitates (IP) of FLAG-tagged SIRT6, but not several other SIRTs, expressed in 293T cells. A very weak interaction with SIRT7 may be observed under these conditions.
(B) Endogenous SIRT6 and RELA proteins interact. Western blot analysis of RELA following co-IP with endogenous SIRT6 or SIRT1, or control IP is shown.
(C) SIRT6-RELA interaction is induced upon TNF-α treatment. Endogenous co-IP of RELA with SIRT6 from 293T cells following TNF-α (20 ng/ml) treatment.
(D) In vitro translated RELA binds purified recombinant SIRT6-GST.
(E) In vitro translated SIRT6 binds purified recombinant GST-RELA (aa1-431).
SIRT6 is Recruited to NF-κB Target Gene Promoters
The TNF-α-inducible physical interaction between SIRT6 and RELA suggested that SIRT6 might associate with promoters of RELA target genes. To test this possibility, HeLa cells stably expressing FLAG-tagged SIRT6 protein, or empty vector control, were stimulated with TNF-α, and chromatin immunoprecipitation (ChIP) was performed with anti-FLAG antibodies. In response to TNF-α, FLAG-SIRT6 was detected at the promoters of several NF-κB target genes, including IAP2, MnSOD, ICAM, and NFKBIA (Figure 2A, data not shown). We then assessed occupancy of endogenous SIRT6 at these promoters by ChIP analysis using SIRT6 antibodies, compared to IgG negative control background signals. Again, TNF-α led to increased levels of SIRT6 occupancy at these RELA target promoters (Figure 2B). We note that SIRT6 expression levels do not change under these conditions, and thus cannot account for the TNF-α dependent increase in the SIRT6 ChIP (Figure S1). To determine whether SIRT6 is recruited to these genomic targets via its interaction with RELA, we used short hairpin RNAs (shRNAs) to knockdown RELA expression (Figure 2C). This depletion of RELA abolished the TNF-α induced occupancy of SIRT6 at the RELA target promoters (Figure 2D). Similar results were observed when RELA was inactivated using siRNAs targeting an independent RELA sequence (Figure S2) and in RelA knockout mouse cells (Figure S3). Together, these data suggest that SIRT6 is recruited to chromatin at the promoters of RELA target genes, via its physical interaction with RELA.
Figure 2. RELA Recruits SIRT6 to Target Promoters.

(A) FLAG-SIRT6 is recruited to promoters of NF-κB target genes. HeLa cells stably expressing either pBabe-FLAG-SIRT6 or empty pBabe vector were treated with TNF-α (20 ng/ml) for 45 minutes. ChIP with α-FLAG antibodies was performed, and FLAG-SIRT6 occupancy (mean ± s.e) is shown relative to background signals in ChIPs from pBabe control cells.
(B) Endogenous SIRT6 is recruited to the promoters of NF-κB target genes. HeLa cells were treated with TNF-α (20 ng/ml) for 45 minutes. ChIP with α-SIRT6 antibodies was performed, and SIRT6 occupancy (mean ± s.e) is shown relative to background signals in IgG negative control ChIPs.
(C) Western analysis of RELA protein in HeLa cells stably expressing RELA shRNA (RELA sh).
(D) RELA is required for recruitment of SIRT6 to the promoters of NF-κB target genes. Cells were transduced with either RELA shRNA or control pSR vector and treated with TNF-α (20 ng/ml) for 1 hour. ChIP with α-SIRT6 antibodies was performed and SIRT6 occupancy relative to untreated pSR control cells is shown.
SIRT6 Represses NF-κB Target Gene Expression
The association of SIRT6 with RELA target promoters suggested that SIRT6 levels might influence RELA-dependent transcription. Therefore, we tested the effects of over-expressing SIRT6 or several other SIRTs on expression of a NF-κB-luciferase reporter gene. Only SIRT6 and SIRT1 significantly repressed NF-κB reporter gene activity, whereas catalytically inactive SIRT6 and SIRT1 mutant proteins did not (Figure 3A). To determine whether endogenous SIRT6 regulates NF-κB transcriptional activity, we stably knocked down SIRT6 expression in HeLa cells using two independent short hairpin RNAs that were previously validated for specific targeting of SIRT6 (Figure 3B) (Michishita et al., 2008). SIRT6 depletion led to constitutive NF-κB reporter gene activity, which, upon TNF-α treatment, was further enhanced to levels considerably higher than in SIRT6-proficient control cells (Figure 3C). SIRT6 depletion also endowed exogenous RELA with higher transcriptional potency (Figure 3D).
Figure 3. SIRT6 Represses RELA Target Gene Expression.
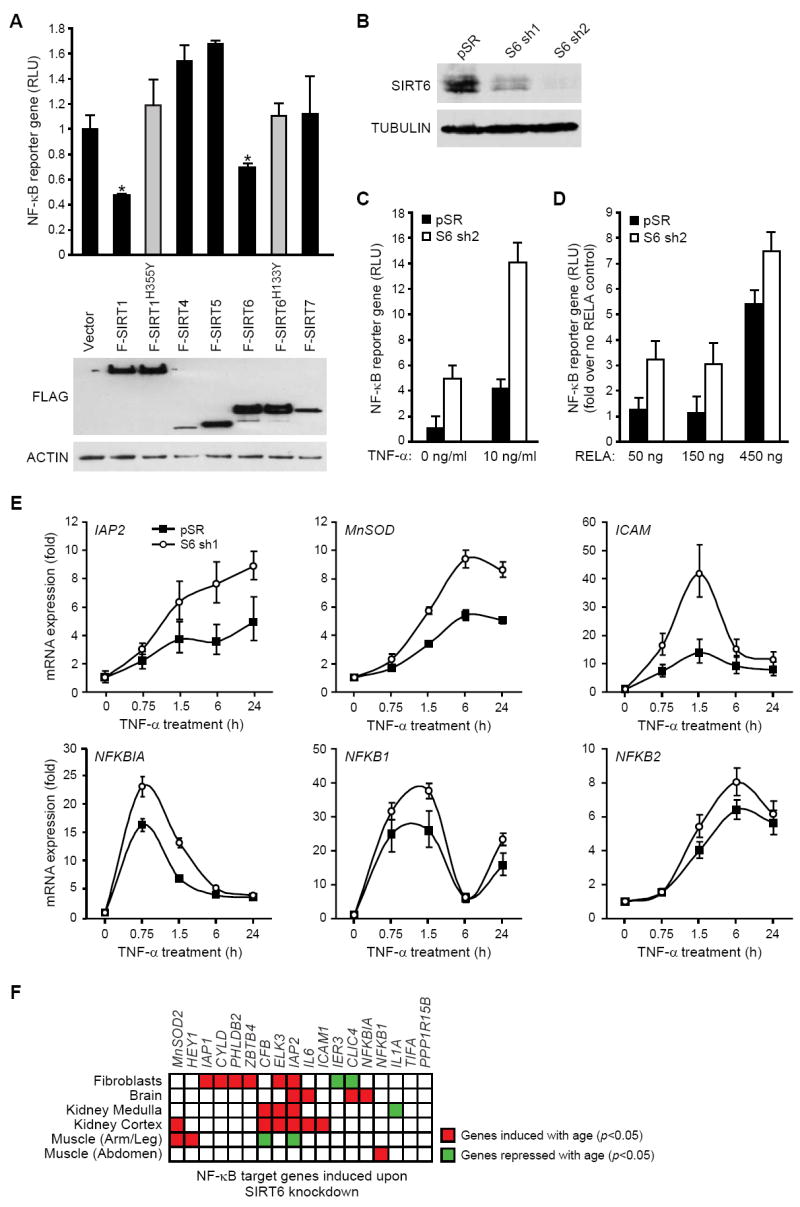
(A) SIRT6 and SIRT1 repress NF-κB transcriptional activity. Top: Relative luciferase units (RLU) of RELA mediated transcription of IL1-Luc (NF-κB reporter gene) activity upon transfection of FLAG-tagged SIRT proteins or vector control (mean ± s.e.). *, p<0.05, Student’s t-test. Bottom: Immunoblot showing expression of FLAG-tagged SIRT proteins.
(B) Western analysis of SIRT6 protein in HeLa cells stably expressing two independent SIRT6 shRNAs (S6 sh1 and S6 sh2).
(C, D) SIRT6 inhibits TNF-α-induced (C) and RELA-induced (D) reporter gene expression. RLU of IL1-Luc (NF-κB reporter gene) activity (mean ± s.e.) in control (pSR) and SIRT6-knockdown (S6 sh2) HeLa cells is shown.
(E) Increased expression of NF-κB target genes in SIRT6 knock-down cells. Quantitative TaqMan real-time PCR analysis of the indicated mRNAs is shown normalized to GAPDH levels (mean ± s.e.).
(F) Regulation of 18 SIRT6-dependent NF-κB target genes in six microarray data sets of human aging. Each row is a data set; each column is a gene. Significant induction (red) or repression (green) with age (p<0.05, one-sided t-test) is shown. The preponderance of age-dependent induction (81% observed vs. 50% expected) is significant (p=0.001, hypergeometric distribution).
To determine whether SIRT6 regulates endogenous RELA-dependent gene expression, we compared the expression of multiple NF-κB target genes in SIRT6 knock-down and control cells by quantitative RT-PCR. In control cells, TNF-α treatment led to inducible expression of NF-κB target genes, as expected (Figure 3E). The time-course of this expression varied for specific genes, consistent with previous reports (Hoffmann et al., 2002). SIRT6 knock-down with both shRNAs led to super-induction of a subset of NF-κB target genes, including IAP2, MnSOD2, ICAM1, and NFKBIA (Figures 3E, S4). Moreover, the increased expression of RELA target genes that occurs with SIRT6 knock-down was abolished in cells depleted of RELA, demonstrating that these effects are not due to RELA-independent effects of SIRT6 on gene expression (Figure S4).
Intriguingly, the SIRT6-repressed NF-κB target genes overlap with genes that are increasingly expressed with age, as assessed by a compendium of 294 microarrays of global gene expression changes that occur with age in six human tissues (Adler et al., 2007), (Figure 3F, p=0.001, hypergeometric distribution). For example, IAP2, MnSOD, ICAM1, NFKBIA and NFKB1, are all induced with age in one or more human tissues. Together, our results suggest that SIRT6 directly inhibits expression of a subset of NF-κB target genes, especially those associated with aging.
SIRT6 Deacetylates Histone H3 Lysine 9 on Promoters of NF-κB Target Genes to Destabilize NF-κB
We recently showed that SIRT6 is a specific histone H3K9 deacetylase that modulates chromatin structure at telomeres (Michishita et al., 2008). In the context of gene expression, acetylated H3K9 (H3K9Ac) is associated with actively transcribed genes, whereas H3K9 deacetylation correlates with gene repression (Kouzarides, 2007). Therefore, we asked whether SIRT6 is required for deacetylation of H3K9 at NF-κB target genes. ChIP analysis revealed that H3K9 acetylation is induced following TNF-α treatment in SIRT6-proficient control cells at the promoters of multiple NF-κB target genes (Figure 4B), consistent with transcriptional induction. In cells depleted of SIRT6 (Figure 4A), H3K9 was hyperacetylated at these promoters in response to TNF-α (Figure 4B). A similar hyperacetylation of H3K9 was observed in Sirt6 knockout mouse cells (Figure S3). In contrast, no hyperacetylation was observed at other histone residues in SIRT6-deficient cells, consistent with the specificity of SIRT6 for H3K9Ac (Figure S2). Together, these data suggest that SIRT6 is recruited to promoters of NF-κB target genes to deacetylate H3K9.
Figure 4. SIRT6 Deacetylates Histone H3K9 at promoters of RELA target genes.

(A) Western analysis of SIRT6 protein in HeLa cells stably expressing SIRT6 shRNA (sh2)
(B) SIRT6 is required for H3K9 deacetylation at promoters of RELA target genes. ChIP with α-H3K9Ac and α-H3 antibodies was performed. H3K9 acetylation at RELA target gene promoters (mean ± s.e.) is shown relative to untreated control samples and normalized to total H3 levels.
(C) SIRT6 is required to limit RELA occupancy at the promoter of RELA target gene promoters. ChIP with α-RELA antibodies was performed following a 30 minute TNF-α pulse; RELA occupancy (mean ± s.e.) at promoters relative to untreated control samples is shown.
(D)-(E) SIRT6-mediated deacetylation of nucleosomes inhibits nucleosome binding to RELA. Western analysis of H3K9Ac levels on nucleosomes following incubation with SIRT6 in NAD-dependent deacetylation or mock reactions (D). The acetylated or SIRT6-deacetylated nucleosomes were used in nucleosome binding assays with increasing amounts of GST-RELA protein (E). Extent of nucleosome binding can be estimated by comparing levels of bound (**) and unbound (*) nucleosomes. Reduced binding is observed in the SIRT6-deacetylated samples (compare 10 ug RELA samples)
Because histone deacetylation is proposed to decrease the accessibility of chromatin to DNA binding factors (Kouzarides, 2007), we reasoned that SIRT6-mediated deacetylation of H3K9 might contribute to destabilization of RELA at promoters of NF-κB target genes. Therefore, we examined the effects of SIRT6 depletion on RELA occupancy at target gene promoters following TNF-α stimulation by ChIP. In SIRT6-depleted cells, RELA occupancy at these promoters was significantly enhanced and prolonged (Figure 4C, S2). Similar effects were observed in Sirt6 knockout mouse cells (Figure S3). This effect was not due to delayed synthesis of IκBα because IκBα is a direct NF-κB target gene and in fact re-accumulates more rapidly in SIRT6-depleted cells (Figure S5). To further investigate the possibility that deacetylation of H3K9 by SIRT6 destabilizes RELA at chromatin, we carried out in vitro analyses to compare RELA association with acetylated nucleosomes versus nucleosomes that were deacetylated by SIRT6. In vitro nucleosome binding assays revealed a strong interaction of RELA with purified, acetylated nucleosomes, whereas much less RELA bound to nucleosomes following deacetylation of H3K9 by SIRT6 (Figures 4D, 4E). Collectively, these results suggest that SIRT6-mediated deacetylation of H3K9 promotes RELA destabilization from target gene chromatin and termination of NF-κB signaling.
SIRT6 Depletion Elevates Apoptotic Resistance and Induction of Senescence via NF-κB
We next examined whether known NF-κB cellular functions are affected by SIRT6 depletion, and if so, whether this is dependent on NF-κB. NF-κB activity is associated with increased resistance to apoptosis and increased cellular senescence (Adler et al., 2007; Hardy et al., 2005; Hayden and Ghosh, 2004). Consistent with the observed induction of anti-apoptotic genes by SIRT6 depletion (Figure 3), SIRT6 knock-down cells were more resistant to apoptosis induced by the death adaptor protein FADD (Figure 5A). This effect of SIRT6 depletion was reversed by co-expression of the super repressor IκB-αM (Van Antwerp et al., 1996), indicating that the apoptosis resistance requires NF-κB (Figure 5A). We also probed a potential functional interplay between SIRT6 and NF-κB in stress-induced senescence of primary human keratinocytes, which occurs spontaneously in ex vivo culture (Sherr and DePinho, 2000). SIRT6 knock-down significantly increased senescence of primary keratinocytes, and this effect was reversed by depletion of RELA by RNAi (Figures 5B, 5C, and 5D). In contrast, NF-κB inhibition did not relieve the premature senescence observed in SIRT6 knock-down primary human fibroblasts (data not shown), which is due to telomere dysfunction and H3K9 hyperacetylation at telomeric chromatin (Michishita et al., 2008). Together, these results show that SIRT6 modulates apoptosis resistance and telomere-independent cellular senescence in human cells, via inhibition of NF-κB.
Figure 5. SIRT6 Alters NF-κB-mediated Apoptosis and Senescence.
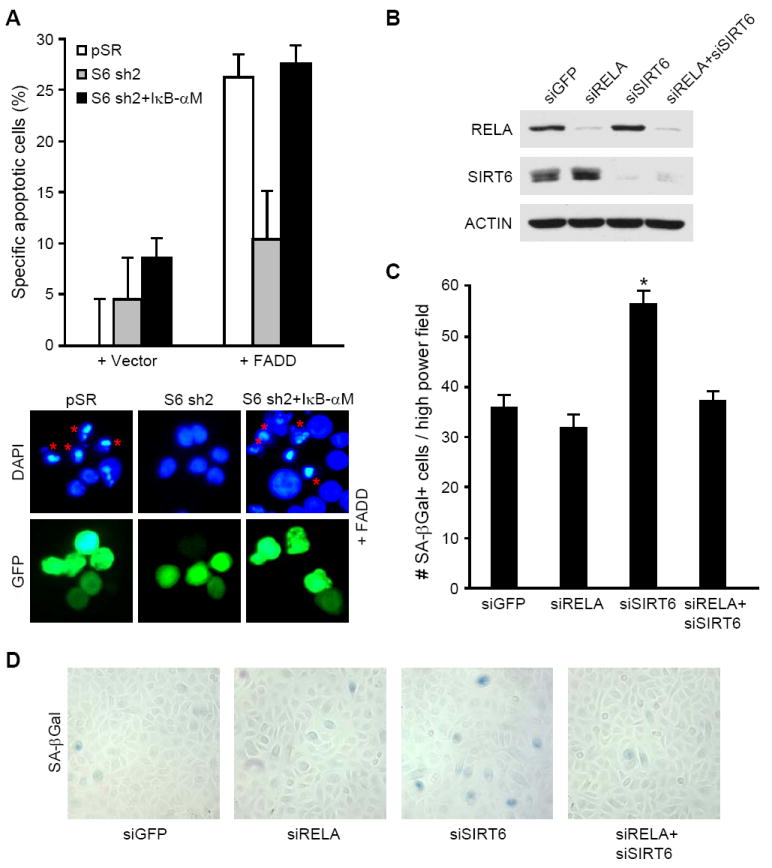
(A) SIRT6 regulates NF-κB-dependent apoptosis resistance. pSR and S6 sh2 cells were transfected with the indicated plasmids and a GFP-expression plasmid. Transfected (GFP-positive) apoptotic cells were identified by nuclear morphology (DAPI-staining; mean ± s.e.). * indicates transfected, apoptotic cells. TUNEL analysis confirmed that cells with pyknotic nuclei were undergoing apoptosis (data not shown).
(B)-(D) RELA depletion reverses cellular senescence in SIRT6-depleted primary human keratinocytes.
(B) Immunoblot of RELA and SIRT6 three days after siRNA nucleofection.
(C) Quantification of SA-β-Gal-positive cells, as shown in (D) (mean ± s.e.). *, p<0.0001 compared to all other samples, Student’s t-test.
Sirt6-/- Tissues Exhibit Ectopic Expression of NF-κB Target Genes
To examine the effects of SIRT6 deficiency on NF-κB and potentially other transcriptional regulators in vivo, we carried out an unbiased screen for transcriptional programs that are perturbed in Sirt6-/- mice compared to wild-type littermate controls (Figure 6A). We harvested liver, spleen, thymus, and skin tissues from 21 to 29 day-old animals, and MEFs from day 13.5 embryos. For each tissue, RNA from wild-type and Sirt6-/- mice was subjected to competitive hybridization to oligonucleotide microarrays. The RNA levels of hundreds of genes were differentially expressed in each Sirt6-/- versus wild-type comparison (data not shown). We next applied cis-regulatory motif analysis to identify candidate transcription factors that are associated with the gene expression changes. Previously, we have identified groups of genes that share specific transcription factor binding motifs in their promoters, termed motif modules (Adler et al., 2007). For instance, the NF-κB motif module comprises genes that contain one or more NF-κB motifs in their promoters. We have constructed similar motif modules for all known cis-regulatory motifs in single, double, or three-way combinations. By comparing the genes showing altered expression in Sirt6-/- tissues with the motif modules (Methods), we identified candidate motifs whose corresponding genes showed a consistent change in expression in the Sirt6-/- tissues compared to wild-type controls (p<0.05, hypergeometric distribution). Many motif modules showed altered expression in two or three Sirt6-/- tissues. However, only 12 motif modules (including the NF-κB module) were induced, and only 5 modules were repressed, in all seven Sirt6-/- tissues (Figure 6B). Three of these modules (AP2, STAT, MAZ) were identified as being both induced and repressed in all tissues, indicating that these are likely “noisy” motifs. Interestingly, four motif modules (NF-κB, CEBP, LFA1, TEF) whose targets were induced in Sirt6-/- tissues were previously identified in a screen for candidate transcription factor motifs associated with gene expression changes in mammalian aging (Adler et al., 2007). Together, these analyses provide in vivo evidence that SIRT6-deficiency leads to increased expression of NF-κB-dependent genes.
Figure 6. Sirt6 Deficiency Leads to Ectopic Expression of NF-κB Target Genes In Vivo.
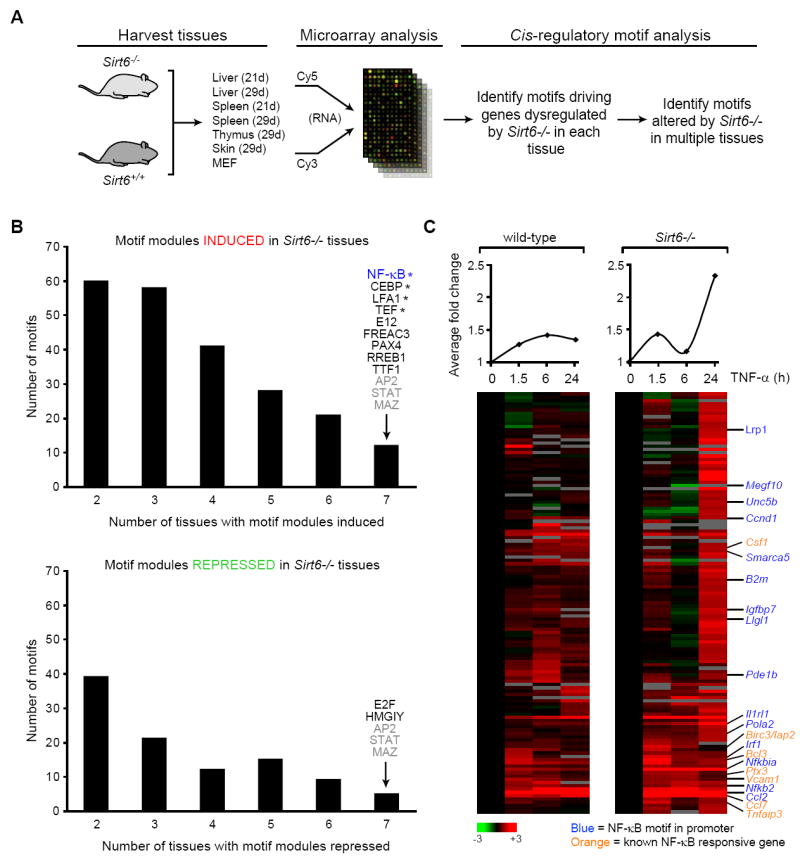
(A) Schematic for unbiased screen for candidate transcriptional regulators of gene expression changes in Sirt6-/- tissues.
(B) Motif modules induced or repressed in Sirt6-/- tissues. Shown is the number of motifs that are induced or repressed in the indicated number of Sirt6-/- tissues. The motif module of NF-κB was induced in all Sirt6-/- tissues examined. Motifs labeled in gray were found to be both induced and repressed in all Sirt6-/- tissues in combination with different motifs. * indicates motifs previously implicated in a screen for transcription factor motifs responsible for driving gene expression changes in mammalian aging (Adler et al., 2007). See Table S1 for motif modules induced or repressed upon Sirt6 knockout.
(C) Sirt6 knockout leads to NF-κB target gene induction. Shown is gene expression analysis in wild-type (WT) or Sirt6-/- MEFs, following TNF-α treatment (20 ng/mL) for the indicated times, of genes enriched for NF-κB motifs in promoters and known NF-κB responsive genes (Hinata et al., 2003; Hinz et al., 2001). Note the increased red (induced expression above control) genes in the SIRT6-/- columns, particularly at 24 hours. Average expression of each column is shown at the top.
We also directly compared gene expression patterns in response to TNF-α in wild-type and Sirt6-/- MEFs (Figure 6C). Unsupervised hierarchical clustering identified a set of genes whose expression differed between wild-type and Sirt6-/- MEFs; many of these genes were enriched for NF-κB binding sites in their promoters, or were previously shown to be induced by NF-κB (Hinata et al., 2003; Hinz et al., 2001). Although the kinetics of expression varied for specific genes, Sirt6-deficiency was associated with an overall increase in NF-κB target gene expression. In Figure 6C, the average expression level of the genes in the cluster at each time-point is shown. The most dramatic increases in expression are observed following 24 hours of TNF-α treatment.
We note that not all putative NF-κB target genes were hyperactive in the SIRT6-deficient cells; rather, the SIRT6-regulated NF-κB target genes were selectively enriched in several Gene Ontology (Harris et al., 2004) terms related to immune response, cell signaling, and metabolism (Figures 6C, S7). Notably, in ChIP analyses, SIRT6 was not detected at several of the NF-κB target genes that were unaffected by SIRT6 depletion (Figure S2). Together, our results suggest that SIRT6 negatively regulates expression of a subset of NF-κB target genes in vivo, by modifying chromatin at the promoters of these genes.
RelA Heterozygosity Attenuates Shortened Lifespan and Aging-related Phenotypes of Sirt6-/- Mice
To test whether excessive activation of NF-κB may contribute to the phenotypes of Sirt6-deficient mice, we examined Sirt6-/- animals in which RelA dosage is reduced. Sirt6+/- and RelA+/- heterozygous mice, both on the 129 genetic background, were crossed to create Sirt6+/-RelA+/- progeny, which were then interbred to obtain Sirt6-/- animals with different dosages of RelA (Figures 7A, S6). We did not obtain any Sirt6-/-RelA-/- animals, as anticipated because homozygous RelA- deficiency is embryonically lethal. Sirt6-/-RelA+/+ animals from these crosses exhibited a phenotype identical to that of Sirt6-/- mice (Mostoslavsky et al., 2006). The Sirt6-/-RelA+/- mice also resembled Sirt6-/- mice for the first 3 weeks of life, with lower body weight, blood glucose, and serum IGF-1 levels compared to Sirt6-proficient littermates (Figures 7D, 7F, S6). However, in approximately 40% of the Sirt6-/-RelA+/- animals, glucose and IGF-1 levels eventually rose, growth resumed, and these individuals survive (Figures 7C, 7F, S6). Thus far, these rescued Sirt6-/-RelA+/- animals have lived beyond 100 days and appear healthy (Figures 7C, 7D, and 7F). In contrast to Sirt6-/- animals, which exhibit profound lymphopenia and a 10-fold reduction of spleen lymphocytes, the rescued Sirt6-/-RelA+/- animals also have relatively normal spleen size and lymphocyte numbers (Figures 7G, 7H), and do not exhibit obvious lordokyphosis (data not shown). Moreover, both male and female Sirt6-/-RelA+/- mice are fertile, suggesting that the hypothalamic-pituitary-gonadal endocrine system and germ-line stem cell compartments are intact. Finally, microarray analysis confirmed that RelA-heterozygosity significantly reduces the increased expression of genes in the NF-κB motif module that was observed in Sirt6- deficient mice (Figures 7E, S7). Together, these data suggest that excessive NF-κB dependent gene expression contributes substantially to the shortened lifespan and degenerative symptoms observed in Sirt6-/- mice.
Figure 7. Haploinsufficiency of RelA Attenuates the Lethality and Aging-like Phenotypes of Sirt6-deficient Mice.
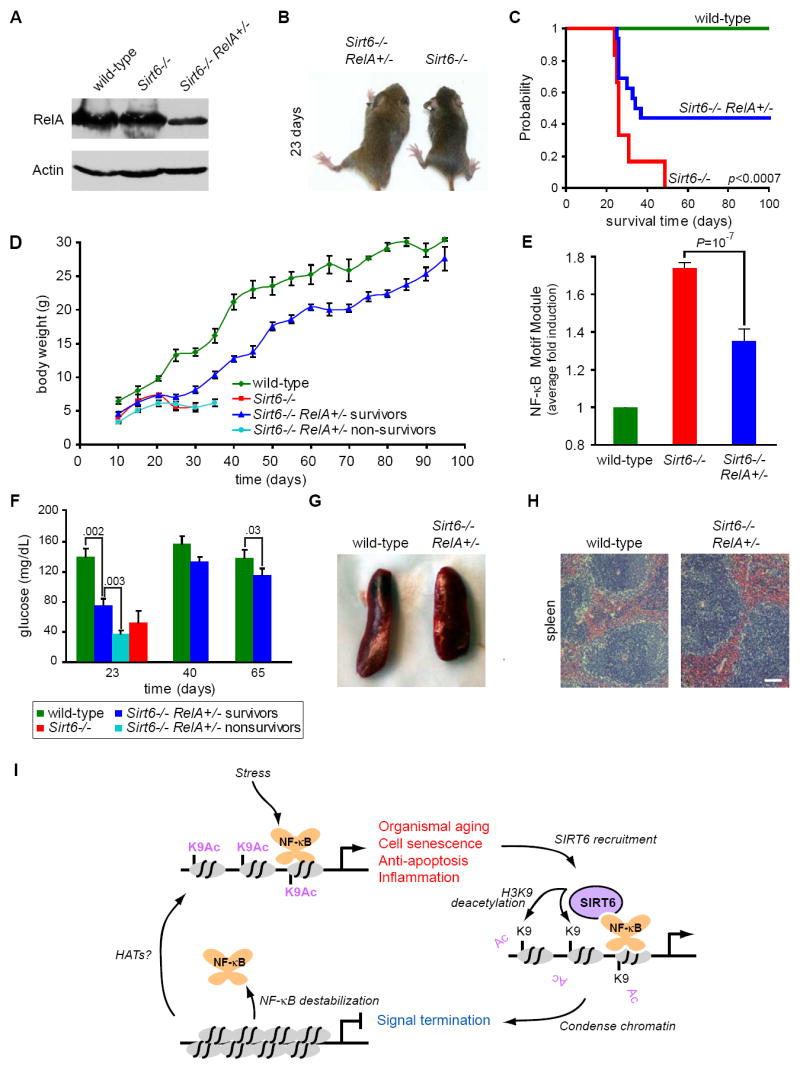
(A) Immunoblot showing reduced RelA protein levels in livers of Sirt6-/-RelA+/- animals compared to RelA+/+ animals.
(B) Photograph of 23 day old Sirt6-/-RelA+/+ and Sirt6-/-RelA+/- littermates.
(C) Kaplan-Meier curve showing the survival of wild-type (n=8), Sirt6-/-RelA+/+ (n=6), and Sirt6-/-RelA+/- (n=16) mice (p<0.0007, Cox-Mantel log-rank test).
(D) Growth curve showing weight (grams; mean ± s.e.) of wild-type (n=6), Sirt6-/-RelA+/+ (n=7), and Sirt6-/-RelA+/- mice versus age in days. Sirt6-/-RelA+/- mice were categorized as survivors (n=13) or non-survivors (n=12) based on their ability to survive past 40 days.
(E) Excessive activation of the NF-κB motif module in Sirt6-/- spleen is attenuated in Sirt6-/-RelA+/- spleen. Shown is the fold induction (mean ± s.e.) of genes in the NF-κB motif module, normalized to wild-type.
(F) Serum glucose levels in wild-type (n=6), Sirt6-/-RelA+/+ (n=4), and Sirt6-/-RelA+/- mice (mean ± s.e.). Sirt6-/-RelA+/- mice were categorized as survivors (n=9) or non-survivors (n=6) based on their ability to survive past 40 days; p-values are indicated.
(G) Gross appearance of spleen from 122-day-old wild-type and Sirt6-/-RelA+/- mice.
(H) Hematoxylin and eosin (H&E) staining of spleen from wild-type and Sirt6-/-RelA+/- mice. Bar, 100μM.
(I) Model. NF-κB is a stress-responsive transcription factor that induces expression of target genes involved in aging-related processes including cell senescence, apoptosis, and inflammation. NF-κB signaling is limited by SIRT6, which is recruited to NF-κB target gene promoters by physical interaction with the NF-κB subunit RELA. SIRT6 deacetylates histone H3 lysine 9 on target gene promoters, thereby altering chromatin structure to facilitate NF-κB destabilization and signal termination.
DISCUSSION
SIRT6 deacetylation of H3K9 modulates NF-κB-dependent gene expression
Despite the dramatic phenotype of Sirt6-deficient mice, the molecular mechanism underlying this phenotype has been unclear, in part because of a lack of understanding of the fundamental mechanistic basis of SIRT6 function. In recent work, we showed that SIRT6 deacetylates H3K9 at telomeric chromatin to prevent telomere dysfunction and cellular senescence (Michishita et al., 2008). However, defective telomeres are unlikely to account for the phenotype of Sirt6 knockout mice, because of the large functional reserve of telomeres in laboratory mouse strains. In this context, no other genomic targets or chromatin regulatory functions of SIRT6 have yet been described. In this study, we have identified a new physiologic context for SIRT6 function as a H3K9 deacetylase. Our data show that SIRT6 negatively regulates NF-κB dependent transcription via histone H3K9 deacetylation. This finding constitutes the first demonstration of a role for SIRT6 in gene expression via the modulation of chromatin. We suggest that SIRT6 likely also regulates gene expression via H3K9 deacetylation in additional physiologic contexts, and indeed, our computational genomic analyses have identified several other gene expression modules that are altered in Sirt6-deficient animals. Future studies should determine whether these effects are also due to direct Sirt6-dependent gene repression.
NF-κB stimuli such as TNF-α promote translocation of RELA to the nucleus, where RELA associates with promoters of its target genes. Our data suggest a model (Figure 7I) in which SIRT6 is recruited to these genomic loci via a physical interaction with RELA, deacetylates histone H3K9, destabilizes RELA from chromatin, and thereby contributes to NF-κB signal termination. As NF-κB signaling controls expression of genes involved in apoptosis, cellular senescence, inflammation, and immunity (Hardy et al., 2005; Hoffmann and Baltimore, 2006), tight regulation of NF-κB activity may be particularly important. Indeed, NF-κB activity is controlled at multiple levels, and several NF-κB regulatory factors are themselves direct transcriptional targets of NF-κB, which contribute to negative feedback. Our findings further highlight the importance of proper control of NF-κB signaling within the nucleus for cell and organismal function, and place SIRT6 in the growing class of negative regulators of NF-κB activity. Many aspects of NF-κB regulation appear to be essential, as inactivation of a single mechanism, as shown here for SIRT6, can lead to inappropriate NF-κB activation and disease. We note that several NF-κB family members are among the RELA target genes that show increased expression in SIRT6-deficient cells (Figure S4), and may contribute to the multiple layers of feedback and complex dynamics of SIRT6-regulated gene expression patterns.
We note that NF-κB RELA itself has been reported to be subject to regulation by acetylation, and is deacetylated by several deacetylases including SIRT1 (Chen and Greene, 2004; Yeung et al., 2004). Although it is possible that SIRT6 might also directly modulate RELA acetylation levels, in our hands SIRT6 did not demonstrate significant deacetylase activity on RELA in vitro or in cells (Figure S5 and data not shown). The fact that the SIRT1-RELA co-IP is relatively weak could reflect the transient enzyme-substrate nature of this interaction, whereas the SIRT6-RELA interaction, which recruits SIRT6 to RELA target promoters, may be more biochemically stable. SIRT1 and other non-Sirtuin HDACs may also contribute to modulation of the complex dynamic patterns of histone acetylation at NF-κB target promoters. However, unlike SIRT6, these deacetylases are not specific for H3K9Ac. In this context, SIRT1 is a relatively promiscuous deacetylase with numerous histone substrates, (Vaquero et al., 2004). Interestingly, the association of SIRT1 with chromatin at RELA target gene promoters (Yeung et al., 2004) occurs with a completely different pattern than SIRT6. Moreover, in contrast to our observations with SIRT6 inactivation, SIRT1 inactivation by RNAi does not lead to changes in NF-κB target gene expression, except when cells are treated with resveratrol (Yeung et al., 2004). Therefore, it appears that SIRT6 and SIRT1 impinge upon NF-κB via distinct molecular mechanisms. It will be interesting to determine whether these (and potentially other) SIRT factors modulate NF-κB activity synergistically, or in distinct non-overlapping cell types or physiologic contexts.
SIRT6, NF-κB, and Longevity
Sirt6-/- mice appear normal at birth, but by three weeks of age exhibit a degenerative syndrome with features of premature aging and succumb to death soon after (Mostoslavsky et al., 2006). Using computational genomics, we have shown that SIRT6 deficiency leads to the activation of many of the same transcriptional programs observed in aged tissues, including genes controlled by NF-κB. This finding supports the idea that the degenerative syndrome in Sirt6 knockout mice provides a physiologically relevant model for aging-associated processes, albeit with accelerated time course and exaggeration of certain features.
Based on several findings, we propose that NF-κB deregulation is a key factor contributing to the degenerative phenotypes in Sirt6-/- mice. First, elevated transcription of NF-κB targets was observed in all Sirt6-/- tissues analyzed, and these gene expression abnormalities were attenuated by reduced RelA dosage. Second, induction of cellular senescence and apoptosis resistance by SIRT6 loss was rescued by simultaneous NF-κB inhibition. Third, haploinsufficiency of RelA can extend the lifespan of Sirt6-deficient mice to more than three months and attenuate the degenerative phenotypes of Sirt6-deficient animals. This genetic rescue is remarkable because Sirt6 or RelA deficiency is individually lethal, and combining these alleles together might be anticipated to make the animal more sick. Instead, the early lethality and degenerative phenotypes caused by Sirt6 deficiency are substantially rescued by haploinsufficiency of RelA. Together, our results suggest that excessive NF-κB activity is required for Sirt6 deficiency to lead to disease.
Studies in yeast, worms, and flies have linked Sir2 factors to the regulation of longevity (Guarente and Picard, 2005; Haigis and Guarente, 2006). Intriguingly, the targets of sirtuins appear to be distinct in different organisms, potentially reflecting different molecular pathways that become limiting for organismal longevity. In S. cerevisiae, Sir2 prevents illegitimate recombination at the ribosomal DNA locus and the formation of presumably toxic extrachromosomal DNA circles (Sinclair and Guarente, 1997). In C. elegans, lifespan extension by Sir2.1 requires the IGF/FOXO pathway (Tissenbaum and Guarente, 2001). Mammalian SIRT6 also regulates metabolism and IGF signaling, and Sirt6-/- mice show severe metabolic defects with hypoglycemia, low IGF1, and low insulin levels (Lombard et al., 2008; Mostoslavsky et al., 2006). Our results show that these effects can be attenuated by haploinsufficiency of NF-κB. Because sirtuins are NAD+-dependent deacetylases, SIRT6 may act as a nutrient- or energy-sensor, and may link NF-κB signaling to the metabolic state of the organism. As NF-κB is a crucial transcription factor implicated in a vast array of disease states (Karin et al., 2004), its interaction with SIRT6 may provide a therapeutic target for these diseases, particularly those associated with aging. We note that we have not observed altered SIRT6 protein expression in young versus old mouse tissues (Figure S8). Given the reported increase in NF-κB target gene activities that has been observed with age, pharmacologic strategies to augment SIRT6 activity may be promising approaches to attenuate excessive NF-κB activity in such contexts.
Our study also highlights many intriguing questions. What are the cell types in which NF-κB signaling orchestrates aging-associated processes in Sirt6-/- animals? How does signal termination by SIRT6 impact on the dynamic pattern and target selection of NF-κB factors on a genome-scale? Will the enhanced survival of Sirt6-/-RelA+/- mice reveal additional aging-related or other pathologic effects of SIRT6 deficiency? The viability of the Sirt6-/-RelA+/- animals may provide a new model to study mammalian aging and address these questions in the future.
EXPERIMENTAL PROCEDURES
Antibodies and Plasmids
Antibodies specific for FLAG (Sigma), RELA (Santa Cruz Biotechnology, Inc. and Biomol International, LP), NF-κB family members (Santa Cruz Biotechnology, Inc.), SIRT6 [(Michishita et al., 2005) and Abcam], SIRT1 (Upstate), T7 (Novagen), H3K9Ac (Sigma), H3 (Abcam), β-ACTIN (Sigma), and TUBULIN (Upstate) are from the indicated sources. FLAG-tagged SIRT4, SIRT5, and SIRT6 are described (Michishita et al., 2005). FLAG-tagged SIRT1 wild-type and HY mutant expression plasmids were generated by subcloning the cDNAs from pYeSIR and pYeSIRHY (Vaziri et al., 2001) into p3x-FLAG (Sigma). GST-SIRT6 plasmid was generated by inserting the SIRT6 cDNA into pGEX-6P1. T7-RELA (Chen et al., 2002) (W. Greene, UCSF), pCMVβ-p300-CHA (Eckner et al., 1994) (Y. Shi, Harvard), pIL1β-Luc, pLZRS-IκB-αM & pCMV-RELA (P. Khavari, Stanford University), and pRK5-FADD (Hsu et al., 1996) are as described and obtained from the indicated investigators. Site-directed mutations of SIRT6 were generated by PCR mutagenesis as previously described (Michishita et al., 2008). The pSR-S6sh1/S6sh2 plasmids are described (Michishita et al., 2008). The RELA shRNA target sequence is GATTGAGGAGAAACGTAAA.
Cell Culture and Retroviral Transduction
MEFs were generated from 13.5-day-old embryos using standard methods. 293T, HeLa, and HT1080 cells were from ATCC and were propagated in DMEM (Invitrogen) plus 10% fetal bovine serum. Primary human keratinocytes were obtained from P. Khavari (Stanford) and cultured in Keratinocyte-SFM media (Invitrogen). Retroviral transduction was performed as previously described (Michishita et al., 2008).
Immunoprecipitation Assays
Cells were lysed in IP-lysis buffer (50 mM Tris-HCl [pH 7.4], 250 mM NaCl, 0.25% Triton X-100, 10% glycerol, and complete protease inhibitor cocktail (Roche)). To reduce non-specific binding, NaCl was added to 500 mM final concentration, and the resulting extracts were incubated overnight with the indicated IP antibodies. IP-antibody complexes were then captured on protein A/G-agarose beads (Pierce), and proteins detected by Western analysis. In the case of FLAG IPs, agarose-conjugated anti-FLAG M2 monoclonal antibodies (Sigma) were used.
GST-pull-down assays
RELA or SIRT6 sequences were in vitro transcribed and translated according to the manufacturers instructions (Promega), and GST-pull down assays were performed as previously described (Shi et al., 2006).
Reporter Gene Assays
293T cells were transfected with IL1-Luc, pRL-Luc, and FLAG-tagged SIRT proteins. pSR control and S6 sh2 HeLa cells were transfected with IL1-Luc and pRL-Luc and treated with TNF-α (10 ng/mL, 8 hours) or transfected with pCMV-RELA or pcDNA3.1 (control). Dual luciferase assays (Promega) were performed as described by the manufacturer.
Microarray Analyses
Total RNA was extracted with TRIzol (Invitrogen) from Sirt6-deficient or control tissues or cells following TNF-α treatment (10 ng/mL and 5 ng/ml, respectively) for the indicated times. RNA from matched wild-type and knockout tissues were labeled with Cy3 and Cy5, respectively, and competitively hybridized to microarrays. Mouse and human Universal Reference RNA (Stratagene) were used for analysis of mouse and human cells, respectively. Construction and array hybridization of human cDNA microarrays were performed as previously described (Perou et al., 2000). Array hybridization of mouse MEEBO arrays is as described (http://www.microarray.org/sfgf/meebo.do).
TNF-α activation time course
Genes that were induced or repressed by at least two fold in any sample during the time course were organized by hierarchical clustering (Eisen et al., 1998). The main cluster that demonstrated increased induction in SIRT6-deficient cells was highly enriched for NF-κB responsive genes (Hinata et al., 2003; Hinz et al., 2001) and genes possessing NF-κB motifs (p<10-3, hypergeometric distribution (Adler et al., 2007)).
Motif module map
To analyze cis-regulatory motifs driving gene expression changes in Sirt6-/- murine tissues, we identified genes that were induced or repressed by at least 1.5-fold change, and tested for their enrichment (p<0.05, hypergeometric distribution) for sets of genes that shared the presence of cis-motifs, termed motif modules (Adler et al., 2007). We then selected for motif modules that were induced or repressed across multiple Sirt6-/- tissues.
Regulation of genes with age
We determined whether 18 SIRT6-dependent NF-κB target genes (from Figure S2) tended to be induced or repressed in an age-associated fashion in six microarray data sets of human aging (compiled in (Adler et al., 2007). We calculated the Pearson correlation of the expression levels of each gene to the age of the samples. Positive/negative correlation values were obtained for genes whose expression level increased/decreased with age, respectively. One-sided t-tests (calculated with Winstat® [R. Fitch Software, Germany]) were used to determine the significance of each Pearson correlation.
Real-Time Quantitative RT-PCR
Total RNA was extracted from cells with TRIzol (Invitrogen). Total RNA (2 ug) was reverse-transcribed with oligo(dT) primer using the SuperScript first-strand synthesis system for RT-PCR (Invitrogen). Each PCR reaction contained first-strand cDNA corresponding to 25 ng of RNA, TaqMan universal PCR master mix, a set of primers & FAM/MGB probe for gene of interest, and a set of primers & VIC/MGB probe for human GAPDH as the endogenous control. All PCR primers and TaqMan probes were from Applied Biosystems. Reactions were in triplicate for each sample and analyzed using the ABI Prism 7300 Sequence Detection System. Data were normalized to GAPDH levels.
Chromatin Immunoprecipitation (ChIP)
Cells were treated with TNF-α (20 ng/ml) for the indicated times. DNA was cross-linked for 10 minutes with 1% formaldehyde and stopped in 0.125 M glycine. Purified chromatin was sonicated to ~300 bp using the Bioruptor (Diagenode, Inc) and incubated with the indicated antibodies. Following reverse cross-linking, ChIP-associated sequences were detected by quantitative Real-Time PCR as described above. PCR primer sequences for promoter regions are provided in Supplemental Table 2.
Nucleosome Binding Assay
Acetylated nucleosomes were obtained by size exclusion chromatography of micrococcal nuclease-digested chromatin isolated from HeLa cells, as previously described (Shi et al., 2006), followed by incubation with PCAF and acetylCoA. These nucleosomes were incubated with purified SIRT6 protein in NAD-dependent deacetylation or mock reactions. Recombinant SIRT6 protein was previously described (Michishita et al., 2005). The acetylated or SIRT6-deacetylated nucleosomes were used in nucleosome binding assays with GST-RELA protein as previously described (Shi et al., 2006).
Apoptosis and Senescence Assays
Cell death
Cells were co-transfected with GFP-expressing plasmid (to identify transfected cells) and either FADD or vector control in the presence or absence of IκB-αM expression. After 30 hours, apoptotic GFP-expressing cells were identified based on nuclear morphology (DAPI-staining); TUNEL analysis confirmed that cells with pyknotic nuclei were undergoing apoptosis (data not shown). The percent apoptotic cells was determined by counting three separate fields for each condition (minimum 100 transfected cells), and the data were normalized to vector-expressing pSR cells.
Cellular Senescence
Primary human keratinocytes were transfected with 0.3 nmol siRNA against SIRT6 (same target sequence as S6 sh2) and/or RELA (siGENOME smart pool; Dharmacon) using the Human Keratinocyte Nucleofect Kit (Amaxa Biosystems). siGFP (Chi et al., 2003) was used as a negative control. Senescence-associated beta-galactosidase (SA-βgal) activity was detected with a SA-β-gal staining kit (Cell Signaling Technology) after six days. Blue cells were counted in ten 10x fields of equally dense cells. Similar results were obtained using 0.75 nmol siRNA (data not shown).
Generation of Sirt6-/-RelA+/- Mice
Sirt6-/- (Mostoslavsky et al., 2006) and RelA+/- (Beg et al., 1995) mice were as described. Sirt6+/- and RelA+/- mice were crossed, and Sirt6+/-RelA+/-progeny were interbred to obtain Sirt6-/-RelA+/- mice. Kaplan-Meier survival curves were compared by the Cox-Mantel log-rank test in Winstat® for Excel (R. Fitch Software, Germany).
Glucose Measurements
Serum glucose was measured from tail blood using the Ascensia Contour Blood Glucose Monitoring System following manufacturer’s instructions.
URLs
Full microarray data are available for download at Stanford Microarray Database (http://genome-www5.stanford.edu/) or Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/).
Supplementary Material
Acknowledgments
We thank O. Gozani, P. Khavari, R. Mostoslavsky, and T. Rando for comments or advice, T. Hong for assistance with primer design, Regeneron Pharmaceuticals for generation of the SIRT6 gene-targeted mouse embryonic stem cell line, and R. Mostoslavsky and F. W. Alt for generation of the SIRT6 mutant mice. Supported by NIH (K.F.C., T.L.A.K., R.A.M), Department of Veterans Affairs Merit Review (K.F.C.), Stanford Graduate Fellowship (T.L.A.K.), California Breast Cancer Research Program (A.S.A.), and American Cancer Society (H.Y.C). H.Y.C. is the Kenneth G. and Elaine A. Langone Scholar of the Damon Runyon Cancer Research Foundation. K.F.C. is a Scholar of the Paul B. Beeson Aging Research Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adler AS, Kawahara TL, Segal E, Chang HY. Reversal of aging by NFkappaB blockade. Cell Cycle. 2008;7:556–559. doi: 10.4161/cc.7.5.5490. [DOI] [PubMed] [Google Scholar]
- Adler AS, Sinha S, Kawahara TL, Zhang JY, Segal E, Chang HY. Motif module map reveals enforcement of aging by continual NF-{kappa}B activity. Genes Dev. 2007;21:3244–3257. doi: 10.1101/gad.1588507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007;5:e201. doi: 10.1371/journal.pbio.0050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- Chen LF, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. Embo J. 2002;21:6539–6548. doi: 10.1093/emboj/cdf660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi JT, Chang HY, Wang NN, Chang DS, Dunphy N, Brown PO. Genomewide view of gene silencing by small interfering RNAs. Proc Natl Acad Sci U S A. 2003;100:6343–6346. doi: 10.1073/pnas.1037853100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckner R, Ewen ME, Newsome D, Gerdes M, DeCaprio JA, Lawrence JB, Livingston DM. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 1994;8:869–884. doi: 10.1101/gad.8.8.869. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- Guarente L, Picard F. Calorie restriction--the SIR2 connection. Cell. 2005;120:473–482. doi: 10.1016/j.cell.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Haigis MC, Guarente LP. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- Hardy K, Mansfield L, Mackay A, Benvenuti S, Ismail S, Arora P, O’Hare MJ, Jat PS. Transcriptional networks and cellular senescence in human mammary fibroblasts. Mol Biol Cell. 2005;16:943–953. doi: 10.1091/mbc.E04-05-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Helenius M, Hanninen M, Lehtinen SK, Salminen A. Changes associated with aging and replicative senescence in the regulation of transcription factor nuclear factor-kappa B. Biochem J. 1996;318(Pt 2):603–608. doi: 10.1042/bj3180603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinata K, Gervin AM, Zhang JY, Khavari PA. Divergent gene regulation and growth effects by NF-kappa B in epithelial and mesenchymal cells of human skin. Oncogene. 2003;22:1955–1964. doi: 10.1038/sj.onc.1206198. [DOI] [PubMed] [Google Scholar]
- Hinz M, Loser P, Mathas S, Krappmann D, Dorken B, Scheidereit C. Constitutive NF-kappaB maintains high expression of a characteristic gene network, including CD40, CD86, and a set of antiapoptotic genes in Hodgkin/Reed-Sternberg cells. Blood. 2001;97:2798–2807. doi: 10.1182/blood.v97.9.2798. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev. 2006;210:171–186. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298:1241–1245. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- Karin M, Yamamoto Y, Wang QM. The IKK NF-kappa B system: a treasure trove for drug development. Nature reviews. 2004;3:17–26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]
- Korhonen P, Helenius M, Salminen A. Age-related changes in the regulation of transcription factor NF-kappa B in rat brain. Neurosci Lett. 1997;225:61–64. doi: 10.1016/s0304-3940(97)00190-0. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Lombard DB, Schwer B, Alt FW, Mostoslavsky R. SIRT6 in DNA repair, metabolism and ageing. J Intern Med. 2008;263:128–141. doi: 10.1111/j.1365-2796.2007.01902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008 doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 2005;16:4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, DePinho RA. Cellular senescence: mitotic clock or culture shock? Cell. 2000;102:407–410. doi: 10.1016/s0092-8674(00)00046-5. [DOI] [PubMed] [Google Scholar]
- Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, Carney D, Pena P, Lan F, Kaadige MR, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–99. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DA, Guarente L. Extrachromosomal rDNA circles--a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Molecular cell. 2004;16:93–105. doi: 10.1016/j.molcel.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. Embo J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.